Submitted:
26 May 2024
Posted:
27 May 2024
You are already at the latest version
Abstract
Selenium is one of the inorganic compounds in human and animal nutrition and is involved in the proper functioning of the body. As a micronutrient, it actively contributes to the regulation of various metabolic activities, i.e., thyroid hormone, and protection against oxidative stress and cancer. Selenium anion also plays an essential role in the mechanism of immunity to viral infec-tions. However, this element can also be hazardous, especially in high concentrations. The toxicity of selenium is strongly associated with its chemical form. Se exhibits a narrow concentration window from having a positive to exerting a toxic effect. The daily uptake for humans is in the range of 55-400 µg/day. In higher doses (of the order of 900-1000 µg/day), it negatively affects the human body and causes DNA damage through the formation of free radicals. Increased reactivity of Se anion can also disrupt the integrity and function of DNA-repairing proteins. As the permis-sible concentration of Se in drinking water is 10 µg/L, it is vital to develop sensitive and robust methods of Se detection in aqueous samples.
Keywords:
aptamer
; selenate ion
; electrochemistry
; aptasensor
; methylene blue
1. Introduction
Selenium exists in all the components of the environment, including rocks, soil, plants, and water. Wastewater discharged from mining, petrochemical agricultural, or metallurgical activities contributes to elevated Se levels in groundwater, which can quickly reach levels toxic to fish and wildlife [1]. Selenium can be found in four different oxidation states as elemental selenium Se(0), as selenide Se(-II) in organic form and in inorganic forms as oxyanions, selenite (SeO32-) (-IV) and selenate (SeO42-) (-VI). Inorganic forms of selenium species are considered more toxic than their organic forms due to substantially better water solubility and environmental mobility [2]. Selenite Se(IV) is known as the most toxic and selenate Se(VI) as the most bioavailable and soluble compound in the oxidizing environment [2,3]. However, because of its high capacity for bioaccumulation, the substantial harmfulness of selenate anion is also described [4,5].
For the speciation analysis and determination of anionic selenium forms in water so far, many spectroscopic techniques have been developed, including inductively coupled plasma-mass-spectrometry (ICP-MS) [6,7,8], inductively coupled plasma-optical emission spectroscopy (ICP-OES) [9], hydride generation atomic absorption spectrometry (HGAAS) [10,11], nuclear magnetic resonance Imaging (NMRI) [12], neutron activation analysis [13] or fluorometry [14]. Also, separation techniques such as high-performance liquid chromatography (HPLC) [15], solid-phase extraction (SPE) [16], ion-exchange chromatography (IEC) [17] or capillary electrophoresis [18] have been employed to improve the performance of the existing selenium detection methods. Unfortunately, a laboratory equipped with expensive analytical devices is indispensable for carrying out such analyses and labor-intensive sample preparation. These methods are also time-consuming and require highly trained personnel.
An alternative to the approaches mentioned above is the employment of electrochemical techniques, which are characterized by simplicity, high sensitivity, and the capacity of miniaturization for on-site applications. To date, stripping voltammetry with the use of modified solid electrodes has been the most widely used approach. Various electrode modifiers, including mercury films [19], boron-doped gold-modification of diamond electrodes [20] as well as direct detection with platinum and gold electrodes, microband electrode array [21] and a rotating gold electrode [22], were typically employed for Se determination. Also, the use of nanomaterials opens up new possibilities in the detection of selenium anions. For this purpose, nitrogen-doped graphene [23], reduced graphene oxide [24], Au/ZnO nanocomposites decorated ITO electrodes [25], poly(1-aminoanthraquinone)/multiwall carbon nanotubes [26] and Mn3O4- chitosan nanocomposite [27] were used. The methods mentioned above offered the possibility of determination at satisfactorily low concentration levels, i.e., below 10 µg/L (WHO limit [28]). However, the selectivity of voltammetric methods can often be unsatisfactory. Therefore, it is desirable to develop electrochemical sensors with enhanced selectivity, e.g., by designing and introducing selective bioreceptors with an affinity for selenium species. There are only a few such attempts described in the literature [29,30]. One of them is the approach proposed by Motlagh et al., who developed enzymatic gold nanodendrite biosensor. Selenate reductase immobilized on the electrode surface reduces selenate to selenite ion, which, as an electroactive compound [31], is then detectable by CV and DPV voltammetry [29]. A similar approach is described by the same author in [30], where instead of a pure enzyme, the bacterial strains capable of selenate reduction were used. A different approach in biosensors development, instead of reaction catalyzed by specific enzymes, is the use of receptors that will express high affinity toward given analyte, selenate ion (SeO42-), and the same its binding in the receptor layer. One of such receptors are nucleic acids strands or, more precisely specifically, selected sequences of nucleic acids called aptamers. Thanks to the abundance of DNA nucleotide functional groups as aptamer building blocks, aptamers can interact according to various mechanisms with a wide range of targets, including small molecules such as inorganic anions. Another advantage of oligonucleotide receptors is the possibility of their design and synthesis in vitro, opposite to protein receptors, and their activity insensitivity even toward toxic compounds. What is more, analyte-triggered conformational changes can be a source of analytical signal, which opens vast possibilities for the employment of aptamers in various detection strategies, including electrochemical detection. Despite the apparent advantages of aptamers as molecular receptors, the design of aptasensors for selenate (SeO42-) anion has not yet been described. This may be explained by the fact that the selection of aptamers for ions and small molecules is challenging. The small molecular weight of such targets and a huge difference in size in comparison to the oligonucleotides significantly hinder the SELEX process. Particular difficulties arise at the step of the separation of unbound DNA sequences from target-aptamer complexes that differ only slightly in mass and general properties [32]. Another challenge in the selection of aptamers for small molecules is the lack of epitopes or functional groups available for strong aptamer binding and the same lower affinity of the aptamers to small target molecules in comparison to larger analytes [33]. Nevertheless, after identification of the above hindrances, there are also several approaches to solving such problems (e.g., reversible aptamers candidates immobilization on magnetic nanoparticles what increases the efficiency in separation of unbound nucleic acids strands) [32] and allowing for the selection of new aptamers offering high affinity toward ions.
In this study, for the first time, we report the sequence of DNA aptamer as novel receptors towards SeO42- ion, chosen after the SELEX process, and we describe their introduction for the construction of an electrochemical aptasensor towards selenate ion. The developed biosensor allows for the direct detection of selenate ions in aqueous samples. We believe that the high selectivity and sensitivity of the developed aptasensor are determined by the high-affinity aptamer to the given selenium ion, as well as the proposed detection strategy based on the use of methylene blue as an electrochemical redox marker. The performed studies also cover the optimization of the sensing conditions, with a focus on the selection of measurement medium composition suitable for the detection of the biosensor response. Conditions for biosensor regeneration and storage were also investigated.
2. Results and Discussion
To date, several aptamers toward inorganic ions and electrochemical aptasensors using such aptamers within receptor layers were described [34,35,36]. This included mercury (Hg2+), lead (Pb2+), potassium (K+), uranyl (UO22+), silver (Ag+) or cadmium (Cd2+). In all cases, the interactions between given ion and ssDNA aptamer(s) resulted in its binding and retention in the biosensing layer. There are proven several mechanisms, which are responsible for it, including G-quadruplex stabilization by Pb2+ and K+ [37], cytosine-cytosine mismatch stabilization by Ag+ [38], covalent bonds formation between thymine- and guanine-rich probes and Cd2+ [39], coordinate bond between phosphate backbone and UO22+ [40] or thymine-thymine mismatch stabilization by Hg2+ [41]. Despite of above, the standard procedure for new aptamer selection is SELEX (Systematic Evolution of Ligands by EXponential enrichment), where in subsequent rounds (between 6 to 12), the pool of nucleic acids sequences of increasing affinity toward given analyte is obtained [42]. The above procedure, however, was developed for proteins, which, compared to inorganic ions, are analytes of rather sizable dimensions, respectively 1–100 nm [43] and 0.027–0.3 nm [44]. To be able to obtain aptamer sequences for such small analytes as inorganic ions, the VENNMultiplex™ mode of SELEX can be used [45]. In the presented studies, the aptamer selection process, consisting of 12 rounds, allowed for defining a sequence of high specificity towards SeO42- ions. It was, however, necessary to further prove that this sequence could be used in the biosensor layer of electrochemical aptasensor as a receptor towards selenate ions. The biosensor construction was based on a typical self-assembled monolayers setup [46]. In order to be able to immobilize the investigated aptamer on the gold disc electrode surface it was modified with thiol group. After its deposition at the gold substrate, the blocking 6-mercapto-1-hexanol was immobilized, which fulfilled unoccupied by the ssDNA electrode area [46,47]. For the electrochemical signal generation the redox marker was used, freely available in the solution. The mechanism of detection of developed electrochemical aptasensor, confirmed during presented studies, is shown in Figure 1, together with the equation for biosensor response calculation.
