Submitted:
30 April 2024
Posted:
02 May 2024
Read the latest preprint version here
Abstract
Obstructive sleep apnea (OSA) is a common sleep disorder that is associated with a wide variety of health conditions, including cardiovascular, cerebrovascular, metabolic, neoplastic, and neurocognitive manifestations. OSA is mainly characterized by repeated upper airway obstructions during sleep that cause episodes of chronic intermittent hypoxia (IH), resulting in tissue hypoxia-reoxygenation. Decreased arterial oxygen pressure (PaO2) and hemoglobin saturation (SatO2) stimulate reflex responses to overcome the obstruction. The prevalence of OSA is significant worldwide, particularly in women during pregnancy, due to the physiological changes associated with this stage of life, especially in its later stages. It is associated with an increased risk of hypertension, pre-eclampsia and diabetes, among others. OSA during pregnancy can interfere with normal fetal development and is associated with growth retardation, preterm birth, or low term weight. Carotid body stimulation and hypoxia-reoxygenation episodes contribute to cardiovascular disease and oxidative stress, that can harm both, mother and the fetus, even into the adulthood. Because gestational intermittent hypoxia (GIH) is the hallmark of OSA in pregnancy, this review examines the impact of GIH on maternal, fetal, and offspring respiratory outcomes. Most adverse outcomes can be prevented by continuous positive airway pressure (CPAP) therapy and lifestyle changes if gestational OSA is detected early and treated appropriately.
Keywords:
gestational intermittent hypoxia
; intermittent hypoxia
; pregnancy
; obstructive sleep apnea
; respiratory system
; placenta
; fetus
; offspring
; oxidative stress
1. Introduction
Obstructive sleep apnea (OSA) is the most common sleep disorder worldwide [1]. It is associated with cardiovascular, cerebrovascular, metabolic, neoplastic and neurocognitive diseases [2,3]. OSA is characterized by repeated complete (apnea) or partial (hypoapnea) obstructions of the upper airway during sleep. With each obstruction, arterial oxygen pressure (PaO2) and hemoglobin saturation for oxygen (SatO2) decrease, making OSA a chronic recurrent intermittent hypoxia (IH) event, the severity of which is related to changes in SatO2 [4]. The decrease in PaO2 activates the carotid body (CB), which activates motor neurons of the inspiratory and dilator muscles of the upper airways and sleep-wake control areas at of the central nervous system [5]. Muscle action and sudden arousals overcome the obstruction, PO2 is restored, and the cycle repeated, resulting in IH, hypercapnia and sleep fragmentation [6]. CB stimulation produces respiratory reflexes and cardiovascular responses to minimize the deleterious effects of hypoxia. Repeated stimulation sensitizes CB chemoreceptors, increasing sympathetic tone, leading to hypertension and cardiovascular and metabolic pathology [7].
Epidemiological data on OSA indicate the magnitude of the social, health and economic problem. Its prevalence is estimated to be 9-24% in men and 4-9% in women over 30 years of age, increasing with age [8]. In pregnant women, the prevalence is estimated to be around 8% in the first trimester and increases to nearly 20% in the third trimester of pregnancy [9,10].
Anatomical and physiological changes during pregnancy, or the presence of the disease prior to pregnancy, increase the prevalence of gestational OSA [11]. This pathology is associated with increased morbidity and mortality and can cause hypertension, pre-eclampsia and gestational diabetes mellitus [12]. Episodes of hypoxia-reoxygenation during pregnancy can affect fetal development and postnatal development, resulting in growth retardation, preterm delivery, low birth weight and even fetal death [13]. Adult changes, such as metabolic and epigenetic alterations in male offspring, have also been described in animal models where intermittent hypoxia exposure during pregnancy mimics the hypoxia-reoxygenation cycles of gestational OSA [14]. These episodes of hypoxia-reoxygenation increase the production of reactive oxygen species (ROS), causing tissue damage and an influx of inflammatory cells to the injured sites and into the fetus via the placenta [15].
The repeated episodes of IH that occur during sleep, and especially during pregnancy, can have significant respiratory effects on both, the mother, and the developing fetus. The following sections highlight the major respiratory effects of gestational intermittent hypoxia (GIH), and how they affect the pregnant woman, the fetus, and the developing offspring into adulthood.
2. Maternal Respiratory Disturbances
2.1. Hormonal changes
Pregnancy induces hormonal alterations, which are the main cause of changes in the respiratory pattern. Progesterone, which acts as a trigger for the primary respiratory center, gradually increases its plasma levels during pregnancy, going from 27 ± 1.5 ng/mL at week 13 to 146 ± 17.2 ng/mL at week 37, decreasing after delivery (3.5 ± 1.5 ng/mL) [16]. This remarkable increase is accompanied by an increase in the sensitivity of the respiratory center to CO2 [17]. In addition, progesterone acts as a bronchodilator lessening airway smooth muscle tone and, by acting directly on the CB, increase the peripheral ventilatory response to hypoxia.
