Submitted:
26 June 2024
Posted:
26 June 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Oxidative stress (OS) occurs from excessive reactive oxygen species or a deficiency of antioxidants – primarily endogenous glutathione (GSH). There are many illnesses, from acute and post-COVID-19, diabetes, myocardial infarction, to Alzheimer’s disease, that are associated with OS. These dissimilar illnesses are in order, a viral infection, a metabolic disorder, an ischemic event, and a neurodegenerative disorder. OS may be an early initiator or a significant promotor of their progressive pathophysiologic processes. Consequently, addressing initial OS-related pathology may avoid subsequent severe and irreversible complications. Early intervention would modulate multiple factors, including cytokine stimulation, complement dysregulation, immuno-thrombosis, fibrosis, and autoimmunity. Past clinical studies that addressed OS have had equivocal results. It may be due to the use of ineffective antioxidants, an untimely delay in their use, or an inefficient delivery system. Experimental studies have used antioxidants that are not available for clinical trials. Although GSH can significantly reduce OS, its exogenous use is hampered by the lack of an effective delivery system. By incorporating GSH into cyclodextrin (CD) through nanotechnology, there is recent evidence that exogenous GSH can be delivered more effectively to prevent or mitigate injury from OS [1]. Future studies using the GSH/cyclodextrin (GC) complex will be needed to confirm its effectiveness and, hopefully, offer a path to treat many sub-optimally managed or currently untreatable illnesses.
Keywords:
Oxidative stress
; reactive oxygen species
; antioxidants
; glutathione
; glutathione-cyclodextrin complex
1. Introduction
As our primary intracellular antioxidant, GSH is integral in maintaining our health and well-being against oxidative stress (OS) [2]. OS results from a high level of reactive oxygen species (ROS) relative to antioxidants. Although ROS is beneficial at physiologic levels, its excess leads to OS. OS plays a role in our immune response (innate, adaptive, and autoimmune) that may be physiologic or pathologic [3,4,5]. OS and GSH deficiency are associated with advanced age and diseases of the brain, heart, lungs, eyes, liver, kidneys, and the nervous and vascular systems [6,7,8,9,10,11]. OS-related injuries can be acute (infections, systemic inflammatory response syndrome, ischemia-reperfusion injury) or chronic (idiopathic pulmonary fibrosis, atherosclerosis, diabetes mellitus, and neurodegenerative diseases) [2,6,10,12,13,14,15]. Physiologically, OS is suppressed by antioxidants, primarily endogenous GSH, our body’s “master antioxidant” [2,10,16]. OS may not be a marker but an initiator or promoter of these diseases. Thus, inhibiting the development of OS would be simpler and more effective than addressing each specific products of the subsequent complex and dynamic inflammatory response. Finally, providing therapeutic levels of exogenous GSH could address the pathophysiologic processes of these diseases and insults before irreversible injury occurs [9,12,17]. Evidence has been provided that a novel agent, the GSH/cyclodextrin (G/C) complex, can effectively increase GSH levels and counter OS [1]. This complex is incorporated in gamma-cyclodextrin (γ-CD), is easily administered topically, and avoids the limitations of oral or IV GSH, and the need for enzymatic conversion of N-acetyl cysteine (NAC) [6,12,16,17,18]. In contrast to the G/C complex, NAC does not increase GSH levels unless there is a depletion of GSH and a functional enzymatic pathway for de novo synthesis of GSH [19]. Hopefully, delivering GSH directly as the complex will prompt studies to determine the efficacy of exogenous GSH to reverse OS. Significantly, GHS deficiency (and the associated elevated GGT as a biomarker) may not only related to aging and diseases, but restoring GSH could be a potential therapeutic target [20].
2. Oxidative Stress (OS)
2.1. Reactive Oxygen Species and OS
At a physiologic level, ROS regulates aging and activates the innate inflammatory response. 3 This innate response, in turn augments the secondary adaptive immune response as shown in Figure 1 [5]. ROS and reactive nitrogen species (RNS) include both radical forms (e.g. superoxide, hydroxyl radical, and nitric oxide) and non-radical forms (e.g. hydrogen peroxide, hypochlorous acid, and peroxynitrite) [5,15]. Although the non-radical species are not directly damaging to cells, they cause oxidative damage when converted to their radical form (usually after reacting with metal ions). As signaling molecules, ROS/RNS regulate or mediate many physiologic functions. These include gene activation, cellular growth, and blood pressure control [15]. The duality of ROS in diseases, whether physiologically beneficial or pathologically harmful, has been outlined [8]. The “Goldilocks” principle of adaptive vs deleterious ROS response using an exercise model has been presented [21]. The level of ROS, which in excess leads to OS, appears to be the determining factor.
OS occurs when the biological system creates excessive ROS, and to a lesser extent RNS, that alters the normal homeostatic balance between pro-oxidant and anti-oxidant molecules as shown in Figure 2. ROS/RNS are produced in response to external stimuli. OS damage to DNA and other biomolecules may impair normal functions of tissue cells and lead to human aging and disease [2,10]. Under adaptive physiologic conditions, excessive ROS can be controlled by enzymatic (superoxide dismutase, catalase, glutathione peroxidase) and non-enzymatic (glutathione) antioxidants. GSH is well-established as the most important cellular antioxidant [2,10,12,16,22]. ROS is primarily generated in mitochondria, but it is also produced in peroxisomes, endoplasmic reticulum, and lysosomes [15].
There are many human disorders clinically linked to OS [2,6,10,11,15]. These include those of the brain (e.g. Alzheimer’s, Parkinson’s, amyotrophic lateral sclerosis, multiple sclerosis, autism, encephalitis, and both thrombotic and hemorrhagic stroke), the liver (non-alcoholic steato hepatitis – NASH and hepatic encephalopathy), the pancreas (pancreatitis, diabetes, and its complications), the lungs (COVID-associated ARDS, idiopathic pulmonary fibrosis, cystic fibrosis), the eyes (macular degeneration, retinitis pigmentosa), pathologic aging (Huntington-Gilford progeria syndrome, Werners syndrome), certain cancers (skin, kidney, bowel, and breast) and the skin (melanin abnormalities). Additional disorders include organ failure (renal, hepatic), age-related frailty (“inflammaging”), and disorders of the cardiovascular system (hypertension, dyslipidemia, atherosclerosis, myocardial infarction, restenosis, myocarditis, ischemia/reperfusion injury, and thrombosis), autoimmunity (rheumatoid arthritis, fibromyalgia), and the inhibition of viral replication (HIV, influenza and herpes simplex) [2,4,6,9,10,11,12,13,14,23,24,25]. Examples of the influence of OS on diseases is illustrated in Figure 3.
There is an inverse relationship between GSH level and OS in diseases [10,16,17]. Congenital GSH synthetase deficiency leads to severe con sequences (recurrent bacterial infections, mental retardation, motor disturbances, retinal pigmentation) that highlights the pathologic sequelae of GSH deficiency. The brain is highly susceptible to OS, likely due to high oxygen consumption and a low GSH level compared to other tissues. However, the limitations of current therapies have been highlighted [10].
2.2. Immunothrombosis and Thromboinflammation and OS
A significant response to OS is immunothrombosis, and its inflammatory consequence known as thromboinflammation. Although activation of the coagulation cascade is our natural hemostatic defense mechanism, pathologic immune activation can lead to numerous thrombotic diseases of the macrovascular and microvascular systems [26]. Diseases associated with pathologic thrombosis include those of the veins, coronary arteries, and lungs [17,23,26,27,28,29,30,31]. Immunothrombosis is activated as a defense response to bacterial or viral infection and other immune response triggers [26,31]. Macrovascular events include myocardial infarction, stroke, venous thromboembolism, and pulmonary embolism. Microvascular thrombosis is involved in sepsis, ischemia-reperfusion injury (IRI), organ transplantation rejection, major trauma, severe burns, antiphospholipid syndrome, preeclampsia, and sickle cell disease [26,30].
The consequent thromboinflammation causes the activation of platelets and innate immune cells that initiates a coagulation cascade involving the complement system (C’) and tissue factor (TF) [17,23,29,30,31]. This process is a prominent feature of ARDS from severe COVID-19 [17]. Severe respiratory symptoms of COVID-19 illness are associated with elevated D-dimer and von Willebrand Factor (vWF) levels, complement activation, and antiphospholipid antibodies [17,23]. Additionally, in arterial thrombosis, OS-induced endothelial injury initiates a coagulation cascade (involving tissue factor, collagen, and (vWF) that leads to thrombin formation [23,32].
Thromboinflammation occurs in both acute and long-COVID diseases of the liver, lungs, and the cardiovascular system [23,33,34,35,36,37,38,39,40]. The developmental sequence has been presented [33,34,41]. It is triggered by external insults (e.g. tobacco, asbestos/silica, radiation, viruses, or bleomycin) followed by macrophage activation, CS, pneumocyte apoptosis, fibroblast recruitment, and collagen deposition. OS is a significant molecular component of fibrosis [35,41,42]. Idiopathic pulmonary fibrosis (IPF), a rare disease without a known initiator, has been noted to have similarities with lung fibrosis from severe COVID-19 and post-COVID pulmonary fibrosis (PCPF) [33,34,42].
The risk of venous thromboembolism increases with age and is influenced by OS in red blood cells (RBC). High levels of ROS disturb RBC membrane structure and function, resulting in loss of membrane integrity and decreased deformity that produces a hypercoagulable state marked by enhanced RBC aggregation, RBC binding to endothelial cells (causing impairment of nitric oxide (NO)-dependent vasodilatation), RBC-induced activation of platelets, complement, and TF [13,17,29,31].
In addition to the process of atherosclerosis, ROS is involved in other pathologic cardiovascular events, including the neointima formation after balloon angioplasty with stenting and possibly the development of abdominal aortic aneurysm [43]. The International Society on Thrombosis and Haemostasis Congress presented an overview of the distinction between immunothrombosis and thrombo-inflammation [44]. Although both involve platelet aggregation, the former primarily involves the venous system (e.g. cerebral venous thrombosis) and the latter the arterial system (e.g. ischemic stroke). However, it appears that they are clinical “bookends” of an integrated pathophysiologic continuum.
2.3. The Complement System and OS
3. OS-Associated Diseases
3.1. COVID-19
As previously mentioned, GSH plays a significant role in the immunothrombotic process involving multiple organs and the vascular system, particularly COVID-19 [10,16,17,46]. GSH deficiency and OS are involved in the pathologic consequences of acute and chronic COVID-19 illness [16,17,47,48]. Acute COVID induces an unchecked cytokine cascade leading to the cytokine storm (CS) that is driven by OS [49,50]. Although not firmly established, post-COVID complications apparently involve a persistent antigenic stimulus leading to what is commonly known as long-COVID and officially as post-acute sequelae of COVID-19 (PASC), arterial thrombosis, and microvascular thrombosis associated with activation of vWF [17,49,51]. VWF is not only a biomarker of severe acute pulmonary injury but also of persistent respiratory symptoms in long-COVID [52]. Finally, complement dysregulation has been associated with long-COVID [47].
Using COVID-19 as a model, the significance of GSH as a critical antioxidant against OS, the association of its depletion by oxidation, and its decreased synthesis have been reviewed [16,17]. This decrease is inversely related to the severity of acute and chronic illnesses. GSH deficiency is associated with TGFβ activity in severe COVID-19 illness. TGFβ causes low levels of GSH by repressing the enzyme γ-glutamyl-cysteine ligase (GCL) required for de novo synthesis of GSH [16,17,53]. Impaired synthesis is also driven by hypoxia that decreases the two enzymes that catalyze the de novo synthesis of GSH (GCL and GS) [16]. Significantly, the lack of studies using GSH was recognized.[54] This exposes the current inability to clinically deliver GSH safely, efficiently, and therapeutically. The efficacy of a novel topical GSH delivery system that appears to overcome this problem has been published [1]. Through nanotechnology, GSH has been incorporated into cyclodextrin (CD) to produce the GSH/CD (G/C) complex. This preliminary study with the complex demonstrated its efficacy in young, healthy individuals to 1) deliver GSH into RBCs, 2) effect OS as demonstrated to lower lipid peroxidation as measured by malondialdyhyde (MDA), and 3) inhibit Mycobacterium avium in vitro. However, its use in large, rigorous clinical trials is needed to confirm if GSH, using the G/C complex, can address OS in current untreatable or sub-optimally treated illnesses. Significantly, it has been confirmed that cyclodextrin can cross the blood-brain barrier (BBB) [55]. As previously stated, the place of the complex in disease management would be its early use in acute illness to address OS before it leads to irreversible tissue damage and its continued use in chronic illness.
3.2. Acute COVID-19
The complex nature of the immuno-thrombotic process has been detailed in previous reviews [16,17,44,56]. Additionally, the inverse relationship of OS with a low glutathione level has been well acknowledged [16,17,48,57,58]. A low GSH level is associated with corresponding COVID-19 disease severity in elderly individuals and those with diabetes, hypertension, and obesity [2,26,46,53,59]. SARS-CoV-2 virus stimulates ROS production through multiple pathways, leading to enhanced production of pro-inflammatory cytokines and the subsequent cytokine storm (CS) [60,62]. Pro-inflammatory cytokines stimulate the inflammatory process by activating the nuclear kappa B (NF- κB) signaling pathway, resulting in a cyclical positive feedback loop [60].
The severe symptoms of acute COVID-19 illness, particularly ARDS, are a consequence of thromboinflammation resulting from immune dysregulation, inflammation, and endothelial cell damage [16,17]. The dysregulated immune response leads to a hyperactive innate immune and hypercoagulable state that involves endothelial damage followed by platelet aggregation and a coagulation cascade involving fibrin and thrombin that is initiated by activated von Willebrand Factor (vWF). A fibrin degradation product, D-dimer, is used as a marker of COVID-19 severity. The hyperinflammatory state from CS often leads to profound pulmonary inflammation and subsequent endothelial damage and platelet aggregation. This thromboinflammation process leads to ARDS in acute COVID-19 and likely contributes to respiratory symptoms of long-COVID [17]. As mentioned earlier, a higher level of TGFβ present in COVID-associated ARDS would contribute to lower levels of GSH in COVID-19 illness [16]. Current management of COVID-19-related ARDS is ventilatory support, either baricitinib or tocilizumab, and dexamethasone.[62] Significantly, the concentration of endogenous GSH in the alveolar epithelial lining fluid of ARDS patients is 5% of comparable healthy individuals [9]. Immunothrombosis biomarkers (specifically levels of tissue factor and vWF) could predict the development of ARDS in moderate-to-severe COVID-19 [51]. Clinically available laboratory markers identifying inflammation, coagulopathy, or tissue injury include neutrophil-lymphocyte ratio, ferritin, C-reactive protein, D-dimer, aspartate aminotransferase, and troponin [58,63,64,65]. Hence, preventing ARDS by avoiding OS-induced cytokine release with early administration of GSH may be an effective strategy to prevent severe consequences of COVID-19 multi-organ injury.
Ideally, COVID-19 illness should be prevented or attenuated with vaccines. Next, to halt the progression of the illness with severe complications requiring hospitalization. Finally, to prevent organ damage in seriously ill high-risk individuals, particularly respiratory failure from ARDS, that can lead to death. Vaccination usefulness is limited by the constant appearance of new COVID-19 variants, its diminished efficacy in those immune-suppressed, and avoidance by those adverse to vaccines. Once COVID-19 is contracted, Paxlovid is available to high-risk individuals. Although it has been shown to decrease the risk for severe disease progression and may prevent long-COVID, it exhibits drug-drug interactions. It is known to occasionally have a peculiar “rebound” phenomenon of COVID-19 positivity after the recommended 5-day course. Its use has been recently reviewed [66]. Using the GSH-CD complex overcomes many of these limitations and presumably may be administered with or instead of Paxlovid, baricitinib or tocilizumab, and dexamethasone. The complex is an immunomodulator and does not cause immunosuppression. Significantly, the timing and duration of its use is not a critical factor.
Although cyclodextrin (CD) is the transport vehicle in the G/C complex, there is evidence that it may inhibit cellular entry of the SARS-CoV-2 virus. CD may inhibit the viral attachment to the ACE-2 receptor and its subsequent endocytosis entry by altering cholesterol microdomains on the cellular membrane [67].