Briefly, as the redox indicator (the only electroactive compound in the used potential range, approximately from -0.2 to 0.8 V) is freely available in the sample, the electrochemical biosensor response will depend on the change in the efficiency of its oxidation or reduction. This efficiency, in turn, depends on the possibility and easiness of reaching the electrode surface. As it changes together with binding the analyte by the receptor layer, the observed changes in redox current are translated into biosensor response (Figure 1). In the presented studies, ultimately, the cationic marker, methylene blue, was chosen and the observed tendency in redox current changes is shown in Figure 1. However as there are available several such indicators, which differ in the structure, charge and the same in mechanism of its interaction with DNA strands what ultimately translates into smaller or larger changes in redox reaction efficiency triggered by the binding the analyte (e.g., selenate ion) by the receptor layer, initially two such indicators were investigated as the source of the signal for biosensor response [46]. These were (i) methylene blue, an aromatic compound exhibiting a positive charge, and (ii) an equimolar mix of ferro-/ferricyanide with a negative charge. Moreover, evaluation of the biosensor response registered for such different redox markers may point to its source.
The initial experiments were conducted at pH 7.0 (Figure 2), as optimal for DNA strands [48,49] and of rather negligible influence on the selenate ion [50]. It should be remembered that depending on the medium composition and properties (e.g., pH), the analyzed ion could be present in a different than initially assumed form. One of the best example of the measurement environment’s influence on the form of analyzed anion, and the same on the obtained results, could be mercury ion (Hg2+) [51,52,53] where depending on medium pH and its composition, mercury could be present as HgCl2, Hg(OH)2, HgClOH, HgOHCO3-, or be complexed by the components of the measurement solution [52]. Luckily, selenate anion (SeO42-) is quite stable in the broad pH range (pH between 3.5 and 14.0). Only below pH 3.0 is protonated and changes into HSeO4- [50]. Similar changes could also be observed for the receptor layer. DNA molecules are stable in the pH range from 4.0 to 9.0 [48,49], but in more extreme values are susceptible to pH-dependent destabilization. Below pH 3.5 DNA loses purine bases (adenine and guanine) in the so called depurination process, and below pH 2.0, it lose also its polyanionic character due to the protonation of the phosphate backbone [48,49,54]. In turn, for pH higher than 9.0, dsDNA is prone to alkaline denaturation due to the abundance of hydroxide ions, which break the hydrogen bonds between DNA strands (remove hydrogen ions from the base pairs of DNA). Nonetheless, as could be seen in Figure 2, the biosensor response in pH 7.0 toward selenate ion (calculated according to the equation presented in Figure 1, was obtained only when methylene blue was used as the redox marker (square wave voltammetry anodic or cathodic scans were chosen based on higher current changes).
This might result from the electrostatic interactions present in the receptor layer. In the case of the cationic redox marker (methylene blue), its attraction by negatively charged phosphate DNA backbone moves the net receptor layer charge toward more positive, which may make the selenate anion approach the electrode surface. However, binding SeO42- by aptamer strands and retention it in the biosensing layer should lead rather diminish the current registered for aptasensor after recognition process because of the possible steric hindrances formation for redox marker approaching the electrode surface [55], not increase as was observed and shown in Figure 2. Nonetheless, in the literature there are also several examples of electrochemical signal increase after analyte binding by the receptor layer [56,57]. Similarly to these reports, we conclude (which is also pictured in Figure 1) that the change in spatial aptamer shape after selenate ion binding leads to more space in the receptor layer for the redox marker to approach the electrode surface. We also conclude that because of exactly the same electrostatic hindrances, no aptasensor response was registered when anionic redox marker, equimolar mix of ferro-/ferricyanide, was used (even for substantially higher selenate ion concentration, 100 μM, compared to assays with methylene blue, 1-12 μM). As described above, electrostatic mechanism in attraction or repulsion of investigated redox marker could also be confirmed by increased reversibility of registered redox reaction after SeO42- ion binding by the receptor layer (ΔE changes from 54 to 38 mV), Figure 3.
Because of the registered aptasensor response for selenate ion in an assay with methylene blue as a redox marker, we evaluated the biosensor response for a given concentration range of selenate ion (Figure 4).
Although it was possible to observe the dependency between biosensor response and the SeO42- concentration in the sample (Figure 4), the obtained LOD at the level of 1 μM (linear range from 1 to 12 μM) was not satisfactory as the limit for selenium presence in water samples specified by WHO is at the level of 10 µg/L [28], what gives approximately 0.127 μM. As the specified WHO limit and the obtained LOD differs 10 times, an attempt was made to lower the detection limit. Based on the above results we attempted to move the overall receptor layer charge toward more positive, which should increase the efficiency in SeO42- anion approximation to the electrode surface. In the presented studies, it was realized by lowering pH of the measurement medium to pH 4.0. Such a change results in the protonation of the last element of the receptor layer, 6-mercapto-1-hexanol, which in higher pH values exhibits a partially negative charge (originating from –OH moieties) and is protonated near pH 5.0 [58]. As was already stated, the selenate ion is stable in the broad pH range [50] and retains its form, SeO42-. Single-stranded DNA, our aptamer used as a receptor, depurination takes place below pH 3.5, which allowed us to conduct experiments at pH 4.0 (Figure 5).
The change in the overall receptor layer charge (toward more positive values) could be observed already for the redox reaction of cationic methylene blue. As its concentration was not changed (50 μM) between assays conducted in pH 4.0 and 7.0, and the registered currents were moreover twice smaller than for pH 7.0, it can be concluded that its electrostatic attraction by receptor layer was of significantly lower efficiency. Also, methylene blue redox potential was moved toward more positive values, from ~ -0.25 V for pH 7.0 to ~ -0.025 V for pH 4.0, which additionally points to changes in its interactions with the receptor layer. However, what is significantly more important is the fact that the calculated biosensor response was high even for significantly lower selenate ion concentration than during measurements in pH 7.0. This, in turn, could originate from the fact that in these conditions the receptor layer negative charge originates only from the immobilized ssDNA aptamer strands, which approximates, at least theoretically, conditions where the aptamer sequences were selected in the depth of solution during the SELEX process. Nonetheless, the change in the pH value of the measurement environment resulted not only in the change of registered voltammograms (methylene blue redox reaction) but also in the obtained analytical parameters of the aptasensor toward analyte (SeO42-). Although we expected increased aptasensor response toward the analyzed ion, thus the obtained LOD (approximately 1 nM) was far below the above mentioned detection limits specified by WHO [28] (Figure 6A).
This made us evaluate the strength of the binding affinity between the obtained receptor layer with developed aptamer and the selenate ion by calculation of the dissociation constant (KD). Assuming a simple aptamer-analyte interaction at equilibrium according to the “one-to-one” kinetic model and good interaction stability, it was possible to determine numerically the equilibrium dissociation constant (KD) [59]. This parameter reflects the binding affinity of the target to the aptamer molecule. As can be seen in Figure 6D, the KD value at the level of 13.9 nM indicates the high ability of aptamer binding sites to form complexes with the detected analyte even at its low concentrations in the sample.
The calculated dissociation constant proofed the high affinity of the as prepared biosensing layers toward selenate ions in the used measurement conditions. This was further confirmed by the selectivity studies (Figure 7), where all analyzed ions were at the level of 100 nM.
As could be seen (Figure 7), the highest aptasensor response was obtained for selenate ion (-52.48 ± 6.00 %) followed by Cd2+ (-15.04 ± 6.80 %) and Fe3+ (-14.69 ± 0.63 %). The lowest biosensor responses were registered for Ni2+ (2.80 ± 4.14 %) and Sb- (3.50 ± 0.00 %). The clear diminish in the registered methylene blue reduction current after biosensor incubation in the sample (different biosensor response) was registered for UO22+ (20.41 ± 3.91 %). These could point out that strong binding in the receptor layer anions or cations changes the overall charge of such a layer. In the case of UO22+ cation, which forms coordination bonds with phosphates present in the DNA backbone, the charge becomes more positive which diminishes the electrostatic attraction of the positive redox marker, methylene blue, the same reducing the registered current. As we can see in Figure 7, a different biosensor response is observed for selenate anions. In this case, the current increases, which can suggest the increase in the negative charge deposited at the electrode surface (increased attraction of cationic redox marker, methylene blue) and increased efficiency in its redox reaction.
Both the presented selectivity and the low dissociation constant made us evaluate the developed biosensor response also for selenate ion in the real sample. This was tap water, as we initially followed by WHO regulations related to the selenate limits in drinking water (Figure 8).
The tap water was diluted 1:1 (v/v) with measurement solution (twice concentrated) and spiked with 100 nM SeO42-. As was previously mentioned, the limit specified by WHO for selenium concentration in drinking water is at the level of 0.01 mg/L (0.127 μM) [28]. As could be seen in Figure 8, the developed aptasensor response was only slightly higher for the spiked real sample (-57.19 ± 0.03 %) than for the laboratory sample (-52.48 ± 0.06 %).
Further studies were dedicated to (i) the evaluation of the possibility of biosensor regeneration and its subsequent use in selenate ion concentration determination (Figure 9A) and (ii) the evaluation of the biosensor stability during one week of storage in chosen conditions (Figure 9B). The first mentioned experiments were conducted by 20 minutes aptasensor incubation in one of the following solutions, 5 mM EDTA pH 7.5 (as a standard complexing agent which proved its applicability in multiple studies [51]), 1 M KH2PO4 pH 4.5 (used in the presented studies as solution for DNA immobilization), solution of 10 mM TRIS, 50 mM KCl and 20 mM MgCl2 with pH 7.0 (used in various studies as a typical DNA/DNA hybridization buffer [47]) and 0.5 M Na2CO3 pH 10.5 (with significantly changed pH and presence of CO32- ions which according to literature can compete with selenate ions during its removal with magnetite from granitic groundwater [60]).
