Submitted:
17 May 2024
Posted:
17 May 2024
You are already at the latest version
Abstract
Prochlorococcus, a cyanobacteria genus of the smallest and most abundant oceanic phototrophs, encompasses ecotype strains adapted to high-light (HL) and low-light (LL) niches. To elucidate the adaptive evolution of this genus, we analyzed 40 Prochlorococcus marinus ORFeomes, including two cornerstone strains, MED4 and NATL1A. Employing deep learning with robust statistical methods, we detected new protein family distributions in the strains and identified key genes differentiating HL and LL strains. HL strains harbor genes (ABC-2 transporters) related to stress resistance, such as DNA repair and RNA processing, while LL strains exhibit unique chlorophyll adaptations (ion transport proteins, HEAT repeats). We report variable, depth-dependent endogenous viral elements in the 40 strains. We constructed the ORFeomes of MED4 and NATL1A covering 99 % of the annotated protein-coding sequences of the two species, totaling 3976 cloned, sequence-verified ORFs. These comparative genomics analyses, paired with MED4 and NATL1A ORFeomes, will facilitate future genotype-to-phenotype mappings and systems biology exploration of Prochlorococcus ecology.
Keywords:
Prochlorococcus
; comparative genomics
; deep learning
; artificial neural networks
; ORFeomes
Introduction
Cyanobacteria are major primary producers of aquatic environments[1,2]. They are model organisms for studying photosynthesis, carbon and nitrogen assimilation, evolution, adaptation to environmental conditions, and specialized biotechnology application[3]. Among cyanobacteria, Prochlorococcus and Synechococcus are the dominant primary producers in marine ecosystems and fix massive quantities of atmospheric carbon[4,5].
Species belonging to Prochlorococcus are genus are adapted to thrive in oceanic conditions with high oxygen and low nutrient levels[6]. Members of this genus are adapted to radiation and can grow at a broad depth in the ocean, with a primary distinction between two major classifications for high-light and low-light adaptive strains[7]. Gene gain and loss frequently occur in P. marinus[8]. Comparing the genomes of 12 isolated strains of P. marinus, a set of 1273 shared genes were identified as a core, conserved gene set. These genes underlie processes essential for P. marinus in any environment, while the remaining genes likely play roles in niche adaptation[9]. Fifty different P. marinus strains have been isolated and sequenced[10], with the strains (SS120, MED4)[11], and MIT9313[12] being the first to be sequenced in 2003. Current genome annotations for P. marinus provide a detailed description of genes, proteins, sequences, and functions, providing a base dataset for bioinformatics analyses; however, most predicted gene products from currently available genome annotations remain experimentally uncharacterized. Thus, a lack of resources limits experimental explorations of genotype-to-phenotype associations.
Here, we describe comparative analyses of P. marinus genomes and their protein family domains (Pfams) to interpret their adaptation to high-light (HL) and low-light (LL) conditions. Our approach involves dimensional reductions to detect clusters of differential gene content among the strains and using robust statistical methods and artificial neural networks to identify sets of key Pfams that can distinguish features of HL and LL strains. Using this approach to explore P. marinus adaptation can provide insights into aquatic ecology, carbon, and climate impacts. Furthermore, we report the construction of nearly complete MED4 and NATL1A ORFeomes through de novo chemical DNA synthesis, generating the first set of available cyanobacteria ORFeome resources. The synthesized ORFs were sequence-verified, and selected ORFs were tested in recombinational cloning into expression vectors available for systematic experimental assessment. Together, the computational and biological resources generated in this work will substantially advance cyanobacterial and variable depth studies and support for the multifaceted ecological and evolutionary fields of research using cyanobacteria as model organisms.
Results
Decoding High-Light and Low-Light Associated Gene Sets from P. marinus Genomes
Prochlorococcus species inhabiting different ocean depths deal with drastically different temperatures and light and nutrient availability[6]. Additionally, their microbial cohorts, including grazers, viruses, and other planktonic species, drastically differ with depth[13]. Understanding the genomic basis for depth and light adaption in Prochlorococcus can provide valuable insight into plant stress tolerance, marine ecology, and global biogeochemistry. Genomic differences between strains inhabiting different depths were hypothesized to potentially include variations in genes for photosynthesis, nutrient acquisition, and abiotic and biotic stress responses, which reflect their adaptation to specific environmental conditions[14].
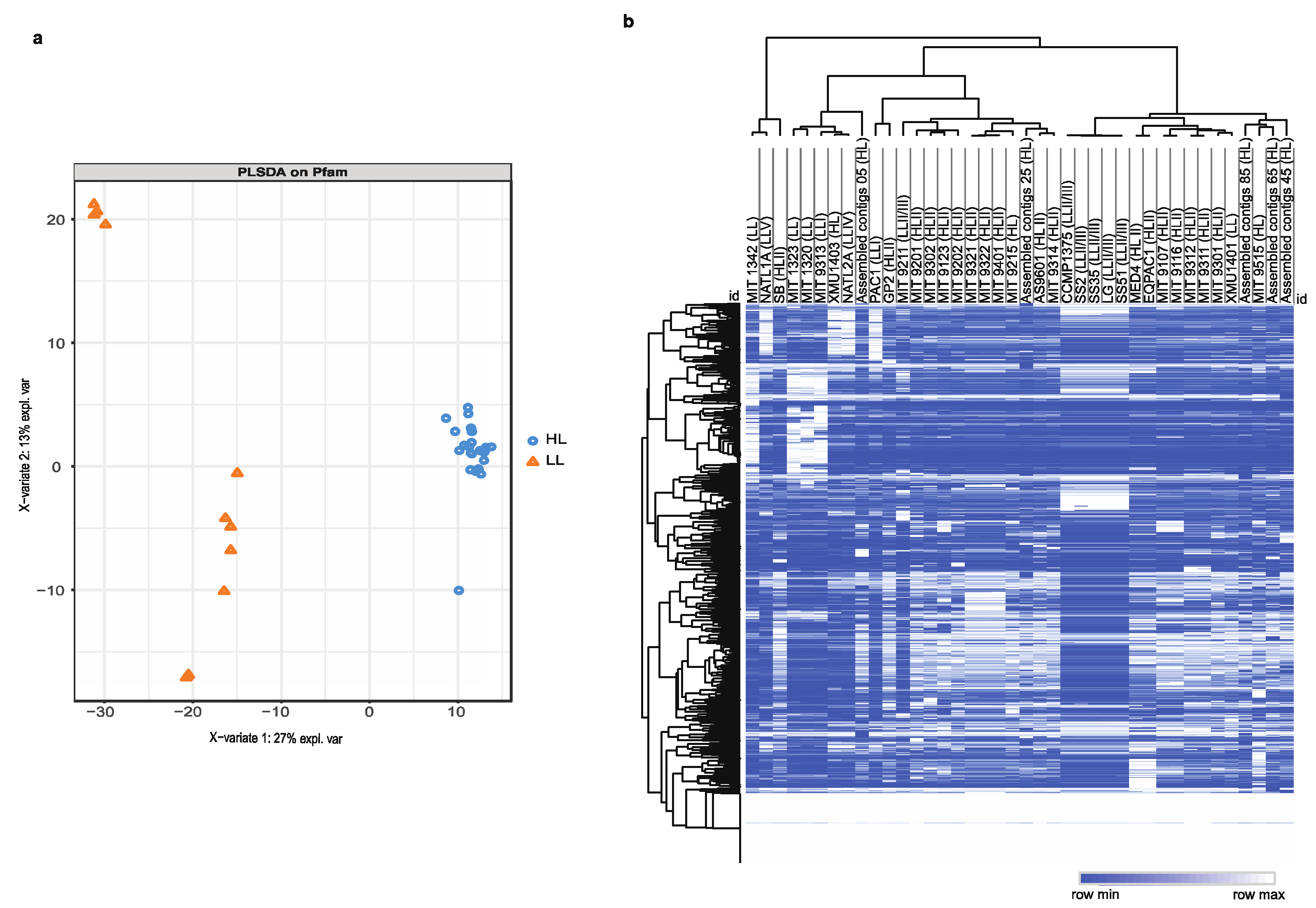
We generated protein family domain (Pfam) annotations for 40 sequenced Prochlorococcus genomes (supplemental data S1). We obtained 1196 Pfam domains associated with the encoded P. marinus proteins and calculated their normalized abundance from sum bit scores for each Pfam among the species (Table S1). Grouping the strains using dimensionality reduction analysis (the partial least squares method, PLS) showed a clear separation between the HL and LL strains based on their Pfams (Figure 1A). Hierarchical bi-clustering of Pfam count arrays revealed distinct clustering of LL and HL P. marinus strains (Figure 1B). Both analyses indicate that environmental adaptation of these species can be recapitulated by the differential presence and copy number variation of their encoded functional domains. Key genes containing these Pfams were extracted and analyzed for their differential representation among the strains. We observed an average ratio ~ 5-fold higher for LL strains compared to HL strains of a GHMP kinase domain (inc. homoserine kinases, galactokinases, and mevalonate kinases (PF00288)). The N-terminal domain of GHMP kinases family and the photosystem II protein domains have high representation in LL strains. Photosystem II (PSII) is critical for photosynthesis and function to increase the efficiency of light capture, therefore maximizing energy production in low light conditions. In addition, protein kinases are essential in the process of phosphorylation and photosynthetic acclimation in the core of photosystem II[15].
We observed a ferrous iron transport protein B at ~ 2-fold higher copy number compared to HL strains. This Pfam is involved in the electron transport chain processes (e.g., photosynthesis and respiration)[16]. In low iron conditions, cyanobacteria adapt by modifying electron transport and metabolic pathways and managing iron levels through specific uptake, transport, and storage mechanisms[17]. In Synechocystis sp. PCC 6803, the FitB protein, a ferrous iron transporter, is key in internal iron transport, becoming more active under iron scarcity to aid in this adaptation[18]. In HL strains, PF16881; the N-terminal domain of lipoyl synthase of radical_SAM family, found in lipoyl synthase enzymes, was found in ~10-fold higher copy numbers in HL compared to LL strains (Supplemental Figure S1). Lipoyl synthase enzymes are responsible for catalyzing the incorporation of two sulfur atoms, the C-6 and C-8 positions of the octanoyl moiety attached to lipoyl domains of proteins[19]. These enzymes play crucial roles in lipoic acid biosynthesis as co-factors for several enzyme complexes involved in central metabolism. Lipoic acid is essential for the proper functioning of various metabolic pathways demonstrated in bacteria (E. coli and B. subtilis)[20]. Although the specific role of lipoyl synthase in HL-adapted cyanobacteria has not been defined, their function suggests they may contribute to metabolic adaptation under high-light conditions.
We found that an ABC-2 transporter (PF06182) was present in HL strains at > 3-fold copy numbers compared to LL strains (p= 1.59 x 10-24, Tables S1 and S4). In Prochlorococcus, ABC transporters promote cellular viability in high-light, upper photic zone conditions by facilitating the transport of essential nutrients, antioxidants, and other molecules[11]. In the upper photic zone, nutrient concentrations (such as nitrogen, phosphorus, and iron) can be limiting for growth[21]. ABC transporters can facilitate sparse nutrient uptake by actively transporting them into the cell against a concentration gradient. Efficient nutrient acquisition can give Prochlorococcus a competitive advantage in these nutrient-poor environments. High-light conditions can lead to the production of reactive oxygen species (ROS), which can damage cellular components. These HL-specific ABC transporter domains may also be involved in the transport of antioxidants and other protective compounds, helping to maintain cellular redox balance and protect against oxidative stress[22].