3.3. Long COVID
New, persistent, or relapsing symptoms following an initial acute COVID-19 illness are officially known as post-acute sequelae of COVID-19 (PASC) or, more commonly, as long-COVID (LC). Due to its complex nature, it is not rigorously defined. Still, it is understood to be a constellation of persistent symptoms that occur more than three weeks after the initial acute exposure to the SARS-CoV-2 virus. The risk of developing PASC is unrelated to the severity of the initial COVID-19 illness. It can occur after mild illness and to those without acute symptoms [68]. The most prominent symptoms include post-exertional malaise (PEM), fatigue, brain fog, dizziness, palpitations, headache, gastrointestinal disturbance, diminished sexual desire or capacity, loss or disturbance in smell or taste, dyspnea, and chronic cough [68,69,70,71]. In addition, there was an increased risk of neuropsychiatric and autoimmune disorders associated with OS [41,71,72,73]. Due to the persistence of these debilitating symptoms, it has become recognized as a second or “rebound” epidemic. It affects the lives and livelihoods of tens of millions of individuals [74]. A review of the presumptive pathophysiologic mechanisms of PASC has included the sequelae of direct viral injury, immune system dysregulation, endothelial dysfunction, mitochondrial metabolic dysregulation, reactivation of harbored pathogens, autoimmunity, and functional changes in blood cells has brought into focus the risks of developing neurologic symptoms similar to myalgic encephalitis/chronic fatigue syndrome (ME/CFS) and post-treatment Lyme disease syndrome (PTLDS) [68,71,75]. Unlike ME/CFS and PTLDS, the sequelae of LC are more complex since they involve the added consequence of tissue injury from acute COVID-19 illness and its influence on exacerbating or precipitating underlying diseases (e.g., diabetes, cardiovascular, and pulmonary) [75]. The similarities and differences between ME/CFS and COVID have been covered in comprehensive reviews [68,75]. Overlapping symptoms with ME/CFS include fatigue, PEM, headaches, impaired reasoning or “brain fog”, myalgia/arthralgia, orthostatic intolerance, nausea/diarrhea, and cough [69]. Symptoms peculiar to long-COVID include decreased or distorted smell and taste, rash, and hair loss. Those peculiar to ME/CFS include painful lymph nodes and tinnitus [75]. Likewise, a comparison of ME/CFS to PTLDS has been presented [76]. There is evidence to support that they all follow a trigger of autoimmunity that stems from the effects of persistent T-cell activation, and in COVID, may additionally reflect the antigenic stimulation by residual intact viruses or viral remnants [68,72,77,78].
Further elucidation of the presumptive mechanism of long-COVID was reported in an exhaustive prospective clinical analysis of 113 COVID-19 vs 39 healthy controls followed for up to a year after initial confirmation of acute SARS-CoV-2 illness. Over 6500 biomarkers were analyzed. This study that long-COVID patients exhibited an enhanced complement activation during the acute illness that persisted at 6-month follow-up [47]. Furthermore, long-COVID patients had evidence of persistent thromboinflammation with its characteristic endothelial activation of vWF and thrombin cascade, platelet aggregation, and prolonged T-cell activation [52,77]. Finally, these patients had an associated increased IgG antibody level against xxx, suggesting a predisposing risk factor. Similar to ME/CFS, the presence of cytomegalovirus or Epstein-Barr virus antibodies is a possible risk factor of long-COVID [79]. Significantly, the role of GSH in inhibiting antibody and complement-mediated immunologic cellular injury has been presented [80].
4. Diabetes and Its Complications
4.1. Diabetes Management
The treatment goals of diabetes management are to avoid the personal and healthcare burden of its neurovascular complications. These complications result from hyperglycemia (both acute and chronic) that leads to OS and the subsequent interconnected inflammation, endothelial dysfunction, and hypercoaguability [82,83,84]. The evolving view is that diabetes is a metabolic disease resulting from chronic inflammation driven by OS [84,85]. This chronic inflammation has been implicated in the development of insulin resistance and diabetes complications (DC) [86,87]. The pathological basis of DC has historically been dichotomized as micro- or macro-vascular. These include microvascular (retinopathy, nephropathy, and neuropathy) and macrovascular (coronary heart disease and stroke). A more holistic concept of the disease processes as “panvascular” was proposed. It recognized that micro- and macro-vascular pathologies often coexist and are interactive [84].
The current target of optimal glucose control is an A1c of <7, which is higher than the A1c of <5.4 of non-diabetic individuals. The higher A1c treatment goal is to avoid severe and potentially fatal episodes of hypoglycemia. Continuous glucose monitoring has reduced this risk for those using insulin. Episodes of hypoglycemia in type 2 diabetes have been reduced by the substitution of sulfonylurea/meglitinide medications with newer agents (e.g., dipeptidyl peptidase four inhibitors (DPP4i), glucagon-like peptide-1 receptor agonists (GLP1ra), and sodium/glucose transporter two inhibitors (SGLT2i). However, the goal of normalizing A1c is often difficult to attain – primarily due to insulin resistance (IR).
OS has been implicated in the development of IR and β-cell dysfunction [86,88,89]. Of note, oscillating glycemic levels from high post-prandial glucose from IGT is strongly linked to both β-cell dysfunction, IR, and cardiovascular disease (CVD) [90]. IR itself is an independent risk factor for CVD that is associated with mitochondrial dysfunction [83]. From the longitudinal studies on diabetes care, it was determined that once diabetes is established, A1c control alone may not prevent complications of diabetes due to long-lasting epigenetic changes that drive persistent expression of proinflammatory genes after glycemia is normalized, known as “hyperglycemic memory” [83].
4.2. Diabetes Complications
Without normalization of hyperglycemia, protein glycation leads to the formation of advanced glycation end-products (AGEs), which induces OS in a positive feedback loop. AGEs are heterogeneous molecules formed from irreversible, non-enzymatic glycation of proteins, lipids, and nucleic acids [83,88,91,92].
AGEs contribute significantly to the vasculopathies associated with diabetes. On a macrovascular level, this includes impaired vasculogenesis in response to ischemia, resulting in defective generation of collateral blood flow. This impaired adaptive response increases the risk of limb amputations and myocardial infarction [83]. Upon binding to receptors for AGEs (RAGE), an inflammatory and pro-coagulant state is initiated, which causes endothelial activation. The resultant expression of inflammatory cytokines and adhesion molecules amplify the inflammatory response. Among the cytokines are TNF-a, IL-1, and IL-6, which stimulate the release of pro-coagulant molecules and inhibit the expression of anti-coagulant molecules. This leads to microvascular disturbances, including lipid deposition, sclerosis, and impaired vasodilation. Similar to the processes of ARDS in COVID-19, there is an activation of vWF from damaged endothelial cells that stimulates platelet aggregation [82]. Significantly, high levels of vWF in patients with diabetes appear to be a predictive biomarker of diabetic nephropathy. A more detailed treatise of the role of AGEs in causing the various neuro-vascular diabetes complications has been presented [91]. The authors reviewed the role of the transcription factor – nuclear factor kappa B (NF-κB) in triggering the development of OS, cytokines and chemokines, cell adhesion molecules, interleukins, tissue growth factor (TGF), pro-inflammatory proteins, and pro-apoptotic genes [91].
The definition of the metabolic syndrome (MetS) and its CVD risks has been outlined [93]. These include insulin resistance, obesity, abnormal lipid profile, hypertension, and impaired glucose tolerance. Clinical criteria for diagnosing MetS were also presented. Furthermore, evidence has been observed that OS links IR and the pathology associated with the MetS [85]. These include OS due to lipid peroxidation, AGE formation, and decreased antioxidant protection, resulting in CVD complications from monocyte activation, vascular smooth muscle proliferation, and endothelial cell dysfunction. The latter additionally leads to arterial hypertension and plaque formation [85].
MetS is associated with nonalcoholic fatty liver disease (NAFLD) and its inflammatory form, nonalcoholic steatohepatitis (NASH). NASH appears to follow the “two hit” phenomenon where the initial hepatic lipid infiltration is followed by OS which leads to tissue injury and subsequent fibrosis, which is associated with enhanced lipid peroxidation [85].
Analogous to the concept expressed by Grattagliano, a detailed presentation by Giacco hypothesizes that OS plays a pivotal role in micro- and macro-vascular diabetes complications [83,85]. ROS, particularly superoxide production by mitochondria, is involved in the pathogenesis of polyneuropathic and nephropathic complications [83,94,95]. These include activation of five major pathways: increased flux of glucose and other sugars via the polyol pathway, increased formation of AGEs, increased expression of RAGE and its activating ligands, activation of protein kinase C (PKC) isoforms, and overactivity of the hexosamine pathway. It has been summarized as a being generated by “glycoxidation”, a combination of glycation and oxidation driven by OS [92]. Microvascular pathology is a consequence of intracellular hyperglycemia and macrovascular from increased oxidation of fatty acids [83]. It appears that a common upstream event triggers all five mechanisms – hypothetically, OS from overproduction of ROS by mitochondria. Significantly, in experimental animal models, treatment with agents directly opposing SOD normalized all five pathways. GSH, as being the most essential and abundant low molecular intracellular ROS scavenger, has been noted [92]. The efficacy of exogenous administration of GSH with the GSH/CD complex remains to be determined.
The role of OS in diabetic nephropathy, neuropathy, and retinopathy has been reviewed [84,94,96,97,98,99]. Presumably, if GSH can prevent the pathologic consequence of glycation resulting from OS, it can be used in conjunction with glucose control to prevent diabetes complications. In essence, to simultaneously treat the cause and effect of hyperglycemia [84]. The dose and cost of insulin and oral medications could be reduced by addressing insulin resistance. Prevention or a delay in the onset of diabetes complications could be achieved by preventing protein glycation. The incidence of neurovascular complications, including macrovascular (stroke, myocardial infarction) and microvascular (blindness, chronic kidney disease) should decline. The socio-economic impact of reducing the burden of caring for individuals with diabetes complications and the avoidance of their impaired quality of life would be substantial. OS plays a critical role in the progression of diabetes complications. What is lacking is an effective antioxidant to address it [89,100,101].
4.3. Neurologic/Neurodegenerative Disorders
The healthcare impact of neurodegenerative disorders can not be understated. The burden of Alzheimer’s disease (AD) and related dementias (including AD, vascular dementia, Lewey body dementia, and frontotemporal dementia) is present in an estimated 6.5 million individuals in the US. This incidence is projected to increase to 13.8 million individuals by 2060 [102]. GSH deficiency and OS have been associated with many degenerative neurologic illnesses, including AD, Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), multiple sclerosis (MS), and autism spectrum disorders (ASD) [103,104,105,106,107,108,109,110,111]. Aoyama et. al. reviewed the association of neural GSH deficiency with Alzheimer’s disease (AD), Parkinson’s disease (PD), and (ALS) [105]. Algahtani et.al., described the association of mitochondrial dysfunction and OS in AD, PD, HD, and ALS [112]. Underscored was the role of GSH in counterbalancing OS. Using proton magnetic resonance spectroscopy (1H-MRS) to detect GSH levels in vivo, evidence was presented of GSH deficiency in the brains of AD, PD, and ALS individuals. Expressly, a lower level of GSH was noted in the temporal and parietal lobes of older adults, with a negative correlation of GSH levels to Aβ levels as assessed with positron-emission tomography (PET) imaging. In PD, there is post-mortem evidence of decreased GSH levels in the substantia nigra of the midbrain. Finally, again using 1H-MRS, a decrease in GSH levels was noted in the motor cortex and corticospinal tract of individuals with ALS [105].
4.4. Alzheimer’s Disease
Alzheimer’s disease is a neurodegenerative disorder with neuropathologic hallmarks that include amyloid beta (Aβ) plaques, neurofibrillary tangles (NFT, synapse and neuronal loss, neuroinflammation, neurotransmitter deficiencies, and reactive astrogliosis that eventually lead to cognitive impairment [113]. Although classical AD comprises 85% of cases, there are other phenotypes, including the lipogenic variant of primary progressive aphasia, posterior cortical atrophy, corticobasal syndrome, and frontal AD [113]. There is growing evidence of the involvement of mitochondrial dysfunction and OS early in the disease progression, including elevated levels of OS markers [114,115]. This includes increased malondialdehyde (MDA), a lipid peroxidation product, early in AD [116]. The treatment of Alzheimer’s disease (AD) with disease-modifying therapies (DMT) has been disappointing [117,118,119]. In addition to neurotransmitter enhancement, disease management has been mainly directed against Aβ deposition and NFT formation. This approach may address AD at a point of the disease process that is difficult to modify or reverse. That is, Ab plaques and NFT formation may be the result and not the cause of AD pathology [120]. Rather than treating AD- associated damage to neurons, avoiding the pathologic insults of Aβ and neurofibrillary tangles may be more effective by preventing their formation. Cheong et al. highlighted the complex and multifactorial disease process and outlined in Figure 1 (p. ) the current disease hypotheses (amyloid cascade, tau formation, excitatory, and cholinergic) that direct current therapeutic interventions. DMT treatments are directed against Ab and tau, and memory deficits are addressed with neurotransmitter therapy [121].
Current established treatments address neurotransmitter imbalance. Anti-amyloid therapies have been disappointing. It has been proposed that this approach may be too little and too late [117,118,122]. This may require addressing AD early, at its pre-clinical stage, preceding mild cognitive impairment directed against OS [28,120,123,124,125]. The obvious barrier to this approach is having methods to identify those at-risk individuals before symptoms of dementia are evident [113]. Encouragingly, early markers and imaging methods have been identified. These include PET scans, functional and proton MRI, AD associate gene markers, ocular imaging, and MDA (a biomarker of lipid peroxidation) [105,106,113,126,127,128,129,130,131,132]. The measurement of a particular combination of CSF biomarkers has a sensitivity of 95% and specificity of 83% and is used in clinical trials to detect individuals who will develop AD [117]. Unfortunately, these interventions are still research tools and are not yet available in routine clinical practice due to their cost, access, or invasiveness. Ocular examination as a non-invasive method for early AD detection has been proposed [128,133,134,135]. OS has been recognized as the initial and a driving force in Aβ deposition, and Aβ in turn, may be a compensatory response to OS [113,116,136]. The scientific conundrum of which came first, Aβ or OS, would be a moot issue if early reversal of OS prevents Aβ deposition and neurofibrillary tangle formation before their burden causes clinical symptoms [28,115,137]. The pathophysiology of AD is complex, is not fully understood, and warrants the studies of alternate disease mechanisms and the use of combination therapies [120,121,132,138,139,140,141,142,143].
In a detailed review, Walker et al. described in detail the role of peripheral inflammatory insults that increase the future risk of developing Alzheimer’s disease [144]. The insults that cause peripheral inflammation could be an acute infection (e.g. COVID-19, pneumonia, septicemia) or a sterile event (e.g., surgery, orthopedic injury, acute coronary syndrome). If the insult leads to the complication of hypoxia, it would trigger additional IRI-associated inflammation. Another risk factor is age-related immunosenescence, which leads to upregulation of inflammation. The group further describes the subsequent deterioration of the blood-brain barrier (BBB) that would allow cross-talk of the peripheral with the central nervous system inflammatory events. They propose that Alzheimer’s disease can be a result of an acute or chronic inflammatory process. Acutely by triggering innate immune activation and chronically through chronic cytokine expression [144]. Through several meetings of the Alzheimer’s Association Research Roundtable, it was affirmed that clinically meaningful treatments need to either slow the decline or prevent future impairment [145]. The current disappointing outcomes of treatment to slow the decline may prompt a greater focus on its prevention. Nevertheless, providing medication to prevent the onset of dementia raises not only the medico-economic challenge of establishing an accurate and cost-effective method of identifying future dementia risk but also the determining the cost vs benefit of treating an asymptomatic individual [144,146,147].
4.5. Parkinson’s Disease
After AD, PD is the second most common neurodegenerative disorder. Its symptoms are attributed to the selective degeneration of dopamine (DA) neurons in the substantia nigra (SN). It has been proposed that chronic OS drives the pathogenesis of the disease [148,149]. Individuals with PD have higher serum levels of OS markers with a simultaneous low level of GSH in the SN that enhances PD vulnerability [150]. Mitochondrial injury from 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) exposure produces OS. MPTP was initially an illicit drug that produced PD in its users. It is now used experimentally in animal studies to induce PD. Key evidence of the significance of OS in PD was 1) an increased ROS and heightened OS driven by endoplasmic reticulum (ER) stress, 2) alpha-synuclein (α-SN) accumulation that is affected by and contributes to OS, and 3) that extracellular DA itself promotes oxidation and increases the vulnerability to OS of DA-producing neurons in the SN, thus concluding that OS is the common denominator driving the development of PD.[148] PD demonstrates the loss of dopaminergic neurons accompanied by OS and preceded by GSH depletion [127]. Interestingly, OS may have a critical role in the pathogenesis of Huntington’s disease, amyotrophic lateral sclerosis, and multiple sclerosis [109,112,151,152,153].