From the analyzed condition (data not shown) the regeneration process took place only for sodium carbonate and the appropriate current change after subsequent selenate ion determination was obtained (Figure 9A). The investigations on biosensor storage were conducted in 1 M KH2PO4 pH 4.5, 5 mM EDTA pH 7.5, (used in the presented studies as solution for DNA immobilization), 0.5 M Na2CO3 pH 10.5 and water solution of 2 mM 6-mercapto-1-hexanol (MCH). From the analyzed conditions, only biosensor which was kept in 2 mM MCH allowed to obtain similar results for the determination of 50 nM selenate ion at asimilar level to the biosensor freshly prepared (Figure 9B).
Nonetheless, taking into account the obtained results, aptasensor response dependency to selenate ion concentration (Figure 6A) and to real sample (Figure 8), low dissociation constant of as prepared receptor layer (Figure 6B) (KD at the level of 13.9 nM) and obtained selectivity (Figure 7), we believe that there is the possibility of using described aptamer sequence in the biosensing layers construction toward SeO42- anion.
3. Materials and Methods
Reagents
2-Amino-2-(hydroxymethyl)-1,3-propanediol—Trizma® base (TRIS), 3-(N-morpholino)propanesulfonic acid (MOPS), sodium dihydrogen phosphate (NaH2PO4), potassium dihydrogen phosphate (KH2PO4), potassium chloride (KCl), sodium hydroxide (NaOH), 6-mercapto-1-hexanol (MCH), methylene blue (MB), lead(II) nitrate (Pb(NO3)2), cadmium chloride (CdCl2), copper(II) nitrate (Cu(NO3)2), iron(III) chloride (FeCl3), uranyl acetate (UO2(CH3COO)2), cobalt(II) sulfate hexahydrate (CoSO4·6H2O), potassium chromate (K2CrO4), manganese(II) chloride (MnCl2), nickel nitrate (Ni(NO3)2), sodium selenate (Na2SeO4), potassium antimony(III) tartrate hydrate (C8H4K2O12Sb2·H2O) were purchased from Merck (Germany). Sulfuric acid (H2SO4) and hydrogen peroxide (H2O2) were purchased from POCh (Poland). Potassium ferricyanide (K3[Fe(CN)6]) and potassium ferrocyanide (K4[Fe(CN)6]) were purchased from Fluka (Switzerland). All reagents were used without further purification. All solutions were prepared with Milli-Q water.
DNA aptamer specific towards selenate ions was identified through SELEX process by Basepair Biotechnologies (USA) with the use of VENNMultiplex™ technique dedicated to low molecular targets. In this SELEX process type aptamer candidates were hybridized to the complementary sequences immobilized on the surfaces of magnetic nanoparticles. After each round, aptamer candidates, which bound with the selenate ion were removed from the nanoparticles and stayed in the supernatant after the magnetic sample separation. Applies SELEX procedure included the use of VENNMultiplex™ for at least 12 rounds, which was followed by next generation sequencing and bioinformatics studies. Based on results, the 32 – nucleotide single-stranded DNA aptamer probe E1 (desalted, HPLC purified) specific towards selenate ions and additionally containing a disulfide group was purchased from Metabion, Germany. The sequence was as follows: 5′ - OH–C6–S–S–C6- TAT GAC ATT GTG ACG AAC TCC TCA CTA GAC CG. Aptamer stock solutions (100 μM) were prepared with a water-molecular biology reagent (Merck, Germany) and stored in a − 20 °C freezer before use.
Solutions
The solutions used in the presented experiments were as follows: 50 mM MOPS pH 7.0; 5 mM ferri/ferrocyanide in 50 mM MOPS, pH 7.0; 50 μM MB in 50 mM MOPS pH 7.0; 50 μM MB in 50 mM TRIS pH 4.0; twice concentrated measurement solution in studies conducted for real sample analysis (100 μM MB in 100 mM TRIS pH 4.0); 5 mM ferri/ferrocyanide in 0.1 M KCl; 1 M H2SO4, 0.1 M H2SO4, 1 M NaOH, piranha solution (H2SO4:H2O2 (3:1) (v/v)), 1 M KH2PO4, pH 4.5; water solution of 2 mM 6-mercapto-1-hexanol. If necessary, redox indicator solutions were spiked with appropriate salts.
Gold Electrodes Cleaning and Modification
The electrochemical measurements were carried out with a conventional three-electrode system consisting of a gold disk electrode (CH Instruments, USA), a gold wire as an auxiliary electrode (Sigma Aldrich, Germany) and Ag/AgCl/1 M KCl reference electrode (Mineral, Poland). The gold disc electrodes were prepared according to [47]. After the electrode cleaning, the given concentration (usually 4 μM) of thiolated DNA probe in immobilized buffer solution (1 M KH2PO4 (pH 4.5)) was deposited at the surface of previously cleaned electrodes and immobilized for a given period of time (e.g., 30 min). Then, the electrodes were washed in DI water and incubated for 2 h in 2 mM MCH. Next, electrodes were rinsed with DI water and were kept for 2 min. in 100 mM NaOH with 1 M NaCl, to remove from weakly bound DNA/MCH moieties before the electrochemical measurements.
Electrochemical Measurements
Voltammetric measurements were performed with CHI660A electrochemical workstation (CH Instruments, USA). The cyclic voltammetry (CV) was conducted at a sweep rate of 100 mV· s−1, while the square wave voltammetry (SWV) was recorded at a pulse amplitude of 25 mV, an increment of 4 mV and a frequency of 15 Hz. Depending on the pH of the measurement solution, cathodic or anodic scans were taken into account during qualitative and quantitative analysis. This was based on calculations made after each set of experiments. Before measurements electrodes were incubated in an analyzed measurement buffer for 10 min. Then, a series of SWV measurements were conducted to stabilize the initial receptor layer signal (I0).
4. Conclusions
Because of human activities, mostly related to natural environment degradation and changes in the global temperature values, the distribution of various pollutants or toxins in the environment changes. This may result in groundwater contamination, bioaccumulation of hazardous compound for human health in agricultural crops, animals consumed or just in the air we breathe. This, in turn, entails the need for elaboration of mobile and easy in use analytical devices, which could be operated by non-professionals directly in the place of need. One of such approach are electrochemical sensors and biosensors, which because of their dimensions, low cost and easiness of integration in more advanced microfluidic devices make such solutions possible. However, one of the most important part of such biosensors is the receptor layer and more precisely, the receptor, which could selectively bind the analyte of choice. The progress in new receptor development is directly connected to the progress in development of the abovementioned sensors and biosensors and ultimately, also to the mobile devices capable of precise analysis of the sample in the place of sampling. One of such receptors are aptamers, single- stranded DNA or RNA oligonucleotides selected during SELEX process. Herein we present the 32-nucleotide aptamer sequences, identified during the VENNMultiplex™ mode of systematic evolution of ligands by exponential enrichment. It was further modified with the thiol group at its 5′ end and applied for the first time in the receptor layer of electrochemical aptasensor toward selenate ion. During the presented studies, the measurement conditions were chosen especially the redox marker used and pH value of the measurement solution. This, however, makes it possible to elucidate, at least at the very beginning step, the mechanism of SeO42- ion interaction with the receptor layer and to obtain interesting analytical parameters of developed electrochemical aptasensor. Especially from the point of view of the level of detection, which was far below the limits described by WHO (10 μg/L) and selectivity. For elaborated receptor layer and measurement conditions the dissociation constant was calculated (13.93 ± 5.54 nM), which farther indicates the high ability of aptamer binding sites to form complexes with the detected analyte even at its low concentrations in the sample. It should be, however, emphasized that this is by far the first attempt to utilize aptamer-based layers for electrochemical detection of selenate ions, and we do not exclude the possibility that any further studies with this aptamer could lead to elaboration of even better analytical solutions which will offer significantly more interesting analytical parameters.
6. Patents
Authors have patent “DNA aptamer sequence, its use for selective detection of selenium ions, a method of manufacturing an electrochemical aptamer sensor and a method of measurement using it, and an electrochemical aptamer sensor containing an oligonucleotide DNA aptamer sequence”, P.448520 pending to Assignee.
Author Contributions
A.Sz.: writing—original draft preparation, writing—review and editing, methodology, investigation, validation; M.P.: investigation; D.B.: investigation; M.O.: supervision, funding acquisition, writing—review & editing; R.Z.: writing—original draft preparation, writing—review and editing, conceptualization, methodology, formal analysis, visualization; E.M.: supervision, writing—review and editing.
Funding
This work was financially supported by the Warsaw University of Technology and The National Centre for Research and Development under the III program TECHMATSTRATEG—Strategic research and development program “Modern material technologies—TECHMATSTRATEG” no. TECHMATSTRATEG-III/0042/2019-00 and acronym ASTACUS, “Biopolymer materials with chemically and genetically programmed heavy metals selectivity for new generation of ultra-sensitive biosensors”.
Data Availability Statement
Data will be available on request.
Acknowledgments
We would like to express our gratitude to PhD Marcin Drozd for his valuable contribution to dissociation constant (KD) calculation.
