Submitted:
28 July 2024
Posted:
30 July 2024
You are already at the latest version
Abstract
Localized protein translation occurs through trafficking of mRNAs and protein translation machineries to different compartments of the cell, leading to rapid on-site synthesis of proteins in response to signaling cues. The spatiotemporally precise nature of the local translation process necessitates continual developments of technologies reviewed herein to visualize and map biomolecular components and the translation process with better spatial and temporal resolution, and with fewer artifacts. We also discuss approaches to control local translation, which can serve as a design paradigm for subcellular genetic devices for eukaryotic synthetic biology.
Keywords:
local translation
; translatome
; ribosome
; genetic code expansion
; imaging
Introduction
In nature, locale-specific production of proteins—or local translation—generates protein concentration gradients within cells and drives asymmetric developmental processes such as cell migration [1], synaptic maturation [2], and establishment of embryonic polarity [3]. In synthetic biology, the messenger RNA and the protein translation apparatus can be reprogrammed to diversify outputs from a single transcript [4,5,6]; encode non-canonical building blocks [7,8,9]; or respond to synthetic trans-acting switches [10]. Inspired by nature, such engineered translation outputs can potentially be compartmentalized within cells to drive complex yet subcellularly defined functions. Technologies to observe localized protein-synthesis machinery and locally translated protein output are therefore critical to the engineering and manipulation of biological systems at the subcellular and translational level.
Many foundational technologies for local translation studies were developed to probe local protein synthesis in highly compartmentalized neurons [11], whose neuropil can be manipulated via microfluidic [12] and compartmentalized transfilter chambers [13] or physically severed via manual [14,15] and laser capture microdissection [16]. Conclusive evidence of local protein synthesis in physically manipulated neuropil was obtained not only from localization studies of cellular components needed for protein synthesis, but also from direct detection of actively translated mRNAs and newly synthesized proteins. Key recent biological findings—such as translatome mapping in neuron subregions [17]; robust translation activities by monosomes in neuronal processes [18]; local remodeling of ribosomes in neuronal processes [19,20]; signal-specific remodeling of nascent proteomes in axons [13]; and characterizations of disease-associated localized translatomes mediated by fragile X messenger ribonucleoprotein (FMRP) [21,22]—were all enabled by neuropil microdissection and compartmentalization technologies.
Local protein synthesis in non-neuronal cells is investigated much less frequently [23], partly due to the inability to dissect or isolate relevant subcellular regions for investigations, thereby demanding imaging, mapping, and controlling technologies of protein translation components to be innately highly precise without requiring physical separation of subcellular parts. Herein we review recent technical advances in the visualization and curation of the local presence of key cellular components—mRNAs and their post-transcriptional modifications; ribosomes and their different compositions—required for protein synthesis. We also discuss recent variations of ribosome-linked technologies for charting spatial translatomes and updated methods to detect newly & locally synthesized proteins. Last, we examine co-translational labeling techniques based on non-canonical amino acids and genetic code expansion used to not only observe but control local protein translation, as well as translational reprogramming approaches based on control of ribosome compositions.
Imaging Actively Translated Individual mRNAs
Asymmetric distribution of mRNAs within the cell are known to influence their translation, processing, storage and degradation [24,25]. Techniques to image individual RNAs, particularly via the use of bacteriophage-derived RNA hairpins and their cognate coat proteins [26,27] to attach genetically encoded labels such as fluorescent proteins (FPs) to individual RNAs (Figure 1A), are well-established and continually developed (recent reviews here [28,29,30]). In particular, the MS2 and MS2-coat protein system has recently been engineered by the Singer group to minimally alter RNA stability [31] and reduce destabilization effects from the nonsense-mediated mRNA decay pathway [32]. To avoid introducing exogenous transcripts, the RNA stem loop repeats along with other transcriptional regulatory elements can be knocked into endogenous loci using CRISPR-based genome editing tools [33]. To address concerns that cis-modified stem-loop-based tagging may perturb mRNA metabolism, trans-acting labeling modalities such as antisense oligonucleotide probes with a molecular beacon design [34,35] (Figure 1B) and RNA-targeted dCas13 variants [36] fused to FPs can be used to image endogenous RNAs without genetic modification to target transcripts. To circumvent off-target signals from excess coat protein- or dCas13-linked FPs, split variants of FPs or other reporter systems, whose signal generation are contingent upon reconstitution of split fragments on the target RNA, can be employed [37].
Modifications to stem loop-based RNA tagging have enabled visualization of actively translated mRNAs down to single-molecule levels. On one hand, the translating RNA imaging by coat protein knock-off (TRICK) technique used orthogonal MS2 and PP7 stem loops to tag the coding and 3′-untranslated regions of mRNAs with two FPs [38] (Figure 1C). Upon mRNA export from the nucleus, the initial round of translation displaces the FP-fused coat protein associated with the coding region, resulting in ratiometric changes upon dual-colored FP fluorescence imaging. On the other hand, stem loop-based tagging of mRNAs can be combined with nascent chain tagging, the latter via the SunTag [39], for single-molecule imaging of translation events [40,41,42] (Figure 1D). While there are caveats related to perturbations to mRNA localization and translation rates upon using array-based tags, TRICK and SunTag-based imaging of actively translated mRNAs have cumulatively revealed much insight into local translation during Drosophila oocyte development [38]; heterogeneity in translation rates due to transient binding of translation factors [42]; kinetics of translation regulation in stress responses [40] and in neuronal processes [41]; and pervasive translation of stress granule-associated mRNAs commonly thought to be translation-inhibitive [43].
Mapping Local Translatomes
Instead of imaging-based methods which limit throughput to assess mRNA translation, ribosome-protected mRNAs can be purified and mass-sequenced to provide snapshots of cellular translatomes in ribosome profiling/Ribo-seq experiments [44]. Coupled with process dissection and compartmentalization techniques, ribosome profiling has provided strong evidence of local translation of localized mRNAs—particularly of ribosomal protein-coding ones—in neuropils [19]. To capture subcellularly localized translatomes in non-neuronal cells where physical isolation of parts cannot be performed, the Weissman group developed proximity-specific ribosome profiling, in which acceptor peptide-tagged ribosomes are biotinylated with spatially restricted biotin ligase variants [45,46]. mRNAs bound to ribosomes from defined subcellular locations such as the endoplasmic reticulum (ER) membrane [45] and the outer mitochondrial membrane (OMM) [46] can therefore be isolated and analyzed. The spatial resolution of proximity-specific ribosome profiling in mapping local translatomes is limited by the movement of the ribosome from the labeling site during the short biotinylation window (2-7 min); the application of the method to primary cells and tissues may be further limited by the need to affinity-purify ribosomes (which requires large amount of biological material) and the toxic biotin starvation step. To circumvent the need to purify ribosomes in conventional ribosome profiling setups, ribosomal subunits can be genetically fused to adenosine (ADAR) or cytosine (APOBEC) deaminases, enabling preferential base editing of ribosome-bound mRNAs over total RNA (Figure 1E). ADAR-based RiboTRIBE [47] and APOBEC-based RiboSTAMP [48] could be extended to local translatomic studies, provided that issues raised regarding low ribosome association of base editor-fused ribosomal proteins [47] and sequence-specific biases are mitigated.