4.6. Autism Spectrum Disorder
Autism spectrum disorder (ASD) is a clinically diagnosed developmental disorder usually determined at 4.5 years of age. It is characterized by impaired social communication and interactions, repetitive behavior, and limited interests or activities. Its etiology and pathogenesis are poorly understood. There are no accepted biomarkers for ASD. It has been proposed that OS (and associated GSH deficiency) is a vital and integral factor in a multifaceted pathological process [154,155,156,157,158,159]. The level of GSH may be monitored in the serum by a ratio of reduced GHS to its oxidized dimer GSSG (the GSH:GSSG ratio) and with 1H-MRI [154,159,160,161]. Serum markers of OS include elevated homocysteine level (and its associated folate and cyanocobalamin deficiency), lipid peroxidation with elevated MDA level, numerous inflammatory cytokines (e.g., interferon-gamma (IFN-γ), interleukin 6 (Il-6), and tumor necrosis factor (TNF) [155,156]. Current treatments address the symptoms of ASD but not the underlying pathophysiology that may be a result of OS [157,162]. Hopefully, studies using the G/C complex will provide a definitive assessment of the influence of OS and GSH in ASD.
5. Encephalopathy
Evidence was presented that OS in Wernicke’s encephalopathy may be addressed by administering antioxidants in addition to thiamine supplementation [163]. In addition, OS appears to play a critical role in the pathogenesis and progression of hypoxic-ischemic encephalopathy in neonates [164].
The pathophysiology of neurologic diseases is complex, and the significance of OS in modifying its development remains to be established. Gribkoff reviewed possible reasons for current treatment failures, including 1) ineffective transport across the blood-brain barrier (BBB), 2) finding animal models that recreate the corresponding human disease, and 3) addressing biomarkers that may not influence the disease [141]. Despite recognizing the utility of GSH to address OS, a lack of an effective delivery method has hindered its use in clinical studies [156]. Oral administration of GSH faces its degradation by gastrointestinal peptidase. Intravenous GSH is rapidly metabolized with an elimination half-life of around 7 minutes. (Ayoama) Transporting GSH across the blood-brain barrier (BBB) has also been an issue for brain diseases. Significantly, it has been demonstrated that γ-cyclodextrin crosses the BBB [55].
6. Fibrosis
Fibrosis is a pathologic consequence of chronic inflammation that involves multiple organs (heart, lungs, liver, kidneys, skin) [165]. It is a sequela of tissue injury that leads to an excessive buildup of fibrous connective tissue in the extra-cellular matrix. Among the illnesses that cause chronic inflammation leading to fibrosis include organ transplantation, high cholesterol, myocardial infarction, idiopathic pulmonary fibrosis, poorly controlled diabetes, obesity, and hypertension [54,165]. The fibrotic process is triggered and mediated by transforming growth factor beta (TGF-β). Other mediators include fibroblast growth factor (FGF), connective tissue growth factor (CTGF), nuclear erythroid 2-related factor 2 (nrf2), and the renin-angiotensin-aldosterone system. (RAAS) Finally, the critical role of OS in the pathogenesis of fibrosis its interaction with TGF-β in a positive feedback loop is detailed by Antar et al [165]. Chronic inflammation is strongly implicated in fibrosis that leads to diabetes complications, renal fibrosis, pulmonary fibrosis, hepatic cirrhosis, and cardiac remodeling [165]. The role of innate and adaptive immune cells in the fibrotic process has also been covered. In particular, a detailed description of the influence of cytokines produced by adaptive immune cells in the pathogenesis of fibrosis was presented [166]. An exhaustive literature review on OS in pulmonary diseases highlighted the top thirty drugs used in 31,373 publications [54]. The most studied drug was NAC. GSH was not on the list, likely reflecting the current absence of an effectively administered compound.
7. OS-Associated Diseases
7.1. Pulmonary Fibrosis
The main parenchyma of the lung is comprised of alveolar epithelial cells (AEC) - formerly known as pneumocytes. Of the two subtypes, AEC1 form the alveoli where gas exchange occurs. ACE2, adjacent to AEC1, has a more complex role. It supplies a surfactant that maintains alveolar integrity, transforms into AEC1 in adaptive tissue repair, and is involved in the inflammatory response. Sustained insults result in a maladaptive response of AEC2 to recruit myofibroblasts, activation of collagen synthesis, AEC2 apoptosis, and ultimately pulmonary fibrosis (PF) [33,35]. This process is stimulated by pro-inflammatory cytokines, particularly tissue growth factor beta (TGF-β), which increases alveolar secretion of fibroblast growth factor 2 (FGF-2). Other cytokines include tumor necrosis factor-alpha (TNF-α) and platelet-derived growth factor (PDGF). OS appears to be implicated in the fibrotic process by inducing epithelial apoptosis, increasing cytokine levels, and promoting the differentiation of fibroblasts to myofibroblasts. Effective prevention of PF should target early intervention of the immunothrombotic/fibrotic process and the development of pulmonary fibrosis associated with the depletion of GSH [35,36]. Murine animal studies with inhaled NAC appears to attenuate bleomycin-induced PF.[33] However, human studies with NAC have been disappointing. 36 Prior studies with antioxidants have shown either toxicity or lack of efficacy in human subjects. Most of the studied compounds are natural flavonoids or polyphenols that have demonstrated poor efficacy in humans. It could be due to an intrinsic lack of efficacy, inadequate dosage, or inadequate duration of use [36]. However, none have used GSH via the G/C complex. Finally, like addressing dementia, the role of treatment may be more effective in preventing their onset and progression rather than reversing the pathological processes of these illnesses.
7.2. Cystic Fibrosis
Another chronic, progressive lung disease associated with high OS is cystic fibrosis (CF) [167]. It is a disease that occurs due to mutations in the gene encoding for the CF transmembrane conductance regulator (CFTR). The primary function of CFTR is to regulate the efflux of chloride and bicarbonate anions. Its dysfunction leads to a water-electrolyte imbalance on the surface of numerous organs and organ systems, including the upper and lower airways, intestine, skeletal muscles, pancreas, biliary tree, cervix, vas deferens, and sweat glands. Clinically, it results in inflammation, recurrent airway infection, and a progressive decline in lung function leading to death [37,168]. Recurrent pulmonary infections lead to a self-amplifying process of cellular damage that involves OS. Given the complexity and variability of CF, it was proposed that the disease be managed with a combination of antioxidant and anti-inflammatory agents plus CFTR-targeted therapies to address the interrelationship of CFTR and OS [38]. It may be due to an inability to increase GSH in the epithelial lining fluid (due to the deficiency of CFTR) may contribute to the poor response to infection in CF patients [10].
7.3. Hepatic Cirrhosis
Hepatic cirrhosis is a major worldwide health problem. Its etiologies include alcohol abuse, hepatitis B and C infection, and nonalcoholic fatty liver disease (NAFLD). With the success of vaccination against hepatitis B and the treatment of hepatitis C, the risk of developing cirrhosis from NAFLD is becoming a significant problem. It is the most common cause of liver disease worldwide ans is influenced by the growing epidemic of obesity and its associated MetS [169]. The incidence of NAFLD, which is up to 75% in obese individuals, and even higher in those with T2DM, has been noted [40]. As reported in 2018, NASH-related cirrhosis accounted for 5% of all young patients listed for liver transplantation in the United States 169]. Finally, the risks of complications (hepatorenal syndrome, variceal hemorrhage, hepatic encephalopathy, and hepatocellular carcinoma) from NASH and hepatitis C cirrhosis were similar.
Although the pathway from NAFLD to cirrhosis is not firmly established, there is strong evidence of OS influence in its process. This association should not be surprising, given the role of OS in IR and MetS, as mentioned earlier. It has been proposed that hepatic dysfunction is the expression of MetS in the liver [40]. This process is initiated simultaneously by fat infiltration of hepatocytes, IR, and mitochondrial beta-oxidation, leading to increased ROS and eventual OS due to mitochondrial dysfunction, ER stress, derangement of iron metabolism, dysbiosis in the gut (including heightened ethanol-producing bacteria), and endothelial dysfunction (ED) [40,170]. As described by Masarone et.al., ED is a result of excessive production of nitric oxide (NO), which in turn contributes to and promotes the structural and functional changes of liver circulation, impairs regeneration after liver injury, and thereby leads to the progression of liver disease [40]. This is a modification of Day and James’s “two-hit” hypothesis. They proposed that steatosis is the first “hit” that requires modifying factors (primarily linked to OS) as a second “hit” to produce the fibrosis and cirrhosis resulting from nonalcoholic steatohepatitis (NASH) [40]. The pathologic sequence was summarized as a consequence of 1) excessive fatty acids in hepatocytes, 2) their energy depletion and resultant mitochondrial dysfunction, 3) an increase in OS, and finally, 4) cellular damage. The association of concurrent decreased antioxidants, particularly of GSH depletion, was also noted [39]. The overall complexity of the pathogenesis of NASH-cirrhosis was diagramed by Bovi – Figure 1. Modest benefits after four months with the administration of a modified oral GSH supplement have been reported [39,170].
The importance of OS in the pathogenesis of cirrhosis in hereditary hemochromatosis (HHC), chronic hepatitis C virus infection, and primary biliary cirrhosis has been reviewed [171,172]. It has been proposed that early interventions against, including maintaining GSH levels, may protect hepatocytes from damage [172]. Notably, the presence of cirrhosis is associated with a lower-than-normal life expectancy and a significantly increased risk for the development of hepatocellular carcinoma.
7.4. Cardiovascular Disease
Cardiovascular disease (CVD) is a group of disorders of the heart and blood vessels. The interrelated disease entities described are atherosclerosis heart disease (ASHD), coronary artery disease (CAD), arterial hypertension (AH), and restenosis [43,173,174]. ASHD is a chronic progressive thickening, hardening, and narrowing of the systemic arterial walls primarily due to elevated plasma cholesterol. CAD is a narrowing of coronary arteries primarily caused by the formation of unstable plaques within the intima of the vessel wall. A rupture of unstable plaques results in acute thrombosis, which is a significant cause of acute myocardial infarction and strokes. The role of the immunothrombotic process leading to thromboinflamation was discussed previously in hepatic and COVID-related coagulopathies – specifically, the cyclic role of the coagulation cascade involving platelet and innate immune cell activation due to cytokine dysregulation from OS [11,23,30,175]. The role of OS from SARS-CoV-2 infection and its subsequent pro-atherogenic CVD complications (heart failure, stroke, venous thrombosis, and pulmonary embolism) after COVID-19 illness has been presented [36,176]. The current lack of an effective antioxidant therapy was noted [175]. Finally, the role of ROS generation by statins in producing tissue toxicity of the liver and muscles was discussed [11].
7.5. Viral Illnesses
7.5.1. Encephalitis/Thrombosis
The pathologic consequence of viral illness from SARS-CoV-2 and hepatitis C has been reviewed. The role of OS infections from the Flaviviridae family was discussed [177]. It highlighted processes by which OS facilitates viral replication and the pathologic consequences of the resultant illnesses. These viruses include dengue virus (DENV), Zika virus (ZIKV), yellow fever virus (YFV), Japanese encephalitis virus (JEV), West Nile virus (WNV), tick-borne encephalitis virus (TBEV), and hepatitis C virus (HCV). The major clinical manifestations are hemorrhagic syndromes (DENV), (YFV) neurologic (ZIKV), (JEV), (WNV), and hepatitis (HCV). Growing evidence supports the link between OS and viral pathogenesis – particularly HCV and DENV [177]. Although direct-acting antiviral agents (DAA) against HCV have significantly avoided its serious complications, the high cost and limited availability of DAA keep it beyond the reach of many individuals – particularly those in third-world countries. Analogous to SARS-CoV-2 infection, Hantavirus appears to cause a cytokine storm that leads to pulmonary and renal injury [178]. The role of GSH treatment in these viral illnesses remains to be determined.
In addition, the role of neutrophil extracellular trap (NET) formation and complement activation in response to bacterial and virus infections is significant. NETs and complement significantly contribute to thrombosis, atherosclerosis, and autoimmune diseases, and they have been described in detail [17,23].
7.5.2. Myocarditis
The pathology of viral cardiomyopathy is complex. A thorough review of virus-induced myocarditis and cardiomyopathy has been presented [179]. The distinction between the direct, immediate virus-induced (cardiotropic) cytotoxicity and the indirect, delayed virus-associated, immune-mediated (vasculopropic and lymphotropic) inflammatory cardiomyopathy was discussed [179]. The numerous viruses include adenoviruses, enteroviruses (e.g., coxsackievirus), parvovirus B19, cytomegalovirus, Epstein-Barr virus, hepatitis C, human immunodeficiency virus (HIV), and coronavirus (including SARS-CV-2). The role of the innate and adaptive immune response, the neutrophil, eosinophil, T and B cell response, and autoimmunity was also reviewed. In this context, the influence and treatment of OS with GSH also remain to be determined.
7.5.3. Hemorrhagic Fever
Many enveloped RNA viruses cause a syndrome known as viral hemorrhagic fever (VHF) that is driven by cytokine dysregulation [180,181,182]. These include the Ebola virus (EBOV), Marburg virus (MARV), Lassa virus (LAV), and the previously mentioned YFV and DENV. Until a successful vaccine or antiviral agent against these viruses is developed, the current treatment is supportive care. In the interim, addressing the cytokine dysregulation with the G/C complex will hopefully reduce the incidence of mortality and severe morbidity from those viruses.
7.5.4. Respiratory Dysfunction
Respiratory syncytial virus (RSV) is a significant cause of pneumonia in children. Infants experience the most severe complications requiring hospitalization. Although a vaccine against RSV is available, current treatment for those contracting the illness is supportive. The pathogenesis of serious illness appears due to the inflammatory response of the lungs to OS. Markers of oxidative damage correlated with the severity of damage [183,184]. Since the G/C complex is applied topically, future studies on infants and neonates will not require typically difficult IV access.
7.6. ISCHEMIA-Reperfusion Injury
The role of OS in ischemia-reperfusion injury (IRI) is another area for future studies. These include sequelae of myocardial infarction (MI), stroke (ischemic and hemorrhagic), shock, and organ transplantation. It has become recognized that organ injury occurs not only as a consequence of initial ischemia but also of subsequent reperfusion [185,186,187]. Reperfusion injury occurs paradoxically upon reoxygenation with the restoration of blood flow. Cellular dysfunction and death result from OS and its associated inflammatory response [187]. The inflammatory response is necessary for tissue repair and includes neutrophil formation, cytokine production (including TNF-α, IL-1β, and IL-6), and other proinflammatory stimuli [185]. OS can usually be identified by measuring malondialdehyde (MDA), a byproduct of OS of lipid peroxidation. The IRI process may involve not only the initial organ but also cause systemic damage to distant organs and potentially lead to multi-system organ failure [187].
7.6.1. Myocardial Ischemia
Prompt reperfusion after acute myocardial infarction (AMI) is a critical cardiovascular intervention. Nevertheless, the subsequent reperfusion may lead to dysfunction more severe than the initial ischemia. These include myocardial stunning, reperfusion arrhythmia, myocyte death, endothelial and microvascular dysfunction that includes the no-reflow phenomenon, and an inflammatory response. Detailed reviews of the process of cardiomyocyte death that follows mitochondrial dysfunction, calcium overload, and a maladaptive inflammatory response that OS drives has been presented [188,189,190]. A prospective clinical study of OS in myocardial infarction-reperfusion injury (MIRI) confirmed the diagnostic value of OS markers The authors compared including levels of superoxide dismutase (SOD), malondialdehyde (MDA), myeloperoxidase (MPO), ischemia-modified albumin (IMA), and heat shock protein 70 (HSP70) after reperfusion with percutaneous intervention (PCI) in elderly patients with AMI and type 2 diabetes [191]. The presence of those markers predicted a higher risk of IRI injury. Presumably measures to prevent or minimize OS could result in more favorable clinical outcomes [192].
7.6.2. Central Nervous System Injuries
The IRI injuries following OS from acute ischemic stroke (IS) have been reviewed [193,194]. OS drives the initial endothelial dysfunction in IS with subsequent platelet aggregation and a coagulation cascade initiated by activated vWF [194]. Successful recanalization procedures, with prompt cerebral blood flow reestablishment, have improved patient outcomes. Paradoxically, reperfusion may worsen neurologic function through cerebral edema and thrombosis [193]. The critical role of OS and subsequent mitochondrial dysfunction leading to apoptosis in potentially salvageable ischemic surrounding areas was also noted.
Additionally, OS appears to have a significant role in the secondary outcomes of the initial injury from intracerebral hemorrhage (ICH), which is similar to IS. Following the initial insult from the mass effect of the hematoma, the secondary brain injury (SBI) is attributed to neuroinflammation, excitatory amino acid, cytotoxicity of blood, hypermetabolism, and spreading depression driven by OS [195,196]. The brain is highly susceptible to OS damage since it is rich in lipids and iron but is deficient in antioxidants. The interplay of OS and the secondary outcomes of ICH (neuroinflammation, glutamate excitotoxicity, cell death, and disruption of the blood-brain barrier (BBB) has been reviewed [194,195]. A higher level of OS, usually measured with MDA, is associated with more severe clinical harm. Current interventions include treating the cause or source of the bleed and reducing its hemorrhagic load with either a surgical or minimally invasive decompression procedure [197]. Addressing SBI should further improve the current neurologic outcomes after ICH.
