Submitted:
03 June 2024
Posted:
05 June 2024
You are already at the latest version
Abstract
This study investigates the valorization potential of yellowfin tuna tails for the production of high-value commercial products. Muscle and skins were firstly mechanically separated by using a specifically designed device. The muscle was then used as a nitrogen source for the growth of the proteolytic enzymes-producer Bacillus subtilis, whereas the skins were employed for gelatin extrac-tion. The proteases from Bacillus subtilis were partially purified and used to produce antioxidant peptides from gelatin obtained. The gelatin obtained formed a gel upon cooling, with gelling and melting temperatures at 16 ºC and 22 ºC, respectively, and Bloom Strength of ≈ 160. RSM was used to determine the optimal hydrolysis conditions to obtain the highest antioxidant activity (35.96%, measured as DPPH radical scavenging activity), which were 50°C / 6.5 IU of enzyme. The findings emphasize the importance of an integrated approach to maximize the value of tuna by-products, promoting sustainability in the frame of circular bioeconomy. Overall, these results contribute to the efficient utilization of tuna by-products, waste reduction, and enhance economic viability of the tuna industry.
Keywords:
yellowfin tuna
; valorization
; by-products
; proteolytic enzymes
; gelatin
; antioxidant activity
1. Introduction
The tuna industry holds great global significance, providing substantial employment opportunities and making significant contributions to the global economy [1]. Global catches of tunas and related species (including Thunnus albacares, Thunnus maccoyii, Thunnus obesus, Thunnus thynnus, Thunnus alalunga, and Katsuwonus pelamis) reached a volume of 7.8 million tons in 2020 [2]. The industrial processing of tuna generates a substantial quantity of solid waste, including muscle trimmings, viscera, gills, dark muscle, heads, tails, bones, skins, and fins, which constitutes up to 70% of the initial raw material [3]. Although these by-products are useful in the production of fertilizers or fishmeal/fish oil for aquaculture fish feed or pet food, there is untapped potential to create higher value products due to their protein-rich composition, thereby enhancing the economic sustainability of this industry [4]. However, it is a stark reality persists that significant quantities of these by-products are discarded, resulting in considerable environmental damage [5]. Consequently, there is an urgent need to develop strategies for the valorization of these by-products, with a primary focus on generating of high-value products. One interesting approach involves converting protein-rich by-products into protein hydrolysates with bioactive and nutritional characteristics. Alternatively, collagen/gelatin can be extracted from the skin for various purposes [6,7,8], and represents a good alternative to mammalian gelatin. It can be utilized as a food ingredient as an emulsifier, for the production of edible films, or even for the development of functional foods [9,10].
The utilization of protein-rich by-products as a substrate for protease production is of great interest, due to the abundance and underutilization of these resources. It has been demonstrated that several microorganisms are able to utilize these by-products as a source of carbon, nitrogen, and energy in the enzyme production process, which has generated scientific and industrial interest. This interest is based on the fact that nearly 50% of the cost associated with enzyme production is attributed to the raw material, namely the substrate for the growth of enzyme-producing microorganisms [11]. Microorganisms secrete proteolytic enzymes into the culture medium during the growth phase, which have excellent properties for application in hydrolysis processes [12]. This strategic approach presents a promising avenue for the efficient production of proteolytic enzymes with industrial relevance, leveraging economically accessible resources and contributing to the valorization of by-products [13]. Proteases of microbial origin offer distinct advantages over those of plant and animal origin, including ease of large-scale production, rapid growth, and lower production costs, among others [14].
Bioactive peptides are present within the sequence of native proteins and must be released by hydrolysis to exert their biological functions. Among the array of hydrolysis methods available, utilizing proteases from animal, plant, or microbial sources is feasible. However, the use of the latest is preferable due to their rapid reaction and enhanced stability [15]. In comparison to chemical hydrolysis, enzymatic hydrolysis offers several advantages. These include the preservation of the nutritional value, the absence of residual organic or chemical solvents, and its rapid, safe, and easily regulated nature [16,17].
Protein hydrolysates consist of free amino acids and peptides with varying molecular weights, which exhibit distinct technological and functional properties. Due to their smaller size, the constituent amino acids are more easily absorbed in the small intestine, fulfilling diverse physiological functions in the human body [18]. Peptides offer a range of health benefits, including the potential treatment of certain diseases. They may also possess specific activities of technological interest, such as antioxidant and antimicrobial properties [19].
Oxidative reactions represent a primary cause of food spoilage, resulting in the formation of free radicals and compounds that can cause chronic diseases by damaging cell membranes and biomacromolecules. These diseases include diabetes mellitus, cancer, and liver disease [20]. In recent years, there has been a trend towards utilizing additives of natural origin. As a result, marine sources have garnered attention for the extraction of antioxidant peptides. For instance, the muscle of the Corvina fish (Miichthys miiuy) has been hydrolyzed using different enzymes (alcalase, trypsin, papain and pepsin), resulting in the release of 10 peptides with strong antioxidant activity [18]. The viscera of tilapia (Oreochromis spp) have also been employed as a source of antioxidant peptides, with positive results [21]. Furthermore, carp skins have been tested as a raw material for the release of antioxidant peptides by hydrolysis with alcalase [22]. In practical applications, antioxidant peptides have demonstrated effectiveness in various contexts. For instance, they have shown promising results in reducing lipid oxidation in meat [23]. Additionally, incorporating antioxidant peptides into the formulation of flour for biscuit production has led to notable improvements in both the nutritional value and antioxidant capacity of the final product [24].
The primary objective of this study is to valorize yellowfin tuna (Thunnus albacares) tails through an integrated approach involving the separate exploitation of different components of the by-product. Initially, the muscle protein was evaluated as a nitrogen source for the production of proteases by B. subtilis. These proteases were then used to release antioxidant peptides from gelatin extracted from the skin. The evaluation of gelatin quality and the optimization of hydrolysis conditions with the objective to maximize antioxidant activity represent additional objectives of this study.
2. Materials and Methods
2.1. Sample Collection and Preparation
The tails of yellowfin tuna (Thunnus albacares) were obtained from a local market in Quito, Ecuador. The tails were carefully selected to ensure freshness and promptly transported on ice to the laboratory for further processing. To separate the muscle tissue from the skin, six tails were placed into a specially designed perforated stainless-steel cylinder. This cylinder allowed liquid to pass through while retaining solid components, achieved through low pressure magnified by fluid pressure effects. Hydraulic pressure was applied to the tails using a Sematech Engineering press (Quito, Ecuador), reaching pressures of up to 6,000 pounds per square inch (PSI). This method effectively extracted the desired muscle tissue, leaving the skin inside the cylinder. The extracted muscle was subsequently freeze-dried, minced, and stored at -80°C until further use.
2.2. Chemicals and Reagents
All experimental procedures were conducted using analytical-grade reagents, including potassium phosphate monobasic and dibasic, sodium chloride, Tris-HCl, and trichloroacetic acid, obtained from Thermo Fisher Scientific (Waltham, MA, USA). Additionally, BHI-Agar, dextrose, azocasein, SDS, TEMED, Tricine, Sephadex G-100, and Sephadex G-25 resin were obtained from Sigma Aldrich (St. Louis, MO, USA), while ammonium sulfate, 2-Mercaptoethanol, glycerol, sodium hydroxide, and BHI broth were purchased from Merck KGaA (Darmstadt, Germany). Ammonium persulfate, Bromophenol brilliant blue, Coomassie Brilliant Blue, acrylamide, and bisacrylamide were obtained from Bio-Rad (Hercules, CA, USA).
2.3. Enzymatic Extract Preparation
The enzymatic extract was obtained by cultivation of Bacillus subtilis in a culture medium prepared with 0.5% sodium chloride, 0.2 % dextrose and 1% freeze-dried yellowfin tuna muscle. The components were dissolved in 0.1 M phosphate buffer (pH 7) and then sterilized (121 ºC, 15 min) in a Trident EA-632 autoclave (Taiwan). The strain of Bacillus subtilis was isolated and identified in a previous study [25]. Bacillus subtilis strain stored at -80 °C in BHI broth was thawed under refrigeration. Then, 100 µl of bacteria were inoculated into BHI broth and incubated at 37 °C for 24 hours. It was then inoculated into the broth prepared with yellowfin tuna muscle and incubated for 3 days at 37 °C under aerobic conditions with continuous shaking at 180 rpm. After the incubation period, the enzyme extract was obtained by precipitation with ammonium sulfate. For this, the fermented culture medium was mixed with ammonium sulfate at a concentration of 40% (w/v) and stirred continuously on ice for 1 hour. The mixture was then centrifuged at 5,500 x g for 15 minutes at 25°C to separate the precipitate (containing the enzymes) from the supernatant (containing the bacterial cells).
2.4. Purification of the Enzymatic Extract
The enzymatic extract obtained after precipitation was desalted using a pre-hydrated Sephadex G-25 gel filtration column [26], pre-equilibrated in 0.1 M phosphate buffer. The enzymatic extract was then purified using a column packed with Sephadex G-100 resin (7.5 mm diameter, 10 cm height), previously hydrated in 0.1 M phosphate buffer at pH 7 for 24 hours. [27]. During this process, 15 fractions of 1 ml each were collected under a constant flow of 0.1M phosphate buffer, at pH 7.
2.5. Determination of Proteolytic Activity of Alkaline Proteases:
The proteolytic activity of the collected samples was determined using azocasein as a substrate [28]. For that, a reaction mixture was prepared by mixing 200 µL of Tris-HCl buffer (0.1 M, pH 8), 200 µL of the extract, and 200 µL of 1% (w/v) azocasein. A blank solution was prepared by combining 200 µL of 1% azocasein, 1 mL of 10% trichloroacetic acid (TCA), 200 µL of Tris-HCl buffer, and 200 µL of distilled water. The reaction mixture was then incubated at 37°C for 30 minutes. Then, 1 mL of TCA solution was added, followed by centrifugation at 5,500 x g for 15 minutes. After centrifugation, 400 µL of 1.8 N NaOH was added to the supernatant. The absorbance of the resulting solution was measured at a wavelength of 420 nm using a UV/Vis spectrophotometer (UV-160A, Shimadzu, Kyoto, Japan)
Under the specified measurement conditions, one unit of proteolytic activity (U) was defined as the amount of enzymatic extract (mL) that resulted in a 0.1 increase in absorbance per minute. Azocaseinolytic activity was quantified by expressing the proteolytic activity in units per milliliter (U/mL).