To drastically increase the multiplexity in assessing localized mRNA translation, the Wang group developed in situ sequencing-based RIBOmap [49]. RiBOmap uses a three-probe system and splint ligation to provide specificity of rolling circle amplification (RCA) in amplifying only the ribosome-bound mRNA of interest (Figure 1F). After amplification, the gene-specific barcodes on RCA products are decoded through cyclic in situ sequencing. The high throughput and multiplexity of RIBOmap—achieving translation profiling of 981 genes in single HeLa cells and 5,413 genes in 105 cells from mouse brain tissues—enabled discoveries of co-regulated as well as co-localized translation modules, suggesting the key role of local coordinated translation in synthesizing components of large protein complexes. In combination with spatial transcriptome mapping technologies [50,51], translationally regulated gene modules during oligodendrocyte maturation were identified. Similar to other in situ hybridization approaches, RIBOmap is limited to the detection of previously known RNA molecules; its SEDAL ligation-based sequencing also has a complex workflow and requires a large number of probes.
Mapping Global Turnover and Associated Regulatory Elements of Localized mRNAs
Barring specific examples of RNA localization motifs with known subcellular binding partners [52], systems-wide investigations of cis-regulatory motifs present in a group of RNAs, and subsequent profiling of RNA-binding proteins capable of association with such motifs can lead to discovery of functional zipcodes and associated binding partners [20]. Moreover, it has now emerged that gross physicochemical properties of mRNAs which increase their stability may be primary enablers of mRNA trafficking to distal sites [53]. Such studies are enabled by foundational methods such as transcriptional shut-off (reviewed here [54]) and RNA-based metabolic labeling assays. A recent addition to mRNA turnover characterization methods is SLAM-seq [55] (Figure 1G), which detect metabolic incorporation of 4-thiouridine in RNAs and provide systems views of expression dynamics and global stability of RNAs. Known mRNA destabilization features such as the m6A post-transcriptional modification can be mapped transcriptome-wide via m6A/Me-RNA-immunoprecipitation-based methods [56] or via antibody-free DART-seq [57] (Figure 1H), which uses APOBEC1 fused to the m6A-binding YTH domain to identify m6A sites. mRNAs trafficked to distal neurites of cortical and mESC-derived neurons were found to be more globally stable and contained stability signatures such as low levels of m6A and AU-rich elements, and good codon optimality [53].
Local Translation of Ribosomal Proteins and Local Remodeling of Ribosomes
Stable housekeeping transcripts associated with translation were identified with SLAM-seq to be enriched in neuronal branches [53]. The finding is consistent with known localizations[58] and local translation [20] of mRNAs encoding ribosomal proteins (RPs) in neurites. As demonstrated by the Holt and Schuman groups, locally translated ribosomal proteins can remodel local pools of ribosomes in axons [20] and dendrites [19], and are needed for the maintenance of axons in vivo [20]. The Holt group identified a cis-element upstream of the initiation codon (CUIC) motif which regulates translation of mRNAs encoding ribosomal proteins in axons, and used crosslinking immunoprecipitation-seq datasets to identify the eIF3 complex as potentially capable of recognizing CUIC motifs on RP-coding mRNAs [20]. Newly synthesized RPs at axons can physically associate with axonal ribosomes, and axonal synthesis of Rps4x/eS4 was shown to be crucial for in vivo branching of retinal ganglion cell axons [20]. The Schuman group further showed that RP-coding mRNAs are translated within dendrites, and a subset of locally translated RPs with short protein half-lives rapidly incorporated into dendritic ribosomes [19]. Fast RP synthesis and exchange is shown to facilitate ribosome repair in response to oxidative stress [19]. In yeast, the Karbstein group has demonstrated that intracellular salts and pH can modulate release and re-association of RPS26 from ribosomes [59].
While there are examples of specific compositions of the ribosome leading to translation of selective mRNAs (key examples being RPL10A/uL1-containing ribosomes promoting translation of IRES-containing mRNAs [60] and changed stoichiometries of RACK1, RPS26, and RPL38 affecting translational preferences of mRNAs [61,62,63], it remains to be seen whether local ribosome remodeling through local synthesis of RPs can lead to differential translation profiles of local ribosomes. Such ribosome heterogeneity can be mapped via translating ribosome affinity purification (TRAP) [64] or endogenously tagged Ribo-FLAG immunoprecipitation [65] (Figure 1I), but these strategies require exogenous genes or gene editing, can lead to altered ribosome concentrations and RP mispositioning [66], and do not preserve weakly bound ribosome-associated proteins (RAPs), many of which are capable of further reprogramming ribosomal translation.
Recently, a label-free biophysical/chromatographical-based method RAPIDASH was developed by the Barna and Ruggero labs which preserve the interactions between RAPs and native ribosomes [67] (Figure 1J). RAPIDASH relies on a cysteine-charged resin, a chromatographic technique used to isolate active bacterial and yeast ribosomes via rRNA binding. Careful modifications to the classic protocol led to the preservation of hundreds of RAPs on ribosomes isolated from the developing mouse forebrain. Two newly identified RAPs, Dhx30 and LLPH, are implicated in providing translation specificity toward mRNAs with specific structural and sequence features which may be linked to neural development. Beyond changes in incorporation of RPs and associations of RAPs, ribosome heterogeneity can arise from rRNA modifications which are thought to further alter RP/RAP associations, but their functional consequences remain to be clearly elucidated [68].
Imaging Newly Synthesized Individual Proteins
Fluorescent reporters have been mainstay tools for imaging of local protein synthesis, since the Schuman group first developed GFP reporters synthesized from mRNAs bearing appropriate untranslated regions to study dendritic local translation [14]. The use of fast-folding, bright FPs such as Venus and temporal gating with photoconvertible FPs has provided incredible insight into stimulation-induced local translation in neuronal processes and at synapses [69,70]. Innovations in even brighter and more photostable FPs such as monomeric StayGold variants [71,72,73] and fluorogenic chemogenetic labeling tags [74,75] should enable real-time reporting of protein synthesis down to second timescales. To permit imaging of protein turnover, reversible ligand-based labeling methods have also been developed. One such example is a class of fluorescence-activating and absorption-shifting tags (FASTs) developed by the Gautier group [76,77,78] (Figure 2A). FASTs were engineered from photoactive yellow protein to be binders of hydroxydenzylidene rhodanine fluorophores. They have several suitable properties for local protein synthesis monitoring: fluorogenic labeling (permitting real-time monitoring with no wash steps); second timescales for quantitative labeling, which should be fast enough for minute-scale monitoring of protein synthesis-dependent plasticity [79,80]; excellent brightness and photostability; and small size (11-14 kD) [81]. Importantly, fluorescence from a polyspecific variant of FAST [78] can be switched off rapidly (in seconds) and with high efficiency (~80% fluorescence removed) through chase labeling with a dark quencher ligand, permitting dynamic imaging of protein turnover.
Mapping Local Nascent Proteomes and Imaging Nascent Proteins
Metabolic labeling approaches with bioorthogonal methionine and puromycin analogs are well-established and have been used to profile proteome dynamics in diverse cells and organisms (recent reviews here [82,83]). Foundational azidohomoalanine (AHA) and homopropargylglycine (HPG)-based BONCAT/FUNCAT labeling approaches [84,85] can be performed in neuronal processes within 15 min, and could be coupled to highly sensitive, super-resolution imaging modalities such as DNA-PAINT for quantitative imaging of nascent proteins at single-molecule resolution [86]. Multiplexed imaging of nascent proteomes and ribosome location, both via DNA-PAINT, enabled the Schuman group to quantify distributions of locally synthesized proteomes from individually stimulated dendritic spines to neighboring spines and dendritic segments. Concurrent measurements of single-spine synaptic activity critically link the amount of locally synthesized proteins at single spines to their activity levels [86]. In vivo, cell-type specific metabolic labeling has also been demonstrated by the Schuman group using a mutant methionyl-tRNA synthetase genetically targeted to defined neuron types in transgenic mice [87].
