
Submitted:
24 July 2024
Posted:
25 July 2024
You are already at the latest version
Abstract
Hereditary cardiomyopathies (CMPs), including Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC), Dilated Cardiomyopathy (DCM), and Hypertrophic Cardiomyopathy (HCM), represent a group of heart disorders that significantly contribute to cardiovascular morbidity and mortality and are often driven by genetic factors. Recent advances in Next-Generation Sequencing (NGS) technology have enabled the identification of rare variants in both well-established and minor genes associated to CMPs. Nowadays, a set of core genes is included in diagnostic panels for ARVC, DCM, HCM. On the other hand, despite their lesser-known status, variants in the minor genes may contribute to disease mechanisms and influence prognosis. This review evaluates the current evidence supporting the involvement of the minor genes in CMPs, considering their potential pathogenicity and clinical significance. A comprehensive analysis of databases such as ClinGen, ClinVar, and GeneReviews, along with recent literature and diagnostic guidelines, provides a thorough overview of the genetic landscape of minor genes in CMPs and offers insights for future research and translation to clinical practice. Given the increasing knowledge on these less-understood genetic factors, future studies are essential to clearly assess their roles, ultimately leading to improved diagnostic precision and therapeutic strategies in hereditary CMPs.
Keywords:
cardiomyopathies
; genetics
; ARVC
; DCM
; HCM
1. Introduction
Cardiomyopathies (CMPs) represent a heterogeneous group of heart disorders characterized by structural and functional abnormalities of the myocardium, often stemming from genetic factors. Among these, Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC), Dilated Cardiomyopathy (DCM), and Hypertrophic Cardiomyopathy (HCM) stand out as significant contributors to cardiovascular morbidity and mortality. Understanding the intricate genetic underpinnings of these conditions is paramount for advancing diagnostic precision, prognostic assessment, and targeted therapeutic interventions. Over the years, substantial progress has been made in unraveling the complex genetic landscape associated with cardiomyopathies [1]. Indeed, recent advances in next generation sequencing (NGS) technology have enabled the identification of rare genetic variants both in genes clearly associated with the disease and in minor genes that show a lower correlation to the clinical phenotype [2,3]. Panel genes, whole-exome sequencing (WES), and whole-genome sequencing (WGS) represent significant advancements in the genetic evaluation of CMPs. Panel genes and WES, which focus on protein-coding regions, effectively identify pathogenic variants and incidental findings. In contrast, WGS provides comprehensive genome coverage, detecting both intronic and structural variations. Despite their relevance for early diagnosis and personalized treatment, variables linked to data interpretation, high costs, and clinical parameters still limit their full application [3].
Nowadays, thanks to the NGS technology which has enabled the identification of pathogenic variants in various genes, and to functional and genetic evidence that has clarified their clinical significance, several genes have been confirmed as definitively associated with ARVC, HCM, and DCM [4,5,6]. ARVC is a cardiac disorder characterized by the progressive replacement of myocardial tissue with fibrous and fatty deposits, particularly affecting the right ventricle [7]. The pathogenesis of ARVC involves a complex interplay of genetic and environmental factors. Pathogenic variants affecting desmosomal proteins, crucial for cell-to-cell adhesion in the myocardium, are commonly implicated in ARVC [8]. According to the above cited sources, four genes are classified as having definitive evidence for ARVC: DSC2, DSG2, PKP2, and TMEM43. DCM is a cardiac disorder characterized by the enlargement and weakened pumping ability of the left ventricle, leading to decreased contractility and impaired systolic function [9]. Genetic factors play a significant role in DCM, with pathogenic variants in different genes associated with the regulation of myocardial structure and function [10]. While there is not a fixed set of "core genes" for DCM, several genes have been associated, according to the above-cited sources, as having definitive evidence for DCM: BAG3, DES, FLNC, LMNA, RBM20, SCN5A, TNNC1 and TTN. HCM is a primary, often inherited, cardiac disease characterized by abnormal hypertrophy of the heart, primarily affecting the left ventricle. The fundamental pathogenesis of HCM involves a genetic mutation leading to the abnormal hypertrophy of cardiomyocytes, resulting in increased cardiac mass [11]. HCM is primarily linked to pathogenic variants in genes encoding sarcomere proteins, playing a crucial role in cardiac muscle contraction. Each gene serves as critical elements in the sarcomere, influencing muscle contraction, structural integrity, and the overall physiological balance within the heart. Nowadays, according to the above-cited sources, eight core genes are classified as having definitive evidence for HCM: ACTC1, MYBPC3, MYH7, MYL2, MYL3, TNNI3, TNNT2, TPM1 [12].
Based on the literature, minor genes in CMPs can be considered genes of uncertain significance (GUSs) due to limited evidence of pathogenicity, difficulties in interpreting variants, phenotype variability, and lack of robust functional and segregation studies. Therefore, GUSs raise several challenges in cardiogenetic, as they often harbor variants whose effects on protein function and disease risk are not yet well understood. In fact, even though current research has begun to explore the potential involvement of these minor genes (e.g. TRIM63, FHOD3) in CMPs [13,14], their potential contribution to disease mechanisms is still unclear. Functional studies and segregation analysis are critical to determine their potential pathogenic role and clinical significance in CMPs.
This review aims to provide a comprehensive synthesis of current knowledge on the less understood, non-definitive genes, devoid of a clear association with syndromic disorders, involved in CMPs. Our effort fulfills the need to unveil the potential role of the rare genetic contributors to ARVC, DCM and HCM.
1.1. Identification of Reported Minor CMPs Genes
The genes not yet definitively associated with CMPs were selected through a systematic review of the literature and querying different sources in the context of the three CMP phenotypes (ARVC, DCM and HCM). We focused on genes considered to have moderate or limited association with CMPs according to the questioned sources. As of today, there is a lack of concordance for JUP and PLN. Whereas they are considered definitive genes for ARVC and HCM/DCM, respectively, in the European Molecular Genetics Quality Network (EMQN) [15,16], they are not reported as definitive in ClinGen [17] because their “gene-disease validity” has not been fully evaluated. However, the literature is overwhelmingly supportive of their role in CMPs [18]. Since this review focuses on minor genes, JUP and PLN will not be discussed. The same holds true for ALPK3. Although ALPK3 is definitive for HCM in ClinGen and GeneReviews, this gene is not considered in the other sources. Therefore, after excluding genes with clearly definitive associations to CMPs and genes associated with syndromic conditions, 36 genes will be discussed as minor genes in this article.
1.2. Gathering Sources
To gain a comprehensive view of the minor genes associated with the three forms of CMPs discussed herein, we used as a main source ClinGen [17]. The latter provides an authoritative and centralized resource, which defines the clinical relevance of the genes and variants to be used in precision medicine and research. ClinGen uses various methodologies to define the association between rare variants and Mendelian diseases such as CMPs. These methods include segregation of variants with disease in large family pedigrees, case-control studies, and functional studies to analyze the consequences of variants. ClinGen’s approach to determining “gene-disease validity” involves a meticulous review process to assess the strength of evidence linking rare variants to CMPs. These classifications are based on the careful analysis of published genetic and experimental data by data curators and expert panels. They generate strong, moderate or limited levels of evidence that can be further modified to definitive or even refuted once new evidence becomes available.
The second source used was ClinVar [19]. ClinVar is a public repository maintained by the National Center for Biotechnology Information (NCBI) [20], as part of the National Library of Medicine (NLM) [21] at the National Institutes of Health (NIH) [21,22]. ClinVar accepts and archives genetic variants identified in different clinical phenotypes by diagnostic laboratories, researchers, clinicians, and organizations, allowing the re-evaluation of classifications over time.
GeneReviews [23] , a journal edited by the University of Washington, Seattle, was also used as a resource. This source aims to provide clinically relevant and actionable information for hereditary conditions, to associate genes to specific diseases, by collecting information from different sources, including peer-reviewed and research studies, data from clinical experts and researchers, genetic databases such as OMIM (Online Mendelian Inheritance in Man) [24] and other genomic databases.
To complete the evaluation of minor genes, we also considered the positions of the EMQN (European Molecular Genetics Quality Network) [15,16], as outlined in their 2023 recommendations for genetic testing in hereditary CMPs and arrhythmias [15]. Additionally, the 2022 Expert Consensus Statement on the State of Genetic Testing for Heart Disease provides valuable insights and guidance [25].
1.3. Literature Review
A comprehensive review of articles was conducted using PubMed as the primary database, searching for the publication period from 1990 to 2024. The Keywords used were: "arrhythmogenic right ventricular cardiomyopathy", "dilated cardiomyopathy", "hypertrophic cardiomyopathy", and specific gene names. These terms were used in various combinations to maximize the retrieval of relevant articles. A filter was applied to narrow down the search results to articles written in English only, to ensure that the review article was based on widely accessible sources and accepted by the international research community. The selection process involved reviewing the titles and abstracts of the retrieved articles to assess their relevance to the topic. Priority was given to articles that provided novel insights into the genetic underpinnings of ARVC, DCM, and HCM, based on functional studies, segregation in families, and clinical implications of genetic findings.
2. Genes Classification
In this review the reported genes were classified as definitive, moderate, limited, not associated, not curated, according to ClinGen terminology. The associations with different forms of CMPs (ARVC, DCM, HCM) are reported in Table 1 for definitive genes, and in Table 2 for minor genes based on the classification provided by the cited sources. Genes considered “strong” in GeneReviews and the Expert Consensus were classified as definitive, those considered “moderate intrinsic” in the Expert Consensus were classified moderate, according to the Clingen classification.
2.1. Definitive Genes Associated with ARVC, DCM and HCM2.2. Minor genes in ARVC, DCM and HCM
ABCC9
The ATP-binding cassette, sub-family C member 9 gene (ABCC9), which encodes the cardiac sulfonylurea receptor subunit SUR2A of the cardiac KATP channel, plays a critical role in linking cellular metabolism to electrical activity through the regulation of potassium flow. These channels are vital in adapting the membrane potential to reflect the cellular ATP/ADP ratio, influencing cardiac excitability and protection under metabolic stress [26,27]. Mutations in ABCC9 were directly associated with DCM. Initial discoveries highlighted two specific mutations in exon 38 based on a genetic screening of 323 DCM patients. One is a frameshift mutation (c.4570_4572delinsAAAT, p.(Leu1524fs)) and the second one is a missense mutation (c.4537G>A, p.(Ala1513Thr)), both altering KATP channel function in vitro and suggesting a mechanistic link to DCM pathogenesis [28]. Further research identified additional variants: the c.4517_4527del, p.(Arg1506fs) in Mexican DCM patients, and p.(Lys976Ile) and p.(Arg1197Cys) in a Chinese cohort, underscoring the global relevance of these mutations in DCM pathology [28,29,30]. In-depth studies demonstrated that these mutations disrupt the regulatory mechanisms of the KATP channel, leading to aberrant channel gating and impaired response to metabolic changes, which are crucial during cardiac stress [31]. Moreover, a recent case highlighted a loss-of-function variant in ABCC9 associated with severe ventricular arrhythmias and DCM, further validating the significant role of this gene in cardiac electrophysiology and its impact on cardiac disease phenotypes [32]. This evidence suggests that both gain-of-function and loss-of-function mutations in ABCC9 can lead to diverse cardiac abnormalities, from Cantu syndrome to Brugada syndrome, emphasizing the complex role of KATP channels in heart diseases.
ACTN2
The alpha-actinin-2 gene (ACTN2) encodes a crucial sarcomere protein involved in linking and crosslinking actin filaments within the cardiac Z-disc, playing a significant role in the structural and functional integrity of the heart muscle. ACTN2 mutations were associated with various forms of CMPs, including DCM and HCM, underscoring its vital role in cardiac pathophysiology [33,34]. Genomic studies identified several ACTN2 mutations linked to CMPs. Notably, a missense mutation p.(Ala119Thr) was found to segregate with HCM in an Australian family, and additional missense variants p.(Thr495Met), p.(Glu583Ala), p.(Glu628Gly) were identified in other HCM families, suggesting a genetic predisposition to HCM [35]. In the context of DCM, a specific ACTN2 variant p.(Gln9Arg) was associated with early mortality, emphasizing the gene impact on cardiac function [36]. Another study identified a missense variant p.(Leu320Arg) in all affected members of a Chinese family with DCM, further implicating ACTN2 in the familial form of the disease [35,37]. Functional studies using hiPSC-derived cardiomyocytes from a HCM patient carrying a missense variant p.(Thr247Met) demonstrated cellular hypertrophy and sarcomere disorganization. These findings provide compelling evidence in favor of a role of ACTN2 in the HCM etiology [34]. Additionally, recent structural analyses showed that ACTN2 mutations like p.(Ala119Thr), p.(Met228Thr), and p.(Thr247Met) disrupt actin-binding interactions, which are crucial for maintaining Z-disc structural integrity and, by extension, normal cardiac muscle function [38]. Moreover, variants in ACTN2 not only lead to structural CMPs but they have implications also for cardiac arrhythmias, likely through mechanisms involving disrupted ion channel interactions within cardiomyocytes [39]. In summary, ACTN2 mutations are significantly associated with both DCM and HCM with a complex phenotype spectrum.
ANKRD1
The Ankyrin Repeat Domain 1 gene (ANKRD1) encodes the cardiac ankyrin repeat protein (CARP), which is involved in maintaining sarcomere integrity, myofibrillar signaling, and stretch sensing in the heart. This highly conserved protein interacts with other sarcomere proteins such as titin/connectin and myopalladin, playing a pivotal role in cardiomyocyte function [40]. Studies have demonstrated the involvement of ANKRD1 in both HCM and DCM, with distinct genetic underpinnings that suggest a complex mechanism of action. In HCM, specific ANKRD1 mutations, such as p.(Pro52Ala), p.(Thr123Met), and p.(Ile280Val), increase the binding affinity of CARP for titin/connectin and myopalladin. This alteration may disrupt normal myofibrillogenesis and sarcomere function, contributing to the hypertrophic response seen in HCM patients [41]. A study identified significant repressor activity alterations and hypertrophic responses in engineered heart tissues carrying the above mentioned HCM-associated ANKRD1 mutations, supporting the pathogenic involvement of these variants [42]. ANKRD1 appears to be more involved in DCM. ANKRD1 coding regions were sequenced in DCM patients. As a result, a total of eight variants in 9 patients were identified (three of them with family history) [43,44]. Expression studies performed in myoblastoid cell lines resulted in loss of CARP binding with Talin 1 and altered mechanical stretch-based signaling [44]. Mutant CARP proteins transfected in rat neonatal cardiomyocytes led to both significantly less repressor activity and greater phenylephrine-induced hypertrophy, suggesting altered function of CARP mutant protein [43].
CALR3
The calreticulin 3 gene (CALR3), encoding a member of the calcium-binding chaperones localized in the endoplasmic reticulum, has a controversial role in HCM. It is primarily expressed in the reproductive system, and its involvement in CMPs is based on limited and inconclusive evidence. Two heterozygous missense variants, p.(Arg73Gln) and p.(Lys82Arg), were identified in a study involving 252 unrelated HCM patients. However, the presence of multiple potentially disease-causing variants in other genes within the same patient’s cohort did not allow the interpretation of the CALR3 variants in this context [45]. Subsequent studies have sought to clarify the role of CALR3, with one notable study assessing the gene influence in a large Dutch cohort of CMP patients. This study identified 17 rare heterozygous CALR3 variants in 48 probands, and initially supported a higher prevalence of CALR3 variants in CMP patients compared to controls. However, after accounting for a potential Dutch founder variant, the statistical significance disappeared, indicating that CALR3 variants might not uniquely influence CMP risk [46]. Moreover, functional studies did not support a pathogenic role for CALR3 in HCM. Knockout and knockdown studies in mice and zebrafish, respectively, did not demonstrate any cardiac abnormalities, suggesting that the primary functions of CALR3 are not cardiac-related [47,48]. Additionally, there is an absence of calreticulin-3 protein expression in myocardial tissue, further questioning its involvement in direct cardiac functions [46]. Taken together, these findings argue against a significant or direct role of CALR3 in causing HCM as a monogenic determinant. The presence of CALR3 variants in CMP patients appears coincidental rather than causal, given the lack of segregation with the disease in family studies and the absence of functional evidence supporting a detrimental effect on cardiac structure and function.
CDH2
The cadherin 2 gene (CDH2) encodes N-cadherin, an essential protein in cellular adhesion in the brain and muscle tissues, including cardiac muscle. This adhesion is calcium-ion dependent and is critical for the structural integrity and function of the heart, particularly in the context of ARVC [49]. Disruption in the function of N-cadherin can lead to impaired cellular cohesion and stability in the cardiac muscle, potentially triggering or exacerbating the myocardial remodeling typical of ARVC [50]. Initial investigations through WES identified a variant in CDH2, p.(Gln229Pro), in a 3-generation family affected by ARVC. In the same study other 73 genotype-negative ARVC probands were tested with the identification of a likely pathogenic variant in CDH2, p.(Asp407Asn). These variants pointed to a possible role of CDH2 in the disease. However, these findings have not been robustly supported by additional genetic or functional studies [49]. On the other hand, immunohistochemistry studies showed a reduced content of N-cadherin in ARVC patients, suggesting a degradation of cell-cell adhesion integrity, which is crucial for normal cardiac function. These studies underscore the potential significance of CDH2 in ARVC. They also highlight the need for more extensive research to fully understand the implications of CDH2 mutations in this condition [51].
CSRP3
The Cysteine and Glycine Rich Protein 3 gene (CSRP3) encodes a muscle LIM protein that is crucial for myogenesis, maintenance of the myocyte cytoskeleton, mechano-signaling and transduction, and actin cytoskeleton assembly. Based on human and animal studies, CSRP3 is implicated in both HCM and DCM [52,53]. CSRP3 mutations were associated with both DCM and HCM since the early 2000s, with variants like p.(Cys58Gly) found in German HCM families, and p.(Leu44Pro) and p.(Cys150Tyr) variants found significantly enriched in HCM cohorts. These mutations lead to functional disruption in protein studies, suggesting a strong causal relationship with HCM [54,55,56]). Interestingly, the role of CSRP3 in DCM appears to be less direct. Although knockout studies in mice show a phenotype resembling DCM, and several missense mutations in DCM patients have been documented, the overall evidence pointing to a significant role of CSRP3 in DCM is still considered limited [36,57,58,59]. Recent studies also highlight the role of synonymous variants in CSRP3 that might influence mRNA stability, splicing, and miRNA binding sites, which could contribute to the pathogenesis of DCM. However, a functional validation through in vitro and in vivo studies is required to assess the impact of these synonymous mutations in DCM [60]. This complex picture is further complicated by research indicating that CSRP3 functional impact may vary significantly between species. For instance, studies using high-resolution proton magnetic resonance spectroscopy showed that CSRP3 knockout in neonatal cardiomyocytes results in metabolic changes that could predispose to CMP, suggesting a potentially broader part in metabolic regulation within the cardiac disease [60]. Finally, whereas the evidence strongly supports a role for CSRP3 in HCM, its involvement in DCM remains less clear, necessitating further research to clarify its mechanistic contribution and potential as a therapeutic target in CMPs.
CTF1
The cardiotrophin-1 gene (CTF1) encodes a member of the interleukin-6 superfamily that has been implicated in the DCM pathophysiology. CTF1 is known for its potent induction of cardiomyocyte hypertrophy and enhancement of myocyte survival, potentially implicating it in the myocardial responses observed in DCM. Despite initial genetic studies suggested a role of CTF1 variants in DCM, new variants, identified in studies from 2000 onwards, were found prevalent in the general population, generating some confusion when trying to assess a direct contribution to DCM [61,62,63]. Increased expression levels of RNA and CTF1 protein were observed in failing left ventricular myocardium, pointing to a contributory role in heart failure. Immunohistochemistry studies confirmed the elevated presence of CTF1 in cardiac myocytes from failing hearts [64]. These observations suggest that while CTF1 is upregulated in heart failure, its direct causative link to DCM remains elusive due to the lack of conclusive functional evidence and consistent genetic segregation data.
