Submitted:
13 June 2024
Posted:
14 June 2024
You are already at the latest version
Abstract
Background: The clinical spectrum of fibrotic interstitial lung diseases (ILD) is highly heterogeneous. We aimed to evaluate the prognostic value of widely available baseline biomarkers for improvement of lung function in patients with fibrotic ILD. Methods: This registry-based study included 142 patients with fibrotic ILD as defined by presence of reticulation, traction bronchiectasis or honeycombing on initial high-resolution computed tomography (HRCT). Functional improvement at 1 year was defined as relative increase of 5% in forced vital capacity (FVC) or of 10% in diffusion capacity for carbon monoxide (DLCO). The prognostic value of baseline biomarkers was evaluated for all patients and the subgroup with anti-inflammatory treatment. Results: At one year, 44 patients showed improvement while 73 progressed. Multivariate analyses found prognostic significance for age <60 years (OR 5.4; 95%CI 1.9-15.4; p=0.002), lactate dehydrogenase (LDH) >250U/L (OR 2.5; 95%CI 1.1-5.8; p=0.043) and blood monocyte count <0.8G/L (OR 3.5; 95%CI 1.1-11.3; p=0.034). In 84 patients undergoing anti-inflammatory treatment, multivariate analysis revealed age <60 years (OR 8.5 (95%CI 2.1-33.4; p=0.002) as the only significant variable. Conclusion: Younger age, higher LDH and lower blood monocyte count predicted functional improvement in fibrotic ILD patients, while in those treated with anti-inflammatory drugs, only age had significant implications.
Keywords:
Interstitial Lung Disease
; Biomarker
; Prognostication
1. Introduction
Interstitial lung diseases (ILD) encompass a wide spectrum of pulmonary disorders characterized by inflammation and fibrosis of the pulmonary alveolar, vascular and interstitial domains.[1] Clinical outcomes vary considerably depending on the specific ILD subtypes and individual patient characteristics. ILD with an assumed inflammatory etiology are more likely to response to immunosuppressive treatment, resulting in improved survival outcomes.[2,3,4,5] In contrast, fibrotic ILD carry a higher risk of progressive and irreversible loss of lung function ultimately leading to respiratory failure and death.[6] Recently, the official ATS/ERS/JRS/ALAT guideline introduced the term “progressive pulmonary fibrosis” (PPF) to describe the progressive subgroup of fibrotic ILDs other than idiopathic pulmonary fibrosis (IPF).[7] This novel classification emphasizes the fact that a heterogenous group of conditions may resemble the disease course of IPF.[8] Consequently, the available literature supports early detection of both IPF and PPF, as antifibrotic agents including pirfenidone or nintedanib can reduce loss of lung function leading to a more favorable outcome.[9] However, only a variable fraction of 18 to 32% of non-IPF ILD will progress significantly, making the identification of PPF challenging.[10] Moreover, evidence for use of antifibrotic therapy in patients presenting with fibrotic non-IPF ILD at first diagnosis is limited.[11] In fact, in those patients, immunosuppression still is the most common initial therapeutic approach.[3]
Over the last years, much research effort has been dedicated to identifying predictive biomarkers that enable personalized risk assessment in ILD. Significant impact on disease course was observed for age, sex, body mass index, specific gene variants like MUC5B, blood monocyte count and pulmonary function tests (PFT).[12] Recent research shows promising results for proteomics-based molecular biomarkers, but these are not commonly available in clinical practice yet.[13,14] In addition, presence and extent of fibrosis on HRCT have been identified as highly significant risk factors for disease progression in various ILD subtypes.[10,15] However, not all patients presenting with fibrotic ILD will experience progression, but a significant proportion may even demonstrate functional improvement over time, reflecting the heterogeneous nature of the disease, like shown in SSC-ILD.[16] Limited evidence exists regarding baseline biomarkers that could predict such improvement of lung function despite fibrotic CT pattern. Moreover, considering that the majority of non-IPF ILD patients receives immunomodulatory or immunosuppressant drugs for initial treatment, there is also a lack of data on the impact of this therapeutic approach both in terms of beneficial effects but also concerning the risk of developing PPF.
In the present study, our objective was to examine the correlation between widely available baseline biomarkers and improvement of lung function after one year in patients with fibrotic ILD of various causes. Furthermore, we assessed the possible impact of anti-inflammatory therapies in the study cohort.
2. Materials and Methods
This retrospective study is conducted using data obtained from patients included in an institutional ILD registry at Kepler University Hospital in Linz, Austria, between 2017 and 2021. It was performed in accordance with the Declaration of Helsinki and received approval as well as reassessment on a yearly basis by the ethics committee of the Medical Faculty of Linz (study number I-26-17). Furthermore, the study was conducted in adherence to the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) guidelines for reporting observational studies.[17]
All patients included for the present evaluation were provided written informed consent, and discussed by a multidisciplinary team in the local ILD board. Before that, a standardized ILD examination was performed including physical examination, HRCT imaging, laboratory analyses, PFT including spirometry, body plethysmography and measurement of diffusion capacity (JAEGER Master Screen PFT/Body/Diffusion, CareFusion, San Diego, United States of America). To be eligible for this study, all patients were required to have at least one of the typical CT hallmarks of pulmonary fibrosis, including reticular abnormalities, traction bronchiectasis, or honeycombing on initial HRCT. One-year survival data as well as at least one follow-up PFT assessment after initial examination had to be available. Treatment with systemic corticosteroids, immunomodulatory or immunosuppressant drugs was categorized as “anti-inflammatory”, when its duration was at least 6 weeks, and it had been primarily prescribed for the management of ILD and not for treating other conditions.