Pregnancy also affects estrogens levels, which rise in parallel with progesterone levels. It acts by increasing the number and sensitivity of progesterone receptors in the areas of the central nervous system associated with respiration, mainly the hypothalamus and medulla oblongata [18]. The prostaglandins have also been described to have a variety of various effects, with PGF2α causing bronchial smooth muscle constriction and PGE1 and PGE2 causing bronchodilation [18].
2.2. Anatomical changes
Pregnancy-induced uterine enlargement increases abdominal pressure, displacing the diaphragm upwards. As a result, pleural pressure increases, leading to small airways obstruction, which reduces functional residual capacity FRC (20-30%), expiratory reserve volume ERV (15-20%) and residual volume RV (20-25%). The shortening in chest height is accompanied by an increase in other dimensions to maintain total lung capacity (TLC) [19]. Along with these changes in FRC and ERV, pregnancy is associated with an increase in the tidal volume (30-50%) and minute ventilation (20-50%), with only a small increase in respiratory frequency.
As the diaphragm moves upwards, the thorax also undergoes significant changes during pregnancy. There is an increase in the subcostal angle of the rib cage (from 68 to 103 degrees) due to relaxation of the lower rib cage ligaments, and an increase in the circumference of the lower chest wall (5 to 7 cm). These anatomical changes peak at week 37, and largely return to normal by week 24 after delivery (although the subcostal angle remains 20% wider than at baseline) [16]. This increase in chest wall size also helps to compensate for the upward displacement of the diaphragm mentioned above.
In addition, pregnant women have significant upper airway narrowing during the third trimester of pregnancy [20]. This upper airway narrowing may be related to the increased incidence of snoring and sleep-disordered breathing in pregnancy. Alongside this observation, women with pre-eclampsia have also been described to have narrower upper airways in supine position than pregnant and non-pregnant women [21]. Besides this narrowing, they have larger neck circumferences, which may be due to differences in fat deposition around the neck, or tissue oedema, which is present in most women with pre-eclampsia. All these factors would certainly increase the severity of OSA.
Besides these effects, the mucosa of the upper airways is also affected during pregnancy, with an increase in mucopolysaccharide content and phagocytic activity, resulting in nasal congestion. This common nasal congestion is known as pregnancy rhinitis, and affects up to 65% of pregnant women, lasting 3 or more weeks [22] and resolving spontaneously 2 weeks after delivery. Although the etiology is unclear, some contributing factors may be smoking, nasal allergy or infections, with the potential of further complications. Nasal obstruction would exacerbate snoring and therefore sleep-disordered breathing.
2.3. Respiratory changes during labour and delivery
The respiratory muscles play a key role in pushing during labour. It has been described in animal models that a long period of stretching causes muscle fibers to add sarcomeres in the longitudinal direction, thereby increasing tension [23]. This may well be what happens to the diaphragm and abdominal muscles during the long stretching period of pregnancy, to improve their efficiency in developing expulsive force during labor.
Intrauterine pressure is increased by contracting the diaphragm during inspiration and the abdominal muscles during expiration. In the latter stages of labor, women may be asked to take deep inspirations and hold their breath while pushing. This maneuver, known as “closed-glottis” (or Valsalva) pushing, fixes the diaphragm in a low position. Alternatively, women may be instructed to maintain a more spontaneous pushing technique, emitting sounds and pushing with a slow exhalation without holding their breath. This second approach is called “open-glottis” pushing technique. Among first-time mothers with singleton pregnancies at term, there was no difference in the risk of assisted vaginal delivery between the two pushing techniques [24].
To conclude this section on maternal respiratory disorders associated with intermittent hypoxia, we mention a retrospective analysis of 4326 pregnant women diagnosed with obstructive sleep apnea in the United States [25]. This study found a direct association between sleep apnea and an increased likelihood of maternal-fetal complications. Along with higher rates of pre-eclampsia and cesarean delivery, there was also a higher prevalence of pulmonary edema, although no significant association with a higher risk of mortality.
3. Effects of Intermittent Hypoxia in the Placenta
The placenta is the first organ to form in mammals and is essential for feeding, breathing, protection and hormone production during the fetal development [26]. During pregnancy, the placenta is subject to a variety of environmental cues that can have an impact on the placental development, fetal growth, and postnatal phenotype of the offspring. These factors include low oxygen levels, which can be caused by maternal anemia, umbilical cord occlusion, or poor placental vascularization, as well as by all high-altitude pregnancies or maternal OSA [27-29]. Attempts to experimentally mimic such conditions in animals, including mice, rats, and guinea pigs, have shown that placental and fetal weights are altered (see [29]). In general, these studies show that the specific effects on placental appear to depend on the type and the severity of the challenge, and the duration and timing of the challenge in relation to placentation. However, an adverse maternal environment has a greater effect on fetal growth than on placental growth, suggesting that the placenta may be spared relative to other organs [30]. In addition, although placental growth and function are similar in females and males, sexually dimorphic phenotypes are observed, as reported by Kalisch-Smith et al. [31].