CTNNA3
The Catenin Alpha 3 gene (CTNNA3) encodes αT-catenin, a protein crucial for cellular adhesion in cardiac tissues, particularly through its interactions with N-cadherins and desmosomal cadherins like plakophilin-2 [65]. Whereas αT-catenin is integral to cardiac structure and function, its involvement in ARVC has been difficult to establish due to conflicting evidence [66]. Initial investigations revealed a significant decrease in αT-catenin expression alongside plakophilin-2 in autopsied ARVC patients compared to controls. Furthermore, knocking down CTNNA3 in cardiomyocytes resulted in reduced plakophilin-2 expression, mirroring ARVC’s pathological features [67]. Early research identified a rare p.(Ala689Val) variant in CTNNA3 among Danish ARVC patients. However, this variant was not deemed disease-causing due to the lack of functional validation and segregation analysis [68]. Subsequent screening in a larger ARVC cohort identified two novel heterozygous variants within important domains of αT-catenin, which were proposed to potentially weaken interactions with β-catenin or plakoglobin. However, these findings alone were not sufficient to conclusively link CTNNA3 mutations directly to ARVC [69]. Moreover, the role of CTNNA3 in cardiac pathology is further complicated by its involvement in multiple cellular functions beyond the desmosomal interactions. As observed in knockout mouse models, the lack of this protein regulating gap junction remodeling does not directly cause ARVC, but it rather leads to progressive dilated cardiomyopathy [70].
DTNA
The dystrobrevin gene (DTNA) encodes dystrobrevin-α, a scaffold protein for signaling molecules at the sarcolemma of cardiac muscle. This protein is involved in maintaining the structural integrity of muscle fibers by linking the extracellular matrix to the subsarcolemmal cytoskeleton [71]. DTNA was reported in relation to autosomal dominant DCM in a study analyzing sequence data from 7,855 CMP cases compared to Exome Aggregation Consortium (ExAC) database where DTNA variants were found in DCM patients [72]. This gene appears to associate more frequently to the left-ventricular non-compaction (LNVC) phenotype. Overexpression of the LNVC-associated DTNA p.(Asn49Ser) variant in a mouse heart resembled a phenotype of deep trabeculation, DCM and cardiac dysfunction [71]. There is still a lack of genetic and functional evidence to assess a possible involvement of this gene in DCM.
EYA4
The EYA Transcriptional Coactivator and Phosphatase 4 gene (EYA4) encodes a factor playing a critical role in both embryonic development and inner ear function, but it has also been implicated in cardiovascular disorders, particularly in DCM [73]. This association between EYA4 and cardiac dysfunction has begun to unfold as more genetic and functional studies were conducted. A study identified a heterozygous truncating mutation in EYA4, which presented with sensorineural hearing loss followed by late-onset DCM, suggesting a novel syndromic linkage between these two conditions and underscoring the importance of EYA4 beyond the auditory system with a potential role in cardiac function. Subsequent functional assays, including knockdown experiments in zebrafish, demonstrated that reduced EYA4 expression leads to cardiovascular dysfunction [74]. Further studies using transgenic mouse models elaborated on the mechanisms through which EYA4 mutations could lead to DCM. For instance, overexpression of a rare EYA4 variant in mice revealed a phenotype characteristic of DCM, linked to an overexpression of p27 in cardiomyocytes, suggesting a complex regulatory role of EYA4 in cell cycle control. Additional research hypothesizes the interaction between EYA4 and the Six1 transcription factor, which together regulate crucial pathways involved in cardiac hypertrophy and cardiomyopathy [75].
FHOD3
Formin homology 2 domain-containing 3 gene (FHOD3) encodes one of the formins, a group of proteins that are ubiquitously expressed due to their role in the actin polymerization. In particular, FHOD3 is expressed almost exclusively in the myocardium, where it contributes to the development and maintenance of the thin sarcomere filament [76]. FHOD3 has emerged as one of the non-sarcomere genes implicated in HCM. In fact, few recent studies documented the identification of FHOD3 variants causing HCM [14,77,78,79,80]. The most recent study evaluated FHOD3 variants by NGS in 22806 consecutive unrelated probands from 2014 to 2021 [14]. The FHOD3 p.(Arg637Gln) variant was enriched in this HCM cohort. This variant affects a highly conserved residue localized in a supercoiled alpha helix considered a clustering site for HCM variants; it can act as a HCM predisposing factor in the heterozygous configuration. Additionally, seven patients carrying the p.(Arg637Gln) were homozygous. All but one (unaffected) showed an early presentation and a more severe HCM phenotype. Another study performed a comparative depth-of-coverage strategy by NGS in 5493 HCM probands and 2973-controls [80]. Three deletions involving exons 15-16 were identified in three HCM families. These exons are part of the FHOD3 diaphanous inhibitory domain. The clinical features of the affected individuals were consistent with those reported in previous studies. In addition to deletions caused by structural variants affecting exons 15-16, splice donor site variants in exon 12 that might lead to exon skipping were also observed [81,82]; . This exon seems to be essential for cardiac myosin-binding protein C-mediated localization of the FHOD3 protein to the sarcomere C-zone [83]. Due to the very recent genetic evidence, functional studies demonstrating a FOHD3 impact in HCM pathogenesis are yet to be performed.
GATAD1
The GATA Zinc Finger Domain Containing 1 gene (GATAD1) encodes a ubiquitously expressed protein involved in all stages of embryonic mouse heart development. GATAD1 interacts with chromatin complexes for histone modification that is crucial in heart failure [84,85]. A genetic study identified a missense mutation, p.(Ser102Pro), in GATAD1 within a consanguineous family with autosomal recessive DCM. The variant segregated with the disease, marking a significant advance in understanding its contribution to DCM. Immunohistochemistry analysis illustrated abnormal subcellular and nuclear localization of GATAD1 in DCM-affected individuals, contrasting with its normal distribution in healthy tissues. These observations indicate the potential impact of the mutation on cellular disease mechanisms [86]. Additionally, zebrafish embryos with altered GATAD1 expression displayed cardiac defects, emphasizing the role of the gene in heart structure and function [87].
ILK
The integrin-linked kinase gene (ILK) encodes a major focal adhesion protein essential for maintaining cardiomyocyte shape and ventricular morphology during embryonic development. This protein has been linked to various cardiac diseases, particularly DCM, through its role in cellular signaling and structural integrity [88]. Research has shown that mutations in ILK, such as p.(Ala262Val) and p.(Pro70Leu) found in DCM patients, impact cardiac function by leading to transcript destabilization and nonsense-mediated decay, particularly in model organisms like zebrafish [89]. These studies suggest a genetic predisposition to the pathogenesis of DCM linked to ILK mutations, which disrupt normal cellular processes and may lead to heart failure. Experimental models further underscored the therapeutic potential of ILK in treating heart diseases. In rat models of DCM, for instance, targeted ILK therapy using adenoviral vectors demonstrated a significant reduction in inflammatory cell infiltration, cardiomyocyte degeneration and overall mortality [90]. The interactions of ILK with other proteins, such as LIM-only adaptor PINCH-1 and α-parvin, are crucial for the assembly of focal adhesions, which are complexes that facilitate cell signaling and survival through the activation of the Akt kinase pathway. This pathway is particularly significant in cardiac physiology as it supports cell survival under stress. Mice deficient in ILK or its associated proteins exhibit severe cardiac abnormalities, underscoring the vital role of ILK in maintaining cardiac structure and function [91]. Moreover, the loss of ILK in murine models leads to spontaneous development of cardiomyopathy and heart failure, characterized by a significant disruption in cell adhesion and a decrease in Akt phosphorylation, crucial for cardiac stress response [92].
JPH2
The Junctophilin 2 gene (JPH2) plays a crucial role in cardiac physiology since its product, junctophilin-2, is essential in forming junctional complexes that bridge the plasma membrane and the endoplasmic/sarcoplasmic reticulum. This function is critical for controlling calcium signaling, which is pivotal for normal cardiac contractility and rhythm stability [93]. Functional studies underscored the involvement of JPH2 in various cardiac pathologies, notably HCM and DCM. For instance, induced pluripotent stem cell-derived cardiomyocytes harboring JPH2 mutations exhibit cellular hypertrophy, sarcomere disarray, and arrhythmias [94]. A founder mutation, JPH2:c.482C>A, p.(Thr161Lys), has been observed across several Finnish families, exhibiting autosomal dominant inheritance and co-segregation with HCM, suggesting a robust link between this particular mutation and the disease [95]. Different modes of inheritance can influence the clinical presentation and severity of the cardiac conditions associated with JPH2 mutations. Loss-of-function mutations transmitted via an autosomal recessive pathway are linked to severe, early-onset DCM, characterized by significant cardiac dysfunction starting at a young age. Conversely, autosomal dominant missense mutations are typically associated with a broader spectrum of cardiac abnormalities, including HCM and various arrhythmias, which may also predispose to sudden cardiac death [5,96,97,98].
KLF10
The Krüppel-like factor 10 gene (KLF10) plays a significant role in various biological processes, including TGF-β signaling, which influences cellular proliferation and differentiation. KLF10 is a transcription factor that has been studied primarily for its regulatory function in the immune response and its expression in response to TGF-β signaling. It acts as a potent modulator of cellular growth and has been implicated in myocardial response to stress and pathological remodeling [99]. TGF-β signaling pathways are significantly involved in the myocardial fibrotic process, contributing to the phenotypic expression of hypertrophy seen in HCM patients. This gene has been linked to HCM in a cohort study that discovered six missense variants in six individuals with HCM diagnosis [99,100]. Nevertheless, the mechanisms by which KLF10 may contribute to HCM remain to be fully delineated.
KLHL24
The ubiquitin ligase substrate receptor Kelch-like protein 24 gene (KLHL24) encodes a member of the Kelch-like protein family which acts as substrate-specific adaptor to Cullin E3 ubiquitin ligases. Genome-wide linkage analysis and exome sequencing identified two KLHL24 homozygous mutations, p.(Glu350*) and p.(Arg306His), in two consanguineous HCM families originated from Iraq and Iran, demonstrating the gene involvement in CMP [100]. Endomyocardial and skeletal muscle samples from individuals of both families showed distinct changes, such as the accumulation of desmin intermediate filaments. Additionally, when the zebrafish counterpart klhl24a is reduced, it leads to heart abnormalities like those seen in other genes associated with HCM. A recent study of consanguineous early-onset CMP families from Saudi Arabia reported a 14-year-old with HCM carrying a KLHL24 homozygous loss of function (LOF) variant, p.(Trp387*) [101]. This line of evidence supports a role for KLHL24 LOF variants in HCM, suggesting the inclusion of this gene in diagnostic panels particularly for consanguineous populations [102]. Interestingly, KLHL24 gain-of-function variants are associated with epidermolysis bullosa simplex, a hereditary skin fragility disorder [103]. Of note, in a cohort of 20 patients with epidermolysis bullosa simplex carrying these gain-of-function variants, 40% had DCM and were negative for screening of pathogenic variants in known DCM-associated genes [104]. This evidence provides an example of a gene with distinct LOF versus gain-of-function effects in HCM and DCM, pointing to the diverse roles of KLHL24 in different tissues and diseases [105].
LAMA4
The Laminin Subunit Alpha 4 gene (LAMA4) is implicated in the structural integrity of the heart through the formation of laminins 8 and 9 parts, which are major components of the basement membranes in cardiac tissues. Mutations in LAMA4 have been associated with various cardiovascular abnormalities, including DCM. Mice deficient in LAMA4 exhibited significant cardiovascular defects, such as endothelial disruptions, hemorrhages, and subsequent cardiac hypertrophy, leading to heart failure [106]. This phenotype mirrors the findings reported in human studies where specific mutations like p.(Pro943Leu) and p.(Arg1073X) were identified in DCM patients. These mutations, located within the integrin-interacting domain, disrupt the interaction between laminins and integrin receptors, thereby affecting the cellular adhesion and signal transduction pathways that are critical for cardiac function [89]. The profound impact of LAMA4 on cardiovascular pathology has also been further illustrated in zebrafish models, where LAMA4 knockdown led to severe cardiac dysfunction and hemorrhages, recapitulating some aspects of the human DCM condition [89]. Additionally, the interaction of LAMA4 with ILK, another protein implicated in cardiac disease, suggests a complex network of protein interactions that are vital for maintaining the structural and functional integrity of the heart. Mutations in both LAMA4 and ILK were shown to lead to significant cardiac defects, underlying the importance of their interplay in maintaining cardiac integrity [89].
LDB3
The LIM domain binding 3 gene (LDB3), encoding the protein Cypher or ZASP, plays a crucial role in cardiac and skeletal muscle structure and function. LDB3 is integral to the Z-line of sarcomeres, where it interacts with α-actinin-2 to maintain structural integrity during muscle contraction [107]. Several LDB3 mutations were first identified in patients with late-onset DCM [108,109]. These findings were supported by animal models, where LDB3 knockout in mice led to severe DCM and heart failure, mirroring the human disease phenotype and underscoring the gene functional relevance [110]. Functional investigations in zebrafish models demonstrated that LDB3 knockdown leads to cardiac dilation and significant thinning of the ventricular walls, typical features of DCM [111]. Moreover, a recent study highlighted the severe phenotype associated with homozygous or compound heterozygous LOF variants of LDB3, found in multiple families [112]. In-depth functional analyses in cell models revealed that LDB3 mutations lead to decreased cell viability, increased cardiomyocyte apoptosis, and disruptions in key signaling pathways such as Akt and p38 MAPK in cardiac models. These molecular changes provide a direct link between genetic mutations and the cascade of cellular events leading to DCM [113].
MYH6
The α-cardiac myosin heavy chain gene (MYH6) encodes the α isoform of cardiac myosin heavy chain. The α isoform is abundant in both atria and ventricles during embryogenesis. After birth, the β isoform becomes predominantly expressed [114]. The first genetic evidence of MYH6 involvement in HCM was published in 2002, where a variant p.(Arg795Gln) was found in one HCM patient presenting symptoms after middle age [115]. This study corroborated previous functional data demonstrating that the α isoform transcripts are expressed in adult ventricular myosin (about 30%), though the abundance of αMyHC protein is very low [116]. The authors suggested that lower abundance of the α versus β isoform accounts for the late onset of HCM. Other four missense variants were then identified: one in a patient diagnosed with HCM p.(Q1065H), and the others in three patients diagnosed with DCM p.((P830L, p.(A1004S), p.(E1457K) [117]. The HCM patient had an early-onset severe phenotype with death occurring in the fifth decade of life, while the three DCM patient carriers had a late-onset phenotype. All MYH6 mutations occurred within highly conserved residues and were predicted to change either the structure or the chemical bonds of the protein. Other studies reported additional rare missense variants in DCM patients [118,119]. A study showed that degenerated myocardial cells from HCM patients had immunoreactivity for MYH6. These cells also exhibited cytoplasmic vacuolation with vacuoles occupying a significant proportion of cell volume [120]. Functional studies on MYH6/DCM-associated variants showed altered myocyte contractility in ventricular myocytes from rats [121]. Notwithstanding this evidence, the relatively small amount of αMyHC protein present in healthy left ventricles has called into question the role of MYH6 as a gene contributing to CMP [117].
MYLK2
The light chain kinase 2 gene (MYLK2) encodes a protein expressed in adult skeletal muscle [122]. A few mutations in MYLK2 were reported in association with digenic forms of HCM, where patients also carried additional variants in MYBPC3, MYH7 and FLNC [123,124]. Unfortunately, no segregation data was available to draw any conclusions of a possible pathogenic involvement of these MYLK2 variants. Only one sequencing study associated MYLK2 to DCM phenotype [72]. Moreover, there is a paucity of functional studies investigating the role of this gene in CMPs.
MYOM1
The myomesin 1 gene (MYOM1) encodes a protein located in the M-band which is found in all types of vertebrate striated muscle. The N-terminal domain 1 of myomesin interacts with the a-helical tail domain of Myosin, whereas the myomesin domains 4–6 bind to the C-terminus of titin, stabilizing the contractile apparatus during striated muscle contraction [125]. MYOM1 was first reported in relation to HCM in 2011 [126]. The identified p.(Val1490Ile) variant led to reduced thermal stability of the myomesin My11–13 and My12–13 dimers and a significantly decreased dimerisation affinity. This variant segregated in two affected family members, although the analysis of the other disease-causing genes was not performed. Additional MYOM1 variants were identified in HCM probands, although these variants have high allele frequencies in the Genome Aggregation Database (GnomAD). Moreover, additional variants in other HCM genes were found in the same patients [127,128,129].
MYOZ2
The Myozenin 2 gene (MYOZ2) encodes a protein important for sarcomere organization which interacts directly with crucial signaling pathways in cardiac and skeletal muscles. MYOZ2 interacts with calcineurin, a key phosphatase in calcium-dependent signaling, tethering it to alpha-actinin at the Z-line of cardiac and skeletal muscle cells via the calcium signaling [130]. The role of MYOZ2 as a modifier gene in modulating cardiomyopathy variability has been demonstrated by showing a significant correlation between its expression and myocardial contractile function in mouse congenic strains [131]. Initial studies highlighted the presence of mutations in MYOZ2, such as p.(Ser48Pro) and p.(Ile246Met), which were found to co-segregate with HCM, suggesting a direct genetic link to the disease phenotype [132]. In a 5-multi-generational Chinese family, a missense variant in MYH7 p.(Ala719His) co-segregated with a variant in MYOZ2 p.(Leu169Gly). Individuals carrying both mutations displayed more severe symptoms than those with the MYH7 mutation only, implying a modifying role of MYOZ2 in HCM [133]. These findings are supported by mouse models demonstrating cardiac hypertrophy like human HCM phenotype, even when calcineurin activity is not altered. This evidence indicates that the contribution of MYOZ2 to HCM phenotype may operate through mechanisms independent of this pathway [132,134].
MYPN
The Myopalladin gene (MYPN) encodes a protein which plays a pivotal role in the structural organization of the sarcomere, particularly at the Z-line and I-band, interacting with essential Z-line proteins. It is increasingly clear that mutations in MYPN contribute to both HCM and DCM, affecting cardiac muscle integrity and function through the disruption of sarcomere and myofibrillar architecture [135,136,137]. Functional studies in mice demonstrated that MYPN knockout showed mild cardiac dilation and systolic dysfunction, which mirrors some aspects of the DCM phenotype in humans [138]. Moreover, the broader impact of MYPN mutations has been noted in diverse populations. A study involving Lebanese and Chinese cohorts highlighted the presence of MYPN variations in DCM patients, further emphasizing the role of this gene in CMPs across different ethnicities [139,140].
NEBL
NEBL encodes the cardiac-specific isoform nebulette, a crucial member of the nebulin family, predominantly involved in the structural integrity of the sarcomere within heart muscle. Highly expressed in cardiac muscle, nebulette binds actin, interacts with thin filaments, and associates with Z-line proteins in striated muscle, playing an essential role in cardiac myofibril assembly. It acts as a pivotal link between sarcomere actin and desmin intermediate filaments within the heart muscle sarcomeres, being integral for maintaining heart muscle integrity and functionality [141]. Mutations in NEBL were associated with various cardiac pathologies, including DCM [142]. Functional studies using transgenic mice revealed that NEBL mutations can significantly affect sarcomere ultrastructure, disrupt cellular contractile function, and impair calcium homeostasis, underscoring the gene role in DCM pathogenesis [143,144]. For instance, mutations such as p.(Lys60Asn), p.(Gln128Arg), p.(Gly202Arg), and p.(Ala592Glu), were observed to cause various alterations in cardiac structure and function, ranging from embryonic lethality to progressive heart failure in adulthood [144]. Moreover, a knockout mouse model for NEBL exhibited widened Z-lines and increased expression of cardiac stress markers, suggesting that disease-causing mutations in NEBL likely exert dominant gain-of-function effects [145]. Diversity in phenotypic outcomes were observed across seven patients in a cohort of 389 patients affected by DCM, HCM and LVNC: four missense mutations were identified in DCM and HCM patients and one in a LVNC patient. Whereas HCM and DCM related mutations were within the nebulin-like repeats, responsible for actin binding, the associated LVNC mutation was in the C-terminal serine-rich linker region. This evidence suggests that specific locations and nature of NEBL mutations can variably influence cardiac muscle function leading to a distinct pathological cardiac phenotype [146].
