
Submitted:
02 May 2024
Posted:
03 May 2024
You are already at the latest version
Abstract
Transient receptor potential melastatin-8 (TRPM8) is a cation channel that is activated by cold and “cooling agents” such as menthol and icilin, which induce a cold sensation. Stimulation of TRPM8 activates an intracellular signaling cascade that ultimately leads to a change in the gene expression pattern of the cells. Here, we investigated the TRPM8-induced signaling pathway that links TRPM8 channel activation to gene transcription. Using a pharmacological approach, we show that inhibition of phosphatidylinositol 4-phosphate 5 kinase alpha (PIP5K), an enzyme essential for the biosynthesis of phosphatidylinositol 4,5-bisphosphate, attenuated TRPM8-induced gene transcription. Analyzing the link between TRPM8 and Gq proteins, we show that pharmacological inhibition of the beta gamma subunits impairs TRPM8 signaling. In addition, genetic studies showed that TRPM8 requires an activated Galpha subunit for signaling. In the nucleus, the TRPM8-induced signaling cascade triggers an activation of the transcription factor AP-1, a complex consisting of a dimer of basic region leucine zipper (bZIP) transcription factors. Here, we identified the bZIP protein c-Jun as an essential component of AP-1 within the TRPM8-induced signling cscade. In summary, with PIP5K, Gq subunits and c-Jun we have identified key molecules in the TRPM8-induced signaling from the plasma membrane to the nucleus.
Keywords:
c-Jun
; G-protein
; Gαq-coupled receptor
; galanin
; ISA-2011B
; phosphatidylinositol 4-phosphate 5 kinase
; RGS2
; TRPM3
; TRPM8
1. Introduction
TRPM8 (transient receptor potential melastatin-8) was identified as a menthol receptor using an expression cloning approach [1]. Menthol, a p-menthane-3-ol derived from the oil of peppermint, is known to induce a cold sensation. Analysis of the primary structure revealed that the receptor belongs to the TRP family of cation channels. The TRPM8 cDNA was simultaneously identified using a genomic DNA database search and PCR from a dorsal root ganglion cDNA library [2]. The TRPM8 channel is activated by cold temperature and by cooling substances such as menthol, eucalyptol, and the synthetic “super-cooling agonist” icilin [3]. TRPM8, like other TRP channels, is a polymodal sensor that integrates temperature and chemical sensations and play an essential role in thermosensation, as shown by the analysis of TRPM8-deficient mice [4,5,6]. TRPM8 is found in sensory neurons, where it functions as a cold nociceptor which mediates nocifensive responses to noxious cold [5,6]. In addition, TRPM8 channels are involved in cold hypersensitivity triggered by nerve injury and inflammation. TRPM8 channels are also thought to be involved in the development of migraine, the development of tumors and other diseases [7,8,9]. An anti-inflammatory role has also been proposed for TRPM8 [10,11].
Stimulation of TRPM8 channels triggers an intracellular signaling pathway that leads to a change in the gene expression pattern of the cells. We are interested in identifying the signaling molecules that are essential for the link between TRPM8 stimulation and gene transcription. Previous studies have shown that an influx of Ca2+ ions is essential for the continuation of the signaling cascade after TRPM8 stimulation [12]. The extracellular signal-regulated protein kinase ERK1/2 was identified as an intracellular signal transducter [13]. In addition, calmodulin, calcineurin, and phospholipase C (PLC) β have been identified as important molecules that enable signal transduction from the plasma membran to the nucleus after TRPM8 channel stimulation [14,15]. However, the TRPM8-induced signaling cascade is far from being described in detail.
In this study, we focused our attention on the role of phosphatidylinositol 4,5-bisphosphate and trimeric G-protein subunits within the TRPM8-induced signaling cascade. Several reports have described the regulation of TRPM8 channels by phosphatidylinositol 4,5-bisphosphate [16,17,18,19,20,21], based on genetic tools that induced dephosphorylation of phosphatidylinositol 4,5-bisphosphate. We explored whether we could attenuate the signaling pathway of TRPM8 by interfering with the biosynthesis of phosphatidylinositol 4,5-bisphosphate.
In has been suggested that G-proteins modulate TRPM8 signaling, although this issue remained controversial. Stimulation of Gαq-coupled receptors activates phospholipase C, which catalyzes the hydrolysis of phosphatidylinositol 4,5-bisphosphate, thereby reducing phosphatidylinositol 4,5-bisphosphate levels. However, stimulation of Gαq-coupled receptors also leads to a rise in cytosolic Ca2+ and activation of protein kinase C and other protein kinases that could influence channel activity. Direct binding of the Gαq subunit to TRPM8 has been shown [22], but it is controversial whether the activated Gαq stimulates or inhibits TRPM8 [22,23] and whether phospholipase C is involved or not. In this study, we used pharmacological and genetic strategies to elucidate the roles of the α and βγ subunits of Gq-coupled receptors in TRPM8-mediated signaling.
Recently, we have shown that stimulation of TRPM8 channels activates the transcription factor AP-1 [13,24]. AP-1 is composed of two basic region leucine zipper (bZIP) transcription factors of the Jun, Fos and ATF families of transcription factors. We asked, which bZIP proteins is involved in the genetic changes in the nucleus after stimulation of TRPM8 channels.
2. Results
2.1. Biosynthesis of Phosphatidylinositol 4,5-Bisphosphate
Phosphatidylinositol 4,5-bisphosphate, an important lipid involved in the control of numerous signaling pathways, is mainly synthesized from phosphatidylinositol 4-phosphate [25], a reaction catalyzed by the enzyme PIP5K, which performs the transfer of a phosphate group to the 5´-position of the inositol ring (Figure 1). Phospholipase C enzymes catalyze the hydrolysis of phosphatidylinositol 4,5-bisphosphate to generate the second messengers IP3 and diacylglycerol. Phosphatidylinositol 4,5-bisphosphate further functions as a substrate for phosphatidylinositol 3-kinases, which catalyze the transfer of a phosphate group to the 3´-position of the inositol ring, thus generating phosphatidylinositol 3,4,5-trisphosphate (Figure 1), a metabolite which is essential for the activation of the phosphoinositide-dependent protein kinase AKT.
2.2. Pharmacological Inhibition of Phosphatidylinositol 4,5-Bisphosphate Biosynthesis Interferes with Signaling via the TRPM8 Channel
Figure 2a shows the modular structure of TRPM8 channels, revealing the typical architecture of TRP channels with 6 transmembrane domains and both N- and C-termini in the cytoplasm. The cartoon also shows the proposed interaction sites of phosphatidylinositol 4,5-bisphosphate with the TRPM8 ion channel [26].
A variety of approaches have been used to manipulate the levels of phosphatidylinositol 4,5-bisphosphate in the plasma membrane, including Gαq-coupled receptor-mediated activation of phospholipase C, expression of a voltage-dependent lipid phosphatase, or administration of various compounds designed to reduce phosphatidylinositol 4,5-bisphosphate levels. In this study, we used a pharmacological approach to inhibit the biosynthesis of phosphatidylinositol 4,5-bisphosphate by incubating the cells with the PIP5Kα inhibitor ISA-2011B (Figure 2b). This compound has been shown to significantly inhibit PIP5K activity and blocks the subsequent activation of AKT [27,28].
Stimulation of various TRP channels (TRPC6, TRPM3, TRPM8, TRPV1) leads to activation of the transcription factor AP-1 [24,29,30,31]. In addition, AP-1 is activated by stimulation of voltage-gated Cav1.2 Ca2+-channels and Gαq-coupled receptors [32,33,34]. We investigate the signaling pathway from the plasma membran to the nucleus and use the activation of AP-1 as a measure for the nuclear response to TRPM8 stimulation. Frequently, calcium imaging techniques and/or patch-clamp electrophysiology are used as an indicator for TRPM8 activation. Our chosen approach has the advantage that we have traced a TRPM8-induced intracellular signaling cascade from the plasma membrane to the nucleus. A collagenase promoter/luciferase reporter gene, shown in Figure 2c, was used as a sensor to detect changes in AP-1 activity [35]. This transcription unit contains an AP-1 binding site, also known as an 12-O-tetradecanoylphorbol-13-acetate (TPA)-responsive element (TRE), in the proximal promoter region. The reporter gene was integrated into the chromatin of the cells using lentiviral gene transfer. This strategy ensured that the reporter gene was embedded into a nucleosomal structure. HEK293-M8 cells were infected with a lentivirus containing the Coll.luc reporter gene. Cells were preincubated with the compound ISA-2011B for 3 hours and then stimulated for 24 hours with icilin in the presence of the PIP5K inhibitor. Figure 2d shows that administration of ISA-2011B strongly inhibited TRPM8 intracellular signaling. AP-1 activity was reduced by 81 % in the presence of ISA-2011B
2.3. Pharmacological Inhibition of Phosphatidylinositol 4,5-Bisphosphate Biosynthesis Interferes with Signaling via the TRPM3 Channel
As a control, we examined the effect of ISA-2011B application on the activation of another TRPM cation channel, TRPM3. TRPM3, similar to TRPM8, is a polymodal channel that can be activated by heat and by chemicals such as the steroid pregnenolone sulfate [36,37]. Activation of TRPM3 has been linked to heat and pain sensation, gene transcription, vascular smooth muscle contraction, insulin secretion, and tumorigenesis [38]. Recent analysis of gain-of-function mutations of TRPM3 have revealed a role of this ion channel in chronic fatigue syndrome/myalgic encephalomyelitis and in the development of neuronal disorders [39,40]. Using cell-free inside-out patches, TRPM3 activity has been shown to increase in response to phosphoinositides, and was reduced after expression of a voltage-sensing phosphatase [21,41]. Figure 3a shows the modular structure of TRPM3, including the proposed interaction sites with phosphatidylinositol 4,5,-bisphosphate. Figure 3b shows that incubation of T-Rex-TRPM3 cells, HEK293 cells expressing a tetracyclin-inducible TRPM3 expression casette, with the PIP5K inhibitor ISA-2011B significantly reduced intracellular signaling after stimulation of TRPM3 channels with pregnenolone sulfate. Quantification revealed that administration of ISA-2011B resulted in 88% inhibition of TRPM3 signaling as measured with the Coll.luc sensor.
2.4. Pharmacological Inhibition Of Phosphatidylinositol 4,5-Bisphosphate Biosynthesis Interferes with Signaling via the Voltage-Gated Cav1.2. Ca2+ Channel
The activity of voltage-gated Ca2+ channels depends on the presence of phosphatidylinositol 4,5-bisphosphate in the plasma membrane, as shown by experiments resulting in a dephosphorylation of phosphatidylinositol 4,5-bisphosphate by a inositol lipid 5´-phosphatase [42], which convert phosphatidylinositol 4,5-bisphosphate into phosphatidylinositol 4-phosphate. Figure 3c shows the modular structure of Cav1.2 L-type voltage-gated Ca2+ channels, which consist of five subunits, the pore-forming α1 subunit and three auxiliary subunits α2δ, β and γ. The cartoon clearly shows that the modular structure of Cav1.2 channels is completely different from that of the TRP channels TRPM8 and TRPM3. Nevertheless, Cav1.2 and the TRPM8 and TRPM3 channels require phosphatidylinositol 4,5-bisphosphate for activation. As a further control for the previous experiments, we analyzed whether inhibition of PIP5Kα impairs Cav1.2 Ca2+ channel signaling. As a measure of Cav1.2 Ca2+ channel activity, we measured the activation of AP-1 in Cav1.2 channel-expressing insulinoma cells [32,33]. Figure 3d shows that administration of KCl and the compound FPL64176 to INS-1 832/13 insulinoma cells resulted in a strong activation of Cav1.2 Ca2+ channels. Inhibition of phosphatidylinositol 4,5-bisphosphate biosynthesis with the PIP5Kα inhibitor ISA-2011B significantly reduced signaling via the Cav1.2 Ca2+ channel in insulinoma cells. Administration of ISA-2011B resulted in 84% inhibition of Cav1.2 Ca2+ channel signaling. We conclude that inhibition of PIP5K strongly interferes with signaling of TRPM8, TRPM3 and Cav1.2 channel.