HRCT chest images were obtained in prone position whenever clinically feasible, following protocols proposed by the relevant guidelines.[18] The presence of parenchymal nodules, reticulation, honeycombing, consolidations, ground glass opacities, emphysema, mosaic attenuation, and traction bronchiectasis was assessed by a specialist ILD radiologist during the respective ILD board session. For semi-quantitative analysis of the ILD findings, the lungs were divided into an upper-, middle- and lower-lung area, as defined by thirds of the largest cranio-caudal diameter in the sagittal reconstructions. This approach resulted in a simple scoring system from one to six, as described in our previous publications.[15,19]
Blood samples were analyzed using a Sysmex® XN-3000 hematology analyzer (Sysmex Europe GmbH, Norderstedt, Germany) for cell counts and a Cobas® 8,000 modular analyzer (Roche Diagnostics International AG, Rotkreuz, Switzerland) for C-reactive protein (CRP), lactate dehydrogenase (LDH), and rheumatoid factor measurement. Autoimmune serology testing was performed using a EuroPatternMicroscope®, a Dynex® , and a EuroBlotOne® platform by Euroimmun (EUROIMMUN Medizinische Labordiagnostik AG, Lübeck, Germany) for anti-nuclear (ANA), anti-neutrophil cytoplasmatic (ANCA) and other disease-specific antibodies.
Pulmonary function tests including spirometry, body plethysmography, and measurement of diffusion capacity (JAEGER MasterScreen PFT/Body/Diffusion® , CareFusion, San Diego, United States of America) were performed to assess forced vital capacity (FVC, L/% predicted), forced expiratory volume in 1 s (FEV1, % predicted), FEV1/FVC ratio and diffusion capacity for carbon monoxide (DLCO, single breath method, mmol/(min x kPa) /%predicted). Normal values for spirometry were based on the GLI-2012 equations and those for body plethysmography and diffusion capacity on the 1993 ERS/ECCS regressions.
Functional improvement was defined as a relative increase of either >5% in FVC (% predicted) or, in case of stable FVC, a relative increase of 10% in DLCO within the first year after primary evaluation, regardless of the timing of the event during that time span. Disease progression was defined by a composite endpoint, including a relative decrease of 5% in FVC, of 10% in DLCO, lung transplant or death with the first year. After a follow-up period of one year, two cohorts were formed, with 44 patients showing functional improvement and 73 patients showing disease progression (Figure 1).
All statistical analyses were performed using R (version 4.3.0). Descriptive analyses included calculations of measures such as the mean, standard error, and proportions. Statistical tests, both parametric and non-parametric, were employed depending on the respective hypotheses and normal distribution of the data. These tests included the T-Test, Kruskal-Wallis Test, Mann-Whitney U Test, Spearman’s rank correlation test, Chi-Square Test, and Fisher’s Exact Test.
In preparation for the multivariate analysis, continuous variables such as age, LDH and monocytes were dichotomized into two categories. This dichotomization was based on a third variable, the presence or absence of functional improvement, with the aim of finding a cut-off value with high selectivity. For instance, the age cutoff of 60 years was identified as the optimal threshold for predicting improvement. Although dichotomization can lead to a loss of information, binary variables are often easier to interpret and more practical for implementation in medical practice.
To determine the optimal cutoff value, the CUTPOINTR-package in R (R: A language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria; Version 3.6.0) was utilized. Significant predictors of improvement were identified using logistic regression models.
3. Results
Out of 209 patients enrolled in the ILD registry between 2017 and 2022, 142 patients were eligible for the present analysis. Most patients had been diagnosed with autoimmune-associated ILD (24%), followed by idiopathic nonspecific interstitial pneumonia (NSIP) (21%), and IPF (16%). During the first year, six patients died, 50 experienced significant worsening in FVC and 36 patients showed improvement. Of the remaining 50 patients with stable FVC, 17 experienced significant worsening of DLCO at one year, while eight showed improvement. Thus, two cohorts were established, consisting of 73 patients with disease progression and 44 patients with functional improvement.
3.1. Primary Outcome
Respective baseline characteristics, PFTs, BAL fluid, peripheral blood- and HRCT findings for all patients, as well as separately for the progressive, stable and improved cohorts are shown in Table 1. Significant association with functional improvement could be observed for younger age (p=0,023), lower FVC (p=0,017) and FEV-1 % of predicted (p=0,018), lower peripheral blood monocyte count (p=0,046), elevated proportions of lymphocytes in BAL fluid (p=0,038), as well as for higher extent of GGO (p=0,047) and less TBR (p=0,019).
Univariate analysis demonstrated a significant association with functional improvement at one year for age (<60 vs. ≥60; OR 4.6; 95%CI 1.8-12.1; p=0.002), honeycombing score (0 vs. ≥0; OR 4.2; 95%CI 1.1-15.1; p=0,031), GGO score (0 vs. ≥0; OR 0,4; 95%CI 0.2-0.8; p=0,021), DLCO% of predicted (<43% vs. ≥43%; OR 2.9; 95%CI 1.3-6.7; p=0.010) and FVC % of predicted (<95% vs. ≥95%; OR 4.6; 95%CI 1.5-14.4; p=0.009), as shown by Table 2 and Figure 2a. In multivariate analysis excluding FVC% and DLCO% of predicted, improvement of lung function at one year was associated with age (<60 vs. ≥60; OR 5.4; 95%CI 1.9-15.4; p=0.002), LDH (>250 U/L vs. ≤250 U/L; OR 2.5; 95%CI 1.1-5.8; p=0.043), and monocyte count (<0.8G/L vs. ≥0.8G/L; OR 3.5; 95%CI 1.1-11.3; p=0.034), as shown in Table 2.
Functional improvement was observed in 66,7% of the patients aged <60 as compared to only 30,1% of the patients aged ≥60. While 48,3% of patients with blood monocyte count <0,8G/L experienced improvement of lung function, it was only 18,5% of patients with monocyte count >0,8G/L. Similarly, 44,7% of patients with LDH >250U/L had functional improvement, in contrast to 30,3% of patients with LDH <250U/L.
3.2. Secondary Outcome
Out of the total of 142 patients, 84 had received anti-inflammatory treatment as previously defined within the first year. Among them, 38 patients (45%) experienced disease progression, whereas 31 (37%) improved, and 15 patients (18%) remained stable.