NEXN
The Nexilin gene (NEXN) encodes a protein critical for the structural and functional integrity of cardiac and skeletal muscles. NEXN is involved in regulating the actin cytoskeleton and sarcomere assembly, key components in maintaining cardiomyocyte stability and responding to mechanical stress [147]. Mutations in NEXN were associated with a spectrum of cardiac pathologies, including DCM, HCM, and sudden cardiac death [148,149,150]. Disruption of nexilin can lead to severe cardiac abnormalities. Constitutive knockout models in mice demonstrated that the absence of NEXN leads to rapid progression of cardiomyopathy, characterized by left ventricular dilation, thinning of the ventricular walls, and a decline in cardiac function [148,151]. Genetic investigations revealed the involvement of NEXN in cardiac diseases. For example, a NEXN variant p.(Glu575*) was found together with a novel SCN5A variant in a family with progressive DCM and cardiac arrhythmias, suggesting a synergistic effect of the two mutations on disease pathogenesis [152]. Targeted NGS of 102 genes in Han Chinese patients with idiopathic DCM revealed TTN truncating variants as predominant, followed by variants in LMNA, RBM20, and NEXN, providing molecular diagnosis in 34.7% of patients and highlighting insights into genotype-phenotype correlations [153]. Pediatric and fetal cases further emphasized the impact of NEXN mutations, with conditions ranging from transient to severe DCM and cardiomegaly in patients carrying heterozygous and homozygous mutations [154,155]. Moreover, an evaluation of NEXN variants in patients with cardiomyopathy or sudden cardiac death showed a predominance of DCM, with particularly severe and early-onset phenotypes in those with double NEXN variants [156]. Pathogenic variants were also detected in patients with HCM and sudden infant death syndrome/idiopathic ventricular fibrillation, further complicating the phenotypic spectrum of NEXN mutations.
NKX2-5
The NK2 homeobox 5 (NKX2-5) gene encodes a homeobox transcription factor essential for heart development and function, specifically in regulating genes involved in cardiac morphogenesis, including sarcomere organization and the development of the conduction system [157,158,159]. Mutations in NKX2-5 were linked to various congenital heart diseases, such as atrial septal defects, ventricular septal defects, and anomalies in the conduction system [160,161]. The critical role of NKX2-5 in cardiac health was highlighted in 2013, marking the first association of its mutations with adult-onset DCM. These mutations were suggested to contribute to DCM by altering protein degradation and transcriptional activity, establishing a new avenue for understanding the molecular basis of this disease [162]. Subsequent research in 2014 identified a novel heterozygous mutation, p.(Ser146Trp), in a family displaying an autosomal dominant DCM pattern. This mutation was found among 130 unrelated patients with idiopathic DCM and demonstrated complete penetrance, with the DCM phenotype also associated with arrhythmias. Functional analyses indicated that this variant resulted in significantly reduced transcriptional activity compared to the wild-type protein, suggesting a mechanism through which NKX2-5 mutations disrupt heart function [163]. Further studies identified additional variants in NKX2-5 among patients with sporadic adult-onset DCM. These mutations lead to reduced transcriptional activity and disrupted interactions with key cardiac transcription factors. This evidence supports a model where NKX2-5 mutations contribute to DCM pathogenesis through altered gene regulation and impaired transcriptional networks, highlighting their significant impact on cardiac structural and functional integrity [164,165].
OBSCN
The obscurin gene (OBSCN) encodes a giant sarcomere signaling protein crucial for myofibrillogenesis, cytoskeletal organization, and cell adhesion. These characteristics facilitate the interactions between the sarcoplasmic reticulum and myofibrils, which are essential for the structural integrity and function of muscle cells. The physiological roles of obscurin were initially elucidated through the knockout mouse model, which revealed significant implications in skeletal muscle but less understood effects in cardiac muscle [166,167,168,169]. OBSCN mutations were associated with both HCM and DCM. In HCM, the p.(Arg4344Gln) and p.(Ala4484Thr) variants affect the binding of obscurin with titin domains, which is crucial for the structural organization of the sarcomere. In particular, the p.(Arg4344Gln) mutation was shown to impair obscurin localization to the Z-band, suggesting a potential mechanism by which this mutation contributes to the pathogenesis of HCM [170]. Recent studies also implicated OBSCN mutations in DCM. A study identified five potentially disease-causing OBSCN mutations in patients with familial DCM, suggesting a significant role for obscurin, potentially through mechanisms like haploinsufficiency which affects protein expression levels and disrupts normal cardiac function [171].
PDLIM3
The PDZ And LIM Domain 3 gene (PDLIM3) encodes a protein with both a PDZ domain and a LIM domain, suggesting its potential role in cytoskeletal organization. This protein has been shown to bind to the spectrin-like repeats of alpha-actinin-2 and to co-localize with alpha-actinin-2 at the Z lines in skeletal muscle [172]. This gene is also referred to as ALP. Research involving ALP knockout mice revealed alterations in regional systolic function and hypertrophy, primarily due to its specific expression in the right ventricular outflow tract. These changes suggest a reduction in contractile function and an increase in wall thickness as a response to chronic hypoxia [173]. Variants in PDLIM3 were identified in two unrelated HCM patients. In the specific, a non-synonymous VUS p.(Glu106Ala) was found in one HCM patient [174]. A large deletion involving the first four exons of PDLIM3 was identified in a patient from a cohort of 505 unrelated HCM patients screened for copy number variations (CNVs) [175]. Functional characterization of these variants in HCM phenotype are yet to be performed to further investigate the involvement of this gene in CMPs.
PLEKHM2
The Pleckstrin Homology and RUN Domain Containing M2 gene (PLEKHM2) encodes a protein predominantly involved in lysosomal trafficking [176,177,178]. In a large Bedouin family with severe recessive DCM and LVNC, WES revealed a novel PLEKHM2:c.2156_2157delAG variant, causing a frameshift and exon skipping. PLEKHM2 regulates endosomal trafficking, and this mutation led to abnormal subcellular distribution of endosomes and lysosomes, as well as impaired autophagy flux in patients’ fibroblasts. Transfection with wild-type PLEKHM2 cDNA restored lysosomal distribution, implicating PLEKHM2 in DCM and LVNC pathogenesis via autophagy disruption [179].
PRDM16
The PR-domain-containing-16 gene (PRDM16) encodes a zinc-finger-containing transcription factor that promotes or represses tissue-specific gene expression [180]. PRDM16 is expressed in mouse and human hearts [181,182]. Missense variants in PRDM16 were primarily associated with DCM, whereas nonsense and frameshift mutations were associated with LVNC [183]. Subsequently, PRDM16 has also been associated with pediatric DCM, a disorder that typically remains clinically silent until adulthood. Genetic screening in families with pediatric DCM cases identified a de novo frameshift PRDM16 variant in a proband diagnosed at four months of age [184]. This variant resulted in the addition of an anomalous peptide tail consisting of 48 residues and prematurely truncating the protein product. PRDM16 is predicted to be highly intolerant to LOF. In zebrafish, PRDM16 was shown to have a dominant-positive effect on cardiomyocyte proliferation, with overexpression or knockdown resulting in impaired cardiomyocyte proliferation [183]. On the other hand, functional studies linked this gene to HCM. In fact, PRDM16 KO mice develop cardiac hypertrophy, excessive fibrosis, and mitochondrial and metabolic defects, leading to heart failure as they age [180]. Further studies are therefore essential to assess PRDM16 role in CMPs.
PSEN2
The Presenilin 2 gene (PSEN2) encodes a transmembrane key protein implicated in Alzheimer’s disease (AD), particularly in the inherited forms of the disease. Mutations in PSEN2, along with mutations in PSEN1 and the amyloid precursor protein (APP), are known to cause familial AD. PSEN2, along with PSEN1, plays a critical role in regulating the processing of APP through its involvement in gamma-secretase, an enzyme responsible for cleaving APP and the cleavage of the Notch receptor. PSEN1 and PSEN2 are expressed in the heart with a potential role in cardiac development. Studies conducted in 132 families with DCM and 183 individuals with idiopathic DCM revealed a novel PSEN1 mutation in one family and a single PSEN2 missense mutation in two other families. Importantly, both mutations were found to be present in all clinically affected individuals and showed segregation with DCM and heart failure within these families. Cultured skin fibroblasts from individuals carrying mutations in PSEN1 and PSEN2 exhibited changes in calcium signaling [185]. PSEN2 knockout mice displayed normal cardiac development without hypertrophy or fibrosis. Notably, they exhibited increased cardiac contractility, supported by higher Ca2+ transients and tension in isolated papillary muscles. PSEN2 was found to interact with sorcin and ryanodine receptor 2, both crucial for cardiac function, indicating a role of this gene in excitation-contraction coupling [186].
RPS6KB1
This gene encodes a protein (ribosomal protein S6 kinase beta-1 or S6K1) belonging to the ribosomal S6 kinase family, which comprises serine/threonine kinases. It is activated by mTOR signaling, facilitating protein synthesis, cell growth, and proliferation [187]. Exome and targeted sequencing of 401 Indian HCM patients revealed a novel heterozygous missense variant in RPS6KB1 in two unrelated families p.(Glu47Trp), co-segregating with the phenotype. Replication study in a UK Biobank cardiomyopathy cohort (n=190) identified other two RPS6KB1 heterozygotes variants: p.(Gln49Lys) and p.(Tyr62His). Functional analysis showed that mutant proteins activated signaling cascades, suggesting a gain-of-function effect. Additionally, the same authors observed a RPS6KB1 variant p.(P445S) in a HCM patient from Arabia [188].
RYR2
The ryanodine receptor 2 gene (RYR2) covers 107 exons and encodes a protein involved in calcium signaling in cardiac muscle cells. It is predominantly found in the sarcoplasmic reticulum (SR). RYR2 plays a crucial role in regulating the release of calcium ions from the sarcoplasmic reticulum into the cytoplasm of cardiac muscle cells during excitation-contraction coupling, a process essential for cardiac muscle contraction [189,190,191]. A functional link between RyR2 and cMyBP-C (encoded by MYBPC3) is known, and it suggests a potential mechanistic association between cytosolic soluble cMyBP-C and SR-triggered Ca2+ release through RyR2. This interaction could be clinically significant in CMPs such as HCM [192]. RYR2 was first associated with HCM when a missense mutation p.(Thr1107Met) was identified in an affected proband, co-segregating with asymmetric septal hypertrophy in the family members [193]. Functional evidence showed that the p.(Ala1107Met) mutation raises the level at which calcium release stops and reduces the proportion of released calcium [194]. Three patients with apical hypertrophy carried two missense mutations [p.(Glu3809Gly) and p(.Arg929His)] and one splicing mutation (c.7966-2A>T) in RYR2. The mutations were predicted to be damaging and affecting highly conserved residues, possibly contributing to HCM pathogenesis [195]. Another novel mutation p.(Pro1124Leu) was identified in a HCM patient. Homozygous mice for the variant showed a clinical phenotype like the human patient, with overexpression of calmodulin as a potential hypertrophic mediator [191,196]. A recent study performed in an Indian cohort of 22 patients with CMPs identified three patients as carriers of RYR2 variants of uncertain significance [197].
SGCD
The SGCD gene encodes the δ-sarcoglycan protein. This protein is part of the sarcoglycan complex, consisting of four components, within the larger dystrophin-glycoprotein complex (DGC) that connects the F-actin cytoskeleton to the extracellular matrix. Its highest expression occurs in skeletal and cardiac muscle, and it is involved in maintaining the structural integrity of muscle fibers [198,199]. This gene has been first associated with autosomal recessive Limb Girdle Muscular Dystrophy and then with DCM when a missense variant and a deletion were identified in three DCM patients [200]. Subsequently, the p.(Arg71Thr) variant was identified in two members of a small DCM family in Finland [201]. Functional studies reveal that this mutation exhibits a dominant negative effect on the function of the dystrophin-glycoprotein complex, resulting in myocardial mechanical instability that could contribute to DCM development [202].
TBX20
The T-box transcription Factor 20 gene (TBX20) encodes a member of the T-box family of transcription factors. These factors play crucial roles in the regulation of developmental processes. TBX20 is particularly involved in heart development since it regulates the expression of genes essential for cardiac morphogenesis and function. TBX20 mutations were associated with various congenital heart diseases, highlighting its relevance in cardiac development [203,204,205,206]. The association between TBX20 and DCM was initially documented in 2007 [203]. A novel heterozygous mutation in TBX20 p.(Phe256Ile) was found in a family with autosomal dominant DCM, showing complete penetrance. This variant impaired TBX20 transcriptional activity and reduced the synergistic activation of TBX20 with NKX2-5 or GATA4 [207]. Endomyocardial biopsies obtained from idiopathic DCM patients showed elevated TBX20 mRNA levels, similarly to what reported in DCM rats [208]. A recent study investigated the association between TBX20 truncating variants (TBX20tv) and DCM or LVNC. TBX20tv was found to be enriched in DCM/LVNC cases compared to controls, with strong co-segregation and a non-aggressive clinical phenotype characterized by low incidence of major cardiovascular events [209].
TCAP
This gene spans 2 exons and encodes a protein (titin-cap) found in both striated and cardiac muscle that binds to the titin Z1-Z2 domains. Titin-cap is a substrate of titin kinase, thought to be critical for sarcomere assembly [210]. TCAP mutations were identified in a cohort of 346 HCM and 136 DCM patients. Functional assays revealed that HCM-linked mutations enhanced Tcap interaction with titin and calsarcin-1, whereas DCM-linked mutations impaired interactions with muscle LIM-protein, titin, and calsarcin-1. This evidence suggested a correlation between mutation effects and clinical phenotypes [211]. Among a cohort of 30 unrelated DCM patients, one novel variant was found in a patient. However, no family data were available for the segregation analysis [212]. In 2021, a study of 230 unrelated DCM patients of Vietnamese origin allowed them to identify the p.(Arg158Cys) variant classified as likely pathogenic according to ACMG [213].
TGFB3
The TGFB3 encodes a protein called transforming growth factor beta 3 (TGF-β3), which is a member of the TGF-β superfamily. TGF-β3 is involved in various cellular processes, including cell growth, differentiation, apoptosis, and immune regulation. It plays a critical role in embryonic development, tissue repair, and wound healing [214]. This gene was first associated with ARVC in 1994. The study was conducted when linkage was established with markers on chromosome 14q42.3 within a sizable Italian family, with a critical interval containing 40 known genes. Subsequently, this region was linked to various other Italian families. Sequence analysis of candidate genes within this region was conducted [215,216,217]. A nucleotide substitution (c.-36G>A) was identified in the 5’ UTR of TGFB3, consistently associated with the typical ARVC clinical features in affected family members, as per established diagnostic criteria. Further examination, involving 30 unrelated ARVC patients, conducted using denaturing high-performance liquid chromatography, revealed an additional mutation (c.1723C>T) in the 3’ UTR of one proband [218]. In another study, two coding variants of uncertain significance were detected in two ARVC probands [219]. Functional studies are critical to further establish a possible pathogenic involvement of this gene in ARVC pathophysiology.
TJP1
The zonula occludens-1 gene (TJP1) encodes a scaffolding protein localized at the inter-calated discs of cardiomyocytes. It interacts with key cardiac proteins like connexin43, N-cadherin, αT-catenin, and actin, playing a significant role in cardiac function and structure [220,221]. Initial identification of TJP1 mutations in ARVC was reported in 2018, marking a significant advancement in the understanding of ARVC pathogenesis. Patients cohorts resulting negative for mutations in known ARVC-associated genes were analyzed with WES and led to the identification of TJP1 variants, such as p.(Arg265Trp) in Italian cohorts and p.(Ser329Leu) and p.(Asp360Val) in a Dutch/German cohort. These variants, particularly the p.(Tyr669Cys), are located in critical regions of the gene and are predicted to be deleterious [220,221]. Molecular dynamic simulations suggested that these mutations could significantly impact the structural stability and functional interaction of the protein within the cardiac tissue [222].
TNNI3K
The troponin I interacting kinase protein was first identified as a cardiac-specific protein kinase that interacts with cardiac Troponin I [223]. The overall domain structure of TNNI3K resembles that of ILK (see above), suggesting similar functions. TNNI3K is suggested to probably interact with additional sarcomere proteins such as cardiac α-actin and myosin binding protein C, proposing a role for TNNI3K in the modulation of sar-comere function through interactions with key components of the sarcomere complex [224]. The first association of TNNI3K with DCM was reported in a study of a multi-generational family having a particular cardiac phenotype characterized by variably expressed atrial tachyarrhythmia, conduction system disease and DCM [225]. Linkage analysis and WES were used to identify a novel TNNI3K variant that resulted in abnormal peptide aggregation in vitro. Of note, ventricular tissue obtained from a mutation carrier displayed histopathological features of DCM. In parallel, a reduced TNNI3K protein staining with unique amorphous nuclear and sarcoplasmic inclusions was observed. Other two important studies reported TNNI3K novel variants in families with DCM phenotype, segregating with the disease [224,226]. In one study, conducted on three different families with phenotype predominantly consisting of supraventricular tachycardia occurring together with cardiac conduction disease and/or DCM, the same TNNI3K variant was identified, p.(Glu768Lys). It co-segregated with disease features in all affected individuals (n= 23) from all three families [226]. This variant displayed enhanced kinase activity consistent with previous mouse studies that demonstrated increased conduction indices and cardiomyopathy development with increased TNNI3K levels [224]. In another study, a splice site variant (c.333+2 T>C) was identified and co-segregated with the disease in affected family members. The variant was predicted to result in a premature stop codon falling in exon 4 and to induce nonsense-mediated mRNA decay. Real-time qPCR also confirmed that TNNI3K mRNA expression decreased significantly compared with the controls [227]. Lastly, an increased burden of rare coding TNNI3K variants in DCM patients was reported, with two additional new likely pathogenic TNNI3K variants associated with increased autophosphorylation, suggesting that enhanced autophosphorylation might be the pathogenic mechanism caused by TNNI3K variants [228].
TRIM63
The tripartite motif-containing protein 63 gene (TRIM63) encodes the E3 ubiquitin-protein ligase, also known as muscle-specific RING finger protein 1, which is involved in the degradation of sarcomere proteins through ubiquitylation [229]. This protein is crucial in maintaining cardiac muscle integrity and function. Homozygous or compound heterozygous variants in TRIM63 were associated to HCM. Evaluations of affected families showed that only homozygous and compound heterozygous carriers exhibited signs of the disease, while heterozygous carriers remained healthy. A study proposed a modifier role for heterozygous missense variants in patients with HCM [230]. Consistent with a LOF mechanism for TRIM63 variants, mice with double knockouts for both TRIM63 and TRIM55 exhibited severe cardiac hypertrophy [13,231]. One study found that 15 cases of HCM were linked to rare homozygous or compound heterozygous variants in TRIM63, and these cases often presented with left ventricular hypertrophy and, in some instances, left ventricular systolic dysfunction. Additionally, these patients frequently exhibited non-sustained ventricular tachycardia, and some suffered adverse cerebrovascular events [13]. Further studies confirmed TRIM63 as an uncommon cause of HCM inherited in an autosomal-recessive manner, associated with significant cardiac manifestations including concentric left ventricular hypertrophy and a high rate of left ventricular dysfunction [232].
VCL
The vinculin gene (VCL) encodes a mechanosensitive protein incorporated in Z-disks, of which two splicing isoforms are produced. Metavinculin is 68 residues longer than vinculin, with exon 19 additionally spliced in. Both isoforms are expressed in the human heart. Few studies reported variants in VCL associated with DCM and HCM [233,234]. One study sequenced VCL exon 19 only in 350 DCM patients and detected two VCL variants (one missense and one inframe deletion) in three patients. Viscometry analysis and electron microscopy demonstrated that both variants altered actin filament organization [235]. In addition, the missense variant also causes disruption of the intercalated disks. The missense variant segregated in a DCM family together with another candidate variant in MYBPC3. In 2020, a large cohort-based study performed in 2538 patients highlighted a significant enrichment of predicted protein-truncating and missense VCL variants compared with the ExAC population database [236]. A second large cohort-based study demonstrated again significant enrichment of rare, predicted protein-truncating VCL variants in DCM patients [237]. Functional studies were performed in mice, zebrafish, and Drosophila, and they supported a role of VCL in myocardial function pointing to LOF as a disease mechanism [238,239,240,241,242,243,244]. VCL variants were associated with HCM. The latter finding is based on the role of VCL in maintaining cellular structure and transmitting mechanical forces necessary for normal heart function. In fact, when these functions are disrupted by mutations, VCL may contribute to HCM. Reduced VCL protein expression is lethal in germline homozygous knockout mice and leads to stress-induced HCM in heterozygotes. An induced pluripotent stem cell line was recently generated from a DCM patient carrying a heterozygous VCL variant and was differentiated into cardiomyocytes. The latter might serve as a disease model to better understand the molecular mechanisms and pathogenesis of VCL in DCM [245].