Beyond AHA and HPG, a recent addition to the repertoire of non-canonical amino acids (ncAAs) for nascent proteomic profiling is the threonine-based THRONCAT [88] by the Bonger group (Figure 2B). THRONCAT co-opts cells to incorporate β-ethynylserine (βES), a clickable alkynyl analog of Thr, into newly synthesized proteomes. Due to good incorporation efficiency of βES in the presence of Thr, metabolic labeling could be performed in complete growth media. THRONCAT can likely be used in combination with BONCAT and other sense codon-mediated incorporation of ncAAs such as SORT-M [89] to improve proteomic coverage. Given faster incorporation efficiency of βES relative to HPG, THRONCAT may also improve the temporal resolution when used in conjunction with proximity ligation assay (PLA)—in the same vein as FUNCAT-PLA [90]—to detect newly synthesized protein targets. Fast ncAA-based tagging of specific nascent proteins can complement puromycin-based Puro-PLA [90], which has excellent temporal resolution but due to the ribosome-trapping nature of puromycin-based labeling, cannot be used to study distribution of locally synthesized proteins over time.
Beyond co-translational labeling approaches, post-translational proximity labeling can be used to differentiate protein pools originated from and sorted to different subcellular compartments. In TransitID developed by the Ting group, origins and destinations of proteomes can be sequentially marked using two orthogonal proximity labeling enzymes TurboID and APEX2, the latter with a new alkynyl substrate [91] (Figure 2C). Dual biotin- and alkyne-labeled proteins therefore have their source and destination information encoded and decipherable by multiplexed mass spectrometry-based proteomic experiments. Candidates of proteins locally synthesized at the OMM then imported into the mitochondrial matrix were identified with TransitID; their synthesis at the OMM were further verified by puromycin-based tagging.
Controlling and Monitoring Local Translation via Orthogonally Translating Organelles and Genetic Code Expansion
Protein translation machineries used in genetic code expansion (GCE) can now be engineered to exist in phase-separated compartments within the cell, enabling incorporation of ncAAs into specific proteins within the compartments (Figure 2D). Developed by the Lemke lab, such orthogonally translating organelles (OTOs [92]) bring together critical components of GCE to specific locales through protein fusions containing four components: 1) an aminoacyl-tRNA synthetase with strong affinity for suppressor tRNAs; 2) a cognate coat protein with strong affinity for stem loop-tagged mRNAs; 3) a phase-separating domain; and 4) a localization moiety which tethers the protein fusion to different organelles [93], including the ER and Golgi apparatus membranes, the OMM, the plasma membrane, and the microtubule plus end [94]. OTOs can mediate local translation of mRNAs artificially targeted to the organelles and insertion of ncAA labels in the resulting proteins. OTOs can likely be repurposed to translate mRNAs naturally targeted to these compartments and incorporate ncAAs at sense codons of locally synthesized proteomes, provided that the temporal resolution of the labeling (currently at hour timescales) is improved to match minute-level timescales of protein synthesis-dependent remodeling of most cellular processes.
Another intriguing prospect is the potential use of OTOs to monitor local translation of specific endogenous transcripts via RNA editing-mediated ncAA protein tagging (RENAPT) [95], developed by the Liu lab (Figure 2E). RENAPT employs dCas13-based targeting of ADAR base editors to introduce an amber (UAG) blank codon as the ncAA incorporation site directly on endogenous transcripts, obviating the need to transfect artificial constructs bearing the gene of interest. Compared to protein-based labeling tags like FASTs, single-residue tags used in GCE have a clear advantage of much smaller tag size and minimal perturbation to protein function. However, current common fluorophores for GCE- and click chemistry-based labeling [96,97,98] are not sufficiently fluorogenic to permit no-wash labeling, rendering real-time observation of protein synthesis off-limits. Design principles from the development of highly fluorogenic substrates for protein- and RNA-based labeling approaches [99,100]—particularly ways to reduce non-radiative decay pathways upon fluorophore attachment to proteins—can likely guide the development of next-generation labels for GCE, OTOs, and RENAPT, for real-time monitoring of locally translated proteins from endogenous transcripts.
Controlling Translation via mRNA-Specific Ribosomes in Eukaryotes
In bacteria, orthogonal ribosomes can be engineered through mutations in the anti-Shine-Dalgarno sequence of the small ribosomal subunit (SSU), enabling selective pairing with orthogonal ribosome-binding, Shine-Dalgarno sequences on mRNAs [101]. Through circular permutation of the large ribosomal RNA, the orthogonal SSU can be covalently attached to an engineered large ribosomal subunit [102,103,104,105] with a mutated peptidyl transferase center capable of new polymerizing functions [103,106,107]. In eukaryotes, translation preferences by the ribosome can be tuned by specific compositions of ribosomal proteins such as RPL38 and RPS26, and ribosome-associated proteins such as RACK1. Notably, the Karbstein group discovered that RPS26-mediated recognition of the Kozak sequence can be reprogrammed for translation of selective mRNAs in yeast [108]. Through understanding of Kozak sequence preferences of RPS26-containing vs RPS26-deficient ribosomes [61], several signaling pathways in yeast were programmed to be RPS26-responsive, via mutations to Kozak sequences of relevant genes and salt stress-triggered depletion of RPS26-containing ribosomes [108]. Such translational control mediated by distinct ribosome compositions may be extended to local protein synthesis control, provided that the RP mediator of ribosome specificity can be locally synthesized—for example, through the aforementioned orthogonally translating organelles and genetic code expansion.
Outlook of Local Translation Imaging and Controlling Tools for Mammalian Synthetic Biology
Continual developments of tools described in this review as well as new innovations will define and clarify further roles of local protein translation in various biological processes. On translational regulation at the subcellular level, key unanswered questions center on the nature of spatiotemporal signaling cues controlling on-site protein production, as well as biomolecular players facilitating these cues. Such biological insight can shed light on the complexity of the mammalian cell context, where heterogenous distributions of resources in different compartments serve to optimize outputs needed for local demand. As demonstrated with OTOs, principles underlying local translation can inform the design of genetic devices to produce specific protein outputs while maintaining orthogonality to the host machinery. Better designs of context-aware synthetic devices for mammalian synthetic biology [109] therefore go hand in hand with better understanding of complex settings the synthetic devices are housed in. The reprogramming of translation already has practical uses in creating novel therapeutics [110], new-to-nature polymers for diverse applications [8], and methods to control cell and organism behavior [111]. Enhanced understanding of how cells manage resources and regulate translation could further enrich these applications.
Acknowledgements
The authors were supported by VISTEC, Swiss National Science Foundation [IZSTZ0_193915], and Thailand Science Research and Innovation (TSRI), Fundamental Fund, fiscal year 2024 (grant number FRB670026/0457), all to C.U. W.O. and C.A. were supported by student and research assistant funds from VISTEC respectively.
Conflict of Interest Statement
The authors declare no competing interests.
References and Recommended Reading
- Papers of particular interest, published within the period of review (2021-2024), have been highlighted as: * of special interest ** of outstanding interest.
- 1. Liao G, Mingle L, Van De Water L, Liu G: Control of cell migration through mRNA localization and local translation. Wiley Interdiscip Rev RNA 2015, 6:1-15.
- 2. Wang DO, Martin KC, Zukin RS: Spatially restricting gene expression by local translation at synapses. Trends Neurosci 2010, 33:173-182.
- 3. Medioni C, Mowry K, Besse F: Principles and roles of mRNA localization in animal development. Development 2012, 139:3263-3276.
- 4. Mountford PS, Smith AG: Internal ribosome entry sites and dicistronic RNAs in mammalian transgenesis. Trends Genet 1995, 11:179-184.
- 5. de Felipe P, Hughes LE, Ryan MD, Brown JD: Co-translational, intraribosomal cleavage of polypeptides by the foot-and-mouth disease virus 2A peptide. J Biol Chem 2003, 278:11441-11448.