Respective baseline characteristics, PFTs, peripheral blood, BAL fluid and HRCT findings for all patients undergoing anti-inflammatory treatment, as well as separately for the progression, stable and improvement cohorts are shown in Table 3. Significant associations with functional improvement could be observed for lower FVC% of predicted (p=0,008), FEV-1% of predicted (p=0,018) and lower TBR score (p= 0,043).
Univariate analysis demonstrated a significant association with function improvement at one year for age (<60 vs. ≥60; OR 8.5; 95%CI 2.1-33.4; p=0.002), DLCO% of predicted (<43% vs. ≥43%; OR 3.6; 95%CI 1.2-10.8; p=0.019) and FVC% of predicted (<95% vs. ≥95%; OR 9.3; 95%CI 1.1-78.2; p=0.040), as shown by Table 2 and Figure 2b. Of interest, the multivariate analysis excluding FVC% and DLCO% of predicted identified younger age (<60 vs. ≥60; OR 8.5; 95%CI 2.1-33.4; p=0.002) as the only significant variable associated with functional improvement.
4. Discussion
The results of this retrospective, registry-based evaluation suggest that younger age, higher LDH level and lower peripheral blood monocyte count were associated with improvement of lung function within one year in fibrotic ILD, irrespective of the underlying diagnosis or treatment. Among patients undergoing anti-inflammatory treatment, only younger age showed significant implications.
These findings are in accordance with the existing prognostic models for fibrotic ILD, with the gender-age-physiology (GAP)-score being the most commonly used.[12,20] In this staging system derived from IPF patients, older age was a significant predictor of mortality. Our observation of an association of younger age with functional improvement may be attributed to the higher prevalence of PPF and IPF in older individuals, as reported in several epidemiological studies.[21,22,23,24] In one study conducted by Hopkins et al., the prevalence for IPF increased twentyfold from the age group 50-59 to the > 90 age group for both genders, men and women.[23] In contrast, younger patients tend to experience ILD with autoimmune pathogenesis that carry a more favorable prognosis than IPF.[25,26] In line with this, we found LDH, a well-established indicator of inflammation, to be significantly higher in the subgroup with functional improvement. Conversely, elevation of LDH was found to be associated with progression of ILD in connective-tissue-diseases as well as in other autoimmune diseases, including Sjögren’s and anti-synthetase-syndrome.[27,28,29] Next to a possible role of LDH as a marker of generally increased disease activity leading to worse prognosis, these conflicting results may be because approximately half of our study population received anti-inflammatory treatment. In the MVA of this subgroup, the protective effect of elevated LDH diminished, possibly reflecting its potential to serve as an indicator of inflammatory disease activity and also treatment response.
Numerous studies have reported on the prognostic significance of monocytes for disease progression in IPF and other fibrotic ILD subtypes.[19,30,31] On the cellular level, monocytes are recruited to the lung following tissue injury, where they differentiate into profibrotic alveolar-macrophages, contributing to pulmonary fibrosis.[32,33] In contrast, depletion of monocytes was shown to reduce the extent of fibrosis in a mouse model and IPF patients with normal monocyte counts were reported to have a significantly improved survival.[34] Consistent with this pathophysiological model, our findings suggest that a lower level of circulating monocytes is predictive for improvement of lung function in fibrotic ILD. Interestingly, the prognostic value of a lower monocyte count diminished in the anti-inflammatory therapy subgroup. Results of previous studies reported on alterations in monocyte count and activity following the use of immunosuppressants, such as glucocorticoids or methotrexate.[35,36] However, the influence of these treatment effects on our findings is unlikely, as the monocyte count was assessed prior to receiving any ILD specific treatment in the majority of the patients. Thus, in our study cohort, elevated monocyte count probably indicated the risk of progressive fibrosis in untreated ILD, whereas lower levels of monocytes were associated with improved lung function. However, in fibrotic ILD patients receiving anti-inflammatory treatment, baseline monocyte count showed no predictive value in terms of functional improvement.
To our knowledge, this is the first study assessing prognostic biomarkers to predict functional improvement in fibrotic ILD. The growing knowledge of risk factors associated with IPF and PPF has led to an improved recognition of patients who are likely to progress and can benefit from antifibrotic therapy. However, a significant number of fibrotic ILD patients is still treated with immunosuppressants either due to an assumed underlying inflammation-driven disease or for not meeting criteria for diagnosing IPF or disease progression allowing prescription of antifibrotics.[3] In the appropriate clinical context, the findings of the present study support a trial of immunosuppressive treatment in patients with younger age, elevated LDH level and lower monocytes count, even despite fibrotic appearance on HRCT and irrespective of the underlying diagnosis. On the other hand, patients lacking these characteristics have a high risk of progression and should be treated with antifibrotic agents with a low threshold when appropriate and possible. If they still receive immunosuppressive agents, they should be monitored for progression very closely.
Limitations of this study include its retrospective, single-center design potentially causing bias and restricting the generalizability of the findings to a broader population. Due to the registry-based and retrospective methodology, no formal sample size calculation was performed, which poses another possible limitation to the presented data. In addition, the study population was heterogeneous, consisting of patients with different fibrotic ILD subtypes and treatment regimens. However, it represents the clinical practice of an university hospital ILD clinic with an emphasis for patients with autoimmune diseases. Our analyses concerning patients treated with anti-inflammatory agents are limited by the lack of a standard treatment leading to the use of a multitude of treatment options for various indications. The definition of “anti-inflammatory” treatment used in our study represents a minimal consensus primarily defined to enable statistical analyses. However, as therapeutic decisions were solely based on expert knowledge and clinical presentation of the patient, they may not be generalizable to other populations and differ from other centers. A major clinical advantage of the prognostic biomarkers identified in the current analysis is that they can be easily obtained in a minimally invasive and inexpensive fashion as part of the primary ILD evaluation. While age is readily available through medical records, LDH and monocyte count are typically included in routine blood tests. Although these biomarkers are unspecific when used alone, they may offer clinicians guidance in management decisions for fibrotic ILD patients, especially when several biomarkers are applied together in the context of the respective clinical data. In addition, they could allow for a decision support independent of the underlying ILD diagnosis and without having to await progression before antifibrotic therapy can be initiated.