3. Gene-Disease Validity and Clinical Evidence
Based on the evaluated sources (ClinGen, Genereview, EMQN, and Expert Consensus Statement), the genes listed in table 1 are considered definitive for their respective pathological conditions. The table 2 provides an overview of the minor genes and highlights how different sources classify genes in relation to their association with specific genetic diseases. Despite some discrepancies, there is generally a consensus on assessing the definitive role of these genes, supported by current evidence. This joint evaluation from various sources provides a clear and updated view of the gene-disease validity for CMPs.In Table 3, the counts of VUS, likely pathogenic (LP), and pathogenic (P) variants reported in ClinVar for these minor genes are detailed (entry May 2024). Notably, few genes such as KLF10 and OBSCN have very few recorded entries, while other genes like RPS6KB1 and TJP1 have no entries at all. In Figure 1, the charts show the distribution of variants for ARVC, DCM and HCM. In ARVC, CTNNA3 has the highest total number of variants, predominantly VUS. CDH2 and TGFB3 show a notable number of pathogenic variants. In DCM, ABCC9 and MYH6 have a high total number of variants, predominantly VUS. MYPN and TCAP have a significant number of pathogenic variants. Instead, in HCM genes like MYH6 and MYOM1 have the highest total number of variants, with a predominance of VUS. Genes such as ALPK3 and MYPN show a significant number of pathogenic variants.
4. Conclusions
CMPs are genetically heterogeneous diseases, meaning that they can be caused by mutations in multiple genes (such as double mutations or polygenic risk score) [246,247], some of which may have minor or additive effects. The minor genes discussed in this review may not be the primary drivers of disease, such as MYH7 and TNNT2 in HCM or DSP and PKP2 in ARVC, but their involvement may contribute to the clinical phenotype and influence the prognosis. For instance, a mutation in a minor gene might not cause a clinically observable effect when present alone, but, when combined with another pathogenic variant, can exert a synergistic effect leading to a more severe form of cardiomyopathy [10,248,249]. For this reason, we felt the need to report all current knowledge on these less-understood genetic factors involved in CMPs. Future studies are needed to clearly assess their role. Whereas current guidelines provide a well-established framework for genetic testing in inherited CMPs and arrhythmias, the increasing knowledge on minor genes may lead to their practical use in clinical applications.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used Conceptualization, C.M. and F.P.; methodology, S.P, S.R., M.P.; software, C.S., M.F., S.C., A.G.; validation, A.P., V.V. and S.P.; formal analysis, C.S.; investigation, M.P.; resources, C.M.; data curation, C.S.; writing—original draft preparation, C.M.; writing—review and editing, S.R.; visualization, S.C.; supervision, S.R.; project administration, M.P.; funding acquisition, S.R. All authors have read and agreed to the submitted version of the manuscript.
Funding
This work was partially funded by Progetto PRIN 2022 (from the Italian Ministry of Instruction, University and Research, no.2022E75TWB) to S.R. and S.N. The research leading to these results has also received funding from the European Union-NextGenerationEU through the Italian Ministry of University and Research under PNRR–M4C2-I1.3 Project PE_00000019 “HEAL ITALIA” to S.R., CUP B53C22004000006. The views and opinions expressed are those of the authors only and do not necessarily reflect those of the European Union or the European Commission. Neither the European Union nor the European Commission can be held responsible for them.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The authors are available to share the dataset collected for this review article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Wilcox JE, Hershberger RE. Genetic cardiomyopathies. Curr Opin Cardiol. 2018;33: 354–362. [CrossRef]
- Walsh, R.; Mazzarotto, F.; Whiffin, N.; Buchan, R.; Midwinter, W.; Wilk, A.; Li, N.; Felkin, L.; Ingold, N.; Govind, R.; et al. Quantitative approaches to variant classification increase the yield and precision of genetic testing in Mendelian diseases: the case of hypertrophic cardiomyopathy. Genome Med. 2019, 11, 1–18. [CrossRef]
- Aiyer, S.; Kalutskaya, E.; Agdamag, A.C.; Tang, W.H.W. Genetic Evaluation and Screening in Cardiomyopathies: Opportunities and Challenges for Personalized Medicine. J. Pers. Med. 2023, 13, 887. [CrossRef]
- Ingles, J.; Goldstein, J.; Thaxton, C.; Caleshu, C.; Corty, E.W.; Crowley, S.B.; Dougherty, K.; Harrison, S.M.; McGlaughon, J.; Milko, L.V.; et al. Evaluating the Clinical Validity of Hypertrophic Cardiomyopathy Genes. Circ. Genom. Precis. Med. 2019, 12, e002460–e002460. [CrossRef]
- Jordan, E.; Peterson, L.; Ai, T.; Asatryan, B.; Bronicki, L.; Brown, E.; Celeghin, R.; Edwards, M.; Fan, J.; Ingles, J.; et al. Evidence-Based Assessment of Genes in Dilated Cardiomyopathy. Circulation 2021, 144, 7–19. [CrossRef]
- Garcia, J.; Tahiliani, J.; Johnson, N.M.; Aguilar, S.; Beltran, D.; Daly, A.; Decker, E.; Haverfield, E.; Herrera, B.; Murillo, L.; et al. Clinical Genetic Testing for the Cardiomyopathies and Arrhythmias: A Systematic Framework for Establishing Clinical Validity and Addressing Genotypic and Phenotypic Heterogeneity. Front. Cardiovasc. Med. 2016, 3, 20. [CrossRef]
- de Oca, M.M.; Varela, M.V.L.; Jardim, J.; Stirvulov, R.; Surmont, F. Bronchodilator treatment for COPD in primary care of four Latin America countries: The multinational, cross-sectional, non-interventional PUMA study. Pulm. Pharmacol. Ther. 2016, 38, 10–16. [CrossRef]
- Vimalanathan, A.K.; Ehler, E.; Gehmlich, K. Genetics of and pathogenic mechanisms in arrhythmogenic right ventricular cardiomyopathy. Biophys. Rev. 2018, 10, 973–982. [CrossRef]
- Mahmaljy H, Yelamanchili VS, Singhal M. Dilated Cardiomyopathy. StatPearls. Treasure Island (FL): StatPearls Publishing; 2023.
- McNally EM, Mestroni L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ Res. 2017;121: 731–748. [CrossRef]
- Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet. 2013;381: 242–255.
- Abbas, M.T.; Ali, N.B.; Farina, J.M.; Mahmoud, A.K.; Pereyra, M.; Scalia, I.G.; Kamel, M.A.; Barry, T.; Lester, S.J.; Cannan, C.R.; et al. Role of Genetics in Diagnosis and Management of Hypertrophic Cardiomyopathy: A Glimpse into the Future. Biomedicines 2024, 12, 682. [CrossRef]
- Salazar-Mendiguchía, J.; Ochoa, J.P.; Palomino-Doza, J.; Domínguez, F.; Díez-López, C.; Akhtar, M.; Ramiro-León, S.; Clemente, M.M.; Pérez-Cejas, A.; Robledo, M.; et al. Mutations in cause an autosomal-recessive form of hypertrophic cardiomyopathy. Heart 2020, 106, 1342–1348. [CrossRef]
- Piqueras-Flores J, Villacorta-Argüelles E, Galvin J, Climent-Payá V, Escobar-López LE, Amor-Salamanca A, et al. Intermediate-effect size p.Arg637Gln in increases risk of HCM and is associated with an aggressive phenotype in homozygous carriers. J Med Genet. 2024;61: 423–427. [CrossRef]
- Hayesmoore, J.B.; Bhuiyan, Z.A.; Coviello, D.A.; du Sart, D.; Edwards, M.; Iascone, M.; Morris-Rosendahl, D.J.; Sheils, K.; van Slegtenhorst, M.; Thomson, K.L. EMQN: Recommendations for genetic testing in inherited cardiomyopathies and arrhythmias. Eur. J. Hum. Genet. 2023, 31, 1003–1009. [CrossRef]
- Homepage. In: EMQN [Internet]. 29 Jan 2024 [cited 17 Jul 2024]. Available: https://www.emqn.org/.
- Clinical Genome Resource. Welcome to ClinGen. [cited 17 Jul 2024]. Available: https://clinicalgenome.org/.
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.G.; Vatta, M.; Ware, S.M.; et al. Genetic evaluation of cardiomyopathy: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2018, 20, 899–909. [CrossRef]
- ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 3 March 2020).
- National Center for Biotechnology Information. [cited 17 Jul 2024]. Available: https://www.ncbi.nlm.nih.gov/.
- National Library of Medicine - National Institutes of Health. 1993 [cited 17 Jul 2024]. Available: https://www.nlm.nih.gov/.
- National Institutes of Health (NIH). In: National Institutes of Health (NIH) [Internet]. [cited 17 Jul 2024]. Available: https://www.nih.gov/.
- Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, et al. GeneReviews®. 2024 [cited 17 Jul 2024]. Available: https://www.ncbi.nlm.nih.gov/books/NBK1116/.
- Home - OMIM. [cited 17 Jul 2024]. Available: https://www.omim.org/.
- Wilde AAM, Semsarian C, Márquez MF, Sepehri Shamloo A, Ackerman MJ, Ashley EA, et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the State of Genetic Testing for Cardiac Diseases. Heart Rhythm. 2022;19: e1–e60.
- Kane, G.C.; Liu, X.-K.; Yamada, S.; Olson, T.M.; Terzic, A. Cardiac KATP channels in health and disease. J. Mol. Cell. Cardiol. 2005, 38, 937–943. [CrossRef]
- Solbach, T.F.; König, J.; Fromm, M.F.; Zolk, O. ATP-Binding Cassette Transporters in the Heart. Trends Cardiovasc. Med. 2005, 16, 7–15. [CrossRef]
- Bienengraeber, M.; Olson, T.M.; A Selivanov, V.; Kathmann, E.C.; O’Cochlain, F.; Gao, F.; Karger, A.B.; Ballew, J.D.; Hodgson, D.M.; Zingman, L.V.; et al. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat. Genet. 2004, 36, 382–387. [CrossRef]
- Carnevale, A.; Rosas-Madrigal, S.; Rosendo-Gutiérrez, R.; López-Mora, E.; Romero-Hidalgo, S.; Avila-Vazzini, N.; Jacobo-Albavera, L.; Domínguez-Pérez, M.; Vargas-Alarcón, G.; Pérez-Villatoro, F.; et al. Genomic study of dilated cardiomyopathy in a group of Mexican patients using site-directed next generation sequencing. Mol. Genet. Genom. Med. 2020, 8. [CrossRef]
- Shen, C.; Xu, L.; Sun, X.; Sun, A.; Ge, J. Genetic variants in Chinese patients with sporadic dilated cardiomyopathy: a cross-sectional study. Ann. Transl. Med. 2022, 10, 129–129. [CrossRef]
- Fahrenbach, J.P.; Stoller, D.; Kim, G.; Aggarwal, N.; Yerokun, B.; Earley, J.U.; Hadhazy, M.; Makielski, J.C.; McNally, E.M.; Shi, N.-Q. Abcc9is required for the transition to oxidative metabolism in the newborn heart. FASEB J. 2014, 28, 2804–2815. [CrossRef]
- Zaytseva, A.; Tulintseva, T.; Fomicheva, Y.; Mikhailova, V.; Treshkur, T.; Kostareva, A. Case Report: Loss-of-Function ABCC9 Genetic Variant Associated With Ventricular Fibrillation. Front. Genet. 2022, 13, 718853. [CrossRef]
- Lornage, X.; Romero, N.B.; Grosgogeat, C.A.; Malfatti, E.; Donkervoort, S.; Marchetti, M.M.; Neuhaus, S.B.; Foley, A.R.; Labasse, C.; Schneider, R.; et al. ACTN2 mutations cause "Multiple structured Core Disease" (MsCD). Acta Neuropathol. 2019, 137, 501–519. [CrossRef]
- Prondzynski, M.; Lemoine, M.D.; Zech, A.T.; Horváth, A.; Di Mauro, V.; Koivumäki, J.T.; Kresin, N.; Busch, J.; Krause, T.; Krämer, E.; et al. Disease modeling of a mutation in α-actinin 2 guides clinical therapy in hypertrophic cardiomyopathy. EMBO Mol. Med. 2019, 11, e11115. [CrossRef]
- Chiu C, Bagnall RD, Ingles J, Yeates L, Kennerson M, Donald JA, et al. Mutations in alpha-actinin-2 cause hypertrophic cardiomyopathy: a genome-wide analysis. J Am Coll Cardiol. 2010;55: 1127–1135.
- Mohapatra, B.; Jimenez, S.; Lin, J.H.; Bowles, K.R.; Coveler, K.J.; Marx, J.G.; A Chrisco, M.; Murphy, R.T.; Lurie, P.R.; Schwartz, R.J.; et al. Mutations in the muscle LIM protein and α-actinin-2 genes in dilated cardiomyopathy and endocardial fibroelastosis. Mol. Genet. Metab. 2003, 80, 207–215. [CrossRef]
- Fan, L.-L.; Huang, H.; Jin, J.-Y.; Li, J.-J.; Chen, Y.-Q.; Xiang, R. Whole-Exome Sequencing Identifies a Novel Mutation (p.L320R) of Alpha-Actinin 2 in a Chinese Family with Dilated Cardiomyopathy and Ventricular Tachycardia. Cytogenet. Genome Res. 2019, 157, 148–152. [CrossRef]
- Atang, A.E.; Rebbeck, R.T.; Thomas, D.D.; Avery, A.W. Cardiomyopathy-associated variants alter the structure and function of the α-actinin-2 actin-binding domain. Biochem. Biophys. Res. Commun. 2023, 670, 12–18. [CrossRef]
- Hou, C.R.; Cortez, D. Novel ACTN2 missense variant is associated with idiopathic ventricular fibrillation: a case report. Eur. Hear. J. - Case Rep. 2022, 6, ytac229. [CrossRef]
- Zhang, N.; Xie, X.-J.; Wang, J.-A. Multifunctional protein: cardiac ankyrin repeat protein. J Zhejiang Univ Sci 2016, 17, 333–341. [CrossRef]
- Arimura, T.; Bos, J.M.; Sato, A.; Kubo, T.; Okamoto, H.; Nishi, H.; Harada, H.; Koga, Y.; Moulik, M.; Doi, Y.L.; et al. Cardiac Ankyrin Repeat Protein Gene (ANKRD1) Mutations in Hypertrophic Cardiomyopathy. Circ. 2009, 54, 334–342. [CrossRef]
- Crocini, C.; Arimura, T.; Reischmann, S.; Eder, A.; Braren, I.; Hansen, A.; Eschenhagen, T.; Kimura, A.; Carrier, L. Impact of ANKRD1 mutations associated with hypertrophic cardiomyopathy on contraction parameters of engineered heart tissue. Basic Res. Cardiol. 2013, 108, 349. [CrossRef]
- Duboscq-Bidot, L.; Charron, P.; Ruppert, V.; Fauchier, L.; Richter, A.; Tavazzi, L.; Arbustini, E.; Wichter, T.; Maisch, B.; Komajda, M.; et al. Mutations in the ANKRD1 gene encoding CARP are responsible for human dilated cardiomyopathy. Eur. Hear. J. 2009, 30, 2128–2136. [CrossRef]
- Moulik, M.; Vatta, M.; Witt, S.H.; Arola, A.M.; Murphy, R.T.; McKenna, W.J.; Boriek, A.M.; Oka, K.; Labeit, S.; Bowles, N.E.; et al. ANKRD1, the Gene Encoding Cardiac Ankyrin Repeat Protein, Is a Novel Dilated Cardiomyopathy Gene. Circ. 2009, 54, 325–333. [CrossRef]
- Chiu, C.; Tebo, M.; Ingles, J.; Yeates, L.; Arthur, J.W.; Lind, J.M.; Semsarian, C. Genetic screening of calcium regulation genes in familial hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2007, 43, 337–343. [CrossRef]
- Verhagen, J.M.A.; Veldman, J.H.; van der Zwaag, P.A.; von der Thüsen, J.H.; Brosens, E.; Christiaans, I.; Dooijes, D.; Enden, A.T.J.M.H.-V.D.; Deprez, R.H.L.; Michels, M.; et al. Lack of evidence for a causal role of CALR3 in monogenic cardiomyopathy. Eur. J. Hum. Genet. 2018, 26, 1603–1610. [CrossRef]
- Ikawa, M.; Tokuhiro, K.; Yamaguchi, R.; Benham, A.M.; Tamura, T.; Wada, I.; Satouh, Y.; Inoue, N.; Okabe, M. Calsperin Is a Testis-specific Chaperone Required for Sperm Fertility. J. Biol. Chem. 2011, 286, 5639–5646. [CrossRef]
- Hung, I.-C.; Cherng, B.-W.; Hsu, W.-M.; Lee, S.-J. Calnexin is required for zebrafish posterior lateral line development. Int. J. Dev. Biol. 2013, 57, 427–438. [CrossRef]
- Mayosi, B.M.; Fish, M.; Shaboodien, G.; Mastantuono, E.; Kraus, S.; Wieland, T.; Kotta, M.-C.; Chin, A.; Laing, N.; Ntusi, N.B.; et al. Identification of Cadherin 2 ( CDH2 ) Mutations in Arrhythmogenic Right Ventricular Cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10. [CrossRef]
- Tiwari P, Mrigwani A, Kaur H, Kaila P, Kumar R, Guptasarma P. Structural-Mechanical and Biochemical Functions of Classical Cadherins at Cellular Junctions: A Review and Some Hypotheses. Adv Exp Med Biol. 2018;1112: 107–138. [CrossRef]
- Kwon, Y.-S.; Park, T.I.; Cho, Y.; Bae, M.H.; Kim, S. Clinical usefulness of immunohistochemistry for plakoglobin, N-cadherin, and connexin-43 in the diagnosis of arrhythmogenic right ventricular cardiomyopathy. Int J Clin Exp Pathol. 2013, 6, 2928–35.