To gain insight into the functional characteristics and biological processes associated with annotated P. marinus proteins, we performed Pfam enrichment analysis. This approach helped identify protein families and GO terms that were significantly overrepresented in HL and LL strains. Some of the enriched molecular functions unique to low-light are important for energy generation under low-light conditions such as the oxidoreductase activity (acting on CH-OH group of donors) (p= 7.02e-[6]; Z-score = 6.16), 3-oxoacyl-[acyl-carrier-protein] reductase (NADPH) activity for fatty acid biosynthesis (p = 1.13e-[4]; z = 8.31) (Table S2). Other functions related to protein synthesis, such as aminoacyl-tRNA editing activity (p = 5.38e-[4]; z= 6.64), are needed for translation. Aminoacyl-tRNA synthetases (AARSs) are enzymes that synthesize aminoacyl-tRNAs to facilitate translation. Quality control by AARS proofreading and other mechanisms maintains translational accuracy, which promotes cellular viability[23]. Comparative genomic analysis revealed that the gene complement of AARSs and their synthesis routes differ among cyanobacterial species. AARSs domains have undergone evolutionary adaptation to ensure accurate and efficient aminoacetylation under different environmental conditions[24], therefore suggesting their crucial role in modifying proteins under low light conditions. We also observed enrichment in Pfams for the GO term ‘ATPase activity’ (coupled to transmembrane movement of ions) (p = 3.9e-3; z = 4.64) (Table S2) that helps maintain ion homeostasis and optimize photosynthesis in deep waters[25]. We saw unique GO-terms enriched in molecular functions related to different metabolic pathways involved in response to high-light stress in the HL strains. Stress from changes in light intensity alters gene expression patterns, including genes for RNA-metabolism and processing (such as RNA helicases).
DNA repair was one of the top GO-terms in biological processes in HL strains (p= 7.78e-3; z= 2.86); this mechanism is essential for this cyanobacterium to recover from UV damage and maintain genome integrity[26]. Due to their exposure to high UV radiation levels from sunlight, DNA damage is a highly challenging factor for this genus, especially the HL strains[27,28]. Cyanobacteria possess large numbers of genes homologous to E. coli DNA recombination and repair genes, such as uvrABCD, recA, and recG, known to be part of their set of core genes[26,29]. One of the primary responses to UV stress is postponing cell division until completion of the DNA repair SOS system, which is a common stress response pathway also found in E. coli[27]. Another enriched term is the nitrogen cycle metabolic process (p= 5.64e-6; z= 8.32) (Table S2), which is important for nitrogen cycling, transforming nitrogen compounds into amino acids and nucleotides, a mechanism essential for growth and survival. Some of the nitrogen uptake pathways are encoded by flexible genes found in some but not all Prochlorococcus. Few HLII strains had nitrate assimilation genes (in addition to strains from the LLI clade). The frequency of cells capable of assimilating nitrate is positively correlated with decreased nitrogen availability, which is a limiting factor in surface waters within the highlight strains[30]. In LL strains, we observed enrichment in GO terms related to metabolic processes and compound biosynthesis, and translation elongation essential for optimizing protein synthesis under low light conditions. The enrichment of these high-level terms indicates that the ability of P. marinus strains to adapt to different habitats (light intensity and nutrient concentration) has inferred the differences in the evolution of these ecotypes.
Identification of Minimal Pfam Sets Distinguishing HL and LL Strains
To delineate the essential Pfam features that differentiate high-light (HL) and low-light (LL) adaptive strains, we performed false-discovery rate (FDR) and outlier-corrected batch t-tests using the bit scores from Pfam calls in the P. marinus strains (n = 40). We aimed to identify a minimal Pfam set that serves as distinguishing characteristics for HL and LL adaptation. We compared high-light (HL, n = 25) and low-light (LL, n = 15) Prochlorococcus strains to determine the main genetic differences facilitating adaptation to HL and LL conditions. We determined the most significantly differing Pfams in the two groups (FDR p < 0.05, Table S3) to provide a training set for an artificial neural network (ANN) model. Considering the CO2 gradient that also differentiates HL and LL strains, we expected some of the most varying Pfams to function in the carbon concentration mechanism (CCM). However, none of the highest variance Pfams had direct roles in carbon sequestration, indicating that CCM in HL and LL strains was generally more conserved than other processes.
Figure 2.
Protein families significantly differing between the HL and LL groups in FDR- and outlier-corrected batch t-tests. Data points are labeled with their Pfam symbol (https://www.ebi.ac.uk/interpro). For example, the LL cluster of points with large practical differences from the HL group contains various tetratricopeptide repeat domains (TPR). These Pfams are consistent with a more heterotrophic lifestyle [32]. The left panel shows Pfams with significantly higher abundance in HL strains, and the right panel shows Pfams with more abundance in LL strains.
Figure 2.
Protein families significantly differing between the HL and LL groups in FDR- and outlier-corrected batch t-tests. Data points are labeled with their Pfam symbol (https://www.ebi.ac.uk/interpro). For example, the LL cluster of points with large practical differences from the HL group contains various tetratricopeptide repeat domains (TPR). These Pfams are consistent with a more heterotrophic lifestyle [32]. The left panel shows Pfams with significantly higher abundance in HL strains, and the right panel shows Pfams with more abundance in LL strains.
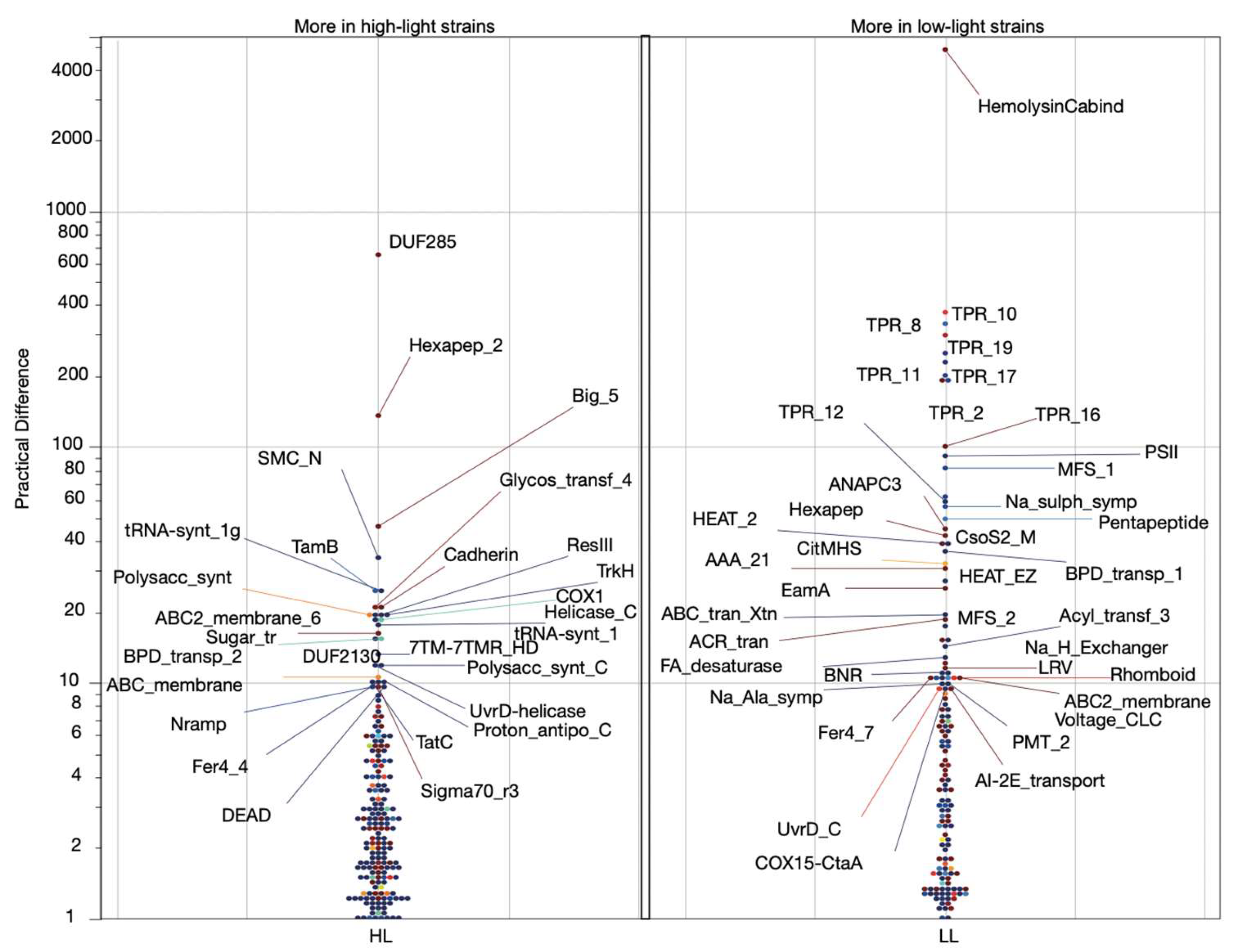
We carried out supervised training of an ANN model to distinguish between HL and LL groups based on the defined Pfam set. The Pfams were selected from the batch t-tests as the top 20 significantly different domains (Table S4). This approach aimed to establish statistical integrity for batch comparisons while generating conclusions distinguishing LL and HL strains with ANNs. Figure 4 displays the top 10 Pfams (of the selected 20) contributing most to the ANN decision-making algorithm. These Pfams can discriminate the P. marinus genomes based on their depth adaptation, with six Pfams indicating a high likelihood (> 99%) of a strain residing in HL conditions and four Pfams suggesting an LL lifestyle. Most of these protein domains have established or predicted roles in photosynthesis.
The most influential Pfam for the ANN was PF06182, an ABC-2 transporter (p = 1.34 x 10-35) (Table S4). Our ANN shows that levels of ABC-2 transporters in Prochlorococcus genomes are highly predictive of an HL environment. The ABC-type transporters are upregulated under stress conditions (such as low temperature), comprising a defense system against accumulated toxic metabolites[31]. Considering the other HL-predictive proteins found to be highly influential in our ANN model, a reasonable explanation for higher levels of ABC transporters in HL strains is that at least one of their functions is to remove and replace damaged proteins, aiding in the maintenance and recovery of optimal photosynthetic efficiency under high-light stress.
Among the most influential Pfams were PF00271 and PF00270, representing helicase conserved C-terminal domain and DEAD box helicase, respectively. Helicases use ATP to bind and remodel nucleoproteins-DNA or -RNA complexes[32,33]. DNA and RNA helicases unwind double-stranded DNA/RNA regions and are involved in DNA replication, recombination, repair, and overall genome stability[32], and RNA-dependent helicases participate in various aspects of RNA metabolism[33]. CrhR-like RNA helicases coordinate the expression of genes required to maintain oxygenic photosynthesis in cyanobacteria, and the C-terminal domain of CyanoP is involved in carotenoid binding and is a paralog of the C-terminal domain of helical carotenoid proteins (HCPs), which are involved in energy transfer and photoprotection in cyanobacteria[34]. DEAD box domains are found in cold shock protein, transcription repair, DNA ligase, and DNA helicase in HL strains.