5. Conclusions
In summary, we conclude that younger age, higher LDH level and lower monocyte count predict functional improvement in fibrotic ILD, whereas in patients treated with anti-inflammatory drugs only younger age prevailed as significant predictor. Further studies with a prospective design, larger sample size, defined patient subgroups and standardized treatment are required to investigate the impact of such biomarkers more accurately.
Author Contributions
Conceptualization, G.S. and D.L.; methodology, G.S., B.K. and D.L.; formal analysis, G.S., B.K. and D.L.; investigation, G.S., B.L., D.L.; writing—original draft preparation, G.S.; writing—review and editing, B.L., P.T. and D.L.; supervision, B.L. and D.L.; project administration, G.S., B.L. and D.L.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the ethics committees of the Medical Faculty of Linz (study number I-26-17.
Informed Consent Statement
Participants received written and verbal information explaining the study and written consent was obtained from all participants.
Data Availability Statement
As mandated by the ethics committee of Medical Faculty Linz, publication or dissemination of any possibly identifiable patient data from the Kepler University Hospital ILD registry is strictly prohibited. The dataset used for the present analyses contains very detailed and thus possibly identifiable patient data, so that a publication of the database is not possible. However, upon reasonable request to the Authors and if permitted by the Ethics Committee of Medical Faculty of Linz in an amendment to the study protocol, anonymized data may under certain circumstances be shared.
Acknowledgments
The authors would like to acknowledge the contribution of the patients who volunteered to participate in this study.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Wijsenbeek, M.; Suzuki, A.; Maher, T.M. Interstitial Lung Diseases. Lancet 2022, 400, 769–786. [Google Scholar] [CrossRef] [PubMed]
- George, P.M.; Spagnolo, P.; Kreuter, M.; Altinisik, G.; Bonifazi, M.; Martinez, F.J.; Molyneaux, P.L.; Renzoni, E.A.; Richeldi, L.; Tomassetti, S.; et al. Progressive Fibrosing Interstitial Lung Disease: Clinical Uncertainties, Consensus Recommendations, and Research Priorities. Lancet Respir Med 2020, 8, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Wijsenbeek, M.; Kreuter, M.; Olson, A.; Fischer, A.; Bendstrup, E.; Wells, C.D.; Denton, C.P.; Mounir, B.; Zouad-Lejour, L.; Quaresma, M.; et al. Progressive Fibrosing Interstitial Lung Diseases: Current Practice in Diagnosis and Management. Curr Med Res Opin 2019, 35, 2015–2024. [Google Scholar] [CrossRef]
- Yamano, Y.; Kataoka, K.; Takei, R.; Sasano, H.; Yokoyama, T.; Matsuda, T.; Kimura, T.; Mori, Y.; Furukawa, T.; Fukuoka, J.; et al. Interstitial Pneumonia with Autoimmune Features and Histologic Usual Interstitial Pneumonia Treated with Anti-Fibrotic versus Immunosuppressive Therapy. Respir Investig 2023, 61, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; du Bois, R. Interstitial Lung Disease in Connective Tissue Disorders. Lancet 2012, 380, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Wijsenbeek, M.; Cottin, V. Spectrum of Fibrotic Lung Diseases. N Engl J Med 2020, 383, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Richeldi, L.; Thomson, C.C.; Inoue, Y.; Johkoh, T.; Kreuter, M.; Lynch, D.A.; Maher, T.M.; Martinez, F.J.; et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med 2022, 205, e18–e47. [Google Scholar] [CrossRef]
- Wells, A.U.; Brown, K.K.; Flaherty, K.R.; Kolb, M.; Thannickal, V.J. What’s in a Name? That Which We Call IPF, by Any Other Name Would Act the Same. Eur. Respir. J. 2018, 51, 1800692. [Google Scholar] [CrossRef]
