
Submitted:
09 July 2024
Posted:
10 July 2024
You are already at the latest version
Abstract
Abstract: Transcription factors are crucial pre-transcriptional regulatory mechanisms that can modulate the expression of downstream genes by binding to their promoter regions. DOF (DNA binding with One Finger) proteins are a unique class of transcription factors with extensive roles in plant growth and development. Our previous research indicated a decrease in iron content in chlorotic bamboo leaves. However, to our knowledge, genes related to iron metabolism pathways in bamboo species have not yet been studied. Therefore, in the current study, we identified iron metabolism-related (IMR) genes in bamboo and determined the transcription factors that significantly influence them. Among these, DOFs were found to have widespread effects and potentially significant impacts on their expression. We identified specific DOF members in Dendrocalamus latiflorus with binding abilities through homology with Arabidopsis DOF proteins, and established connections between some of these members and IMR genes using RNA-seq data. Additionally, molecular docking confirmed the binding interactions between these DlDOFs and the DOF binding sites in the promoter regions of IMR genes. The co-expression relationship between the two gene sets was further validated using q-PCR experiments. This study paves the way for research into iron metabolism pathways in bamboo and lays the foundation for understanding the role of DOF transcription factors in D. latiflorus.
Keywords:
DOF transcription factors
; Iron Metabolism Related Genes
; gene regulation
; gene expression
; Dendrocalamus latiflorus
1. Introduction
Transcription factors have a broad range of functions in gene regulation, influencing various physiological and developmental processes in plants [1,2,3]. Among these, the DOF (DNA binding with One Finger) transcription factor (TF) family is unique to plants and plays critical biological roles [4]. Members of the DOF family contain a highly conserved DOF DNA-binding domain, consisting of 52 amino acids with a C2-C2 type zinc finger structure, allowing them to specifically bind to DNA sequences containing AAAG motif or its reversibly orientated motif CTTT [5,6,7]. Notably, DOF TFs have been found to regulate gene expression related to nutrient metabolism, stress responses, and developmental processes by binding to DNA sequences in target gene promoter regions [5,8,9,10,11,12]. This means that they have the potential to modulate the expression of target genes involved in iron uptake, transport, and homeostasis through the same mechanism, thereby influencing iron metabolism. To date, no studies have identified or characterized DOF members in Dendrocalamus latiflorus.
Iron plays a crucial role in plant metabolism and is closely related to cellular processes, including photosynthesis, respiration, and DNA synthesis [13,14,15]. Its deficiency or excess can lead to significant metabolic disturbances, making its homeostasis vital for plant health and growth. In the formal study, we discovered that the iron ion content varies among different colored leaves of Bambusa multiplex f. silverstripe [16]. Additionally, since iron acts as a cofactor in the early stages of chlorophyll biosynthesis [17,18] , it may cause thylakoid malformation and lead to changes in cell structure [19]. This led us to further explore the reasons behind this difference in iron accumulation and the regulatory mechanisms. However, as B. multiplex is a horticultural plant that has received little attention, there is currently no reference genome available for it, which hinders our ability to understand the gene expression and regulatory mechanisms related to iron metabolism [20]. Fortunately, our laboratory has previously assembled a chromosome-level genome for D. latiflorus, which has the potential to become a model for clumping bamboo plants [21]. This advancement makes it possible to explore the molecular mechanisms of gene regulation involved in iron metabolism in bamboo plants.
In this study, our aim was to elucidate the regulation of iron metabolism related (IMR) genes by DOF TFs in D. latiflorus. Firstly, we identified genes associated with iron metabolism in D. latiflorus and established the potential regulatory role of the DOF TF family in their expression. Subsequently, reliable regulatory relationships were confirmed through protein-DNA binding models, transcriptome data, and q-PCR experiments. This study provides new insights into the regulatory mechanisms of IMR genes in D. latiflorus and paves the way for understanding the role of DOF TFs in this context.
2. Results
2.1. IMR Genes in Dendrocalamus Latiflorus
Plants have evolved efficient iron-uptake mechanisms and sophisticated internal iron-transport mechanisms, regulating gene expression in response to iron availability to maintain iron homeostasis [15]. In total, we identified 311 Iron Metabolism Related Genes in D. latiflorus (Figure 1; Table S1). The genes related to siderophore synthesis were the most numerous, totaling 177. This was followed by 74 genes related to iron transport. Additionally, there were 24 genes related to iron reduction and 11 genes associated with siderophore transport. The number of genes related to magnetosome formation and iron storage was the same, with 8 genes each. Furthermore, the detected genes related to heme oxygenase, heme transport, and iron gene regulation were fewer, numbering 4, 3, and 1 respectively.
2.2. TF Binding Sites in IMR Genes Promoters
In the promoter regions of IMR genes, we identified 11,857 TF binding sites belonging to 35 transcription factor families (Figure 2A; Table S2). The ERF family had the highest number of binding sites, with 4,821, followed by Dof and BBR-BPC with 1,173 and 1,105 binding sites respectively. These are the only three TF families with more than 1,000 binding sites. The C2H2, TALE, and LBD families had between 500 and 1,000 binding sites each, with 726, 580, and 560 binding sites respectively. Additionally, 24 families had between 20 and 500 binding sites each. Only five families had fewer than 20 binding sites: FAR1, ARR-B, YABBY, CAMTA, and E2F/DP and LFY, with 17, 15, 14, 10, 7, and 2 binding sites respectively.
Upon further examination, we found significant variability in the types and numbers of transcription factor binding sites (TFBS) within the promoters of IMR genes. To identify the transcription factor families that potentially have the broadest impact on IMR genes, we counted the number of IMR genes in each family's promoter regions that have binding sites (Figure 2B). The results show that the DOF family has the highest number of IMR genes with binding sites in their promoter regions, with 209 genes, followed by the ERF family with 176 genes. Additionally, the ERF, MYB, TALE, BBR-BPC, CPP, and MIKC_MADS families each have over 100 genes, with 158, 153, 128, 122, 106, and 102 genes respectively. Seven families have fewer than 20 IMR genes occupying their promoter regions, namely EIL, FAR1, AP2, YABBY, ARR-B, CAMTA, E2F/DP, and LFY, with LFY having the least impact with only 1 IMR gene in its promoter regions.
Overall, the ERF transcription factor family has significantly more TF binding sites than any other family, with over four times the number of the second-ranking DOF family. However, it occupies a smaller proportion of promoter region sequences compared to the DOF and C2H2 families. This suggests that ERF may exert stronger influence on a smaller subset of genes. On the other hand, while the DOF family has relatively fewer TF binding sites compared to ERF, it occupies the largest proportion of promoter region sequences. We believe this indicates a broader regulatory role of the DOF family in IMR genes. Therefore, we will focus on analyzing this family in subsequent analyses.
2.3. DOF Members in D. latiflorus
In the present study, we identified 33, 32, and 28 new DOF members in the A, B, and C genomes of D. latiflorus, respectively (Table S3). We renamed them according to their phylogenic relationship (Figure 3). The proteins encoded by these genes ranged in size from 143 to 554 amino acids, and the theoretical molecular weight of D. latiflorus DOF members (DlDOFs) varied from 14,956.73 to 60,809.6 Da, with isoelectric points (pI) ranging from 4.76 to 10.28. All DlDOF proteins were characterized as unstable and hydrophilic proteins. Signal peptide prediction analysis revealed that all DlDOF members contained signal peptide sequences. Trans-membrane domain (TM) prediction showed that all DlDOFs contained one TMs. Subcellular localization prediction indicated that DlDOFs are located in the nucleus, cytoplasm, mitochondria, and extracellular regions (including the cell wall). The majority of DlDOFs, 69 in total, are distributed in the nucleus. Additionally, 16 DlDOFs are located in the cytoplasm, 6 in the mitochondria, and only 2 are found in the extracellular regions.