7.6.3. Organ Transplantation
IRI is an inevitable consequence of organ transplantation. Effective organ preservation and successful transplantation is critical since the need for organs continues to exceed its supply. By reducing OS, increased donor pool and transplantation success have benefitted from machine perfusion (MP) technology [198]. IRI caused by OS during early reperfusion negatively affects both short and long-term outcomes following transplantation [199]. This reflects the significance of mitochondrial dysfunction and immunothrombosis produced by ischemia and reperfusion [186]. Evidence has been summarized that OS-induced IRI is a crucial event during the full spectrum of transplantation from retrieval, preservation, and reperfusion [199].
7.6.4. Sickle Cell Disease
Sickle cell disease (SCD) is a global health burden affecting over 40 million individuals with the disease or trait. Of those, 3.2 million live with the disease, a vaso-occlusive consequence of impaired blood rheology, hemolysis, increased adhesiveness of erythrocytes to vascular endothelium with co-existing inflammation, and hemostasis activation [200,201,202]. OS, and its subsequent inflammatory response, is driven by IRI through the Fenton reaction resulting from iron release due to chronic hemolysis [200,202,203]. Similar to diabetes, AGEs appear to have a role in the pathology of SCD [203]. Likewise, the similarity of thomboinflammation in COVID-19 and SCD has been noted [204]. Finally, advanced lipoxidation end-products (ALEs) and lipid peroxidation (as measured by high levels of MDA) have been identified [203]. A low level of GSH in RBCs and the impairment of its de novo synthesis has been recognized [203]. The administration of GSH directly via the G/C complex may overcome these deficiencies. As a relatively inexpensive topical product, the complex would be readily available for chronic use in rural and undeveloped regions.
7.7. Pathologic Aging
Pathologic premature aging, or progeria, is a group of rare inherited genetic disorders associated with OS [205,206,207]. These include Hutchinson-Gilford progeria syndrome (HGPS) and the Werner syndrome (WS). Although the underlying molecular mechanisms of disease differ, they both lead to premature pathologic aging. HGPS is characterized by accelerated aging with cardiovascular dysfunction that leads to death at an average age of 13 years, usually from myocardial infarction or stroke. Unlike HGPS and atypical WS, the onset of symptoms in classic WS occurs after puberty [208]. Although there are several hallmarks of progeria (including telomere dysfunction, increased DNA damage, cell cycle dysregulation, and stem cell exhaustion), inflammaging appears to play an integral role [205]. Antioxidants are widely promoted and used for anti-aging purposes [7]. The use of antioxidants in pathologic premature aging has not been well studied [6]. The effect of early intervention with antioxidants, including GSH, remains to be determined [209].
7.8. Toxicities
In addition to illnesses, OS is induced by toxic substances – both from an overdose and as a side effect. A well-known overdose is from acetaminophen (also known as paracetamol) [210]. Toxicities can occur from exposure to heavy metals (mainly lead and mercury), contrast agents, and some chemotherapeutic agents (anthracyclines, bleomycin, and cisplatin) [211,212,213].
7.8.1. Chemotherapy
Chemotherapeutic toxicity can occur acutely (during the treatment course) or delayed (several years after administration). These adverse effects are associated with OS, including anthracyclines, bleomycin, and cisplatin.[211] Acute renal toxicity of cisplatin is complex [214]. The current reno-protective protocol includes saline hydration and mannitol administration [215]. The cardioprotective effect against anthracyclines with enalapril and carvedilol was effective if initiated within two months of completion of chemotherapy but not if initiated after six months [216]. Carrasco et al. suggested the use of antioxidants against anthracycline-induced cardiotoxicity [217]. However, addressing OS from chemotherapeutic agents must be balanced between their antineoplastic activity and their toxic adverse side effects; hence, the use and timing of antioxidants remain to be established [212,215,217,218,219]. Finally, treating chemotherapy-associated toxicity is complicated by the fact that most cancer treatments involve a “cocktail” of two or more agents
7.8.2. Antiretroviral Therapy
The possible role of OS in toxic adverse effects from highly active antiretroviral therapy (HAART) for human immunodeficiency virus (HIV) infections has been presented [11]. The toxic effects on the liver from mitochondrial dysfunction (steatosis, steatohepatitis, and disorders of lipid regulation) were discussed.
7.8.3. Contrast-Induced Nephropathy
Contrast-induced nephropathy (CIN) is multifaceted and includes direct tubular toxicity, intrarenal vasoconstriction, and excessive production of ROS [213]. Although CIN usually resolves, 15% may require temporary dialysis, and 4% progress to end-stage renal disease. High-risk individuals include those with prior chronic kidney disease, congestive heart failure, volume depletion, advanced age, hypertension, and hyperuricemia. In vitro and animal studies have supported using antioxidants for CIN prevention [213]. The clinical use of GSH to prevent CIN remains to be investigated
7.8.4. Acetaminophen Hepatotoxicity
Acetaminophen (APAP) hepatotoxicity is the most common cause of acute liver failure in the United States, with unintentional overdose (OD) comprising the majority of episodes. APAP is typically eliminated through conjugation through glucuronidation and sulfonation. A secondary, hepatotoxic metabolite (N-acetyl-p-benzoquinone imine (NAPQI) is normally rendered harmless by conjugation with GSH. However, when the level of APAP overwhelms the primary glucuronidation pathway, the GSH stores are consumed by a generation of the excess NAPQI metabolites and by the formation of mitochondrial NAPQI protein adducts, leading to hepatocellular injury [220]. The standard treatment for APAP overdose is the use of NAC. Early administration of NAC is critical [210]. While this is possible with intentional OD, it is not practical with unintentional OD. The latter instance is usually associated with the overuse of narcotic/APAP pain medications [210]. For intentional OD, direct administration of GSH using the G/C complex would overcome the inherent delay NAC requires to promote de novo GSH synthesis and extend the ability to prevent hepatotoxicity beyond the current 72-hour window. Furthermore, the concurrent administration of the complex when APAP use is excessive may prevent unintentional OD
7.8.5. Isoniazid and Rifampicin Hepatotoxicity
As the primary medications for the treatment of Mycobacterium tuberculosis, hepatotoxicity occurs directly from isoniazid (INH) and indirectly from rifampicin (RMP) metabolites [221]. Evidence has been presented that the hepatic injury and subsequent fibrosis involve OS. The in vivo animal study demonstrated that activation of hepatic stellate cells and liver fibrosis depends on OS
7.8.6. Amanitin Hepatotoxicity
Hepatotoxicity and death from mushroom poisoning mainly occur from the ingestion of Amanita phalloides. The toxic amanitins cause direct and indirect (through OS) hepatorenal injury. Currently, there is no definitive treatment for amanita poisoning [222]. Except for forced diuresis, care is essentially supportive. Perhaps future treatment studies will examine a combined approach – forced diuresis to hasten the renal clearance of amanitins, cholestyramine to block its entero-hepatic reuptake, and GSH to target OS.
7.9. Occular Diseases
OS has been implicated in the pathogenesis of many ocular diseases. These retinopathies may be of degenerative, genetic, senile, or inflammatory origin. Among the resulting diseases are age-related macular degeneration (AMD), diabetic retinopathy (DM), retinitis pigmentosa (RP), retinitis of prematurity, retinal degeneration, and Stargardt disease [223].
7.9.1. Diabetic Retinopathy
Diabetic retinopathy (DR) is the most common microvascular complication in diabetic patients and is the primary cause of blindness in people between 27 and 75 years of age [224,225]. It results from retinal vascular abnormalities, including hyperpermeability, hypoperfusion, and neoangiogenesis. Kusuhara et al. have outlined the classification and management of DM in Table 1 of their article [224]. Interestingly, a subpopulation of diabetic patients does not develop DR despite having the risk factors of sustained hyperglycemia, hypertension, hyperlipidemia, and pregnancy [224]. This suggests the presence of an additional unidentified risk factor. Finally, if OS initiates and promotes inflammation associated with DR, early antioxidant intervention may prevent its onset or slow its progression [225].
7.9.2. Retinitis Pigmentosum
Retinitis pigmentosum (RP) is an inherited, progressive retinal degeneration of the eyes. The involvement of OS in the complex pathologic progression of RP has been detailed [226]. This includes damage to cone photoreceptor morphology, the blood-retinal barrier, and upregulation of retinal expression of vascular endothelial growth factor (VEGF). In addition, a significant loss of GSH and associated high levels of MDA in the aqueous humor was noted. The high level of MDA reflects lipid peroxidation that promotes cytokine-associated inflammation and angiogenesis [227].
7.9.3. Age-Related Macular Degeneration
Age-related macular degeneration (AMD) is a progressive and irreversible ocular disease that is a significant cause of visual impairment in elderly individuals and is the fourth leading cause of blindness worldwide [228]. The Beckman clinical classification of AMD was reviewed. The retina’s high oxygen metabolism compared to any other tissue and its generation of ROS make it susceptible to OS. Animal studies have supported this premise. Ruan et al. made a detailed presentation of OS in the development of AMD [228]. Although the incidence has declined with the introduction of anti-VEGF agents, OS appears to be the initial trigger of AMD and influences late-stage neovascularization [229].
7.10. Systemic Inflammatory Response Syndrome/Sepsis
Systemic inflammatory response syndrome (SIRS) and its infection-associated variant known as sepsis involves a complex pathologic process that can progress to multi-organ failure (MOF) [230,231]. In addition to antibiotics, current sepsis management is supportive (mechanical ventilation, fluid administration, and pressors). Bar-Or et.al. hypothesized that tissue damage may be a consequence of IRI. If so, treating OS could be an additional treatment modality in SIRS/sepsis management.
7.11. Pancreatitis
7.11.1. Acute Pancreatitis
Acute pancreatitis (AP) is usually caused by excessive alcohol consumption or bile stone occlusion of the pancreatic duct and, to a lesser extent, by extremely high triglyceridemia. The initial insult causes injury to pancreatic acinar cells, leading to increased ROS and OS production. This subsequently results in a dysregulated cytokine response and potentially SIRS [232]. The Atlanta Classification identifies two types of AP. There is an initial interstitial edematous AP and a severe necrotizing AP. Although beneficial, the current management of acute pancreatitis is supportive care. Antioxidant therapy has produced mixed results. However, studies with experimental models have demonstrated that OS is present from the early stage of disease, and its degree (as measured by elevated MDA and c-reactive protein levels and inversely by low antioxidant levels) is associated with the severity of AP [233,234]. Interestingly, pretreatment with current antioxidants to prevent AP from endoscopic retrograde cholangiopancreatography (ERCP) has been unsuccessful. Given the uncertainty of the efficacy of numerous antioxidants (including NAC and vitamin C), the role of GSH remains to be determined
7.11.2. Chronic Pancreatitis
In a comparison of 35 healthy subjects with 35 chronic pancreatitis (CP) patients, multiple measures of both OS and antioxidant capacity were used [235]. Although the presence of OS was demonstrated, its significance in initiating and maintaining CP also remains to be determined.
8. Discussion
The evidence has been presented that supports the association of OS with numerous seemingly disparate illnesses. The association of OS with illnesses presents more questions than answers. The unanswered questions are: 1) is OS only an associative biomarker? 2) is OS a significant initiator and promoter? 3) is OS present as a result of GSH deficiency? 4) Can GSH be restored and reverse OS? 5) as illustrated in Figure 4, can the early use of the G/C complex prevent the onset of progressive illnesses and avoid irreversible pathology? Due to the inverse relationship of OS with GSH levels, either due to age, metabolic status, or acute illness, a deficiency of GSH appears to result in OS. Given this premise, as an antioxidant, restoring GSH should reverse OS. Except for NAC, studies with numerous other antioxidants have been problematic to administer, equivocal, or disappointing [73,236]. However, most are weak antioxidants, and NAC is a GSH precursor that requires endogenous enzymatic conversion into GSH. Experimental antioxidants are not available for clinical use. Many immunotherapeutic agents are costly and are not readily administered. Delivery of GSH orally or intravenously is inefficient. It is proposed that the transdermal G/C complex can deliver exogenous GSH effectively. The complex has the following advantages over other antioxidants: 1) through nanotechnology, it delivers GSH directly in a measured amount, 2) it avoids the delivery problems of oral and IV GSH, 3) it avoids enzymatic conversion required by NAC, 4) as a topical agent, it is easily administered, 5) it has been an available product for over ten years, 6) it does not require special storage, 7) relative to immunotherapeutic agents, it is a low-cost product.
Given the complex pathophysiology of the many illnesses discussed in this review, the unanswered question is, what influence does OS have in the various disease processes? Is it a marker or a participant? If it is a participant, what effect will the G/C complex have in preventing or modifying OS? The complex should promote studies to establish the efficacy of GSH in managing OS and to support or refute the role of OS in those illnesses. Significantly, CD can cross the blood-brain barrier. If future studies support the efficacy of the G/C complex, it could be used as an adjunct or alternative product to prevent or treat illnesses associated with OS that currently lack optimal intervention. Among the proposed studies are, can the complex: 1) treat the rare individuals with congenital absence of the enzymes GCL or GS? 2) modify the rapid aging process of H-G progeria or Werner’s syndrome? 3) prevent or reduce the consequences of IRI injury? 4) prevent the onset of progressive neurodegeneration? 5) attenuate the process of thromboinflammation in ARDS, cirrhosis, and CAD? 6) improve the management of diabetes and its complications? 7) improve the treatment of Mycobacterium infections? Finally, can the complex contribute to the avoidance or management of severe acute insults from COVID-19, SIRS, pancreatitis, ischemia, toxicities, and virus-associated complications?
9. Conclusions
There are numerous disparate acute and chronic illnesses associated with OS. Essentially, acute insults overwhelm cellular antioxidants, and chronic ones exhaust them, leading to excessive ROS, which results in OS. Although many illnesses have a documented association with OS and antioxidant deficiency, the efficacy of antioxidant replacement, including GSH, has been varied. Preliminary evidence has been presented of a compound, the G/C complex, that appears to deliver GSH effectively and therapeutically. Since the pathophysiology of illnesses is inherently complex and variable, presenting GSH administered via the G/C complex as the therapeutic “answer” is evocative but unrealistic. However, rigorous studies with the G/C complex may answer the many questions about the role and efficacy of GSH in addressing OS and the intrinsic influence of OS in illnesses. It should be used in studies to determine if OS is solely an associated marker of illnesses or is an initiator and promoter of those illnesses. Hopefully, the complex will find its place as an adjunct or alternative to current therapeutic regimens in the management of untreatable or sub-optimally treated illnesses.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sasaninia K, Kelley M, Abnousian A, et al. Topical Absorption of Glutathione-Cyclodextrin Nanoparticle Complex in Healthy Human Subjects Improves Immune Response against Mycobacterium avium Infection. Antioxidants (Basel). Jul 2 2023;12(7). [CrossRef]
- Patel N. The glutathione revolution : fight disease, slow aging, and increase energy with the master antioxidant. First edition. ed. Hachette Go, an imprint of Hachette Books; 2020:xix, 266 pages.