- Ghazipura, M.; Mammen, M.J.; Herman, D.D.; Hon, S.M.; Bissell, B.D.; Macrea, M.; Kheir, F.; Khor, Y.H.; Knight, S.L.; Raghu, G.; et al. Nintedanib in Progressive Pulmonary Fibrosis: A Systematic Review and Meta-Analysis. Ann Am Thorac Soc 2022, 19, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Rajan, S.K.; Cottin, V.; Dhar, R.; Danoff, S.; Flaherty, K.R.; Brown, K.K.; Mohan, A.; Renzoni, E.; Mohan, M.; Udwadia, Z.; et al. Progressive Pulmonary Fibrosis: An Expert Group Consensus Statement. Eur Respir J 2023, 61. [Google Scholar] [CrossRef]
- Distler, O.; Brown, K.K.; Distler, J.H.W.; Assassi, S.; Maher, T.M.; Cottin, V.; Varga, J.; Coeck, C.; Gahlemann, M.; Sauter, W.; et al. Design of a Randomised, Placebo-Controlled Clinical Trial of Nintedanib in Patients with Systemic Sclerosis-Associated Interstitial Lung Disease (SENSCISTM). Clin Exp Rheumatol 2017, 35 Suppl 106, 75–81. [Google Scholar]
- Ryerson, C.J.; Vittinghoff, E.; Ley, B.; Lee, J.S.; Mooney, J.J.; Jones, K.D.; Elicker, B.M.; Wolters, P.J.; Koth, L.L.; King, T.E.; et al. Predicting Survival across Chronic Interstitial Lung Disease: The ILD-GAP Model. Chest 2014, 145, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Flaherty, K.R.; Lederer, D.J.; Lynch, D.A.; Colby, T.V.; Myers, J.L.; Groshong, S.D.; Larsen, B.T.; Chung, J.H.; Steele, M.P.; et al. Use of a Molecular Classifier to Identify Usual Interstitial Pneumonia in Conventional Transbronchial Lung Biopsy Samples: A Prospective Validation Study. Lancet Respir Med 2019, 7, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Bowman, W.S.; Newton, C.A.; Linderholm, A.L.; Neely, M.L.; Pugashetti, J.V.; Kaul, B.; Vo, V.; Echt, G.A.; Leon, W.; Shah, R.J.; et al. Proteomic Biomarkers of Progressive Fibrosing Interstitial Lung Disease: A Multicentre Cohort Analysis. Lancet Respir Med 2022, 10, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Lang, D.; Akbari, K.; Walcherberger, S.; Hergan, B.; Horner, A.; Hepp, M.; Kaiser, B.; Pieringer, H.; Lamprecht, B. Computed Tomography Findings as Determinants of Pulmonary Function Tests in Fibrotic Interstitial Lung Diseases-Network-Analyses and Multivariate Models. Chron Respir Dis 2020, 17, 1479973120967025. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann-Vold, A.M.; Allanore, Y.; Alves, M.; Brunborg, C.; Airó, P.; Ananieva, L.P.; Czirják, L.; Guiducci, S.; Hachulla, E.; Li, M.; et al. Progressive Interstitial Lung Disease in Patients with Systemic Sclerosis-Associated Interstitial Lung Disease in the EUSTAR Database. Ann Rheum Dis 2021, 80, 219–227. [Google Scholar] [CrossRef] [PubMed]
- von Elm, E.; Altman, D.G.; Egger, M.; Pocock, S.J.; Gøtzsche, P.C.; Vandenbroucke, J.P. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) Statement: Guidelines for Reporting Observational Studies. J Clin Epidemiol 2008, 61, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Gruden, J.F.; Naidich, D.P.; Machnicki, S.C.; Cohen, S.L.; Girvin, F.; Raoof, S. An Algorithmic Approach to the Interpretation of Diffuse Lung Disease on Chest CT Imaging: A Theory of Almost Everything. Chest 2020, 157, 612–635. [Google Scholar] [CrossRef] [PubMed]
- Shao, G.; Hawle, P.; Akbari, K.; Horner, A.; Hintenberger, R.; Kaiser, B.; Lamprecht, B.; Lang, D. Clinical, Imaging, and Blood Biomarkers to Assess 1-Year Progression Risk in Fibrotic Interstitial Lung Diseases-Development and Validation of the Honeycombing, Traction Bronchiectasis, and Monocyte (HTM)-Score. Front Med Lausanne 2022, 9, 1043720. [Google Scholar] [CrossRef]
- Ley, B.; Ryerson, C.J.; Vittinghoff, E.; Ryu, J.H.; Tomassetti, S.; Lee, J.S.; Poletti, V.; Buccioli, M.; Elicker, B.M.; Jones, K.D.; et al. A Multidimensional Index and Staging System for Idiopathic Pulmonary Fibrosis. Ann Intern Med 2012, 156, 684–691. [Google Scholar] [CrossRef]
- Dhooria, S.; Agarwal, R.; Sehgal, I.S.; Prasad, K.T.; Garg, M.; Bal, A.; Aggarwal, A.N.; Behera, D. Spectrum of Interstitial Lung Diseases at a Tertiary Center in a Developing Country: A Study of 803 Subjects. PLoS One 2018, 13, e0191938. [Google Scholar] [CrossRef]