- 6. Orr MW, Mao Y, Storz G, Qian S-B: Alternative ORFs and small ORFs: shedding light on the dark proteome. Nucleic Acids Research 2019, 48:1029-1042.
- 7. Shandell MA, Tan Z, Cornish VW: Genetic Code Expansion: A Brief History and Perspective. Biochemistry 2021, 60:3455-3469.
- 8. de la Torre D, Chin JW: Reprogramming the genetic code. Nat Rev Genet 2021, 22:169-184.
- 9. Liu CC, Schultz PG: Adding New Chemistries to the Genetic Code. Annual Review of Biochemistry 2010, 79:413-444.
- 10. Anzalone AV, Lin AJ, Zairis S, Rabadan R, Cornish VW: Reprogramming eukaryotic translation with ligand-responsive synthetic RNA switches. Nature Methods 2016, 13:453-458.
- 11. Holt CE, Martin KC, Schuman EM: Local translation in neurons: visualization and function. Nature Structural & Molecular Biology 2019, 26:557-566.
- 12. Taylor AM, Wu J, Tai HC, Schuman EM: Axonal translation of β-catenin regulates synaptic vesicle dynamics. J Neurosci 2013, 33:5584-5589.
- 13. Cagnetta R, Frese CK, Shigeoka T, Krijgsveld J, Holt CE: Rapid Cue-Specific Remodeling of the Nascent Axonal Proteome. Neuron 2018, 99:29-46.e24.
- 14. Aakalu G, Smith WB, Nguyen N, Jiang C, Schuman EM: Dynamic visualization of local protein synthesis in hippocampal neurons. Neuron 2001, 30:489-502.
- 15. Cajigas IJ, Tushev G, Will TJ, tom Dieck S, Fuerst N, Schuman EM: The local transcriptome in the synaptic neuropil revealed by deep sequencing and high-resolution imaging. Neuron 2012, 74:453-466.
- 16. Perez JD, Dieck ST, Alvarez-Castelao B, Tushev G, Chan IC, Schuman EM: Subcellular sequencing of single neurons reveals the dendritic transcriptome of GABAergic interneurons. Elife 2021, 10.
- 17. Glock C, Biever A, Tushev G, Nassim-Assir B, Kao A, Bartnik I, Tom Dieck S, Schuman EM: The translatome of neuronal cell bodies, dendrites, and axons. Proc Natl Acad Sci U S A 2021, 118.
- *18. Biever A, Glock C, Tushev G, Ciirdaeva E, Dalmay T, Langer JD, Schuman EM: Monosomes actively translate synaptic mRNAs in neuronal processes. Science 2020, 367.
- Polysome profiling of microdissected regions to determine association of axonal, dendritic, and somatic transcripts with monosomes or polysomes. Discovery that monosomes actively translate synaptic mRNAs in neuronal processes.
- **19. Fusco CM, Desch K, Dörrbaum AR, Wang M, Staab A, Chan ICW, Vail E, Villeri V, Langer JD, Schuman EM: Neuronal ribosomes exhibit dynamic and context-dependent exchange of ribosomal proteins. Nature Communications 2021, 12:6127.
- Local synthesis of ribosomal proteins and local ribosome remodeling in dendrites. Ribosomal protein incorporation into ribosomes is regulated in part by oxidative stress.
- **20. Shigeoka T, Koppers M, Wong HH, Lin JQ, Cagnetta R, Dwivedy A, de Freitas Nascimento J, van Tartwijk FW, Ströhl F, Cioni JM, et al.: On-Site Ribosome Remodeling by Locally Synthesized Ribosomal Proteins in Axons. Cell Rep 2019, 29:3605-3619.e3610.
- Local synthesis of ribosomal proteins and local ribosome remodeling in axons, which are needed for the maintenance of axon branching in vivo. Identification of a sequence motif for axonal ribosomal protein translation.
- 21. Clifton NE, Lin JQ, Holt CE, O’Donovan MC, Mill J: Enrichment of the Local Synaptic Translatome for Genetic Risk Associated With Schizophrenia and Autism Spectrum Disorder. Biol Psychiatry 2024, 95:888-895.
- 22. Hale CR, Sawicka K, Mora K, Fak JJ, Kang JJ, Cutrim P, Cialowicz K, Carroll TS, Darnell RB: FMRP regulates mRNAs encoding distinct functions in the cell body and dendrites of CA1 pyramidal neurons. Elife 2021, 10.
- 23. Meservey LM, Topkar VV, Fu MM: mRNA Transport and Local Translation in Glia. Trends Cell Biol 2021, 31:419-423.
- 24. Berkovits BD, Mayr C: Alternative 3′ UTRs act as scaffolds to regulate membrane protein localization. Nature 2015, 522:363-367.
- 25. Fasken MB, Corbett AH: Mechanisms of nuclear mRNA quality control. RNA Biology 2009, 6:237-241.
- 26. Bertrand E, Chartrand P, Schaefer M, Shenoy SM, Singer RH, Long RM: Localization of ASH1 mRNA particles in living yeast. Mol Cell 1998, 2:437-445.
- 27. Chao JA, Patskovsky Y, Almo SC, Singer RH: Structural basis for the coevolution of a viral RNA–protein complex. Nature Structural & Molecular Biology 2008, 15:103-105.
- 28. Le P, Ahmed N, Yeo GW: Illuminating RNA biology through imaging. Nature Cell Biology 2022, 24:815-824.
- 29. Yin P, Kuang S, Nie Z: Fluorescent RNA Tags for In Situ RNA Imaging in Living Cells. Analysis & Sensing 2023, 3:e202200090.
- 30. Das S, Vera M, Gandin V, Singer RH, Tutucci E: Intracellular mRNA transport and localized translation. Nat Rev Mol Cell Biol 2021, 22:483-504.
- 31. Tutucci E, Vera M, Biswas J, Garcia J, Parker R, Singer RH: An improved MS2 system for accurate reporting of the mRNA life cycle. Nature Methods 2018, 15:81-89.
- 32. Li W, Maekiniemi A, Sato H, Osman C, Singer RH: An improved imaging system that corrects MS2-induced RNA destabilization. Nature Methods 2022, 19:1558-1562.
- 33. Ohishi H, Shimada S, Uchino S, Li J, Sato Y, Shintani M, Owada H, Ohkawa Y, Pertsinidis A, Yamamoto T, et al.: STREAMING-tag system reveals spatiotemporal relationships between transcriptional regulatory factors and transcriptional activity. Nature Communications 2022, 13:7672.
- 34. Tyagi S, Kramer FR: Molecular Beacons: Probes that Fluoresce upon Hybridization. Nature Biotechnology 1996, 14:303-308.
- 35. Donlin-Asp PG, Polisseni C, Klimek R, Heckel A, Schuman EM: Differential regulation of local mRNA dynamics and translation following long-term potentiation and depression. Proceedings of the National Academy of Sciences 2021, 118:e2017578118.
- 36. Abudayyeh OO, Gootenberg JS, Essletzbichler P, Han S, Joung J, Belanto JJ, Verdine V, Cox DBT, Kellner MJ, Regev A, et al.: RNA targeting with CRISPR-Cas13. Nature 2017, 550:280-284.
- 37. Han Y, Branon TC, Martell JD, Boassa D, Shechner D, Ellisman MH, Ting A: Directed Evolution of Split APEX2 Peroxidase. ACS Chemical Biology 2019, 14:619-635.
- 38. Halstead JM, Lionnet T, Wilbertz JH, Wippich F, Ephrussi A, Singer RH, Chao JA: Translation. An RNA biosensor for imaging the first round of translation from single cells to living animals. Science 2015, 347:1367-1671.
- 39. Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, Vale RD: A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 2014, 159:635-646.