2.5. Overexpression of Regulator of G-Protein Signaling-2 (RGS2) Blocks the Activation of AP-1 after Stimulation of TRPM8 Channels
It has been proposed that the activated GTP-bound Gαq subunit of trimeric Gq proteins binds directly to TRPM8 channels, resulting in an inhibition of channel activity [22]. To test this assumption, we expressed regulator of G protein signaling-2 (RGS2) in HEK293-M8 cells, stimulated the cells with icilin, and measured the AP-1 activity using the Coll.luc sensor. RGS2 (Figure 4a) accelerates the rate of hydrolysis of GTP-Gαq to GDP-Gαq. The inactive GDP-Gαq then binds to Gβγ and forms an inactive trimeric G protein [43]. Figure 4b shows that expression of RGS2 impairs icilin-induced activation of AP-1. Expression of RGS2 reduced AP-1 activity in icilin-stimulated cells by 82%. These results suggest that stimulation of TRPM8 channels with icilin requires an activated Gαq subunit. As a positive control, we show that overexpression of RGS2 reduced signaling of Gαq-coupled receptors on the order of 82% (Figure 4c). Overexpression of RGS2 did not alter signaling pathways following stimulation of either TRPM3 channels (Figure 4d) or voltage-gated Cav1.2 Ca2+ channels (Figure 4e).
2.6. Pharmacological Inhibition of Gβγ Interferes with Signaling via the TRPM3 and TRPM8 Channels
TRPM3 ion channels are regulated by the Gβγ subunits of trimeric Gq proteins [41,44,45]. Other Ca2+ channels also rely on Gβγ for activation [46,47]. We used a pharmacological approach as suggested [45] to confirm this observation. Pregnenolone sulfate-induced AP-1 activity was reduced in the presence of the Gβγ inhibitor gallein (Figure 5a) by 40 % (Figure 5b). For TRPM8, Gαq but not Gβγ was suggested to induce an inhibition in excised patches [22]. We examined the effect of gallein on icilin-induced TRPM8 signaling. Figure 5C shows that signaling via TRPM8 is attenuated in the presence of gallein. Icilin-induced AP-1 activity was reduced by 60 %.
2.7. The Transcription Factor c-Jun or c-Jun-Dimeriziation Proteins are Essential for the Activation of AP-1 after Stimulation of TRPM8 Channels with Icilin
After stimulation of TRPM8 cells with icilin, Ca2+ ions enter the cells through the channel. The extracellular signal-regulated protein kinase ERK1/2 acts as a signal transducer to extend the signaling cascade through the cytoplasm into the nucleus, where gene regulatory proteins are regulated via phosphorylation [12,13]. Recently, we showed that stimulation of HEK293-M8 cells with icilin increased expression of c-Fos [12], one of the bZIP transcription factors that form the AP-1 transcription factor complex. AP-1 was originally described as a dimer of the bZIP proteins c-Fos and c-Jun [48].
Therefore, we investigated whether the c-Fos dimerization partner c-Jun is also involved within the TRPM8-induced signaling cascade to generate an active AP-1 complex. We expressed a dominant-negative mutant of c-Jun termed c-JunΔN in HEK293-M8 cells. The c-Jun mutant had no transcriptional activation domain and was therefore unable to activate gene transcription. c-JunΔN retained the bZIP domain, which is responsible for DNA binding and dimerization (Figure 6a). The biological activity of c-JunΔN has been demonstrated in various control experiments [29]. Figure 6b shows that expression of c-JunΔN significantly reduced AP-1 activity in HEK293-M8 cells after stimulation of the cells with icilin. Expression of c-JunΔN reduced the signaling of TRPM8 in the order of 89 %. We conclude that c-Jun or a c-Jun dimerization partner is an essential signaling molecule required for the activation of AP-1 within the TRPM8-induced signaling cascade.
3. Discussion
In this study, we analyzed the TRPM8-induced signaling pathway that induces a change in gene transcription. Several signaling molecules have been identified in recent years [12,13,14,15], but the description of TRPM8 signaling still contained several gaps and open questions. We focused on the role of phosphatidylinositol 4,5-bisphosphate and trimeric G protein subunits in the regulation of TRPM8 signaling. Finally, we analyzed the impact of c-Jun, a bZIP transcription factor, on TRPM8-induced activation of AP-1.
It has been proposed that most TRP channels, along with other ion channels, are regulated by the lipid signaling molecule phosphatidylinositol 4,5-bisphosphate [49]. A variety of experimental strategies have been used to test this concept, including pharmacological and genetic methods. The most convincing results were obtained with the use of sophisticated electrogenetic and chemical genetic tools to reduce plasma membrane phosphatidylinositol 4,5-bisphosphate levels by dephosphorylation, including the use of the rapamycin-inducible 4,5-phosphoinositide phosphatase pseudojanin and the voltage-activatable phosphatase ci-VSP. These experiments conclusively demonstrated that activation of TRPM8 and TRPM3 require phosphatidylinositol 4,5-bisphosphate [19,20,21,41,50]. Similar results were shown for the voltage-gated Ca2+ channel Cav1.2 [42]. In contrast, chemical tools might produce questionable results, due to their nonspecific activities. The application of lipids to patch membranes can lead to non-specific physicochemical changes. The addition of MgATP to excised inside-out patches has been used to activate phosphatidylinositol 4-kinase [41,50], but may also lead to the activation of other kinases. The compound wortmannin, known as an inhibitor of phosphatidylinositol-3-kinase and myosin light chain kinase, has been used as a phosphatidylinositol 4-kinase inhibitor to decrease the concentration of phosphatidylinositol 4,5-bisphosphate [3,16,21,41,50]. In our hands, application of wortmannin did not inhibit TRPM3 signaling at all. Rather, we observed an increase in AP-1 activity after stimulation of TRPM3 channels with pregnenolone sulfate in the presence of 35 μM wortmannin (G.Thiel, unpublished observations). In this study, we used the compound ISA-2011B for inhibiting PIP5K [27,28], the main phosphatidylinositol 4,5-bisphosphate-synthesizing enzyme. The results show that administration of this compound strongly reduced signaling mediated by TRPM8 and TRPM3 channels. Administration of ISA-2011B to T611 cells expressing TRPC6 channels also significantly inhibited hyperforin-induced activation of AP-1 (G.Thiel, unpublished observations), supporting the view that PIP5K-catalyzed biosynthesis of phosphatidylinositol 4,5-bisphosphate is essential for the activation of numerous TRP channels. Finally, in this study, we demonstrated that administration of ISA-2011B also strongly reduced signaling through the voltage-gated Cav1.2 Ca2+ channel, confirming the previous suggestion that phosphatidylinositol 4,5-bisphosphate is a cofactor required for full Cav1.2 channel activity [42]. These results put the spotlight on PIP5K as an important regulator of TRP channel signaling via the regulation of phosphatidylinositol 4,5-bisphosphate biosynthesis.
Phosphatidylinositol 4,5-bisphosphate is thought to interact with ion channels via electrostatic interactions or by direct binding to specific binding sites within the channel proteins. Recently published structural data provide a detailed view into the binding of phosphatidylinositol 4,5-bisphosphate to the TRPM8 and TRPM3 channels. Structural data suggest a phosphatidylinositol 4,5-bisphosphate binding site involving the TRP domain, the pre-S1 domain, and the melastatin homology region-4 (MHR4) of the adjacent subunit [26]. A similar binding site has been proposed for the TRPM3 channel, involving amino acid residues within the pre-S1 segment, the S4-S5 linker and the TRP domain [50,51].
Stimulation of Gαq-coupled receptors has been suggested to impair activation of TRPM8 and TRPM3 [17,21] via activation of PLCβ, leading to hydrolysis of phosphatidylinositol 4,5-bisphosphate. Similarly, stimulation of TrkA or PDGBβ receptors, which stimulates PLCγ, has been shown to inhibit TRPM8 current [16,17]. In these studies, indirect evidence for a direct relationship between receptor stimulation and phosphatidylinositol 4,5-bisphosphate hydrolysis was provided by using translocation of the PLCγ-PH domain from the plasma membrane to the cytoplasm as a biosensor. This translocation assay, i.e. the loss of membrane localization of the biosensor, should be treated with caution [52]. The PLCγ-PH domain does not specifically bind to phosphatidylinositol 4,5-bisphosphate, but interacts 20-fold more strongly to IP3 [52,53]. Thus, the PLCγ-PH domain could act as an IP3 sponge and attenuate IP3-mediated downstream signaling. Much stonger binding to phosphatidylinositol 3,4,5-trisphosphate than to phosphatidylinositol 4,5-bisphosphate has also been reported [54]. Another study showed that increased intracellular Ca2+ concentrations, as occurring after stimulation of Gαq-coupled receptors or TRP channels, interfere with PLCγ-PH binding to the membrane [55]. Decorating the plasma membrane with a PLCγ-PH domain protein may sequester its targets and interfere with the binding of other phosphatidylinositol 4,5-bisphosphate-binding proteins. As a result, intracellular signaling pathways downstream of TRP channels would be disrupted and off-target effects may occur. For example, expression of the PH domain of PLCβ inhibits the activation of PLCβ by Gβγ [56], the expression of the PH domain of PLCγ1 inhibits the stimulation of PLC by platelet-derived growth factor [54], and the expression of the PH domain of PLCδ1 reduced the concentration of PIP5K in the plasma membrane [57].
In contrast to the hypothesis that activation of Gαq-coupled receptors inhibit TRPM8 activation via a transient reduction of the phosphatidylinositol 4,5-bisphosphate levels, direct binding of the inactive and activated Gαq subunit to TRPM8 channels has been proposed. Activated Gαq forms a complex with TRPM8 channels and in this way directly inhibits activation of TRPM8 after stimulation of Gαq-coupled receptors independently of the downstream PLC pathway [22]. Overexpression experiments showed that a Gαq mutant very actively inhibited TRPM8 current that lacked intrinsic GTPase activity and was therefore in its active, GTP-bound conformation. Direct binding of TRPM8 and Gαq has also been reported by others [23]. Furthermore, this study demonstrated that stimulation of TRPM8 leads to dissociation, i.e. activation of trimeric G-proteins and the subsequent activation of PLC.
In this study, we used a genetic approach to inhibit the activity of Gαq. We expressed the regulator of G-protein signaling-2 (RGS2) in the cells, which stimulates the GTPase activity of Gαq and thus inactivates Gαq, which in its GDP-bound state forms a complex with the Gβγ subunits. Expression of RGS2 strongly inhibited the signaling of a Gαq-coupled designer receptor, clearly demonstrating its activity. Similarly, expression of RGS2 strongly inhibited TRPM8 signaling, suggesting that stimulation of TRPM8 requires an activated Gαq subunit. Thus, it is not the activated Gαq that inhibits TRPM8 as suggested [22], but rather the inactivation of Gαq. Overexpression of RGS2 had no effect on TRPM3 or Cav1.2 signaling, indicating clear differences in the regulation of TRPM8 and TRPM3/Cav1.2 signaling. Similarly, overexpression of RGS2 was shown to reduce the Ca2+ and Na+ current of TRPV6, whereas the activity of TRPV5 channels was unaffected by RGS2 [58]. Thus, a subset of TRP channels is regulated by an activated Gαq subunit and therefore responds to overexpression of RGS2 (TRPM8, TRPV6), while others are completely inert to RGS2 overexpression and do not require Gαq (TRPM3, TRPV5)
Recently, the TRPM3 ion channel has been shown to be regulated by the Gβγ subunits of trimeric Gq proteins [41,44,45], and a binding site has been proposed [59]. In this study, we used a pharmacological approach to confirm that the Gβγ subunits modulate the activity of TRPM3 channels. Furthermore, we demonstrated that TRPM8 channels also respond to the Gβγ-inhibitor gallein. However, the proposed Gβγ binding site for TRPM3 is absent in TRPM8, suggesting that the regulatory role of Gβγ to TRPM8 is mediated by other interactions. It is possible that the Gβγ subunits do not interact directly with TRPM8, but regulate the activity of TRPM8 by manipulating the activity of PLCβ. We conclude that TRPM8 channel activity depends on both an activated Gαq subunit and Gβγ subunits.