The fraction containing the enzymes was supplemented with 1% glycerol to improve stability and then freeze-dried to obtain a stable form.
2.6. Molecular Sizes Determination
The fraction containing the enzymes was subjected to a polyacrylamide gel electrophoresis analysis [29], using 10% acrylamide gels prepared from a 49.5%T, 3%C acrylamide-bisacrylamide mixture (T denotes the total percentage concentration of both monomers (acrylamide and bisacrylamide) and C denotes the percentage concentration of the cross-linking agent relative to the total concentration of T). A constant voltage of 110 V was used to run the samples. To prepare the sample, 20 mg of lyophilized samples were dissolved in 1 mL of a denaturing solution (50 mM Tris, 4% SDS, 2% mercaptoethanol, 12% glycerol, and 0.01% bromophenol blue) adjusted to pH 6.8 with 1 N HCl. The solution was boiled for 10 minutes and then centrifuged at 10,000 x g. The protein bands were subsequently stained with Coomassie blue. A molecular weight marker ranging from 6.5 to 200 kDa (SigmaMarker S8445-10VL) was used to determine the tentative molecular weight of the proteins in the stained bands.
2.7. Gelatin Extraction from Yellowfin Tuna:
Firstly, the skins were washed with two volumes of a 5% NaCl solution for 30 minutes to remove any non-collagenous adhered proteins. The process was repeated twice. Subsequently, the skins were immersed in 0.1N NaOH solution for one hour to eliminate any remaining lipids. To ensure complete lipid removal, the samples were afterwards treated thrice with two volumes of 10% isobutyl alcohol for 30 minutes. Finally, the samples were thoroughly washed with distilled water and treated with 0.05 M acetic acid for 21 hours at room temperature. The final extraction of gelatin was performed twice with distilled water at 60°C overnight. The extracts were filtered using gauze, then dried in a forced-air oven at 45°C for 12 hours. The gelatin was finally ground into a fine powder and stored at -20°C until use [30].
2.8. Amino Acid Analysis
Gelatin was dissolved (1 mg/mL) in ultrapure water and further hydrolyzed in vacuum-sealed glass at 110 °C for 24 h in presence of continuously boiling 6 N HCl containing 0.1% phenol and norleucine as internal standard. After hydrolysis, the sample was again vacuum-dried, dissolved in application buffer, and injected onto a Biochrom 30 amino acid analyser (Biochrom Ltd., Cambridge, UK) equipped with a LKB Ultropack 8 resin column (Pharmacia LKB Biotechnology, Inc. Pascataway, NJ, USA). Results were expressed as grams of amino acids per 100 grams.
2.9. Gel-Forming Properties
Gelatin (6.67 g/100 mL) was dissolved in distilled water at 40ºC for 20 minutes. The viscoelastic properties of the gelatin were evaluated using a rotational rheometer (Advanced Rheometer AR 2000, TA Instruments Ltd, Newcastle, England) equipped with a 2° cone angle and a 40 mm plate distance. A dynamic temperature sweep was performed by subjecting the gelatin to a temperature sweep from 35 to 5°C and then returning it to 35°C. The sweep rate was 1°C per minute, with a frequency of 0.5 Hz, an initial stress of 3 Pa, and a deformation of 0.2 [31].
The gelation and subsequent melting points of the gelatin were evaluated by analyzing the elastic modulus, viscous modulus, and phase angle as a function of temperature. To study the dynamic frequency sweep, the gelatin was maintained at 4°C, and oscillatory measurements were performed over a frequency range of 0.1 to 10 Hz, with an oscillation amplitude (strain) within the linear range (0.005). The elastic and viscous moduli were measured as a function of frequency, providing valuable insight into the mechanical properties of the gelatin under varying deformation rates.
2.10. Gel Strength
The gel strength of the gelatin extract was measured as described by Boran and Regenstein [32] with some modifications, employing a Perten Instruments TVT 6700 texturometer (PerkinElmer Company, Sydney, Australia) equipped with a 40 mm diameter flag compression. Gelatin (6.67%) was dissolved in distilled water at 45°C, introduced into 100 mL containers measuring 60 mm in height and 50 mm in diameter and then cooled to 7°C over a period of 15 hours. Gel strength was measured at 7°C, as determined by the maximum force (in grams) required to penetrate 4 mm of gelatin with the plunger. Results were the average of at least five determinations [31].
2.11. Optimization of Gelatin Hydrolysis Conditions
The optimization of gelatin hydrolysis to achieve the optimal degree of hydrolysis was performed using a response surface experimental design. This design included three temperature levels (50°C, 60°C, and 70°C) and three enzyme concentrations, expressed in enzymatic units per gram of dehydrated gelatin (1.5, 4, and 6.5). Two replicates were performed at the central point. For the optimization process, the degree of hydrolysis (DH) was chosen as the response variable. The hydrolysis reaction was initiated by dissolving one gram of dehydrated gelatin in 100 mL of distilled water and adjusting the pH of the mixture to 8. The previously obtained lyophilized enzyme extract was then added to the mixture [33].
The degree of hydrolysis (DH) was evaluated by the pH-stat method [34]. The amount of NaOH 0.1N consumed to maintain a pH value of 8 during the protein hydrolysis was recorded at fixed intervals throughout the hydrolysis process. An 848 Titrino plus complete titrator (Metrohm Inc., Tampa, FL) was used to maintain a constant pH and obtain the NaOH consumption data. Upon completion of the hydrolysis process, the enzyme was inactivated by heating at 95 ± 0.3°C for 10 minutes in a thermostatic bath followed by cooling it to room temperature [35]. Results were expressed as the percentage of cleaved peptides (h) relative to the total number of peptide bonds available for proteolysis as a percentage (Eq 1):
Where, VNaOH is the volume of NaOH consumed (mL). Nb is the Normality of the base. MP is the protein mass (g). ℎtotal is the total number of peptide bonds in gelatin (10.82 mEq/ g protein) and ∝ is the dissociation degree of the ∝NH2 groups released during the hydrolysis.
The hydrolysate was freeze-dried and stored at -20°C until use.
2.12. Determionation of Protein Content
The Kjeldahl method (AOAC, 2005) [36] was used to determine the nitrogen content using the conversion factor of 5.55 for nitrogen-to-protein. The results were expressed as g/100g dried sample.
2.13. DPPH Radical Scavenging Activity
The antioxidant activity of the gelatin hydrolysates was determined according to Mosquera, Gómez, Montero and Giménez (2016) [19]. A solution was prepared by dissolving 3.5 mg of DPPH+ (1,1-diphenyl-2-picrylhydrazyl) in 10 mL of methanol. The solution was then diluted with more methanol until the absorbance at 515 nm was 1 ± 0.009. Next, 1.95 mL of the diluted DPPH+ solution was mixed with 0.05 mL of the hydrolysate (previously diluted to 0.06 g/mL with distilled water). The resulting mixture was stirred and allowed to stand for 30 minutes at room temperature. After centrifugation at 6,000 x g for 10 minutes, the absorbance of the supernatant was measured at 515 nm using a spectrophotometer (UV-160A, Shimadzu, Kyoto, Japan). The results were expressed as percentages.
3. Results
3.1. Molecular Size Determination of enzymatic Extract
The molecular weight profile of the enzymatic extract is shown in Figure 1, lane b. Several bands of different molecular sizes were observed, indicating the presence of diverse proteins in the extract. The most abundant component had a molecular weight of approximately 150 kDa, while the smallest one had a molecular weight of approximately 24 kDa, as determined by the reference pattern used (lane a). Notably, the molecular weights of the last three bands were below 30 kDa, while the second and third bands were in the range of 55 to 50 kDa. It is important to consider the possibility that these bands may correspond to enzymes as well as other proteins present in the extract.
3.2. Amino acid Composition
Table 1 shows the amino acid composition of gelatin extracted from yellowfin tuna skin. It is noteworthy that glycine emerges as the predominant amino acid, with a remarkable abundance of 332 residues per 1000. Furthermore, a significant content of proline and hydroxyproline was detected, with values of 115 and 73 residues per 1000, respectively.
Furthermore, the significant presence of arginine, along with glutamic acid and glutamine, has been identified as relevant amino acids in the composition of the obtained gelatin. It is noteworthy that the composition and sequence of amino acids in gelatin showed specific variations depending on the species studied. However, the predominant presence of glycine and proline is consistently observed across different species [37]. Alanine was the most abundant hydrophobic amino acid. These results contribute to the characterization of yellowfin tuna gelatin and provide valuable insights for its potential applications in various scientific and technological fields. The amino acid composition plays a crucial role in determining the functional properties of both gelatin and its hydrolysates, especially in terms of antioxidant activity.
3.3. Viscoelastic Properties and Gel Strength
Figure 2a shows the viscoelastic properties of yellowfin tuna skin-derived gelatin as a function of temperature. Analysis of the phase angle indicated that gelation began at 16°C, and the onset of gelation was observed at this temperature. In addition, the melting temperature was determined to be 22°C. It is noteworthy that the gelatin exhibited a higher melting point than the gelation point, suggesting an energy absorption phenomenon during the melting process [38].
Figure 2b depicts the relationship between the elastic modulus (G') and the viscous modulus (G") as a function of angular frequency, offering insights into the rheological behavior of the gelatin gel. In particular, G' exceeded G", indicating the formation of a gel. Moreover, the gel strength of the sample was determined to be 425±11 g, as illustrated in Figure 3a. For comparison purposes, the gel strength of a commercial gelatin sample (260 °Bloom) was also determined, yielding a value of 701±7 g (Figure 3b). Based on this comparison, an approximate gel strength of 160 °Bloom can be inferred for the extracted yellowfin tuna gelatin.