Conflicts of Interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Marcin Olszewski reports financial support was provided by The National Centre for Research and Development. Robert Ziółkowski and Marcin Olszewski have patent “DNA aptamer sequence, its use for selective detection of selenium ions, a method of manufacturing an electrochemical aptamer sensor and a method of measurement using it, and an electrochemical aptamer sensor containing an oligonucleotide DNA aptamer sequence”, P.448520 pending to Assignee. No other Authors declare that they know competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Somogyi, Z.; Kádár, I.; Kiss, I.; Juríková, T.; Szekeres, L.; Balla, Š.; Nagy, P.; Bakonyi, G. Comparative toxicity of the selenate and selenite to the potworm Enchytraeus albidus (Annelida: Enchytraeidae) under laboratory conditions. Eur. J. Soil Biol. 2012, 50, 159–164. [Google Scholar] [CrossRef]
- Wake, B.D.; Bowie, A.R.; Butler, E.C.V.; Haddad, P.R. Modern preconcentration methods for the determination of selenium species in environmental water samples. TrAC Trends Anal. Chem. 2004, 23, 491–500. [Google Scholar] [CrossRef]
- Bleiman, N.; Mishael, Y.G. Selenium removal from drinking water by adsorption to chitosan–clay composites and oxides: Batch and columns tests. J. Hazard. Mater. 2010, 183, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Ečimović, S.; Velki, M.; Vuković, R.; Štolfa Čamagajevac, I.; Petek, A.; Bošnjaković, R.; Grgić, M.; Engelmann, P.; Bodó, K.; Filipović-Marijić, V.; et al. Acute toxicity of selenate and selenite and their impacts on oxidative status, efflux pump activity, cellular and genetic parameters in earthworm Eisenia andrei. Chemosphere 2018, 212, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Palmer, I.S.; Olson, O.E. Relative Toxicities of Selenite and Selenate in the Drinking Water of Rats. J. Nutr. 1974, 104, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Wallschläger, D.; Roehl, R. Determination of inorganic selenium speciation in waters by ion chromatography-inductively coupled plasma-mass spectrometry using eluant elimination with a membrane suppressor. J. Anal. At. Spectrom. 2001, 16, 922–925. [Google Scholar] [CrossRef]
- Jackson, B.P.; Miller, W.P. Soluble Arsenic and Selenium Species in Fly Ash/Organic Waste-Amended Soils Using Ion Chromatography−Inductively Coupled Plasma Mass Spectrometry. Environ. Sci. Technol. 1999, 33, 270–275. [Google Scholar] [CrossRef]
- Yang, R.; Li, Q.; Zhou, W.; Yu, S.; Liu, J. Speciation Analysis of Selenium Nanoparticles and Inorganic Selenium Species by Dual-Cloud Point Extraction and ICP-MS Determination. Anal. Chem. 2022, 94, 16328–16336. [Google Scholar] [CrossRef] [PubMed]
- Panhwar, A.H.; Tuzen, M.; Kazi, T.G. Ultrasonic assisted dispersive liquid-liquid microextraction method based on deep eutectic solvent for speciation, preconcentration and determination of selenium species (IV) and (VI) in water and food samples. Talanta 2017, 175, 352–358. [Google Scholar] [CrossRef]
- Goldberg, S.; Martens, D.A.; Forster, H.S.; Herbel, M.J. Speciation of Selenium(IV) and Selenium(VI) using Coupled Ion Chromatography—Hydride Generation Atomic Absorption Spectrometry. Soil Sci. Soc. Am. J. 2006, 70, 41–47. [Google Scholar] [CrossRef]
- Huang, P.M.; Fujii, R. Selenium and Arsenic. 2018; pp. 793–831. [Google Scholar] [CrossRef]
- Finley, J.W.; Sigrid-Keck, A.; Robbins, R.J.; Hintze, K.J. Selenium Enrichment of Broccoli: Interactions between Selenium and Secondary Plant Compounds. J. Nutr. 2005, 135, 1236–1238. [Google Scholar] [CrossRef]
- Lavi, N.; Alfassi, Z.B. Determination of trace amounts of cadmium, cobalt, chromium, iron, molybdenum, nickel, selenium, titanium, vanadium and zinc in blood and milk by neutron activation analysis. Analyst 1990, 115, 817. [Google Scholar] [CrossRef] [PubMed]
- Utterback, P.L.; Parsons, C.M.; Yoon, I.; Butler, J. Effect of supplementing selenium yeast in diets of laying hens on egg selenium content. Poult. Sci. 2005, 84, 1900–1901. [Google Scholar] [CrossRef]
- Ando, M.; Takizawa, M.; Suwabe, S.; Yamato, S.; Shimada, K. Determination of Selenium in Human Serum by Liquid Chromatography/Electron Capture Atmospheric Pressure Chemical Ionization Mass Spectrometry after Acid Digestion and Derivatization Using 2,3-Diaminonaphthalene. Eur. J. Mass Spectrom. 2003, 9, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Kalantari, H.; Manoochehri, M. A nanocomposite consisting of MIL-101(Cr) and functionalized magnetite nanoparticles for extraction and determination of selenium(IV) and selenium(VI). Microchim. Acta 2018, 185, 196. [Google Scholar] [CrossRef] [PubMed]
- Karaś, K.; Zioła-Frankowska, A.; Frankowski, M. New Method for Simultaneous Arsenic and Selenium Speciation Analysis in Seafood and Onion Samples. Molecules 2021, 26, 6223. [Google Scholar] [CrossRef]
- Zhao, Y.; Zheng, J.; Yang, M.; Yang, G.; Wu, Y.; Fu, F. Speciation analysis of selenium in rice samples by using capillary electrophoresis-inductively coupled plasma mass spectrometry. Talanta 2011, 84, 983–988. [Google Scholar] [CrossRef]
- Rubinskaya, T.B.; Kovaleva, S.V.; Kulagin, E.M.; Gladyshev, V.P. Determination of Selenium(IV) by Stripping Voltammetry at a Mercury-Film Electrode. J. Anal. Chem. 2003, 58, 165–170. [Google Scholar] [CrossRef]
- Ivandini, T.A.; Einaga, Y. Electrochemical Detection of Selenium (IV) and (VI) at Gold-Modified Diamond Electrodes. Electrocatalysis 2013, 4, 367–374. [Google Scholar] [CrossRef]
- Beni, V.; Collins, G.; Arrigan, D.W.M. Investigation into the voltammetric behaviour and detection of selenium(IV) at metal electrodes in diverse electrolyte media. Anal. Chim. Acta 2011, 699, 127–133. [Google Scholar] [CrossRef]
- Andrews, R.W.; Johnson, D.C. Voltammetric deposition and stripping of selenium(IV) at a rotating gold-disk electrode in 0.1M perchloric acid. Anal. Chem. 1975, 47, 294–299. [Google Scholar] [CrossRef]
- Fakude, C.T.; Arotiba, O.A.; Moutloali, R.; Mabuba, N. Nitrogen-doped Graphene Electrochemical Sensor for Selenium (IV) in Water. Int. J. Electrochem. Sci. 2019, 14, 9391–9403. [Google Scholar] [CrossRef]
- Idris, A.O.; Mabuba, N.; Nkosi, D.; Maxakato, N.; Arotiba, O.A. Electrochemical detection of selenium using glassy carbon electrode modified with reduced graphene oxide. Int. J. Environ. Anal. Chem. 2017, 97, 534–547. [Google Scholar] [CrossRef]
- Jain, R.; Thakur, A.; Kumar, P.; Pooja, D. Au/ZnO nanocomposites decorated ITO electrodes for voltammetric sensing of selenium in water. Electrochim. Acta 2018, 290, 291–302. [Google Scholar] [CrossRef]
- Ali, A.G.; Altahan, M.F.; Beltagi, A.M.; Hathoot, A.A.; Abdel-Azzem, M. Voltammetric and impedimetric determinations of seleniumiv by an innovative gold-free poly(1-aminoanthraquinone)/multiwall carbon nanotube-modified carbon paste electrode. RSC Adv. 2022, 12, 4988–5000. [Google Scholar] [CrossRef]
- John, N.; Abraham, K.E. Detection of Selenium and Nickel Metal Ion in Water Using Mn3O4-Cn-Modified Electrode. Int. J. Electrochem. 2021, 2021, 1–9. [Google Scholar] [CrossRef]
- Guidelines for drinking-water quality: Fourth edition incorporating the first and second addenda [Internet]; World Health Organization: Geneva, 2022; ISBN 9789240045064.