What role does PLC play in regulating TRPM8 activity and signaling? It has been proposed that many TRP channels “are either activated downstream of the PLC pathway, or modulated by it” [60]. An influx of Ca2+ ions through the TRPM8 channel has been shown to activate PLCδ, which regulates TRPM8 activity by inducing a depletion of the phosphatidylinositol 4,5-bisphosphate levels in the plasma membrane [16]. In contrast, it has been suggested that an activated Gαq-subunit inhibits TRPM8 independently of PLC [22]. The fact that activation of TRPM8 requires Gβγ and an activated Gαq suggests that PLCβ is subsequently activated, leading to a reduction of the phosphatidylinositol 4,5-bisphosphate concentration. The Gq subunits can simultaneously and independently bind to PLCβ and modulate its activity. Gαq changes the autoinhibition mediated by the X-Y linker of PLCβ, leading to increased kcat of PLCβ. Gαq could also support the orientation of the catalytic core of the enzyme at the membrane [61]. The Gβγ subunits activate PLCβ by recruiting it to the membrane, i.e. bringing it closer to its lipid substrate [62]. However, due to experimental problems and the use of indirect assay systems, it is difficult to quantify the reduction of the phosphatidylinositol 4,5-bisphosphate concentration after PLC activation and to determine the time frame until the original phosphatidylinositol 4,5-bisphosphate concentration is restored by resynthesis from phosphatidylinositol 4-phosphate. Stimulation of Gαq-coupled receptors has been shown to cause only a small, transient decrease in the total amount of phosphatidylinositol 4,5-bisphosphate, which is efficiently replenished by phosphatidylinositol 4,5-bisphosphate-synthesizing enzymes [63,64]. Recently, we showed that the C-terminal domain of PLCβ1 and PLCβ3 interacts with plasma membrane targets, most likely phosphatidylinositol 4,5-bisphosphate, and blocks the biological activation of TRPM8 channels [15.]. It has been suggested that about two thirds of the phosphatidylinositol 4,5-bisphosphate pool is sequestered by binding proteins and is not freely available for phosphatidylinositol 4,5-bisphosphate effector proteins [65]. Therefore, PLCβ enzymes might regulate TRPM8 activation by masking phosphatidylinositol 4,5-bisphosphate with its C-terminal domain. It is tempting to speculate that the binding of Gβγ and Gαq to PLCβ enzymes induces a conformational switch of PLCβ that removes this blockade and allows activation of TRPM8 channels.
The pathway from the plasma membrane to the nucleus ends with the activation of stimulus-responsive transcription factors. In this study, we were able to show that the transcription factor c-Jun is essential for the formation of an active AP-1 complex within the TRPM8-induced signaling. Recently, we have shown that expression of the c-Jun dimerization parter c-Fos is upregulated upon stimulation of TRPM8 [12]. AP-1 controls numerous biological activities, including the regulation of proliferation, differentiation and cell death [66]. The outcome often depends on the cell type. TRPM8 is associated with tumor development, for example prostate cancer, colon cancer, and squamous cell carcinoma [8,9]. In this context, it is of particular interest to investigate the oncogenic role of the c-Jun within the TRPM8-induced signaling cascade. This also provides an indication of where future studies on c-Jun-regulated genes could be directed.
4. Materials and Methods
4.1. Cell Culture and Reagents
HEK293 cells expressing either TRPM3 (T-REx-TRPM3 cells) or TRPM8 (HEK293-M8 cells) have been described elsewhere [67,68]. HEK293T/17 cells were infected with a lentivirus to express of Rαq, a Gαq-coupled designer receptor, as described [34]. HEK293-M8 cells, T-REx-TRPM3 cells, and HEK293 cells expressing Rαq were incubated in DMEM containing 0.05% fetal bovine serum for 24 hours prior to stimulation. Stimulation was performed with icilin (PubChem CID: 161930; 1 μM, Santa Cruz Biotechnology, Heidelberg, Germany, # sc-201557), pregnenolone sulfate (PubChem CID: 105074; PregS, 20 μM, dissolved in DMSO, Sigma-Aldrich GmbH, Taufkirchen, Germany, # P162), or clozapine-N-oxide (PubChem CID: 135445691; 1 μM CNO, dissolved in ethanol, Enzo Life Sciences, Lörrach, Germany, # NS-105-0005), respectively, for 24 hours in medium containing 0.05% fetal bovine serum. INS-1 832/13 insulinoma cells were a kind gift of Hindrik Mulder [69], Lund University, Sweden, with the permission of Hans-Ewald Hohmeier and Christopher Newgard, Duke University, USA. Cells were cultured in RPM1 medium containing 10 % fetal calf serum, 2 mM glucose, 10 mM HEPES, 2 mM L-glutamine, 1 mM sodium pyruvate, 50 µM β-mercaptoethanol, 100 units/ml penicillin and 100 µg/ml streptomycin. Cells were incubated for 24 hours in DME medium containing 0.5 % fetal calf serum, 2 mM glucose, 10 mM HEPES, 2 mM L-glutamine, 1 mM sodium pyruvate, 50 µM β-mercaptoethanol, 100 units/ml penicillin and 100 µg/ml streptomycin before stimulation. Stimulation of INS-1 832/13 cells with KCl (PubChem CID: 4873; 55 mM) and FPL64176 (PubChem CID: 3423; 2.5 μM, dissolved in DMSO, a kind gift of Alomone Labs, Israel, Cat #: F-160) was performed in the same medium for 24 hours in the presence of the inhibitory compound. Cells were preincubated for 3 hours with the PIP5α inhibitor ISA-2011B, a diketopiperazine fused C-1 indol-3-yl substituted tetrahydroisoquinoline (PubChem CID: 49853637; MedChemExpress, Monmouth, NJ, USA, Cat.No. HY-16937, dissolved in DMSO) at a concentration of 10 μM, or gallein (PubChem CID: 73685, Santa Cruz Biotechnology, Inc., Heidelberg, Germany Ct.-No. sc-202631, dissolved in DMSO) at a concentration of 20 μM. Cells were stimulated for 24 hours in the presence of these compounds.
4.2. Lentiviral Gene Transfer
The lentiviral transfer vectors pFUW-HA-Rαq which encodes the Gαq-coupled designer receptor Rαq, pFUW-HA-RGS2, encoding HA-tagged RGS2, and pFUW-c-JunΔN and pFUW-ATF2ΔN, encoding dominant-negative mutants of c-Jun or ATF2, respecively, have been described elsewhere [24,34,70]. The lentiviral transfer vector pFWColl.luc, has been described [35]. Viral particles were produced by triple transfection of HEK293-TN cells with the gag-pol-rev packaging plasmid, the pCMVG plasmid encoding glycoprotein of vesicular stomatitis virus, and a lentiviral transfer vector.
4.3. Reporter Gene Assay
Infected cells were maintained in medium containing 0.05% fetal bovine serum for 24 hours prior to stimulation for 24 hours. Cell extracts were prepared using reporter lysis buffer (Promega, Mannheim, Germany) and assayed for luciferase activities that were normalized to the protein concentrations of the extracts. Luciferase activities were measured using a luminometer (Berthold Detection Systems, Pforzheim, Germany). Protein concentrations of the extracts were determined using a BCA protein assay kit.
4.4. Statistics
Data shown are means +/− SD of at least three independent experiments performed in quadruplicate. The two-tailed Student ́s t-test was used for the statistical analyses. Statistical probability is expressed as *** p < 0.001; ** p < 0.01, and * p < 0.05. We considered values significant when p < 0.05.
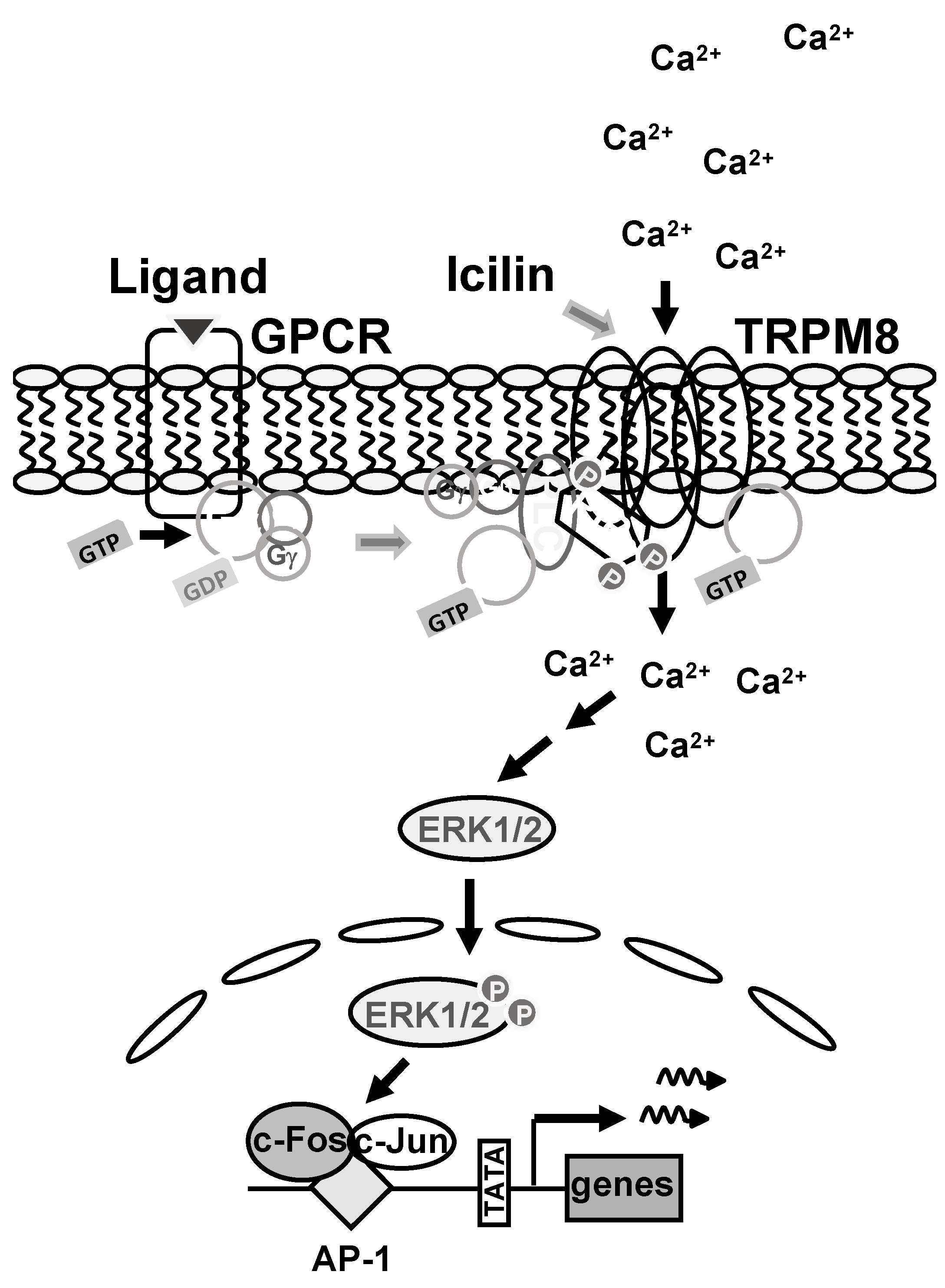
The TRPM8 channel is a tetramer embedded into the plasma membrane. The channel interacts with phosphatidylinositol 4,5-bisphosphate, which is essential for its activation. Reducing phosphatidylinositol 4,5-bisphosphate levels by blocking its biosynthesis with a PIP5K inhibitor impairs TRPM8 activation. PIP5K is therefore an important regulator of TRPM8 signaling. Stimulation of Gαq-coupled receptors triggers the dissociation of the trimeric G proteins into a GTP-bound α-subunit and the βγ subunits. The Gα subunit interacts with TRPM8 channels and is essential for TRPM8 activation. The α-subunit and the βγ subunits bind to PLCβ and activate the enzyme by increasing kcat and changing the orientation of the enzyme in the membrane to its substrate. This conformational change may remove the blockade of PLCβ to TRPM8 channels. After activation of TRPM8, Ca2+ ions flow into the cells through the channel and trigger the activation of ERK1/2, which acts as signal transducer The kinase translocates into the cell nucleus and activates AP-1, which is composed by the bZIP proteins c-Jun and c-Fos.