3.4. Optimization of Gelatin Hydrolysis Conditions
Table 2 presents the degree of hydrolysis as a function of the temperature and enzyme extract concentration, both recognized factors known to influence the extent of proteolysis. According to the experimental design, the maximum degree of hydrolysis was achieved under conditions of 50°C and 6.5 IU, resulting in a value of 19.81%.
The response surface experimental design predicted a maximum antioxidant activity of 35.70% at 70°C and 1.5 enzyme units. The analysis of variance yielded a high R-squared value of 98.35%, confirming the reliability of the prediction. The formula derived from the analysis is as follows:
The results suggest that an increase in temperature causes a decrease in the degree of hydrolysis, possibly due to thermal denaturation of the enzymes. This denaturation affects the tertiary structure and reduces the ability to cleave large proteins into peptides or induces protein aggregation [39]. The lowest degree of hydrolysis was recorded under conditions of 70°C and 1.5 IU, reaching 12.77%. The decrease in hydrolysis may be due to a lack of catalytic sites [40]. The results also suggest a relationship between a low degree of hydrolysis and a high antioxidant capacity, highlighting the importance of longer peptides on this activity. This is supported by Figure 4, which shows a statistically significant (P<0.05) positive effect on antioxidant activity. Furthermore, it was found that an increase in the enzyme units used had a negative effect on the antioxidant activity, confirming that extensive hydrolysis is not required to produce a potent antioxidant hydrolysate. Moreover, both temperature and enzyme units had a statistically significant quadratic effect (P<0.05). The statistical analysis confirms the evidence of an optimal range for both temperature and enzyme units, beyond which further changes may result in a decrease in activity.
4. Discussion
4.1. Molecular Sizes Determination
Analysis of the electrophoretic profile for the enzymatic extract obtained from yellowfin tuna provided significant insight into the molecular sizes of the proteins detected. Among the observed proteins, the highest recorded molecular weight was 150 kDa, surpassing the molecular weights reported in the "Brenda" enzyme database for proteases from other strains of Bacillus subtilis. Notably, the highest molecular weight (86.7 kDa) corresponded to thermolysin (EC 3.4.21.53) [41]. Additionally, the electrophoresis showed other bands corresponding with varying molecular sizes, probably corresponding to proteases. For example, a band around 53 kDa may correspond to a C-terminal processing enzyme (EC 3.4.21.102), which is consistent with a similar molecular weight reported for a metalloprotease from Bacillus stearothermophilus [41]. Comparison with the "Brenda" enzyme database at NCBI for Bacillus subtilis suggested the presence of other enzymes in the electrophoretic bands. The fourth band (37 kDa) could correspond to both endopeptidase La (EC 3.4.21.53) and subtilisin (EC 3.4.21.62), consistent with previous research identifying a metalloprotease (37 kDa) in B. subtilis [42]. Additionally, bands with molecular weights of approximately 27 and 29 kDa were observed, consistent with previous studies of proteases from Bacillus strains, typically ranging from 28 to 70 kDa [43]. Interestingly, an alkaline protease from Bacillus subtilis was identified at 27 kDa in the use of industry by-products as a fermentation medium, consistent with the penultimate band in this study [44]. Moreover, a comparable molecular weight of approximately 30 kDa was reported in a study utilizing Bacillus subtilis and agricultural waste as a substrate [45]. The sixth band may be associated with a fibrinolytic enzyme identified at 29 kDa, which was isolated and characterized in previous research [46]. Further investigation identified an extracellular protease with a molecular weight of 29 kDa, characterized as subtilisin, which may correspond to the sixth band [47].
Finally, the Brenda database provided additional insight, indicating that the Repressor LexA enzyme (registered as 3.4.21.88) with a molecular weight of 24 kDa could correspond to the smallest molecular weight molecule observed in the electrophoresis profile, while a keranolytic protease from Bacillus subtilis with a molecular weight of 25.4 kDa, similar to the last band in Figure 4, was also found [48]. These results, together with the protease activity of the extract, deepen the understanding of the potential of Bacillus subtilis for enzyme production using yellowfin tuna muscle as a nitrogen and carbon source in the culture media.
4.2. Amino Acid Composition
As expected, the gelatin had a high glycine content. This is because glycine is required in every third position of the protein to achieve a compact packing of the triple helix of the native protein (collagen) [49]. The presence of proline and hydroxyproline contributes significantly to the thermal stabilization of the collagen triple helix and enhances its ordered conformation by forming a gel-like network [50]. The content of imino acids was found to be 188 residues per 1000, which is comparable to the findings of a study on tuna skin gelatin that reported 185 residues per 1000. These values are within the expected range for imino acid content in gelatins derived from warm-blooded animal tissues [51]. Moreover, the tuna gelatin had low levels of methionine, cysteine, and tyrosine, with values of 15, 2, and 3 residues per 1000, respectively, which is consistent with previous research [49].
4.3. Gel Strength
The rheological properties of gelatin primarily determine its quality, with the gel strength (measured in Bloom degrees) being a key parameter for evaluation. The main factors influencing the rheological properties of gelatin are the extraction conditions and the characteristics of the raw material, which affect both the amino acid composition and the molecular weight profile [52]. Gelatins that more closely resemble the triple helix structure upon cooling, resulting in improved rheological properties, are associated with higher average molecular weights and increased content of imino acids (Pro+Hyp) [53,54]. The gelatin obtained in this study had a favorable composition of these imino acids (115 Pro and 73 Hyp). Gel strength measurements typically use a 'bloom jar' container that requires approximately 155 ml of gelatin solution, which is approximately 10 g of gelatin. However, the use of bloom jars is not practical for many scientific studies due to the large sample volumes required. As a result, researchers often use alternative containers of different sizes and shapes, leading to significant variation in the results obtained. This lack of standardization makes it difficult to compare data from different studies [55].
To ensure a meaningful comparison, the same procedure was performed using a commercially available bovine gelatin with a Bloom value of 260°. The resulting gel strength was then used to estimate the Bloom strength of the yellowfin tuna gelatin. Based on this comparison, it can be inferred that the yellowfin tuna skin gelatin may have a Bloom strength of approximately 160°. This finding is consistent with a previous study in which the gel strength of tuna skin gelatin was reported to be 191.0° Bloom, which is lower than the commercial gelatins analyzed in the same study [37]. Previous research has shown that yellowfin tuna gelatin tends to have a lower gel strength than commercial gelatins. Gel formation and strength can vary due to factors such as amino acid composition, particularly proline and hydroxyproline, and the methods used for extraction, filtration, and drying [56]. However, a previous study found that yellowfin tuna gelatin had a higher Bloom value (426) than bovine gelatin. This difference may be attributed to the extraction method, which involved alkaline hydrolysis over a period of 5 days [57]. The prolonged exposure to alkaline hydrolysis facilitated the extraction and solubilization of collagen proteins, resulting in a gelatin with an 89% of hydroxyproline content. This value is comparable to that observed in mammals, which typically ranges from 90.1% to 90.7%, as reported in the literature [58]. Therefore, research suggests that maximizing gel strength can be achieved by extending the extraction time [57].
The approximate Bloom value obtained for the yellowfin tuna gelatin extracted in the present work is comparable to that of other fish skin gelatins. For example, snapper skin gelatin has a value of 108 [49], unicorn leather jacket (Aluterus monoceros) skin gelatin has a value of 150 [59], gray triggerfish (Balistes capriscus) skin gelatin has a value of 168.3 [60], and cod skin gelatin has a value of 180 [61]. While the gelatin in the present study has a lower gel strength than bovine gelatin, it is important to note that gelatin with different Bloom strengths serves different commercial applications. High gel strength gelatins, such as bovine gelatin (260g Bloom), are ideal for products that require a firmer texture and higher water retention and are commonly used in the production of molded gelatins, mousses, gummy bears, and marshmallows [62]. Gelatins with lower gel strength, such as those obtained from fish, are more suitable for specific applications in bakery products, food additives, coatings for drug capsules, as well as technical applications such as the production of gelatin films for coatings in the photographic industry [53]. Another advantage of fish gelatin over bovine or porcine gelatins is the absence of religious restrictions (such as those in Hinduism, Islam, Adventists, and others) and the absence of health concerns such as the foot-and-mouth disease, bovine spongiform encephalopathy, and transmissible encephalopathy [53].
4.4. Viscoelastic Properties
The gelation and melting temperature values obtained in this study fall within the typical range observed for fish skin gelatins, which are generally reported to be between 8-25°C and 11-28°C, respectively [63]. Previous research showed that gelatin derived from warm-water fish species, may have a thermal stability closer to that of mammalian gelatin than that of cold-water species [53]. However, it is imperative to consider variables such as fish species, raw material and processing conditions, which have a significant impact on thermal stability [64]. These results demonstrate the suitability of the gelatin obtained for food applications.
Previous studies using yellowfin tuna skin gelatin reported gelation and melting points similar to those found in this research (18.7°C and 24.3°C, respectively) [57]. In contrast, other authors reported lower gelation and melting points of tuna skin gelatin (10°C and 13.4°C, and 18°C and 20.6°C respectively [65]). In another study, gelatins with melting points ranging from 23.3°C to 28.4°C were obtained, concluding that the longer the extraction time, the lower the thermostability (melting point) of the derived gelatins [66]. These variations can be attributed to the different extraction conditions and tuna species, resulting in different molecular weight profiles and amino acid compositions [67]. The gelling properties of gelatin are known to be positively influenced by the proportion of alpha and beta chains whereas they are negatively affected by the presence of low molecular weight fragments [68].