2.4. Potential Regulatory IMR Genes of DOFs
The predictions of TFBS suggest that numerous DOFs might influence the expression of IMR genes. To further identify potential DOFs regulating IMR gene expression, we first identified the conserved domains of DOF binding sites. The results showed that these sequences contain two conserved domains, referred to as Motif1 and Motif2, which are present in the promoter regions of 95 and 126 IMR genes, respectively (Table S4). This indicates that DlDOFs capable of binding to these motifs in the promoter regions might regulate the expression of the corresponding IMR genes. Moreover, searches of these motif sequences in the JASPAR database revealed that in Arabidopsis, 14 DOFs can bind to both motifs, 14 DOFs can only bind to Motif1, and 23 DOFs can only bind to Motif2 (Table S5). The protein-DNA interactions of these AtDOFs have been validated through DAP-Seq experiments [22], making them highly reliable. To identify DOFs in D. latiflorus capable of binding to these motifs, we compared them with these AtDOFs based on sequence similarity. We identified 53 DlDOFs homologous to these genes (Table S6). Subsequently, these DlDOFs were linked with IMR genes containing Motif1 or Motif2, resulting in the identification of 7,862 potential interactions (Table S7). These DlDOFs might regulate IMR gene expression by binding to their promoters.
2.5. Expression of DlDOFs and IMR Genes in D. latiflorus Leaves
We examined the expression of DlDOFs and IMR genes in D. latiflorus leaves. The results indicated that 39 out of 93 DlDOFs had an average Transcripts Per Million (TPM) value greater than 1 across all leaf samples (Figure 4A), while 252 out of 272 IMR genes had an average TPM value greater than 1 (Figure 4B). To delve deeper into the impact of DOFs on IMR gene expression, we generated a co-expression network between the two gene sets using a threshold of Pearson's Correlation Coefficients (PCCs) > 0.9 (Figure 4C). We identified 36 connections between the two gene sets, involving 18 DlDOFs and 27 IMR genes. Among these, DlDOF3 and DlDOF66 had the most connections with IMR genes, each regulating the expression of four IMR genes. Following them, DlDOF56, DlDOF7, and DlDOF73 each had three connections with IMR genes. Five DlDOFs had two connections with IMR genes: DlDOF55, DlDOF6, DlDOF89, DlDOF91, and DlDOF92. Lastly, eight DlDOFs had only one connection with IMR genes.
2.6. Molecular Docking of Key DlDOFs and Binding Sites in IMR Gene Promoters
Despite predicting 7,862 potential regulatory relationships between DlDOFs and IMR genes based on data, transcriptome data supported only 35 co-expression relationships between the two gene sets. To determine reliable gene interactions and select key DlDOFs, we examined the overlapping relationships between the two datasets (Figure 5A). The results revealed that only eight relationships were present in both datasets, involving six DlDOFs (DlDOF3, DlDOF37, DlDOF55, DlDOF56, DlDOF6, DlDOF8) and eight IMR genes (Dl7AG000538, Dl12AG001562, Dl13AG000670, Dl23AG001841, Dl3AG000581, Dl23AG001068, Dl15AG001499, Dl28AG000366). Except for two genes, DlDOF3 and DlDOF56, which each had connections with two IMR genes, the other DlDOFs were linked to only one IMR gene. This suggests that DlDOF3 and DlDOF56 may play relatively important roles in regulating IMR gene expression. Subsequently, we performed molecular docking calculations for six intersecting DlDOFs with the DOF binding sites in the promoter regions of the target IMR genes (Table S8).
Typically, lower binding energies indicate stronger binding interactions. The docking results showed that among the interactions between three pairs of DlDOFs and the binding sites in the promoter regions of IMR genes, six interactions had binding energies less than -4 kcal/mol, and three had binding energies between -3 and -4 kcal/mol. The binding energy between DlDOF56 and the binding sites in the promoter of the Dl23AG001068 gene was the lowest at -5.8 kcal/mol, followed by DlDOF3-Dl7AG000538 at -4.9 kcal/mol. Additionally, the strongest interactions were found between DlDOF3-Dl7AG000538, DlDOF3-Dl12AG001562, and DlDOF56-Dl23AG001068 (Figure 5B). The promoter regions of Dl12AG001562 and Dl23AG001068 each contained three DOF binding sites, indicating that they are highly influenced by DlDOFs. However, the binding energies of DlDOF6-Dl15AG001499 and DlDOF56-Dl3AG000581 were -3.7 and -3.5 kcal/mol, respectively, suggesting weaker interactions. Notably, their DOF binding sites both contained Motif1. Coincidentally, we found that among the entries containing Motif1, only DlDOF3-Dl7AG000538 had a binding energy less than -4 kcal/mol, indicating that DlDOFs may have weaker binding interactions with sites containing Motif1 compared to sites containing Motif2. Further, the binding energy between DlDOF55 and the TFBS of Dl23AG001841, which contains Motif1, reached as high as 12.9 kcal/mol, suggesting a weak interaction. Therefore, the previously observed co-expression relationship between these two genes might be coincidental or influenced by other genes.
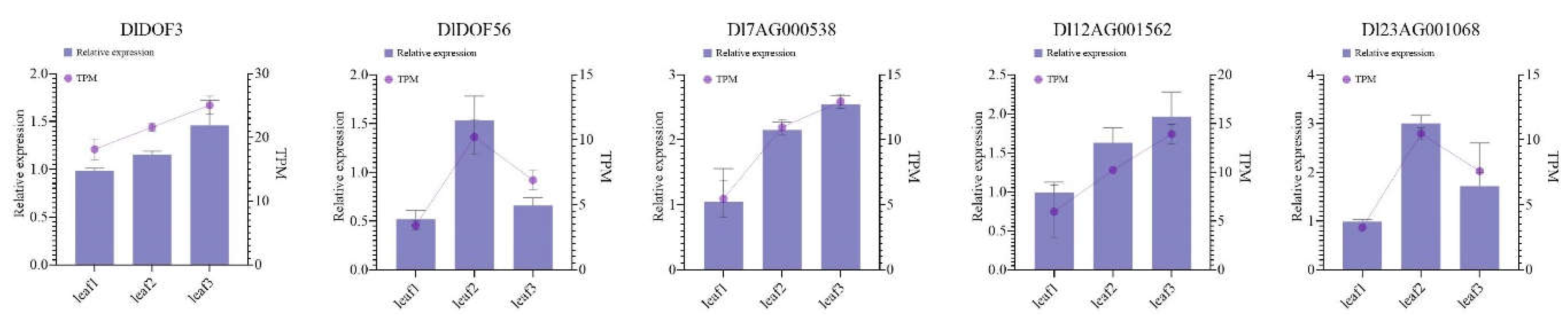
2.7. q-PCR Vaditiation
Using q-PCR, we determined the relative expression levels of two DlDOFs and three IMR genes potentially regulated by them in leaf samples identical to those used in RNA-seq (Figure 6). Results indicated that all five genes showed the lowest expression in sample leaf1. DlDOF3, Dl7AG000538, and Dl12AG001562 exhibited the highest expression in sample leaf3, while DlDOF56 and Dl23AG001068 showed the highest expression in sample leaf2. Furthermore, correlation analysis of the relative expression patterns of these five genes supported high correlations between DlDOF3-Dl7AG000538, DlDOF3-Dl12AG001562, and DlDOF56-Dl23AG001068 (Figure S1). We also compared the fold changes in relative expression and TPM values between different samples (Figure S2), using Sample leaf1 as a control, which demonstrated a strong correlation between q-PCR and transcriptome data, validating the reliability of our sequencing data.