The Pfams PF00133 and PF09334, annotated as aminoacyl tRNA synthetases, have increased copy numbers in HL strains (p = 1.73 e-17 and 1.25e-16) (Table S4). These play central roles in cell physiology and may have influenced the origin and evolution of the genetic code. In MED4, tRNA domains are found in different tRNA ligases (methionine, leucine, isoleucine, valine, and cysteine). AARS genes show evidence of horizontal gene transfer (HGT); it has been shown that the set of AARS-encoding genes varies from one cyanobacterium to another due to gene gain, loss, or duplication[24]. The higher copy number of some of the Pfams encoding proteins involved in RNA processing and DNA repair in the ANN indicated that a strain was HL. Our results suggest that HL conditions endured by upper photic cyanobacteria require more copies of the helicase and tRNA synthetases. This can be associated with the regulation of genes involved in stress responses, including responses to light and nutrient conditions in HL zones.
The most represented Pfams in LL strains are TPR repeats (PF13414, PF00515, and PF13424 representing TPR_11, TPR_1, and TPR_12 (p = 0.000821054; 0.003134574; 0.00053802) (Table S4). The tetratricopeptide repeats (TPR) structural motif is present in a wide variety of proteins in procaryotes and eukaryotes[35]. Repeat proteins are known for their extended modular nature, allowing interactions with different ligands to facilitate the formation of functional complexes[36]. In cyanobacteria, TPR repeats play an important role in photosystem II (PSII) assembly and repair[37]. The difference in these TPR repeats between LL and HL strains suggests different adaptations used by PSII under different light conditions. In Synechocystis, slr0151 (TPR protein) mutants showed reduced photoautotrophic growth and oxygen production rates under high-light stress conditions[37]. We hypothesize that due to their wide variety and ability to bind to diverse ligands, the TPRs specific to LL strains may regulate the assembly and stability of PSII under low light conditions[38]. Interestingly, a TPR-family membrane protein gene is required for light-activated heterotrophic growth of the cyanobacterium Synechocystis sp. PCC 6803[39]. Thus, the many proteins we observed with TPR repeats, especially in LL strains, may be important for P. marinus heterotrophy and adaptation to the lower oceanic photic zone.
We observed relatively high levels of the HEAT (Huntingtin, elongation factor 3, protein phosphatase 2A, and TOR1) domain (PF13646) in the LL group (p =7.22e-22). HEAT repeats are protein motifs found in a wide spectrum of eukaryotic and prokaryotic proteins and are involved in many cellular processes. While their functions are diverse, a common function of HEAT repeats is thought to mediate protein-protein interactions[40]. In NATL1A, HEAT repeats are found in a hypothetical protein and PBS (phycobilisome) lyase (Table S4). Phycobilisomes are large light-harvesting complexes attached to the stromal side of thylakoids in cyanobacteria and red algae. Lyases can modulate lyase-catalyzed binding and detachment of chromophores in a complex fashion[41].
The third most influential Pfam was PF00421, representing the intrinsic antenna proteins CP43 (PsbC) and CP47 (PsbB) found in the reaction center of PSII. Higher copy numbers of this Pfam indicated an LL lifestyle (p = 1.26e-61) (Table S4). PsbC and PsbB antennas transport the excitation energy to the core of the photosystem II. Some Cyanobacteria adapt to low-light environments by altering their photosynthetic machinery to absorb far-red light (FRL, 700-800 nm). This process (Far-Red Light Acclimation process, FaRLiP) requires the activation of PsbA, PsbB, PsbC, and PsbD subunits[42]. The observation that PSII is highly predictive in the ANN for LL status suggests the decisive role of photosystem light harvesting processes in Prochlorococcus’ adaptation to LL conditions.
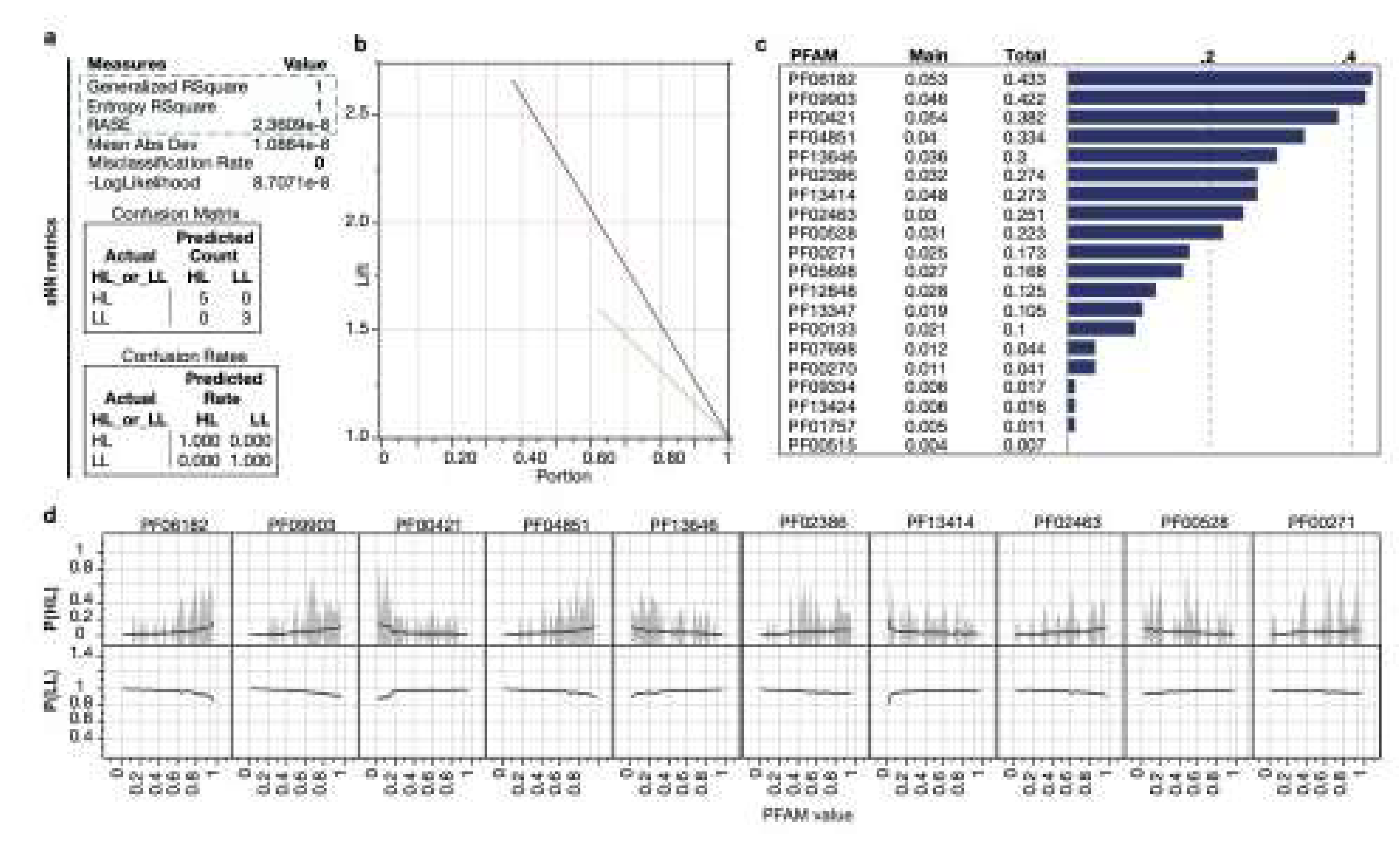
Figure 3.
An artificial neural network (ANN) reconstructed from the top ten differing Pfams in HL and LL strains (n=40). (a) Performance of training model using three TanH nodes in a single-layer neural network using a squared penalty and transformed covariates. The input dataset consisted of normalized sum bit scores of Pfams. These metrics describe the performance of the model using a randomly selected 20% holdout set of genomes that were removed before model creation. The model demonstrates a misclassification rate of zero on the unknown set of Prochlorococcus genomes. (b) Lift curves for the training and validation (holdout) sets. The lift curves show how well the variables perform at prediction per sample size. (c) Assessment of variable importance in the creation of the ANN model. The influence is shown regarding the total effect and main effect for classification. (d) Marginal model plots for the variables used to create the model. The y-axes represent probabilities for predicting whether a strain is HL or LL as a function of each Pfam. As the copy number of a Pfam increases, the probability of either HL or LL is determined.
Figure 3.
An artificial neural network (ANN) reconstructed from the top ten differing Pfams in HL and LL strains (n=40). (a) Performance of training model using three TanH nodes in a single-layer neural network using a squared penalty and transformed covariates. The input dataset consisted of normalized sum bit scores of Pfams. These metrics describe the performance of the model using a randomly selected 20% holdout set of genomes that were removed before model creation. The model demonstrates a misclassification rate of zero on the unknown set of Prochlorococcus genomes. (b) Lift curves for the training and validation (holdout) sets. The lift curves show how well the variables perform at prediction per sample size. (c) Assessment of variable importance in the creation of the ANN model. The influence is shown regarding the total effect and main effect for classification. (d) Marginal model plots for the variables used to create the model. The y-axes represent probabilities for predicting whether a strain is HL or LL as a function of each Pfam. As the copy number of a Pfam increases, the probability of either HL or LL is determined.
Variable, Depth-Dependent Accumulation of Endogenous Viral Elements
Cyanobacterial viruses[43] exhibit considerable effects on the evolution, lifecycle[44], and ecosystem dynamics[45] of widespread cyanobacterial populations. Viral sequences can integrate into their hosts’ genomes[46]; thus, we examined the P. marinus genomes for viral family sequence differences in strains (Figure 5). Their abundances were compared with response screening. We observed many viral families (VFams) from viruses infecting eukaryotic hosts[10] in P. marinus genomes (Table S4). Although these eukaryotic viruses were not expected to infect prokaryotes, the exchange of genetic material may have still occurred in ancestors of P. marinus, especially with giant viruses[46,47,48]. The genomes of giant DNA viruses contain up to 1.5 Mbp of DNA. In the open ocean, they constitute large populations that, through lysis, release enormous amounts of DNA, which can be then taken up by caynobacteria.