4.5. Degree of Hydrolysis
The degree of hydrolysis (DH) plays a crucial role in determining the extent of protein fractionation, resulting in the release of peptides with various biological activities such as antioxidant, antihypertensive, anti-inflammatory, nootropic, hypoglycemic, antimicrobial, and antiproliferative, among others [69]. Under the experimental conditions of this study (temperatures ranging from 55 to 65°C and enzyme concentrations ranging from 1.5 to 6.5 IU), the highest degree of hydrolysis obtained was 19.81%.
In a previous study, yellowfin tuna meat was used as raw material to produce a hydrolysate with emulsifying and foaming properties using papain, resulting in a DH of 14.35% [70]. This relatively low DH resulted in the prevalence of components above 10 kDa (60%) and may be related to the substrate specificity of papain, which may different from the enzyme used in the present research. Conversely, another study investigated the effects of different enzymes and hydrolysis times using tuna skin gelatin as the raw material, resulting in DHs ranging from 76% to 85% [71]. Notably, a treatment time of approximately 3 hours showed the highest efficiency, which is consistent with previous findings suggesting that a stationary phase, characteristic of protease-induced protein hydrolysis, is reached around this time. In a separate study, yellowfin tuna skin gelatin subjected to hydrolysis with 2% alcalase showed a significantly higher DH of 45.29% [56]. The discrepancy between the DH obtained in that study and the present study (15-24%) can be attributed to the high proteolytic activity of alcalase, resulting in more extensive protein breakdown. Furthermore, hydrolysis of flounder skin gelatin with alcalase for 2 hours yielded DH values of 23.6%, similar to those obtained in this study. The variation in DH can be attributed not only to differences in hydrolysis conditions and enzyme concentration, but also to variations in enzymatic affinity for different substrates [72]. The degree of hydrolysis achieved during protein hydrolysis is crucial as it directly influences the biological activities of the resulting peptides.
4.6. Antioxidant Activity
An inverse relationship between DH and antioxidant activity was observed: as DH decreases, antioxidant activity increases, with the highest value being 35.96 ± 0.22%. Longer peptides resulting from limited hydrolysis may retain specific secondary structures important for antioxidant activity [72]. Loss of these structures due to more extensive hydrolysis could reduce antioxidant potency. The radical scavenging capacity of hydrolysates depends on several factors, including peptide size, peptide sequence, amino acid composition, enzyme used, and degree of hydrolysis. Among these factors, both amino acid composition and peptide sequence play a crucial role in defining the antioxidant potency [72].
Peptides generated through limited hydrolysis, which involves breaking down protein molecules into shorter peptide chains without completely degrading the protein, have enhanced electron and hydrogen donating capabilities, allowing them to effectively scavenge free radicals [73]. It is important to note that the use of B. subtilis enzymatic extract, by affecting the size of peptides, also influences their sequence and ultimately the antioxidant activity of the resulting hydrolysates [56]. That is, the hydrolytic capacity of this enzymatic extract results in the generation of peptides with antioxidant properties. Furthermore, these results are consistent with a previous study in which hydrolysates obtained after 3 hours of hydrolysis showed superior antioxidant activity compared to those obtained using extended hydrolysis times (4 and 5 hours). This suggests the existence of an optimal hydrolysis time, beyond which the antioxidant activity may decrease due to the degradation of high MW peptides [74]. In addition to DH, the presence of specific residues within the peptide sequence may contribute to their antioxidant potential. Amino acids such as glutamic acid, glutamine and arginine have been associated with increased antioxidant activity [75]. In the context of the present study, the gelatin obtained was found to have a significant content of these amino acids. The amino acid composition was particularly enriched in arginine (8.17 g/100 g) and glutamic acid + glutamine (9.81 g/100 g). In previous research, the gelatin extracted from yellowfin tuna skin has an amino acid composition particularly enriched in arginine (9.16 g/100 g) and glutamic acid (10.31 g/100 g), demonstrating excellent antioxidant activity [56]. Similarly, in another study, arginine and glutamic acid were measured at levels of 7.06 g/100g and 8.81g/100g, respectively [76]. In another study focusing on the amino acid composition of yellowfin tuna skin gelatin, it was found that after the prominent presence of glycine, proline and hydroxyproline, the levels of arginine and glutamic acid were found to be significant [71]. Furthermore, in this study, values of hydroxyproline (8.80 g/100 g) and hydroxylysine (0.97 g/100 g) were obtained. These amino acids are known to confer additional antioxidant properties. Hydroxyproline enhances the stability of the triple helix structure of collagen, which plays a crucial role in the antioxidant activity of gelatin-derived peptides. These amino acids provide additional stability and enhance the radical scavenging capacity of the peptides. Hydroxyproline and hydroxylysine are predominantly found in collagen and its derivatives, such as gelatin, and are typically present in lower amounts in other protein sources. [77].
In a recent study, the antioxidant activity of a yellowfin tuna skin hydrolysate obtained using alcalase was investigated, and high scavenging rates around 90% were found [37]. The efficacy in DPPH radical scavenging activity was lower when the enzyme used was neutrase (51.8%) and flavourzyme (44.1%). These values are higher than those obtained in the present study (35.96%). The differences are attributed to the type of enzyme used, with different enzymatic cleavage sites. However, when comparing the results obtained with a previous study in which alcalase was used, a similar scavenging effect (33%) was observed [71]. This highlights the potential of yellowfin tuna residues as a valuable source of antioxidants. Similarly, a protein hydrolysate obtained from by-products of bigeye tuna (Thunnus obesus) showed remarkable antioxidant activity, reaching 57.08% [78]. In another study, the DPPH radical scavenging activity of hydrolyzed collagen from yellowfin tuna (Thunnus albacares) skin ranged from 36.54% to 69.39% [75]. An additional study demonstrated the release of antioxidant peptides from protein of the same tuna species using papain, showing an activity level of 56.74% [73]. It is noteworthy that while the aforementioned studies used commercially available enzymes, the values obtained in this study were obtained using enzymes extracted directly from Bacillus subtilis using tuna muscle tissue as a growth medium. This highlights the remarkable antioxidant activity that can be achieved by this method, which is characterized by its economic viability and ease of acquisition. Importantly, the results obtained are in close agreement with those reported in previous research, as discussed above. In addition, various by-products of marine species, including round scad (Decapterus maruadsi) [79], red snapper (Lutjanus vitta) [80], skipjack tuna (Katsuwonus pelamis) [81], and yellowstripe scad (Selaroides leptolepis) [82], have been investigated for the release of antioxidant peptides by protein hydrolysis. These studies have highlighted the potential of such by-products to act as electron donors for free radicals. Therefore, the exploration of low-value marine products, particularly the peptide fractions of collagen by-products from the seafood industry, offers a vast and promising field of research.
5. Conclusions
In this study, a methodology was developed to optimize the valorization of a by-product abundant in the industrial processing of yellowfin tuna, such as tails. This involved obtaining a proteolytic enzymatic extract of B. subtilis using the tuna by-products as a source of nitrogen and carbon, followed by extraction of gelatin from the tails and release of antioxidant peptides through controlled enzymatic hydrolysis of this gelatin by optimizing the conditions. These results underline the efficacy and potential applicability of the methodology developed for the valorization of yellowfin tuna tails, thus contributing to the sustainable use of marine by-products for the production of ingredients.
Funding
This research was performed under the i-COOP project ref. COOPA22013, financed by the Spanish National Research Council (CSIC) and the Project PIJ 16–12 entitled “Analysis of the toxicity of peptide hydrolysates with biological activities obtained from agricultural industry by-products," conducted at the Department of Food Science and Biotechnology (EPN - DECAB).
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Miyake, M.P.; Guillotreau, P.; Sun, C.-H.; Ishimura, G. Recent Developments in the Tuna Industry: Stocks, Fisheries, Management, Processing, Trade and Markets; Food and Agriculture Organization of the United Nations Rome, Italy, 2010; ISBN 9251066205.
- FAO The State of World Fisheries and Aquaculture 2022. Towards Blue Transformation; FAO: Rome. 2022; ISBN 978-92-5-136364-5.
- Yao, L.; Lu, J.; Qu, M.; Jiang, Y.; Li, F.; Guo, Y.; Wang, L.; Zhai, Y. Methodology and Application of PCR-RFLP for Species Identification in Tuna Sashimi. Food Sci. Nutr. 2020, 8, 3138–3146. [Google Scholar] [CrossRef]
- Herpandi, N.H.; Rosma, A.; Wan Nadiah, W.A. The Tuna Fishing Industry: A New Outlook on Fish Protein Hydrolysates. Compr. Rev. Food Sci. Food Saf. 2011, 10, 195–207. [Google Scholar] [CrossRef]
- Idowu, A.T.; Igiehon, O.O.; Idowu, S.; Olatunde, O.O.; Benjakul, S. Bioactivity Potentials and General Applications of Fish Protein Hydrolysates. Int. J. Pept. Res. Ther. 2020. [CrossRef]
- Olsen, R.L.; Toppe, J.; Karunasagar, I. Challenges and Realistic Opportunities in the Use of By-Products from Processing of Fish and Shellfish. Trends Food Sci. Technol. 2014, 36, 144–151. [Google Scholar] [CrossRef]
- Ardiansyah, A.; Sahubawa, L. Restructuring Steak from Flakes of Yellowfin Tuna Meat Using Low Salt Microbial Transglutaminase (MTGase). In Proceedings of the IOP Conference Series: Earth and Environmental Science; IOP Publishing, 2020; Vol. 404, p. 12073.