3. Discussion
Transcriptional regulation is a key process in defining cell characteristics, growth, differentiation, and development [1,23]. As an important pre-transcriptional regulatory mechanism, transcription factors can regulate the expression of target genes by binding to specific DNA sequences [24,25]. However, this binding is influenced by various factors. For example, chromatin accessibility determines whether specific regions can be bound by DNA-binding proteins [26,27], and this accessibility is constrained by many epigenetic conditions. For instance, cytosine methylation at the fifth carbon (5mC) is a modification present in the DNA of all known vertebrates and terrestrial plants [28,29], plays a crucial role in genome defense [30], and can influence chromatin accessibility [31,32]. Additionally, DNA is typically wrapped around chromatin nucleosomes. Changes in their chromatin structure result in pleiotropic developmental phenotypes [33,34], impacting the growth and development of plants [35,36], and playing a crucial role in their adaptation to environmental stresses [37,38]. This implies that chromatin structure or DNA methylation can affect chromatin accessibility, thereby regulating the binding of transcription factors and subsequently influencing downstream gene expression. This might explain why we predicted a large number of potential DOF transcription factors regulating the IMR gene in this study, yet only a few were supported by transcriptional level data in bamboo leaves (Figure 5A). Based on these reasons, we speculate that more relationships between the two gene groups might be observed in other tissues of bamboo. Overall, the current research on the role of transcription factors in bamboo is still in its early stages. To gain deeper insights into the functions of DOF family members, future research could explore strategies focusing on transcription factor binding influenced by epigenetic factors. This may be an important driver for bamboo and other ecological species to quickly adapt to evolution through self-regulation of transcriptional levels.
For transcription factors themselves, their binding affinity is influenced by protein structure and various interfering factors such as enzymes [39], metal ions [40], and hormones [41]. Although molecular docking calculations indicate that some DOF transcription factors have strong binding affinity to the IMR gene promoter sites, the number is much lower than expected (Table S8). Current research on plant protein structures is limited and mainly focused on model species [42,43]. In the protein sequences of DlDOF family members, aside from the docking sites in the central region, most residues do not show reliable protein structure predictions (Figure 5B). This may result in some DOF transcription factors exhibiting weak binding affinity to the binding site sequences in molecular docking. Furthermore, while there are currently no reports of other DOF transcription factors interacting with metal ions or enzymes to cause changes in their activity, the limited understanding of the DlDOF protein structure suggests potential interactions with these substances. Additionally, DOF, as a broadly acting family of transcription factors [5], suggests that more DOF proteins are expected to exhibit stronger binding capabilities to target sequences. However, this speculation currently lacks empirical evidence to support it. Observing the structure of DlDOF through cryo-electron microscopy represents an optimal approach for directly targeting protein studies [44], but these studies are costly and constrained by researchers' economic conditions. Therefore, to further explore the molecular functions of DOF transcription factors, our laboratory plans to proceed with testing antibodies suitable for DlDOF members and using CHI-Seq and DAP-seq to specifically determine the binding sequences of DOF proteins across the entire bamboo genome.
In previous studies, we observed higher iron content in chlorotic bamboo leaves [16]. Building on these findings, in the current study, we investigated the potential regulatory role of DOF transcription factors on IMR genes. We believe that knocking out or overexpressing some DlDOF members will affect the expression of the IMR gene, and due to the broad role of transcription factor proteins [1], it may have more significant effects on plant cell. However, the specific impact of IMR protein abundance on iron metabolism is currently unclear. Both heme and chlorophyll synthesis depend on the common precursor protoporphyrin [45], which have antagonistic effects on each other [46]. The genes in the tetrapyrrole synthesis pathway are downregulated in chlorotic bamboo leaves [16], indicating reduced accumulation of protoporphyrin, which may lead to decreased synthesis of iron-containing heme and chlorophyll. However, it is still unclear whether the decrease in iron content is due to reduced protoporphyrin levels leading to the inability to synthesize and accumulate iron-containing heme, or directly due to changes in cell structure caused by chlorophyll deficiency, resulting in decreased IMR gene expression and thus affecting the iron metabolism pathway. Therefore, we believe that the next step in studying the function of the IMR gene should further explore the relationship between iron metabolism and other related biological pathways from both physiological and genetic perspectives.
4. Methods
4.1. Identification of IMR Genes and DOFs in D. latiflorus
The haplotype genome of D. latiflorus were downloaded from BambooBase [47]. We selected the longest transcript as the representative transcript for each gene and generated CDS and translated protein sequences using SeqKit v0.15.0 [48]. Based on these protein sequences, the FeGenie v1.2 [49] was employed to identify IMR genes in D. latiflorus. DOF members (DOFs) in D. latiflorus were initially identified DOFs from Arabidopsis thaliana and Oryza sativa genome-wide using blastp function in Blast v2.10.1 [50]. Additionally, D. latiflorus protein sequences were screened using the DOF domain model (PF02701) from the Pfam [51] database via the hmmsearch function of HMMER v3.3.2 [52] with p-value < 1e-5. Subsequently, the search results were used with the hmmbuild function to construct species-specific hidden Markov model. The newly generated model was then used to re-identify DlDOFs using hmmersearch with p-value < 1e-20. The union of results from both methods was examined using Interproscan v5.63-95.0 to verify the conserved domains of DOF and remove sequences lacking the conserved DOF domain, resulting in the reliable identification of DlDOFs. We used the ProtParam tool on the ExPASy [53] platform for protein physicochemical property analysis, and the WoLF PSORT [54] server for predicting subcellular localization.
4.2. Mutiple Algnment and Phylogenic Analysis
To investigate the phylogenetic relationship of DlDOFs, we aligned these genes using Muscle v5.1 [55] and trimmed the alignment result automatically using trimAL v1.4 [56]. We constructed a maximum likelihood (ML) tree using IQ-TREE v2.1.2 [57] with MFP model and 1000 times bootstrap replicates and visualized the phylogenetic tree and alignment result using custom python script with ETE v3.1.3 toolkit [58].
4.2. Identifying TF Binding Sites in IMR Gene Promoters and Predicting Potential Regulatory Transcription Factors
We used the 1500 bp upstream region from the transcription start site of IMR genes as the promoter region. The DOF binding sites in this promoter region were scanned using the PlantRegMap [59] server. Next, the MEME [60] suite was used to identify potential DOFs that regulate the expression of IMR genes. Specifically, all potential binding sites sequences were scanned with meme function, setting the parameter to the number of motifs 3 to identify the conserved domains of these sequences. Finally, these domain sequences were searched using TOMTOM function in the JASPAR [61] non-redundant database to find the Arabidopsis DOF genes (AtDOFs) that bind to these domains.