- Motlagh, M.K.; Noroozifar, M.; Kraatz, H.-B. Highly sensitive and selective detection of selenate in water samples using an enzymatic gold nanodendrite biosensor. Can. J. Chem. 2023, 101, 440–448. [Google Scholar] [CrossRef]
- Motlagh, M.K.; Noroozifar, M.; Sodhi, R.N.S.; Kraatz, H. Development of a Bacterial Enzyme-Based Biosensor for the Detection and Quantification of Selenate. Chem. – A Eur. J. 2022, 28. [Google Scholar] [CrossRef]
- Kolliopoulos, A.V.; Metters, J.P.; Banks, C.E. Electroanalytical sensing of selenium(iv) utilising screen printed graphite macro electrodes. Anal. Methods 2013, 5, 851. [Google Scholar] [CrossRef]
- Ruscito, A.; DeRosa, M.C. Small-Molecule Binding Aptamers: Selection Strategies, Characterization, and Applications. Front. Chem. 2016, 4. [Google Scholar] [CrossRef]
- Carothers, J.M.; Goler, J.A.; Kapoor, Y.; Lara, L.; Keasling, J.D. Selecting RNA aptamers for synthetic biology: investigating magnesium dependence and predicting binding affinity. Nucleic Acids Res. 2010, 38, 2736–2747. [Google Scholar] [CrossRef]
- Ziółkowski, R.; Górski, Ł. Electrochemical metal sensors with DNA receptor layers. Curr. Anal. Chem. 2014, 10, 600–608. [Google Scholar] [CrossRef]
- Jarczewska, M.; Szymczyk, A.; Zajda, J.; Olszewski, M.; Ziółkowski, R.; Malinowska, E. Recent Achievements in Electrochemical and Optical Nucleic Acids Based Detection of Metal Ions. Molecules 2022, 27, 7481. [Google Scholar] [CrossRef]
- Guo, W.; Zhang, C.; Ma, T.; Liu, X.; Chen, Z.; Li, S.; Deng, Y. Advances in aptamer screening and aptasensors’ detection of heavy metal ions. J. Nanobiotechnology 2021, 19, 166. [Google Scholar] [CrossRef]
- Bhattacharyya, D.; Mirihana Arachchilage, G.; Basu, S. Metal Cations in G-Quadruplex Folding and Stability. Front. Chem. 2016, 4. [Google Scholar] [CrossRef]
- Wang, Y.; Luan, B.-Q.; Yang, Z.; Zhang, X.; Ritzo, B.; Gates, K.; Gu, L.-Q. Single Molecule Investigation of Ag+ Interactions with Single Cytosine-, Methylcytosine- and Hydroxymethylcytosine-Cytosine Mismatches in a Nanopore. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef]
- Hossain, Z.; Huq, F. Studies on the interaction between Cd2+ ions and DNA. J. Inorg. Biochem. 2002, 90. [Google Scholar] [CrossRef]
- Rossberg, A.; Abe, T.; Okuwaki, K.; Barkleit, A.; Fukuzawa, K.; Nakano, T.; Mochizuki, Y.; Tsushima, S. Destabilization of DNA through interstrand crosslinking by UO22+. Chem. Commun. 2019, 55. [Google Scholar] [CrossRef]
- Torigoe, H.; Miyakawa, Y.; Ono, A.; Kozasa, T. Positive cooperativity of the specific binding between Hg2+ ion and T:T mismatched base pairs in duplex DNA. Thermochim. Acta 2012, 532. [Google Scholar] [CrossRef]
- Kohlberger, M.; Gadermaier, G. SELEX: Critical factors and optimization strategies for successful aptamer selection. Biotechnol. Appl. Biochem. 2022, 69, 1771–1792. [Google Scholar] [CrossRef]
- Edsall, J.T. The size and shape of protein molecules. In Fortschritte der Chemischen Forschung Volume 1; Springer Berlin Heidelberg: Berlin, Heidelberg; pp. 119–174. [CrossRef]
- Rahm, M.; Hoffmann, R.; Ashcroft, N.W. Atomic and Ionic Radii of Elements 1 –96. Chem. – A Eur. J. 2016, 22, 14625–14632. [Google Scholar] [CrossRef]
- https://basepairbio.com/multiplex-selex-venn-multiplex-selex/.
- Sankar, K.; Kuzmanović, U.; Schaus, S.E.; Galagan, J.E.; Grinstaff, M.W. Strategy, Design, and Fabrication of Electrochemical Biosensors: A Tutorial. ACS Sensors 2024. [Google Scholar] [CrossRef]
- Szymczyk, A.; Soliwodzka, K.; Moskal, M.; Różanowski, K.; Ziółkowski, R. Further insight into the possible influence of electrode blocking agents on the stem-loop based electrochemical DNA sensor parameters. Sensors Actuators B Chem. 2022, 354, 131086. [Google Scholar] [CrossRef]
- Wong, K.L.; Liu, J. Factors and methods to modulate DNA hybridization kinetics. Biotechnol. J. 2021, 16. [Google Scholar] [CrossRef]
- Krishnamurthy, R. Role of pK a of Nucleobases in the Origins of Chemical Evolution. Acc. Chem. Res. 2012, 45, 2035–2044. [Google Scholar] [CrossRef]
- Lichtfouse, E.; Morin-Crini, N.; Bradu, C.; Boussouga, Y.-A.; Aliaskari, M.; Schäfer, A.I.; Das, S.; Wilson, L.D.; Ike, M.; Inoue, D.; et al. Methods for selenium removal from contaminated waters: a review. Environ. Chem. Lett. 2022, 20, 2019–2041. [Google Scholar] [CrossRef]
- Szymczyk, A.; Popiołek, M.; Krzemiński, J.; Olszewski, M.; Ziółkowski, R.; Malinowska, E. Identification of medium- and mechanism-related pitfalls towards improved performance and applicability of electrochemical mercury(II) aptasensors. Microchim. Acta 2024, 191, 189. [Google Scholar] [CrossRef]
- Ferreira, C.M.H.; Pinto, I.S.S.; Soares, E.V.; Soares, H.M.V.M. (Un)suitability of the use of pH buffers in biological, biochemical and environmental studies and their interaction with metal ions—A review. RSC Adv. 2015, 5, 30989–31003. [Google Scholar] [CrossRef]
- Powell, K.J.; Brown, P.L.; Byrne, R.H.; Gajda, T.; Hefter, G.; Sjöberg, S.; Wanner, H. Chemical speciation of environmentally significant heavy metals with inorganic ligands. Part 1: The Hg2+ Cl−, OH−, CO32−, SO42−, and PO43− aqueous systems (IUPAC Technical Report). Pure Appl. Chem. 2005, 77. [Google Scholar] [CrossRef]
- Zhang, X.; Servos, M.R.; Liu, J. Surface Science of DNA Adsorption onto Citrate-Capped Gold Nanoparticles. Langmuir 2012, 28, 3896–3902. [Google Scholar] [CrossRef]
- Ziółkowski, R.; Górski, Ł.; Oszwałdowski, S.; Malinowska, E. Electrochemical uranyl biosensor with DNA oligonucleotides as receptor layer. Anal. Bioanal. Chem. 2012, 402, 2259–2266. [Google Scholar] [CrossRef] [PubMed]
- Jarczewska, M.; Kierzkowska, E.; Ziółkowski, R.; Górski, T.; Malinowska, E. Electrochemical oligonucleotide-based biosensor for the determination of lead ion. Bioelectrochemistry 2015, 101, 35–41. [Google Scholar] [CrossRef]
- Ziółkowski, R.; Uścińska, A.; Mazurkiewicz-Pawlicka, M.; Małolepszy, A.; Malinowska, E. Directly-thiolated graphene based electrochemical sensor for Hg(II) ion. Electrochim. Acta 2019, 305, 329–337. [Google Scholar] [CrossRef]
- Cristiano, E.; Hu, Y.-J.; Sigfried, M.; Kaplan, D.; Nitsche, H. A Comparison of Point of Zero Charge Measurement Methodology. Clays Clay Miner. 2011, 59, 107–115. [Google Scholar] [CrossRef]
- Thevendran, R.; Navien, T.N.; Meng, X.; Wen, K.; Lin, Q.; Sarah, S.; Tang, T.-H.; Citartan, M. Mathematical approaches in estimating aptamer-target binding affinity. Anal. Biochem. 2020, 600. [Google Scholar] [CrossRef]
- Kim, S.S.; Min, J.H.; Lee, J.K.; Baik, M.H.; Choi, J.-W.; Shin, H.S. Effects of pH and anions on the sorption of selenium ions onto magnetite. J. Environ. Radioact. 2012, 104, 1–6. [Google Scholar] [CrossRef]
Figure 1.
Schematic illustration of the biosensor response mechanism and the equation used for biosensor response calculation.
Figure 1.
Schematic illustration of the biosensor response mechanism and the equation used for biosensor response calculation.