- Ramakrishnan, S.R.; Jeong, C.-R.; Park, J.-W.; Cho, S.-S.; Kim, S.-J. A Review on the Processing of Functional Proteins or Peptides Derived from Fish By-Products and Their Industrial Applications. Heliyon 2023. [Google Scholar] [CrossRef]
- Nguyen, H.T.M.; Sylla, K.S.B.; Randriamahatody, Z.; Donnay-Moreno, C.; Moreau, J.; Tran, L.T.; Bergé, J.P. Enzymatic Hydrolysis of Yellowfin Tuna (Thunnus Albacares) by-Products Using Protamex Protease. Food Technol. Biotechnol. 2011, 49, 48–55. [Google Scholar]
- Guérard, F.; Dufosse, L.; De La Broise, D.; Binet, A. Enzymatic Hydrolysis of Proteins from Yellowfin Tuna (Thunnus Albacares) Wastes Using Alcalase. J. Mol. Catal. B Enzym. 2001, 11, 1051–1059. [Google Scholar] [CrossRef]
- Silva, E.V.C. da; Lourenço, L. de F.H.; Pena, R.S. Optimization and Characterization of Gelatin from Kumakuma ( Brachyplatystoma Filamentosum ) Skin. CyTA - J. Food 2017, 15. [CrossRef]
- Zulkaidah Siburian, W.; Raya Bandung, J.; Java, W.; Rochima, E.; Raya Bandung-Sumedang, J.; Yuli Andriani, I.; Praseptiangga, D.; Author, C.; Andriani, Y. Fish Gelatin (Definition, Manufacture, Analysis of Quality Characteristics, and Application): A Review. Int. J. Fish. Aquat. Stud. 2020, 8, 90–95. [Google Scholar]
- Ben Rebah, F.; Miled, N. Fish Processing Wastes for Microbial Enzyme Production: A Review. 3 Biotech 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- dos Santos Aguilar, J.G.; Sato, H.H. Microbial Proteases: Production and Application in Obtaining Protein Hydrolysates. Food Res. Int. 2018, 103. [Google Scholar] [CrossRef]
- Clerici, N.J.; Lermen, A.M.; Daroit, D.J. Agro-Industrial by-Products as Substrates for the Production of Bacterial Protease and Antioxidant Hydrolysates. Biocatal. Agric. Biotechnol. 2021, 37. [Google Scholar] [CrossRef]
- Singh, S.; Bajaj, B.K. Medium Optimization for Enhanced Production of Protease with Industrially Desirable Attributes from Bacillus Subtilis K-1. [CrossRef]
- Contesini, F.J.; de Melo, R.R.; Sato, H.H. An Overview of Bacillus Proteases: From Production to Application. Crit. Rev. Biotechnol. 2018, 38. [Google Scholar] [CrossRef] [PubMed]
- Doan, C.T.; Tran, T.N.; Nguyen, V.B.; Nguyen, A.D.; Wang, S.L. Utilization of Seafood Processing By-Products for Production of Proteases by Paenibacillus Sp. TKU052 and Their Application in Biopeptides’ Preparation. Mar. Drugs 2020, 18. [CrossRef]
- Sathishkumar, R.; Ananthan, G.; Arun, J. Production, Purification and Characterization of Alkaline Protease by Ascidian Associated Bacillus Subtilis GA CAS8 Using Agricultural Wastes. Biocatal. Agric. Biotechnol. 2015, 4. [Google Scholar] [CrossRef]
- Contesini, F.J.; De Melo, R.R.; Sato, H.H. Critical Reviews in Biotechnology An Overview of Bacillus Proteases : From Production to Application An Overview of Bacillus Proteases : From Production to Application. Crit. Rev. Biotechnol. 2017, 0. [Google Scholar]
- Cruz-Casas, D.E.; Aguilar, C.N.; Ascacio-Valdés, J.A.; Rodríguez-Herrera, R.; Chávez-González, M.L.; Flores-Gallegos, A.C. Enzymatic Hydrolysis and Microbial Fermentation: The Most Favorable Biotechnological Methods for the Release of Bioactive Peptides. Food Chem. Mol. Sci. 2021, 3. [Google Scholar] [CrossRef] [PubMed]
- Greyling, N.; Bordoloi, A.; Goosen, N.J. Optimising Enzymatic Conditions of Monkfish (Lophius Vomerinus) Heads Hydrolysis towards Potential Waste Biomass Valorisation. Biomass Convers. Biorefinery 2020. [Google Scholar] [CrossRef]
- Iosageanu, A.N.D.R.E.E.A.; Ilie, D.A.N.I.E.L.A.; Anton, E.D.; Craciunescu, O. The Effect of Fish Bone Bioactive Peptides on the Wound Healing Process: An in Vitro Study on Keratinocytes. Rom. Biotechnol 2021, 2692–2699. [Google Scholar] [CrossRef]
- He, Y.; Pan, X.; Chi, C.F.; Sun, K.L.; Wang, B. Ten New Pentapeptides from Protein Hydrolysate of Miiuy Croaker (Miichthys Miiuy) Muscle: Preparation, Identification, and Antioxidant Activity Evaluation. LWT 2019, 105. [Google Scholar] [CrossRef]
- Mosquera, M.; Giménez, B.; Ramos, S.; López-Caballero, M.E.M.E.; Gómez-Guillén, M.D.C.M. del C.; Montero, P. Antioxidant, ACE-Inhibitory, and Antimicrobial Activities of Peptide Fractions Obtained From Dried Giant Squid Tunics. J. Aquat. Food Prod. Technol. 2016, 25. [CrossRef]
- Chen, Y.; Jin, H.; Yang, F.; Jin, S.; Liu, C.; Zhang, L.; Huang, J.; Wang, S.; Yan, Z.; Cai, X.; et al. Physicochemical, Antioxidant Properties of Giant Croaker (Nibea Japonica) Swim Bladders Collagen and Wound Healing Evaluation. Int. J. Biol. Macromol. 2019, 138. [Google Scholar] [CrossRef]
- Sepúlveda, C.T.; Zapata, J.E.; Martínez-Álvarez, O.; Alemán, A.; Montero, M.P.; Gómez-Guillén, M.C. The Preferential Use of a Soy-Rapeseed Lecithin Blend for the Liposomal Encapsulation of a Tilapia Viscera Hydrolysate. LWT 2021, 139. [Google Scholar] [CrossRef]
- González-Serrano, D.J.; Hadidi, M.; Varcheh, M.; Jelyani, A.Z.; Moreno, A.; Lorenzo, J.M. Bioactive Peptide Fractions from Collagen Hydrolysate of Common Carp Fish Byproduct: Antioxidant and Functional Properties. Antioxidants 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Bougatef, H.; Krichen, F.; Kobbi, S.; Martinez-Alvarez, O.; Nedjar, N.; Bougatef, A.; Sila, A. Physicochemical and Biological Properties of Eel By-Products Protein Hydrolysates: Potential Application to Meat Product Preservation. Waste and Biomass Valorization 2020, 11. [Google Scholar] [CrossRef]
- Kumar, A.; Elavarasan, K.; Hanjabam, M.D.; Binsi, P.K.; Mohan, C.O.; Zynudheen, A.A.; Kumar K, A. Marine Collagen Peptide as a Fortificant for Biscuit: Effects on Biscuit Attributes. LWT 2019, 109. [Google Scholar] [CrossRef]
- Sisa, A.; Sotomayor, C.; Buitrón, L.; Gómez-Estaca, J.; Martínez-Alvarez, O.; Mosquera, M. Evaluation of By-Products from Agricultural, Livestock and Fishing Industries as Nutrient Source for the Production of Proteolytic Enzymes. Heliyon 2023, 9, e20735. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Mo, X.; Chen, X.; Han, D.; Zhao, C. Purification and Enzymatic Identification of an Acid Stable and Thermostable A-amylase from Rhizopus Microsporus. J. Inst. Brew. 2012, 118, 309–314. [Google Scholar] [CrossRef]
- El-Bendary, M.A.; Moharam, M.E.; Ali, T.H. Purification and Characterization of Milk Clotting Enzyme Produced by Bacillus Sphaericus. J. Appl. Sci. Res. 2007, 3, 695–699. [Google Scholar]
- Rieger, T.J.; de Oliveira, C.T.; Pereira, J.Q.; Brandelli, A.; Daroit, D.J. Proteolytic System of Bacillus Sp. CL18 Is Capable of Extensive Feather Degradation and Hydrolysis of Diverse Protein Substrates. Br. Poult. Sci. 2017, 58. [Google Scholar] [CrossRef] [PubMed]
- Schägger, H.; von Jagow, G. Tricine-Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis for the Separation of Proteins in the Range from 1 to 100 KDa. Anal. Biochem. 1987. [CrossRef] [PubMed]
- Boran, G.; Regenstein, J.M. Fish Gelatin. Adv. Food Nutr. Res. 2010, 60, 119–143. [Google Scholar] [PubMed]
- Sifuentes-Penagos, G.; León-Vásquez, S.; Castillo, A. Hydrolysis of Proteins from Anchovy (Engraulis Ringens) Whole by Action of the ProtamexTM Enzyme. Sci. Agropecu. 2018, 9. [Google Scholar] [CrossRef]
- Salazar-Posada, C.; López-Padilla, A.; Cano-Salazar, J.A. Efecto Del PH y La Temperatura En La Hidrólisis Enzimática de Subproductos de La Industria Bovina. Rev. Lasallista Investig. 2013, 9. [Google Scholar]
- AOAC Association of Official Analytical Chemists 2005. Official Methods of Analysis 2005.