5. Conclusions
Stimulation of TRPM8 channels induces an intracellular signaling pathway that leads to the activation of stimulus-responsive transcription factors that alter the gene expression pattern of the cells. In this study, we have analyzed signaling molecules required for the link between stimulation of TRPM8 channels and gene transcription and a summary of the results is depicted in Figure 7. We focused on the role of phosphatidylinositol 4,5-bisphosphate and trimeric G-protein subunits within the TRPM8-induced signaling cascade. The TRPM8 channel interact with the lipid mediator phosphatidylinositol 4,5-bisphosphate, which is essential for its activation. Reducing phosphatidylinositol 4,5-bisphosphate levels by blocking its biosynthesis with a PIP5K inhibitor impaired TRPM8 activation. These data puts PIP5K into the limelight as an impprtant regulator of TRPM8 signaling. It has been suggested that stimulation of G protein-coupled receptors can modulate TRPM8 activation, although this issue is controversial. Stimulation of Gαq-coupled receptors triggers the dissociation of the trimeric G proteins into a GTP-bound α-subunit and the βγ subunits. The Gα subunit has been shown to interact with TRPM8 channels. This study showed that the activated GTP-bound form of Gαq is essential for TRPM8 signaling. Similarly, we showed that inhibition of the βγ subunits also impairs TRPM8 signaling. The βγ subunits could directly interact with the TRPM8 channel or carry out its activity via binding to PLCβ. The α-subunit and the βγ subunits bind simultaneously to PLCβ and activate the enzyme by recruiting it to the membrane, increasing kcat and changing the orientation of the enzyme to its substrate. The C-terminal domain of PLCβ1 and PLCβ3 blocks the biological activation of TRPM8 channels. We propose that the binding of Gβγ and Gαq to PLCβ enzymes causes a conformational change of the enzyme that removes this blockade and enables the activation of TRPM8 channels. After stimulation of TRPM8, Ca2+ ions flow through the channel into the cytoplasm and trigger the activation of the signal transducer ERK1/2. The phosphorylated and activated form of ERK1/2 migrates into the cell nucleus and activates AP-1, which consists of the bZIP proteins c-Jun and c-Fos.
Author Contributions
“Conceptualization, G.T.; methodology, G.T..; software, O.G.R.; validation, G.T. and O.G.R..; formal analysis, G.T. and O.G.R..; investigation, G.T.; resources, G.T..; data curation, G.T.; writing—original draft preparation, G.T.; writing—review and editing, G.T. and O.G.R..; visualization, O.G.R.; supervision, G.T.; project administration, G.T..; funding acquisition, G.T.. All authors have read and agreed to the published version of the manuscript.”
Funding
This research was funded by the University of Saarland, grant number LOM-T201000492.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgment
We thank Sabine Plant for excellent technical support and Libby Guethlein for critical reading of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- McKemy, D.D.; Neuhausser, W.M.; Julius, D. Identification of a cold receptor reveals a general role of TRP channels in thermosensatio. Nature 2002, 416, 52–58. [Google Scholar] [CrossRef]
- Peier, A.M.; Moqrich, A.; Hergarden, A.C.; Reeve, A.J.; Andersson, D.A.; Story, G.M.; Earley, T.J.; Dragoni, I.; McIntyre, P.; Bevan, S.; Patapoutian, A. A TRP channel that senses cold stimuli and menthol. Cell 2002, 108, 705–715. [Google Scholar] [CrossRef]
- Liu, Y.; Qin, N. TRPM8 in health and disease: Cold sensing and beyond. Adv. Exp. Med. Biol. 2011; 704, 185–208. [Google Scholar]
- Bautista, D.M.; Siemens, J.; Glazer, J.M.; Tsuruda, P.R.; Basbaum, A.I.; Stuck, C.L.; Jordt, S.E.; Julius, D. The menthol receptor TRPM8 is the principal detector of environmental cold. Nature 2007, 448, 204–208. [Google Scholar] [CrossRef]
- Colburn, R.W.; Lubin, M.L.; Stone, D.J. Jr.; Wang, Y.; Lawrence, D.; D´Andrea, M.R.; Brandt, M.R.; Liu, Y.; Flores, C.M.; Qin, N. Attenuated cold sensitivity in TRPM8 null mice. Neuron 2007, 54, 379–386. [Google Scholar] [CrossRef]
- Dhaka, A.; Murray, A.N.; Mathur, J.; Earley, T.J.; Petrus, M.J.; Patapoutian, A. TRPM8 is required for cold sensation in mice. Neuron 2007, 54, 371–378. [Google Scholar] [CrossRef]
- Almaraz, L.; Manenschijn, J.-A.; de la Peña, E. ; Viana, F. TRPM8, In: Mammalian transient receptor potential (TRP) cation channels (Eds., B. Nilius, V. Flockerzi), Handbook of Experimental Pharmacology 2014, 222, 547–579.
- Hantute-Ghesquier, A.; Haustrate, A.; Prevarskaya, N.; Lehen´kyi, V. TRPM family channels in cancer. Pharmaceuticals 2018, 11, 58. [Google Scholar] [CrossRef]
- Izquierdo, C.; Martín-Martínez, M.; Gómez-Monterrey, I.; González-Muñiz, R. TRPM8 channels: Advances in structural studies and pharmacological modulation. Int. J. Mol. Sci. 2021, 22, 8502. [Google Scholar] [CrossRef]
- Ramachandran, R.; Hyun, E.; Zhao, L.; Lapointe, T.K.; Chapman, K.; Hirota, C.L.; Ghosh, S.; McKemy, D.D.; Vergnolle, N.; Beck, P.L.; Altier, C.; Hollenberg, M.D. TRPM8 activation attenuates inflammatory responses in mouse models of colitis. Proc. Natl. Acad. Sci. USA 2013, 110, 7476–7481. [Google Scholar] [CrossRef]
- Caceres, A.I.; Liu, B.; Jabba, S.V.; Achanta, S.; Morris, J.B.; Jordt, S.-E. Transient receptor potential cation channel subfamily M member 8 channels mediate the anti-inflammatory effects of eucalyptol. Brit. J. Pharmacol. 2017, 174, 867–879. [Google Scholar] [CrossRef]
- Ulrich, M.; Wissenbach, U.; Thiel, G. The super-cooling compound icilin stimulates c-Fos and Egr-1 expression and activity involving TRPM8 channel activation, Ca2+ ion influx and activation of the ternary complex factor Elk-1. Biochem. Pharmacol. 2020, 177, 113936. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G.; Backes, T.M.; Welck, J.; Steinhausen, S.; Fischer, A.-L.; Langfermann, D. S.; Ulrich, M.; Wissenbach, U.; Rössler, O.G. Pharmacological inhibition of TRPM8-induced gene transcription. Biochem. Pharmacol. 2019, 170, 113678. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G.; Rössler, O.G. Calmodulin regulates transient receptor potential TRPM3 and TRPM8-induced gene transcription. Int. J. Mol. Sci. 2023, 24, 7902. [Google Scholar] [CrossRef]
- Thiel, G.; Rössler, O.G. Expression of the C-terminal domain of phospholipase Cβ3 inhibits signaling via Gαq-coupled receptors and transient receptor potential channels. Int. J. Mol. Sci. 2022, 23, 9590. [Google Scholar] [CrossRef]
- Rohacs, T.; Lopes, C.M.; Michailidis, I.; Logothetis, D.E. PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nature Neurosci. 2005, 8, 626–634. [Google Scholar] [CrossRef]
- Liu, B.; Qin, F. Functional control of cold- and menthol-sensitive TRPM8 ion channels by phosphatidylinositol 4,5-bisphosphate. J. Neurosci. 2005, 25, 1674–1681. [Google Scholar] [CrossRef]
- Daniels, R.L.; Takashima, Y.; McKemy, D.D. Activity of the neuronal cold sensor TRPM8 is regulated by phospholipase C via the phospholipid phosphoinositol 4,5-bisphosphate. J. Biol. Chem. 2009, 284, 1570–1582. [Google Scholar] [CrossRef]
- Yudin, Y.; Lukacs, V.; Cao, C.; Rohács, T. Decrease in phosphatidylinositol 4,5-bisphosphate levels mediates desensitization of the cold sensor TRPM8 channels. J. Physiol. 2011, 589, 6007–6027. [Google Scholar] [CrossRef]
- Hammond, G.R.V.; Fischer, M.J.; Anderson, K.E.; Holdich, J.; Koteci, A.; Balla, T.; Irvine, R.F. PI4P and PI(4,5)P2 are essential but independent lipid determinants of membrane identity. Science 2012, 337, 727–730. [Google Scholar] [CrossRef]
- Tóth, B.I.; Konrad, M.; Ghosh, D.; Mohr, F.; Halaszovich, C.R.; Leitner, M.G.; Vriens, J.; Oberwinkler, J.; Voets, T. Regulation of the transient receptor potential channel TRPM3 by phosphoinositides. J. Gen. Physiol. 2015, 146, 51–63. [Google Scholar] [CrossRef]
- Zhang, X.; Mak, S.; Li, L.; Parra, A.; Denlinger, B.; Belmonte, C.; McNaughton, P.A. Direct inhibition of the cold-activated TRPM8 ion channel by Gαq. Nat. Cell. Biol. 2012, 14, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Klasen, K.; Hollatz, D.; Zielke, S.; Gisselmann, G.; Hatt, H.; Wetzel, C.H. The TRPM8 ion channel comprises direct Gq protein-activating capacity. Pflügers Arch. – Eur. J. Physiol. 2012, 463, 779–797. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G.; Welck, J.; Wissenbach, U.; Rössler, O.G. Dihydrotestosterone activates AP-1 in prostate cancer cells. Int. J. Biochem. Cell Biol. 2019, 110, 9–20. [Google Scholar] [CrossRef]
- Van den Bout, I.; Divecha, N. PIP5K-driven Ptd(4,5)P2 synthesis: regulation and cellular functions. J. Cell. Sci. 2009, 122, 3837–3850. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Wu, M.; Zubcevic, L.; Borschel, W.F.; Lander, G.C.; Lee, S.-Y. Structure of the cold- and menthol-sensing ion channel TRPM8. Science, 1126. [Google Scholar]
- Semenas, J.; Hedblom, A.; Miftakhova, R.R.; Sarwar, M.; Larsson, R.; Shcherbina, L.; Johansson, M.E.; Härkönen, P.; Sterner, O.; Persson, J.L. The role of PI3K/AKT-related PIP5K1α and the discovery of its selective inhibitor for treatment of advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2014, 111, E3689–E3698. [Google Scholar] [CrossRef] [PubMed]