4.3. Plant Materials, RNA-Seq, SequencingReads Alignment and Quantification
The samples were collected from the Bamboo Botanical Garden of Fujian Agriculture and Forestry University, located in Cangshan District, Fuzhou City, Fujian Province, China (N26°05′, E119°14′). The plant experiments and field studies conducted in this study, including the collection of plant material, comply with relevant institutional, national, and international guidelines and legislation. We collected fresh leaves from three D. latiflorus cutting seedlings, each propagated from stems cut from different 20-year-old bamboo plants. Each cutting seedling was over one year old, with stems exceeding 1 meter in height. These seedlings were grown under partial shade in a forest setting, without any artificial light control. We randomly selected three of these seedlings and collected mature leaves from each. Before collecting the leaves, alcohol was uniformly sprayed on both sides of the leaves to ensure complete surface coverage. The alcohol was allowed to act on the leaf surfaces for approximately 30 s to 1 minute for thorough disinfection. Subsequently, the alcohol-sprayed leaves were gently placed in deionized water to remove any residual alcohol and potential dead microorganisms from the surface, for a duration of about 2 min. The collected leaves were promptly mixed and then flash-frozen in liquid nitrogen. Subsequently, they were then stored at −80°C. Total RNA from the leaves was extracted using the RNA prep Pure Plant Kit (Tiangen, Beijing, China). The quality of total RNA was assessed by 2% agarose gel electrophoresis. RNA concentration was measured using a NanoPhotometer® spectrophotometer (IMPLEN, Westlake Village, CA, USA) and a Qubit® RNA Assay Kit with a Qubit® 2.0 fluorometer (Life Technologies, Carlsbad, CA, USA). RNA integrity was evaluated using an RNA Nano 6000 Assay Kit on an Agilent® Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, USA), setting a benchmark RNA Integrity Number of 7 as the standard for quality assessment. Library construction was carried out using the NEB-Next® Ultra™ RNA Library Prep Kit for Illumina® (NEB, USA). All samples were sequenced on the Illumina® 6000 platform, generating 150 bp paired-end reads.
We performed the initial processing of raw RNA-Seq reads using fastp v0.23.2 [24], removing sequences with adapters and sequences where more than 50% of the total length had bases with a Qphred ≤ 20, as well as sequences with an N-base percentage greater than 15%. The remaining clean reads were aligned to the reference genome using STAR v2.7.8a [62] and quantified using featureCounts v2.0.1 [63]. Then TPM value were calculated by custom python script. log2(TPM+1) was used generate heat map by TBtools v2.097 [64].
4.5. Key DlDOFs Selection and Molecular Docking
To identify the DOF TFs in D. latiflorus that potentially regulate the expression of IMR genes, we first obtained AtDOFs from the JASPAR [61] database with experimental evidence supporting their binding to the promoter regions of target genes. Subsequently, based on Blastp sequence similarity results with thresholds set at bit score > 90, p-value < 1e-30, and identity > 75%, we identified homologous proteins of these AtDOFs in D. latiflorus. Additionally, using GCEN v0.6.3 [65], we applied a PCCs threshold of ≥ 0.9 between TPM values to identify DlDOFs that co-express with IMR genes. The intersection of these two sets of genes was taken to obtain DlDOFs that are most likely to regulate the expression of IMR genes in D. latiflorus by binding to their promoter regions.
We used AlphaFold 3 [66] server to construct protein-protein and protein-DNA interaction models, Avogadro v1.2.0 [67] to build DNA models, and AutoDock Tools v1.5.7 [68] for receptor and ligand preprocessing including desolation and hydrogenation. Protein binding sites were identified using the DeepSite [69] server. For proteins exceeding 1000 residues, binding sites were manually determined. AutoDock Vina v1.1.2 [70] was employed to compute binding energies.
4.4. q-PCR Vadiation
Collected leaves used for transcriptome sequencing were placed into a mortar and ground with liquid nitrogen. Total RNA was isolated from the ground material using the RNA prep Pure Plant Kit (Tiangen, Beijing, China), and then reverse transcribed into cDNA using PrimeScript™ RT reagent Kit (Perfect Real Time, Takara, Japan). q-PCR analysis of selected genes was performed Applied Biosystems® 7500 Real-Time System (Applied Biosystems, Foster City, CA, USA) using Hieff® qPCR SYBR Green Master Mix (Low Rox Plus) kit (Yisheng, Shanghai, China). The q-PCR amplification program was: 95°C pre-denaturation for 5 minutes, 95°C denaturation for 10 seconds, 60°C annealing for 20 seconds; 72°C extension for 20 seconds, 40 cycles. The GAPDH was used as an internal reference gene [71], and the relative expression of each gene was calculated using the 2-ΔΔCT method. The primers used in this study are shown in Table S10.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1. Information of IMR Genes in D. latiflorus. Table S2. Transcription factor binding sites (TFBS) in the Promoters of IMG Genes. Table S3. Basic information for the D. latiflorus DOF members. Table S4. Motifs in the DOF binding sites. Table S5. Arabidopsis DOFs binding to sequences containing Motif1 or Motif2. Table S6. Homologous DlDOFs of AtDOFs. Table S7. Potential interactions between DlDOFs and IMR genes containing binding motif. Table S8. Molecular docking results of DlDOFs with DOF binding sites in IMR gene Promoters. Table S9. q-PCR data. Table S10. Primers used for q-PCR. Figure S1. The relative expression correlation of two DlDOFs and three IMR genes in leaves. Figure S2. The linear regression between transcriptome data and q-PCR data.
Author Contributions
P.-K. Z.: Conceptualization, Formal Analysis, Visualization, Writing - Original Draft. M.-X. L.: Data Curation, Investigation. M.-Y. Z.: Investigation, Writing- Reviewing and Editing. Y. T.: Investigation. X.-R. L.: Investigation. T.-Y. H.: Resources. Y.-S. Z.: Project Administration. L.-Y. C.: Supervision.