- Nurilmala, M.; Adinugraha, S.C.; Jacoeb, A.M.; Susilawati, S.; Ochiai, Y. Evaluation of the Properties of Tuna Skin Gelatin as a Hard Capsule Material. Fish. Sci. 2020, 86. [Google Scholar] [CrossRef]
- Kaewdang, O.; Benjakul, S.; Prodpran, T.; Kaewmanee, T.; Kishimura, H. Characteristics of Gelatin Extracted from the Swim Bladder of Yellowfin Tuna (Thunnus Albacores) as Affected by Alkaline Pretreatments. J. Aquat. Food Prod. Technol. 2016, 25. [Google Scholar] [CrossRef]
- Alahmad, K.; Noman, A.; Xia, W.; Jiang, Q.; Xu, Y. Influence of the Enzymatic Hydrolysis Using Flavourzyme Enzyme on Functional, Secondary Structure, and Antioxidant Characteristics of Protein Hydrolysates Produced from Bighead Carp (Hypophthalmichthys Nobilis). Molecules 2023, 28. [Google Scholar] [CrossRef] [PubMed]
- Noman, A.; Xu, Y.; AL-Bukhaiti, W.Q.; Abed, S.M.; Ali, A.H.; Ramadhan, A.H.; Xia, W. Influence of Enzymatic Hydrolysis Conditions on the Degree of Hydrolysis and Functional Properties of Protein Hydrolysate Obtained from Chinese Sturgeon (Acipenser Sinensis) by Using Papain Enzyme. Process Biochem. 2018, 67. [Google Scholar] [CrossRef]
- Wang, S.L.; Yeh, P.Y. Production of a Surfactant- and Solvent-Stable Alkaliphilic Protease by Bioconversion of Shrimp Shell Wastes Fermented by Bacillus Subtilis TKU007. Process Biochem. 2006, 41. [Google Scholar] [CrossRef]
- Patel, A.R.; Mokashe, N.U.; Chaudhari, D.S.; Jadhav, A.G.; Patil, U.K. Production Optimisation and Characterisation of Extracellular Protease Secreted by Newly Isolated Bacillus Subtilis AU-2 Strain Obtained from Tribolium Castaneum Gut. Biocatal. Agric. Biotechnol. 2019, 19. [Google Scholar] [CrossRef]
- Mahmoud, A.; Kotb, E.; Alqosaibi, A.I.; Al-Karmalawy, A.A.; Al-Dhuayan, I.S.; Alabkari, H. In Vitro and in Silico Characterization of Alkaline Serine Protease from Bacillus Subtilis D9 Recovered from Saudi Arabia. Heliyon 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, I.; Zia, M.A.; Iqbal, H.M.N. Purification and Kinetic Parameters Characterization of an Alkaline Protease Produced from Bacillus Subtilis through Submerged Fermentation Technique. World Appl. Sci. J. 2011, 12. [Google Scholar]
- Ramalingam, K.; Nandhi, P.; Murugan, R.; Venkatesan, R. Physical and chemical characterization of alkaline protease from bacillus subtilis vbc7 using agro waste as substrate. J. Microbiol. Biotechnol. Food Sci. 2022, 12. [Google Scholar] [CrossRef]
- Hu, Y.; Yu, D.; Wang, Z.; Hou, J.; Tyagi, R.; Liang, Y.; Hu, Y. Purification and Characterization of a Novel, Highly Potent Fibrinolytic Enzyme from Bacillus Subtilis DC27 Screened from Douchi, a Traditional Chinese Fermented Soybean Food. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Choi, N.S.; Chung, D.M.; Ryu, C.H.; Yoon, K.S.; Maeng, P.J.; Kim, S.H. Identification of Three Extracellular Proteases from Bacillus Subtilis KCTC 3014. J. Microbiol. Biotechnol. 2006, 16. [Google Scholar]
- Suh, H.J.; Lee, H.K. Characterization of a Keratinolytic Serine Protease from Bacillus Subtilis KS-1. J. Protein Chem. 2001, 20. [Google Scholar] [CrossRef] [PubMed]
- Binsi, P.K.; Shamasundar, B.A.; Dileep, A.O.; Badii, F.; Howell, N.K. Rheological and Functional Properties of Gelatin from the Skin of Bigeye Snapper (Priacanthus Hamrur) Fish: Influence of Gelatin on the Gel-Forming Ability of Fish Mince. Food Hydrocoll. 2009, 23. [Google Scholar] [CrossRef]
- S, J.; H, S.; S.M., R.; H, A.; F, K. Extraction and Evaluation of Gelatin from Yellow Fin Tuna (Thunnus Albacares) Skin and Prospect as an Alternative to Mammalian Gelatin. Iran. J. Fish. Sci. 2016, 18.
- Gómez-Estaca, J.; Montero, P.; Fernández-Martín, F.; Gómez-Guillén, M.C. Physico-Chemical and Film-Forming Properties of Bovine-Hide and Tuna-Skin Gelatin: A Comparative Study. J. Food Eng. 2009, 90. [Google Scholar] [CrossRef]
- Tümerkan, E.T.A.; Cansu, Ü.; Boran, G.; Mac Regenstein, J.; Özoğul, F. Physiochemical and Functional Properties of Gelatin Obtained from Tuna, Frog and Chicken Skins. Food Chem. 2019, 287, 273–279. [Google Scholar] [CrossRef]
- Karim, A.A.; Bhat, R. Fish Gelatin: Properties, Challenges, and Prospects as an Alternative to Mammalian Gelatins. Food Hydrocoll. 2009, 23, 563–576. [Google Scholar] [CrossRef]
- Gómez-Guillén, M.C.; Turnay, J.; Fernández-Díaz, M.D.; Ulmo, N.; Lizarbe, M.A.; Montero, P. Structural and Physical Properties of Gelatin Extracted from Different Marine Species: A Comparative Study. Food Hydrocoll. 2002, 16. [Google Scholar] [CrossRef]
- Nurilmala, M.; Hizbullah, H.H.; Karnia, E.; Kusumaningtyas, E.; Ochiai, Y. Characterization and Antioxidant Activity of Collagen, Gelatin, and the Derived Peptides from Yellowfin Tuna (Thunnus Albacares) Skin. Mar. Drugs 2020, 18. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.M.; Gu, Y.S.; Kim, S.B. Extracting Optimization and Physical Properties of Yellowfin Tuna (Thunnus Albacares) Skin Gelatin Compared to Mammalian Gelatins. Food Hydrocoll. 2005, 19. [Google Scholar] [CrossRef]
- Ningrum, A.; Widyastuti Perdani, A.; Supriyadi; Siti Halimatul Munawaroh, H.; Aisyah, S.; Susanto, E. Characterization of Tuna Skin Gelatin Edible Films with Various Plasticizers-essential Oils and Their Effect on Beef Appearance. J. Food Process. Preserv. 2021, 45, e15701.
- Ahmad, M.; Benjakul, S. Characteristics of Gelatin from the Skin of Unicorn Leatherjacket (Aluterus Monoceros) as Influenced by Acid Pretreatment and Extraction Time. Food Hydrocoll. 2011, 25. [Google Scholar] [CrossRef]
- Jellouli, K.; Balti, R.; Bougatef, A.; Hmidet, N.; Barkia, A.; Nasri, M. Chemical Composition and Characteristics of Skin Gelatin from Grey Triggerfish (Balistes Capriscus). LWT 2011, 44. [Google Scholar] [CrossRef]
- Gudmundsson, M.; Hafsteinsson, H. Gelatin from Cod Skins as Affected by Chemical Treatments. J. Food Sci. 1997, 62. [Google Scholar] [CrossRef]
- Gómez-Guillén, M.C.; Giménez, B.; al López-Caballero, M.E.; Montero, M.P. Functional and Bioactive Properties of Collagen and Gelatin from Alternative Sources: A Review. Food Hydrocoll. 2011, 25, 1813–1827. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Huang, M.; Cao, G.; Tao, F.; Shen, Q.; Zhou, G.; Yang, H. Repurposing Fish Waste into Gelatin as a Potential Alternative for Mammalian Sources: A Review. Compr. Rev. Food Sci. Food Saf. 2022, 21, 942–963. [Google Scholar] [CrossRef]
- Alfaro, A. da T.; Balbinot, E.; Weber, C.I.; Tonial, I.B.; Machado-Lunkes, A. Fish Gelatin: Characteristics, Functional Properties, Applications and Future Potentials. Food Eng. Rev. 2015, 7.