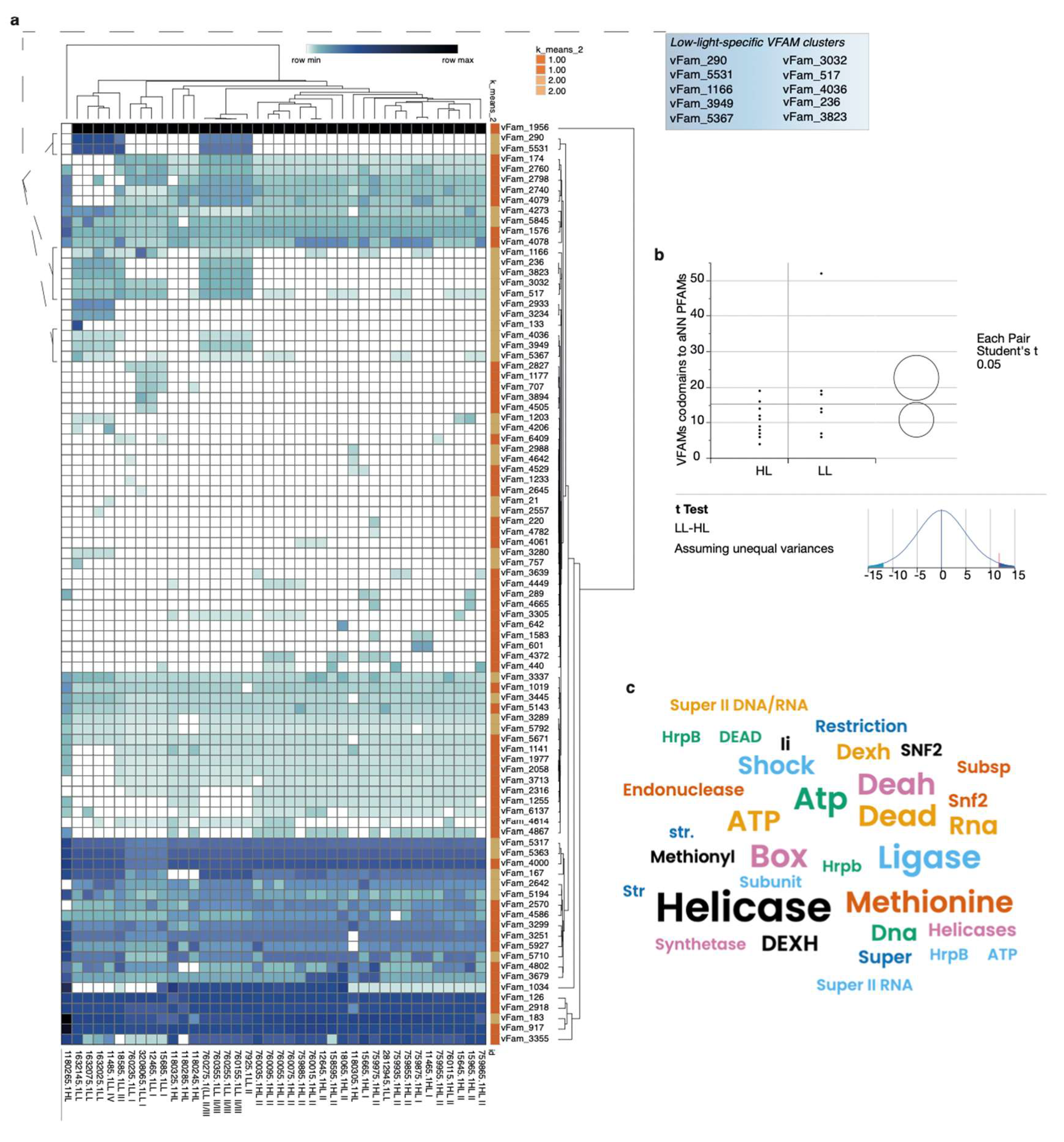
P. marinus genomes showed several highly conserved VFams, including VFams 5317 (methionyl-tRNA synthetase; Megavirus lba[49] and Acanthamoeba polyphaga mimivirus[50]), 5363 (phospho-2-dehydro-3-deoxyheptonate aldolase; Pandoraviruses), 4000 (adenosylhomocysteinase; Pandoraviruses[51]), 126 (HSP70, conserved in plant viruses, including Ampelovirus, Closterovirus, and Crinivirus), 2918 (AAA family ATPase; Acanthamoeba polyphaga moumouvirus and Megavirus chiliensis), 183 (DNA topoisomerase II; Marseillevirus[52], Wiseana iridescent virus, Lausannevirus), 917 (Glutamine:fructose-6-phosphate amidotransferase; Paramecium bursaria Chlorella virus 1, Phaeocystis globosa virus, Megavirus lba), and 3355 (Fucose synthetase; Paramecium bursaria Chlorella virus 1[53]). These conserved VFams in P. marinus are likely necessary for the survival of the species worldwide.
Two clusters of VFams were unique to low-light strains, including strains 760155.1LL II/III (LG), 760255.1LL II/III (SS2), 760355.1LL II/III (SS51), 11485.1LL IV (MIT9313). These unique VFams included VFam_5531 (Rad5-like protein; Pandoraviruses), VFam_1166 (2OG-Fe(II) oxygenase family protein; Phaeocystis globosa virus[54], Ostreococcus viruses[55]), VFam_236 (ATPase; Ectocarpus siliculosus virus 1[56]), VFam_3032 (putative DNA helicase, Megaviruses), VFam_517 (NAD-dependent DNA ligase; Betaentomopoxviruses[57], Iridoviruses), VFam_5367 (guanine deaminase; Pandoraviruses), and VFam_3949 (SAM-dependent methyltransferase; Megaviruses). The selective uptake or retention of genes with these VFams in low-light species suggests they facilitate cyanobacterial survival at depth.
We extracted all ORFs from the 40 sample P. marinus genomes containing VFams. These ORFs were examined for the Pfams used in our ANN model. We observed a mean of 15.3 VFams/genome on ORFs that also had ANN Pfams. The Pfams with VFam codomains mainly consisted of ORFs annotated as methionine tRNA ligases, ATP-dependent and DEAD/DEAH-box helicases, and cold-shock proteins. These Pfams significantly varied among HL and LL groups and were included in the ANN set. Interestingly, LL strains had an average of 11.8 ANN-linked VFams more than HL strains (t ratio = 2.4 and p = 0.0319 in a two-tailed t-test). The significantly higher level of VFams from these genes suggests their involvement in distinguishing HL and LL groups. The putative viral (phage) origin for their codomain Pfams outlines a scenario where phage-acquired genes were instrumental in the adaption of HL and LL Prochlorococcus strains to their respective niches.
Figure 4.
Viral families (VFams) in P. marinus strains (n = 40), including NATL1A and MED4. Most VFams were highly conserved in all P. marinus strains except for two distinct clusters unique to some low-light strains. (a) Hierarchical bi-clustering of HMM match bit scores. The LL group had unique clusters of VFams (shown in Box). (b) VFams from genes in the aNN Pfam set (Figure 4). We found that LL strains had significantly more VFams related to these influential Pfams. (c) Word cloud generated from the annotations of genes containing both VFams and aNN Pfams. Several types of helicases were overrepresented in this gene set.
Figure 4.
Viral families (VFams) in P. marinus strains (n = 40), including NATL1A and MED4. Most VFams were highly conserved in all P. marinus strains except for two distinct clusters unique to some low-light strains. (a) Hierarchical bi-clustering of HMM match bit scores. The LL group had unique clusters of VFams (shown in Box). (b) VFams from genes in the aNN Pfam set (Figure 4). We found that LL strains had significantly more VFams related to these influential Pfams. (c) Word cloud generated from the annotations of genes containing both VFams and aNN Pfams. Several types of helicases were overrepresented in this gene set.
The Prochlorococcus Strains MED4 and NATL1A as HL and LL Representatives
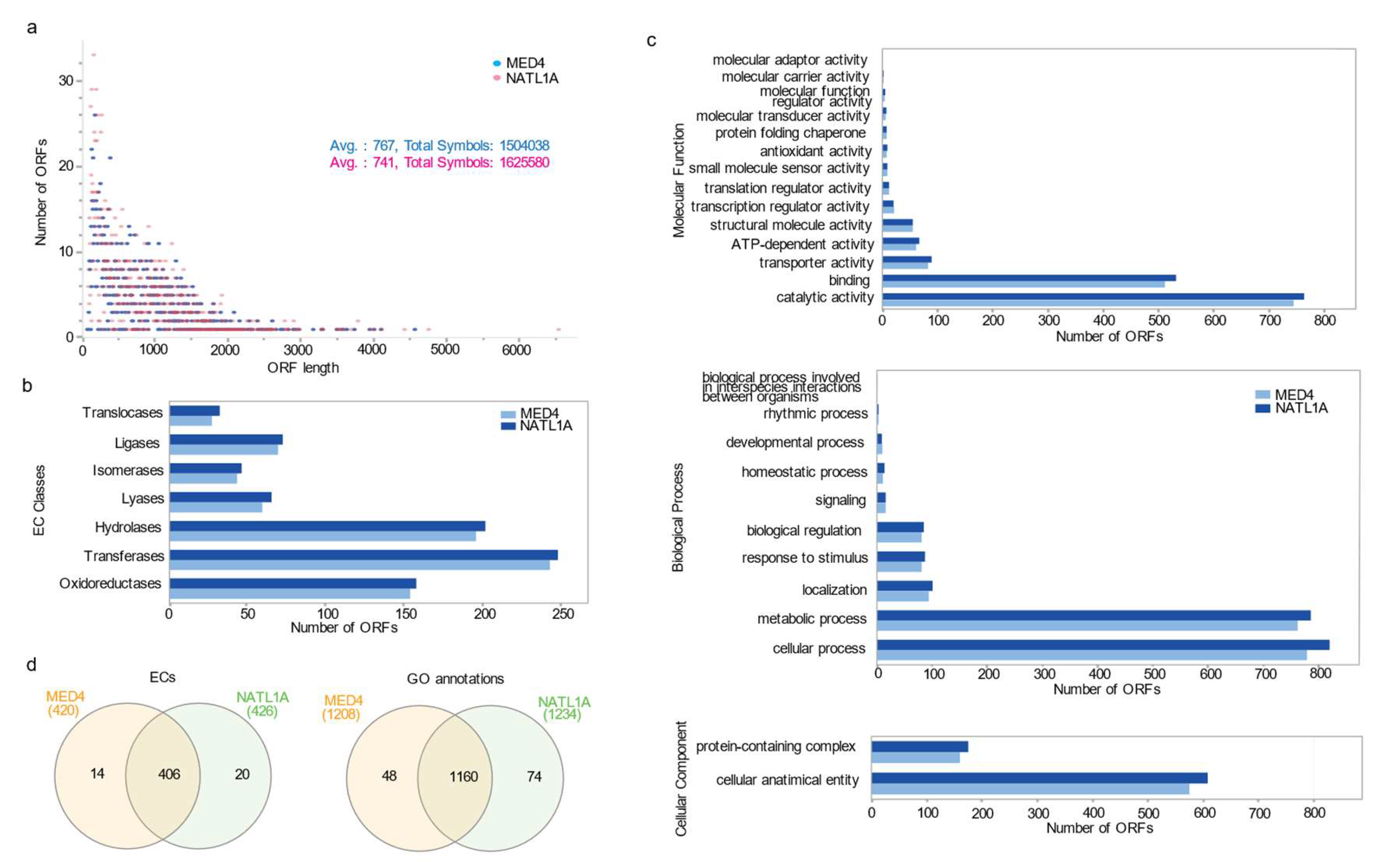
To explore the differences between the HL and LL P. marinus, we invested in analyzing the genomes of the MED4 (HL) and NATL1A (LL). We extracted functional annotation using Blast2GO[58], with complete gene sequences available from EnsemblBacteria. The average length distribution for genes is 767 bp and 741 bp for MED4 and NATL1A, respectively (Figure 6 A and Table S6), with 92% of the cDNA sequences smaller than 1.5 kb (1809/1960 for MED4 and 2024/2193 of NATL1A cDNA sequences). B2GO analysis assigned available annotations for 1514 and 1593 sequences for MED4 and NATL1A respectively (446 and 600 sequences could not be annotated) with 1123/1177 (MED4/NATL1A) assigned gene ontology terms (GO-terms), with 869 and 902 for assigned enzyme commissions (ECs) corresponding for 724 and 751 sequences among the available annotated genes for MED4 and NATL1A respectively (Table S6). EC distribution analyses showed similar distributions among both strains for different enzymatic classes and sub-classes (Figure 6B). We analyzed the EC overlap using iPath3 (Interactive Pathway Explorer version 3), and similar to what we show in the Venn diagram, the ECs map mostly to common metabolic pathways (supplemental Figure 1). The unique metabolic pathways differed in specific modules or enzymatic reactions. An example is the amino sugar and nucleotide metabolism pathway that is part of the carbohydrate pathway shown to be unique in some modules in MED4 and NATL1A (highlighted in blue and red respectively, the metabolic map in supplemental Figure 1). Though a purine metabolism pathway is common to both high- and low-light strains (highlighted in green), UDP-N-acetyl-D-glucosamine 2-epimerase is unique to NATL1A, while UTP:N-acetyl-alpha-D-glucosamine-1-phosphate uridylyltransferase is specific to MED4.