- Vafiadaki, E.; Arvanitis, D.A.; Sanoudou, D. Muscle LIM Protein: Master regulator of cardiac and skeletal muscle functions. Gene 2015, 566, 1–7. [CrossRef]
- Liang, S.; Zhou, Y.; Chang, Y.; Li, J.; Zhang, M.; Gao, P.; Li, Q.; Yu, H.; Kawakami, K.; Ma, J.; et al. A novel gene-trap line reveals the dynamic patterns and essential roles of cysteine and glycine-rich protein 3 in zebrafish heart development and regeneration. Cell. Mol. Life Sci. 2024, 81, 1–14. [CrossRef]
- Geier, C.; Gehmlich, K.; Ehler, E.; Hassfeld, S.; Perrot, A.; Hayess, K.; Cardim, N.; Wenzel, K.; Erdmann, B.; Krackhardt, F.; et al. Beyond the sarcomere: CSRP3 mutations cause hypertrophic cardiomyopathy. Hum. Mol. Genet. 2008, 17, 2753–2765. [CrossRef]
- Walsh, R.; Buchan, R.; Wilk, A.; John, S.; Felkin, L.E.; Thomson, K.L.; Chiaw, T.H.; Loong, C.C.W.; Pua, C.J.; Raphael, C.; et al. Defining the genetic architecture of hypertrophic cardiomyopathy: re-evaluating the role of non-sarcomeric genes. Eur. Hear. J. 2017, 38, 3461–3468. [CrossRef]
- Salazar-Mendiguchía, J.; Barriales-Villa, R.; Lopes, L.R.; Ochoa, J.P.; Rodríguez-Vilela, A.; Palomino-Doza, J.; Larrañaga-Moreira, J.M.; Cicerchia, M.; Cárdenas-Reyes, I.; García-Giustiniani, D.; et al. The p.(Cys150Tyr) variant in CSRP3 is associated with late-onset hypertrophic cardiomyopathy in heterozygous individuals. Eur. J. Med Genet. 2020, 63, 104079. [CrossRef]
- Arber, S.; Hunter, J.J.; Ross, J., Jr.; Hongo, M.; Sansig, G.; Borg, J.; Perriard, J.-C.; Chien, K.R.; Caroni, P. MLP-Deficient Mice Exhibit a Disruption of Cardiac Cytoarchitectural Organization, Dilated Cardiomyopathy, and Heart Failure. Cell 1997, 88, 393–403. [CrossRef]
- Hershberger RE, Parks SB, Kushner JD, Li D, Ludwigsen S, Jakobs P, et al. Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy. Clin Transl Sci. 2008;1: 21–26. [CrossRef]
- Zimmerman, R.S.; Cox, S.; Lakdawala, N.K.; Cirino, A.; Mancini-Dinardo, D.; Clark, E.; Leon, A.; Duffy, E.; White, E.; Baxter, S.; et al. A novel custom resequencing array for dilated cardiomyopathy. Genet Med. 2010, 12, 268–278. [CrossRef]
- Giri, P.; Jain, D.; Kumar, A.; Mohapatra, B. Identification and in silico characterization of CSRP3 synonymous variants in dilated cardiomyopathy. Mol. Biol. Rep. 2023, 50, 4105–4117. [CrossRef]
- Erdmann, J.; Hassfeld, S.; Kallisch, H.; Fleck, E.; Regitz-Zagrose, V. Genetic variants in the promoter (g983G>T) and coding region (A92T) of the human cardiotrophin-1 gene (CTF1) in patients with dilated cardiomyopathy. Hum. Mutat. 2000, 16, 448. [CrossRef]
- Pugh, T.J.; Kelly, M.A.; Gowrisankar, S.; Hynes, E.; Seidman, M.A.; Baxter, S.M.; Bowser, M.; Harrison, B.; Aaron, D.; Mahanta, L.M.; et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med. 2014, 16, 601–608. [CrossRef]
- Akinrinade, O.; Ollila, L.; Vattulainen, S.; Tallila, J.; Gentile, M.; Salmenperä, P.; Koillinen, H.; Kaartinen, M.; Nieminen, M.S.; Myllykangas, S.; et al. Genetics and genotype–phenotype correlations in Finnish patients with dilated cardiomyopathy. Eur. Hear. J. 2015, 36, 2327–2337. [CrossRef]
- Zolk, O.; Ng, L.L.; O’brien, R.J.; Weyand, M.; Eschenhagen, T. Augmented Expression of Cardiotrophin-1 in Failing Human Hearts Is Accompanied by Diminished Glycoprotein 130 Receptor Protein Abundance. Circulation 2002, 106, 1442–1446. [CrossRef]
- Goossens S, Janssens B, Bonné S, De Rycke R, Braet F, van Hengel J, et al. A unique and specific interaction between alphaT-catenin and plakophilin-2 in the area composita, the mixed-type junctional structure of cardiac intercalated discs. J Cell Sci. 2007;120: 2126–2136. [CrossRef]
- van Tintelen, J.; Entius, M.M.; Bhuiyan, Z.A.; Jongbloed, R.; Wiesfeld, A.C.; Wilde, A.A.; van der Smagt, J.; Boven, L.G.; Mannens, M.M.; van Langen, I.M.; et al. Plakophilin-2 Mutations Are the Major Determinant of Familial Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Circulation 2006, 113, 1650–1658. [CrossRef]
- Hung, P.-F.; Chung, F.-P.; Hung, C.-L.; Lin, Y.-J.; Kuo, T.-T.; Liao, J.-N.; Chen, Y.-Y.; Pan, C.-H.; Shaw, K.-P.; Chen, S.-A. Decreased Expression of Plakophilin-2 and αT-Catenin in Arrhythmogenic Right Ventricular Cardiomyopathy: Potential Markers for Diagnosis. Int. J. Mol. Sci. 2022, 23, 5529. [CrossRef]
- Christensen, A.H.; Benn, M.; Tybjærg-Hansen, A.; Haunso, S.; Svendsen, J.H. Screening of Three Novel Candidate Genes in Arrhythmogenic Right Ventricular Cardiomyopathy. Genet. Test. Mol. Biomarkers 2011, 15, 267–271. [CrossRef]
- van Hengel J, Calore M, Bauce B, Dazzo E, Mazzotti E, De Bortoli M, et al. Mutations in the area composita protein αT-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2013;34: 201–210. [CrossRef]
- Li J, Goossens S, van Hengel J, Gao E, Cheng L, Tyberghein K, et al. Loss of αT-catenin alters the hybrid adhering junctions in the heart and leads to dilated cardiomyopathy and ventricular arrhythmia following acute ischemia. J Cell Sci. 2012;125: 1058–1067. [CrossRef]
- Cao, Q.; Shen, Y.; Liu, X.; Yu, X.; Yuan, P.; Wan, R.; Liu, X.; Peng, X.; He, W.; Pu, J.; et al. Phenotype and Functional Analyses in a Transgenic Mouse Model of Left Ventricular Noncompaction Caused by a DTNA Mutation. Int. Hear. J. 2017, 58, 939–947. [CrossRef]
- Walsh, R.; Thomson, K.L.; Ware, J.S.; Funke, B.H.; Woodley, J.; McGuire, K.J.; Mazzarotto, F.; Blair, E.; Seller, A.; Taylor, J.C.; et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 2017, 19, 192–203. [CrossRef]
- Abe, S.; Takeda, H.; Nishio, S.-Y.; Usami, S.-I. Sensorineural hearing loss and mild cardiac phenotype caused by an EYA4 mutation. Hum. Genome Var. 2018, 5, 23. [CrossRef]
- Schönberger, J.; Wang, L.; Shin, J.T.; Kim, S.D.; Depreux, F.F.S.; Zhu, H.; Zon, L.; Pizard, A.; Kim, J.B.; A MacRae, C.; et al. Mutation in the transcriptional coactivator EYA4 causes dilated cardiomyopathy and sensorineural hearing loss. Nat. Genet. 2005, 37, 418–422. [CrossRef]
- Williams, T.; Hundertmark, M.; Nordbeck, P.; Voll, S.; Muehlfelder, M.; Schraut, S.; Elsner, I.; Schoenberger, J.; Ritter, O. Abstract 080: Eya4 Induces Hypertrophy Via Regulation Of p27kip1. Circ Cardiovasc Genet. 2013, 113. [CrossRef]
- Ushijima, T.; Fujimoto, N.; Matsuyama, S.; Kan-O, M.; Kiyonari, H.; Shioi, G.; Kage, Y.; Yamasaki, S.; Takeya, R.; Sumimoto, H. The actin-organizing formin protein Fhod3 is required for postnatal development and functional maintenance of the adult heart in mice. J. Biol. Chem. 2018, 293, 148–162. [CrossRef]
- Ochoa, J.P.; Sabater-Molina, M.; García-Pinilla, J.M.; Mogensen, J.; Restrepo-Córdoba, A.; Palomino-Doza, J.; Villacorta, E.; Martinez-Moreno, M.; Ramos-Maqueda, J.; Zorio, E.; et al. Formin Homology 2 Domain Containing 3 (FHOD3) Is a Genetic Basis for Hypertrophic Cardiomyopathy. J Am Coll Cardiol. 2018, 72, 2457–2467. [CrossRef]
- Vodnjov, N.; Toplišek, J.; Maver, A.; Čuturilo, G.; Jaklič, H.; Teran, N.; Višnjar, T.; Pušenjak, M..; Hodžić, A.; Miljanović, O.; et al. A novel splice-site FHOD3 founder variant is a common cause of hypertrophic cardiomyopathy in the population of the Balkans–A cohort study. PLOS ONE 2023, 18, e0294969. [CrossRef]
- Wu, G.; Ruan, J.; Liu, J.; Zhang, C.; Kang, L.; Wang, J.; Zou, Y.; Song, L. Variant Spectrum of Formin Homology 2 Domain-Containing 3 Gene in Chinese Patients With Hypertrophic Cardiomyopathy. J. Am. Hear. Assoc. 2021, 10, e018236. [CrossRef]
- Ochoa, J.P.; Lopes, L.R.; Perez-Barbeito, M.; Cazón-Varela, L.; de la Torre-Carpente, M.M.; Sonicheva-Paterson, N.; De Uña-Iglesias, D.; Quinn, E.; Kuzmina-Krutetskaya, S.S.; Garrote, J.A.; et al. Deletions of specific exons of FHOD3 detected by next-generation sequencing are associated with hypertrophic cardiomyopathy. Clin. Genet. 2020, 98, 86–90. [CrossRef]
- Semsarian, C.; Ingles, J.; Bagnall, R.D. Revisiting Genome Sequencing Data in Light of Novel Disease Gene Associations. Journal of the American College of Cardiology 2019, 73, 1365–1366. [CrossRef]
- Lesurf, R.; Said, A.; Akinrinade, O.; Breckpot, J.; Delfosse, K.; Liu, T.; Yao, R.; Persad, G.; McKenna, F.; Noche, R.R.; et al. Whole genome sequencing delineates regulatory, copy number, and cryptic splice variants in early onset cardiomyopathy. npj Genom. Med. 2022, 7, 1–17. [CrossRef]
- Matsuyama, S.; Kage, Y.; Fujimoto, N.; Ushijima, T.; Tsuruda, T.; Kitamura, K.; Shiose, A.; Asada, Y.; Sumimoto, H.; Takeya, R. Interaction between cardiac myosin-binding protein C and formin Fhod3. Proc. Natl. Acad. Sci. 2018, 115, E4386–E4395. [CrossRef]
- Vermeulen, M.; Eberl, H.C.; Matarese, F.; Marks, H.; Denissov, S.; Butter, F.; Lee, K.K.; Olsen, J.V.; Hyman, A.A.; Stunnenberg, H.G.; et al. Quantitative Interaction Proteomics and Genome-wide Profiling of Epigenetic Histone Marks and Their Readers. Cell 2010, 142, 967–980. [CrossRef]
- Kaneda, R.; Takada, S.; Yamashita, Y.; Choi, Y.L.; Nonaka-Sarukawa, M.; Soda, M.; Misawa, Y.; Isomura, T.; Shimada, K.; Mano, H. Genome-wide histone methylation profile for heart failure. Genes Cells 2008, 14, 69–77. [CrossRef]
- Theis, J.L.; Sharpe, K.M.; Matsumoto, M.E.; Chai, H.S.; Nair, A.A.; Theis, J.D.; de Andrade, M.; Wieben, E.D.; Michels, V.V.; Olson, T.M. Homozygosity Mapping and Exome Sequencing Reveal GATAD1 Mutation in Autosomal Recessive Dilated Cardiomyopathy. Circ. Cardiovasc. Genet. 2011, 4, 585–594. [CrossRef]
- Yang, J.; Shah, S.; Olson, T.M.; Xu, X. Modeling GATAD1-Associated Dilated Cardiomyopathy in Adult Zebrafish. J. Cardiovasc. Dev. Dis. 2016, 3, 6. [CrossRef]
- Bendig, G.; Grimmler, M.; Huttner, I.G.; Wessels, G.; Dahme, T.; Just, S.; Trano, N.; Katus, H.A.; Fishman, M.C.; Rottbauer, W. Integrin-linked kinase, a novel component of the cardiac mechanical stretch sensor, controls contractility in the zebrafish heart. Genes Dev. 2006, 20, 2361–2372. [CrossRef]
- Knöll, R.; Postel, R.; Wang, J.; Krätzner, R.; Hennecke, G.; Vacaru, A.M.; Vakeel, P.; Schubert, C.; Murthy, K.; Rana, B.K.; et al. Laminin-α4 and Integrin-Linked Kinase Mutations Cause Human Cardiomyopathy Via Simultaneous Defects in Cardiomyocytes and Endothelial Cells. Circulation 2007, 116, 515–525. [CrossRef]
- Gu, R.; Bai, J.; Ling, L.; Ding, L.; Zhang, N.; Ye, J.; Ferro, A.; Xu, B. Increased Expression of Integrin-Linked Kinase Improves Cardiac Function and Decreases Mortality in Dilated Cardiomyopathy Model of Rats. PLOS ONE 2012, 7, e31279. [CrossRef]
- Sopko, N.; Qin, Y.; Finan, A.; Dadabayev, A.; Chigurupati, S.; Qin, J.; Penn, M.S.; Gupta, S. Significance of Thymosin β4 and Implication of PINCH-1-ILK-α-Parvin (PIP) Complex in Human Dilated Cardiomyopathy. PLOS ONE 2011, 6, e20184. [CrossRef]
- Roura, S.; Gálvez-Montón, C.; Pujal, J.M.; Casani, L.; Fernández, M.A.; Astier, L.; Gastelurrutia, P.; Domingo, M.; Prat-Vidal, C.; Soler-Botija, C.; et al. New insights into lipid raft function regulating myocardial vascularization competency in human idiopathic dilated cardiomyopathy. Atherosclerosis 2013, 230, 354–364. [CrossRef]
- Lehnart, S.E.; Wehrens, X.H.T. The role of junctophilin proteins in cellular function. Physiol. Rev. 2022, 102, 1211–1261. [CrossRef]
- Valtonen, J.; Prajapati, C.; Cherian, R.M.; Vanninen, S.; Ojala, M.; Leivo, K.; Heliö, T.; Koskenvuo, J.; Aalto-Setälä, K. The Junctophilin-2 Mutation p.(Thr161Lys) Is Associated with Hypertrophic Cardiomyopathy Using Patient-Specific iPS Cardiomyocytes and Demonstrates Prolonged Action Potential and Increased Arrhythmogenicity. Biomedicines 2023, 11, 1558. [CrossRef]
- Vanninen, S.U.M.; Leivo, K.; Seppälä, E.H.; Aalto-Setälä, K.; Pitkänen, O.; Suursalmi, P.; Annala, A.-P.; Anttila, I.; Alastalo, T.-P.; Myllykangas, S.; et al. Heterozygous junctophilin-2 (JPH2) p.(Thr161Lys) is a monogenic cause for HCM with heart failure. PLOS ONE 2018, 13, e0203422. [CrossRef]
- Landstrom, A.P.; Weisleder, N.; Batalden, K.B.; Bos, J.M.; Tester, D.J.; Ommen, S.R.; Wehrens, X.H.; Claycomb, W.C.; Ko, J.-K.; Hwang, M.; et al. Mutations in JPH2-encoded junctophilin-2 associated with hypertrophic cardiomyopathy in humans. J. Mol. Cell. Cardiol. 2007, 42, 1026–1035. [CrossRef]
- Parker, L.E.; Kramer, R.J.; Kaplan, S.; Landstrom, A.P. One gene, two modes of inheritance, four diseases: A systematic review of the cardiac manifestation of pathogenic variants in JPH2-encoded junctophilin-2. Trends Cardiovasc. Med. 2022, 33, 1–10. [CrossRef]
- Lahiri, S.K.; Jin, F.; Zhou, Y.; Quick, A.P.; Kramm, C.F.; Wang, M.C.; Wehrens, X.H. Altered myocardial lipid regulation in junctophilin-2–associated familial cardiomyopathies. Life Sci. Alliance 2024, 7, e202302330. [CrossRef]
- Wu T, Li X, Jia X, Zhu Z, Lu J, Feng H, et al. Krüppel like factor 10 prevents intervertebral disc degeneration via TGF-β signaling pathway both and. J Orthop Translat. 2021;29: 19–29. [CrossRef]
- Bos JM, Subramaniam M, Hawse JR, Christiaans I, Rajamannan NM, Maleszewski JJ, et al. TGFβ-inducible early gene-1 (TIEG1) mutations in hypertrophic cardiomyopathy. J Cell Biochem. 2012;113: 1896–1903. [CrossRef]
- Al-Hassnan, Z.N.; Almesned, A.; Tulbah, S.; Alakhfash, A.; Alhadeq, F.; Alruwaili, N.; Alkorashy, M.; Alhashem, A.; Alrashdan, A.; Faqeih, E.; et al. Categorized Genetic Analysis in Childhood-Onset Cardiomyopathy. Circ. Genom. Precis. Med. 2020, 13, 504–514. [CrossRef]
- Maurer, C.; Boleti, O.; Torbati, P.N.; Norouzi, F.; Fowler, A.N.R.; Minaee, S.; Salih, K.H.; Taherpour, M.; Birjandi, H.; Alizadeh, B.; et al. Genetic Insights from Consanguineous Cardiomyopathy Families. Genes 2023, 14, 182. [CrossRef]
- Lin, Z.; Li, S.; Feng, C.; Yang, S.; Wang, H.; Ma, D.; Zhang, J.; Gou, M.; Bu, D.; Zhang, T.; et al. Stabilizing mutations of KLHL24 ubiquitin ligase cause loss of keratin 14 and human skin fragility. Nat. Genet. 2016, 48, 1508–1516. [CrossRef]
- Schwieger-Briel, A.; Fuentes, I.; Castiglia, D.; Barbato, A.; Greutmann, M.; Leppert, J.; Duchatelet, S.; Hovnanian, A.; Burattini, S.; Yubero, M.J.; et al. Epidermolysis Bullosa Simplex with KLHL24 Mutations Is Associated with Dilated Cardiomyopathy. J. Investig. Dermatol. 2018, 139, 244–249. [CrossRef]
- Walsh, R.; Offerhaus, J.A.; Tadros, R.; Bezzina, C.R. Minor hypertrophic cardiomyopathy genes, major insights into the genetics of cardiomyopathies. Nat. Rev. Cardiol. 2021, 19, 151–167. [CrossRef]
- Wang, J.; Hoshijima, M.; Lam, J.; Zhou, Z.; Jokiel, A.; Dalton, N.D.; Hultenby, K.; Ruiz-Lozano, P.; Ross, J.; Tryggvason, K.; et al. Cardiomyopathy Associated with Microcirculation Dysfunction in Laminin α4 Chain-deficient Mice. J. Biol. Chem. 2006, 281, 213–220. [CrossRef]
- Lopez-Ayala, J.M.; Ortiz-Genga, M.; Gomez-Milanes, I.; Lopez-Cuenca, D.; Ruiz-Espejo, F.; Sanchez-Munoz, J.J.; Oliva-Sandoval, M.J.; Monserrat, L.; Gimeno, J.R. A mutation in the Z-line Cypher/ZASP protein is associated with arrhythmogenic right ventricular cardiomyopathy. Clin. Genet. 2014, 88, 172–176. [CrossRef]
- Arimura, T.; Hayashi, T.; Terada, H.; Lee, S.-Y.; Zhou, Q.; Takahashi, M.; Ueda, K.; Nouchi, T.; Hohda, S.; Shibutani, M.; et al. A Cypher/ZASP Mutation Associated with Dilated Cardiomyopathy Alters the Binding Affinity to Protein Kinase C. J. Biol. Chem. 2004, 279, 6746–6752. [CrossRef]
- Vatta M, Mohapatra B, Jimenez S, Sanchez X, Faulkner G, Perles Z, et al. Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. J Am Coll Cardiol. 2003;42: 2014–2027.