- 40. Wang C, Han B, Zhou R, Zhuang X: Real-Time Imaging of Translation on Single mRNA Transcripts in Live Cells. Cell 2016, 165:990-1001.
- 41. Wu B, Eliscovich C, Yoon YJ, Singer RH: Translation dynamics of single mRNAs in live cells and neurons. Science 2016, 352:1430-1435.
- 42. Yan X, Hoek TA, Vale RD, Tanenbaum ME: Dynamics of Translation of Single mRNA Molecules In Vivo. Cell 2016, 165:976-989.
- 43. Mateju D, Eichenberger B, Voigt F, Eglinger J, Roth G, Chao JA: Single-Molecule Imaging Reveals Translation of mRNAs Localized to Stress Granules. Cell 2020, 183:1801-1812.e1813.
- 44. Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS: Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 2009, 324:218-223.
- 45. Jan CH, Williams CC, Weissman JS: Principles of ER cotranslational translocation revealed by proximity-specific ribosome profiling. Science 2014, 346:1257521.
- 46. Williams CC, Jan CH, Weissman JS: Targeting and plasticity of mitochondrial proteins revealed by proximity-specific ribosome profiling. Science 2014, 346:748-751.
- 47. Xu W, Abruzzi K, Rosbash M: RiboTRIBE: Monitoring Translation with ADAR-meditated RNA Editing. bioRxiv 2021:2021.2006.2020.449184.
- 48. Brannan KW, Chaim IA, Marina RJ, Yee BA, Kofman ER, Lorenz DA, Jagannatha P, Dong KD, Madrigal AA, Underwood JG, et al.: Robust single-cell discovery of RNA targets of RNA-binding proteins and ribosomes. Nat Methods 2021, 18:507-519.
- **49. Zeng H, Huang J, Ren J, Wang CK, Tang Z, Zhou H, Zhou Y, Shi H, Aditham A, Sui X, et al.: Spatially resolved single-cell translatomics at molecular resolution. Science 2023, 380:eadd3067.
- A massively multiplexed platform to assess translation, based on clever probe design that captures only ribosome-bound mRNAs of interests and barcode-based detection with in situ sequencing.
- 50. Zeng H, Huang J, Zhou H, Meilandt WJ, Dejanovic B, Zhou Y, Bohlen CJ, Lee S-H, Ren J, Liu A, et al.: Integrative in situ mapping of single-cell transcriptional states and tissue histopathology in a mouse model of Alzheimer’s disease. Nature Neuroscience 2023, 26:430-446.
- 51. Wang X, Allen WE, Wright MA, Sylwestrak EL, Samusik N, Vesuna S, Evans K, Liu C, Ramakrishnan C, Liu J, et al.: Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 2018, 361:eaat5691.
- 52. Kislauskis EH, Zhu X, Singer RH: Sequences responsible for intracellular localization of beta-actin messenger RNA also affect cell phenotype. J Cell Biol 1994, 127:441-451.
- *53. Loedige I, Baranovskii A, Mendonsa S, Dantsuji S, Popitsch N, Breimann L, Zerna N, Cherepanov V, Milek M, Ameres S, et al.: mRNA stability and m(6)A are major determinants of subcellular mRNA localization in neurons. Mol Cell 2023, 83:2709-2725.e2710.
- The paper used SLAM-seq to characterize expression dynamics and global stability of RNAs, and found correlation between mRNA stability and their propensity to trafficking to distal neuronal processes.
- 54. Chen CYA, Ezzeddine N, Shyu AB: Chapter 17 Messenger RNA Half-Life Measurements in Mammalian Cells. In Methods in Enzymology. Edited by: Academic Press; 2008:335-357. vol 448.].
- 55. Herzog VA, Reichholf B, Neumann T, Rescheneder P, Bhat P, Burkard TR, Wlotzka W, von Haeseler A, Zuber J, Ameres SL: Thiol-linked alkylation of RNA to assess expression dynamics. Nat Methods 2017, 14:1198-1204.
- 56. Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al.: Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485:201-206.
- 57. Meyer KD: DART-seq: an antibody-free method for global m6A detection. Nature Methods 2019, 16:1275-1280.
- 58. Poon MM, Choi SH, Jamieson CA, Geschwind DH, Martin KC: Identification of process-localized mRNAs from cultured rodent hippocampal neurons. J Neurosci 2006, 26:13390-13399.
- 59. Yang YM, Karbstein K: The chaperone Tsr2 regulates Rps26 release and reincorporation from mature ribosomes to enable a reversible, ribosome-mediated response to stress. Sci Adv 2022, 8:eabl4386.
- 60. Shi Z, Fujii K, Kovary KM, Genuth NR, Röst HL, Teruel MN, Barna M: Heterogeneous Ribosomes Preferentially Translate Distinct Subpools of mRNAs Genome-wide. Mol Cell 2017, 67:71-83.e77.
- 61. Ferretti MB, Ghalei H, Ward EA, Potts EL, Karbstein K: Rps26 directs mRNA-specific translation by recognition of Kozak sequence elements. Nat Struct Mol Biol 2017, 24:700-707.
- 62. Xue S, Tian S, Fujii K, Kladwang W, Das R, Barna M: RNA regulons in Hox 5′ UTRs confer ribosome specificity to gene regulation. Nature 2015, 517:33-38.
- 63. Majzoub K, Hafirassou ML, Meignin C, Goto A, Marzi S, Fedorova A, Verdier Y, Vinh J, Hoffmann JA, Martin F, et al.: RACK1 controls IRES-mediated translation of viruses. Cell 2014, 159:1086-1095.
- 64. Heiman M, Kulicke R, Fenster RJ, Greengard P, Heintz N: Cell type–specific mRNA purification by translating ribosome affinity purification (TRAP). Nature Protocols 2014, 9:1282-1291.
- 65. Simsek D, Tiu GC, Flynn RA, Byeon GW, Leppek K, Xu AF, Chang HY, Barna M: The Mammalian Ribo-interactome Reveals Ribosome Functional Diversity and Heterogeneity. Cell 2017, 169:1051-1065.e1018.
- 66. Ferretti MB, Karbstein K: Does functional specialization of ribosomes really exist? Rna 2019, 25:521-538.
- *67. Susanto TT, Hung V, Levine AG, Kerr CH, Yoo Y, Chen Y, Oses-Prieto JA, Fromm L, Fujii K, Wernig M, et al.: RAPIDASH: A tag-free enrichment of ribosome-associated proteins reveals compositional dynamics in embryonic tissues and stimulated macrophages. bioRxiv 2023:2023.2012.2007.570613.
- An enabling technique for the isolation of native ribosomes while preserving weak interactions of ribosome-associated proteins.
- 68. Georgeson J, Schwartz S: The ribosome epitranscriptome: inert-or a platform for functional plasticity? Rna 2021, 27:1293-1301.
- 69. Wang DO, Kim SM, Zhao Y, Hwang H, Miura SK, Sossin WS, Martin KC: Synapse- and stimulus-specific local translation during long-term neuronal plasticity. Science 2009, 324:1536-1540.
- 70. Wong HH, Lin JQ, Ströhl F, Roque CG, Cioni JM, Cagnetta R, Turner-Bridger B, Laine RF, Harris WA, Kaminski CF, et al.: RNA Docking and Local Translation Regulate Site-Specific Axon Remodeling In Vivo. Neuron 2017, 95:852-868.e858.
- 71. Ando R, Shimozono S, Ago H, Takagi M, Sugiyama M, Kurokawa H, Hirano M, Niino Y, Ueno G, Ishidate F, et al.: StayGold variants for molecular fusion and membrane-targeting applications. Nature Methods 2023.