Funding
This research was funded by the National Key Research and Development Program of China (2021YFD2200501), Scientific Research Project of Fujian Province (2023J01478) and Forestry Peak Discipline Construction Project from Fujian Agriculture and Forestry University (72202200205). The funding agency was not involved in the design of the study, collection, analysis, interpretation of data and writing the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data supporting the findings of this work are available within the supplementary material. Please request further information from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Strader, L.; Weijers, D.; Wagner, D. Plant Transcription Factors — Being in the Right Place with the Right Company. Current Opinion in Plant Biology 2022, 65, 102136. [Google Scholar] [CrossRef]
- Ge, Y.; Chen, X.; Nan, N.; Bard, J.; Wu, F.; Yergeau, D.; Liu, T.; Wang, J.; Mu, X. Key Transcription Factors Influence the Epigenetic Landscape to Regulate Retinal Cell Differentiation. Nucleic Acids Research 2023, 51, 2151–2176. [Google Scholar] [CrossRef] [PubMed]
- Suter, D.M. Transcription Factors and DNA Play Hide and Seek. Trends in Cell Biology 2020, 30, 491–500. [Google Scholar] [CrossRef]
- Yanagisawa, S. The Dof Family of Plant Transcription Factors. Trends in Plant Science 2002, 7, 555–560. [Google Scholar] [CrossRef]
- Noguero, M.; Atif, R.M.; Ochatt, S.; Thompson, R.D. The Role of the DNA-Binding One Zinc Finger (DOF) Transcription Factor Family in Plants. Plant Science 2013, 209, 32–45. [Google Scholar] [CrossRef]
- Riechmann, J.L.; Heard, J.; Martin, G.; Reuber, L.; Jiang, C.-Z.; Keddie, J.; Adam, L.; Pineda, O.; Ratcliffe, O.J.; Samaha, R.R.; et al. Arabidopsis Transcription Factors: Genome-Wide Comparative Analysis Among Eukaryotes. Science 2000, 290, 2105–2110. [Google Scholar] [CrossRef]
- Yanagisawa, S.; Schmidt, R.J. Diversity and Similarity among Recognition Sequences of Dof Transcription Factors. The Plant Journal 1999, 17, 209–214. [Google Scholar] [CrossRef]
- Salaria, N.; Siddappa, S.; Thakur, K.; Tomar, M.; Goutam, U.; Sharma, N.; Sood, S.; Bhardwaj, V.; Singh, B. Solanum Tuberosum (CYCLING DOF FACTOR) CDF1.2 Allele: A Candidate Gene for Developing Earliness in Potato. South African Journal of Botany 2020, 132, 242–248. [Google Scholar] [CrossRef]
- Hu, G.; Wang, K.; Huang, B.; Mila, I.; Frasse, P.; Maza, E.; Djari, A.; Hernould, M.; Zouine, M.; Li, Z.; et al. The Auxin-Responsive Transcription Factor SlDOF9 Regulates Inflorescence and Flower Development in Tomato. Nat. Plants 2022, 8, 419–433. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.-X.; Xiao, S.; Chye, M.-L. The Gene Encoding Arabidopsis Acyl-CoA-Binding Protein 3 Is Pathogen Inducible and Subject to Circadian Regulation. J Exp Bot 2012, 63, 2985–3000. [Google Scholar] [CrossRef]
- Liu, J.; Cheng, Z.; Xie, L.; Li, X.; Gao, J. Multifaceted Role of PheDof12-1 in the Regulation of Flowering Time and Abiotic Stress Responses in Moso Bamboo (Phyllostachys Edulis). International Journal of Molecular Sciences 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Ma, Y.; Lu, Y.; Yue, J.; Ming, R. Expression Profiling of the Dof Gene Family under Abiotic Stresses in Spinach. Sci Rep 2021, 11, 14429. [Google Scholar] [CrossRef] [PubMed]
- Briat, J.; Curie, C.; Gaymard, F. Iron Utilization and Metabolism in Plants. Current opinion in plant biology 2007, 10 3, 276–282. [Google Scholar] [CrossRef]
- Dutt, S.; Hamza, I.; Bartnikas, T. Molecular Mechanisms of Iron and Heme Metabolism. Annual review of nutrition 2022. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Nozoye, T.; Nishizawa, N. Iron Transport and Its Regulation in Plants. Free radical biology & medicine 2019, 133, 11–20. [Google Scholar] [CrossRef]
- Peng-Kai, Zhu; Zeng, M.-Y.; Lin, Y.-H.; Tang, Y.; He, T.-Y.; Zheng, Y.-S.; Chen, L.-Y. Peng-Kai Zhu; Zeng, M.-Y.; Lin, Y.-H.; Tang, Y.; He, T.-Y.; Zheng, Y.-S.; Chen, L.-Y. Variability in Leaf Color Induced by Chlorophyll Deficiency: Transcriptional Changes in Bamboo Leaves. Curr. Issues Mol. Biol. 2024. [Google Scholar] [CrossRef]
- Fujita, Y.; Yamakawa, H. Biochemistry of Chlorophyll Biosynthesis in Photosynthetic Prokaryotes. Modern Topics in the Phototrophic Prokaryotes: Metabolism, Bioenergetics, and Omics 2017, 67–122. [Google Scholar]
- Bryant, D.A.; Hunter, C.N.; Warren, M.J. Biosynthesis of the Modified Tetrapyrroles—the Pigments of Life. Journal of Biological Chemistry 2020, 295, 6888–6925. [Google Scholar] [CrossRef] [PubMed]
- Cackett, L.; Luginbuehl, L.H.; Schreier, T.B.; Lopez-Juez, E.; Hibberd, J.M. Chloroplast Development in Green Plant Tissues: The Interplay between Light, Hormone, and Transcriptional Regulation. New Phytologist 2022, 233, 2000–2016. [Google Scholar] [CrossRef]
- Lee, S.-G.; Na, D.; Park, C. Comparability of Reference-Based and Reference-Free Transcriptome Analysis Approaches at the Gene Expression Level. BMC bioinformatics 2021, 22, 1–9. [Google Scholar] [CrossRef]
- Zheng, Y.; Yang, D.; Rong, J.; Chen, L.; Zhu, Q.; He, T.; Chen, L.; Ye, J.; Fan, L.; Gao, Y.; et al. Allele-aware Chromosome-scale Assembly of the Allopolyploid Genome of Hexaploid Ma Bamboo ( Dendrocalamus Latiflorus Munro). JIPB 2022, 64, 649–670. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, R.C.; Huang, S.-S.C.; Song, L.; Lewsey, M.G.; Bartlett, A.; Nery, J.R.; Galli, M.; Gallavotti, A.; Ecker, J.R. Cistrome and Epicistrome Features Shape the Regulatory DNA Landscape. Cell 2016, 165, 1280–1292. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Li, H.; Li, Y.; Yu, G.; Zhang, J.; Huang, B. Characterization and Transcriptional Regulation of Chlorophyll b Reductase Gene NON-YELLOW COLORING 1 Associated with Leaf Senescence in Perennial Ryegrass (Lolium Perenne L.). Environmental and Experimental Botany 2018, 149, 43–50. [Google Scholar] [CrossRef]
- Sigova, A.; Abraham, B.; Ji, X.; Molinie, B.; Hannett, N.; Guo, Y.; Jangi, M.; Giallourakis, C.C.; Sharp, P.; Young, R. Transcription Factor Trapping by RNA in Gene Regulatory Elements. Science 2015, 350, 978–981. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.A.; Jolma, A.; Campitelli, L.; Das, P.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.; Weirauch, M. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef]
- Bubb, K.; Deal, R. Considerations in the Analysis of Plant Chromatin Accessibility Data. Current opinion in plant biology 2020, 54, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W. Chromatin Accessibility and the Regulatory Epigenome. Nature Reviews Genetics 2019, 20, 207–220. [Google Scholar] [CrossRef]
- Bird, A. DNA Methylation Patterns and Epigenetic Memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef]
- Kim, M.; Costello, J. DNA Methylation: An Epigenetic Mark of Cellular Memory. Exp Mol Med 2017, 49, e322–e322. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-Wide Evolutionary Analysis of Eukaryotic DNA Methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef]
- Ngo, T.T.M.; Yoo, J.; Dai, Q.; Zhang, Q.; He, C.; Aksimentiev, A.; Ha, T. Effects of Cytosine Modifications on DNA Flexibility and Nucleosome Mechanical Stability. Nature Communications 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Medvedeva, Y.; Khamis, A.M.; Kulakovskiy, I.; Ba-alawi, W.; Bhuyan, M.S.; Kawaji, H.; Lassmann, T.; Harbers, M.; Forrest, A.; Bajic, V. Effects of Cytosine Methylation on Transcription Factor Binding Sites. BMC Genomics 2014, 15, 119–119. [Google Scholar] [CrossRef] [PubMed]