- Bardaweel SK, Gul M, Alzweiri M, Ishaqat A, HA AL, Bashatwah RM. Reactive Oxygen Species: the Dual Role in Physiological and Pathological Conditions of the Human Body. Eurasian J Med. Oct 2018;50(3):193-201. [CrossRef]
- Pizzino G, Irrera N, Cucinotta M, et al. Oxidative Stress: Harms and Benefits for Human Health. Oxid Med Cell Longev. 2017;2017:8416763. [CrossRef]
- Bassoy EY, Walch M, Martinvalet D. Reactive Oxygen Species: Do They Play a Role in Adaptive Immunity? Front Immunol. 2021;12:755856. [CrossRef]
- Tan BL, Norhaizan ME, Liew WP, Sulaiman Rahman H. Antioxidant and Oxidative Stress: A Mutual Interplay in Age-Related Diseases. Front Pharmacol. 2018;9:1162. [CrossRef]
- Liguori I, Russo G, Curcio F, et al. Oxidative stress, aging, and diseases. Clin Interv Aging. 2018;13:757-772. [CrossRef]
- Yang S, Lian G. ROS and diseases: role in metabolism and energy supply. Mol Cell Biochem. Apr 2020;467(1-2):1-12. [CrossRef]
- Hecker L. Mechanisms and consequences of oxidative stress in lung disease: therapeutic implications for an aging populace. Am J Physiol Lung Cell Mol Physiol. Apr 1 2018;314(4):L642-L653. [CrossRef]
- Ballatori N, Krance SM, Notenboom S, Shi S, Tieu K, Hammond CL. Glutathione dysregulation and the etiology and progression of human diseases. Biol Chem. Mar 2009;390(3):191-214. [CrossRef]
- Garcia-Sanchez A, Miranda-Diaz AG, Cardona-Munoz EG. The Role of Oxidative Stress in Physiopathology and Pharmacological Treatment with Pro- and Antioxidant Properties in Chronic Diseases. Oxid Med Cell Longev. 2020;2020:2082145. [CrossRef]
- Townsend DM, Tew KD, Tapiero H. The importance of glutathione in human disease. Biomed Pharmacother. May-Jun 2003;57(3-4):145-55. [CrossRef]
- Yang J, Xu J, Xu S, et al. Oxidative stress in acute pulmonary embolism: emerging roles and therapeutic implications. Thromb J. Jan 12 2024;22(1):9. [CrossRef]
- Forman HJ, Zhang H. Targeting oxidative stress in disease: promise and limitations of antioxidant therapy. Nat Rev Drug Discov. Sep 2021;20(9):689-709. [CrossRef]
- Di Meo S, Reed TT, Venditti P, Victor VM. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid Med Cell Longev. 2016;2016:1245049. [CrossRef]
- Silvagno F, Vernone A, Pescarmona GP. The Role of Glutathione in Protecting against the Severe Inflammatory Response Triggered by COVID-19. Antioxidants (Basel). Jul 16 2020;9(7). [CrossRef]
- Glassman I, Le N, Mirhosseini M, et al. The Role of Glutathione in Prevention of COVID-19 Immunothrombosis: A Review. Front Biosci (Landmark Ed). Mar 20 2023;28(3):59. [CrossRef]
- Almeida B, Domingues C, Mascarenhas-Melo F, et al. The Role of Cyclodextrins in COVID-19 Therapy-A Literature Review. Int J Mol Sci. Feb 3 2023;24(3). [CrossRef]
- Rushworth GF, Megson IL. Existing and potential therapeutic uses for N-acetylcysteine: the need for conversion to intracellular glutathione for antioxidant benefits. Pharmacol Ther. Feb 2014;141(2):150-9. [CrossRef]
- Hristov BD. The Role of Glutathione Metabolism in Chronic Illness Development and Its Potential Use as a Novel Therapeutic Target. Cureus. Sep 2022;14(9):e29696. [CrossRef]
- Alleman RJ, Katunga LA, Nelson MA, Brown DA, Anderson EJ. The “Goldilocks Zone” from a redox perspective-Adaptive vs. deleterious responses to oxidative stress in striated muscle. Front Physiol. 2014;5:358. [CrossRef]
- Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J Nutr. Mar 2004;134(3):489-92. [CrossRef]
- Stark K, Massberg S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat Rev Cardiol. Sep 2021;18(9):666-682. [CrossRef]
- Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. Sep 2018;15(9):505-522. [CrossRef]
- Perricone C, De Carolis C, Perricone R. Glutathione: a key player in autoimmunity. Autoimmun Rev. Jul 2009;8(8):697-701. [CrossRef]
- Jackson SP, Darbousset R, Schoenwaelder SM. Thromboinflammation: challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood. Feb 28 2019;133(9):906-918. [CrossRef]
- Bai Y, Li K, Li X, et al. Effects of oxidative stress on hepatic encephalopathy pathogenesis in mice. Nat Commun. Jul 24 2023;14(1):4456. [CrossRef]
- Wang W, Zhao F, Ma X, Perry G, Zhu X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol Neurodegener. May 29 2020;15(1):30. [CrossRef]
- Subramaniam S, Jurk K, Hobohm L, et al. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood. Apr 20 2017;129(16):2291-2302. [CrossRef]
- Rawish E, Sauter M, Sauter R, Nording H, Langer HF. Complement, inflammation and thrombosis. Br J Pharmacol. Jul 2021;178(14):2892-2904. [CrossRef]
- Bettiol A, Galora S, Argento FR, et al. Erythrocyte oxidative stress and thrombosis. Expert Rev Mol Med. Aug 26 2022;24:e31. [CrossRef]
- Sagcan G, Konukoglu D, Uzun H, Arseven O, Okumus G, Cuhadaroglu C. Importance of oxidative stress in the evaluation of acute pulmonary embolism severity. BMC Pulm Med. Oct 17 2022;22(1):382. [CrossRef]
- Tran S, Ksajikian A, Overbey J, Li P, Li Y. Pathophysiology of Pulmonary Fibrosis in the Context of COVID-19 and Implications for Treatment: A Narrative Review. Cells. Aug 11 2022;11(16). [CrossRef]
- Patrucco F, Solidoro P, Gavelli F, Apostolo D, Bellan M. Idiopathic Pulmonary Fibrosis and Post-COVID-19 Lung Fibrosis: Links and Risks. Microorganisms. Mar 30 2023;11(4). [CrossRef]
- Cheresh P, Kim SJ, Tulasiram S, Kamp DW. Oxidative stress and pulmonary fibrosis. Biochim Biophys Acta. Jul 2013;1832(7):1028-40. [CrossRef]
- Estornut C, Milara J, Bayarri MA, Belhadj N, Cortijo J. Targeting Oxidative Stress as a Therapeutic Approach for Idiopathic Pulmonary Fibrosis. Front Pharmacol. 2021;12:794997. [CrossRef]
- Causer AJ, Shute JK, Cummings MH, et al. Circulating biomarkers of antioxidant status and oxidative stress in people with cystic fibrosis: A systematic review and meta-analysis. Redox Biol. May 2020;32:101436. [CrossRef]
- Moliteo E, Sciacca M, Palmeri A, et al. Cystic Fibrosis and Oxidative Stress: The Role of CFTR. Molecules. Aug 21 2022;27(16). [CrossRef]
- Honda Y, Kessoku T, Sumida Y, et al. Efficacy of glutathione for the treatment of nonalcoholic fatty liver disease: an open-label, single-arm, multicenter, pilot study. BMC Gastroenterol. Aug 8 2017;17(1):96. [CrossRef]
- Masarone M, Rosato V, Dallio M, et al. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid Med Cell Longev. 2018;2018:9547613. [CrossRef]
- Al-Hakeim HK, Al-Rubaye HT, Al-Hadrawi DS, Almulla AF, Maes M. Long-COVID post-viral chronic fatigue and affective symptoms are associated with oxidative damage, lowered antioxidant defenses and inflammation: a proof of concept and mechanism study. Mol Psychiatry. Feb 2023;28(2):564-578. [CrossRef]
- Alrajhi NN. Post-COVID-19 pulmonary fibrosis: An ongoing concern. Ann Thorac Med. Oct-Dec 2023;18(4):173-181. [CrossRef]
- Chen Q, Wang Q, Zhu J, Xiao Q, Zhang L. Reactive oxygen species: key regulators in vascular health and diseases. Br J Pharmacol. Apr 2018;175(8):1279-1292. [CrossRef]
- Li L, Stegner D. Immunothrombosis versus thrombo-inflammation: platelets in cerebrovascular complications. Res Pract Thromb Haemost. Jan 2024;8(1):102344. [CrossRef]
- Trakkides TO, Schafer N, Reichenthaler M, et al. Oxidative Stress Increases Endogenous Complement-Dependent Inflammatory and Angiogenic Responses in Retinal Pigment Epithelial Cells Independently of Exogenous Complement Sources. Antioxidants (Basel). Nov 13 2019;8(11). [CrossRef]
- Labarrere CA, Kassab GS. Glutathione: A Samsonian life-sustaining small molecule that protects against oxidative stress, ageing and damaging inflammation. Front Nutr. 2022;9:1007816. [CrossRef]
- Cervia-Hasler C, Bruningk SC, Hoch T, et al. Persistent complement dysregulation with signs of thromboinflammation in active Long Covid. Science. Jan 19 2024;383(6680):eadg7942. [CrossRef]
- Labarrere CA, Kassab GS. Glutathione deficiency in the pathogenesis of SARS-CoV-2 infection and its effects upon the host immune response in severe COVID-19 disease. Front Microbiol. 2022;13:979719. [CrossRef]
- Bhaskar S, Sinha A, Banach M, et al. Cytokine Storm in COVID-19-Immunopathological Mechanisms, Clinical Considerations, and Therapeutic Approaches: The REPROGRAM Consortium Position Paper. Front Immunol. 2020;11:1648. [CrossRef]
- Gain C, Song S, Angtuaco T, Satta S, Kelesidis T. The role of oxidative stress in the pathogenesis of infections with coronaviruses. Front Microbiol. 2022;13:1111930. [CrossRef]
- Yiang GT, Wu YK, Tsai KW, et al. Immunothrombosis biomarkers as potential predictive factors of acute respiratory distress syndrome in moderate-to-critical COVID-19: A single-center, retrospective cohort study. Immunol Lett. Feb 2023;254:30-38. [CrossRef]
- Li H, Wu Q, Qin Z, et al. Serum levels of laminin and von Willebrand factor in COVID-19 survivors 6 months after discharge. Int J Infect Dis. Feb 2022;115:134-141. [CrossRef]
- Polonikov A. Endogenous Deficiency of Glutathione as the Most Likely Cause of Serious Manifestations and Death in COVID-19 Patients. ACS Infect Dis. Jul 10 2020;6(7):1558-1562. [CrossRef]
- Liu X, Wang X, Chang J, Zhang H, Cao P. Landscape analysis and overview of the literature on oxidative stress and pulmonary diseases. Front Pharmacol. 2023;14:1190817. [CrossRef]
- Wong KH, Xie Y, Huang X, et al. Delivering Crocetin across the Blood-Brain Barrier by Using gamma-Cyclodextrin to Treat Alzheimer’s Disease. Sci Rep. Feb 27 2020;10(1):3654. [CrossRef]
- Wang Q, Zennadi R. Oxidative Stress and Thrombosis during Aging: The Roles of Oxidative Stress in RBCs in Venous Thrombosis. Int J Mol Sci. Jun 15 2020;21(12). [CrossRef]
- Karkhanei B, Talebi Ghane E, Mehri F. Evaluation of oxidative stress level: total antioxidant capacity, total oxidant status and glutathione activity in patients with COVID-19. New Microbes New Infect. Jul 2021;42:100897. [CrossRef]
- Roessler C, de Oliveira KCS, de Oliveira Portella AX, et al. Evaluation of oxidative stress level: reactive oxygen species, reduced glutathione, and D-dimer in patients hospitalized due to COVID-19. Redox Rep. Dec 2023;28(1):1-6. [CrossRef]
- Marseglia L, Manti S, D’Angelo G, et al. Oxidative stress in obesity: a critical component in human diseases. Int J Mol Sci. Dec 26 2014;16(1):378-400. [CrossRef]
- Meftahi GH, Jangravi Z, Sahraei H, Bahari Z. The possible pathophysiology mechanism of cytokine storm in elderly adults with COVID-19 infection: the contribution of “inflame-aging”. Inflamm Res. Sep 2020;69(9):825-839. [CrossRef]
- Fajgenbaum DC, June CH. Cytokine Storm. N Engl J Med. Dec 3 2020;383(23):2255-2273. [CrossRef]
- COVID-19 updates: NIH outpatient treatment guidelines. Med Lett Drugs Ther. Feb 21 2022;64(1644):32.
- Ponti G, Maccaferri M, Ruini C, Tomasi A, Ozben T. Biomarkers associated with COVID-19 disease progression. Crit Rev Clin Lab Sci. Sep 2020;57(6):389-399. [CrossRef]
- Cappanera S, Palumbo M, Kwan SH, et al. When Does the Cytokine Storm Begin in COVID-19 Patients? A Quick Score to Recognize It. J Clin Med. Jan 15 2021;10(2). [CrossRef]
- Neves FF, Pott-Junior H, Yamashita KMC, et al. Do the oxidative stress biomarkers predict COVID-19 outcome? An in-hospital cohort study. Free Radic Biol Med. Oct 2023;207:194-199. [CrossRef]
- Rubin R. Paxlovid Is Effective but Underused-Here’s What the Latest Research Says About Rebound and More. JAMA. Jan 31 2024. [CrossRef]
- Yutani R, Venketaraman V. The COVID-19 Illness: Addressing the Current Treatment Limitations and Care Gaps with a Novel Alternative and Complementary Agent-the Glutathione-Cyclodextrin Complex. Altern Ther Health Med. May 2023;29(4):28-35.
- Haunhorst S, Bloch W, Wagner H, et al. Long COVID: a narrative review of the clinical aftermaths of COVID-19 with a focus on the putative pathophysiology and aspects of physical activity. Oxf Open Immunol. 2022;3(1):iqac006. [CrossRef]
- Jason LA, Dorri JA. ME/CFS and Post-Exertional Malaise among Patients with Long COVID. Neurol Int. Dec 20 2022;15(1):1-11. [CrossRef]
- Hama Amin BJ, Kakamad FH, Ahmed GS, et al. Post COVID-19 pulmonary fibrosis; a meta-analysis study. Ann Med Surg (Lond). May 2022;77:103590. [CrossRef]
- Xu E, Xie Y, Al-Aly Z. Long-term neurologic outcomes of COVID-19. Nat Med. Nov 2022;28(11):2406-2415. [CrossRef]
- Sharma C, Bayry J. High risk of autoimmune diseases after COVID-19. Nat Rev Rheumatol. Jul 2023;19(7):399-400. [CrossRef]
- Vollbracht C, Kraft K. Oxidative Stress and Hyper-Inflammation as Major Drivers of Severe COVID-19 and Long COVID: Implications for the Benefit of High-Dose Intravenous Vitamin C. Front Pharmacol. 2022;13:899198. [CrossRef]
- Bonner C, Ghouralal SL. Long COVID and Chronic Conditions in the US Workforce: Prevalence, Productivity Loss, and Disability. J Occup Environ Med. Mar 1 2024;66(3):e80-e86. [CrossRef]
- Komaroff AL, Lipkin WI. ME/CFS and Long COVID share similar symptoms and biological abnormalities: road map to the literature. Front Med (Lausanne). 2023;10:1187163. [CrossRef]
- Bai NA, Richardson CS. Posttreatment Lyme disease syndrome and myalgic encephalomyelitis/chronic fatigue syndrome: A systematic review and comparison of pathogenesis. Chronic Dis Transl Med. Sep 2023;9(3):183-190. [CrossRef]
- Santopaolo M, Gregorova M, Hamilton F, et al. Prolonged T-cell activation and long COVID symptoms independently associate with severe COVID-19 at 3 months. Elife. Jun 13 2023;12. [CrossRef]
- Proal AD, VanElzakker MB, Aleman S, et al. SARS-CoV-2 reservoir in post-acute sequelae of COVID-19 (PASC). Nat Immunol. Oct 2023;24(10):1616-1627. [CrossRef]
- Ruiz-Pablos M, Paiva B, Zabaleta A. Epstein-Barr virus-acquired immunodeficiency in myalgic encephalomyelitis-Is it present in long COVID? J Transl Med. Sep 17 2023;21(1):633. [CrossRef]
- Zhang Z, Zhang X, Fang X, et al. Glutathione inhibits antibody and complement-mediated immunologic cell injury via multiple mechanisms. Redox Biol. Aug 2017;12:571-581. [CrossRef]
- Oggianu L, Lancellotti S, Pitocco D, et al. The oxidative modification of von Willebrand factor is associated with thrombotic angiopathies in diabetes mellitus. PLoS One. 2013;8(1):e55396. [CrossRef]
- Domingueti CP, Dusse LM, Carvalho M, de Sousa LP, Gomes KB, Fernandes AP. Diabetes mellitus: The linkage between oxidative stress, inflammation, hypercoagulability and vascular complications. J Diabetes Complications. May-Jun 2016;30(4):738-45. [CrossRef]
- Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. Oct 29 2010;107(9):1058-70. [CrossRef]
- Li Y, Liu Y, Liu S, et al. Diabetic vascular diseases: molecular mechanisms and therapeutic strategies. Signal Transduct Target Ther. Apr 10 2023;8(1):152. [CrossRef]
- Grattagliano I, Palmieri VO, Portincasa P, Moschetta A, Palasciano G. Oxidative stress-induced risk factors associated with the metabolic syndrome: a unifying hypothesis. J Nutr Biochem. Aug 2008;19(8):491-504. [CrossRef]
- Oguntibeju OO. Type 2 diabetes mellitus, oxidative stress and inflammation: examining the links. Int J Physiol Pathophysiol Pharmacol. 2019;11(3):45-63.