Similar to the postnatal role of the lung, the placenta is the organ where fetal blood gases are exchanged with the environment by a concentration gradient-dependent passive extraction of oxygen from maternal blood, similar to the process of diffusion and transfer of oxygen from alveolar gas. The human placenta consists of an extensively branched fetal villous tree that is bathed by maternal blood circulating in the intervillous space. The interface between fetal placental tissue and maternal blood is the syncytiotrophoblast, which acts as the endothelium for the intervillous space [32]. Oxygen transfer across the placental barrier is achieved as fetal blood circulates from the umbilical arteries to the capillaries within the placental villi and back to the umbilical vein [33], whereas maternal blood is filtered through a high-volume intervillous compartment, making the exchange process slower and less efficient than at the alveolar-capillary interface [34]. In addition, the placenta is a high consumer of oxygen, resulting in a PO2 in fetal blood leaving the placenta through the umbilical vein that is significantly lower than the PO2 in the maternal artery [34,35]. This fact, which is not a limitation under normal conditions of oxygenation, may compromise gas exchange during the intermittent hypoxia episodes that occur in OSA. Notably, there is evidence of hypoxia in the human placenta in OSA [36]. Fluctuations in oxygen delivery in the intervillous space following intermittent hypoxia during the process of gas exchange in the placenta result in hypoxia-reoxygenation events that can overwhelm local antioxidant defenses and lead to oxidative stress and pathological placental changes [37]. The effects of hypoxia-reoxygenation are well documented in other organs such as the heart, brain and intestine, where its detrimental effects are mainly due to its ability to generate high levels of ROS, oxidative stress and increased inflammation [32,38,39]. It is therefore biologically plausible that OSA, via one or more of these pathways, may adversely affect placental tissue perfusion and oxygenation, potentially predisposing to the development of placental-mediated adverse outcomes.
The severity of maternal oxygen deprivation seems affect the gross architecture of the placenta. Thus, maternal inspired hypoxia of 12-13% in the last third of pregnancy selectively expanded the labyrinthine zone [40,41]. Conversely, when maternal inspired oxygen is reduced to 10%, the labyrinthine zone is reduced with an associated increase in the volume density of the junctional zone [41,42]. Maternal intermittent hypoxia has been reported to alter the placental area [43]. Switching metabolism from aerobic to anaerobic by inhibiting mitochondrial oxidation early in blastocyst development also alters placentation in rats in the short term [44]. Hypoxia-induced changes in gross placental morphology have been associated with altered expression of genes and proteins involved in proliferation, apoptosis, oxidative stress, and cell lineage differentiation in which the hypoxia-inducible factors (HIF) appear to be essential [29,45].
In the same line, human trophoblasts proliferate in vitro under low O2 conditions, but differentiate at higher O2 levels, mimicking the developmental transition they undergo when invading the placental bed to establish the maternal-fetal circulation in vivo. Using a human trophoblast cell line (HTR8/SVneo) and primary extravillous trophoblast cells (EVTs), Song et al [46] reported that, in contrast to sustained hypoxia, intermittent hypoxia blocks trophoblast proliferation and induces excessive trophoblast apoptosis by activating the ER stress signaling pathway. They found that intermittent hypoxia can also significantly impair the invasive and migratory ability of trophoblasts which they attributed in part to the reduced secretion of matrix metalloproteinase-2. These changes in trophoblast function may have implications for proper pregnancy development [47].
Placental delivery of maternal nutrients and oxygen to the fetus is the primary determinant of intrauterine fetal growth. Therefore, pregnancy is associated with a continuous remodeling of the uteroplacental circulation to ensure adequate blood flow to provide sufficient nutrients and oxygen for fetal and placental growth [48,49]. As a result, human uterine perfusion increases from 50 ml/min at 10 weeks' gestation to as much as 1,300 ml/min at the end of pregnancy [50]. This increased blood flow is directly correlated with fetal growth and survival, as well as neonatal birth weight [49,51].