- Bowler, C.; Benvenuto, G.; Laflamme, P.; Molino, D.; Probst, A.; Tariq, M.; Paszkowski, J. Chromatin Techniques for Plant Cells. The Plant journal : for cell and molecular biology, 2004; 39, 776–789. [Google Scholar] [CrossRef]
- Samo, N.; Ebert, A.; Kopka, J.; Mozgová, I. Plant Chromatin, Metabolism and Development - an Intricate Crosstalk. Current opinion in plant biology 2021, 61, 102002. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, C.; Roqueiro, D.; Grimm, D.; Schwab, R.; Becker, C.; Lanz, C.; Weigel, D. Genome-Wide Analysis of Local Chromatin Packing in Arabidopsis Thaliana. Genome Res. 2015, 25, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Wang, J.; Zhu, Z.; Liu, Y.; Chen, J.; Zhou, Y.; Liu, F.; Lei, J.; Gaut, B.S.; Cao, B.; et al. The 3D Architecture of the Pepper Genome and Its Relationship to Function and Evolution. Nat Commun 2022, 13, 3479. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kaur, S.; Seem, K.; Kumar, S.; Mohapatra, T. Understanding 3D Genome Organization and Its Effect on Transcriptional Gene Regulation Under Environmental Stress in Plant: A Chromatin Perspective. Front. Cell Dev. Biol. 2021, 9, 774719. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Zhang, Q.; Ji, C.; Hu, G.; Zhang, P.; Wang, Y.; Yang, L.; Gu, X. Reorganization of the 3D Chromatin Architecture of Rice Genomes during Heat Stress. BMC Biol 2021, 19, 53. [Google Scholar] [CrossRef] [PubMed]
- Morgunova, E.; Taipale, J. Structural Insights into the Interaction between Transcription Factors and the Nucleosome. Current Opinion in Structural Biology 2021, 71, 171–179. [Google Scholar] [CrossRef]
- Leung, C.; He, H.; Liu, L.-J.; Wang, M.; Chan, D.; Ma, D.-L. Metal Complexes as Inhibitors of Transcription Factor Activity. Coordination Chemistry Reviews 2013, 257, 3139–3151. [Google Scholar] [CrossRef]
- Takeuchi, J.; Fukui, K.; Seto, Y.; Takaoka, Y.; Okamoto, M. Ligand–Receptor Interactions in Plant Hormone Signaling. The Plant Journal 2021, 105, 290–306. [Google Scholar] [CrossRef]
- Rasheed, F.; Markgren, J.; Hedenqvist, M.; Johansson, E. Modeling to Understand Plant Protein Structure-Function Relationships—Implications for Seed Storage Proteins. Molecules 2020, 25, 873. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; An, C.; Lawson, A.W.; Cao, Y.; Sun, Y.; Tan, E.Y.J.; Pan, J.; Jirschitzka, J.; Kümmel, F.; Mukhi, N.; et al. Oligomerization-Mediated Autoinhibition and Cofactor Binding of a Plant NLR. Nature 2024. [Google Scholar] [CrossRef] [PubMed]
- Yip, K.M.; Fischer, N.; Paknia, E.; Chari, A.; Stark, H. Atomic-Resolution Protein Structure Determination by Cryo-EM. Nature 2020, 587, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Jahn, D.; Verkamp, E.; So¨ll, D. Glutamyl-Transfer RNA: A Precursor of Heme and Chlorophyll Biosynthesis. Trends in Biochemical Sciences 1992, 17, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Skotnicová, P.; Sobotka, R.; Shepherd, M.; Hájek, J.; Hrouzek, P.; Tichý, M. The Cyanobacterial Protoporphyrinogen Oxidase HemJ Is a New B-Type Heme Protein Functionally Coupled with Coproporphyrinogen III Oxidase. The Journal of Biological Chemistry 2018, 293, 12394–12404. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Gao, S.-Y.; Jin, G.; Zhou, M.-Y.; Gao, Q.; Guo, C.; Yang, Y.-Z.; Niu, L.-Z.; Xia, E.; Guo, Z.-H.; et al. BambooBase: A Comprehensive Database of Bamboo Omics and Systematics. Molecular Plant 2024, 17, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef] [PubMed]
- Garber, A.I.; Nealson, K.H.; Okamoto, A.; McAllister, S.M.; Chan, C.S.; Barco, R.A.; Merino, N. FeGenie: A Comprehensive Tool for the Identification of Iron Genes and Iron Gene Neighborhoods in Genome and Metagenome Assemblies. Front. Microbiol. 2020, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. Journal of Molecular Biology 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The Protein Families Database in 2021. Nucleic Acids Research 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput Biol 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E. ExPASy: The Proteomics Server for in-Depth Protein Knowledge and Analysis. Nucleic Acids Research 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [PubMed]
- Horton, P.; Park, K.-J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein Localization Predictor. Nucleic Acids Research 2007, 35, W585–W587. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Muscle5: High-Accuracy Alignment Ensembles Enable Unbiased Assessments of Sequence Homology and Phylogeny. Nat Commun 2022, 13, 6968. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A Tool for Automated Alignment Trimming in Large-Scale Phylogenetic Analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Molecular Biology and Evolution 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, Analysis, and Visualization of Phylogenomic Data. Mol Biol Evol 2016, 33, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Yang, D.-C.; Meng, Y.-Q.; Jin, J.; Gao, G. PlantRegMap: Charting Functional Regulatory Maps in Plants. Nucleic Acids Research 2019, gkz1020. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Williams, N.; Misleh, C.; Li, W.W. MEME: Discovering and Analyzing DNA and Protein Sequence Motifs. Nucleic Acids Research 2006, 34, W369–W373. [Google Scholar] [CrossRef]
- Rauluseviciute, I.; Riudavets-Puig, R.; Blanc-Mathieu, R.; Castro-Mondragon, J.A.; Ferenc, K.; Kumar, V.; Lemma, R.B.; Lucas, J.; Chèneby, J.; Baranasic, D.; et al. JASPAR 2024: 20th Anniversary of the Open-Access Database of Transcription Factor Binding Profiles. Nucleic Acids Research 2024, 52, D174–D182. [Google Scholar] [CrossRef]
- Dobin, A.; Gingeras, T.R. Mapping RNA-Seq Reads with STAR. Current protocols in bioinformatics 2015, 51, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Molecular Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Li, J.; Huang, S.; Li, X.; Zhang, X.; Hu, X.; Xiang, S.; Liu, C. GCEN: An Easy-to-Use Toolkit for Gene Co-Expression Network Analysis and lncRNAs Annotation. CIMB 2022, 44, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate Structure Prediction of Biomolecular Interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J Cheminform 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational Protein–Ligand Docking and Virtual Drug Screening with the AutoDock Suite. Nat Protoc 2016, 11, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, J.; Doerr, S.; Martínez-Rosell, G.; Rose, A.S.; De Fabritiis, G. DeepSite: Protein-Binding Site Predictor Using 3D-Convolutional Neural Networks. Bioinformatics 2017, 33, 3036–3042. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Liu, M.; Jiang, J.; Han, X.; Qiao, G.; Zhuo, R. Validation of Reference Genes Aiming Accurate Normalization of qRT-PCR Data in Dendrocalamus Latiflorus Munro. PLoS ONE 2014, 9, e87417. [Google Scholar] [CrossRef]
Figure 1.
Iron Metabolism Related (IMR) Genes in D. latiflorus and the Number of Genes in Different Metabolic Categories.
Figure 1.
Iron Metabolism Related (IMR) Genes in D. latiflorus and the Number of Genes in Different Metabolic Categories.