- Bagnato G, Bitto A, Pizzino G, et al. Propylthiouracil modulates aortic vasculopathy in the oxidative stress model of systemic sclerosis. Vascul Pharmacol. Aug 2015;71:79-83. [CrossRef]
- Ottum MS, Mistry AM. Advanced glycation end-products: modifiable environmental factors profoundly mediate insulin resistance. J Clin Biochem Nutr. Jul 2015;57(1):1-12. [CrossRef]
- Das AK, Kalra S, Punyani H, Deshmukh S, Taur S. ‘Oxidative stress’-A new target in the management of diabetes mellitus. J Family Med Prim Care. Nov 2023;12(11):2552-2557. [CrossRef]
- Ceriello A, Esposito K, Piconi L, et al. Oscillating glucose is more deleterious to endothelial function and oxidative stress than mean glucose in normal and type 2 diabetic patients. Diabetes. May 2008;57(5):1349-54. [CrossRef]
- Mengstie MA, Chekol Abebe E, Behaile Teklemariam A, et al. Endogenous advanced glycation end products in the pathogenesis of chronic diabetic complications. Front Mol Biosci. 2022;9:1002710. [CrossRef]
- Nowotny K, Jung T, Hohn A, Weber D, Grune T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules. Mar 16 2015;5(1):194-222. [CrossRef]
- Grundy SM, Cleeman JI, Daniels SR, et al. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute scientific statement. Curr Opin Cardiol. Jan 2006;21(1):1-6. [CrossRef]
- Sifuentes-Franco S, Pacheco-Moises FP, Rodriguez-Carrizalez AD, Miranda-Diaz AG. The Role of Oxidative Stress, Mitochondrial Function, and Autophagy in Diabetic Polyneuropathy. J Diabetes Res. 2017;2017:1673081. [CrossRef]
- Sifuentes-Franco S, Padilla-Tejeda DE, Carrillo-Ibarra S, Miranda-Diaz AG. Oxidative Stress, Apoptosis, and Mitochondrial Function in Diabetic Nephropathy. Int J Endocrinol. 2018;2018:1875870. [CrossRef]
- Santiago AR, Boia R, Aires ID, Ambrosio AF, Fernandes R. Sweet Stress: Coping With Vascular Dysfunction in Diabetic Retinopathy. Front Physiol. 2018;9:820. [CrossRef]
- Bokhary K, Aljaser F, Abudawood M, et al. Role of Oxidative Stress and Severity of Diabetic Retinopathy in Type 1 and Type 2 Diabetes. Ophthalmic Res. 2021;64(4):613-621. [CrossRef]
- Pang L, Lian X, Liu H, et al. Understanding Diabetic Neuropathy: Focus on Oxidative Stress. Oxid Med Cell Longev. 2020;2020:9524635. [CrossRef]
- Kowluru RA, Chan PS. Oxidative stress and diabetic retinopathy. Exp Diabetes Res. 2007;2007:43603. [CrossRef]
- Johansen JS, Harris AK, Rychly DJ, Ergul A. Oxidative stress and the use of antioxidants in diabetes: linking basic science to clinical practice. Cardiovasc Diabetol. Apr 29 2005;4:5. [CrossRef]
- Ceriello A, Testa R, Genovese S. Clinical implications of oxidative stress and potential role of natural antioxidants in diabetic vascular complications. Nutr Metab Cardiovasc Dis. Apr 2016;26(4):285-92. [CrossRef]
- Nandi A, Counts N, Broker J, et al. Cost of care for Alzheimer’s disease and related dementias in the United States: 2016 to 2060. NPJ Aging. Feb 8 2024;10(1):13. [CrossRef]
- Cobley JN, Fiorello ML, Bailey DM. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. May 2018;15:490-503. [CrossRef]
- Salim S. Oxidative Stress and the Central Nervous System. J Pharmacol Exp Ther. Jan 2017;360(1):201-205. [CrossRef]
- Aoyama K. Glutathione in the Brain. Int J Mol Sci. May 9 2021;22(9). [CrossRef]
- Sidorova Y, Domanskyi A. Detecting Oxidative Stress Biomarkers in Neurodegenerative Disease Models and Patients. Methods Protoc. Sep 24 2020;3(4). [CrossRef]
- Cenini G, Lloret A, Cascella R. Oxidative Stress in Neurodegenerative Diseases: From a Mitochondrial Point of View. Oxid Med Cell Longev. 2019;2019:2105607. [CrossRef]
- Wei Z, Li X, Li X, Liu Q, Cheng Y. Oxidative Stress in Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front Mol Neurosci. 2018;11:236. [CrossRef]
- Kumar A, Ratan RR. Oxidative Stress and Huntington’s Disease: The Good, The Bad, and The Ugly. J Huntingtons Dis. Oct 1 2016;5(3):217-237. [CrossRef]
- Cioffi F, Adam RHI, Bansal R, Broersen K. A Review of Oxidative Stress Products and Related Genes in Early Alzheimer’s Disease. J Alzheimers Dis. 2021;83(3):977-1001. [CrossRef]
- Sanabria-Castro A, Alape-Giron A, Flores-Diaz M, Echeverri-McCandless A, Parajeles-Vindas A. Oxidative stress involvement in the molecular pathogenesis and progression of multiple sclerosis: a literature review. Rev Neurosci. Jan 2 2024;doi:10.1515/revneuro-2023-0091.
- Alqahtani T, Deore SL, Kide AA, et al. Mitochondrial dysfunction and oxidative stress in Alzheimer’s disease, and Parkinson’s disease, Huntington’s disease and Amyotrophic Lateral Sclerosis -An updated review. Mitochondrion. Jul 2023;71:83-92. [CrossRef]
- Dubois B, von Arnim CAF, Burnie N, Bozeat S, Cummings J. Biomarkers in Alzheimer’s disease: role in early and differential diagnosis and recognition of atypical variants. Alzheimers Res Ther. Oct 13 2023;15(1):175. [CrossRef]
- Moreira PI. Sweet Mitochondria: A Shortcut to Alzheimer’s Disease. J Alzheimers Dis. 2018;62(3):1391-1401. [CrossRef]
- Tonnies E, Trushina E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J Alzheimers Dis. 2017;57(4):1105-1121. [CrossRef]
- Zhao Y, Zhao B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid Med Cell Longev. 2013;2013:316523. [CrossRef]
- Yiannopoulou KG, Anastasiou AI, Zachariou V, Pelidou SH. Reasons for Failed Trials of Disease-Modifying Treatments for Alzheimer Disease and Their Contribution in Recent Research. Biomedicines. Dec 9 2019;7(4). [CrossRef]
- Haass C, Selkoe D. If amyloid drives Alzheimer disease, why have anti-amyloid therapies not yet slowed cognitive decline? PLoS Biol. Jul 2022;20(7):e3001694. [CrossRef]
- Kametani F, Hasegawa M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front Neurosci. 2018;12:25. [CrossRef]
- Yiannopoulou KG, Papageorgiou SG. Current and Future Treatments in Alzheimer Disease: An Update. J Cent Nerv Syst Dis. 2020;12:1179573520907397. [CrossRef]
- Cheong SL, Tiew JK, Fong YH, et al. Current Pharmacotherapy and Multi-Target Approaches for Alzheimer’s Disease. Pharmaceuticals (Basel). Dec 14 2022;15(12). [CrossRef]
- Wright AL, Zinn R, Hohensinn B, et al. Neuroinflammation and neuronal loss precede Abeta plaque deposition in the hAPP-J20 mouse model of Alzheimer’s disease. PLoS One. 2013;8(4):e59586. [CrossRef]
- Nunomura A, Perry G. RNA and Oxidative Stress in Alzheimer’s Disease: Focus on microRNAs. Oxid Med Cell Longev. 2020;2020:2638130. [CrossRef]
- Nunomura A, Perry G, Aliev G, et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. Aug 2001;60(8):759-67. [CrossRef]
- Wang X, Wang W, Li L, Perry G, Lee HG, Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta. Aug 2014;1842(8):1240-7. [CrossRef]
- Suppiah S, Didier MA, Vinjamuri S. The Who, When, Why, and How of PET Amyloid Imaging in Management of Alzheimer’s Disease-Review of Literature and Interesting Images. Diagnostics (Basel). Jun 25 2019;9(2). [CrossRef]
- Sultana MA, Hia RA, Akinsiku O, Hegde V. Peripheral Mitochondrial Dysfunction: A Potential Contributor to the Development of Metabolic Disorders and Alzheimer’s Disease. Biology (Basel). Jul 19 2023;12(7). [CrossRef]
- Majeed A, Marwick B, Yu H, Fadavi H, Tavakoli M. Ophthalmic Biomarkers for Alzheimer’s Disease: A Review. Front Aging Neurosci. 2021;13:720167. [CrossRef]
- Klyucherev TO, Olszewski P, Shalimova AA, et al. Advances in the development of new biomarkers for Alzheimer’s disease. Transl Neurodegener. Apr 21 2022;11(1):25. [CrossRef]
- Auso E, Gomez-Vicente V, Esquiva G. Biomarkers for Alzheimer’s Disease Early Diagnosis. J Pers Med. Sep 4 2020;10(3). [CrossRef]
- Collin F, Cheignon C, Hureau C. Oxidative stress as a biomarker for Alzheimer’s disease. Biomark Med. Mar 2018;12(3):201-203. [CrossRef]
- Li W, Zhang M, Huang R, et al. Topographic metabolism-function relationships in Alzheimer’s disease: A simultaneous PET/MRI study. Hum Brain Mapp. Feb 1 2024;45(2):e26604. [CrossRef]
- Fracassi A, Marcatti M, Zolochevska O, et al. Oxidative Damage and Antioxidant Response in Frontal Cortex of Demented and Nondemented Individuals with Alzheimer’s Neuropathology. J Neurosci. Jan 20 2021;41(3):538-554. [CrossRef]
- Grari O, Elmoujtahide D, Sebbar E, Choukri M. The Biochemistry Behind Cognitive Decline: Biomarkers of Alzheimer’s Disease. EJIFCC. Dec 2023;34(4):276-283.
- Koronyo Y, Rentsendorj A, Mirzaei N, et al. Retinal pathological features and proteome signatures of Alzheimer’s disease. Acta Neuropathol. Apr 2023;145(4):409-438. [CrossRef]
- Tamagno E, Guglielmotto M, Vasciaveo V, Tabaton M. Oxidative Stress and Beta Amyloid in Alzheimer’s Disease. Which Comes First: The Chicken or the Egg? Antioxidants (Basel). Sep 16 2021;10(9). [CrossRef]
- Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. Apr 2015;14(4):388-405. [CrossRef]
- Pardo-Moreno T, Gonzalez-Acedo A, Rivas-Dominguez A, et al. Therapeutic Approach to Alzheimer’s Disease: Current Treatments and New Perspectives. Pharmaceutics. May 24 2022;14(6). [CrossRef]
- Plascencia-Villa G, Perry G. Lessons from antiamyloid-beta immunotherapies in Alzheimer’s disease. Handb Clin Neurol. 2023;193:267-292. [CrossRef]
- Plascencia-Villa G, Perry G. Roles of Oxidative Stress in Synaptic Dysfunction and Neuronal Cell Death in Alzheimer’s Disease. Antioxidants (Basel). Aug 17 2023;12(8). [CrossRef]
- Gribkoff VK, Kaczmarek LK. The need for new approaches in CNS drug discovery: Why drugs have failed, and what can be done to improve outcomes. Neuropharmacology. Jul 1 2017;120:11-19. [CrossRef]
- Yao W, Yang H, Yang J. Small-molecule drugs development for Alzheimer’s disease. Front Aging Neurosci. 2022;14:1019412. [CrossRef]
- Mehta RI, Mehta RI. The Vascular-Immune Hypothesis of Alzheimer’s Disease. Biomedicines. Jan 30 2023;11(2). [CrossRef]
- Walker KA, Le Page LM, Terrando N, Duggan MR, Heneka MT, Bettcher BM. The role of peripheral inflammatory insults in Alzheimer’s disease: a review and research roadmap. Mol Neurodegener. Jun 5 2023;18(1):37. [CrossRef]
- Rentz DM, Wessels AM, Annapragada AV, et al. Building clinically relevant outcomes across the Alzheimer’s disease spectrum. Alzheimers Dement (N Y). 2021;7(1):e12181. [CrossRef]
- Si ZZ, Zou CJ, Mei X, et al. Targeting neuroinflammation in Alzheimer’s disease: from mechanisms to clinical applications. Neural Regen Res. Apr 2023;18(4):708-715. [CrossRef]
- Novoa C, Salazar P, Cisternas P, et al. Inflammation context in Alzheimer’s disease, a relationship intricate to define. Biol Res. Dec 23 2022;55(1):39. [CrossRef]
- Puspita L, Chung SY, Shim JW. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol Brain. Nov 28 2017;10(1):53. [CrossRef]
- Aborode AT, Pustake M, Awuah WA, et al. Targeting Oxidative Stress Mechanisms to Treat Alzheimer’s and Parkinson’s Disease: A Critical Review. Oxid Med Cell Longev. 2022;2022:7934442. [CrossRef]
- Watanabe H, Dijkstra JM, Nagatsu T. Parkinson’s Disease: Cells Succumbing to Lifelong Dopamine-Related Oxidative Stress and Other Bioenergetic Challenges. Int J Mol Sci. Feb 7 2024;25(4). [CrossRef]
- Tavassolifar MJ, Vodjgani M, Salehi Z, Izad M. The Influence of Reactive Oxygen Species in the Immune System and Pathogenesis of Multiple Sclerosis. Autoimmune Dis. 2020;2020:5793817. [CrossRef]
- Motataianu A, Serban G, Barcutean L, Balasa R. Oxidative Stress in Amyotrophic Lateral Sclerosis: Synergy of Genetic and Environmental Factors. Int J Mol Sci. Aug 19 2022;23(16). [CrossRef]
- Ortiz GG, Pacheco-Moises FP, Bitzer-Quintero OK, et al. Immunology and oxidative stress in multiple sclerosis: clinical and basic approach. Clin Dev Immunol. 2013;2013:708659. [CrossRef]
- Chauhan A, Audhya T, Chauhan V. Brain region-specific glutathione redox imbalance in autism. Neurochem Res. Aug 2012;37(8):1681-9. [CrossRef]
- Liu X, Lin J, Zhang H, et al. Oxidative Stress in Autism Spectrum Disorder-Current Progress of Mechanisms and Biomarkers. Front Psychiatry. 2022;13:813304. [CrossRef]
- Pangrazzi L, Balasco L, Bozzi Y. Oxidative Stress and Immune System Dysfunction in Autism Spectrum Disorders. Int J Mol Sci. May 6 2020;21(9). [CrossRef]
- Morimoto M, Hashimoto T, Tsuda Y, Suenaga M, Nakamura T, Katoh S. Study on oxidative stress and inflammatory/antioxidant substance levels in autism spectrum disorder. J Chin Med Assoc. May 1 2023;86(5):489-493. [CrossRef]
- Delhey LM, Tippett M, Rose S, et al. Comparison of Treatment for Metabolic Disorders Associated with Autism:Reanalysis of Three Clinical Trials. Front Neurosci. 2018;12:19. [CrossRef]
- Rose S, Melnyk S, Pavliv O, et al. Evidence of oxidative damage and inflammation associated with low glutathione redox status in the autism brain. Transl Psychiatry. Jul 10 2012;2(7):e134. [CrossRef]
- Bjorklund G, Dosa MD, Maes M, et al. The impact of glutathione metabolism in autism spectrum disorder. Pharmacol Res. Apr 2021;166:105437. [CrossRef]
- Wang M, Xu D, Zhang L, Jiang H. Application of Multimodal MRI in the Early Diagnosis of Autism Spectrum Disorders: A Review. Diagnostics (Basel). Sep 22 2023;13(19). [CrossRef]
- Lin X, Wang G, Shen S, Zhan J. Advances in the Diagnosis and Treatment of Autism Spectrum Disorders in Children. Altern Ther Health Med. Oct 27 2023;
- Wei JD, Xu X. Oxidative stress in Wernicke’s encephalopathy. Front Aging Neurosci. 2023;15:1150878. [CrossRef]
- Zhao M, Zhu P, Fujino M, et al. Oxidative Stress in Hypoxic-Ischemic Encephalopathy: Molecular Mechanisms and Therapeutic Strategies. Int J Mol Sci. Dec 10 2016;17(12). [CrossRef]
- Antar SA, Ashour NA, Marawan ME, Al-Karmalawy AA. Fibrosis: Types, Effects, Markers, Mechanisms for Disease Progression, and Its Relation with Oxidative Stress, Immunity, and Inflammation. Int J Mol Sci. Feb 16 2023;24(4). [CrossRef]