- Kunkl, M.; Porciello, N.; Mastrogiovanni, M.; Capuano, C.; Lucantoni, F.; Moretti, C.; Persson, J.L.; Galandrini, R.; Buzzetti, R.; Tuosto, L. ISA-2011B, a phosphatidylinositol 4-phosphate 5-kinase α inhibitor, impairs CD28-dependent costimulatory and pro-inflammatory signals in human T lymphocytes. Front. Immunol. 2017, 8, 502. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G.; Rössler, O.G. Hyperforin activates gene transcription involving transient receptor potential C6 channels. Biochem. Pharmacol. 2017, 129, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Müller, I.; Rössler, O.G.; Thiel, G. Pregnenolone sulfate activates basic region leucine zipper transcription factors in insulinoma cells: role of voltage-gated Ca2+ channels and transient receptor potential melastatin 3 channels. Mol. Pharmacol. 2011, 80, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Backes, T.M.; Rössler, O.G.; Hui, X.; Grötzinger, C.; Lipp, P.; Thiel, G. Stimulation of TRPV1 channels activates the AP-1 transcription factor. Biochem. Pharmacol. 2018, 150, 160–169. [Google Scholar] [CrossRef]
- Langfermann, D.S.; Rössler, O.G.; Thiel, G. Stimulation of B-Raf increases c-Jun and c-Fos expression and upregulates AP-1-regulated gene transcription in insulinoma cells. Mol. Cell. Endocrinol. 2018, 472, 126–139. [Google Scholar] [CrossRef]
- Loviscach, L.; Backes, T.M.; Langfermann, D.S.; Ulrich, M.; Thiel, G. Zn2+ ions inhibit gene transcription following stimulation of the Ca2+ channels Cav1.2 and TRPM3. Metallomics 2020, 12, 1735–1747. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, A.; Keim, A.; Thiel, G. Regulation of immediate-early gene transcription following activation of Gαq-coupled designer receptors. J. Cell. Biochem. 2013, 114, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G.; Backes, T.M.; Guethlein, L.A.; Rössler, O.G. Chromatin-embedded reporter genes: Quantification of stimulus-induced gene transcription. Gene 2021, 787, 145645. [Google Scholar] [CrossRef] [PubMed]
- Vriens, J.; Owsianik, G.; Hofmann, T.; Philipp, S.E.; Stab, J.; Chen, X.; Benoit, M.; Xue, F.; Janssens, A.; Kerselaers, S.; Oberwinkler, J.; Vennekens, R.; Gudermann, T.; Nilius, B.; Voets, T. TRPM3 is a nociceptor channel involved in the detection of noxious heat. Neuron 2011, 70, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Lesch, A.; Rubil, S.; Thiel, G. Activation and inhibition of transient receptor potential TRPM3-induced gene transcription. Br. J. Pharmacol. 2014, 171, 2645–2658. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G.; Rubil, S.; Lesch, A.; Guethlein, L.A.; Rössler, O.G. Transient receptor potential TRPM3 channels: Pharmacology, signaling, and biological functions. Pharmacol. Res. 2017, 124, 92–99. [Google Scholar] [CrossRef]
- Cabanas, H.; Muraki, K.; Balinas, C.; Eaton-Fitch, N.; Staines, D.; Marshall-Gradisnik, S. Validation of impaired transient receptor potential melastatin 3 ion channel activity in natural killer cells from chronic fatigue syndrome/myalgic encephalomyelitis patients. Mol. Med. 2019, 25, 14. [Google Scholar] [CrossRef]
- Roelens, R.; Peigneur, A.N.F.; Voets, T.; Vriens, J. Neurodevelopmental disorders caused by variants in TRPM3. Biochim. Biophys. Acta – Mol. Cell Res. 2024, 1871, 119709. [Google Scholar] [CrossRef] [PubMed]
- Badheka, D.; Borbiro, I.; Rohacs, T. Transient receptor potential melastatin 3 is a phosphoinositide-dependent ion channel. J. Gen. Physiol. 2015, 146, 65–77. [Google Scholar] [CrossRef]
- Suh, B.-C.; Leal, K.; Hille, B. Modulation of high-voltage activated Ca2+ channels by membrane phosphatidylinositol 4,5-bisphosphate. Neuron 2010, 67, 224–238. [Google Scholar] [CrossRef]
- Kehrl, J.H.; Sinnarajah, S. RGS2: a multifunctional regulator of G-protein signaling. Int. J. Biochem. Cell Biol. 2002, 34, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Dembla, S.; Behrendt, M.; Mohr, F. , Goecke, C.; Sondermann, J.; Schneider, F.M.; Schmidt, M.; Stab, J.; Enzeroth, R.; Leitner, M.G.; Nuñez-Badinez, P.; Schwenk, J.; Nürnberg, B.; Cohen, A.; Philipp, S.E.; Greffrath, W.; Bünemann, M.; Oliver, D.; Zakharian, E.; Schmidt, M.; Oberwinkler, J. Ant-nociceptive action of peripheral mu-opioid receptors by G-beta-gamma protein-mediated inhibition of TRPM3 channels. Elife 2017, 6, e26280. [Google Scholar]
- Quallo, T.; Alkhatib, O.; Gentry, C.; Andersson, D.A.; Bevan, S. G protein βγ subunits inhibit TRPM3 ion channel in sensory neurons. Elife 2017, 6, e26138. [Google Scholar] [CrossRef]
- Herlitze, S.; Garcia, D.E.; Mackie, K.; Hille, B.; Scheuer, T.; Catterall, W.A. Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature 1996, 380, 258–262. [Google Scholar] [CrossRef]
- Ikeda, S.R. Voltage-dependent modulation of N-type calcium channels by G-protein betagamma subunits. Nature 1996, 380, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Chiu, R.; Boyle, W.J.; Meek, J.; Smeal, T.; Hunter, T.; Karin, M. The c-Fos protein interacts with c-Jun/AP-1 to stimulate transcription of AP-1 responsive genes. Cell 1988, 54, 541–552. [Google Scholar] [CrossRef]
- Rohacs, T. Phosphoinositide regulation of TRP channels: A functional overview in the structural era. Annu. Rev. Physiol. 2024, 86, 329–355. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Carnevale, V.; Gabrielle, M.; Gianti, E.; Rohacs, T. Computational and functional studies of the PI(4,5)P2 binding site of the TRPM3 ion channel reveal interactions with other regulators. J Biol. Chem. 2022, 298, 102547. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; MacKinnon, R. Structural and functional analyses of a GPCR-inhibited ion channel TRPM3. Neuron 2023, 111, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Wills, R.C.; Goulden, B.D.; Hammond, G.R.V. Genetically encoded lipid biosensors. Mol. Biol. Cell 2018, 29, 1526–1532. [Google Scholar] [CrossRef]
- Hille, B.; Dickson, E.J.; Kruse, M.; Vivas, O.; Suh, B.-C. Phosphoinositides regulate ion channels. Biochim. Biophys. Acta 2015, 1851, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Falasca, M.; Logan, S.K.; Lehto, V.P.; Baccante, G.; Lemmon, M.A.; Schlessinger, J. Activation of phospholipase Cγ by PI 3-kinase-induced PH domain-mediated membrane targeting. EMBO J. 1998, 17, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Kang, J. K.; Kim, O.-H.; Hur, J.; Yu, S.H.; Lamichhane, S.; Lee, J.W.; Ojha, U.; Hong, J. H.; Lee, C. S.; Cha, J.-Y.; Lee, Y. J.; Im, S.-S.; Park, Y. J.; Choi, C. S.; Lee, D. H.; Lee, I.-K.; Oh, B.-C. Increased intracellular Ca2+ concentrations prevent membrane localization of PH domains through the formation of Ca2+-phosphoinositides. Proc. Natl. Acad. Sci. USA 2017, 114, 11926–11931. [Google Scholar] [CrossRef] [PubMed]
- Kadamur, G.; Ross, E.M. Intrinsinc pleckstrin homology (PH) domain motion in phospholipase C-β exposes a Gβγ protein binding site. J. Biol. Chem. 2016, 291, 11394–11406. [Google Scholar] [CrossRef] [PubMed]
- Szmanska, E.; Sobota, A.; Czurylo, E.; Kwiatkowska, K. Expression of PI(4,5)P2-binding proteins lowers the PI(4,5)P2 level and inhibits FcγRIIA-mediated cell spreading and phagocytosis. Eur. J. Immunol. 2008, 38, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Schoeber, J.P.; Topala, C.N.; Wang, X.; Diepens, R.J.; Lambers, T.T.; Hoenderop, J.G.; Bindels, R.J. RGS2 inhibits the epithelial Ca2+ channel TRPV6. J. Biol. Chem. 2006, 281, 29669–29674. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, M.; Gruss, F.; Enzeroth, R.; Dembla, S.; Zhao, S.; Crassous, P.-A.; Mohr, F.; Nys, M.; Louros, N.; Gallardo, R.; Zorzini, V.; Wagner, D.; Economou, A.; Rousseau, F.; Schymkowitz, J.; Philipp, S.E.; Rohacs, T.; Ulens, C.; Oberwinkler, J. The structural basis for an on-off switch controlling Gβγ-mediated inhibition of TRPM3 channels. Proc. Nat. Acad. Sci. USA 2020, 117, 29090–29100. [Google Scholar] [CrossRef]
- Rohács, T. Regulation of transient receptor potential channels by the phospholipase C pathway. Adv. Biol. Reg. 2013, 53, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Falzone, M.E.; MacKinnon, R. The mechanism of Gαq regulation of PLCβ3-catalyzed PIP2 hydrolysis. Proc. Natl. Acad. Sci. USA 2023, 120, e2315011120. [Google Scholar] [CrossRef]
- Falzone, M.E.; MacKinnon, R. Gβγ activates PIP2 hydrolysis by recruiting and orienting PLCβ on the membrane surface. Proc. Natl. Acad. Sci. USA 2023, 120, e2301121120. [Google Scholar] [CrossRef]
- Stephens, L.; Jackson, T.R.; Hawkins, P.T. Activation of phosphatidylinositol 4,5-bisphosphate supply by agonists and non-hydrolysable GTP analogues. Biochem. J. 1993, 296, 481–488. [Google Scholar] [CrossRef]
- Barneda, D.; Janardan, V.; Niewczas, I.; Collins, D. M.; Cosulich, S.; Clark, J.; Stephens, L.R.; Hawkins, P.T. Acyl chain selection couples the consumption and synthesis of phosphoinositides. EMBO J. 2022, 41, e110038. [Google Scholar] [CrossRef]
- Golebiewska, U.; Nyako, M.; Woturski, W.; Zaitseva, I.; McLaughlin, S. Diffusion coefficient of fluorescent phosphatidylinositol 4,5-bisphosphate in the plasma membrane of cells. Mol. Biol. Cell 2008, 19, 1663–1669. [Google Scholar] [CrossRef]
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136. [Google Scholar] [CrossRef]
- Bödding, M.; Wissenbach, U.; Flockerzi, V. Characterization of TRPM8 as a pharmacophore receptor. Cell Calcium 2007, 42, 618–628. [Google Scholar] [CrossRef] [PubMed]
- Naylor, J.; Milligan, C.J.; Zeng, F.; Jones, C.; Beech, D.J. Production of a specific extracellular inhibitor of TRPM3 channels. Brit. J. Pharmacol. 2008, 155, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Hohmeier, H.E.; Mulder, H.; Chen, G.; Henkel-Rieger, R.; Prentki, M.; Newgard, C.B. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin release. Diabetes 2000, 49, 424–430. [Google Scholar] [CrossRef]
- Mayer, S.I.; Dexheimer, V.; Nishida, E.; Kitajima, S.; Thiel, G. (2008) Expression of the transcriptional repressor ATF3 in gonadotrophs is regulated by Egr-1, CREB, and ATF2 after gonadotropin-releasing hormone receptor stimulation. Endocrinology 2008, 149, 6311–6325. [Google Scholar] [CrossRef]
Figure 1.