We found that N-glycan biosynthesis (highlighted in blue in the glycan synthesis and metabolism in the map (supplemental Figure 1), was unique to the high-light strains with no overlap with the low-light strain. Glycans are complex carbohydrates with high structural diversity; methylation, acetylation, or the addition of sulfate groups enhance their diversity. Their presence at the cell surface confers adaptability to a variety of environmental factors[59]. Glycans in the extracellular polysaccharide layer can play a protective role against desiccation, guarding against ROS under UV-A or UV-B irradiation[22]. Another example showing unique modules and reactions to NATL1A strain (highlighted in red within the carbohydrate metabolism shown in the metabolic map, supplemental Figure 1), within the glycolysis, pyruvate metabolism, and carbon fixation pathways.
Gene ontology (GO) hierarchies for HL and LL strains showed mostly similar GO distributions for MED4 and NATL1A represented by three GO terms for biological processes (BP), molecular functions (MF), and cellular components (CC, see Figure 6C and table S6). The GO levels give a broad overview of the ontology content from the minimal GO-Slim ontology set. We found similar GO-term distributions for MED4 and NATL1A among the three different categories (BP, MF, and CC), with a slightly higher number of sequences representing the top 2 GO terms in each category for NATL1A strain (Figure 6 C and Table S6). We compared the number of encoded annotated enzymes for both strains and found that only 14 and 20 ECs are unique for MED4 and NATL1A, respectively. In comparison, most ECs are shared between the two genomes (406 ECs) (supplemental Figure 2). GO terms had similar distributions in MED4 and NATL1A. The majority of GO terms were shared among the strains, while only 48 and 74 were unique for MED4 and NATL1A, respectively (supplemental Figure 2). The common GO terms were related to essential survival functions, while species-specific GO sets were related to processes involved in the adaptation of these strains to their local environmental conditions.
Figure 5.
Sequence length distribution and EC classes GO-term distribution among the MED4 and NATL1A ORFs. Panel (a) represents the sequence length distribution with an average of 767bp and 741bp for MED4 and NATL1A, respectively. Classification of the enzyme commission numbers assigned to the ORF sequences to MED4 and NATL1A (b). Representation of GO annotation level distribution after GO-Slim for MED4 and NATL1A. The number of sequences is represented on the x-axis and the GO-terms on the y-axis for both strains representing Biological Processes, Molecular Functions, and Cellular Compartments as indicated. (d) Functional annotation analysis using Blast2GO. Comparison between the MED4 and NATL1A strains based on GO annotations and Enzyme Commission (ECs).
Figure 5.
Sequence length distribution and EC classes GO-term distribution among the MED4 and NATL1A ORFs. Panel (a) represents the sequence length distribution with an average of 767bp and 741bp for MED4 and NATL1A, respectively. Classification of the enzyme commission numbers assigned to the ORF sequences to MED4 and NATL1A (b). Representation of GO annotation level distribution after GO-Slim for MED4 and NATL1A. The number of sequences is represented on the x-axis and the GO-terms on the y-axis for both strains representing Biological Processes, Molecular Functions, and Cellular Compartments as indicated. (d) Functional annotation analysis using Blast2GO. Comparison between the MED4 and NATL1A strains based on GO annotations and Enzyme Commission (ECs).
ORFeome Resource Development
To complement our computational analyses, we synthesized the complete ORFeomes for the P. marinus strains MED4 and NATL1A to serve as biological resources for downstream studies. Prochlorococcus species have the most compact genomes of free-living, oxygenic phototrophs. MED4, a high-light adaptive strain, is the smallest with 1,65 Mbp, and 30.8 G+C%, whereas NATL1A is a low-light adapted strain with a slightly larger genome of 1.86 Mb and a higher GC content of 35%. The genomes of MED4 and NATL1A consist of a single circular chromosome and encode ~1790 and ~1976 genes, respectively[12]. To synthesize MED4 and NATL1A ORFs, we extracted structural gene annotation and cDNA sequences for complete genomes from the EnsemblBacteria database (https://bacteria.ensembl.org/), resulting in a list of 1960 and 2193 genes for MED4 and NATL1A, respectively. Open reading frame sequences ranged from 80 to 4500 bp with an average length of 760 bp for MED4 and from 100 bp to 6 kb with an average length of 1,260 pb for NATL1A. ORFs were flanked with ATTL1 and ATTL2, allowing recombinational sub-cloning into a variety of Gateway-compatible vectors[60]. The ORFs were synthesized in the pTWIST-ENTR vector (twistbioscience.com), providing a Gateway® cloning compatible system[61]. Stop codons were removed from the cDNA sequence, and codon fitting was applied for optimal synthesis (supplemental Figure 2). Approximately 99% of attempted ORFs were successfully synthesized and sequence-verified, with 1826 out of 1840 ORFs, and 2150 out of 2193 ORFs.
To validate the recombinational transferability of ORFs, a set of 70 ORFs of the MED4 strain were recombinationally cloned into two different yeast two-hybrid Gateway compatible vectors, pAD and pDB[62] (supplemental Figure 3). We randomly selected half of the re-cloned ORFs and their respective parental ORFs, using Sanger sequencing with both forward and reverse vector-specific primers. We observed 90% perfect alignment to the reference ORF sequence.
MED4 and NATL1A ORFeomes are available as bacterial glycerol stocks; their respective distribution and plate maps are in supplemental tables 7A and 7B . Users can find information about each ORF, their plate reference (well and plate number), their corresponding sequence, gene reference, and related information in NCBI.
Discussion
Shifts in phytoplankton communities serve as indicators of climate-induced disruptions in marine ecosysytems[1]. Cyanobacteria are highly sensitive to environmental changes, including light, temperature, and nutrient availability. Studying their adaptation to different environmental conditions helps predict shifts in phytoplankton distributions in marine ecosystems[63]. Despite cyanobacteria being a model for studying essential cellular mechanisms, including photosynthesis, plant evolution, and carbon fixation, the adaptive evolution of the species with respect to depth is not well understood[14,64,65,66]. Cyanobacteria often have multiple, seemingly redundant copies of genes in their genomes; these genes are rarely paralogs. Instead, they are more likely to be independent acquisitions from the ubiquitous clouds of cyanophages surrounding cyanobacteria in their native environment[67]. For this reason, focusing on Pfams, instead of segments of genomic sequences can bring new insights that would not be detected by paralog analyses[68]. To identify the genomic determinants of cyanobacterial adaptation to low- and high-light (LL and HL), we used Prochlorococcus marinus strains as models and explored their 40 sequenced and published genomes. We analyzed domains and functional regions and uncovered differences in genomic signatures between LL and HL. We carried out Pfam analysis, which provides information on the identity and number of coded protein domains. We augmented our analyses by reconstructing ANNs that could accurately distinguish between HL and LL status using a subset of Pfam predictors, thereby identifying the small number of features that are sufficient to distinguish HL and LL strains. The most influential Pfams were quantified, and our results show that the Pfams with the highest predictive capacity were mostly photosynthesis-related (predictive of HL status (pHL)) or involved in redox (i.e., electron transfer) processes (predictive of LL status (pLL)) (Figure 4).
Over the past centuries, human activities related to urbanization, agriculture, and industrial development have led to an excessive influx of nutrients, especially nitrogen and phosphorus[69], in aquatic systems, along with a dramatic increase in atmospheric levels of CO2[70]. This nutrient over-enrichment has resulted in eutrophication (an acceleration in the rate of primary production), which in turn can lead to an increase in the growth of blooms of harmful cyanobacteria[69]. In addition, due to climate change and ocean warming, stratification is leading to an increase in light availability in the upper levels and reduced transport of nutrients[71]. To better understand how phytoplankton may respond to these changes and generate biological resources for downstream wet-bench experiments, we expanded the genome analysis for 40 P. marinus strains and applied bi-clustering to detect differential gene content in the high- and low-light strains of P. marinus and generated genome-wide ORFeome resources for NATL1A and MED4, as representative LL and HL-adapted species of Prochlorococcus genus. Cyanophage genomes abundantly contain accessory metabolic genes (AMGs) presumably used to manipulate their hosts' metabolisms. We hypothesize that cyanophage populations act as genetic reservoirs for Prochlorococcus and are partly responsible for the differential genetic contents in HL and LL strains. The results from our analysis of viral families in Prochlorococcus support this hypothesis with substantial functional overlap with the total differential gene content between HL and LL groups.
Our analyses showed that most VFams are highly conserved among the strains. We also identified distinct clusters in LL strains where the enriched VFams highlight their viral origin. Phages play an essential role in the evolution of microbial communities, including cyanobacteria. Cyanophages are among the key factors determining the rate and direction of evolution in cyanobacterial populations[72]. Among the identified viral domains, some isoforms can also be found in HL strains (involved in DNA repair or light-harvesting system) but were not detected in the VFam analysis. This finding suggests that the transfer of some protein domains has undergone convergent evolution, explaining their differential origins in LL and HL strains[11]. Studies show that marine viruses serve as a potential genetic pool in shaping the evolution of cyanobacterial photosynthesis. Some LL P. marinus strains have acquired many of their genes from other cyanobacteria, such as Synechococcus, placing these LL strains closer to Synechococcus than HL strains[73]. Phage intermediates are involved in the transfer and shuffling of genes encoding photosynthetic proteins (psbA) between Prochlorococcus and Synechococcus[74]. Other studies showed the dynamic co-evolutionary process in both host and phages[75], explaining differences in genes’ evolution and genome differences among P. marinus strains.
Functional annotation allowed us to compare MED4 and NATL1A genomes on a functional and metabolic level. Functional annotation from available genome sequencing for both strains revealed a high degree of similarity when the two strains are compared for their respective encoded enzymes and GO-term functions. Metabolic pathway analyses allowed us to dissect the metabolic pathway implication for available EC annotations, confirming a majority of shared metabolisms for MED4 and NATL1A, including energy metabolism, amino acid metabolism, and fatty acid metabolism (Supplemental Figure 1). In addition to ECs representing comparable distribution among enzymatic classes and sub-classes for the two strains (Figure 6 B). While both strains are characterized by unique pathways that differ in specific enzymatic reactions, they also share metabolic pathways such as glycolysis and galactose metabolism among the unique reactions (supplemental Figure 1).