- Joung, K.I.; Park, H.; Park, S.; Shin, J.Y.; Kim, Y.H. Nationwide Epidemiologic Study for Fibrosing Interstitial Lung Disease (F-ILD) in South Korea: A Population-Based Study. BMC Pulm Med 2023, 23, 98. [Google Scholar] [CrossRef]
- Hopkins, R.B.; Burke, N.; Fell, C.; Dion, G.; Kolb, M. Epidemiology and Survival of Idiopathic Pulmonary Fibrosis from National Data in Canada. Eur Respir J 2016, 48, 187–195. [Google Scholar] [CrossRef]
- Fernández Pérez, E.R.; Daniels, C.E.; Schroeder, D.R.; St Sauver, J.; Hartman, T.E.; Bartholmai, B.J.; Yi, E.S.; Ryu, J.H. Incidence, Prevalence, and Clinical Course of Idiopathic Pulmonary Fibrosis: A Population-Based Study. Chest 2010, 137, 129–137. [Google Scholar] [CrossRef]
- Navaratnam, V.; Fleming, K.M.; West, J.; Smith, C.J.; Jenkins, R.G.; Fogarty, A.; Hubbard, R.B. The Rising Incidence of Idiopathic Pulmonary Fibrosis in the U.K. Thorax 2011, 66, 462–467. [Google Scholar] [CrossRef]
- Duchemann, B.; Annesi-Maesano, I.; Jacobe de Naurois, C.; Sanyal, S.; Brillet, P.Y.; Brauner, M.; Kambouchner, M.; Huynh, S.; Naccache, J.M.; Borie, R.; et al. Prevalence and Incidence of Interstitial Lung Diseases in a Multi-Ethnic County of Greater Paris. Eur Respir J 2017, 50. [Google Scholar] [CrossRef] [PubMed]
- He, S.H.; He, Y.J.; Guo, K.J.; Liang, X.; Li, S.S.; Li, T.F. Risk Factors for Progression of Interstitial Lung Disease in Sjögren’s Syndrome: A Single-Centered, Retrospective Study. Clin Rheumatol 2022, 41, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Sheng, J.; Qiu, X.; Wang, D.; Wang, D.; Wang, Y.; Xiao, Y.; Cai, H. Acute Exacerbations of Fibrosing Interstitial Lung Disease Associated with Connective Tissue Diseases: A Population-Based Study. BMC Pulm Med 2019, 19, 215. [Google Scholar] [CrossRef]
- Fu, H.; Zheng, Z.; Zhang, Z.; Yang, Y.; Cui, J.; Wang, Z.; Xue, J.; Chi, S.; Cao, M.; Chen, J. Prediction of Progressive Pulmonary Fibrosis in Patients with Anti-Synthetase Syndrome-Associated Interstitial Lung Disease. Clin Rheumatol 2023, 42, 1917–1929. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.K.D.; Quinn, K.; Li, Q.; Carroll, R.; Warsinske, H.; Vallania, F.; Chen, S.; Carns, M.A.; Aren, K.; Sun, J.; et al. Increased Monocyte Count as a Cellular Biomarker for Poor Outcomes in Fibrotic Diseases: A Retrospective, Multicentre Cohort Study. Lancet Respir Med 2019, 7, 497–508. [Google Scholar] [CrossRef]
- Kreuter, M.; Lee, J.S.; Tzouvelekis, A.; Oldham, J.M.; Molyneaux, P.L.; Weycker, D.; Atwood, M.; Kirchgaessler, K.U.; Maher, T.M. Monocyte Count as a Prognostic Biomarker in Patients with Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med 2021, 204, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Peng, H.; Sun, H.; Peng, X.; Tang, C.; Gan, Y.; Chen, X.; Mathur, A.; Hu, B.; Slade, M.D.; et al. Chitinase 3-like 1 Suppresses Injury and Promotes Fibroproliferative Responses in Mammalian Lung Fibrosis. Sci Transl Med 2014, 6, 240ra76. [Google Scholar] [CrossRef] [PubMed]
- Misharin, A.V.; Morales-Nebreda, L.; Reyfman, P.A.; Cuda, C.M.; Walter, J.M.; McQuattie-Pimentel, A.C.; Chen, C.I.; Anekalla, K.R.; Joshi, N.; Williams, K.J.N.; et al. Monocyte-Derived Alveolar Macrophages Drive Lung Fibrosis and Persist in the Lung over the Life Span. J Exp Med 2017, 214, 2387–2404. [Google Scholar] [CrossRef]
- Ehrchen, J.M.; Roth, J.; Barczyk-Kahlert, K. More Than Suppression: Glucocorticoid Action on Monocytes and Macrophages. Front Immunol 2019, 10, 2028. [Google Scholar] [CrossRef]
- Elmér, E.; Nived, P.; Pettersson, Å.; Skattum, L.; Hellmark, T.; Kapetanovic, M.C.; Johansson Å, C.M. Methotrexate Treatment Suppresses Monocytes in Nonresponders to Pneumococcal Conjugate Vaccine in Rheumatoid Arthritis Patients. J Immunol Res 2022, 2022, 7561661. [Google Scholar] [CrossRef]
Figure 1.
Derivation of the study cohorts. FVC, forced vital capacity; DLCO, diffusion capacity for carbon monoxide.
Figure 1.
Derivation of the study cohorts. FVC, forced vital capacity; DLCO, diffusion capacity for carbon monoxide.
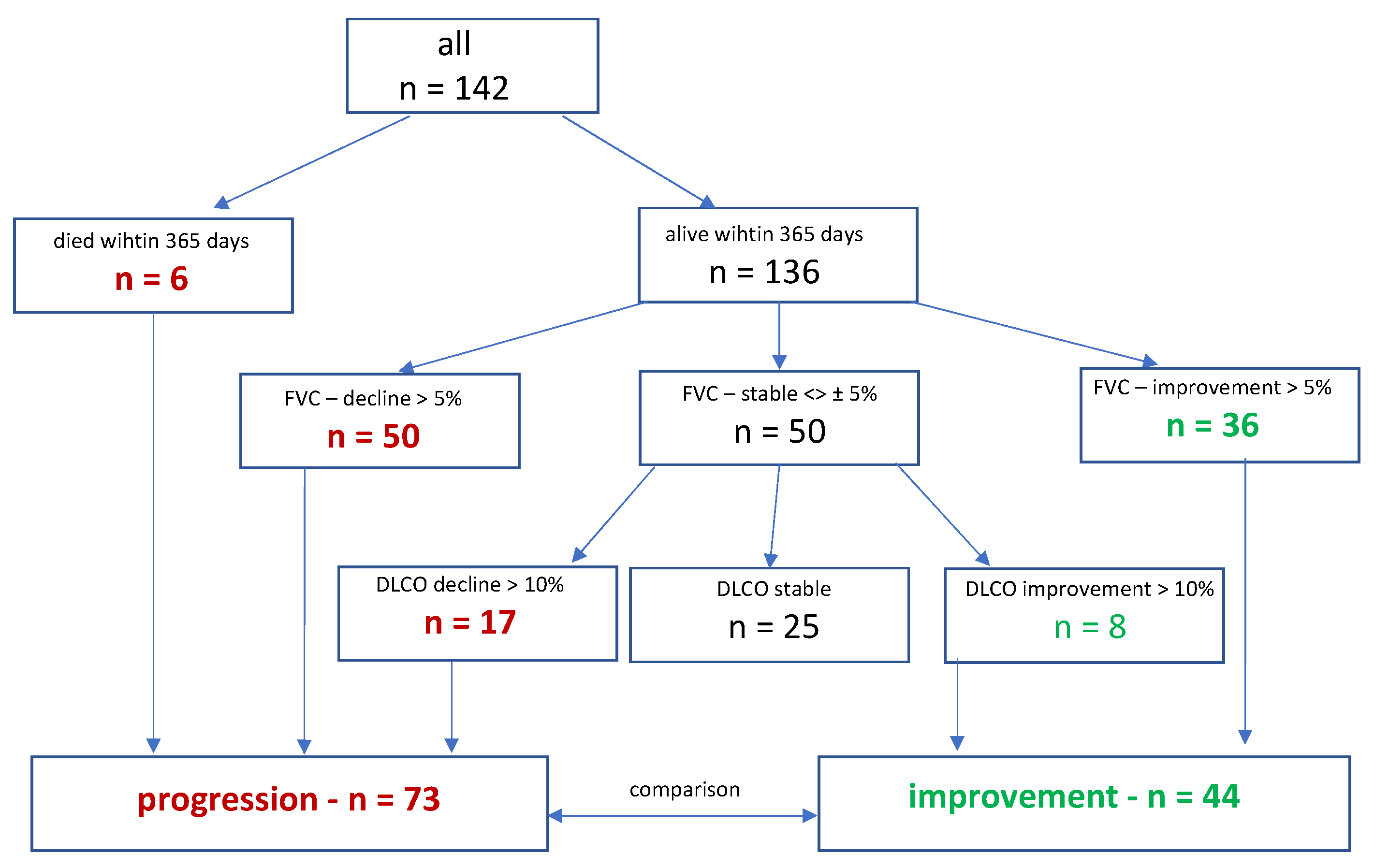
Figure 2.