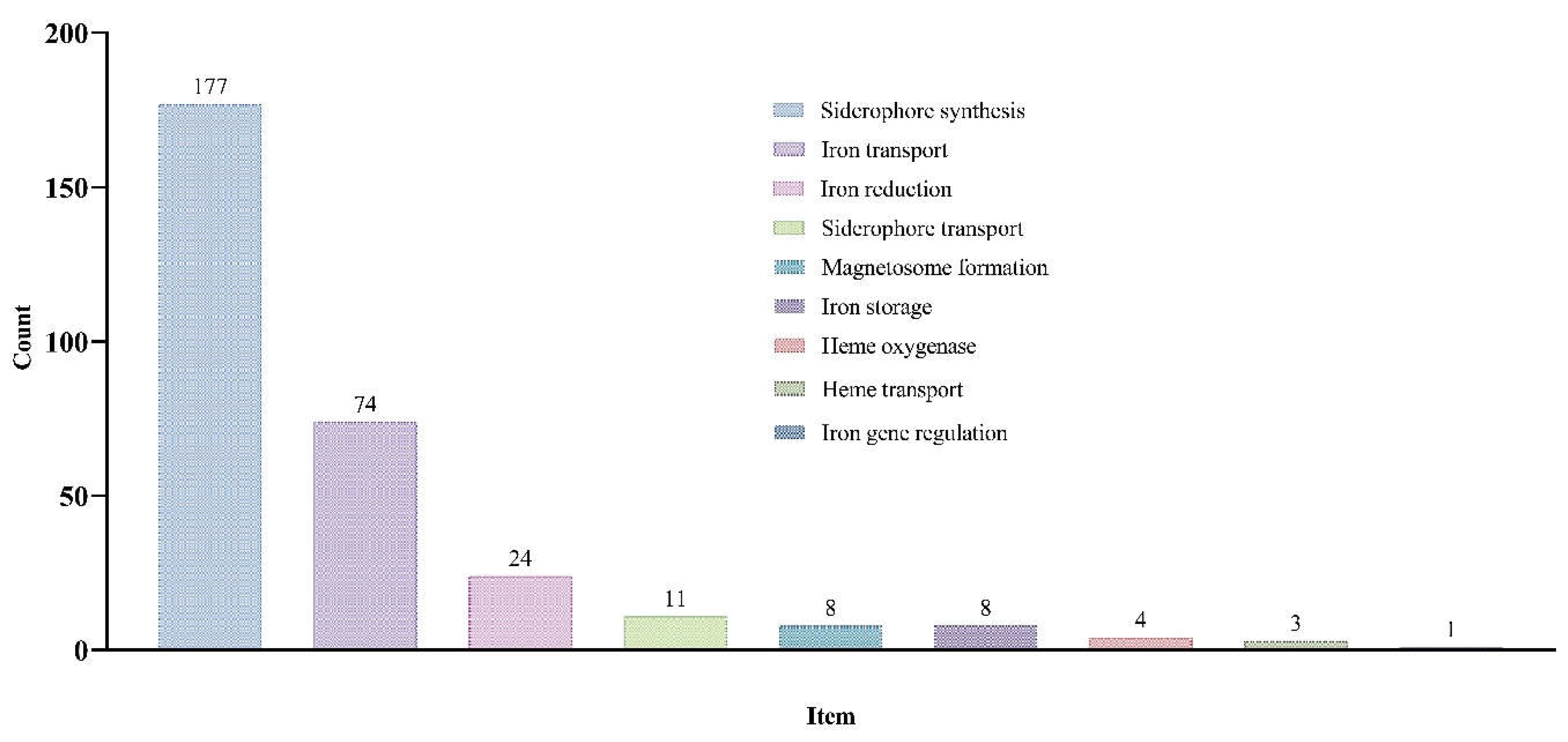
Figure 2.
The number of TF binding sites from different transcription factor (TF) families in IMR gene promoters (A), and the number of IMR genes in the promoter regions of each TF family that have binding sites (B).
Figure 2.
The number of TF binding sites from different transcription factor (TF) families in IMR gene promoters (A), and the number of IMR genes in the promoter regions of each TF family that have binding sites (B).
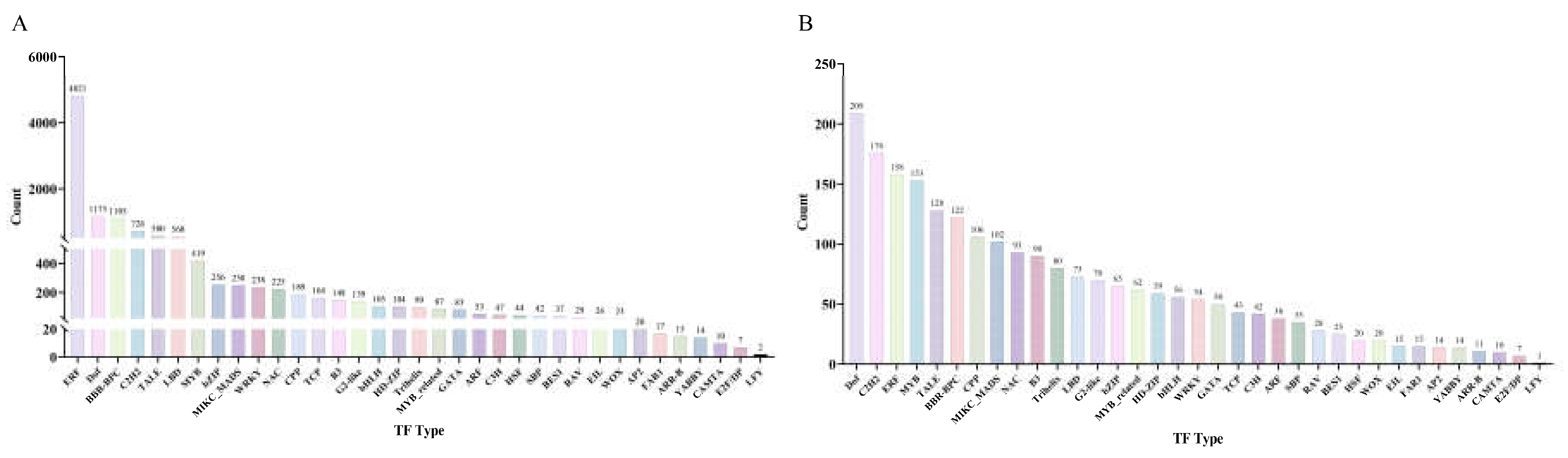
Figure 3.
The phylogenetic relationship, conserved motifs, and multiple alignment diagram of 93 DlDOFs.
Figure 3.
The phylogenetic relationship, conserved motifs, and multiple alignment diagram of 93 DlDOFs.

Figure 4.
Transcriptome analysis of genes in D. latiflorus leaves. Expression heatmap of DlDOFs in leaves (B). Expression heatmap of IMR genes in leaves (B). Co-expression network of DlDOFs and IMR genes.
Figure 4.
Transcriptome analysis of genes in D. latiflorus leaves. Expression heatmap of DlDOFs in leaves (B). Expression heatmap of IMR genes in leaves (B). Co-expression network of DlDOFs and IMR genes.