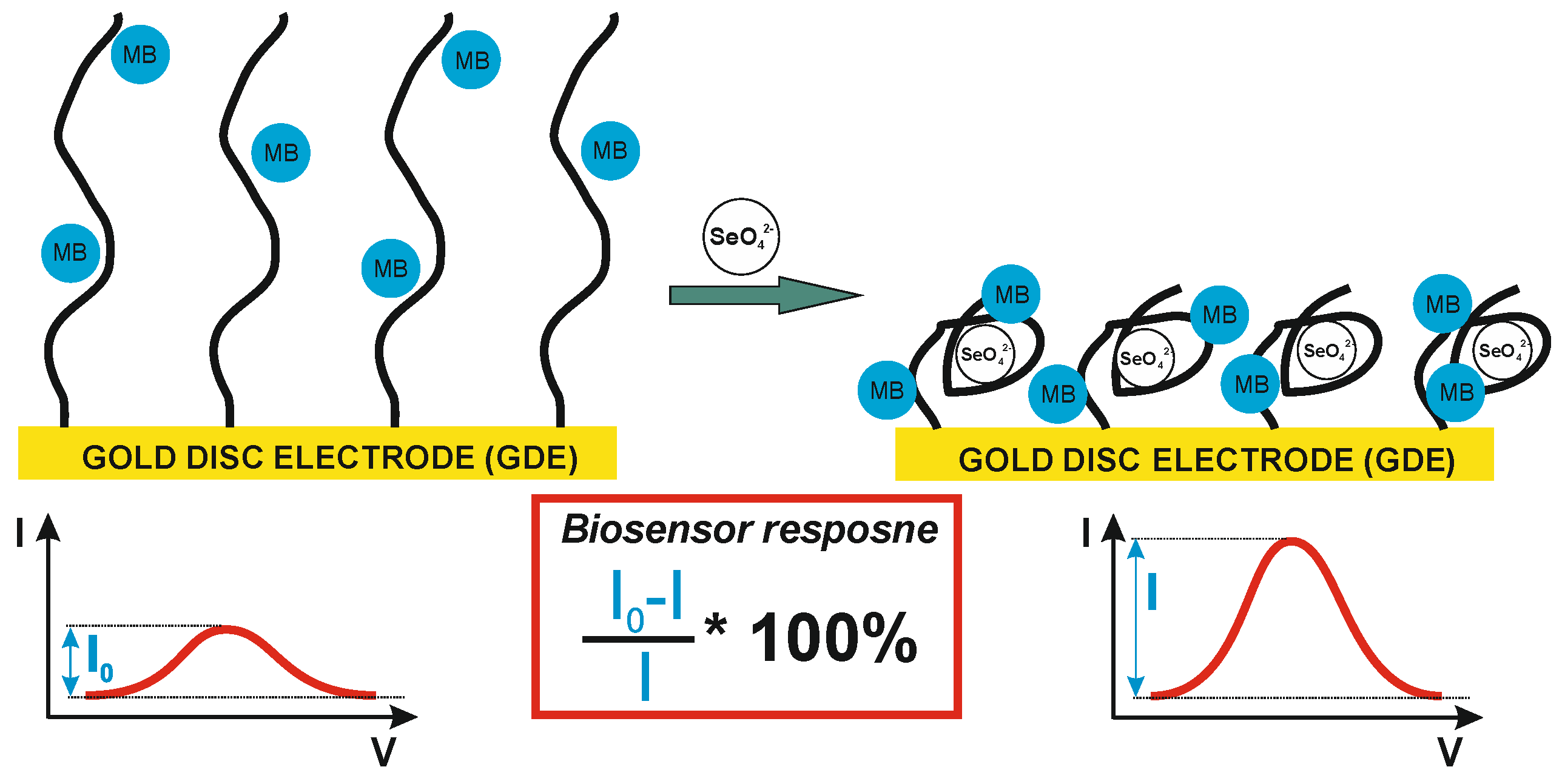
Figure 2.
Example of SWV voltammograms: A)-C) - biosensor response registered with methylene blue as a redox marker with increasing selenate ion concentration (from 1 to 12 μM) and D) - biosensor response registered with ferro-/ferricyanide as a redox marker with selenate ion concentration of 100 μM.
Figure 2.
Example of SWV voltammograms: A)-C) - biosensor response registered with methylene blue as a redox marker with increasing selenate ion concentration (from 1 to 12 μM) and D) - biosensor response registered with ferro-/ferricyanide as a redox marker with selenate ion concentration of 100 μM.
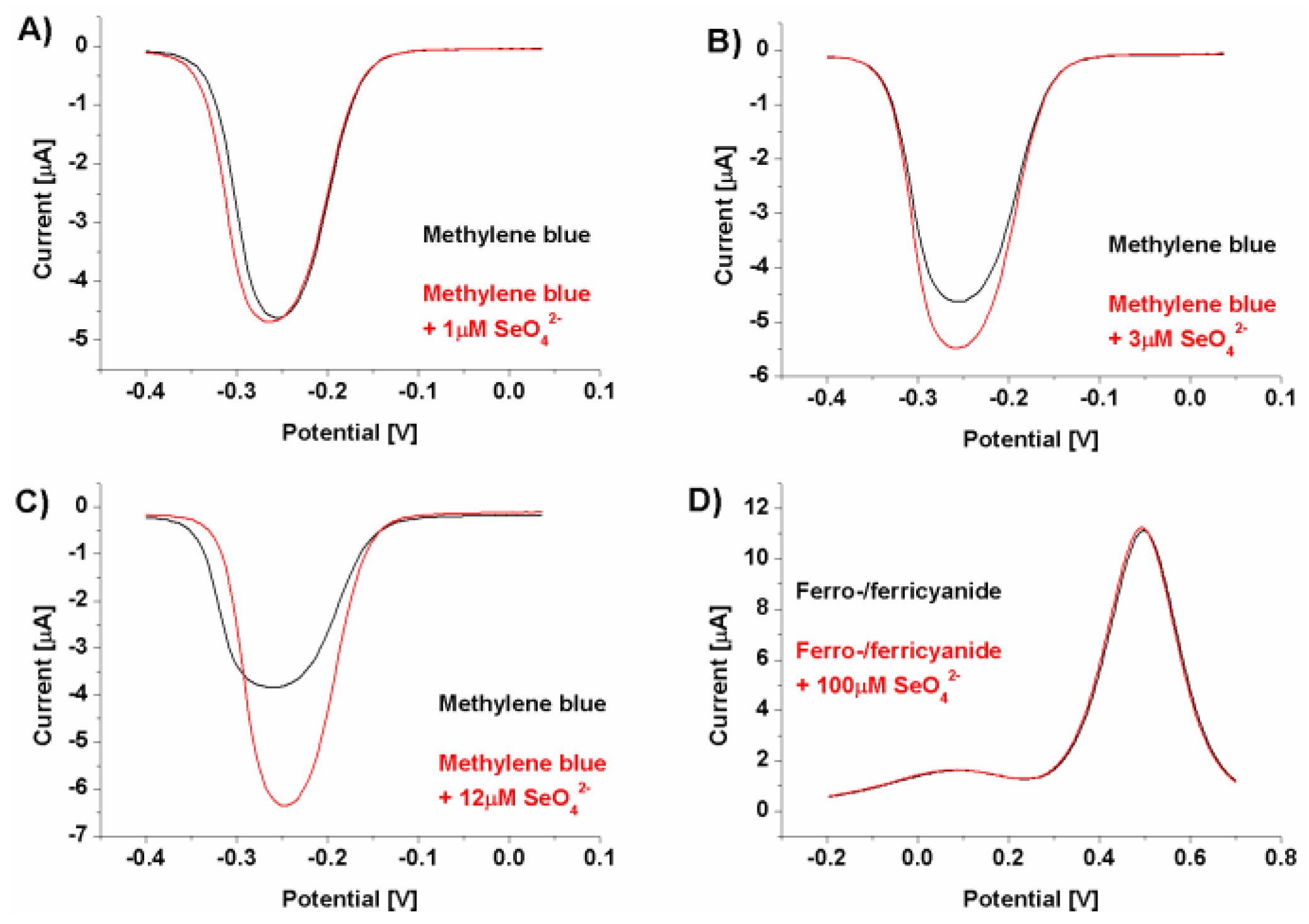
Figure 3.
Example of CV voltammogram for biosensor response registered with methylene blue as a redox marker with selenate ion concentration of 12 μM.
Figure 3.
Example of CV voltammogram for biosensor response registered with methylene blue as a redox marker with selenate ion concentration of 12 μM.
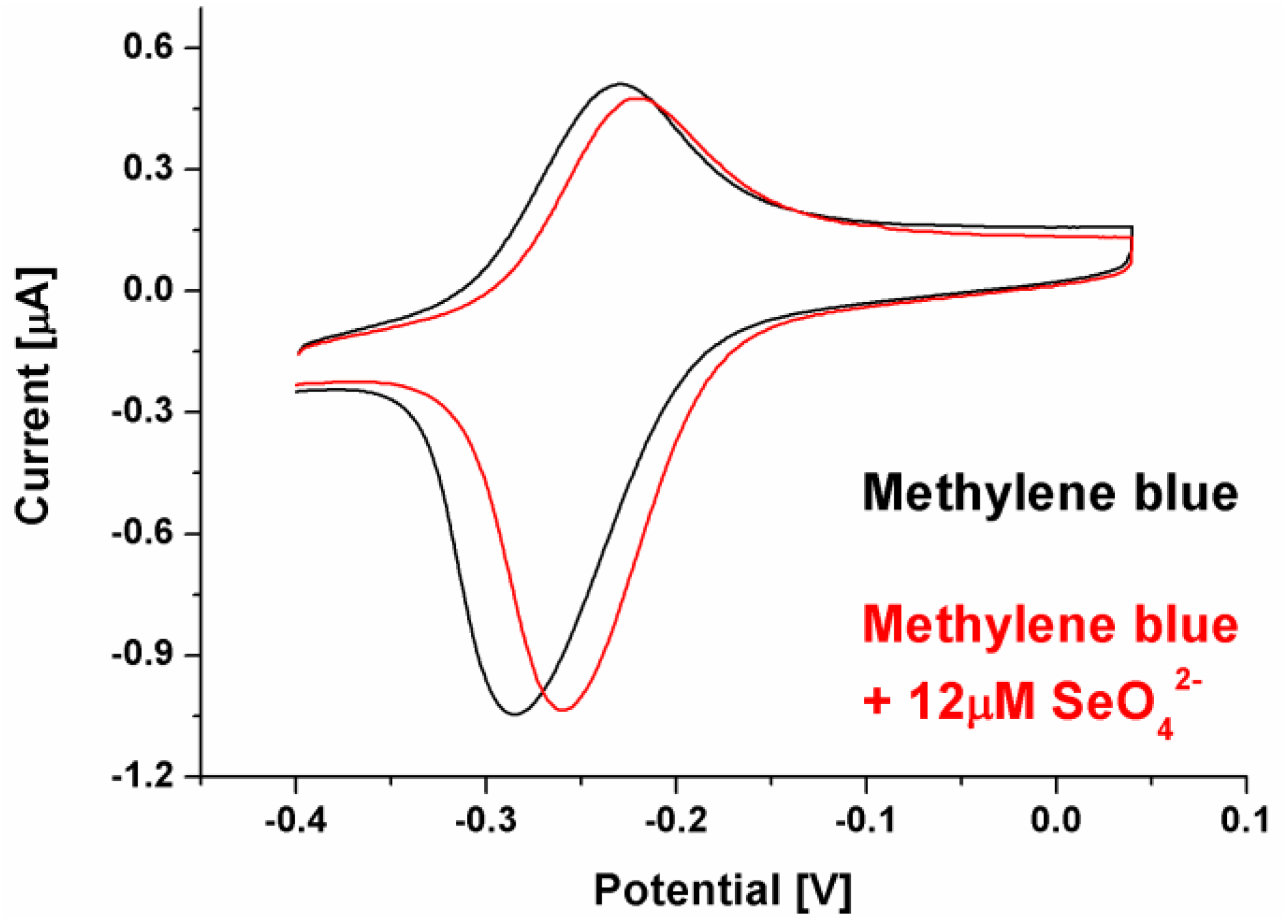
Figure 4.