- Zheng, M.; Cheng, H.; Li, X.; Zhang, J.; Cui, L.; Ouyang, K.; Han, L.; Zhao, T.; Gu, Y.; Dalton, N.D.; et al. Cardiac-specific ablation of Cypher leads to a severe form of dilated cardiomyopathy with premature death. Hum. Mol. Genet. 2008, 18, 701–713. [CrossRef]
- van der Meer, D.L.; Marques, I.J.; Leito, J.T.; Besser, J.; Bakkers, J.; Schoonheere, E.; Bagowski, C.P. Zebrafish cypher is important for somite formation and heart development. Dev. Biol. 2006, 299, 356–372. [CrossRef]
- Koopmann, T.T.; Jamshidi, Y.; Naghibi-Sistani, M.; van der Klift, H.M.; Birjandi, H.; Al-Hassnan, Z.; Alwadai, A.; Zifarelli, G.; Karimiani, E.G.; Sedighzadeh, S.; et al. Biallelic loss of LDB3 leads to a lethal pediatric dilated cardiomyopathy. Eur. J. Hum. Genet. 2022, 31, 97–104. [CrossRef]
- Xuan, T.; Wang, D.; Lv, J.; Pan, Z.; Fang, J.; Xiang, Y.; Cheng, H.; Wang, X.; Guo, X. Downregulation of Cypher induces apoptosis in cardiomyocytes via Akt/p38 MAPK signaling pathway. Int. J. Med Sci. 2020, 17, 2328–2337. [CrossRef]
- Schiaffino, S.; Reggiani, C. Molecular diversity of myofibrillar proteins: gene regulation and functional significance. Physiol. Rev. 1996, 76, 371–423. [CrossRef]
- Niimura, H.; Patton, K.K.; McKenna, W.J.; Soults, J.; Maron, B.J.; Seidman, J.; Seidman, C.E. Sarcomere Protein Gene Mutations in Hypertrophic Cardiomyopathy of the Elderly. Circulation 2002, 105, 446–451. [CrossRef]
- Reiser, P.J.; Portman, M.A.; Ning, X.-H.; Moravec, C.S. Human cardiac myosin heavy chain isoforms in fetal and failing adult atria and ventricles. Am J Physiol Heart Circ Physiol. 2001, 280, H1814–H1820. [CrossRef]
- Carniel E, Taylor MRG, Sinagra G, Di Lenarda A, Ku L, Fain PR, et al. Alpha-myosin heavy chain: a sarcomeric gene associated with dilated and hypertrophic phenotypes of cardiomyopathy. Circulation. 2005;112: 54–59. [CrossRef]
- Hershberger RE, Norton N, Morales A, Li D, Siegfried JD, Gonzalez-Quintana J. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ Cardiovasc Genet. 2010;3: 155–161. [CrossRef]
- Zhao T, Ma Y, Zhang Z, Xian J, Geng X, Wang F, et al. Young and early-onset dilated cardiomyopathy with malignant ventricular arrhythmia and sudden cardiac death induced by the heterozygous LDB3, MYH6, and SYNE1 missense mutations. Ann Noninvasive Electrocardiol. 2021;26: e12840. [CrossRef]
- Ntelios, D.; Meditskou, S.; Efthimiadis, G.; Pitsis, A.; Zegkos, T.; Parcharidou, D.; Theotokis, P.; Alexouda, S.; Karvounis, H.; Tzimagiorgis, G. α-Myosin heavy chain (MYH6) in hypertrophic cardiomyopathy: Prominent expression in areas with vacuolar degeneration of myocardial cells. Pathol. Int. 2022, 72, 308–310. [CrossRef]
- Klos, M.; Mundada, L.; Banerjee, I.; Morgenstern, S.; Myers, S.; Leone, M.; Kleid, M.; Herron, T.; Devaney, E. Altered myocyte contractility and calcium homeostasis in alpha-myosin heavy chain point mutations linked to familial dilated cardiomyopathy. Arch. Biochem. Biophys. 2017, 615, 53–60. [CrossRef]
- Davis, J.S.; Hassanzadeh, S.; Winitsky, S.; Lin, H.; Satorius, C.; Vemuri, R.; Aletras, A.H.; Wen, H.; Epstein, N.D. The Overall Pattern of Cardiac Contraction Depends on a Spatial Gradient of Myosin Regulatory Light Chain Phosphorylation. Cell 2001, 107, 631–641. [CrossRef]
- Qin, X.; Li, P.; Qu, H.-Q.; Liu, Y.; Xia, Y.; Chen, S.; Yang, Y.; Huang, S.; Wen, P.; Zhou, X.; et al. FLNC and MYLK2 Gene Mutations in a Chinese Family with Different Phenotypes of Cardiomyopathy. Int. Hear. J. 2021, 62, 127–134. [CrossRef]
- Burstein, D.S.; Gaynor, J.W.; Griffis, H.; Ritter, A.; Connor, M.J.O.; Rossano, J.W.; Lin, K.Y.; Ahrens-Nicklas, R.C. Genetic variant burden and adverse outcomes in pediatric cardiomyopathy. Pediatr. Res. 2020, 89, 1470–1476. [CrossRef]
- Lange, S.; Himmel, M.; Auerbach, D.; Agarkova, I.; Hayess, K.; Fürst, D.O.; Perriard, J.-C.; Ehler, E. Dimerisation of Myomesin: Implications for the Structure of the Sarcomeric M-band. J. Mol. Biol. 2004, 345, 289–298. [CrossRef]
- Siegert, R.; Perrot, A.; Keller, S.; Behlke, J.; Michalewska-Włudarczyk, A.; Wycisk, A.; Tendera, M.; Morano, I.; Özcelik, C. A myomesin mutation associated with hypertrophic cardiomyopathy deteriorates dimerisation properties. Biochem. Biophys. Res. Commun. 2011, 405, 473–479. [CrossRef]
- Cecconi, M.; Parodi, M.I.; Formisano, F.; Spirito, P.; Autore, C.; Musumeci, M.B.; Favale, S.; Forleo, C.; Rapezzi, C.; Biagini, E.; et al. Targeted next-generation sequencing helps to decipher the genetic and phenotypic heterogeneity of hypertrophic cardiomyopathy. Int. J. Mol. Med. 2016, 38, 1111–1124. [CrossRef]
- Bottillo, I.; D’Angelantonio, D.; Caputo, V.; Paiardini, A.; Lipari, M.; De Bernardo, C.; Giannarelli, D.; Pizzuti, A.; Majore, S.; Castori, M.; et al. Molecular analysis of sarcomeric and non-sarcomeric genes in patients with hypertrophic cardiomyopathy. Gene 2015, 577, 227–235. [CrossRef]
- Guo, X.; Fan, C.; Tian, L.; Liu, Y.; Wang, H.; Zhao, S.; Duan, F.; Zhang, X.; Zhao, X.; Wang, F.; et al. The clinical features, outcomes and genetic characteristics of hypertrophic cardiomyopathy patients with severe right ventricular hypertrophy. PLOS ONE 2017, 12, e0174118. [CrossRef]
- Gontier, Y.; Taivainen, A.; Fontao, L.; Sonnenberg, A.; van der Flier, A.; Carpen, O.; Faulkner, G.; Borradori, L. The Z-disc proteins myotilin and FATZ-1 interact with each other and are connected to the sarcolemma via muscle-specific filamins. J. Cell Sci. 2005, 118, 3739–3749. [CrossRef]
- Ivandic, B.T.; Mastitsky, S.E.; Schönsiegel, F.; Bekeredjian, R.; Eils, R.; Frey, N.; Katus, H.A.; Brors, B. Whole-genome analysis of gene expression associates the ubiquitin-proteasome system with the cardiomyopathy phenotype in disease-sensitized congenic mouse strains. Cardiovasc. Res. 2012, 94, 87–95. [CrossRef]
- Osio, A.; Tan, L.; Chen, S.N.; Lombardi, R.; Nagueh, S.F.; Shete, S.; Roberts, R.; Willerson, J.T.; Marian, A.J. Myozenin 2 Is a Novel Gene for Human Hypertrophic Cardiomyopathy. Circ. Res. 2007, 100, 766–768. [CrossRef]
- Guo, X.; Fan, C.; Wang, Y.; Wang, M.; Cai, C.; Yang, Y.; Zhao, S.; Duan, F.; Li, Y. Genetic anticipation in a special form of hypertrophic cardiomyopathy with sudden cardiac death in a family with 74 members across 5 generations. Medicine 2017, 96, e6249. [CrossRef]
- Ruggiero, A.; Chen, S.N.; Lombardi, R.; Rodriguez, G.; Marian, A.J. Pathogenesis of hypertrophic cardiomyopathy caused by myozenin 2 mutations is independent of calcineurin activity. Cardiovasc. Res. 2012, 97, 44–54. [CrossRef]
- Otey, C.A.; Dixon, R.; Stack, C.; Goicoechea, S.M. Cytoplasmic Ig-domain proteins: Cytoskeletal regulators with a role in human disease. Cell Motil. Cytoskelet. 2009, 66, 618–634. [CrossRef]
- Bang, M.-L.; Mudry, R.E.; McElhinny, A.S.; Trombitás, K.; Geach, A.J.; Yamasaki, R.; Sorimachi, H.; Granzier, H.; Gregorio, C.C.; Labeit, S. Myopalladin, a Novel 145-Kilodalton Sarcomeric Protein with Multiple Roles in Z-Disc and I-Band Protein Assemblies. J. Cell Biol. 2001, 153, 413–428. [CrossRef]
- Purevjav, E.; Arimura, T.; Augustin, S.; Huby, A.-C.; Takagi, K.; Nunoda, S.; Kearney, D.L.; Taylor, M.D.; Terasaki, F.; Bos, J.M.; et al. Molecular basis for clinical heterogeneity in inherited cardiomyopathies due to myopalladin mutations. Hum. Mol. Genet. 2012, 21, 2039–2053. [CrossRef]
- Filomena, M.C.; Yamamoto, D.L.; Carullo, P.; Medvedev, R.; Ghisleni, A.; Piroddi, N.; Scellini, B.; Crispino, R.; D’Autilia, F.; Zhang, J.; et al. Myopalladin knockout mice develop cardiac dilation and show a maladaptive response to mechanical pressure overload. eLife 2021, 10. [CrossRef]
- Cronin, H.; Crinion, D.; Kerins, D.; Fahy, G.; Vaughan, C.J. Inferolateral T wave inversion in athletes: phenotype-genotype correlation. Ir. J. Med Sci. (1971 -) 2020, 189, 1283–1287. [CrossRef]
- Refaat, M.M.; Hassanieh, S.; Ballout, J.A.; Zakka, P.; Hotait, M.; Khalil, A.; Bitar, F.; Arabi, M.; Arnaout, S.; Skouri, H.; et al. Non-familial cardiomyopathies in Lebanon: exome sequencing results for five idiopathic cases. BMC Med Genom. 2019, 12, 1–11. [CrossRef]
- Hernandez, D.A.; Bennett, C.M.; Dunina-Barkovskaya, L.; Wedig, T.; Capetanaki, Y.; Herrmann, H.; Conover, G.M. Nebulette is a powerful cytolinker organizing desmin and actin in mouse hearts. Mol. Biol. Cell 2016, 27, 3869–3882. [CrossRef]
- Arimura, T.; Nakamura, T.; Hiroi, S.; Satoh, M.; Takahashi, M.; Ohbuchi, N.; Ueda, K.; Nouchi, T.; Yamaguchi, N.; Akai, J.; et al. Characterization of the human nebulette gene: a polymorphism in an actin-binding motif is associated with nonfamilial idiopathic dilated cardiomyopathy. Hum. Genet. 2000, 107, 440–451. [CrossRef]
- Maiellaro-Rafferty, K.; Wansapura, J.; Mendsaikhan, U.; Osinska, H.; James, J.; Taylor; Robbins, J.; Kranias, E.; Towbin, J.; Purevjav, E. Altered regional cardiac wall mechanics are associated with differential cardiomyocyte calcium handling due to nebulette mutations in preclinical inherited dilated cardiomyopathy. J. Mol. Cell. Cardiol. 2013, 60, 151–160. [CrossRef]
- Purevjav, E.; Varela, J.; Morgado, M.; Kearney, D.L.; Li, H.; Taylor, M.D.; Arimura, T.; Moncman, C.L.; McKenna, W.; Murphy, R.T.; et al. Nebulette Mutations Are Associated With Dilated Cardiomyopathy and Endocardial Fibroelastosis. Circ. 2010, 56, 1493–1502. [CrossRef]
- Mastrototaro, G.; Liang, X.; Li, X.; Carullo, P.; Piroddi, N.; Tesi, C.; Gu, Y.; Dalton, N.D.; Peterson, K.L.; Poggesi, C.; et al. Nebulette knockout mice have normal cardiac function, but show Z-line widening and up-regulation of cardiac stress markers. Cardiovasc. Res. 2015, 107, 216–225. [CrossRef]
- Perrot, A.; Tomasov, P.; Villard, E.; Faludi, R.; Melacini, P.; Lossie, J.; Lohmann, N.; Richard, P.; De Bortoli, M.; Angelini, A.; et al. Mutations in NEBL encoding the cardiac Z-disk protein nebulette are associated with various cardiomyopathies. Arch. Med Sci. 2016, 2, 263–278. [CrossRef]
- Lee, A.; Hakuno, F.; Northcott, P.; Pessin, J.E.; Adcock, M.R. Nexilin, a Cardiomyopathy-Associated F-Actin Binding Protein, Binds and Regulates IRS1 Signaling in Skeletal Muscle Cells. PLOS ONE 2013, 8, e55634. [CrossRef]
- Chopra, N.; Knollmann, B.C. Genetics of sudden cardiac death syndromes. Curr. Opin. Cardiol. 2011, 26, 196–203. [CrossRef]
- Liu, C.; Spinozzi, S.; Chen, J.-Y.; Fang, X.; Feng, W.; Perkins, G.; Cattaneo, P.; Guimarães-Camboa, N.; Dalton, N.D.; Peterson, K.L.; et al. Nexilin Is a New Component of Junctional Membrane Complexes Required for Cardiac T-Tubule Formation. Circulation 2019, 140, 55–66. [CrossRef]
- Spinozzi S, Liu C, Chen Z ’e, Feng W, Zhang L, Ouyang K, et al. Nexilin Is Necessary for Maintaining the Transverse-Axial Tubular System in Adult Cardiomyocytes. Circ Heart Fail. 2020;13: e006935. [CrossRef]
- Aherrahrou, Z.; Schlossarek, S.; Stoelting, S.; Klinger, M.; Geertz, B.; Weinberger, F.; Kessler, T.; Aherrahrou, R.; Moreth, K.; Bekeredjian, R.; et al. Knock-out of nexilin in mice leads to dilated cardiomyopathy and endomyocardial fibroelastosis. Basic Res. Cardiol. 2015, 111, 6. [CrossRef]
- Kean, A.C.; Helm, B.M.; Vatta, M.; Ayers, M.D.; Parent, J.J.; Darragh, R.K. Clinical characterisation of a novel SCN5A variant associated with progressive malignant arrhythmia and dilated cardiomyopathy. Cardiol. Young- 2019, 29, 1257–1263. [CrossRef]
- Zhang, X.-L.; Xie, J.; Lan, R.-F.; Kang, L.-N.; Wang, L.; Xu, W.; Xu, B. Genetic Basis and Genotype-Phenotype Correlations in Han Chinese Patients with Idiopathic Dilated Cardiomyopathy. Sci. Rep. 2020, 10, 1–8. [CrossRef]
- Bruyndonckx, L.; Vogelzang, J.L.; Bugiani, M.; Straver, B.; Kuipers, I.M.; Onland, W.; Nannenberg, E.A.; Clur, S.; van der Crabben, S.N.&. Childhood onset nexilin dilated cardiomyopathy: A heterozygous and a homozygous case. Am. J. Med Genet. Part A 2021, 185, 2464–2470. [CrossRef]
- Johansson, J.; Frykholm, C.; Ericson, K.; Kazamia, K.; Lindberg, A.; Mulaiese, N.; Falck, G.; Gustafsson, P.; Lidéus, S.; Gudmundsson, S.; et al. Loss of Nexilin function leads to a recessive lethal fetal cardiomyopathy characterized by cardiomegaly and endocardial fibroelastosis. Am. J. Med Genet. Part A 2022, 188, 1676–1687. [CrossRef]
- Hermida, A.; Ader, F.; Millat, G.; Jedraszak, G.; Maury, P.; Cador, R.; Catalan, P.-A.; Clerici, G.; Combes, N.; De Groote, P.; et al. NEXN Gene in Cardiomyopathies and Sudden Cardiac Deaths: Prevalence, Phenotypic Expression, and Prognosis. Circ. Genom. Precis. Med. 2024, 17, e004285. [CrossRef]
- Schott, J.-J.; Benson, D.W.; Basson, C.T.; Pease, W.; Silberbach, G.M.; Moak, J.P.; Maron, B.J.; Seidman, C.E.; Seidman, J.G. Congenital Heart Disease Caused by Mutations in the Transcription Factor NKX2-5. Science 1998, 281, 108–111. [CrossRef]
- Jay PY, Harris BS, Maguire CT, Buerger A, Wakimoto H, Tanaka M, et al. Nkx2-5 mutation causes anatomic hypoplasia of the cardiac conduction system. J Clin Invest. 2004;113: 1130–1137.
- Brody, M.J.; Cho, E.; Mysliwiec, M.R.; Kim, T.-G.; Carlson, C.D.; Lee, K.-H.; Lee, Y. Lrrc10 is a novel cardiac-specific target gene of Nkx2-5 and GATA4. J. Mol. Cell. Cardiol. 2013, 62, 237–246. [CrossRef]
- McElhinney, D.B.; Geiger, E.; Blinder, J.; Benson, D.W.; Goldmuntz, E. NKX2.5mutations in patients with congenital heart disease. J Am Coll Cardiol. 2003, 42, 1650–1655. [CrossRef]
- Hanley, A.; Walsh, K.A.; Joyce, C.; McLellan, M.A.; Clauss, S.; Hagen, A.; Shea, M.A.; Tucker, N.R.; Lin, H.; Fahy, G.J.; et al. Mutation of a common amino acid in NKX2.5 results in dilated cardiomyopathy in two large families. BMC Med Genet. 2016, 17, 1–7. [CrossRef]
- Costa, M.W.; Guo, G.; Wolstein, O.; Vale, M.; Castro, M.L.; Wang, L.; Otway, R.; Riek, P.; Cochrane, N.; Furtado, M.; et al. Functional Characterization of a Novel Mutation in NKX2-5 Associated With Congenital Heart Disease and Adult-Onset Cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 238–247. [CrossRef]
- Yuan, F.; Qiu, X.-B.; Li, R.-G.; Qu, X.-K.; Wang, J.; Xu, Y.-J.; Liu, X.; Fang, W.-Y.; Yang, Y.-Q.; Liao, D.-N. A novel NKX2-5 loss-of-function mutation predisposes to familial dilated cardiomyopathy and arrhythmias. Int. J. Mol. Med. 2014, 35, 478–486. [CrossRef]
- Xu, J.-H.; Gu, J.-Y.; Guo, Y.-H.; Zhang, H.; Qiu, X.-B.; Li, R.-G.; Shi, H.-Y.; Liu, H.; Yang, X.-X.; Xu, Y.-J.; et al. Prevalence and Spectrum of NKX2-5 Mutations Associated With Sporadic Adult-Onset Dilated Cardiomyopathy. Int. Hear. J. 2017, 58, 521–529. [CrossRef]
- Sveinbjornsson G, Olafsdottir EF, Thorolfsdottir RB, Davidsson OB, Helgadottir A, Jonasdottir A, et al. Variants in NKX2-5 and FLNC Cause Dilated Cardiomyopathy and Sudden Cardiac Death. Circ Genom Precis Med. 2018;11: e002151. [CrossRef]
- Bang, M.-L.; Centner, T.; Fornoff, F.; Geach, A.J.; Gotthardt, M.; McNabb, M.; Witt, C.C.; Labeit, D.; Gregorio, C.C.; Granzier, H.; et al. The Complete Gene Sequence of Titin, Expression of an Unusual ≈700-kDa Titin Isoform, and Its Interaction With Obscurin Identify a Novel Z-Line to I-Band Linking System. Circ. Res. 2001, 89, 1065–1072. [CrossRef]
- Borisov, A.B.; Sutter, S.B.; Kontrogianni-Konstantopoulos, A.; Bloch, R.J.; Westfall, M.V.; Russell, M.W. Essential role of obscurin in cardiac myofibrillogenesis and hypertrophic response: evidence from small interfering RNA-mediated gene silencing. Histochem Cell Biol. 2005, 125, 227–238. [CrossRef]
- Hu, L.-Y.R.; Kontrogianni-Konstantopoulos, A. The kinase domains of obscurin interact with intercellular adhesion proteins. FASEB J. 2013, 27, 2001–2012. [CrossRef]
- Marston, S. Obscurin variants and inherited cardiomyopathies. Biophys. Rev. 2017, 9, 239–243. [CrossRef]
- Arimura, T.; Matsumoto, Y.; Okazaki, O.; Hayashi, T.; Takahashi, M.; Inagaki, N.; Hinohara, K.; Ashizawa, N.; Yano, K.; Kimura, A. Structural analysis of obscurin gene in hypertrophic cardiomyopathy. Biochem. Biophys. Res. Commun. 2007, 362, 281–287. [CrossRef]
- Marston S, Montgiraud C, Munster AB, Copeland O ’neal, Choi O, Dos Remedios C, et al. OBSCN Mutations Associated with Dilated Cardiomyopathy and Haploinsufficiency. PLoS One. 2015;10: e0138568.