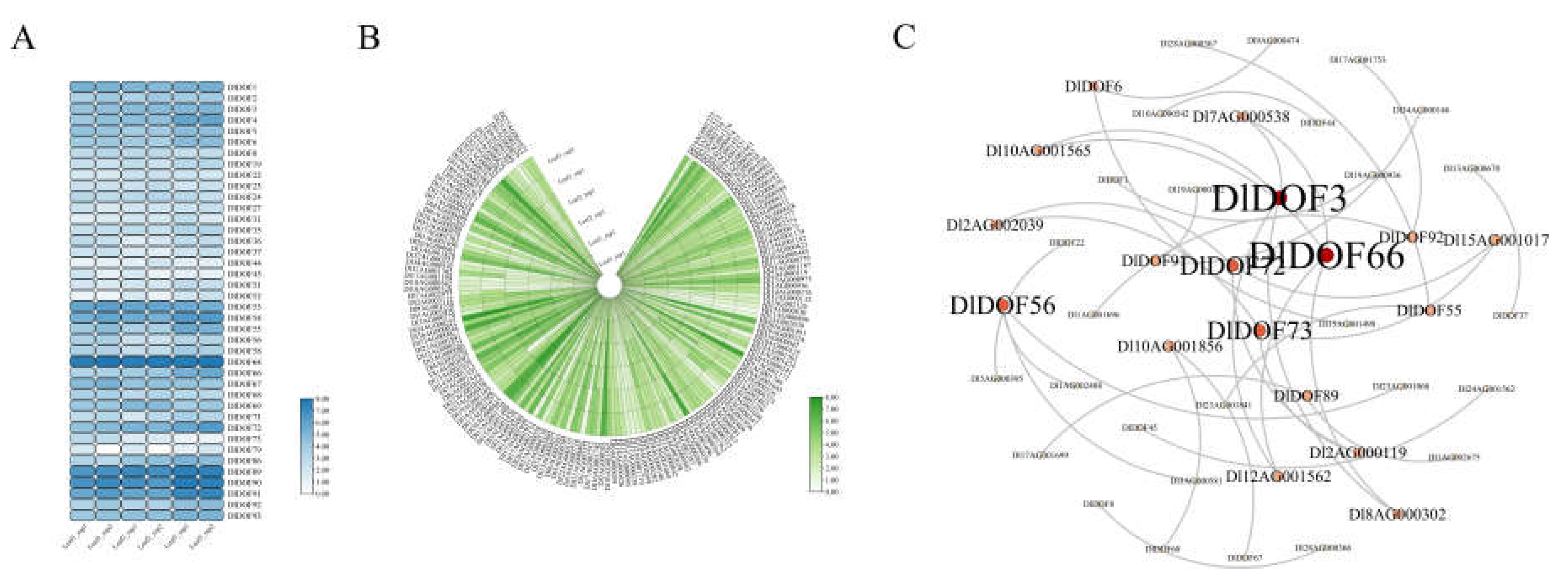
Figure 5.
Key DlDOFs selection and molecular docking. Venn diagram illustrating co-expression and potential regulatory relationships between DlDOFs and IMR genes through promoter binding (A). Green circles represent the number of co-expression relationships, while red circles represent potential regulatory relationships. Protein-DNA interaction models (B). Text below the model indicates the motif types present in transcription factor binding sites (TFBS) and the binding of DlDOFs to IMR gene promoters.
Figure 5.
Key DlDOFs selection and molecular docking. Venn diagram illustrating co-expression and potential regulatory relationships between DlDOFs and IMR genes through promoter binding (A). Green circles represent the number of co-expression relationships, while red circles represent potential regulatory relationships. Protein-DNA interaction models (B). Text below the model indicates the motif types present in transcription factor binding sites (TFBS) and the binding of DlDOFs to IMR gene promoters.
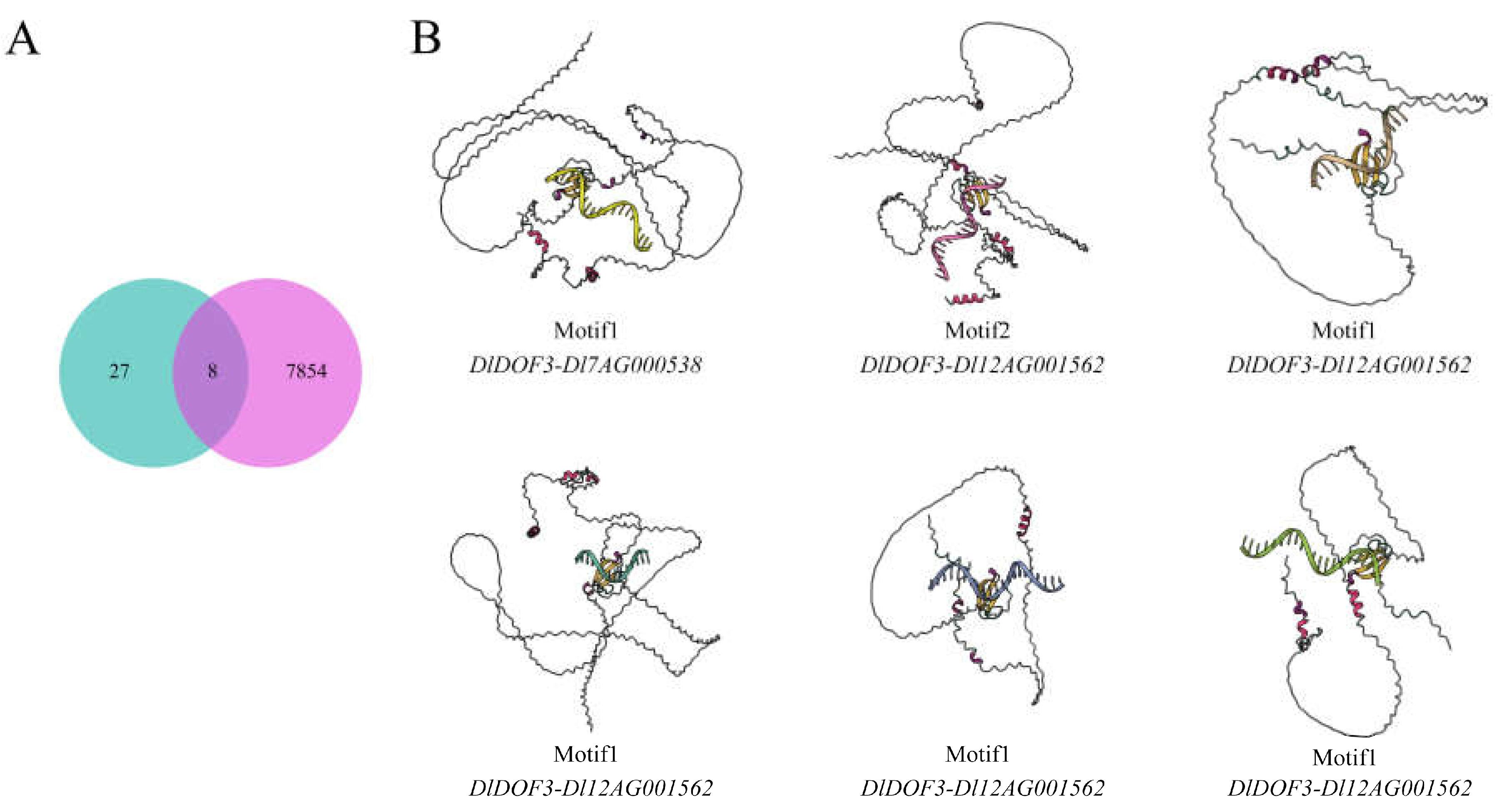
Figure 6.
Relative expression patterns of two DlDOFs and three IMR genes in leaves.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