Computational analyses of the available annotated sequences for P. marinus strains provided a comparative description of their genes, enzymes, and functions but, in many cases, are still lacking experimental validations. ORFeome resources allow a more in-depth examination of the differences and similarities between the P. marinus strains by exploring their potential interactions, wiring protein interactions in signaling pathways, and their implications in cellular and biological functions. Despite being model organisms for studies on photosynthesis and evolution, comprehensive ORFeome resources have not been made available outside the present work. The availability of complete genome sequences, in addition to the reduced cost of DNA synthesis, allowed us to generate the complete ORFeomes of MED4 and NATL1A successfully. The ORFs are available as DNA clones in Gateway-compatible vectors (from TWIST bioscience, San Francisco, USA and Genome Synthesis and Editing Platform, China National GeneBank (CNGB), BGI-Research, Shenzhen, China), offering the advantage of sub-cloning the genes into various expression vectors. Therefore, they can be used in a wide range of studies and for multiple applications. Generation of cDNA libraries has been established for various organisms, from Arabidopsis thaliana[76,77] to Homo sapiens[78,79]. Later, efforts for complete ORFeome synthesis have been described in some model organisms. Clones are archived in Gateway-compatible vectors, allowing the high-throughput transfer of ORFs into a variety of expression vectors. ORFeome collections' usefulness is illustrated by generating successive human ORFeome versions that allowed biophysical proteome-wide protein-protein interactions and related human cellular mechanisms. ORFeome libraries were generated for other model organisms such as E.coli[80] from bacteria, Drosophila melanogaster[81] and Chlamydomonas reinhardtii[82], the algal model organism. These efforts were followed by generating databases and interfaces that provide integrated sets of bioinformatics tools to analyze and clone ORFs. For example, the ViralORFeome (https://omictools.com/viralorfeome-tool) provides a framework to establish an extensive collection of viruses' ORFs[83], with an interface to define ORFs and design ORF-specific cloning primers such as the specific for virus genome sequences. The development of technologies in DNA synthesis methods progressed from the template-based DNA amplification and cloning approaches, to de novo gene synthesis, allowing codon optimization with higher DNA yield, increased oligonucleotide length (up to 5kb), and higher success rate[84]. The synthesized metabolic ORFeome of Chlamydomonas reinhardtii was generated with a 70% success rate (using reverse-transcription PCR, amplification, and cloning)[82]. In comparison, de novo synthesis of MED4 and NATL1A ORFeomes resulted in a 99% success rate. Industrialization of DNA synthesis has also resulted in cost reduction and accelerated experimental process, making large-scale genes’ and ORFeome synthesis accessible and affordable for synthetic biology applications.
The newly synthesized ORFeomes for P. marinus MED4 and NATL1A are enabling resources to interrogate biological mechanisms of adaptation to high- or low-light, which are fundamental processes in photosynthesis evolution. Here, we have presented broad comparative genomic analyses, but the synthesized ORFeomes can be used in more focused biological/biochemical experiments to understand the fundamental roles of the included genes in Prochlorococcus.
Material and Methods
Protein Family Domain Prediction
The P. marinus genomes were downloaded from NCBI/Genome assembly and annotation report for 42 available isolates. Genes’ annotation yielded a set of peptide predictions serving as a base for HMM alignment with the Pfam-A-v31.1 database. The command was: 'hmmsearch --noali -E 0.000000001 --cpu 28 --domtblout $OUT $IN.aa.fa'. The HMMer user guide (http://eddylab.org/software/hmmer/Userguide.pdf) provides in-depth descriptions of the reported values, including sequence coordinates for alignment and matches with HMM models, E-values, percent identity, and bias.
The predicted proteins were also analyzed for similarity to known proteins using BLASTP (v2.2.31).
Hierarchical Bi-Clustering
The assemblies for P. marinus used in the manuscript predicted 1196 Pfams that were used for bi-clustered heatmap visualization in Morpheus[85]: a web tool for visualizing the clustering of multivariate data (https://software.broadinstitute.org/morpheus/). Clustering was performed on Pfams and species (bi-clustering) using Pearson correlation scores.
The complete sets of resulting annotations are available as transcripts and proteins in fasta format (.fa) in the supplemental file (Data S1).
Response Screening
Parsed HMMsearch results (i.e., Pfam matrices) were used in comparative analyses. Briefly, normalized sum bit scores for Pfams in the 40 P. marinus strains surveyed (i.e., ‘input matrix’ see Data S2) were used as input for false-discovery rate (FDR)- and outlier-corrected batch t-tests (response screens). Means were analyzed using Analysis of Variance (ANOVA) tests, where each comparison’s p-value, logworth, False Discovery Rate (FDR, or adjusted) P-value, and FDR logworth are reported. Protein family averages between the HL and LL groups were compared and screened for significant differences and equivalencies (Table S3).
Standardized residues were used to compare means in > 100,000 t-tests while controlling for FDR. Robust Huber M-estimation (using maximum-likelihood type estimators) was used to reduce the sensitivity of the tests to outliers. Briefly, this method reduces the influence of outliers by minimizing their weights for the comparative statistics algorithm. The most informative information from these statistical tests for the general reader is in the ‘Response Screen Compare Means’ table, which includes tests for all possible comparisons and results for practical equivalences and differences. Comparisons not passing either of these tests are labeled as ‘inconclusive’. The practical difference is the difference in means that are considered to be of practical interest. Standard deviation estimates were computed from their interquartile ranges (IQRs). The estimate was σ=(IQR)/(1.3489795). The Practical Difference was computed as 6(σ) multiplied by 0.10 as a proxy for the Practical Difference Proportion. Practical difference p values are given for tests of whether the absolute value of the mean difference in Y between comparison levels is less than or equal to the practical difference. Low p values indicate that the absolute difference exceeds the practical difference, indicating that the difference in the comparison is of practical significance.
Practical equivalence p values were computed using the Two One-Sided Tests (TOST) method to test for practical differences between means. The Practical Difference specifies a threshold difference for which smaller differences are considered practically equivalent. One-sided t-tests are constructed for two null hypotheses: the true difference exceeds the Practical Difference; the true difference is less than the negative of the Practical Difference. If both tests reject, this indicates that the absolute difference in the means falls within the practical difference. Therefore, the groups are considered practically equivalent. The practical equivalence P value is the largest P value obtained on the one-sided t-tests. Low practical equivalence p-values indicate that the mean response for the top comparison level is equivalent, in a practical sense, to the mean for the lower level.
Deep Learning ANN Analysis
We compared high-light (HL, n = 25) and low-light (LL, n = 15) Prochlorococcus strains to determine the main genetic differences facilitating adaptation to HL and LL conditions. We used the top 20 significantly differing Pfams from the aforementioned response screen in the two groups (FDR p < 0.05) to analyze their differential genomic contents and provide a training set for an artificial neural network (ANN) model. We constructed an ANN model using the top 20 significantly differing Pfams to distinguish between HL and LL groups based on their genomic contents. The combined approach aimed to establish statistical integrity for batch comparisons while generating nonintuitive conclusions with ANNs.
We compared high-light (HL, n = 25) and low-light (LL, n = 15) Prochlorococcus strains to determine the main genetic differences facilitating adaptation to HL and LL conditions. We determined significantly differing Pfams in the two groups (FDR p < 0.05) to analyze their differential genomic contents and provide a training set for an artificial neural network (ANN) model. Predictive modeling using neural networks was carried out in the JMP Neural Networks module. Four sequential models were used with three TanH nodes each and a learning rate of 0.1. In fitting, covariates were transformed and a squared penalty method was used. The Python code for the formulae necessary to reproduce the dNN is provided in the supplementary materials (Data S3). Both the training and validation runs yielded R2 values > 0.99 and misclassification rates of zero, indicating that the genomic contents of the HL and LL strains are sufficient to classify them into their respective environments.
VFam Analysis
Viral family sequences within genomes were discovered using HMMs built from Markov clustering and multiple sequence alignments[81] from viral proteins in RefSeq as of 2014. These VFams can detect viral sequences with high accuracy and did not cluster non-viral sequences into the Markov clusters in the published test sets. We used a strict 1e-9 E-value to call VFam domains in the translated CDS (tCDS) from the P. marinus genomes. The command was ‘'hmmsearch --noali -E 0.000000001 --cpu 28 --domtblout $OUT $IN.aa.fa VFam.hmm'.
The assemblies for P. marinus used in the manuscript predicted 3762 VFams that were utilized for bi-clustered heatmap visualization in Morpheus: a web tool for visualizing the clustering of multivariate data (https://software.broadinstitute.org/morpheus/). Clustering was performed on VFams and species (bi-clustering) using Pearson correlation scores, and heatmaps were visualized in Morpheus using sum HMM match scores as a color Z-depth as in the Pfam analyses.
Metabolic Pathway Analysis
Interactive Pathways Explorer 3 (iPath: https://pathways.embl.de) is a web-based tool for the visualization, analysis, and customization of various pathway maps[86]. iPath provides extensive map customization and data mapping capabilities. We used EC numbers (Enzyme Codes) from the BLAST2GO analysis for both MED4 and NATL1A and mapped the metabolic pathways, comparing unique and shared pathways between the 2 HL- and LL-strains.
Functional Annotation Analysis using Blast2GO
cDNA sequences of Prochlorococcus marinus MED4 and NATL1A were obtained from the National Center for Biotechnology Information (NCBI) and the Joint Genome Institute (JGI), respectively. The Blast2GO analysis was performed using Blast2GO Command Line Version 1.5.1 with the following parameters: (1) NCBI NR Database: A custom database was created using the latest NCBI non-redundant (nr) fasta file as of October 1, 2022, totaling 136GB. (2) BLASTX Alignment: Primary sequence alignments were obtained using BLASTX 2.13.0+ with the following parameters: -evalue 0.001, -show_gis, -max_target_seqs 5, -num_threads 60, -mt_mode 0, -outfmt 5. (3) GO Database: The Gene Ontology (GO) database used was go_latest.obo as of October 2022. Blast2GO is a BLAST-based tool used for large-scale functional annotation of novel sequence data of non-model species. B2G development is the creation of a comprehensive, user-friendly, and research-oriented framework for large-scale function assignments[58].
ORFeome Synthesis and Cloning
Information about gene annotation and cDNA sequence were extracted for the complete genomes of MED4 and NATL1A strains from the EnsemblBacteria database (GCA_000011465.1 and GCA_000011485.1 respectively). ORFs were synthesized in collaboration with Twist Bioscience (San Francisco, USA; for the majority of the ORFeomes) and BGI (Shenzhen, China; 99 ORFs for the MED4 strain ranging from 78bp to 4500bp; including the longest gene sequences from 3000bp-4566bp).
The synthesized ORFs are flanked by the Gateway L1 and L2 sites:
ATTL1:caaataatgattttattttgactgatagtgacctgttcgttgcaacacattgatgagcaatgcttttttataatgccaactttgtacaaaaaagcaggctac
ATTL2:ttggacccagctttcttgtacaaagttggcattataagaaagcattgcttatcaatttgttgcaacgaacaggtcactatcagtcaaaataaaatcattatttg
DNA vectors were received in 96-well or 384-well plates as DNA and glycerol stocks. Uni9 primers can be used to amplify the ORFs (outside the ATTL sequences).