Biosynthesis, metabolism and hydrolysis of phosphatidylinositol 4,5,-bisphosphate. Phosphoinositides are phosphorylated metabolites of phosphatidylinositol. Catalyzed by PIP5K, phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2 is synthesisized from phosphatidylinositol 4-phosphate (PI 4-phosphate). Phosphatidylinositol 4,5,-bisphosphate can be phosphorylated by phosphatidylinositol 3-kinases to generate phosphatidylinositol 3,4,5,-trisphosphate (PI(3,4,5)P3), a lipid mediator essential to activate the serin/threonine protein kinase AKT. Phosphatidylinositol 4,5-bisphosphate can also be metabolized by phospholipase C (PLC), which generates IP3 and diacylgycerol.
Figure 1.
Biosynthesis, metabolism and hydrolysis of phosphatidylinositol 4,5,-bisphosphate. Phosphoinositides are phosphorylated metabolites of phosphatidylinositol. Catalyzed by PIP5K, phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2 is synthesisized from phosphatidylinositol 4-phosphate (PI 4-phosphate). Phosphatidylinositol 4,5,-bisphosphate can be phosphorylated by phosphatidylinositol 3-kinases to generate phosphatidylinositol 3,4,5,-trisphosphate (PI(3,4,5)P3), a lipid mediator essential to activate the serin/threonine protein kinase AKT. Phosphatidylinositol 4,5-bisphosphate can also be metabolized by phospholipase C (PLC), which generates IP3 and diacylgycerol.
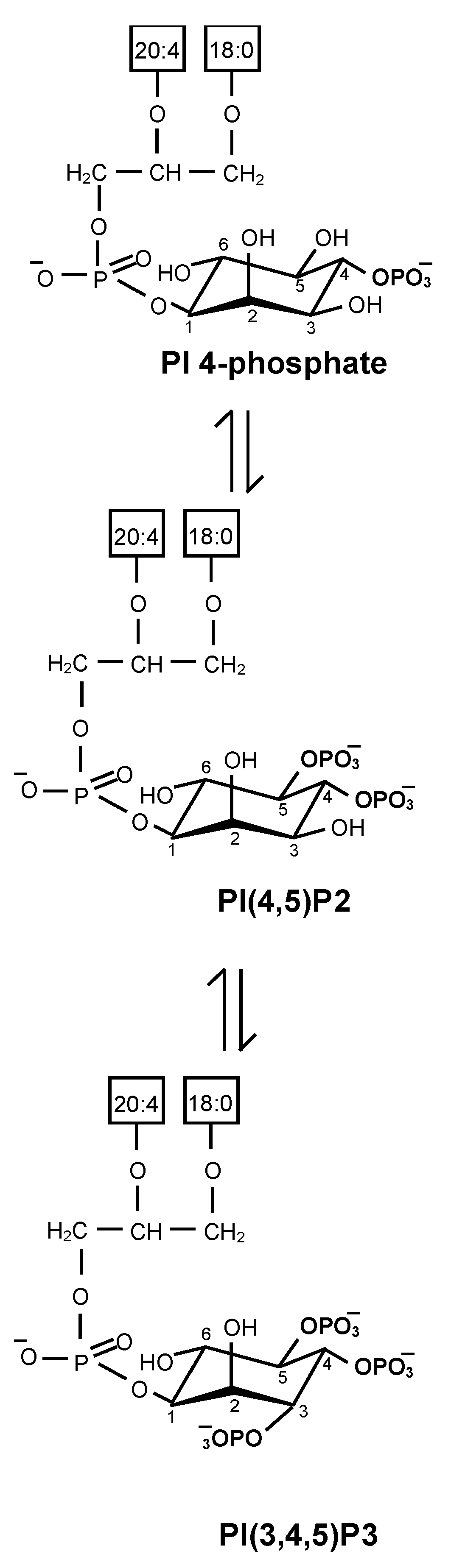
Figure 2.
Pharmacological inhibition of PIP5Kα attenuates TRPM8 intracellular signaling. (a) Modular structure of TRPM8. The interaction sites of phosphatidylinositol 4,5-bisphosphate with the channel are shown (preS1 segment, S4-S5 linker, TRP domain). (b) Chemical structure of the PIP5Kα inhibitor ISA-2011B. (c) Luciferase reporter gene under control of the collagenase promoter (Coll.luc), which serves as a sensor for measuring AP-1 activity. Shown is the chromatin-embedded provirus, which also contains the Woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) and the HIV flap element. The U3 region of the 5´ LTR has been deleted. (d) HEK293-M8 cells were infected with a recombinant lentivirus containing the Coll.luc reporter gene. Cells were serum-starved for 24 hours, preincubated for 3 hours with ISA-2011B (10 μM), and then stimulated with icilin (1 μM) in serum-reduced medium in the presence of the PIP5Kα inhibitor. Cell extracts were prepared, and luciferase activities and protein concentrations were determined. The luciferase activity was normalized to the protein concentration. Data shown are means +/- SD of three experiments performed in quadruplicate (***P < 0.001).
Figure 2.
Pharmacological inhibition of PIP5Kα attenuates TRPM8 intracellular signaling. (a) Modular structure of TRPM8. The interaction sites of phosphatidylinositol 4,5-bisphosphate with the channel are shown (preS1 segment, S4-S5 linker, TRP domain). (b) Chemical structure of the PIP5Kα inhibitor ISA-2011B. (c) Luciferase reporter gene under control of the collagenase promoter (Coll.luc), which serves as a sensor for measuring AP-1 activity. Shown is the chromatin-embedded provirus, which also contains the Woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) and the HIV flap element. The U3 region of the 5´ LTR has been deleted. (d) HEK293-M8 cells were infected with a recombinant lentivirus containing the Coll.luc reporter gene. Cells were serum-starved for 24 hours, preincubated for 3 hours with ISA-2011B (10 μM), and then stimulated with icilin (1 μM) in serum-reduced medium in the presence of the PIP5Kα inhibitor. Cell extracts were prepared, and luciferase activities and protein concentrations were determined. The luciferase activity was normalized to the protein concentration. Data shown are means +/- SD of three experiments performed in quadruplicate (***P < 0.001).
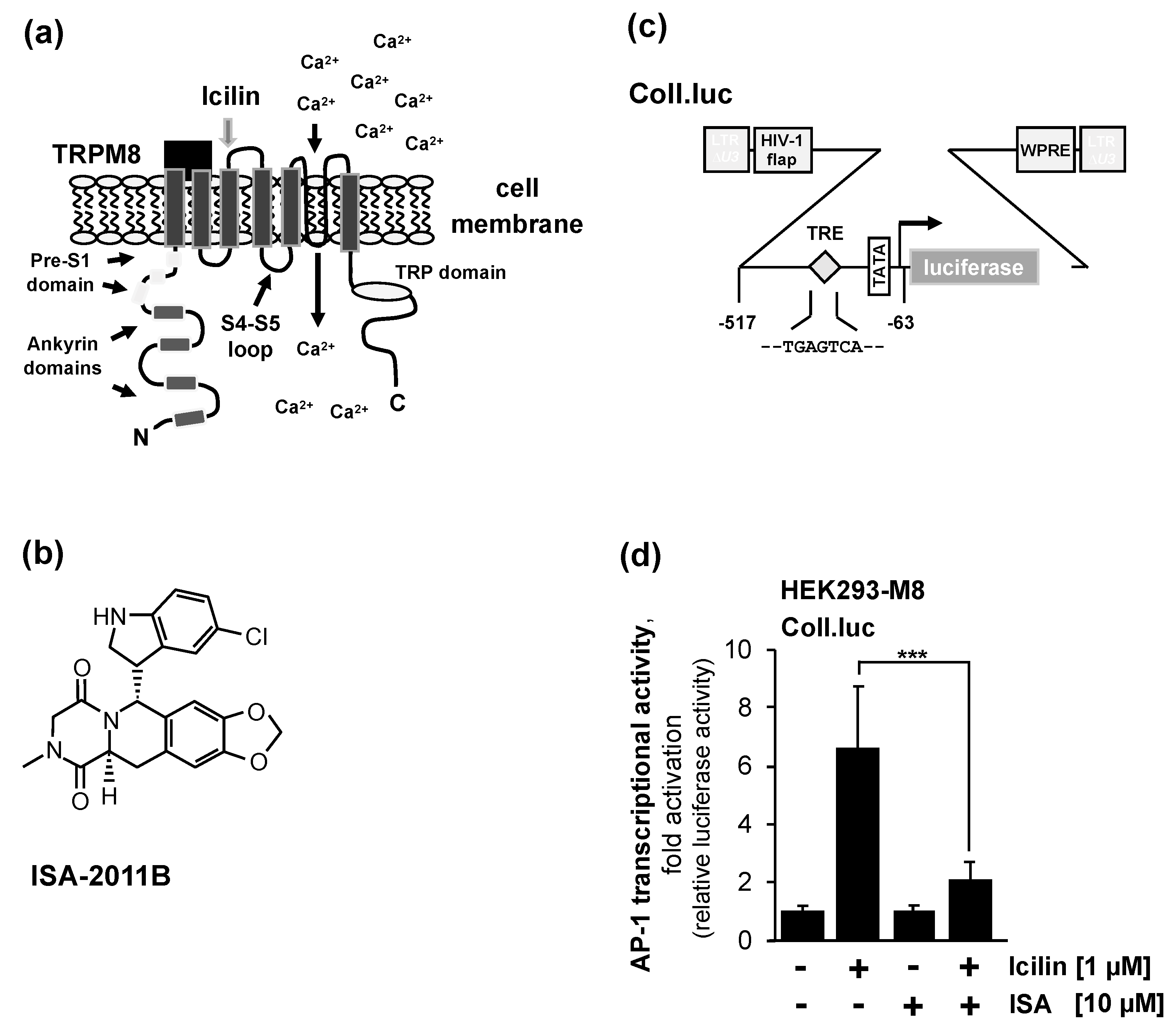
Figure 3.
Pharmacological inhibition of PIP5Kα attenuates intracellular signaling following stimulation of TRPM3 and Cav1.2 Ca2+ channels. (a) Modular structure of TRPM3 showing the interaction sites of phosphatidylinositol 4,5-bisphosphate with the channel (preS1 segment, S4-S5 linker, TRP domain). (b) T-REx-TRPM3 cells containing a Coll.luc reporter gene integrated into the chromatin were serum-starved for 24 hours in the presence of tetracycline (1 μg/ml) to induced TRPM3 expression. The serum-starved cells were preincubated with ISA-2011B (10 μM) for 3 hours, and then stimulated with pregnenolone sulfate (20 μM) for 24 hours in the presence of the inhibitor. Cells were harvested and analyzed as described in the legend to Figure 2 (n=4; ***P < 0.001). (c) Modular structur of Cav1.2 voltage-gated Ca2+ channels, consisting of the α1 subunit, which forms the pore, and the auxiliary subunits α2δ, β and γ. (d) INS-1 832/13 insulinoma cells were infected with a recombinant lentivirus containing the Coll.luc reporter gene. Cells were serum-starved in medium containing 0.5% serum and 2 mM glucose for 24 hours. Cells were preincubated in the same medium with ISA-2011B (10 μM) for three hours. Stimulation of the cells was performed with KCl (25 mM) and the voltage-gated Ca2+ channel activator FPL64176 (2.5 μM) in the presence of the inhibitor for 24 hours. Cells were harvested and analyzed as described in the legend to Figure 2 (n=3; ***P < 0.001).
Figure 3.