Forest-plots illustrating univariate analyses of all patients (a) and the antiinflammatory subgroup (b). LDH, lactate dehydrogenase.
Figure 2.
Forest-plots illustrating univariate analyses of all patients (a) and the antiinflammatory subgroup (b). LDH, lactate dehydrogenase.


Table 1.
Baseline patient, treatment and pulmonary function test characteristics, baseline peripheral blood and BAL markers and baseline computed tomography scores in all patients according to disease course. SE, standard error; FVC, forced vital capacity; FEV-1, forced expiratory volume in 1 second; DLCO, diffusion capacity for carbon monoxide; BAL, bronchoalveolar lavage.
Table 1.
Baseline patient, treatment and pulmonary function test characteristics, baseline peripheral blood and BAL markers and baseline computed tomography scores in all patients according to disease course. SE, standard error; FVC, forced vital capacity; FEV-1, forced expiratory volume in 1 second; DLCO, diffusion capacity for carbon monoxide; BAL, bronchoalveolar lavage.
| All patients (n=142) | |||||
| Variable | All patients (n=142) |
Progression at 1 year (n=73) |
Stable at 1 year (n=25) |
Improvement at 1 year (n=44) |
p-value |
| Baseline characteristics | |||||
| Mean age (SE) | 67.0 (1.1) | 70.4 (1.0) | 64.8 (2.5) | 62.4 (2.4) | 0.023 |
| Age ≥70 years (%) | 47.2 | 54.8 | 36.0 | 40.9 | 0.162 |
| Female sex (%) | 36.6 | 32.9 | 28.0 | 47.7 | 0.167 |
| Treatment characteristics (%) | |||||
| Antiinflammatory | 52.1 | 41.1 | 52.0 | 70.5 | 0.066 |
| Antifibrotic | 12.0 | 15.1 | 12.0 | 6.8 | |
| Antiinflammatory and antifibrotic | 7.0 | 11.0 | 8.0 | 0.0 | |
| No ILD-specific therapy | 28.9 | 32.0 | 28.0 | 22.7 | |
| Pulmonary functions tests; mean (SE) | |||||
| FVC (% pred.) | 81.3 (1.5) | 84.3 (2.1) | 84.6 (3.9) | 74.5 (2.6) | 0.017 |
| FEV1 (% pred.) | 82.8 (1.6) | 86.6 (2.1) | 84.4 (4.1) | 75.9 (2.5) | 0.018 |
| DLCO (% pred.) | 55.2 (1.5) | 57.1 (2.0) | 58.2 (3.4) | 50.3 (2.6) | 0.104 |
| Peripheral blood Biomarkers; mean (SE) | |||||
| Absolute leukocyte count (G/L) | 8.8 (0.3) | 8.6 (0.4) | 8.8 (1.0) | 9.0 (0.4) | 0.442 |
| Absolute monocyte count (G/L) | 0.6 (0.1) | 0.6 (0.1) | 0.5 (0.1) | 0.6 (0.1) | 0.046 |
| Absolute eosinophil count (G/L) | 0.2 (0.1) | 0.2 (0.1) | 0.1 (0.0) | 0.2 (0.1) | 0.397 |
| C-reactive protein (mg/dL) | 1.2 (0.2) | 0.9 (0.2) | 1.1 (0.5) | 1.8 (0.4) | 0.285 |
| Lactate Dehydrogenase (U/L) | 248.3 | 241.4 (9.8) | 224.8 (12.1) | 272.5 (15.7) | 0.095 |
| Bronchoalveolar lavage; mean (SE) n = 81 | |||||
| BAL - macrophage fraction | 57.7 (3.1) | 61.2 (4.2) | 57.5 (8.3) | 52.9 (5.3) | 0.484 |
| BAL – lymphocyte fraction | 17.9 (2.2) | 14.7 (2.4) | 9.3 (2.8) | 26.5 (5.0) | 0.038 |
| BAL – Neutrophile fraction | 15.1 (2.2) | 17.5 (3.5) | 18.9 (7.3) | 10.1 (1.9) | 0.592 |
| BAL – Eosinophile fraction | 4.1 (0.9) | 3.8 (1.4) | 5.1 (1.8) | 4.1 (1.4) | 0.631 |
| Computed tomography finding scores; median, range | |||||
| Reticular abnormalities | 6 (0 – 6) | 6 (0 – 6) | 6 (0 – 6) | 6 (1 – 6) | 0.807 |
| Honeycombing | 0 (0 – 6) | 0 (0 – 6) | 0 (0 – 6) | 0 (0 – 3) | 0.063 |
| Ground Glass Opacities | 0 (0 – 6) | 0 (0 – 6) | 0 (0 – 6) | 2 (0 – 6) | 0.047 |
| Emphysema | 0 (0 – 6) | 0 (0 – 6) | 0 (0 – 6) | 0 (0 – 6) | 0.534 |
| Traction bronchiectasis | 2 (0 – 6) | 2 (0 – 6) | 2 (0 – 6) | 2 (0 – 6) | 0.019 |
Table 2.
Results of the logistic regression for all patients (Improvement n=44 vs. progression n=73) and for the anti-inflammatory subgroup (Improvement n=31 vs. progression =38). CI, confidence interval; LDH, lactate dehydrogenase; FVC, forced vital capacity; DLCO, diffusion capacity for carbon monoxide.