- Xia, H.; Winokur, S.T.; Kuo, W.-L.; Altherr, M.R.; Bredt, D.S. Actinin-associated LIM Protein: Identification of a Domain Interaction between PDZ and Spectrin-like Repeat Motifs. J. Cell Biol. 1997, 139, 507–515. [CrossRef]
- Lorenzen-Schmidt, I.; McCulloch, A.D.; Omens, J.H. Deficiency of Actinin-Associated LIM Protein Alters Regional Right Ventricular Function and Hypertrophic Remodeling. Ann. Biomed. Eng. 2005, 33, 888–896. [CrossRef]
- Bagnall, R.D.; Yeates, L.; Semsarian, C. Analysis of the Z-disc genes PDLIM3 and MYPN in Patients with Hypertrophic Cardiomyopathy. Int. J. Cardiol. 2010, 145, 601–602. [CrossRef]
- Lopes, L.; Murphy, C.; Syrris, P.; Dalageorgou, C.; McKenna, W.; Elliott, P.; Plagnol, V. Use of high-throughput targeted exome-sequencing to screen for copy number variation in hypertrophic cardiomyopathy. Eur. J. Med Genet. 2015, 58, 611–616. [CrossRef]
- Rosa-Ferreira, C.; Munro, S. Arl8 and SKIP Act Together to Link Lysosomes to Kinesin-1. Dev. Cell 2011, 21, 1171–1178. [CrossRef]
- Khatter, D.; Raina, V.B.; Dwivedi, D.; Sindhwani, A.; Bahl, S.; Sharma, M. The small GTPase Arl8b regulates assembly of the mammalian HOPS complex to lysosomes. J. Cell Sci. 2015, 128, 1746–1761. [CrossRef]
- Almacellas, E.; Pelletier, J.; Day, C.; Ambrosio, S.; Tauler, A.; Mauvezin, C. Lysosomal degradation ensures accurate chromosomal segregation to prevent chromosomal instability. Autophagy 2020, 17, 796–813. [CrossRef]
- Muhammad E, Levitas A, Singh SR, Braiman A, Ofir R, Etzion S, et al. PLEKHM2 mutation leads to abnormal localization of lysosomes, impaired autophagy flux and associates with recessive dilated cardiomyopathy and left ventricular noncompaction. Hum Mol Genet. 2015;24: 7227–7240. [CrossRef]
- Cibi, D.M.; Bi-Lin, K.W.; Shekeran, S.G.; Sandireddy, R.; Tee, N.; Singh, A.; Wu, Y.; Srinivasan, D.K.; Kovalik, J.-P.; Ghosh, S.; et al. Prdm16 Deficiency Leads to Age-Dependent Cardiac Hypertrophy, Adverse Remodeling, Mitochondrial Dysfunction, and Heart Failure. Cell Rep. 2020, 33, 108288. [CrossRef]
- Bjork, B.C.; Turbe-Doan, A.; Prysak, M.; Herron, B.J.; Beier, D.R. Prdm16 is required for normal palatogenesis in mice. Hum. Mol. Genet. 2009, 19, 774–789. [CrossRef]
- Liu, C.; Shao, N.-Y. The Differences in the Developmental Stages of the Cardiomyocytes and Endothelial Cells in Human and Mouse Embryos at the Single-Cell Level. Int. J. Mol. Sci. 2024, 25, 3240. [CrossRef]
- Arndt, A.-K.; Schafer, S.; Drenckhahn, J.-D.; Sabeh, M.K.; Plovie, E.R.; Caliebe, A.; Klopocki, E.; Musso, G.; Werdich, A.A.; Kalwa, H.; et al. Fine Mapping of the 1p36 Deletion Syndrome Identifies Mutation of PRDM16 as a Cause of Cardiomyopathy. Am. J. Hum. Genet. 2013, 93, 67–77. [CrossRef]
- Long, P.A.; Evans, J.M.; Olson, T.M. Diagnostic Yield of Whole Exome Sequencing in Pediatric Dilated Cardiomyopathy. J. Cardiovasc. Dev. Dis. 2017, 4, 11. [CrossRef]
- Li, D.; Parks, S.B.; Kushner, J.D.; Nauman, D.; Burgess, D.; Ludwigsen, S.; Partain, J.; Nixon, R.R.; Allen, C.N.; Irwin, R.P.; et al. Mutations of Presenilin Genes in Dilated Cardiomyopathy and Heart Failure. Am. J. Hum. Genet. 2006, 79, 1030–1039. [CrossRef]
- Takeda, T.; Asahi, M.; Yamaguchi, O.; Hikoso, S.; Nakayama, H.; Kusakari, Y.; Kawai, M.; Hongo, K.; Higuchi, Y.; Kashiwase, K.; et al. Presenilin 2 regulates the systolic function of heart by modulating Ca2+signaling. FASEB J. 2005, 19, 2069–2071. [CrossRef]
- Sciarretta, S.; Forte, M.; Frati, G.; Sadoshima, J. New Insights Into the Role of mTOR Signaling in the Cardiovascular System. Circ. Res. 2018, 122, 489–505. [CrossRef]
- Jain, P.K.; Jayappa, S.; Sairam, T.; Mittal, A.; Paul, S.; Rao, V.J.; Chittora, H.; Kashyap, D.K.; Palakodeti, D.; Thangaraj, K.; et al. Ribosomal protein S6 kinase beta-1 gene variants cause hypertrophic cardiomyopathy. J. Med Genet. 2021, 59, 984–992. [CrossRef]
- Grossman, W. The role of sarcoplasmic reticulum proteins in heart disease: introduction.. Ann. New York Acad. Sci. 1998, 853, 207–208. [CrossRef]
- Yano M, Ikeda Y, Matsuzaki M. Altered intracellular Ca2+ handling in heart failure. J Clin Invest. 2005;115: 556–564.
- Kohno, M.; Kobayashi, S.; Yamamoto, T.; Yoshitomi, R.; Kajii, T.; Fujii, S.; Nakamura, Y.; Kato, T.; Uchinoumi, H.; Oda, T.; et al. Enhancing calmodulin binding to cardiac ryanodine receptor completely inhibits pressure-overload induced hypertrophic signaling. Commun. Biol. 2020, 3, 1–15. [CrossRef]
- Stanczyk, P.J.; Seidel, M.; White, J.; Viero, C.; George, C.H.; Zissimopoulos, S.; Lai, F.A. Association of cardiac myosin-binding protein-C with the ryanodine receptor channel – putative retrograde regulation?. J. Cell Sci. 2018, 131, jcs210443. [CrossRef]
- Fujino N, Ino H, Hayashi K, Uchiyama K, Nagata M, Konno T, et al. Abstract 915: A Novel Missense Mutation in Cardiac Ryanodine Receptor Gene as a Possible Cause of Hypertrophic Cardiomyopathy: Evidence From Familial Analysis. Circulation. 2006 [cited 17 Jul 2024]. Available: https://www.ahajournals.org/doi/10.1161/circ.114.suppl_18.II_165-b.
- Tang, Y.; Tian, X.; Wang, R.; Fill, M.; Chen, S.W. Abnormal Termination of Ca 2+ Release Is a Common Defect of RyR2 Mutations Associated With Cardiomyopathies. Circ. Res. 2012, 110, 968–977. [CrossRef]
- Xu, J.; Li, Z.; Ren, X.; Dong, M.; Li, J.; Shi, X.; Zhang, Y.; Xie, W.; Sun, Z.; Liu, X.; et al. Investigation of Pathogenic Genes in Chinese sporadic Hypertrophic Cardiomyopathy Patients by Whole Exome Sequencing. Sci. Rep. 2015, 5, 16609–16609. [CrossRef]
- Alvarado, F.J.; Bos, J.M.; Yuchi, Z.; Valdivia, C.R.; Hernández, J.J.; Zhao, Y.-T.; Henderlong, D.S.; Chen, Y.; Booher, T.R.; Marcou, C.A.; et al. Cardiac hypertrophy and arrhythmia in mice induced by a mutation in ryanodine receptor 2. JCI Insight. 2019, 4. [CrossRef]
- Krishnaswamy, S.M.; Arunachal, G.; Singh, K.G.; Thomson, V.S.; George, P.; Rao, S.; Danda, S. Investigation of mutation spectrum amongst patients with familial primary cardiomyopathy using targeted NGS in Indian population. J. Appl. Genet. 2024, 1–14. [CrossRef]
- Ervasti, J.M.; Campbell, K.P. Membrane organization of the dystrophin-glycoprotein complex. Cell 1991, 66, 1121–1131. [CrossRef]
- Way M, Pope B, Cross RA, Kendrick-Jones J, Weeds AG. Expression of the N-terminal domain of dystrophin in E. coli and demonstration of binding to F-actin. FEBS Lett. 1992;301: 243–245. [CrossRef]
- Tsubata S, Bowles KR, Vatta M, Zintz C, Titus J, Muhonen L, et al. Mutations in the human delta-sarcoglycan gene in familial and sporadic dilated cardiomyopathy. J Clin Invest. 2000;106: 655–662.
- Kärkkäinen S, Miettinen R, Tuomainen P, Kärkkäinen P, Heliö T, Reissell E, et al. A novel mutation, Arg71Thr, in the delta-sarcoglycan gene is associated with dilated cardiomyopathy. J Mol Med . 2003;81: 795–800. [CrossRef]
- Campbell, M.D.; Witcher, M.; Gopal, A.; Michele, D.E. Dilated cardiomyopathy mutations in δ-sarcoglycan exert a dominant-negative effect on cardiac myocyte mechanical stability. Am J Physiol Heart Circ Physiol. 2016, 310, H1140–H1150. [CrossRef]
- Kirk, E.P.; Sunde, M.; Costa, M.; Rankin, S.A.; Wolstein, O.; Castro, M.L.; Butler, T.L.; Hyun, C.; Guo, G.; Otway, R.; et al. Mutations in Cardiac T-Box Factor Gene TBX20 Are Associated with Diverse Cardiac Pathologies, Including Defects of Septation and Valvulogenesis and Cardiomyopathy. Am. J. Hum. Genet. 2007, 81, 280–291. [CrossRef]
- Qian, L.; Mohapatra, B.; Akasaka, T.; Liu, J.; Ocorr, K.; Towbin, J.A.; Bodmer, R. Transcription factor neuromancer/TBX20 is required for cardiac function in Drosophila with implications for human heart disease. Proc. Natl. Acad. Sci. 2008, 105, 19833–19838. [CrossRef]
- Chen, Y.; Xiao, D.; Zhang, L.; Cai, C.-L.; Li, B.-Y.; Liu, Y. The Role of Tbx20 in Cardiovascular Development and Function. Front. Cell Dev. Biol. 2021, 9. [CrossRef]
- Giri, P.; Mukhopadhyay, A.; Gupta, M.; Mohapatra, B. Dilated cardiomyopathy: a new insight into the rare but common cause of heart failure. Hear. Fail. Rev. 2021, 27, 431–454. [CrossRef]
- Zhao C-M, Bing-Sun, Song H-M, Wang J, Xu W-J, Jiang J-F, et al. TBX20 loss-of-function mutation associated with familial dilated cardiomyopathy. Clin Chem Lab Med. 2016;54: 325–332.
- Mittal, A.; Sharma, R.; Prasad, R.; Bahl, A.; Khullar, M. Role of cardiac TBX20 in dilated cardiomyopathy. Mol. Cell. Biochem. 2016, 414, 129–136. [CrossRef]
- Amor-Salamanca, A.; Rodríguez, A.S.; Rasoul, H.; Rodríguez-Palomares, J.F.; Moldovan, O.; Hey, T.M.; Delgado, M.G.; Cuenca, D.L.; Campos, D.d.C.; Basurte-Elorz, M.T.; et al. Role of TBX20 Truncating Variants in Dilated Cardiomyopathy and Left Ventricular Noncompaction. Circ. Genom. Precis. Med. 2024, 17, e004404–e004404. [CrossRef]
- Knöll, R.; Hoshijima, M.; Hoffman, H.M.; Person, V.; Lorenzen-Schmidt, I.; Bang, M.-L.; Hayashi, T.; Shiga, N.; Yasukawa, H.; Schaper, W.; et al. The Cardiac Mechanical Stretch Sensor Machinery Involves a Z Disc Complex that Is Defective in a Subset of Human Dilated Cardiomyopathy. Cell 2002, 111, 943–955. [CrossRef]
- Hayashi, T.; Arimura, T.; Itoh-Satoh, M.; Ueda, K.; Hohda, S.; Inagaki, N.; Takahashi, M.; Hori, H.; Yasunami, M.; Nishi, H.; et al. Tcap gene mutations in hypertrophic cardiomyopathy and dilated cardiomyopathy. Circ. 2004, 44, 2192–2201. [CrossRef]
- Martins E, Sousa A, Canedo P, Leite S, Pinto R, Campelo M, et al. Genetic variants identified by target next-generation sequencing in heart transplant patients with dilated cardiomyopathy. Rev Port Cardiol. 2019;38: 441–447. [CrossRef]
- Nguyen, T.V.; Vu, M.T.T.; Do, T.N.P.; Tran, T.H.N.; Huynh, B.N.T.; Le, L.A.; Pham, N.T.N.; Nguyen, T.M.N.; Le, N.H.P.; Nguyen, V.P.; et al. Genetic Determinants and Genotype-Phenotype Correlations in Vietnamese Patients With Dilated Cardiomyopathy. Circ. J. 2021, 85, 1469–1478. [CrossRef]
- Graycar JL, Miller DA, Arrick BA, Lyons RM, Moses HL, Derynck R. Human transforming growth factor-beta 3: recombinant expression, purification, and biological activities in comparison with transforming growth factors-beta 1 and -beta 2. Mol Endocrinol. 1989;3: 1977–1986. [CrossRef]
- Rampazzo, A.; Nava, A.; Danieli, G.A.; Buja, G.; Daliento, L.; Fasoli, G.; Scognamiglio, R.; Corrado, D.; Thlene, G. The gene for arrhythmogenic right ventricular cardiomyopathy maps to chromosome 14q23–q24. Hum. Mol. Genet. 1994, 3, 959–962. [CrossRef]
- Rampazzo, A.; Beffagna, G.; Nava, A.; Occhi, G.; Bauce, B.; Noiato, M.; Basso, C.; Frigo, G.; Thiene, G.; Towbin, J.; et al. Arrhythmogenic right ventricular cardiomyopathy type 1 (ARVD1): confirmation of locus assignment and mutation screening of four candidate genes. Eur. J. Hum. Genet. 2003, 11, 69–76. [CrossRef]
- Beffagna G, Occhi G, Nava A, Vitiello L, Ditadi A, Basso C, et al. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res. 2005;65: 366–373. [CrossRef]
- Rampazzo A. Regulatory mutations in transforming growth factor-β3 gene involved in arrhythmogenic right ventricular cardiomyopathy: AUTHOR’S RETROSPECTIVE. Cardiovasc Res. 2012;96: 191–194. [CrossRef]
- Bao, J.-R.; Wang, J.-Z.; Yao, Y.; Wang, Y.-L.; Fan, X.-H.; Sun, K.; Zhang, S.; Hui, R.-T.; Song, L. Screening of pathogenic genes in Chinese patients with arrhythmogenic right ventricular cardiomyopathy. Chin Med J. 2013, 126, 4238–41. [CrossRef]
- Vermij, S.H.; Abriel, H.; van Veen, T.A.B. Refining the molecular organization of the cardiac intercalated disc. Cardiovasc. Res. 2017, 113, 259–275. [CrossRef]
- Roberts, J.D. TJP1 Mutations in Arrhythmogenic Cardiomyopathy. Circ. Genom. Precis. Med. 2018, 11, e002337. [CrossRef]
- De Bortoli, M.; Postma, A.V.; Poloni, G.; Calore, M.; Minervini, G.; Mazzotti, E.; Rigato, I.; Ebert, M.; Lorenzon, A.; Vazza, G.; et al. Whole-Exome Sequencing Identifies Pathogenic Variants in TJP1 Gene Associated With Arrhythmogenic Cardiomyopathy. Circ. Genom. Precis. Med. 2018, 11, e002123. [CrossRef]
- Zhao, Y.; Meng, X.-M.; Wei, Y.-J.; Zhao, X.-W.; Liu, D.-Q.; Cao, H.-Q.; Liew, C.-C.; Ding, J.-F. Cloning and characterization of a novel cardiac-specific kinase that interacts specifically with cardiac troponin I. J. Mol. Med. 2003, 81, 297–304. [CrossRef]
- Wheeler, F.C.; Tang, H.; Marks, O.A.; Hadnott, T.N.; Chu, P.-L.; Mao, L.; Rockman, H.A.; Marchuk, D.A. Tnni3k Modifies Disease Progression in Murine Models of Cardiomyopathy. PLOS Genet. 2009, 5, e1000647. [CrossRef]
- Theis, J.L.; Zimmermann, M.T.; Larsen, B.T.; Rybakova, I.N.; Long, P.A.; Evans, J.M.; Middha, S.; de Andrade, M.; Moss, R.L.; Wieben, E.D.; et al. TNNI3K mutation in familial syndrome of conduction system disease, atrial tachyarrhythmia and dilated cardiomyopathy. Hum. Mol. Genet. 2014, 23, 5793–5804. [CrossRef]
- Podliesna, S.; Delanne, J.; Miller, L.; Tester, D.J.; Uzunyan, M.; Yano, S.; Klerk, M.; Cannon, B.C.; Khongphatthanayothin, A.; Laurent, G.; et al. Supraventricular tachycardias, conduction disease, and cardiomyopathy in 3 families with the same rare variant in TNNI3K (p.Glu768Lys). Hear. Rhythm. 2018, 16, 98–105. [CrossRef]
- Fan L-L, Huang H, Jin J-Y, Li J-J, Chen Y-Q, Zhao S-P, et al. Whole exome sequencing identifies a novel mutation (c.333 + 2T > C) of TNNI3K in a Chinese family with dilated cardiomyopathy and cardiac conduction disease. Gene. 2018;648: 63–67. [CrossRef]
- Pham, C.; Andrzejczyk, K.; Jurgens, S.J.; Deprez, R.L.; Palm, K.C.; Vermeer, A.M.; Nijman, J.; Christiaans, I.; Barge-Schaapveld, D.Q.; van Dessel, P.F.; et al. Genetic Burden of TNNI3K in Diagnostic Testing of Patients With Dilated Cardiomyopathy and Supraventricular Arrhythmias. Circ. Genom. Precis. Med. 2023, 16, 328–336. [CrossRef]
- Chen, S.N.; Czernuszewicz, G.; Lombardi, R.; Jin, J.; Willerson, J.; Marian, A.J. Abstract 18430: Human Molecular Genetic and Functional Studies Identify TRIM63, Encoding Muscle RING Finger Protein 1, as a Novel Gene for Human Hypertrophic Cardiomyopathy. Circulation 2012, 126. [CrossRef]
- Su, M.; Wang, J.; Kang, L.; Wang, Y.; Zou, Y.; Feng, X.; Wang, D.; Ahmad, F.; Zhou, X.; Hui, R.; et al. Rare Variants in Genes Encoding MuRF1 and MuRF2 Are Modifiers of Hypertrophic Cardiomyopathy. Int. J. Mol. Sci. 2014, 15, 9302–9313. [CrossRef]
- Witt, C.C.; Witt, S.H.; Lerche, S.; Labeit, D.; Back, W.; Labeit, S. Cooperative control of striated muscle mass and metabolism by MuRF1 and MuRF2. EMBO J. 2007, 27, 350–360. [CrossRef]
- Andreeva, S.; Chumakova, O.; Karelkina, E.; Lebedeva, V.; Lubimtseva, T.; Semenov, A.; Nikitin, A.; Speshilov, G.; Kozyreva, A.; Sokolnikova, P.; et al. Case Report: Two New Cases of Autosomal-Recessive Hypertrophic Cardiomyopathy Associated With TRIM63-Compound Heterozygous Variant. Front. Genet. 2022, 13, 743472. [CrossRef]
- Moiseyeva, E.; Weller, P.; Zhidkova, N.; Corben, E.; Patel, B.; Jasinska, I.; Koteliansky, V.; Critchley, D. Organization of the human gene encoding the cytoskeletal protein vinculin and the sequence of the vinculin promoter.. J. Biol. Chem. 1993, 268, 4318–4325. [CrossRef]
- Vasile VC, Will ML, Ommen SR, Edwards WD, Olson TM, Ackerman MJ. Identification of a metavinculin missense mutation, R975W, associated with both hypertrophic and dilated cardiomyopathy. Mol Genet Metab. 2006;87: 169–174. [CrossRef]
- Wells, Q.S.; Ausborn, N.L.; Funke, B.H.; Pfotenhauer, J.P.; Fredi, J.L.; Baxter, S.; DiSalvo, T.G.; Hong, C.C. Familial Dilated Cardiomyopathy Associated with Congenital Defects in the Setting of a Novel VCL Mutation (Lys815Arg) in Conjunction with a Known MYPBC3 Variant. Cardiogenetics 2011, 1, e10–e10. [CrossRef]
- Mazzarotto, F.; Tayal, U.; Buchan, R.J.; Midwinter, W.; Wilk, A.; Whiffin, N.; Govind, R.; Mazaika, E.; de Marvao, A.; Dawes, T.J.; et al. Reevaluating the Genetic Contribution of Monogenic Dilated Cardiomyopathy. Circulation 2020, 141, 387–398. [CrossRef]
- Hawley, M.H.; Almontashiri, N.; Biesecker, L.G.; Berger, N.; Chung, W.K.; Garcia, J.; Grebe, T.A.; Kelly, M.A.; Lebo, M.S.; Macaya, D.; et al. An assessment of the role of vinculin loss of function variants in inherited cardiomyopathy. Hum. Mutat. 2020, 41, 1577–1587. [CrossRef]
- Xu, W.; Baribault, H.; Adamson, E.D. Vinculin knockout results in heart and brain defects during embryonic development. Development 1998, 125, 327–337. [CrossRef]
- Zemljic-Harpf, A.E.; Ponrartana, S.; Avalos, R.T.; Jordan, M.C.; Roos, K.P.; Dalton, N.D.; Phan, V.Q.; Adamson, E.D.; Ross, R.S. Heterozygous Inactivation of the Vinculin Gene Predisposes to Stress-Induced Cardiomyopathy. Am. J. Pathol. 2004, 165, 1033–1044. [CrossRef]
- Marg S, Winkler U, Sestu M, Himmel M, Schönherr M, Bär J, et al. The vinculin-DeltaIn20/21 mouse: characteristics of a constitutive, actin-binding deficient splice variant of vinculin. PLoS One. 2010;5: e11530.