- Huang E, Peng N, Xiao F, Hu D, Wang X, Lu L. The Roles of Immune Cells in the Pathogenesis of Fibrosis. Int J Mol Sci. Jul 22 2020;21(15). [CrossRef]
- Pinzaru AD, Mihai CM, Chisnoiu T, et al. Oxidative Stress Biomarkers in Cystic Fibrosis and Cystic Fibrosis-Related Diabetes in Children: A Literature Review. Biomedicines. Sep 29 2023;11(10). [CrossRef]
- Grasemann H, Ratjen F. Cystic Fibrosis. N Engl J Med. Nov 2 2023;389(18):1693-1707. [CrossRef]
- Li B, Zhang C, Zhan YT. Nonalcoholic Fatty Liver Disease Cirrhosis: A Review of Its Epidemiology, Risk Factors, Clinical Presentation, Diagnosis, Management, and Prognosis. Can J Gastroenterol Hepatol. 2018;2018:2784537. [CrossRef]
- Delli Bovi AP, Marciano F, Mandato C, Siano MA, Savoia M, Vajro P. Oxidative Stress in Non-alcoholic Fatty Liver Disease. An Updated Mini Review. Front Med (Lausanne). 2021;8:595371. [CrossRef]
- Stickel F, Osterreicher CH, Datz C, et al. Prediction of progression to cirrhosis by a glutathione S-transferase P1 polymorphism in subjects with hereditary hemochromatosis. Arch Intern Med. Sep 12 2005;165(16):1835-40. [CrossRef]
- Grattagliano I, Calamita G, Cocco T, Wang DQ, Portincasa P. Pathogenic role of oxidative and nitrosative stress in primary biliary cirrhosis. World J Gastroenterol. May 21 2014;20(19):5746-59. [CrossRef]
- Doroszko A, Dobrowolski P, Radziwon-Balicka A, Skomro R. New Insights into the Role of Oxidative Stress in Onset of Cardiovascular Disease. Oxid Med Cell Longev. 2018;2018:9563831. [CrossRef]
- Frak W, Wojtasinska A, Lisinska W, Mlynarska E, Franczyk B, Rysz J. Pathophysiology of Cardiovascular Diseases: New Insights into Molecular Mechanisms of Atherosclerosis, Arterial Hypertension, and Coronary Artery Disease. Biomedicines. Aug 10 2022;10(8). [CrossRef]
- Pignatelli P, Menichelli D, Pastori D, Violi F. Oxidative stress and cardiovascular disease: new insights. Kardiol Pol. 2018;76(4):713-722. [CrossRef]
- Eberhardt N, Noval MG, Kaur R, et al. SARS-CoV-2 infection triggers pro-atherogenic inflammatory responses in human coronary vessels. Nat Cardiovasc Res. Oct 2023;2(10):899-916. [CrossRef]
- Zhang Z, Rong L, Li YP. Flaviviridae Viruses and Oxidative Stress: Implications for Viral Pathogenesis. Oxid Med Cell Longev. 2019;2019:1409582. [CrossRef]
- Khaiboullina SF, Levis S, Morzunov SP, et al. Serum Cytokine Profiles Differentiating Hemorrhagic Fever with Renal Syndrome and Hantavirus Pulmonary Syndrome. Front Immunol. 2017;8:567. [CrossRef]
- Tschope C, Ammirati E, Bozkurt B, et al. Myocarditis and inflammatory cardiomyopathy: current evidence and future directions. Nat Rev Cardiol. Mar 2021;18(3):169-193. [CrossRef]
- Messaoudi I, Basler CF. Immunological features underlying viral hemorrhagic fevers. Curr Opin Immunol. Oct 2015;36:38-46. [CrossRef]
- Choi JH, Croyle MA. Emerging targets and novel approaches to Ebola virus prophylaxis and treatment. BioDrugs. Dec 2013;27(6):565-83. [CrossRef]
- van Leur SW, Heunis T, Munnur D, Sanyal S. Pathogenesis and virulence of flavivirus infections. Virulence. Dec 2021;12(1):2814-2838. [CrossRef]
- Hosakote YM, Jantzi PD, Esham DL, et al. Viral-mediated inhibition of antioxidant enzymes contributes to the pathogenesis of severe respiratory syncytial virus bronchiolitis. Am J Respir Crit Care Med. Jun 1 2011;183(11):1550-60. [CrossRef]
- Castro SM, Guerrero-Plata A, Suarez-Real G, et al. Antioxidant treatment ameliorates respiratory syncytial virus-induced disease and lung inflammation. Am J Respir Crit Care Med. Dec 15 2006;174(12):1361-9. [CrossRef]
- Guler MC, Tanyeli A, Ekinci Akdemir FN, et al. An Overview of Ischemia-Reperfusion Injury: Review on Oxidative Stress and Inflammatory Response. Eurasian J Med. Dec 2022;54(Suppl1):62-65. [CrossRef]
- Choi EK, Lim DG. Hepatic ischemia-reperfusion injury with respect to oxidative stress and inflammatory response: a narrative review. J Yeungnam Med Sci. Apr 2023;40(2):115-122. [CrossRef]
- Chazelas P, Steichen C, Favreau F, et al. Oxidative Stress Evaluation in Ischemia Reperfusion Models: Characteristics, Limits and Perspectives. Int J Mol Sci. Feb 27 2021;22(5). [CrossRef]
- Xiang M, Lu Y, Xin L, et al. Role of Oxidative Stress in Reperfusion following Myocardial Ischemia and Its Treatments. Oxid Med Cell Longev. 2021;2021:6614009. [CrossRef]
- He J, Liu D, Zhao L, et al. Myocardial ischemia/reperfusion injury: Mechanisms of injury and implications for management (Review). Exp Ther Med. Jun 2022;23(6):430. [CrossRef]
- Kurian GA, Rajagopal R, Vedantham S, Rajesh M. The Role of Oxidative Stress in Myocardial Ischemia and Reperfusion Injury and Remodeling: Revisited. Oxid Med Cell Longev. 2016;2016:1656450. [CrossRef]
- Zhao C, Liu T, Wei H, Li J. Serum oxidative stress factors predict myocardial ischemia reperfusion injury after percutaneous coronary intervention in patients with acute myocardial infarction and type 2 diabetes mellitus. Postepy Kardiol Interwencyjnej. Dec 2023;19(4):333-342. [CrossRef]
- Wu L, Xiong X, Wu X, et al. Targeting Oxidative Stress and Inflammation to Prevent Ischemia-Reperfusion Injury. Front Mol Neurosci. 2020;13:28. [CrossRef]
- Jurcau A, Ardelean AI. Oxidative Stress in Ischemia/Reperfusion Injuries following Acute Ischemic Stroke. Biomedicines. Mar 1 2022;10(3). [CrossRef]
- Li Z, Bi R, Sun S, et al. The Role of Oxidative Stress in Acute Ischemic Stroke-Related Thrombosis. Oxid Med Cell Longev. 2022;2022:8418820. [CrossRef]
- Zhang Y, Khan S, Liu Y, Wu G, Yong VW, Xue M. Oxidative Stress Following Intracerebral Hemorrhage: From Molecular Mechanisms to Therapeutic Targets. Front Immunol. 2022;13:847246. [CrossRef]
- Shao L, Chen S, Ma L. Secondary Brain Injury by Oxidative Stress After Cerebral Hemorrhage: Recent Advances. Front Cell Neurosci. 2022;16:853589. [CrossRef]
- Gross BA, Jankowitz BT, Friedlander RM. Cerebral Intraparenchymal Hemorrhage: A Review. JAMA. Apr 2 2019;321(13):1295-1303. [CrossRef]
- Simona MS, Alessandra V, Emanuela C, et al. Evaluation of Oxidative Stress and Metabolic Profile in a Preclinical Kidney Transplantation Model According to Different Preservation Modalities. Int J Mol Sci. Jan 5 2023;24(2). [CrossRef]
- Hofmann J, Puhringer M, Steinkellner S, et al. Novel, Innovative Models to Study Ischemia/Reperfusion-Related Redox Damage in Organ Transplantation. Antioxidants (Basel). Dec 24 2022;12(1). [CrossRef]
- Sundd P, Gladwin MT, Novelli EM. Pathophysiology of Sickle Cell Disease. Annu Rev Pathol. Jan 24 2019;14:263-292. [CrossRef]
- Antwi-Boasiako C, Dankwah GB, Aryee R, Hayfron-Benjamin C, Donkor ES, Campbell AD. Oxidative Profile of Patients with Sickle Cell Disease. Med Sci (Basel). Jan 25 2019;7(2). [CrossRef]
- Wang Q, Zennadi R. The Role of RBC Oxidative Stress in Sickle Cell Disease: From the Molecular Basis to Pathologic Implications. Antioxidants (Basel). Oct 13 2021;10(10). [CrossRef]
- Vona R, Sposi NM, Mattia L, Gambardella L, Straface E, Pietraforte D. Sickle Cell Disease: Role of Oxidative Stress and Antioxidant Therapy. Antioxidants (Basel). Feb 16 2021;10(2). [CrossRef]
- Chiang KC, Gupta A, Sundd P, Krishnamurti L. Thrombo-Inflammation in COVID-19 and Sickle Cell Disease: Two Faces of the Same Coin. Biomedicines. Jan 25 2023;11(2). [CrossRef]
- Carrero D, Soria-Valles C, Lopez-Otin C. Hallmarks of progeroid syndromes: lessons from mice and reprogrammed cells. Dis Model Mech. Jul 1 2016;9(7):719-35. [CrossRef]
- Villa-Bellosta R. Redox theory in progeria. Aging (Albany NY). Oct 31 2020;12(21):20934-20935. [CrossRef]
- Trigueros-Motos L, Gonzalez JM, Rivera J, Andres V. Hutchinson-Gilford progeria syndrome, cardiovascular disease and oxidative stress. Front Biosci (Schol Ed). Jun 1 2011;3(4):1285-97. [CrossRef]
- Seco-Cervera M, Spis M, Garcia-Gimenez JL, et al. Oxidative stress and antioxidant response in fibroblasts from Werner and atypical Werner syndromes. Aging (Albany NY). Mar 2014;6(3):231-45. [CrossRef]
- Iskusnykh IY, Zakharova AA, Pathak D. Glutathione in Brain Disorders and Aging. Molecules. Jan 5 2022;27(1). [CrossRef]
- Ben-Shachar R, Chen Y, Luo S, Hartman C, Reed M, Nijhout HF. The biochemistry of acetaminophen hepatotoxicity and rescue: a mathematical model. Theor Biol Med Model. Dec 19 2012;9:55. [CrossRef]
- Deavall DG, Martin EA, Horner JM, Roberts R. Drug-induced oxidative stress and toxicity. J Toxicol. 2012;2012:645460. [CrossRef]
- Conklin KA. Chemotherapy-associated oxidative stress: impact on chemotherapeutic effectiveness. Integr Cancer Ther. Dec 2004;3(4):294-300. [CrossRef]
- Kusirisin P, Chattipakorn SC, Chattipakorn N. Contrast-induced nephropathy and oxidative stress: mechanistic insights for better interventional approaches. J Transl Med. Oct 20 2020;18(1):400. [CrossRef]
- Oh GS, Kim HJ, Shen A, et al. New Therapeutic Concept of NAD Redox Balance for Cisplatin Nephrotoxicity. Biomed Res Int. 2016;2016:4048390. [CrossRef]
- Volarevic V, Djokovic B, Jankovic MG, et al. Molecular mechanisms of cisplatin-induced nephrotoxicity: a balance on the knife edge between renoprotection and tumor toxicity. J Biomed Sci. Mar 13 2019;26(1):25. [CrossRef]
- Narezkina A, Nasim K. Anthracycline Cardiotoxicity. Circ Heart Fail. Mar 2019;12(3):e005910. [CrossRef]
- Carrasco R, Castillo RL, Gormaz JG, Carrillo M, Thavendiranathan P. Role of Oxidative Stress in the Mechanisms of Anthracycline-Induced Cardiotoxicity: Effects of Preventive Strategies. Oxid Med Cell Longev. 2021;2021:8863789. [CrossRef]
- Jiang H, Zuo J, Li B, et al. Drug-induced oxidative stress in cancer treatments: Angel or devil? Redox Biol. Jul 2023;63:102754. [CrossRef]
- Fabiani I, Aimo A, Grigoratos C, et al. Oxidative stress and inflammation: determinants of anthracycline cardiotoxicity and possible therapeutic targets. Heart Fail Rev. Jul 2021;26(4):881-890. [CrossRef]
- Ramachandran A, Jaeschke H. Mitochondria in Acetaminophen-Induced Liver Injury and Recovery: A Concise Review. Livers. Jun 2023;3(2):219-231. [CrossRef]
- Biswas A, Santra S, Bishnu D, Dhali GK, Chowdhury A, Santra A. Isoniazid and Rifampicin Produce Hepatic Fibrosis through an Oxidative Stress-Dependent Mechanism. Int J Hepatol. 2020;2020:6987295. [CrossRef]
- Le Dare B, Ferron PJ, Gicquel T. Toxic Effects of Amanitins: Repurposing Toxicities toward New Therapeutics. Toxins (Basel). Jun 11 2021;13(6). [CrossRef]
- Nebbioso M, Franzone F, Lambiase A, et al. Oxidative Stress Implication in Retinal Diseases-A Review. Antioxidants (Basel). Sep 10 2022;11(9). [CrossRef]
- Kusuhara S, Fukushima Y, Ogura S, Inoue N, Uemura A. Pathophysiology of Diabetic Retinopathy: The Old and the New. Diabetes Metab J. Oct 2018;42(5):364-376. [CrossRef]
- Cecilia OM, Jose Alberto CG, Jose NP, et al. Oxidative Stress as the Main Target in Diabetic Retinopathy Pathophysiology. J Diabetes Res. 2019;2019:8562408. [CrossRef]
- Vingolo EM, Casillo L, Contento L, Toja F, Florido A. Retinitis Pigmentosa (RP): The Role of Oxidative Stress in the Degenerative Process Progression. Biomedicines. Mar 2 2022;10(3). [CrossRef]
- Abokyi S, To CH, Lam TT, Tse DY. Central Role of Oxidative Stress in Age-Related Macular Degeneration: Evidence from a Review of the Molecular Mechanisms and Animal Models. Oxid Med Cell Longev. 2020;2020:7901270. [CrossRef]
- Ruan Y, Jiang S, Gericke A. Age-Related Macular Degeneration: Role of Oxidative Stress and Blood Vessels. Int J Mol Sci. Jan 28 2021;22(3). [CrossRef]
- Toma C, De Cilla S, Palumbo A, Garhwal DP, Grossini E. Oxidative and Nitrosative Stress in Age-Related Macular Degeneration: A Review of Their Role in Different Stages of Disease. Antioxidants (Basel). Apr 23 2021;10(5). [CrossRef]
- Bar-Or D, Carrick MM, Mains CW, Rael LT, Slone D, Brody EN. Sepsis, oxidative stress, and hypoxia: Are there clues to better treatment? Redox Rep. Sep 2015;20(5):193-7. [CrossRef]
- Hsiao SY, Kung CT, Su CM, et al. Impact of oxidative stress on treatment outcomes in adult patients with sepsis: A prospective study. Medicine (Baltimore). Jun 26 2020;99(26):e20872. [CrossRef]
- Makhija R, Kingsnorth AN. Cytokine storm in acute pancreatitis. J Hepatobiliary Pancreat Surg. 2002;9(4):401-10. [CrossRef]
- Robles L, Vaziri ND, Ichii H. Role of Oxidative Stress in the Pathogenesis of Pancreatitis: Effect of Antioxidant Therapy. Pancreat Disord Ther. Apr 1 2013;3(1):112. [CrossRef]
- Padureanu V, Florescu DN, Padureanu R, Ghenea AE, Gheonea DI, Oancea CN. Role of antioxidants and oxidative stress in the evolution of acute pancreatitis (Review). Exp Ther Med. Mar 2022;23(3):197. [CrossRef]
- Verlaan M, Roelofs HM, van-Schaik A, et al. Assessment of oxidative stress in chronic pancreatitis patients. World J Gastroenterol. Sep 21 2006;12(35):5705-10. [CrossRef]
- Madamanchi NR, Hakim ZS, Runge MS. Oxidative stress in atherogenesis and arterial thrombosis: the disconnect between cellular studies and clinical outcomes. J Thromb Haemost. Feb 2005;3(2):254-67. [CrossRef]
Figure 1.
This illustrates the beneficial role of reactive oxygen species (ROS) when it is in physiologic balance with antioxidants. These include activating innate immunity and augmenting adaptive immunity. It should be noted that peroxynitrite and nitric oxide are co-existing reactive nitrogen species (RNS).
Figure 1.
This illustrates the beneficial role of reactive oxygen species (ROS) when it is in physiologic balance with antioxidants. These include activating innate immunity and augmenting adaptive immunity. It should be noted that peroxynitrite and nitric oxide are co-existing reactive nitrogen species (RNS).