- Montero, M.; Acosta, Ó.G.; Bonilla, A.I. Membrane Fractionation of Gelatins Extracted from Skin of Yellowfin Tuna (Thunnus Albacares): Effect on Molecular Sizes and Gelling Properties of Fractions. CYTA - J. Food 2022, 20. [CrossRef]
- Karayannakidis, P.D.; Chatziantoniou, S.E.; Zotos, A. Effects of Selected Process Parameters on Physical and Sensorial Properties of Yellowfin Tuna (Thunnus Albacares) Skin Gelatin. J. Food Process Eng. 2014, 37. [Google Scholar] [CrossRef]
- Lin, L.; Regenstein, J.M.; Lv, S.; Lu, J.; Jiang, S. An Overview of Gelatin Derived from Aquatic Animals: Properties and Modification. Trends Food Sci. Technol. 2017, 68. [Google Scholar] [CrossRef]
- Lv, L.-C.; Huang, Q.-Y.; Ding, W.; Xiao, X.-H.; Zhang, H.-Y.; Xiong, L.-X. Fish Gelatin: The Novel Potential Applications. J. Funct. Foods 2019, 63, 103581. [Google Scholar] [CrossRef]
- Jampilek, J.; Kos, J.; Kralova, K. Potential of Nanomaterial Applications in Dietary Supplements and Foods for Special Medical Purposes. Nanomaterials 2019, 9, 296. [Google Scholar] [CrossRef]
- Parvathy, U.; Binsi, P.K.; Visnuvinayagam, S.; Zynudheen, A.A.; Ninan, G.; Ravishankar, C.N. Functional Peptides from Yellowfin Tuna (Thunnus Albacares): Characterisation and Storage Stability Assessment. Indian J. Fish. 2020, 67, 69–79. [Google Scholar] [CrossRef]
- Han, Y.; Byun, S.H.; Park, J.H.; Kim, S.B. Bioactive Properties of Enzymatic Hydrolysates from Abdominal Skin Gelatin of Yellowfin Tuna (Thunnus Albacares). Int. J. Food Sci. Technol. 2015, 50. [Google Scholar] [CrossRef]
- Viji, P.; Phannendra, T.S.; Jesmi, D.; Madhusudana Rao, B.; Dhiju Das, P.H.; George, N. Functional and Antioxidant Properties of Gelatin Hydrolysates Prepared from Skin and Scale of Sole Fish. J. Aquat. Food Prod. Technol. 2019, 28. [Google Scholar] [CrossRef]
- Wardani, D.W.; Ningrum, A. Manikharda; Vanidia, N.; Munawaroh, H.S.H.; Susanto, E.; Show, P.L. In Silico and in Vitro Assessment of Yellowfin Tuna Skin (Thunnus Albacares) Hydrolysate Antioxidation Effect. Food Hydrocoll. Heal. 2023, 3. [CrossRef]
- Cai, B.; Wan, P.; Chen, H.; Huang, J.; Ye, Z.; Chen, D.; Pan, J. Purification and Identification of Novel Myeloperoxidase Inhibitory Antioxidant Peptides from Tuna (Thunnas Albacares) Protein Hydrolysates. Molecules 2022, 27. [Google Scholar] [CrossRef]
- Nguyen, B.C.; Nguyen, H.M.X.; Nguyen, K.H.N.; Kha, T.C. Functional Properties of Yellowfin Tuna (Thunnus Albacares) Skin Collagen Hydrolysate Fraction Obtained By Ultrafiltration Purification. Curr. Res. Nutr. Food Sci. 2021, 9. [Google Scholar] [CrossRef]
- Thilanja, G.P.D.D.S.; Dissanayake, K.S.M.; Kariyawasam, M.G.T.R.; Abeyrathne, E.D.N.S. Extraction of Crude Collagen from Yellowfin Tuna ( Thunnus Albacares ) Skin and Determination of the Functional Properties of Its Hydrolysates. J. Technol. Value Addit. 2020, 2. [Google Scholar]
- Mendis, E.; Rajapakse, N.; Kim, S.K. Antioxidant Properties of a Radical-Scavenging Peptide Purified from Enzymatically Prepared Fish Skin Gelatin Hydrolysate. J. Agric. Food Chem. 2005, 53. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.I.; Ho, H.Y.; Chu, Y.J.; Chow, C.J. Characteristic and Antioxidant Activity of Retorted Gelatin Hydrolysates from Cobia (Rachycentron Canadum) Skin. Food Chem. 2008, 110. [Google Scholar] [CrossRef]
- Nagano, H.; To, K.A. Purification of Collagenase and Specificity of Its Related Enzyme from Bacillus Subtilis FS-2. Biosci. Biotechnol. Biochem. 2000, 64. [Google Scholar] [CrossRef]
- Khantaphant, S.; Benjakul, S.; Ghomi, M.R. The Effects of Pretreatments on Antioxidative Activities of Protein Hydrolysate from the Muscle of Brownstripe Red Snapper (Lutjanus Vitta). LWT 2011, 44. [Google Scholar] [CrossRef]
- Qiu, Y.T.; Wang, Y.M.; Yang, X.R.; Zhao, Y.Q.; Chi, C.F.; Wang, B. Gelatin and Antioxidant Peptides from Gelatin Hydrolysate of Skipjack Tuna (Katsuwonus Pelamis) Scales: Preparation, Identification and Activity Evaluation. Mar. Drugs 2019, 17. [Google Scholar] [CrossRef]
- Klompong, V.; Benjakul, S.; Yachai, M.; Visessanguan, W.; Shahidi, F.; Hayes, K.D. Amino Acid Composition and Antioxidative Peptides from Protein Hydrolysates of Yellow Stripe Trevally (Selaroides Leptolepis). J. Food Sci. 2009, 74. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
SDS-PAGE gel, a) molecular weight standard b) enzymatic extract.
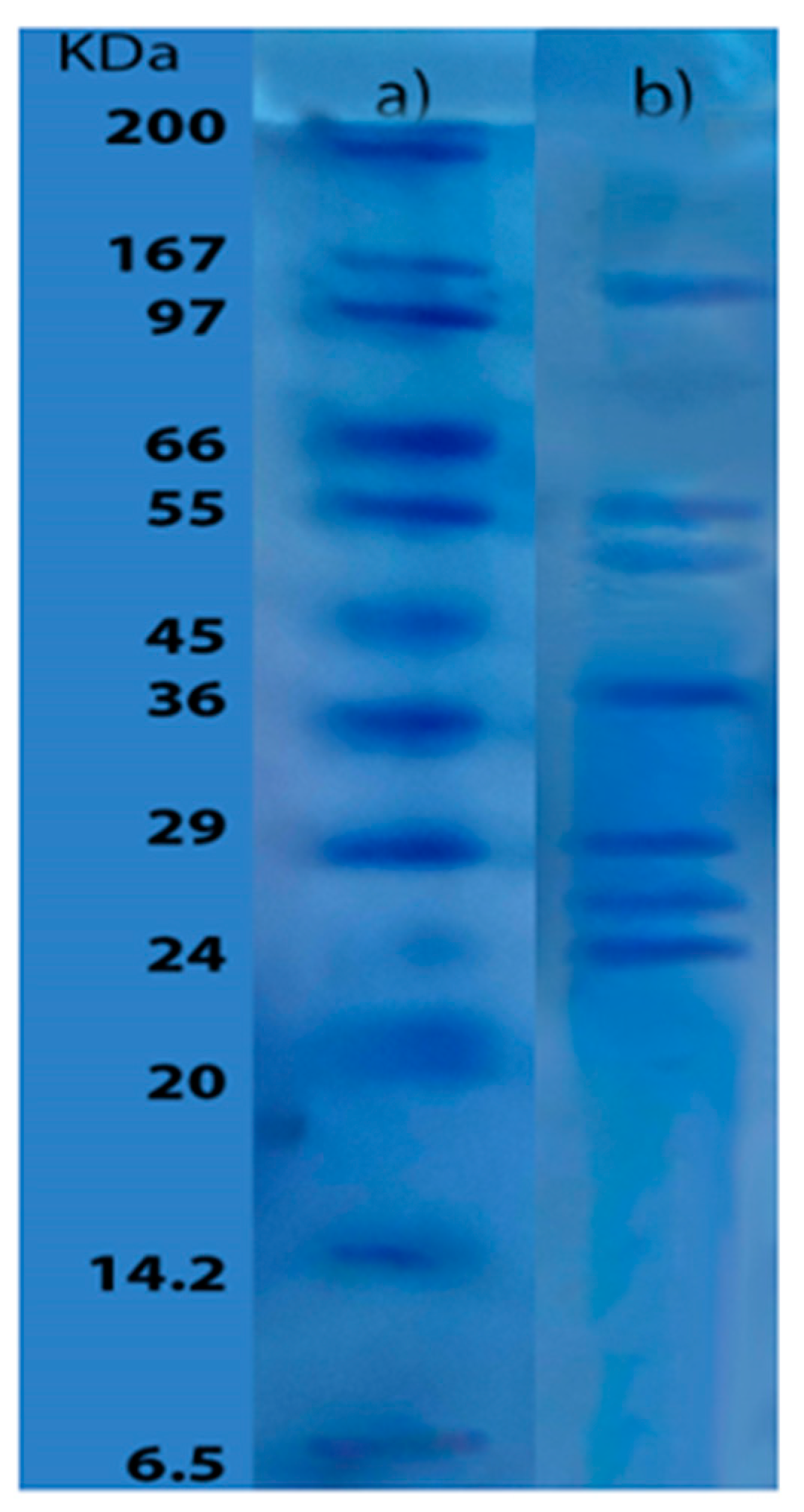
Figure 2.
Viscoelastic properties: a) Changes in elastic modulus (red), viscous modulus (blue), and phase angle (black) as function of temperature; b) Changes as a function of angular frequency in elastic modulus (red) and viscous modulus (blue). The cooling ramp is represented by the open symbols and the heating ramp by the solid symbols. .
Figure 2.
Viscoelastic properties: a) Changes in elastic modulus (red), viscous modulus (blue), and phase angle (black) as function of temperature; b) Changes as a function of angular frequency in elastic modulus (red) and viscous modulus (blue). The cooling ramp is represented by the open symbols and the heating ramp by the solid symbols. .
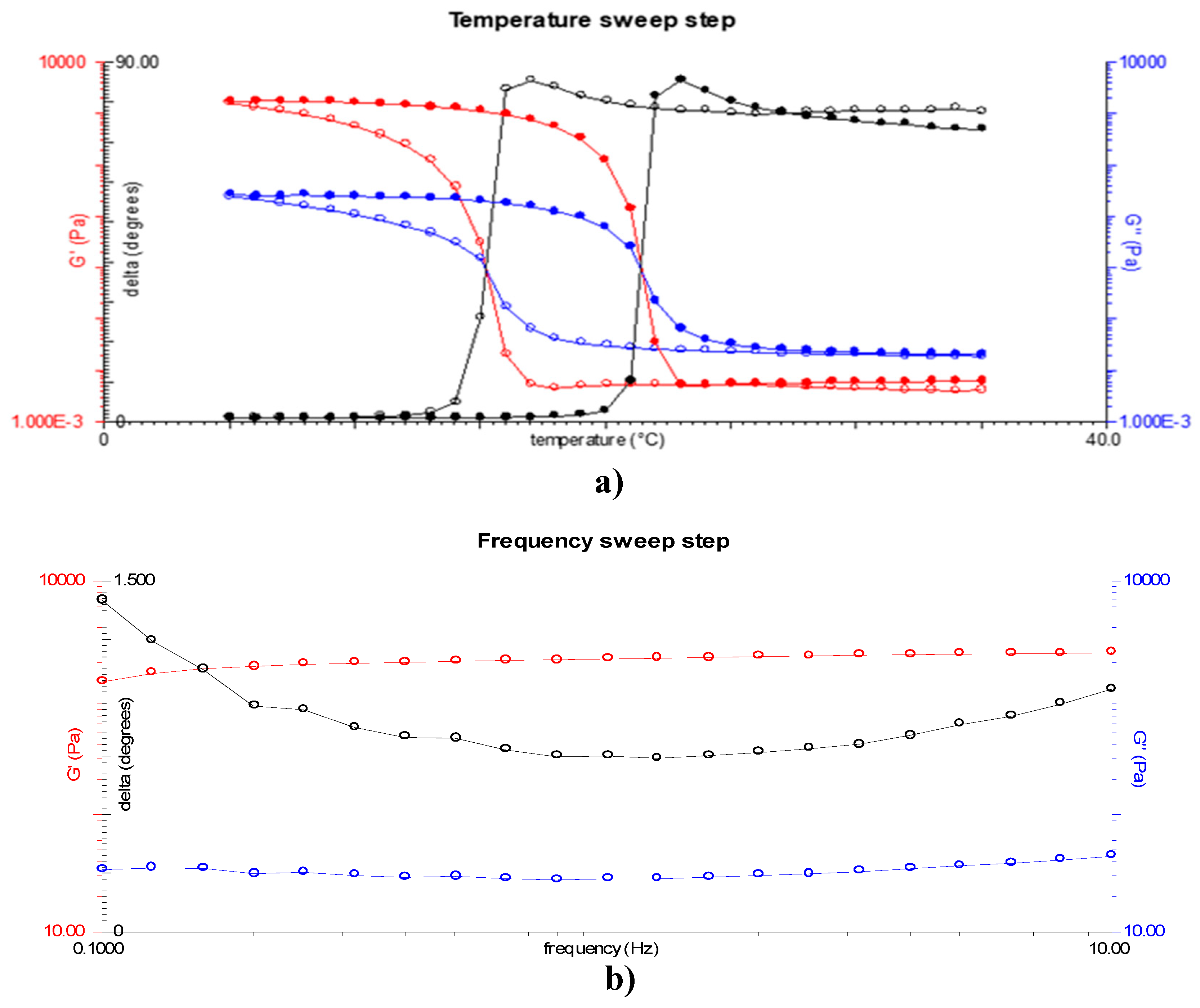
Figure 3.