Dependency between selenate ion concentration and calculated biosensor response (result obtained for SWV experiments, n = 4).
Figure 4.
Dependency between selenate ion concentration and calculated biosensor response (result obtained for SWV experiments, n = 4).
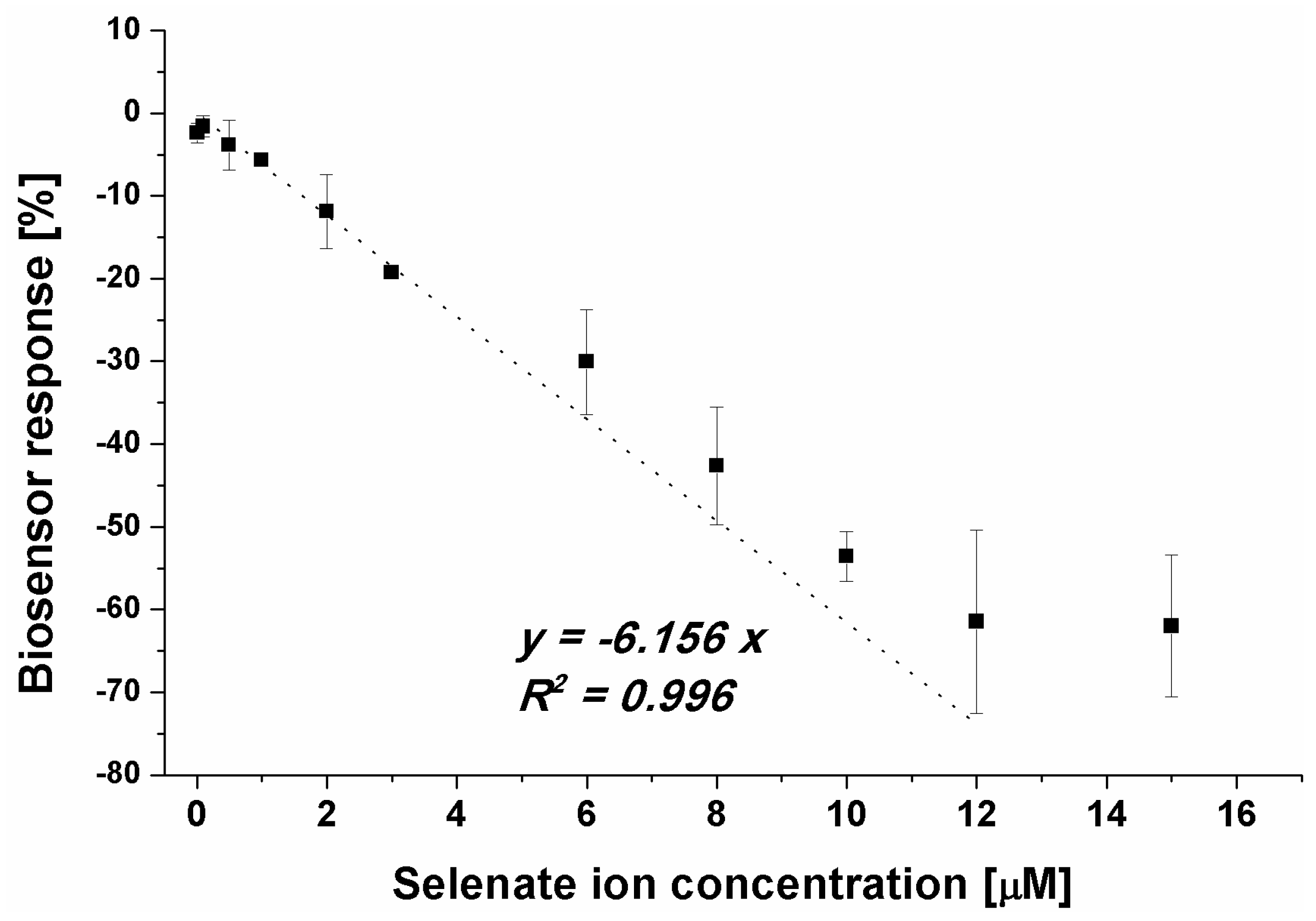
Figure 5.
Example of SWV voltammograms for biosensor response registered with methylene blue (pH 4.0) as a redox marker and for selenate ion concentration at the level of A) 1 nM and B) 50 nM.
Figure 5.
Example of SWV voltammograms for biosensor response registered with methylene blue (pH 4.0) as a redox marker and for selenate ion concentration at the level of A) 1 nM and B) 50 nM.
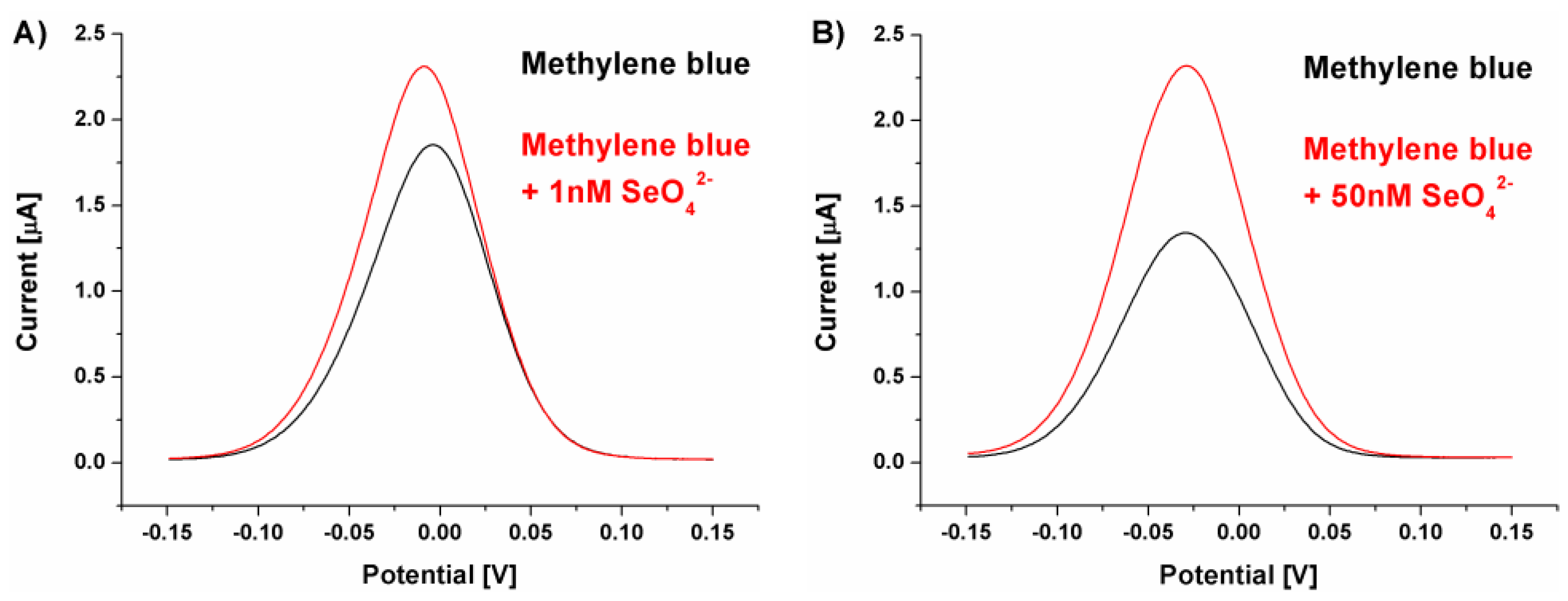
Figure 6.