Uni 9 for (5' – 3'): GAAGTGCCATTCCGCCTGACCT
Uni 9 rev (5' – 3): CACTGAGCCTCCACCTAGCCT
Gateway Transfer and Sequence Validation
A set of MED4 ORFs was recombinationally cloned into yeast expression vectors (pAD and pDB) [87] using LR clonase (Invitrogen) according to the manufacturer’s instructions. Expression clones were subsequently transformed into chemically competent E. coli DH5α (Mix & Go competent cells Zymo 5z). Transformants were cultured as minipools in liquid LB with ampicillin (100mg/l). Following growth in liquid media, the transformed bacteria were used as a source of template for DNA extraction with GeneJET plasmid miniprep kit from Thermo Scientific following manufacturer’s instructions. Cloning was verified with PCR using KOD hot-start DNA polymerase (Sigma-Aldrich) with pAD and pDB-specific primers, followed by gel electrophoresis.
A set of amplified clones was verified by sequencing bi-directionally at 1st base sequencing (Apical Scientific Sdn Bhd). Forward and reverse sequences were mapped to reference using ChromasPro version 2.1.5 http://www.technelysium.com/ChromasPro.html.
AD-forward: 5’-CGCGTTTGGAATCACTACAGGG-3’
DB-forward: 5’-GGCTTCAGTGGAGACTGATATGCCTC-3’
Term-reverse: 5’- GGAGACTTGACCAAACCTCTGGCG-3’
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supplemental Figure 1: Metabolic pathways representation for MED4 and NATL1A using iPath3 showing common and unique pathways for MED4 and NATL1A. Blue and red links represent unique pathways for MED4 and NATL1A, respectively, while green links represent the shared pathways among the two strains. Supplemental Figure 2: GO-term enrichment analysis comparing Pfams distinguishing high- and low-light P. marinus strains. Venn diagrams representing enrichment for biological processes and molecular functions GO-terms are shown in the left and right panels, respectively. High-light (HL) and low-light (LL) are highlighted in the blue and orange circles, respectively. Supplemental Figure 3: Schematic representation of ORFs entry clones and cloning into Y2H expression vectors. MED4 and NATL1A ORFs were synthesized in entry vectors, flanked with ATTL sites compatible with Gateway recombinational cloning. An example of LR Clonase recombination reaction with Y2H pAD and pDB destination vectors is represented. ATT sites, and antibiotic resistance are represented for each vector. In addition, pAD and pDB selectable markers are indicated, where TRP1 and LEU2 enable the growth of specific yeast strains in media lacking tryptophane and leucine, respectively. Supplemental Figure 4: Cloning and transformation of a selected set of ORFs for MED4 strains. A set of 70 ORFs were cloned into pAD and pDB yeast expression vectors and validated with PCR with 85% cloning success rate. Fragments ranging from 302bp to 3030bp for MED4 strain. Expression vectors were transformed into yeast haploid cells and verified with a PCR on transformant yeast colonies. Supplemental tables: Table S1. Bit scores for predicted Pfam domains from the 40 queried strains.Table S2.GO-term enrichment results. Table S3. Response screening results. Table S4. aNN top Pfam contributors. Table S5. VFam scores. Table S6. Blast2GO analyses results. Table S7.A MED4 ORFeome. Table S7.B NATL1A ORFeome. Supplemental data: Data S1. Pfam annotations. Data S2. Response screening JMP project files. Data S3. Python scripts.
Author Contributions
(tentative, to be verified by authors) Conceptualization: K.S.-A., S.D., D.R.N., J.K., and W.F.; experimental validations and gene synthesis: S.D, X.H, and Y.S.; Interpretation of the results: S.D., D.R. N., and K.S.-A.; Gene annotation and bioinformatics: A.J., S.D., D.R.N., and B.D.; Deep learning and artificial neural networks: D.R.N.; writing – original draft, S.D., D.R.N., and K.S.-A.; review and editing, W.F., A.A., A.J., J.K., X.H., and J.-C.T.; supervision of the study: K.S.-A.
Acknowledgments
We thank NYU and NYUAD HPC platforms for providing computational resources. This research was supported by NYUAD Faculty Research Funds (AD060) and Tamkeen under the NYU Abu Dhabi Research Institute Award to the NYUAD Center for Genomics and Systems Biology (73 71210 CGSB9).
References
- Percival, S.L. & Williams, D.W. in Microbiology of Waterborne Diseases (Second Edition). (eds. S.L. Percival, M.V. Yates, D.W. Williams, R.M. Chalmers & N.F. Gray) 79-88 (Academic Press, London; 2014).
- Demoulin, C.F. et al. Cyanobacteria evolution: Insight from the fossil record. Free Radic Biol Med 140, 206-223 (2019). [CrossRef]
- Ruffing, A.M. Engineered cyanobacteria: Teaching an old bug new tricks. Bioengineered Bugs 2, 136-149 (2011). [CrossRef]
- Goericke, R. & Welschmeyer, N.A. The marine prochlorophyte Prochlorococcus contributes significantly to phytoplankton biomass and primary production in the Sargasso Sea. Deep Sea Research Part I: Oceanographic Research Papers 40, 2283-2294 (1993). [CrossRef]
- Scanlan, D.J. et al. Ecological genomics of marine picocyanobacteria. Microbiol Mol Biol Rev 73, 249-299 (2009). [CrossRef]
- Partensky, F., Hess, W.R. & Vaulot, D. Prochlorococcus, a marine photosynthetic prokaryote of global significance. Microbiology and Molecular Biology Reviews 63, 106 (1999). [CrossRef]
- Moore, L.R., Rocap, G. & Chisholm, S.W. Physiology and molecular phylogeny of coexisting Prochlorococcus ecotypes. Nature 393, 464-467 (1998). [CrossRef]
- Luo, H., Huang, Y., Stepanauskas, R. & Tang, J. Excess of non-conservative amino acid changes in marine bacterioplankton lineages with reduced genomes. Nature Microbiology 2, 17091 (2017). [CrossRef]
- Kettler, G.C. et al. Patterns and implications of gene gain and loss in the evolution of Prochlorococcus. PLoS genetics 3, e231-e231 (2007). [CrossRef]
- O'Leary, N.A. et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44, D733-D745 (2015). [CrossRef]
- Dufresne, A. et al. Genome sequence of the cyanobacterium Prochlorococcus marinus SS120, a nearly minimal oxyphototrophic genome. Proc Natl Acad Sci U S A 100, 10020-10025 (2003).
- Rocap, G. et al. Genome divergence in two Prochlorococcus ecotypes reflects oceanic niche differentiation. Nature 424, 1042-1047 (2003). [CrossRef]
- Weitz, J.S. & Wilhelm, S.W. Ocean viruses and their effects on microbial communities and biogeochemical cycles. F1000 Biol Rep 4, 17 (2012). [CrossRef]
- Chen, M.Y. et al. Comparative genomics reveals insights into cyanobacterial evolution and habitat adaptation. ISME J 15, 211-227 (2021). [CrossRef]
- Yang, F. et al. Function of Protein Kinases in Leaf Senescence of Plants. Frontiers in Plant Science 13 (2022). [CrossRef]
- Andrés, G., María, F.F., María-Teresa, B., María-Luisa, P. & Emma, S. in Cyanobacteria. (ed. T. Archana) Ch. 6 (IntechOpen, Rijeka; 2018).
- Jia, A., Zheng, Y., Chen, H. & Wang, Q. Regulation and Functional Complexity of the Chlorophyll-Binding Protein IsiA. Front Microbiol 12, 774107 (2021). [CrossRef]
- Kroh, G.E. & Pilon, M. in International Journal of Molecular Sciences, Vol. 21 (2020).
- Christensen, Q.H. & Cronan, J.E. Lipoic acid synthesis: a new family of octanoyltransferases generally annotated as lipoate protein ligases. Biochemistry 49, 10024-10036 (2010). [CrossRef]
- Cronan, J.E. Assembly of Lipoic Acid on Its Cognate Enzymes: an Extraordinary and Essential Biosynthetic Pathway. Microbiol Mol Biol Rev 80, 429-450 (2016). [CrossRef]
- Bristow, L.A., Mohr, W., Ahmerkamp, S. & Kuypers, M.M.M. Nutrients that limit growth in the ocean. Curr Biol 27, R474-r478 (2017). [CrossRef]
- Latifi, A., Ruiz, M. & Zhang, C.-C. Oxidative stress in cyanobacteria. FEMS Microbiology Reviews 33, 258-278 (2009). [CrossRef]
- Santamaría-Gómez, J., Ochoa de Alda, J.A.G., Olmedo-Verd, E., Bru-Martínez, R. & Luque, I. Sub-Cellular Localization and Complex Formation by Aminoacyl-tRNA Synthetases in Cyanobacteria: Evidence for Interaction of Membrane-Anchored ValRS with ATP Synthase. Frontiers in Microbiology 7 (2016).
- Luque, I., Riera-Alberola, M.L., Andújar, A. & Ochoa de Alda, J.A.G. Intraphylum Diversity and Complex Evolution of Cyanobacterial Aminoacyl-tRNA Synthetases. Molecular Biology and Evolution 25, 2369-2389 (2008).
- Song, K. et al. AtpΘ is an inhibitor of F0F1 ATP synthase to arrest ATP hydrolysis during low-energy conditions in cyanobacteria. Current Biology 32, 136-148.e135 (2022).
- Cassier-Chauvat, C., Veaudor, T. & Chauvat, F. Comparative Genomics of DNA Recombination and Repair in Cyanobacteria: Biotechnological Implications. Front Microbiol 7, 1809 (2016). [CrossRef]
- Kolowrat, C. et al. Ultraviolet stress delays chromosome replication in light/dark synchronized cells of the marine cyanobacterium Prochlorococcus marinus PCC9511. BMC Microbiology 10, 204 (2010). [CrossRef]
- Osburne, M.S. et al. UV hyper-resistance in Prochlorococcus MED4 results from a single base pair deletion just upstream of an operon encoding nudix hydrolase and photolyase. Environ Microbiol 12, 1978-1988 (2010). [CrossRef]
- Cassier-Chauvat, C. & Chauvat, F. Responses to oxidative and heavy metal stresses in cyanobacteria: recent advances. Int J Mol Sci 16, 871-886 (2014). [CrossRef]
- Berube, P.M., Rasmussen, A., Braakman, R., Stepanauskas, R. & Chisholm, S.W. Emergence of trait variability through the lens of nitrogen assimilation in Prochlorococcus. Elife 8 (2019).
- Varkey, D. et al. Effects of low temperature on tropical and temperate isolates of marine Synechococcus. The ISME Journal 10, 1252-1263 (2016). [CrossRef]
- Knoll, A. & Puchta, H. The role of DNA helicases and their interaction partners in genome stability and meiotic recombination in plants. Journal of Experimental Botany 62, 1565-1579 (2010). [CrossRef]
- Hilbert, M., Karow, A.R. & Klostermeier, D. The mechanism of ATP-dependent RNA unwinding by DEAD box proteins. 390, 1237-1250 (2009). [CrossRef]
- Muzzopappa, F. et al. Paralogs of the C-Terminal Domain of the Cyanobacterial Orange Carotenoid Protein Are Carotenoid Donors to Helical Carotenoid Proteins. Plant Physiol 175, 1283-1303 (2017). [CrossRef]
- Cerveny, L. et al. Tetratricopeptide repeat motifs in the world of bacterial pathogens: role in virulence mechanisms. Infect Immun 81, 629-635 (2013). [CrossRef]
- Grove, T.Z., Cortajarena, A.L. & Regan, L. Ligand binding by repeat proteins: natural and designed. Curr Opin Struct Biol 18, 507-515 (2008). [CrossRef]
- Rast, A., Rengstl, B., Heinz, S., Klingl, A. & Nickelsen, J. The Role of Slr0151, a Tetratricopeptide Repeat Protein from Synechocystis sp. PCC 6803, during Photosystem II Assembly and Repair. Frontiers in Plant Science 7 (2016). [CrossRef]
- Biller, S.J., Coe, A. & Chisholm, S.W. Torn apart and reunited: impact of a heterotroph on the transcriptome of Prochlorococcus. The ISME Journal 10, 2831-2843 (2016). [CrossRef]
- Kong, R., Xu, X. & Hu, Z. A TPR-family membrane protein gene is required for light-activated heterotrophic growth of the cyanobacterium Synechocystis sp. PCC 6803. FEMS Microbiol Lett 219, 75-79 (2003).