- 72. Ivorra-Molla E, Akhuli D, McAndrew MBL, Scott W, Kumar L, Palani S, Mishima M, Crow A, Balasubramanian MK: A monomeric StayGold fluorescent protein. Nature Biotechnology 2023.
- 73. Zhang H, Lesnov GD, Subach OM, Zhang W, Kuzmicheva TP, Vlaskina AV, Samygina VR, Chen L, Ye X, Nikolaeva AY, et al.: Bright and stable monomeric green fluorescent protein derived from StayGold. Nature Methods 2024.
- 74. Zhang D, Chen Z, Du Z, Bao B, Su N, Chen X, Ge Y, Lin Q, Yang L, Hua Y, et al.: Design of a palette of SNAP-tag mimics of fluorescent proteins and their use as cell reporters. Cell Discovery 2023, 9:56.
- 75. Frei MS, Tarnawski M, Roberti MJ, Koch B, Hiblot J, Johnsson K: Engineered HaloTag variants for fluorescence lifetime multiplexing. Nature Methods 2022, 19:65-70.
- 76. Gautier A: Fluorescence-Activating and Absorption-Shifting Tags for Advanced Imaging and Biosensing. Accounts of Chemical Research 2022, 55:3125-3135.
- 77. Plamont M-A, Billon-Denis E, Maurin S, Gauron C, Pimenta FM, Specht CG, Shi J, Quérard J, Pan B, Rossignol J, et al.: Small fluorescence-activating and absorption-shifting tag for tunable protein imaging in vivo. Proceedings of the National Academy of Sciences 2016, 113:497-502.
- *78. Benaissa H, Ounoughi K, Aujard I, Fischer E, Goïame R, Nguyen J, Tebo AG, Li C, Le Saux T, Bertolin G, et al.: Engineering of a fluorescent chemogenetic reporter with tunable color for advanced live-cell imaging. Nature Communications 2021, 12:6989.
- The paper described the development of promiscuous FAST variants which can bind to different fluorogenic chromophores covering the visible spectrum.
- 79. Kang H, Schuman EM: A Requirement for Local Protein Synthesis in Neurotrophin-Induced Hippocampal Synaptic Plasticity. Science 1996, 273:1402-1406.
- 80. Martin KC, Casadio A, Zhu H, E Y, Rose JC, Chen M, Bailey CH, Kandel ER: Synapse-Specific, Long-Term Facilitation of Aplysia Sensory to Motor Synapses: A Function for Local Protein Synthesis in Memory Storage. Cell 1997, 91:927-938.
- 81. Mineev KS, Goncharuk SA, Goncharuk MV, Povarova NV, Sokolov AI, Baleeva NS, Smirnov AY, Myasnyanko IN, Ruchkin DA, Bukhdruker S, et al.: NanoFAST: structure-based design of a small fluorogen-activating protein with only 98 amino acids. Chemical Science 2021, 12:6719-6725.
- 82. Tang Q, Chen X: Nascent Proteomics: Chemical Tools for Monitoring Newly Synthesized Proteins. Angewandte Chemie International Edition 2023, 62:e202305866.
- 83. Stone SE, Glenn WS, Hamblin GD, Tirrell DA: Cell-selective proteomics for biological discovery. Current Opinion in Chemical Biology 2017, 36:50-57.
- 84. Beatty KE, Liu JC, Xie F, Dieterich DC, Schuman EM, Wang Q, Tirrell DA: Fluorescence Visualization of Newly Synthesized Proteins in Mammalian Cells. Angewandte Chemie International Edition 2006, 45:7364-7367.
- 85. Dieterich DC, Link AJ, Graumann J, Tirrell DA, Schuman EM: Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). Proceedings of the National Academy of Sciences 2006, 103:9482-9487.
- 86. Sun C, Nold A, Fusco CM, Rangaraju V, Tchumatchenko T, Heilemann M, Schuman EM: The prevalence and specificity of local protein synthesis during neuronal synaptic plasticity. Science Advances 2021, 7:eabj0790.
- *87. Alvarez-Castelao B, Schanzenbächer CT, Hanus C, Glock C, tom Dieck S, Dörrbaum AR, Bartnik I, Nassim-Assir B, Ciirdaeva E, Mueller A, et al.: Cell-type-specific metabolic labeling of nascent proteomes in vivo. Nature Biotechnology 2017, 35:1196-1201.
- Cell-type specific metabolic labeling of nascent proteomes using a genetically targetable mutant methionyl-tRNA synthetase.
- *88. Ignacio BJ, Dijkstra J, Mora N, Slot EFJ, van Weijsten MJ, Storkebaum E, Vermeulen M, Bonger KM: THRONCAT: metabolic labeling of newly synthesized proteins using a bioorthogonal threonine analog. Nature Communications 2023, 14:3367.
- A new metabolic labeling approach for newly synthesized proteins based on a threonine analog.
- 89. Elliott TS, Townsley FM, Bianco A, Ernst RJ, Sachdeva A, Elsässer SJ, Davis L, Lang K, Pisa R, Greiss S, et al.: Proteome labeling and protein identification in specific tissues and at specific developmental stages in an animal. Nat Biotechnol 2014, 32:465-472.
- 90. tom Dieck S, Kochen L, Hanus C, Heumüller M, Bartnik I, Nassim-Assir B, Merk K, Mosler T, Garg S, Bunse S, et al.: Direct visualization of newly synthesized target proteins in situ. Nat Methods 2015, 12:411-414.
- *91. Qin W, Cheah JS, Xu C, Messing J, Freibaum BD, Boeynaems S, Taylor JP, Udeshi ND, Carr SA, Ting AY: Dynamic mapping of proteome trafficking within and between living cells by TransitID. Cell 2023, 186:3307-3324.e3330.
- Orthogonal proximity labeling approaches allow intracellular and intercellular protein trafficking studies.
- 92. Reinkemeier CD, Girona GE, Lemke EA: Designer membraneless organelles enable codon reassignment of selected mRNAs in eukaryotes. Science 2019, 363:eaaw2644.
- **93. Reinkemeier CD, Lemke EA: Dual film-like organelles enable spatial separation of orthogonal eukaryotic translation. Cell 2021, 184:4886-4903.e4821.
- Phase separation of mRNAs and aminoacyl-tRNA synthetases at the membranes of organelles enables orthogonal local translation of the mRNAs.
- 94. Reinkemeier CD, Lemke EA: Condensed, Microtubule-coating Thin Organelles for Orthogonal Translation in Mammalian Cells. Journal of Molecular Biology 2022, 434:167454.
- *95. Hao M, Ling X, Sun Y, Wang X, Li W, Chang L, Zeng Z, Shi X, Niu M, Chen L, et al.: Tracking endogenous proteins based on RNA editing-mediated genetic code expansion. Nat Chem Biol 2024.
- The combination of RNA-targeted base editing with genetic code expansion to incorporate non-canonical amino acids to proteins synthesized from endogenous transcripts.
- 96. Sappakhaw K, Jantarug K, Slavoff SA, Israsena N, Uttamapinant C: A Genetic Code Expansion-Derived Molecular Beacon for the Detection of Intracellular Amyloid-β Peptide Generation. Angew Chem Int Ed Engl 2021, 60:3934-3939.
- 97. Uttamapinant C, Howe JD, Lang K, Beránek V, Davis L, Mahesh M, Barry NP, Chin JW: Genetic Code Expansion Enables Live-Cell and Super-Resolution Imaging of Site-Specifically Labeled Cellular Proteins. Journal of the American Chemical Society 2015, 137:4602-4605.
- 98. Aphicho K, Kittipanukul N, Uttamapinant C: Visualizing the complexity of proteins in living cells with genetic code expansion. Curr Opin Chem Biol 2022, 66:102108.