Table 2.
Results of the logistic regression for all patients (Improvement n=44 vs. progression n=73) and for the anti-inflammatory subgroup (Improvement n=31 vs. progression =38). CI, confidence interval; LDH, lactate dehydrogenase; FVC, forced vital capacity; DLCO, diffusion capacity for carbon monoxide.
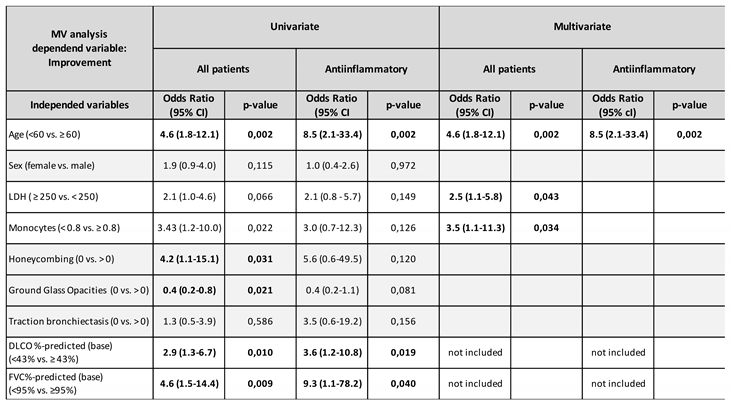
Table 3.
Baseline patient, treatment and pulmonary function test characteristics, baseline peripheral blood and BAL markers and baseline computed tomography scores in the anti-inflammatory-treatment subgroup according to disease course. SSE, standard error; FVC, forced vital capacity; FEV-1, forced expiratory volume in 1 second; DLCO, diffusion capacity for carbon monoxide; BAL, bronchoalveolar lavage.
Table 3.
Baseline patient, treatment and pulmonary function test characteristics, baseline peripheral blood and BAL markers and baseline computed tomography scores in the anti-inflammatory-treatment subgroup according to disease course. SSE, standard error; FVC, forced vital capacity; FEV-1, forced expiratory volume in 1 second; DLCO, diffusion capacity for carbon monoxide; BAL, bronchoalveolar lavage.
| Patients with antiinflammatory treatment (n=84) | |||||
|---|---|---|---|---|---|
| Variable | All patients (n=84) |
Progression at 1 year (n=38) |
Stable at 1 year (n=15) |
Improvement at 1 year( n=31) |
p-value |
| Baseline characteristics | |||||
| Mean age (SE) | 66.2 (1.5) | 70.2 (1.4) | 66.8 (2.9) | 61.1 (3.1) | 0.122 |
| Age ≥70 years (%) | 40 (47.6) | 22 (57.9) | 6 (40.0) | 12 (38.7) | 0.229 |
| Female sex (%) | 35 (41.7) | 17 (44.7) | 4 (26.7) | 14 (45.2 | 0.429 |
| Pulmonary functions tests; mean (SE) | |||||
| FVC (% pred.) | 80.3 (2.1) | 82.9 (3.0) | 89.4 (4.8) | 72.7 (3.0) | 0.008 |
| FEV1 (% pred.) | 81.9 (2.0) | 84.9 (2.9) | 89.3 (4.4) | 74.8 (3.0) | 0.018 |
| DLCO (% pred.) | 52.9 (1.8) | 53.9 (2.1) | 58.9 (5.4) | 48.7 (3.1) | 0.183 |
| Peripheral blood Biomarkers; mean (SE) | |||||
| Absolute leukocyte count (G/L) | 8.9 (0.3) | 8.7 (0.6) | 8.5 (0.9) | 9.2 (0.5) | 0.379 |
| Absolute monocyte count (G/L) | 0.6 (0.1) | 0.6 (0.1) | 0.6 (0.1) | 0.5 (0.1) | 0.638 |
| Absolute eosinophil count (G/L) | 0.2 (0.1) | 0.2 (0.1) | 0.1 (0.0) | 0.2 (0.1) | 0.662 |
| C-reactive protein (mg/dL) | 1.2 (0.2) | 0.7 (0.2) | 0.7 (0.2) | 2.1 (0.6) | 0.308 |
| Lactate Dehydrogenase (U/L) | 259.7 (11.1) | 253.1 (16.2) | 218.9 (12.7) | 288.6 (21.0) | 0.078 |
| Bronchoalveolar lavage; mean (SE) n = 81 | |||||
| BAL – Macrophage fraction | 54.9 (3.5) | 58.7 (4.9) | 59.7 (9.9) | 49.0 (5.7) | 0.429 |
| BAL – Lymphocyte fraction | 22.1 (2.9) | 19.3 (3.3) | 11.9 (3.7) | 28.9 (5.7) | 0.215 |
| BAL – Neutrophile fraction | 12.8 (1.9) | 13.0 (3.1) | 17.4 (6.6) | 10.8 (2.2) | 0.551 |
| BAL – Eosinophile fraction | 4.7 (1.2) | 4.6 (2.1) | 5.6 (2.4) | 4.5 (1.6) | 0.647 |
| Computed tomography finding scores; median, range | |||||
| rReticular abnormalities | 6 (0 – 6) | 6 (0 – 6) | 6 (2 – 6) | 6 (1 – 6) | 0.740 |
| Honeycombing | 0 (0 – 6) | 0 (0 – 6) | 0 (0 – 2) | 0 (0 – 2) | 0.179 |
| Ground glass opacities | 1.5 (0 – 6) | 0.5 (0 – 6) | 0 (0 – 6) | 2 (0 – 6) | 0.118 |
| Emphysema | 0 (0 – 6) | 0 (0 – 6) | 0 (0 – 6) | 0 (0 – 4) | 0.151 |
| Traction bronchiectasis | 2 (0 – 6) | 2 (0 – 6) | 2 (0 – 6) | 2 (0 – 6) | 0.043 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