Gel strength a) yellowfin tuna skin gelatin b) gelatin coil with 260 °Bloom.
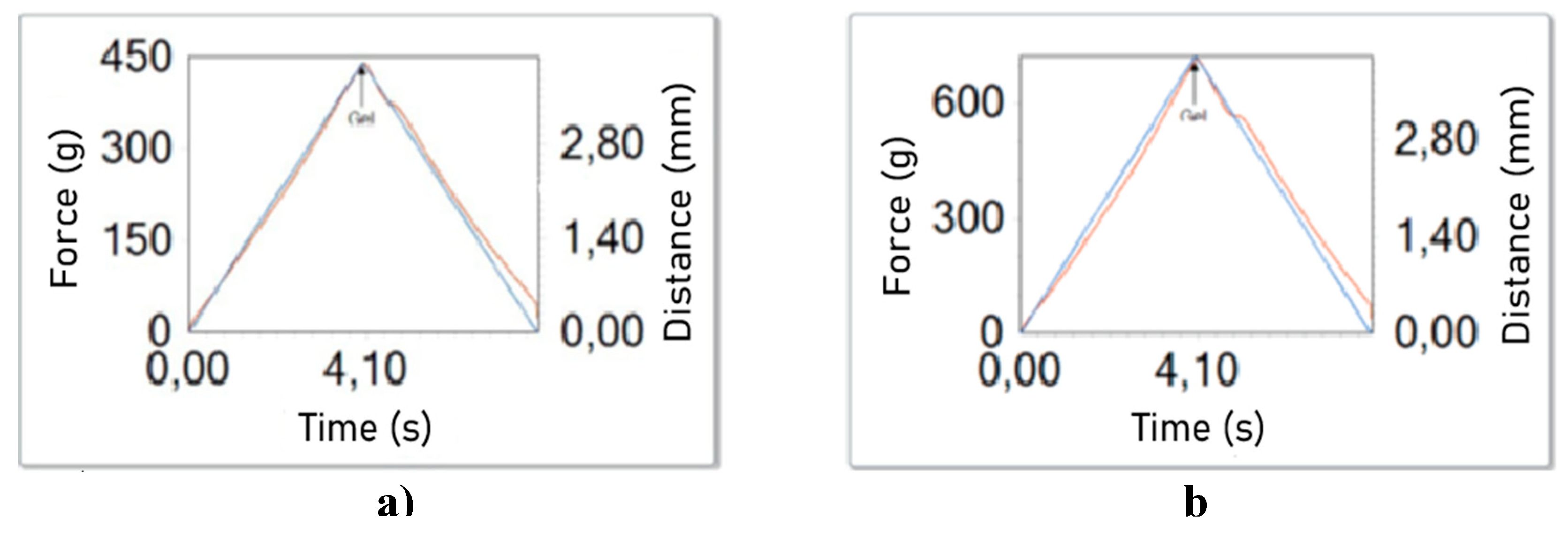
Figure 4.
Pareto chart for antioxidant activity: A represents the linear effect of Temperature, while B denotes the linear effect of enzyme units. AA indicates the quadratic effect of temperature, BB signifies the quadratic effect of enzyme units, and AB represents the interaction between the two variables.
Figure 4.
Pareto chart for antioxidant activity: A represents the linear effect of Temperature, while B denotes the linear effect of enzyme units. AA indicates the quadratic effect of temperature, BB signifies the quadratic effect of enzyme units, and AB represents the interaction between the two variables.
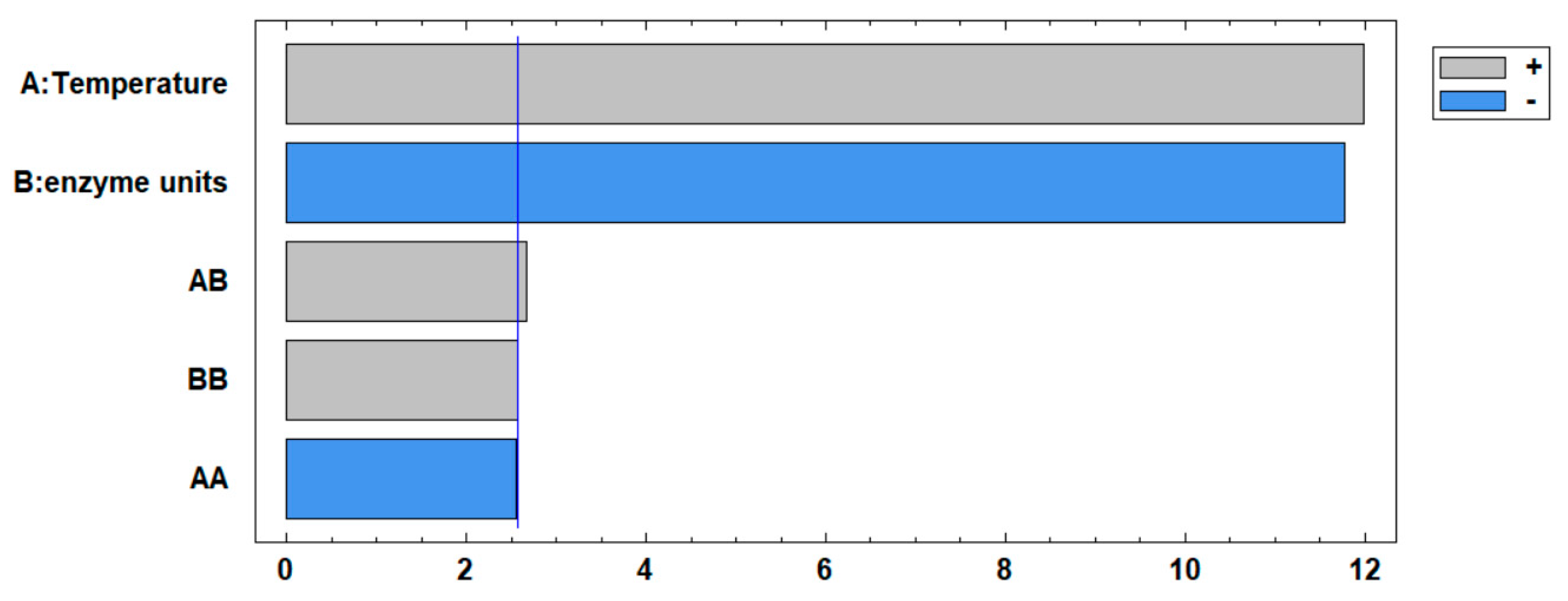

Table 1.
Amino acid composition of gelatin from yellowfin tuna skin.
| Amino acids | g/100g dry sample |
|---|---|
| Asp+Asn | 5.26 |
| Thr | 3.29 |
| Ser | 3.77 |
| Glu+Gln | 9.81 |
| Pro | 12.17 |
| Gly | 22.93 |
| Ala | 10.49 |
| Cys | 0.22 |
| Val | 1.83 |
| Met | 2.06 |
| Ile | 0.84 |
| Leu | 2.53 |
| Tyr | 0.50 |
| Phe | 14 |
| His | 5 |
| Lys | 26 |
| Arg | 51 |
| OH-Pro | 73 |
| OH-Lys | 6 |
| Total | 1000 |
Table 2.
Hydrolysis degree and antioxidant activity of the hydrolysates of gelatin from yellowfin tuna at different temperatures and enzyme Units.
Table 2.
Hydrolysis degree and antioxidant activity of the hydrolysates of gelatin from yellowfin tuna at different temperatures and enzyme Units.
| Temperature (°C) | Enzymatic Units (IU) | Hydrolysis degree (% DH) |
Antioxidant activity (%) |
|---|---|---|---|
| 70 | 6.5 | 16.66 | 28.65 ± 0.16 |
| 70 | 4 | 15.40 | 31.71 ± 0.41 |
| 60 | 4 | 16.06 | 28.01 ± 0.49 |
| 70 | 1.5 | 12.77 | 35.96 ± 0.22 |
| 60 | 6.5 | 18.30 | 26.60 ± 0.55 |
| 60 | 4 | 16.24 | 28.15 ± 0.45 |
| 60 | 1.5 | 13.46 | 33.26 ± 0.32 |
| 50 | 4 | 17.16 | 22.32 ± 0.54 |
| 60 | 4 | 16.10 | 27.65 ± 0.88 |
| 50 | 6.5 | 19.81 | 17.64 ± 0.38 |
| 50 | 1.5 | 16.35 | 29.77 ± 0.22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