- Morimoto, K., Nishio, K. & Nakai, M. Identification of a novel prokaryotic HEAT-repeats-containing protein which interacts with a cyanobacterial IscA homolog. FEBS Lett 519, 123-127 (2002). [CrossRef]
- Hu, P.-P. et al. The role of lyases, NblA and NblB proteins and bilin chromophore transfer in restructuring the cyanobacterial light-harvesting complex‡. The Plant Journal 102, 529-540 (2020).
- Gisriel, C.J. et al. Structure of a dimeric photosystem II complex from a cyanobacterium acclimated to far-red light. J Biol Chem 299, 102815 (2023). [CrossRef]
- Safferman, R.S. et al. Classification and nomenclature of viruses of cyanobacteria. Intervirology 19, 61-66 (1983). [CrossRef]
- Dammeyer, T., Bagby, S.C., Sullivan, M.B., Chisholm, S.W. & Frankenberg-Dinkel, N. Efficient phage-mediated pigment biosynthesis in oceanic cyanobacteria. Curr Biol 18, 442-448 (2008). [CrossRef]
- Voorhies, A.A. et al. Ecological and genetic interactions between cyanobacteria and viruses in a low-oxygen mat community inferred through metagenomics and metatranscriptomics. Environ Microbiol 18, 358-371 (2016). [CrossRef]
- Moniruzzaman, M., Weinheimer, A.R., Martinez-Gutierrez, C.A. & Aylward, F.O. Widespread endogenization of giant viruses shapes genomes of green algae. Nature 588, 141-145 (2020). [CrossRef]
- Rozenberg, A. et al. Lateral Gene Transfer of Anion-Conducting Channelrhodopsins between Green Algae and Giant Viruses. Curr Biol 30, 4910-4920.e4915 (2020). [CrossRef]
- Chelkha, N., Levasseur, A., La Scola, B. & Colson, P. Host-virus interactions and defense mechanisms for giant viruses. Ann N Y Acad Sci (2020). [CrossRef]
- Yoosuf, N. et al. Complete genome sequence of Courdo11 virus, a member of the family Mimiviridae. Virus Genes 48, 218-223 (2014). [CrossRef]
- de Aquino, I.L.M. et al. Diversity of Surface Fibril Patterns in Mimivirus Isolates. J Virol 97, e0182422 (2023).
- . Bisio, H. et al. Evolution of giant pandoravirus revealed by CRISPR/Cas9. Nat Commun 14, 428 (2023). [CrossRef]
- Hikida, H., Okazaki, Y., Zhang, R., Nguyen, T.T. & Ogata, H. A rapid genome-wide analysis of isolated giant viruses using MinION sequencing. Environ Microbiol 25, 2621-2635 (2023). [CrossRef]
- Esmael, A., Agarkova, I.V., Dunigan, D.D., Zhou, Y. & Van Etten, J.L. Viral DNA Accumulation Regulates Replication Efficiency of Chlorovirus OSy-NE5 in Two Closely Related Chlorella variabilis Strains. Viruses 15 (2023)abilis Strains. [CrossRef]
- Fernández-García, J.L., de Ory, A., Brussaard, C.P.D. & de Vega, M. Phaeocystis globosa Virus DNA Polymerase X: a "Swiss Army knife", Multifunctional DNA polymerase-lyase-ligase for Base Excision Repair. Sci Rep 7, 6907 (2017).
- Derelle, E. et al. Diversity of Viruses Infecting the Green Microalga Ostreococcus lucimarinus. J Virol 89, 5812-5821 (2015). [CrossRef]
- Delaroque, N., Maier, I., Knippers, R. & DG, M.l. Persistent virus integration into the genome of its algal host, Ectocarpus siliculosus (Phaeophyceae). J Gen Virol 80 ( Pt 6), 1367-1370 (1999). [CrossRef]
- Haller, S.L., Peng, C., McFadden, G. & Rothenburg, S. Poxviruses and the evolution of host range and virulence. Infect Genet Evol 21, 15-40 (2014). [CrossRef]
- Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674-3676 (2005).
- Kehr, J.C. & Dittmann, E. Biosynthesis and function of extracellular glycans in cyanobacteria. Life (Basel) 5, 164-180 (2015). [CrossRef]
- Walhout, A.J. et al. in Methods in enzymology, Vol. 328 575-IN577 (Elsevier, 2000).
- Hartley, J.L., Temple, G.F. & Brasch, M.A. DNA cloning using in vitro site-specific recombination. Genome Res 10, 1788-1795 (2000). [CrossRef]
- Dreze, M. et al. in Methods in Enzymology, Vol. 470 281-315 (Academic Press, 2010).
- Sánchez-Baracaldo, P. Origin of marine planktonic cyanobacteria. Scientific Reports 5, 17418 (2015). [CrossRef]
- Ulloa, O. et al. The cyanobacterium <i>Prochlorococcus</i> has divergent light-harvesting antennae and may have evolved in a low-oxygen ocean. Proceedings of the National Academy of Sciences 118, e2025638118 (2021).
- El-Seedi, H.R. et al. Review of Marine Cyanobacteria and the Aspects Related to Their Roles: Chemical, Biological Properties, Nitrogen Fixation and Climate Change. Mar Drugs 21 (2023). [CrossRef]
- Flombaum, P. et al. Present and future global distributions of the marine Cyanobacteria <i>Prochlorococcus</i> and <i>Synechococcus</i>. Proceedings of the National Academy of Sciences 110, 9824-9829 (2013).
- Puxty, R.J., Millard, A.D., Evans, D.J. & Scanlan, D.J. Shedding new light on viral photosynthesis. Photosynth Res 126, 71-97 (2015). [CrossRef]
- James, J.E., Nelson, P.G. & Masel, J. Differential Retention of Pfam Domains Contributes to Long-term Evolutionary Trends. Molecular Biology and Evolution 40, msad073 (2023). [CrossRef]
- Godbold, J.A. & Calosi, P. Ocean acidification and climate change: advances in ecology and evolution. Philos Trans R Soc Lond B Biol Sci 368, 20120448 (2013). [CrossRef]
- Beardall, J., Stojkovic, S. & Larsen, S. Living in a high CO2 world: impacts of global climate change on marine phytoplankton. Plant Ecology & Diversity 2, 191-205 (2009). [CrossRef]
- Hallegraeff, G.M. Ocean climate change, phytoplankton community responses, and harmful algal blooms: a formidable predictive challenge 1. Journal of phycology 46, 220-235 (2010).
- Shestakov, S.V. & Karbysheva, E.A. The role of viruses in the evolution of cyanobacteria. Biology Bulletin Reviews 5, 527-537 (2015). [CrossRef]
- Zhaxybayeva, O., Doolittle, W.F., Papke, R.T. & Gogarten, J.P. Intertwined evolutionary histories of marine Synechococcus and Prochlorococcus marinus. Genome Biol Evol 1, 325-339 (2009). [CrossRef]
- Zeidner, G. et al. Potential photosynthesis gene recombination between Prochlorococcus and Synechococcus via viral intermediates. Environ Microbiol 7, 1505-1513 (2005). [CrossRef]
- Lindell, D. et al. Genome-wide expression dynamics of a marine virus and host reveal features of co-evolution. Nature 449, 83-86 (2007). [CrossRef]
- Gong, W. et al. Genome-wide ORFeome cloning and analysis of Arabidopsis transcription factor genes. Plant Physiol 135, 773-782 (2004). [CrossRef]
- Kang, S.E., Breton, G. & Pruneda-Paz, J.L. Construction of Arabidopsis Transcription Factor ORFeome Collections and Identification of Protein-DNA Interactions by High-Throughput Yeast One-Hybrid Screens. Methods Mol Biol 1794, 151-182 (2018).
- Rual, J.-F. et al. Human ORFeome Version 1.1: A Platform for Reverse Proteomics. Genome Research 14, 2128-2135 (2004). [CrossRef]
- Lamesch, P. et al. hORFeome v3.1: a resource of human open reading frames representing over 10,000 human genes. Genomics 89, 307-315 (2007). [CrossRef]
- Rajagopala, S.V. et al. The Escherichia coli K-12 ORFeome: a resource for comparative molecular microbiology. BMC Genomics 11, 470-470 (2010). [CrossRef]
- Özkan, E. et al. An extracellular interactome of immunoglobulin and LRR proteins reveals receptor-ligand networks. Cell 154, 228-239 (2013).
- Ghamsari, L. et al. Genome-wide functional annotation and structural verification of metabolic ORFeome of Chlamydomonas reinhardtii. BMC Genomics 12 Suppl 1, S4-S4 (2011). [CrossRef]
- Pellet, J. et al. ViralORFeome: an integrated database to generate a versatile collection of viral ORFs. Nucleic Acids Res 38, D371-D378 (2010)n integrated database to generate a versatile collection of viral ORFs. [CrossRef]
- Fu, W., Nelson, D.R., Mystikou, A., Daakour, S. & Salehi-Ashtiani, K. Advances in microalgal research and engineering development. Current Opinion in Biotechnology 59, 157-164 (2019). [CrossRef]
- Metsalu, T. & Vilo, J. ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res 43, W566-W570 (2015). [CrossRef]
- Darzi, Y., Letunic, I., Bork, P. & Yamada, T. iPath3.0: interactive pathways explorer v3. Nucleic Acids Res 46, W510-W513 (2018). [CrossRef]
- Rual, J.-F. et al. Towards a proteome-scale map of the human protein–protein interaction network. Nature 437, 1173-1178 (2005). [CrossRef]
Figure 1.
Partial least squares and hierarchical biclustering of Pfams from 40 P. marinus isolates. (A) Cluster analysis using cluster-based partial least squares (PLS) of the 40 P. marinus strains. This method shows separate clustering for high-light and low-light strains represented in blue and orange, respectively. (B) Heatmap of Pfam domains for 40 P. marinus species showing clustering of distinct Pfam groups in high-light and low-light strains on the right and left sides, respectively. The 40 strains are represented on the X-axis with LL for Low-light and HL for High-light; approximately 1000 different Pfams are represented on the Y-axis.
Figure 1.
Partial least squares and hierarchical biclustering of Pfams from 40 P. marinus isolates. (A) Cluster analysis using cluster-based partial least squares (PLS) of the 40 P. marinus strains. This method shows separate clustering for high-light and low-light strains represented in blue and orange, respectively. (B) Heatmap of Pfam domains for 40 P. marinus species showing clustering of distinct Pfam groups in high-light and low-light strains on the right and left sides, respectively. The 40 strains are represented on the X-axis with LL for Low-light and HL for High-light; approximately 1000 different Pfams are represented on the Y-axis.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