- 99. Lampkin BJ, Kritzer JA: Engineered fluorogenic HaloTag ligands for turn-on labelling in live cells††Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc05536a. Chemical Communications 2023, 60:200-203.
- 100. Martin A, Rivera-Fuentes P: A general strategy to develop fluorogenic polymethine dyes for bioimaging. Nature Chemistry 2024, 16:28-35.
- 101. Rackham O, Chin JW: A network of orthogonal ribosome·mRNA pairs. Nature Chemical Biology 2005, 1:159-166.
- 102. Fried SD, Schmied WH, Uttamapinant C, Chin JW: Ribosome Subunit Stapling for Orthogonal Translation in E. coli. Angew Chem Int Ed Engl 2015, 54:12791-12794.
- 103. Schmied WH, Tnimov Z, Uttamapinant C, Rae CD, Fried SD, Chin JW: Controlling orthogonal ribosome subunit interactions enables evolution of new function. Nature 2018, 564:444-448.
- 104. Orelle C, Carlson ED, Szal T, Florin T, Jewett MC, Mankin AS: Protein synthesis by ribosomes with tethered subunits. Nature 2015, 524:119-124.
- 105. Carlson ED, d’Aquino AE, Kim DS, Fulk EM, Hoang K, Szal T, Mankin AS, Jewett MC: Engineered ribosomes with tethered subunits for expanding biological function. Nat Commun 2019, 10:3920.
- 106. Adaligil E, Song A, Cunningham CN, Fairbrother WJ: Ribosomal Synthesis of Macrocyclic Peptides with Linear γ(4)- and β-Hydroxy-γ(4)-amino Acids. ACS Chem Biol 2021, 16:1325-1331.
- 107. Chan AI, Sawant MS, Burdick DJ, Tom J, Song A, Cunningham CN: Evaluating Translational Efficiency of Noncanonical Amino Acids to Inform the Design of Druglike Peptide Libraries. ACS Chem Biol 2023, 18:81-90.
- **108. Ferretti MB, Barre JL, Karbstein K: Translational Reprogramming Provides a Blueprint for Cellular Adaptation. Cell Chem Biol 2018, 25:1372-1379.e1373.
- Translational reprogramming in yeast through modulation of ribosomal protein compositions.
- 109. Shakiba N, Jones RD, Weiss R, Del Vecchio D: Context-aware synthetic biology by controller design: Engineering the mammalian cell. Cell Syst 2021, 12:561-592.
- 110. Huang Y, Liu T: Therapeutic applications of genetic code expansion. Synthetic and Systems Biotechnology 2018, 3:150-158.
- 111. Davis L, Radman I, Goutou A, Tynan A, Baxter K, Xi Z, O’Shea JM, Chin JW, Greiss S: Precise optical control of gene expression in C elegans using improved genetic code expansion and Cre recombinase. eLife 2021, 10:e67075.
Figure 1.
RNA-centric techniques for visualization and mapping of local translation. (A) Stem loop-based tags and (B) molecular beacons for imaging of individual mRNAs. (C) TRICK (translating RNA imaging by coat protein knock-off) technique and SINAP (single-molecule imaging of nascent peptides) can be used to image actively translated individual mRNAs. To map local translatomes, (E) ADAR (Apolipoprotein B mRNA Editing Enzyme)-based RiboTRIBE and APOBEC (Apolipoprotein B mRNA Editing Enzyme, Catalytic Polypeptide-Like)-based RiboSTAMP or (F) RIBOmap (ribosome-bound messenger RNA imaging technique) can be employed. Related STARmap (spatially-resolved transcript amplicon readout mapping technique) is used to map spatial transcriptomes. To map global turnover and associated regulatory elements of localized mRNAs, (G) SLAM-seq (SH-linked alkylation for the metabolic sequencing of RNA) and (H) DART-seq (deamination adjacent to RNA modification targets) can be used. (I) TRAP (Translating ribosome affinity purification)/RiboTag and (J) RAPIDASH can be used to profile ribosome heterogeneity, particularly for characterizations of ribosome-associated proteins (RAPs). scFv: single-chain fragment variable; sfGFP: superfolder green fluorescent protein; SEDAL: sequencing with error-reduction by dynamic annealing and ligation; m6A: N6-methyladenosine; 4SU: 4-thiouridine; IAA: iodoacetamide; YHT: YT521-B homology domain-containing protein which specifically recognizes m6A containing RNAs.
Figure 1.
RNA-centric techniques for visualization and mapping of local translation. (A) Stem loop-based tags and (B) molecular beacons for imaging of individual mRNAs. (C) TRICK (translating RNA imaging by coat protein knock-off) technique and SINAP (single-molecule imaging of nascent peptides) can be used to image actively translated individual mRNAs. To map local translatomes, (E) ADAR (Apolipoprotein B mRNA Editing Enzyme)-based RiboTRIBE and APOBEC (Apolipoprotein B mRNA Editing Enzyme, Catalytic Polypeptide-Like)-based RiboSTAMP or (F) RIBOmap (ribosome-bound messenger RNA imaging technique) can be employed. Related STARmap (spatially-resolved transcript amplicon readout mapping technique) is used to map spatial transcriptomes. To map global turnover and associated regulatory elements of localized mRNAs, (G) SLAM-seq (SH-linked alkylation for the metabolic sequencing of RNA) and (H) DART-seq (deamination adjacent to RNA modification targets) can be used. (I) TRAP (Translating ribosome affinity purification)/RiboTag and (J) RAPIDASH can be used to profile ribosome heterogeneity, particularly for characterizations of ribosome-associated proteins (RAPs). scFv: single-chain fragment variable; sfGFP: superfolder green fluorescent protein; SEDAL: sequencing with error-reduction by dynamic annealing and ligation; m6A: N6-methyladenosine; 4SU: 4-thiouridine; IAA: iodoacetamide; YHT: YT521-B homology domain-containing protein which specifically recognizes m6A containing RNAs.