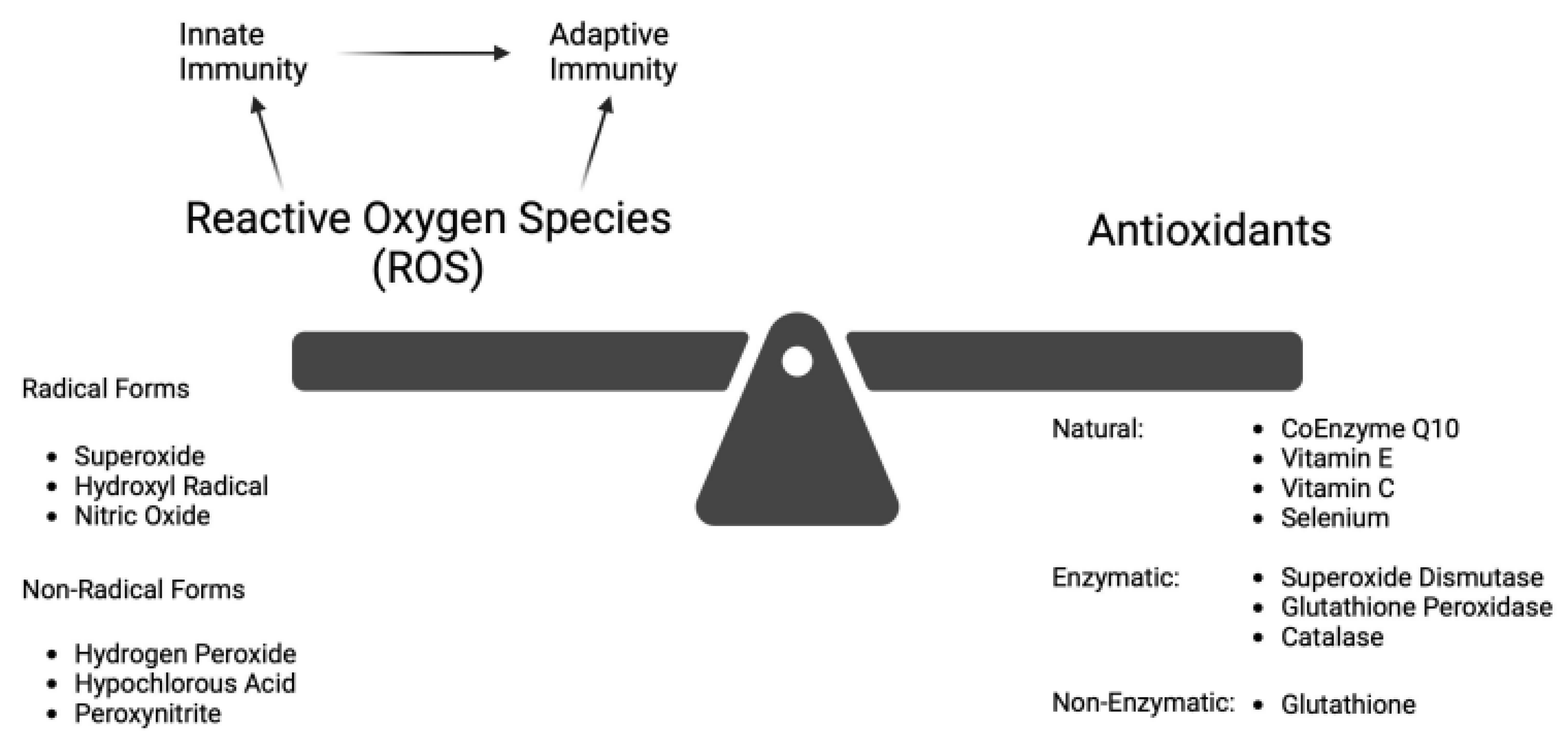
Figure 2.
This illustrates the state of oxidative stress (OS) that occurs from an imbalance of ROS relative to antioxidants. It can result from excess ROS, a deficiency of antioxidants, or a combination of both. Acute insults occur when stimulation of ROS overwhelms available antioxidants. Chronic insults occur when antioxidants are either deficient or exhausted. Presumptively, the development of numerous seemingly disparate acute and chronic diseases may the result of OS. (Created with BioRender).
Figure 2.
This illustrates the state of oxidative stress (OS) that occurs from an imbalance of ROS relative to antioxidants. It can result from excess ROS, a deficiency of antioxidants, or a combination of both. Acute insults occur when stimulation of ROS overwhelms available antioxidants. Chronic insults occur when antioxidants are either deficient or exhausted. Presumptively, the development of numerous seemingly disparate acute and chronic diseases may the result of OS. (Created with BioRender).
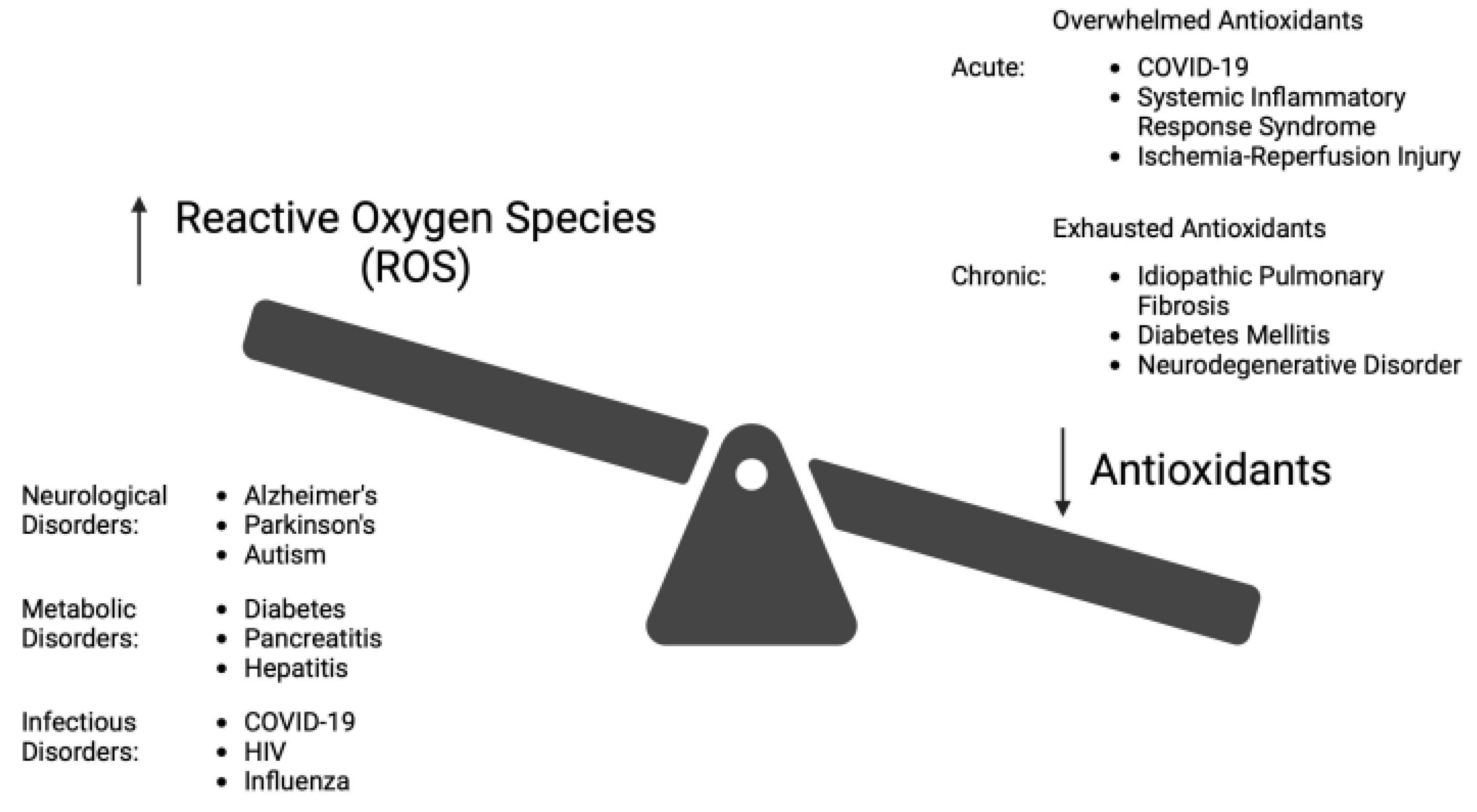
Figure 3.
Illustrated are examples of acute and chronic diseases associated with OS. An acute disease resulting from OS is COVID-19. OS leads to a dysregulation of the cytokine response leading to the cytokine storm (CS). The CS causes multi-organ failure (MOF), including COVID-associated acute respiratory distress syndrome (ARDS). Chronically, OS causes beta-cell dysfunction, insulin resistance leading to diabetes and its many neuro-vascular complications that results from the formation of advanced glycation end-products (AGEs). The onset of neurogenerative disorders appears to be initiated and enhanced by OS. (Created with BioRender).
Figure 3.
Illustrated are examples of acute and chronic diseases associated with OS. An acute disease resulting from OS is COVID-19. OS leads to a dysregulation of the cytokine response leading to the cytokine storm (CS). The CS causes multi-organ failure (MOF), including COVID-associated acute respiratory distress syndrome (ARDS). Chronically, OS causes beta-cell dysfunction, insulin resistance leading to diabetes and its many neuro-vascular complications that results from the formation of advanced glycation end-products (AGEs). The onset of neurogenerative disorders appears to be initiated and enhanced by OS. (Created with BioRender).
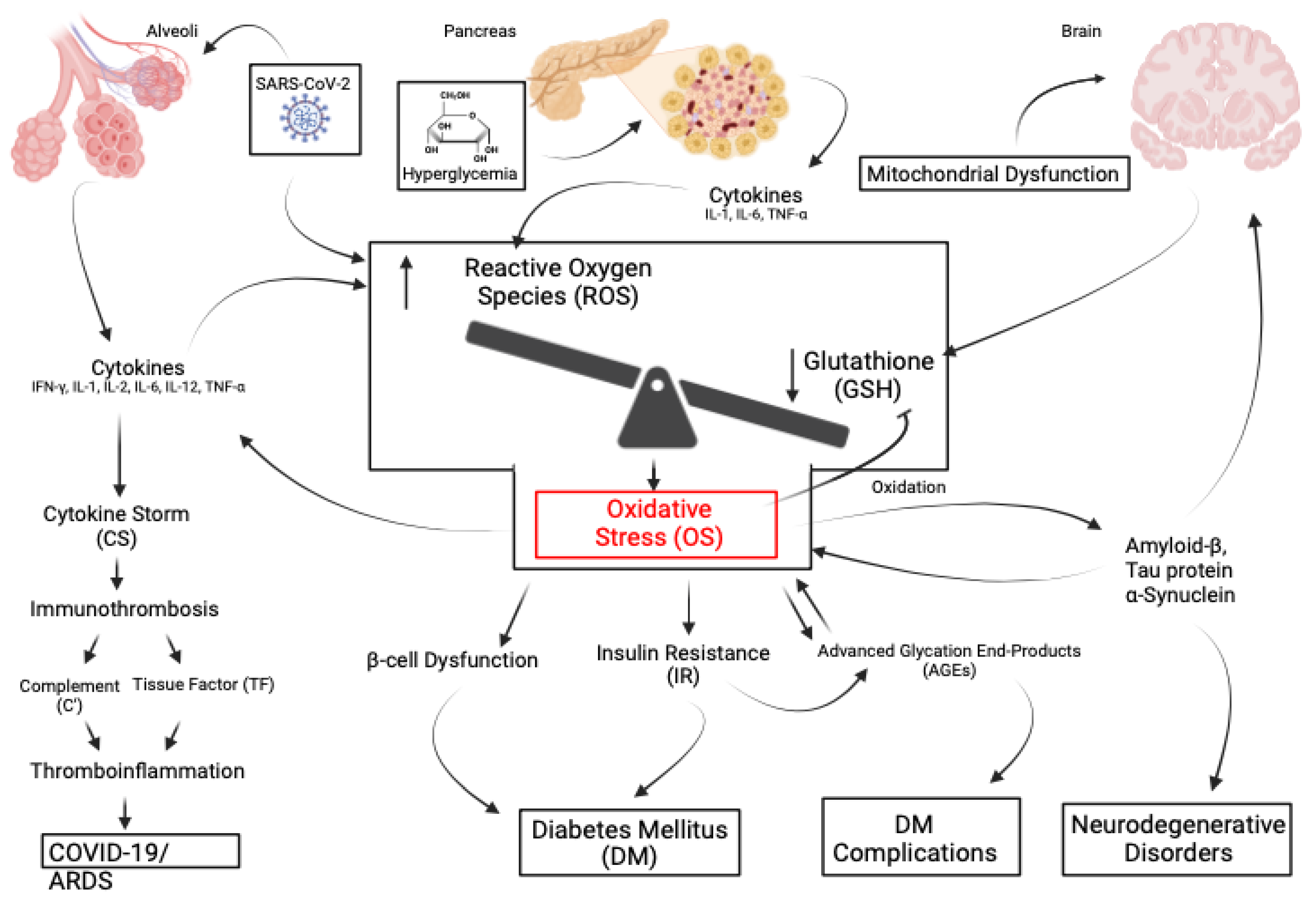
Figure 4.
This illustrates the concept that the prevention or early inhibition of OS is a more effective approach to managing diseases. This should be achieved by targeting diseases early when the pathophysiologic process is more easily controlled and prior to the development of irreversible tissue injury. (Created with BioRender).
Figure 4.
This illustrates the concept that the prevention or early inhibition of OS is a more effective approach to managing diseases. This should be achieved by targeting diseases early when the pathophysiologic process is more easily controlled and prior to the development of irreversible tissue injury. (Created with BioRender).
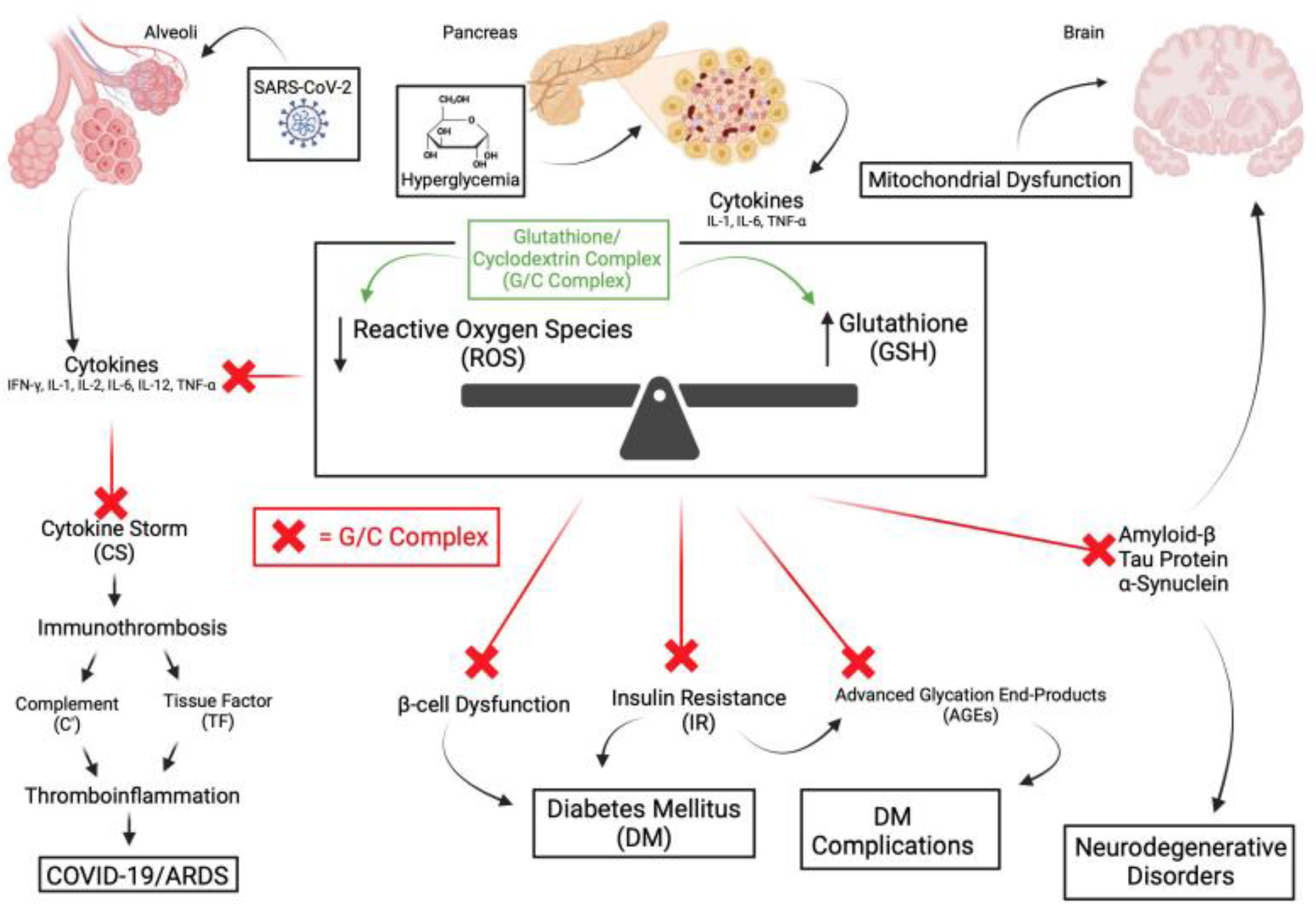
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