- Zemljic-Harpf, A.E.; Miller, J.C.; Henderson, S.A.; Wright, A.T.; Manso, A.M.; Elsherif, L.; Dalton, N.D.; Thor, A.K.; Perkins, G.A.; McCulloch, A.D.; et al. Cardiac-Myocyte-Specific Excision of the Vinculin Gene Disrupts Cellular Junctions, Causing Sudden Death or Dilated Cardiomyopathy. Mol. Cell. Biol. 2007, 27, 7522–7537. [CrossRef]
- Vogel, B.; Meder, B.; Just, S.; Laufer, C.; Berger, I.; Weber, S.; Katus, H.A.; Rottbauer, W. In-vivo characterization of human dilated cardiomyopathy genes in zebrafish. Biochem. Biophys. Res. Commun. 2009, 390, 516–522. [CrossRef]
- Cheng, F.; Miao, L.; Wu, Q.; Gong, X.; Xiong, J.; Zhang, J. Vinculin b deficiency causes epicardial hyperplasia and coronary vessel disorganization in zebrafish. Development 2016, 143, 3522–3531. [CrossRef]
- Maartens AP, Wellmann J, Wictome E, Klapholz B, Green H, Brown NH. Drosophila vinculin is more harmful when hyperactive than absent, and can circumvent integrin to form adhesion complexes. J Cell Sci. 2016;129: 4354–4365. [CrossRef]
- Liu, Y.; Li, X.; Zhao, X.; Dong, J.; Zhang, C.; Lin, T. Establishment of a human iPSC (ZZUNEUi026-A) from a dilated cardiomyopathy patient carrying heterozygous Vinculin (c. 625A > T) mutant. Stem Cell Res. 2022, 62, 102812. [CrossRef]
- Fatkin, D.; Johnson, R. Are Double Mutations Double Trouble?. Circ. Cardiovasc. Genet. 2017, 10. [CrossRef]
- Gray, M.P.; Fatkin, D.; Ingles, J.; Robertson, E.N.; A Figtree, G. Genetic testing in cardiovascular disease. The Medical Journal of Australia 2024, 220, 428–434. [CrossRef]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.; Vatta, M.; Ware, S.M. Genetic Evaluation of Cardiomyopathy—A Heart Failure Society of America Practice Guideline. J. Card. Fail. 2018, 24, 281–302. [CrossRef]
- Monaco, I.; Santacroce, R.; Casavecchia, G.; Correale, M.; Bottigliero, D.; Cordisco, G.; Leccese, A.; Di Biase, M.; Margaglione, M.; Brunetti, N.D. Double de novo mutations in dilated cardiomyopathy with cardiac arrest. J. Electrocardiol. 2018, 53, 40–43. [CrossRef]
Figure 1.
Distribution of VUS and P/LP variants in minor genes reported in ClinVar [19] in ARVC, DCM, and HCM cases. The bars are color-coded, with red indicating P/LP variants and grey indicating variants of uncertain significance. The background colors represent the ClinGen [17,19] classification for each gene: orange for moderate, yellow for limited, and light grey for not associated.
Figure 1.
Distribution of VUS and P/LP variants in minor genes reported in ClinVar [19] in ARVC, DCM, and HCM cases. The bars are color-coded, with red indicating P/LP variants and grey indicating variants of uncertain significance. The background colors represent the ClinGen [17,19] classification for each gene: orange for moderate, yellow for limited, and light grey for not associated.
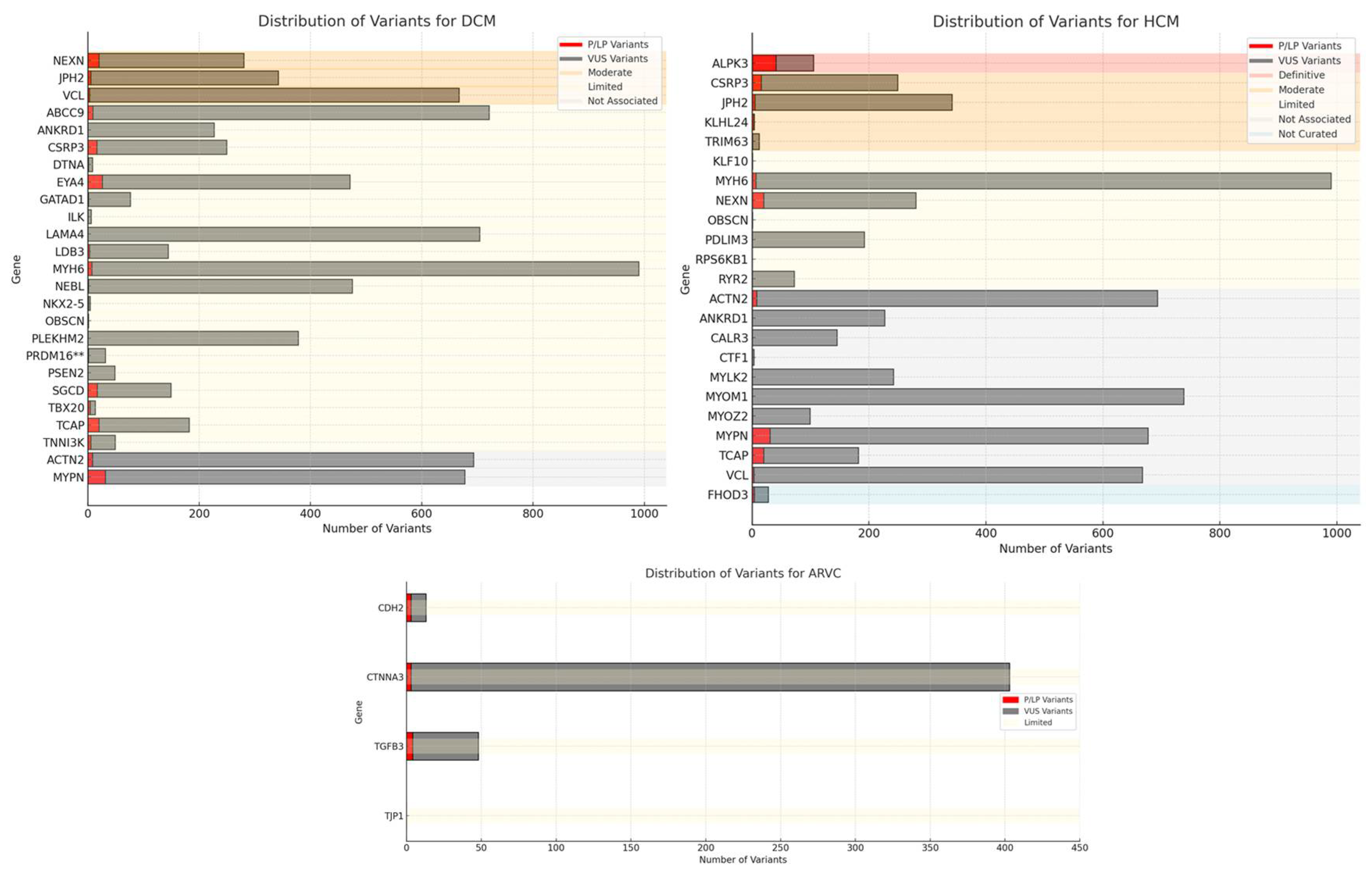

Table 1.
list of genes definitively associated with ARVC, DCM and HCM according to: ClinGen [17], GeneReview [23], EMQN 2023 [15,16] and an Expert Consensus Statement, 2022 [25]. ALPK3, JUP and PLN are not included Definitive genes.
| Gene | Phenotype Association |
|---|---|
| ACTC1 | HCM |
| BAG3 | DCM |
| DES | DCM |
| DSC2 | ARVC |
| DSG2 | ARVC |
| DSP | ARVC |
| FLNC | DCM |
| LMNA | DCM |
| MYBPC3 | HCM |
| MYH7 | DCM; HCM |
| MYL2 | HCM |
| MYL3 | HCM |
| PKP2 | ARVC |
| RBM20 | DCM |
| SCN5A | DCM |
| TMEM43 | ARVC |
| TNNC1 | DCM |
| TNNI3 | HCM |
| TNNT2 | DCM; HCM |
| TPM1 | HCM |
| TTN | DCM |
Table 2.
Genes discussed in this review. They were categorized as definitive, moderate, limited, not associated, or not curated based on ClinGen terminology and on their association with different forms of CMPs. AD= Autosomic Dominant, AR= Autosomic Recessive, SD= Autosomic Semidominant, - = the gene is not curated by the source.
Table 2.
Genes discussed in this review. They were categorized as definitive, moderate, limited, not associated, or not curated based on ClinGen terminology and on their association with different forms of CMPs. AD= Autosomic Dominant, AR= Autosomic Recessive, SD= Autosomic Semidominant, - = the gene is not curated by the source.
| ClinGen [17] | GeneReview [23] | EMQN [15,16] | Expert Consensus Statement [25] | ClinGen [17] | GeneReview [23] | EMQN [15,16] | Expert Consensus Statement [25] | ClinGen [17] | GeneReview [23] | EMQN [15,16] | Expert Consensus Statement [25] | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Hereditary | ARVC | DCM | HCM | |||||||||
| ABCC9 | AD | not associated | not associated | - | not associated | limited | limited | - | not associated | not associated | not associated | - | not associated |
| ACTN2 | AD | not associated | not associated | not associated | not associated | not associated | moderate | moderate | moderate | not associated | moderate | not associated | moderate |
| ANKRD1 | AD | not associated | not associated | - | - | limited | limited | - | - | not associated | limited | - | - |
| CALR3 | AD | not associated | not associated | - | - | not associated | not associated | - | - | not associated | limited | - | - |
| CSRP3 | AD, SD | not associated | not associated | - | not associated | limited | limited | - | not associated | moderate | moderate | - | moderate |
| CTF1 | AD | not associated | not associated | - | - | limited | limited | - | - | not associated | not associated | - | - |
| DTNA | AD | not associated | not associated | - | - | limited | limited | - | - | not associated | not associated | - | - |
| EYA4 | AD | not associated | not associated | - | - | limited | limited | - | - | not associated | not associated | - | - |
| GATAD1 | AR | not associated | not associated | - | - | limited | limited | - | - | not associated | not associated | - | - |
| ILK | AD | not associated | not associated | - | - | limited | limited | - | - | not associated | not associated | - | - |
| JPH2 | AD, SD | not associated | not associated | not associated | not associated | moderate | moderate | moderate | moderate | moderate | moderate | not associated | moderate |
| KLF10 | AD | not associated | not associated | - | - | not associated | not associated | - | - | limited | limited | - | - |
| KLHL24 | AR | not associated | - | - | - | not associated | - | - | - | moderate | - | - | - |
| LAMA4 | AD | not associated | not associated | - | - | limited | limited | - | - | not associated | not associated | - | - |
| LDB3 | AD | not associated | not associated | - | not associated | limited | limited | - | not associated | not associated | not associated | - | not associated |
| MYH6 | AD | not associated | not associated | - | - | limited | limited | - | - | limited | limited | - | - |
| MYLK2 | AD | not associated | not associated | - | - | not associated | not associated | - | - | not associated | limited | - | - |
| MYOM1 | AD | not associated | not associated | - | - | not associated | not associated | - | - | not associated | limited | - | - |
| MYOZ2 | AD | not associated | not associated | - | - | not associated | not associated | - | - | not associated | limited | - | - |
| MYPN | AD | not associated | not associated | - | - | not associated | limited | - | - | not associated | limited | - | - |
| NEBL | AD | not associated | not associated | - | - | limited | limited | - | -- | not associated | not associated | - | - |
| NEXN | AD | not associated | not associated | - | - | moderate | moderate | - | - | limited | limited | - | - |
| NKX2-5 | AD | not associated | not associated | - | - | limited | limited | - | not associated | not associated | - | - | |
| OBSCN | AD | not associated | not associated | - | limited | limited | - | - | limited | not associated | - | - | |
| PDLIM3 | AD | not associated | not associated | - | - | not associated | not associated | - | - | limited | limited | - | - |
| PLEKHM2 | AR | not associated | not associated | - | - | limited | limited | - | - | not associated | not associated | - | - |
| PRDM16 | AD | not associated | not associated | - | limited | limited | - | - | not associated | not associated | - | - | |
| PSEN2 | AD | not associated | not associated | - | - | limited | limited | - | - | not associated | not associated | - | - |
| RPS6KB1 | AD | not associated | not associated | - | - | not associated | not associated | - | - | limited | not associated | - | - |
| RYR2 | AD | not associated | not associated | - | - | not associated | not associated | - | - | limited | limited | - | - |
| SGCD | AD | not associated | not associated | - | - | limited | limited | - | not associated | not associated | - | - | |
| TBX20 | AD | not associated | not associated | - | - | limited | limited | - | - | not associated | not associated | - | - |
| TCAP | AD | not associated | not associated | - | - | limited | limited | - | - | not associated | limited | - | - |
| TNNI3K | AD | not associated | not associated | - | limited | limited | - | - | not associated | not associated | - | - | |
| TRIM63 | AD, AR | not associated | not associated | - | - | not associated | not associated | - | - | moderate | limited | - | - |
| VCL | AD | not associated | not associated | - | - | moderate | moderate | - | - | not associated | limited | - | - |
Table 3.
Minor genes identified in this review. The clinical validity reported in the primary source ClinGen [17] and both pathogenic and VUS variants reported in ClinVar [19] are provided.
| Gene | Protein | ARVC | DCM | HCM | N. P/LP Variants | N. VUS | Phenotype Submission | Hereditary |
|---|---|---|---|---|---|---|---|---|
| ABCC9 | ATP Binding Cassette Subfamily C Member 9 | not associated | limited | not associated | 9 | 712 | DCM | AD |
| ACTN2 | Alpha-actinin-2 | not associated | not associated | not associated | 8 | 685 | DCM, HCM | AD |
| ANKRD1 | Ankyrin Repeat Domain 1 | not associated | limited | not associated | 0 | 227 | DCM, HCM | AD |
| CALR3 | Calreticulin 3 | not associated | not associated | not associated | 0 | 145 | HCM | AD |
| CDH2 | Cadherin 2 | limited | not associated | not associated | 3 | 10 | ARVC | AD |
| CSRP3 | Cysteine And Glycine Rich Protein 3 | not associated | limited | moderate | 16 | 233 | DCM, HCM | AD, SD |
| CTF1 | Cardiotrophin 1 | not associated | limited | not associated | 0 | 3 | HCM | AD |
| CTNNA3 | Catenin Alpha 3 | limited | not associated | not associated | 3 | 400 | ARVC | AD |
| DTNA | Dystrobrevin Alpha | not associated | limited | not associated | 1 | 7 | DCM | AD |
| EYA4 | EYA Transcriptional Coactivator and Phosphatase 4 | not associated | limited | not associated | 26 | 445 | DCM | AD |
| FHOD3 | Formin Homology 2 Domain Containing 3 | not curated | not curated | not curated | 4 | 24 | HCM | na |
| GATAD1 | GATA Zinc Finger Domain Containing 1 | not associated | limited | not associated | 1 | 75 | DCM | AR |
| ILK | Integrin Linked Kinase | not associated | limited | not associated | 0 | 6 | DCM | AD |
| JPH2 | Junctophilin 2 | not associated | moderate | moderate | 5 | 337 | DCM, HCM | AD, SD |
| KLF10 | Kruppel -Like Factor 1 | not associated | not associated | limited | 0 | 1 | HCM | AD |
| KLHL24 | Kelch Like Family Member 24 | not associated | not associated | moderate | 3 | 1 | HCM | AR |
| LAMA4 | Laminin Subunit Alpha 4 | not associated | limited | not associated | 0 | 704 | DCM | AD |
| LDB3 | LIM Domain Binding 3 | not associated | limited | not associated | 3 | 141 | DCM | AD |
| MYH6 | Myosin Heavy Chain 6 | not associated | limited | limited | 7 | 983 | DCM, HCM | AD |
| MYLK2 | Myosin Light Chain Kinase 2 | not associated | not associated | not associated | 0 | 242 | HCM | AD |
| MYOM1 | Myomesin 1 | not associated | not associated | not associated | 0 | 738 | HCM | AD |
| MYOZ2 | Myozenin 2 | not associated | not associated | not associated | 1 | 98 | HCM | AD |
| MYPN | Myopalladin | not associated | not associated | not associated | 31 | 646 | DCM, HCM | AD |
| NEBL | Nebulette | not associated | limited | not associated | 1 | 474 | DCM | AD |
| NEXN | Nexilin F-Actin Binding Protein | not associated | moderate | limited | 20 | 260 | DCM, HCM | AD |
| NKX2-5 | NK2 Homeobox 5 | not associated | limited | not associated | 1 | 3 | DCM | AD |
| OBSCN | Obscurin | not associated | limited | limited | 0 | 1 | DCM, HCM | AD |
| PDLIM3 | PDZ And LIM Domain 3 | not associated | not associated | limited | 0 | 192 | HCM | AD |
| PLEKHM2 | Pleckstrin Homology and RUN Domain Containing M2 | not associated | limited | not associated | 0 | 378 | DCM | AR |
| PRDM16 | PR/SET Domain 16 | not associated | limited | not associated | 1 | 30 | DCM | AD |
| PSEN2 | Presenilin 2 | not associated | limited | not associated | 0 | 48 | DCM | AD |
| RPS6KB1 | Ribosomal Protein S6 Kinase B1 | not associated | not associated | limited | 0 | 0 | HCM | AD |
| RYR2 | Ryanodine Receptor 2 | not associated | not associated | limited | 0 | 72 | HCM | AD |
| SGCD | Sarcoglycan Delta | not associated | limited | not associated | 17 | 132 | DCM | AD |
| TBX20 | T-Box Transcription Factor 20 | not associated | limited | not associated | 4 | 9 | DCM | AD |
| TCAP | Titin-Cap | not associated | limited | not associated | 20 | 162 | DCM, HCM | AD |
| TGFB3 | Transforming Growth Factor Beta 3 | limited | not associated | not associated | 4 | 44 | ARVC | AD |
| TJP1 | Tight Junction Protein 1 | limited | not associated | not associated | 0 | 0 | ARVC | AD |
| TNNI3K | TNNI3 Interacting Kinase | not associated | limited | not associated | 5 | 44 | DCM | AD |
| TRIM63 | Tripartite Motif Containing 63 | not associated | not associated | moderate | 1 | 11 | HCM | AD, AR |
| VCL | Vinculin | not associated | moderate | not associated | 3 | 664 | DCM, HCM | AD |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