Pharmacological inhibition of PIP5Kα attenuates intracellular signaling following stimulation of TRPM3 and Cav1.2 Ca2+ channels. (a) Modular structure of TRPM3 showing the interaction sites of phosphatidylinositol 4,5-bisphosphate with the channel (preS1 segment, S4-S5 linker, TRP domain). (b) T-REx-TRPM3 cells containing a Coll.luc reporter gene integrated into the chromatin were serum-starved for 24 hours in the presence of tetracycline (1 μg/ml) to induced TRPM3 expression. The serum-starved cells were preincubated with ISA-2011B (10 μM) for 3 hours, and then stimulated with pregnenolone sulfate (20 μM) for 24 hours in the presence of the inhibitor. Cells were harvested and analyzed as described in the legend to Figure 2 (n=4; ***P < 0.001). (c) Modular structur of Cav1.2 voltage-gated Ca2+ channels, consisting of the α1 subunit, which forms the pore, and the auxiliary subunits α2δ, β and γ. (d) INS-1 832/13 insulinoma cells were infected with a recombinant lentivirus containing the Coll.luc reporter gene. Cells were serum-starved in medium containing 0.5% serum and 2 mM glucose for 24 hours. Cells were preincubated in the same medium with ISA-2011B (10 μM) for three hours. Stimulation of the cells was performed with KCl (25 mM) and the voltage-gated Ca2+ channel activator FPL64176 (2.5 μM) in the presence of the inhibitor for 24 hours. Cells were harvested and analyzed as described in the legend to Figure 2 (n=3; ***P < 0.001).
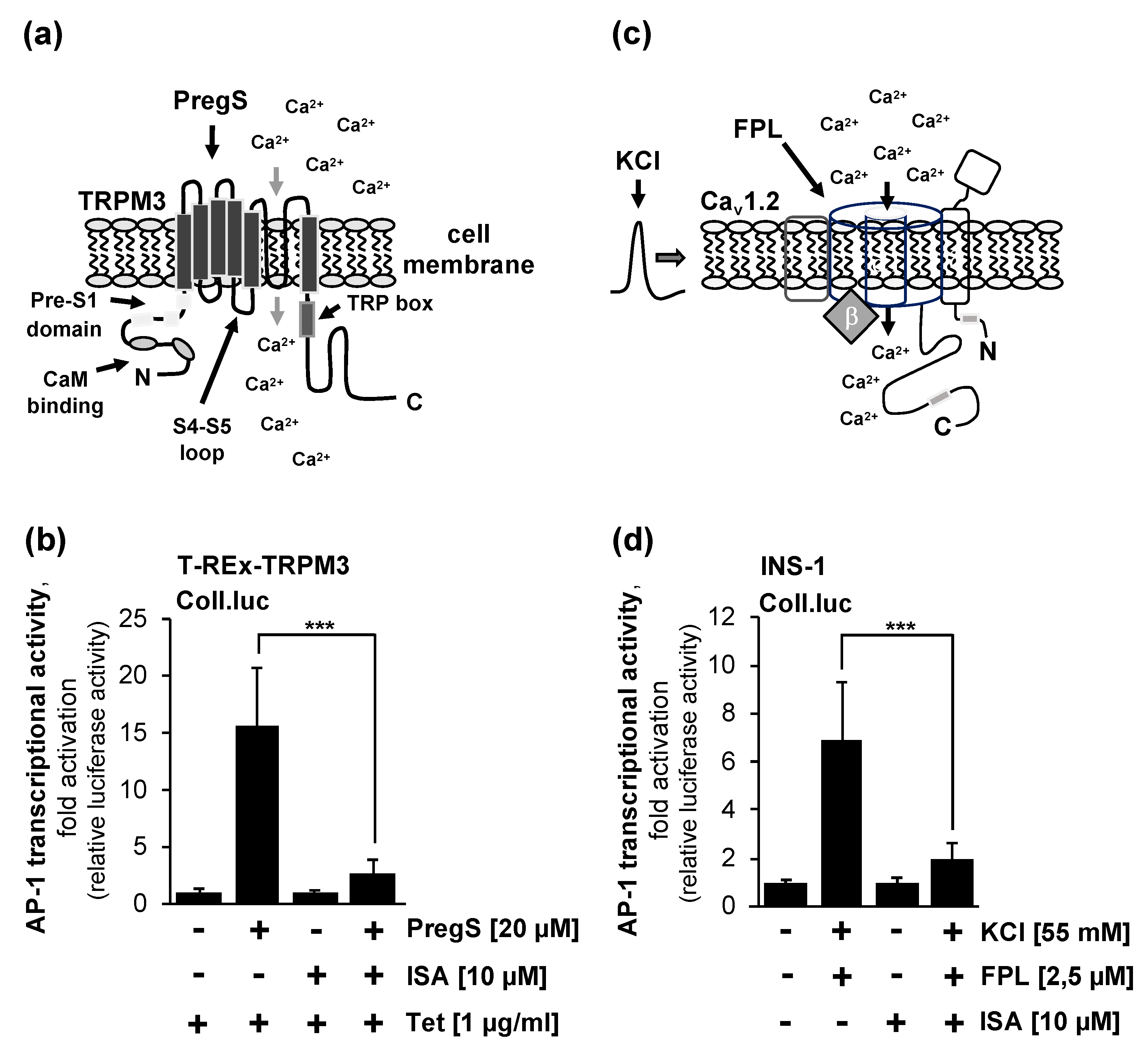
Figure 4.
RGS2 expression reduces signal transduction after stimulation of TRPM8 channels or Gαq-coupled designer receptors. (a) Modular structure of RGS2. The RGS domain is responsible for binding to Gαq, while the N-terminal domain is required for targeting the protein to the plasma membrane. (b) HEK293-M8 cells containing the Coll.luc reporter gene were infected with a recombinant lentivirus encoding either RGS2 or β-galactosidase (mock). Cells were serum-starved for 24 hours, and then stimulated with icilin (1 μM) in serum-reduced medium for 24 hours. Cells were harvested and analyzed as described in the legend to Figure 2 (n=3, ***P < 0.001).(c) HEK293 cells were infected with a lentivirus encoding the Gαq-coupled designer receptor Rαq. Cells were additionally infected with a lentivirus containing the Coll.luc reporter gene. Furthermore, the cells were infected with a lentivirus encoding either RGS2 or β-galactosidase (mock). We incubated the cells in medium containing 0.05 % serum for 24 hours. Stimulation of the cells was performed with CNO (1 μM) for 24 hours in serum-reduced medium. Cells were harvested and analyzed as described in the legend to Figure 2 (n=5, ***P < 0.001). (d) T-REx-TRPM3 cells containing a chromatin-integrated Coll.luc reporter gene were serum-starved for 24 hours in the presence of tetracycline (1 μg/ml). The serum-starved cells were infected with a lentivirus encoding either RGS2 or β-galactosidase (mock). Cells were stimulated with pregnenolone sulfate (20 μM) for 24 hours. Cells were harvested and analyzed as described in the legend to Figure 2 (n=3; n.s. not sifnificant). (e) INS-1 832/13 insulinoma cells were infected with a lentivirus containing the reporter gene Coll.luc. Additionally, we infected the cells with a lentivirus encoding either RGS2 or β-galactosidase (mock). Cells were incubated in medium containing 0.5% serum and 2 mM glucose for 24 hours, and then stimulated with KCl (25 mM) and the FPL64176 (2.5 μM) for 24 hours. Cells were harvested and analyzed as described in the legend to Figure 2 (n=3; n.s., not significant).
Figure 4.
RGS2 expression reduces signal transduction after stimulation of TRPM8 channels or Gαq-coupled designer receptors. (a) Modular structure of RGS2. The RGS domain is responsible for binding to Gαq, while the N-terminal domain is required for targeting the protein to the plasma membrane. (b) HEK293-M8 cells containing the Coll.luc reporter gene were infected with a recombinant lentivirus encoding either RGS2 or β-galactosidase (mock). Cells were serum-starved for 24 hours, and then stimulated with icilin (1 μM) in serum-reduced medium for 24 hours. Cells were harvested and analyzed as described in the legend to Figure 2 (n=3, ***P < 0.001).(c) HEK293 cells were infected with a lentivirus encoding the Gαq-coupled designer receptor Rαq. Cells were additionally infected with a lentivirus containing the Coll.luc reporter gene. Furthermore, the cells were infected with a lentivirus encoding either RGS2 or β-galactosidase (mock). We incubated the cells in medium containing 0.05 % serum for 24 hours. Stimulation of the cells was performed with CNO (1 μM) for 24 hours in serum-reduced medium. Cells were harvested and analyzed as described in the legend to Figure 2 (n=5, ***P < 0.001). (d) T-REx-TRPM3 cells containing a chromatin-integrated Coll.luc reporter gene were serum-starved for 24 hours in the presence of tetracycline (1 μg/ml). The serum-starved cells were infected with a lentivirus encoding either RGS2 or β-galactosidase (mock). Cells were stimulated with pregnenolone sulfate (20 μM) for 24 hours. Cells were harvested and analyzed as described in the legend to Figure 2 (n=3; n.s. not sifnificant). (e) INS-1 832/13 insulinoma cells were infected with a lentivirus containing the reporter gene Coll.luc. Additionally, we infected the cells with a lentivirus encoding either RGS2 or β-galactosidase (mock). Cells were incubated in medium containing 0.5% serum and 2 mM glucose for 24 hours, and then stimulated with KCl (25 mM) and the FPL64176 (2.5 μM) for 24 hours. Cells were harvested and analyzed as described in the legend to Figure 2 (n=3; n.s., not significant).
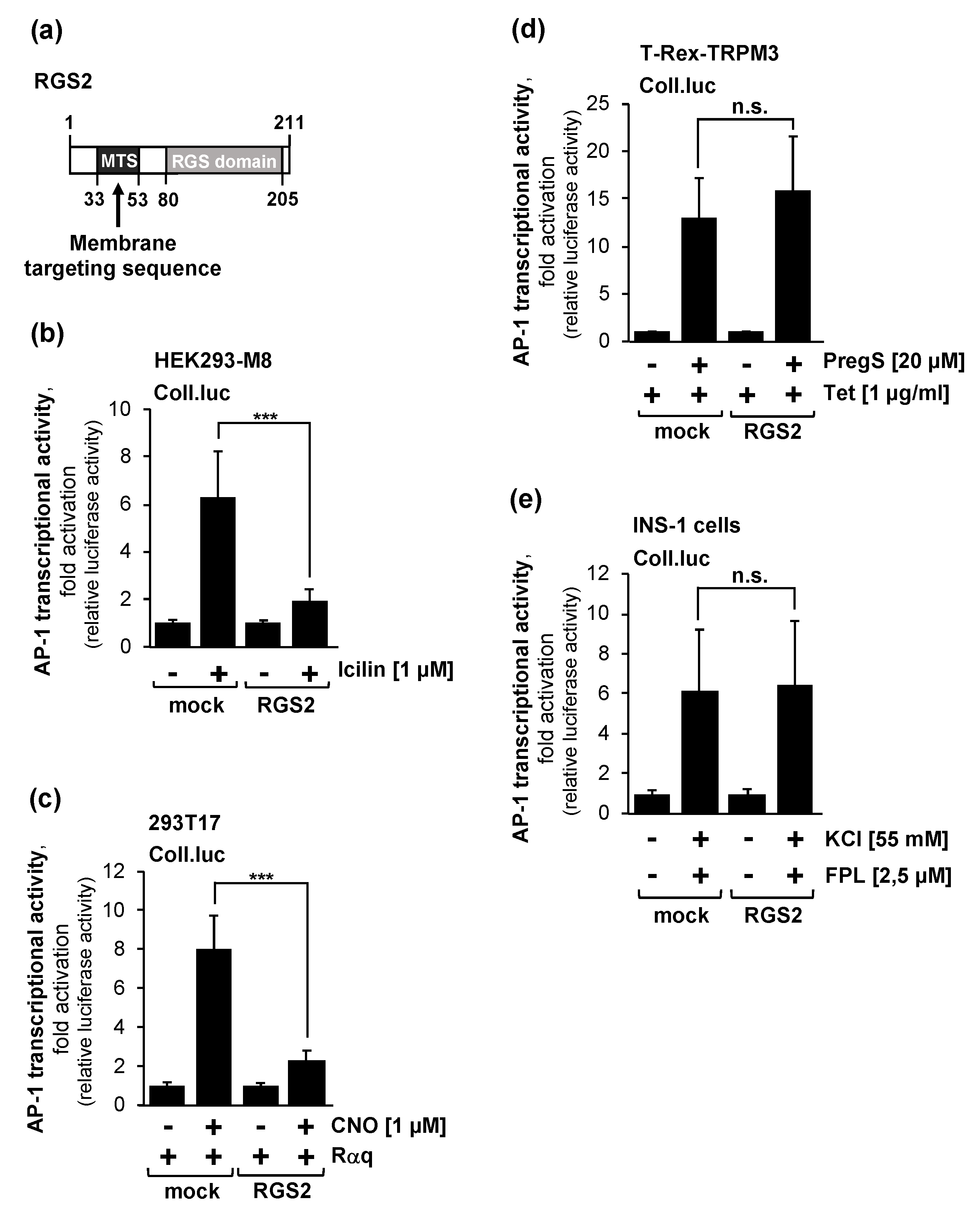
Figure 5.