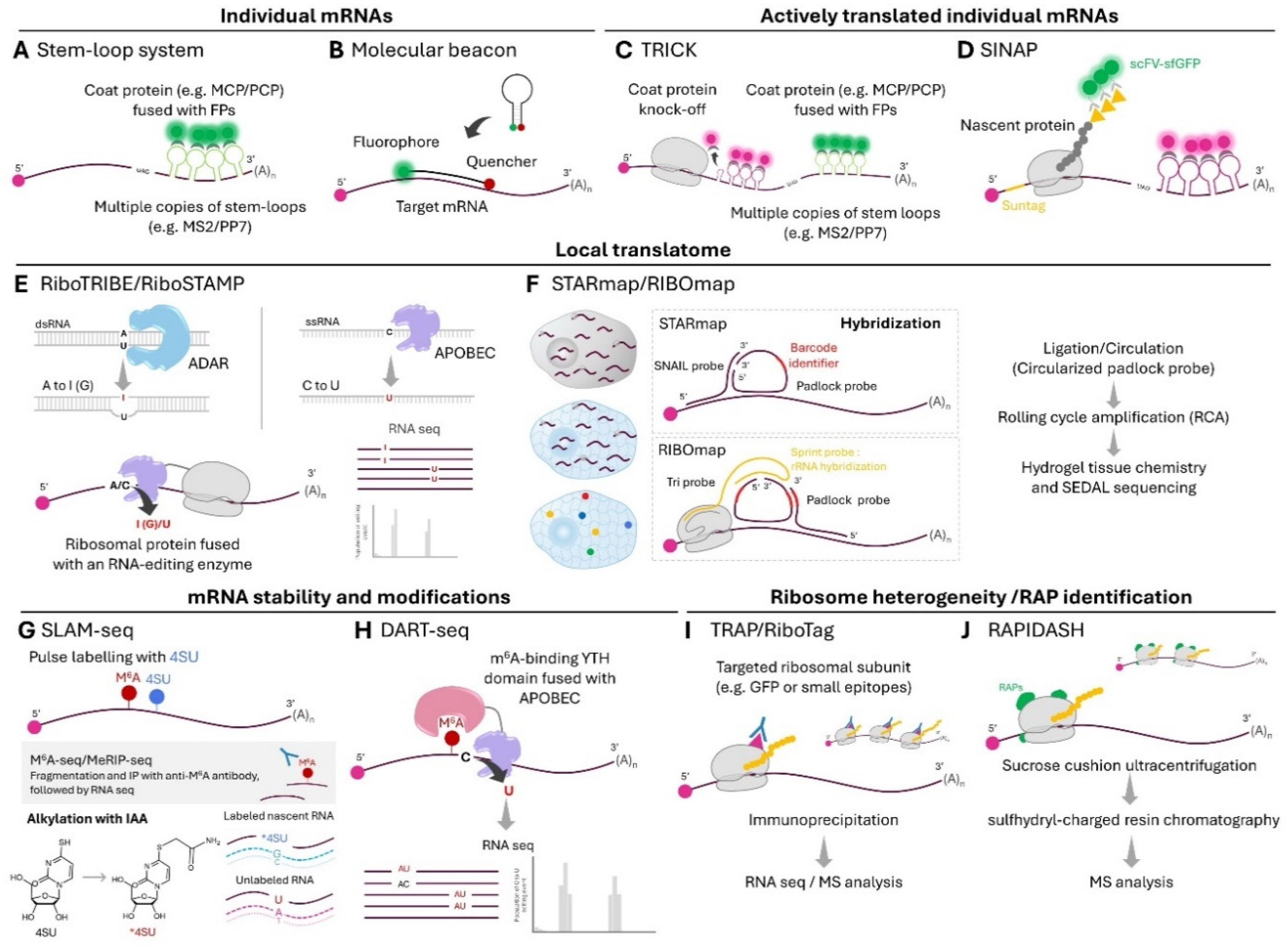
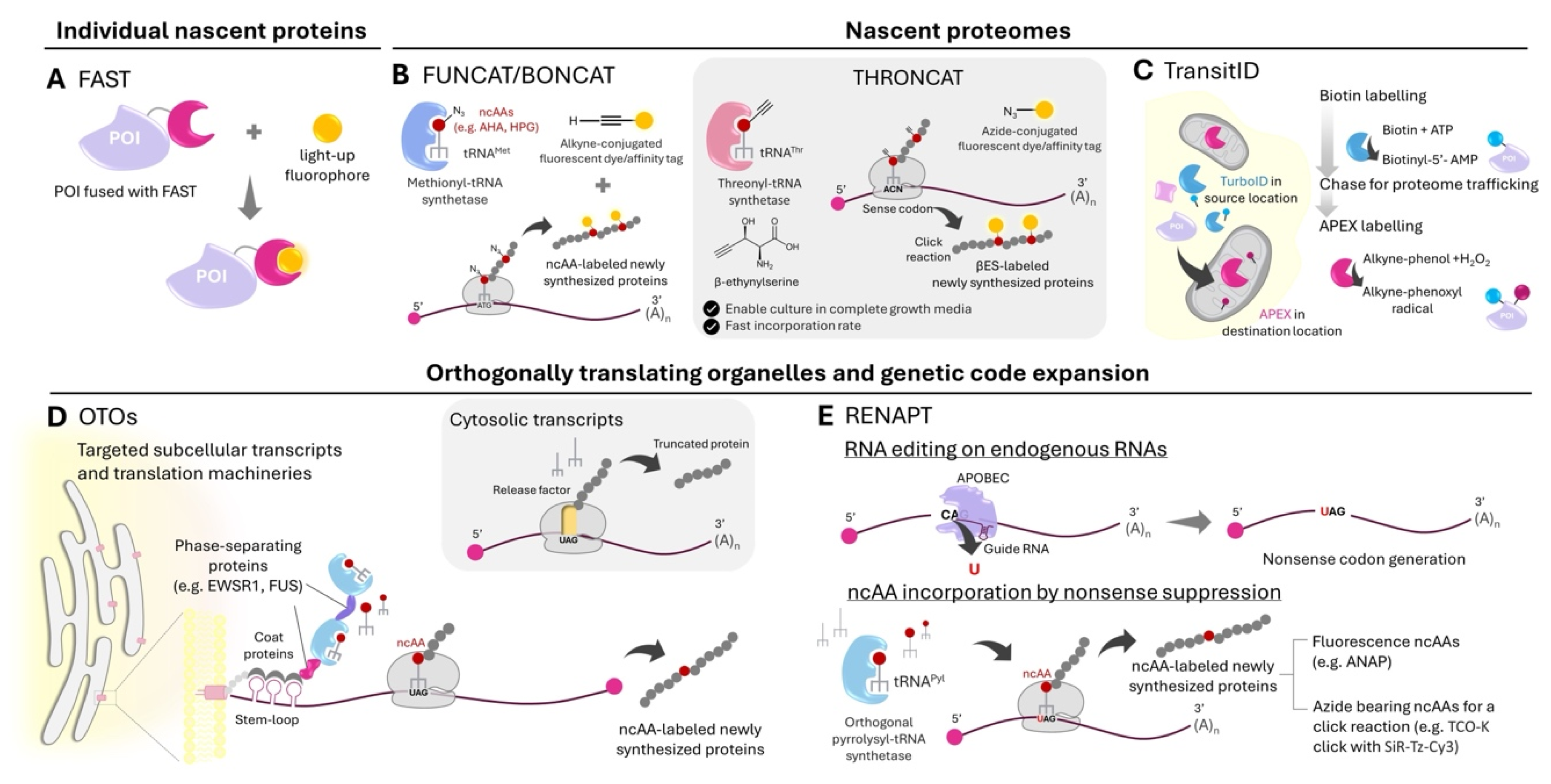
Figure 2.
Protein-centric techniques for imaging, mapping, and controlling of local translation. (A) Fluorescence-activating and absorption-shifting tags (FASTs) for individual protein imaging. (B) Nascent proteomes can be labeled with bio-orthogonal noncanonical amino acid tagging (BONCAT), fluorescent noncanonical amino acid tagging (FUNCAT). Or threonine-derived non-canonical amino acid tagging (THRONCAT). (C) Proximity labeling-based TransitID can map protein pools trafficked to and from different subcellular locations. (D) Genetic code expansion-based OTOs (orthogonally translating organelles) can mediate local translation of mRNAs artificially targeted to the organelles. (E) RENAPT (RNA editing-mediated noncanonical amino acids (ncAAs) protein tagging) introduces the amber stop codon on desired mRNAs, and allows genetic code expansion and ncAA incorporation on endogenous transcripts.
Figure 2.
Protein-centric techniques for imaging, mapping, and controlling of local translation. (A) Fluorescence-activating and absorption-shifting tags (FASTs) for individual protein imaging. (B) Nascent proteomes can be labeled with bio-orthogonal noncanonical amino acid tagging (BONCAT), fluorescent noncanonical amino acid tagging (FUNCAT). Or threonine-derived non-canonical amino acid tagging (THRONCAT). (C) Proximity labeling-based TransitID can map protein pools trafficked to and from different subcellular locations. (D) Genetic code expansion-based OTOs (orthogonally translating organelles) can mediate local translation of mRNAs artificially targeted to the organelles. (E) RENAPT (RNA editing-mediated noncanonical amino acids (ncAAs) protein tagging) introduces the amber stop codon on desired mRNAs, and allows genetic code expansion and ncAA incorporation on endogenous transcripts.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