A) Dependency between selenate ion concentration and calculated biosensor response (result obtained for SWV experiments, n = 4); B) plot of the absolute aptasensor electrochemical responses (transformed to positive values versus calculated with the equation presented in Figure 1) in the function of analyte concentration for the determination of equilibrium dissociation constant (KD). Black points represent experimental points, and red curve represents fitting through non-linear regression according to the “one-to-one” kinetic model.
Figure 6.
A) Dependency between selenate ion concentration and calculated biosensor response (result obtained for SWV experiments, n = 4); B) plot of the absolute aptasensor electrochemical responses (transformed to positive values versus calculated with the equation presented in Figure 1) in the function of analyte concentration for the determination of equilibrium dissociation constant (KD). Black points represent experimental points, and red curve represents fitting through non-linear regression according to the “one-to-one” kinetic model.
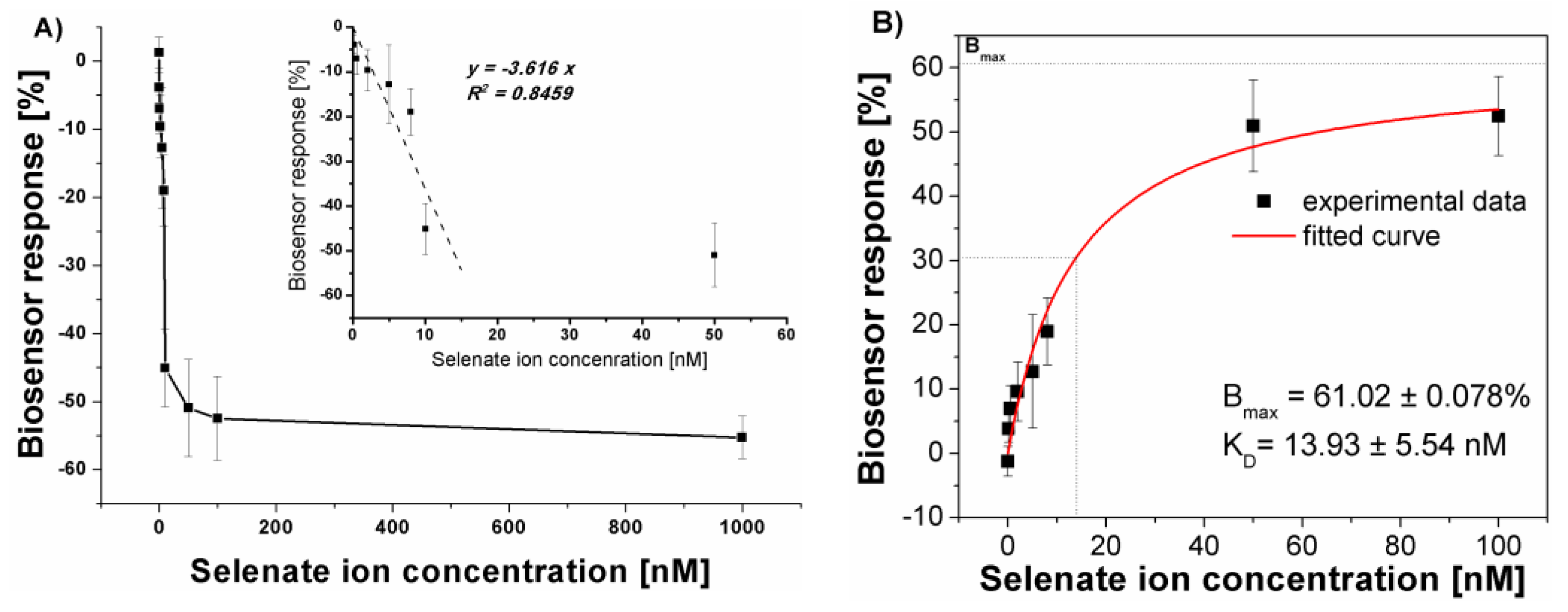
Figure 7.
Selectivity studies of as prepared electrochemical aptasensor. All ions were at the level of 100 nM and the sensor incubation in the sample was conducted for 30 minutes (n = 4).
Figure 7.
Selectivity studies of as prepared electrochemical aptasensor. All ions were at the level of 100 nM and the sensor incubation in the sample was conducted for 30 minutes (n = 4).
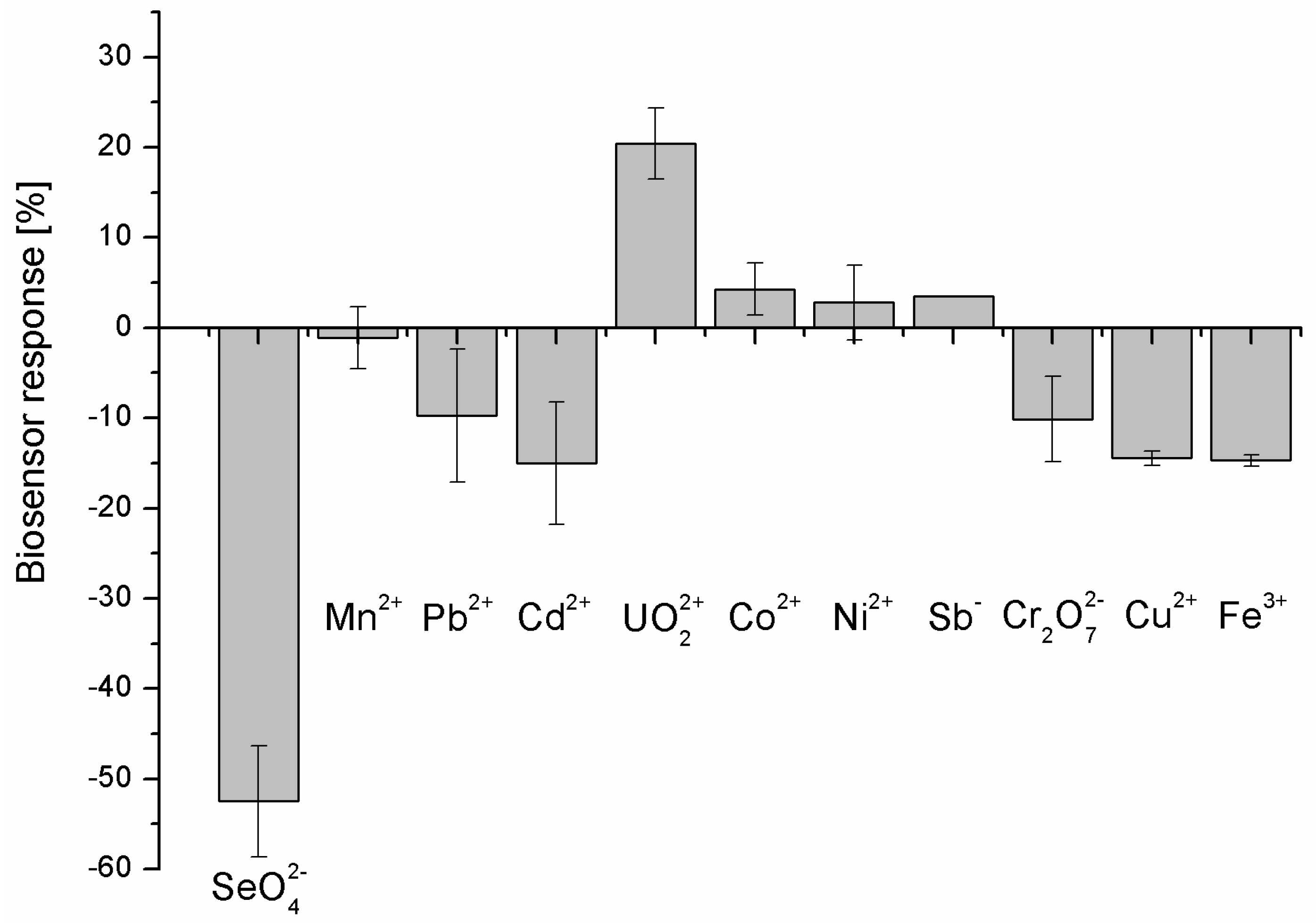
Figure 8.
Real sample analysis with the use of prepared biosensor. The selenate ion concentration was 100 nM (n = 4).
Figure 8.
Real sample analysis with the use of prepared biosensor. The selenate ion concentration was 100 nM (n = 4).
Figure 9.
A) Aptasensor regeneration studies: black line – before selenate ion detection, red line – after 30 minutes electrode incubation in selenate ion solution (100 nM); blue line – after 20 minutes of aptasensor incubation in 0.5 M Na2CO3 pH 10.5 (regeneration); green line – after 30 minutes electrode incubation in selenate ion solution (after regeneration process); B) aptasensor response (50 nM selenate ion concentration) after one week storage in the specific conditions (blue bar – response obtained with freshly prepared biosensor).
Figure 9.
A) Aptasensor regeneration studies: black line – before selenate ion detection, red line – after 30 minutes electrode incubation in selenate ion solution (100 nM); blue line – after 20 minutes of aptasensor incubation in 0.5 M Na2CO3 pH 10.5 (regeneration); green line – after 30 minutes electrode incubation in selenate ion solution (after regeneration process); B) aptasensor response (50 nM selenate ion concentration) after one week storage in the specific conditions (blue bar – response obtained with freshly prepared biosensor).

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