The Gβγ inhibitor gallein attenuates signal transduction mediated by TRPM8 and TRPM3 channels. (a) T-REx-TRPM3 cells containing a chromatin-embedded Coll.luc reporter gene were serum-starved for 24 hours in the presence of tetracycline (1 μg/ml) to induced TRPM3 expression. Serum-starved cells were preincubated with gallein (20 μM) for three hours, and then stimulated with pregnenolone sulfate (20 μM) for 24 hours in the presence of the compound. Cells were harvested and analyzed as described in the legend to Figure 2 (n=4; ***P < 0.001). (b) HEK293-M8 cells containing the Coll.luc reporter gene integrated into the chromatin were serum-starved for 24 hours, preincubated for 3 hours with gallein (20 μM), and then stimulated with icilin (1 μM) in serum-reduced medium in the presence of gallein. Cells were harvested and analyzed as described in the legend to Figure 2 (n=3; ***P < 0.001).
Figure 5.
The Gβγ inhibitor gallein attenuates signal transduction mediated by TRPM8 and TRPM3 channels. (a) T-REx-TRPM3 cells containing a chromatin-embedded Coll.luc reporter gene were serum-starved for 24 hours in the presence of tetracycline (1 μg/ml) to induced TRPM3 expression. Serum-starved cells were preincubated with gallein (20 μM) for three hours, and then stimulated with pregnenolone sulfate (20 μM) for 24 hours in the presence of the compound. Cells were harvested and analyzed as described in the legend to Figure 2 (n=4; ***P < 0.001). (b) HEK293-M8 cells containing the Coll.luc reporter gene integrated into the chromatin were serum-starved for 24 hours, preincubated for 3 hours with gallein (20 μM), and then stimulated with icilin (1 μM) in serum-reduced medium in the presence of gallein. Cells were harvested and analyzed as described in the legend to Figure 2 (n=3; ***P < 0.001).
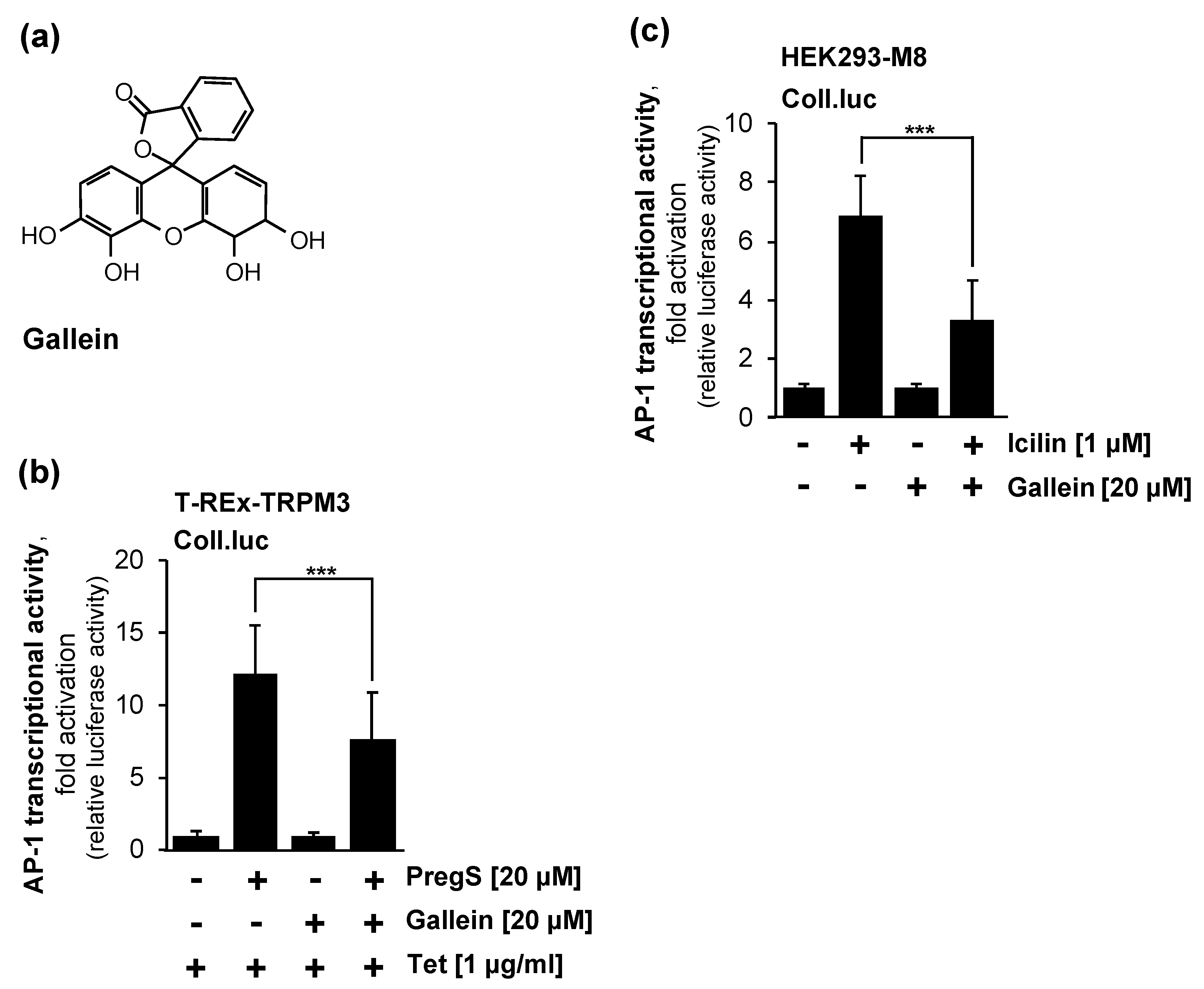
Figure 6.
The transcription factor c-Jun is required to link the stimulation of TRPM8 with the activation of AP-1. (a) Modular structure of the transcription factor c-Jun and its dominant-negative c-Jun mutant c-JunΔN. c-JunΔN contains the C-terminal amino acids 188 to 331 of c-Jun, which comprise the bZIP domain. The mutant lacks the transcriptional activation domain. (b) Expression of c-JunΔN attenuates icilin-induced activation of AP-1 in HEK293-M8 cells. Cells were infected with a lentivirus containing a Coll.luc reporter gene. Cells were additionally infected with a lentivirus encoding either c-JunΔN or β-galactosidase (mock). Serum-starved cells were stimulated with icilin (1 μM) for 24 hours. Cells were harvested and analyzed as described in the legend to Figure 2 (n=3; ***P < 0.001).
Figure 6.
The transcription factor c-Jun is required to link the stimulation of TRPM8 with the activation of AP-1. (a) Modular structure of the transcription factor c-Jun and its dominant-negative c-Jun mutant c-JunΔN. c-JunΔN contains the C-terminal amino acids 188 to 331 of c-Jun, which comprise the bZIP domain. The mutant lacks the transcriptional activation domain. (b) Expression of c-JunΔN attenuates icilin-induced activation of AP-1 in HEK293-M8 cells. Cells were infected with a lentivirus containing a Coll.luc reporter gene. Cells were additionally infected with a lentivirus encoding either c-JunΔN or β-galactosidase (mock). Serum-starved cells were stimulated with icilin (1 μM) for 24 hours. Cells were harvested and analyzed as described in the legend to Figure 2 (n=3; ***P < 0.001).
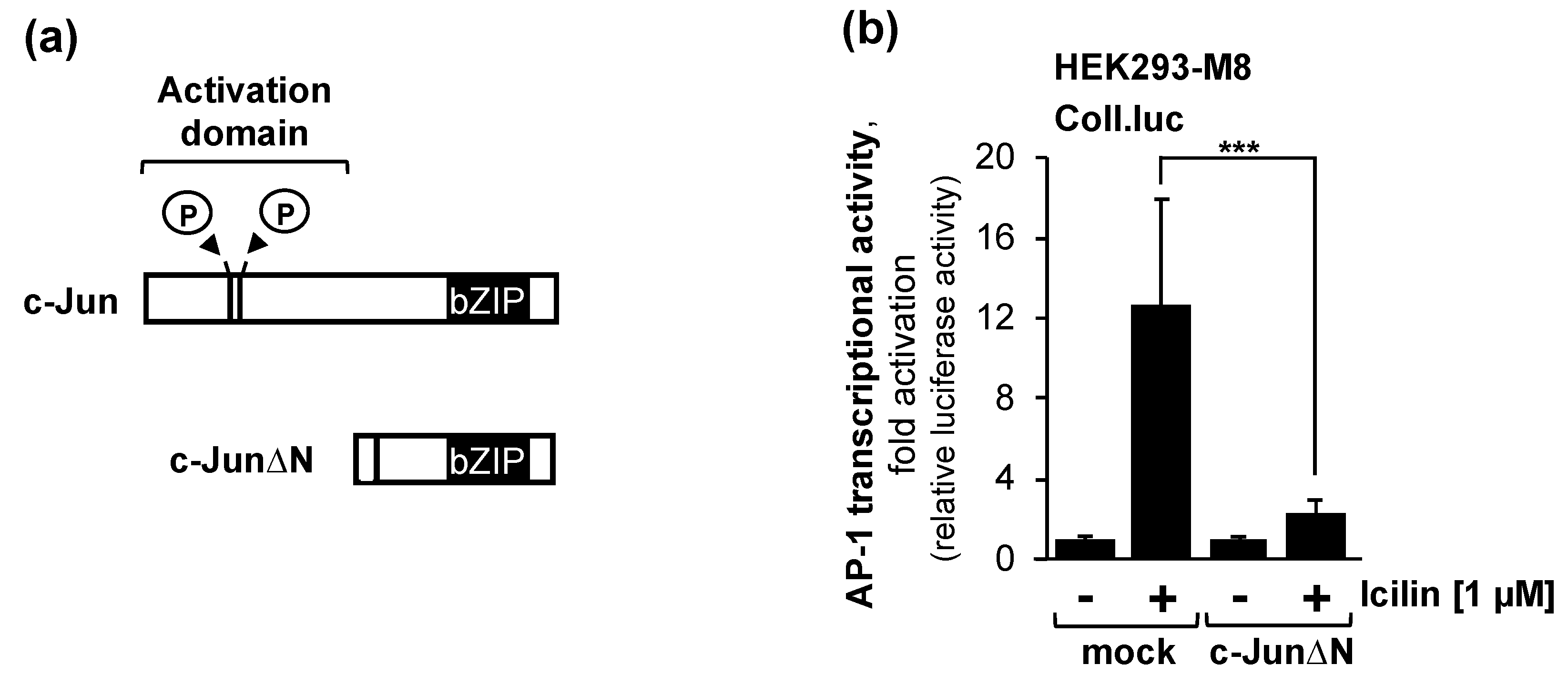
Figure 7.
Signal transduction of TRPM8 channels.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