However, placental development during normal pregnancy occurs in a low-oxygen environment (~2-8 % O2) and this "physiological hypoxia" appears to be critical for early placental development and angiogenesis [52,53]. Vascular endothelial growth factor (VEGF) and fibroblast growth factor-2 (FGF2) are two potent angiogenic factors that have been implicated in placental angiogenesis [54,55]. It is well established that hypoxia increases the expression of VEGF and its receptors VEGFR-1 and VEGFR-2 in endothelial cells [56]. Hypoxia also increases the expression of FGF receptors and alters the distribution of FGF2 in different cellular compartments [57]. It has been reported that hypoxia may differentially modulate angiogenic factor-stimulated placental angiogenesis depending on the severity and duration of hypoxia exposure. Wang et al [58] showed in human placental artery endothelial (HPAE) cells that in vitro exposure to sustained hypoxia (3% O2) enhanced FGF2 and VEGF-stimulated HPAE cell proliferation, but independently of the canonical MEK1/ERK1/2 and PI3K/AKT1 pathways, suggesting that hypoxia may induce a very complex signaling network in the placenta. Whether intermittent hypoxia adds further complexity is currently unknown.
Finally, how intermittent hypoxemia is transmitted from the mother to the fetus in OSA deserves some comment. By simultaneously measuring PO2 in the maternal artery and umbilical vein, Almendros et al [59] showed in a sheep pregnancy model that the PO2 fluctuations resulting from experimental maternal obstructive apnea are significantly smaller in fetal blood than those registered in maternal blood, due to the damping effect of oxygen transfer across the placenta. This work raises an important question about the potential effects of intermittent hypoxia on the fetus during pregnancy, as the placenta may attenuate the severity of hypoxemic changes in the fetus.
In addition to potential differences in the structure and physiology of the placenta in sheep and humans and, therefore, in the values of oxygen swing attenuation through the placenta in both species, the study by Almendros et al [59] rises several considerations when translating the findings to pregnant women. Since the severity of hypoxemic swings is attenuated in the fetus, it could be anticipated that hypoxia-reoxygenation, and thus the oxidative stress induced in the various fetal organs and tissues would be lower than those experienced by adult tissues [60].
It is also possible that the sensitivity and tolerance of fetal tissues to very short intermittent hypoxia, which have so far not been studied, are different from those in adults. Moreover, the impact of intermittent hypoxemia, even of low amplitude, could be different in the fetus and the adult, since fetal mechanisms to regulate the distribution of blood flow among organs are markedly different from those in adults [61,62,63]. These potential differences could modulate the epigenetic changes observed in the offspring after GIH [64-66].
4. Fetal Respiratory Distress
As a result of maternal GIH, and the recurrent episodes of intermittent hypoxia, there is a reduction in oxygen saturation in the maternal blood which could compromise oxygen delivery from the placenta to the fetus, leading to fetal hypoxemia. However, the extent to which the placenta provides protection to the fetus, and therefore the amount of oxygen that reaches the fetus is still unknown.
Although there is not a functional, fully developed gas exchange system in the lungs of the fetus, it doesn´t mean that they remain inactive. There are rhythmic contractions of the diaphragm and other respiratory muscles, known as fetal breathing movements (FBM), which play a key role in lung maturation. They are a distinctive adaptation to the intrauterine environment.
With completely unique characteristics that differ from neonatal and adult breathing, they also differ from maternal respiratory movements. In humans, FBM begin at around week-10 of gestation and increase in frequency with gestational age, reaching up to 35% of the time around week-30 of pregnancy [67,68]. They occur at a frequency of approximately 30-70 contractions/breaths per minute in each bout, followed by apneic periods that can last up to 2 hours near term [69]. Between 28 and 36 weeks, FBM are closely correlated with REM states, with less frequent FBM occurring in non-REM states [70]. The frequency of FBM episodes is also correlated with other factors, such as glucose levels, with a higher incidence described in humans after maternal meals [71]. In terms of respiratory gases, high levels of PaCO2 (hypercapnia) increase the incidence of FBM, while low levels of PaO2 (hypoxia) inhibit FBM in studies in ewes [72] and humans [73].
Based on umbilical cord blood analysis, one study found that maternal snoring during pregnancy was associated with enhanced fetal erythropoiesis [74], suggesting that the fetus becomes hypoxic in utero, at least to some extent, during the repeated bouts of intermittent hypoxia. These episodes of fetal hypoxemia, if prolonged or severe, could impair fetal development and increase the risk of adverse perinatal outcomes.
Due to the increased metabolic demands of the fetus during pregnancy, increases in O2 consumption (20%) and CO2 production (35%) have been described in pregnant individuals when compared with their respective controls. These changes do not correlate exactly with the changes in minute ventilation described in the previous section which were around 20 and 50%. This greater increase in minute ventilation translates into an increase in the alveolar (PAO2) and arterial partial pressures of O2 (PaO2) and a decrease in the corresponding partial pressures of CO2 (PACO2 and PaCO2). These increases in oxygen tension are important to facilitate oxygen transfer across the placenta. Thus, measurements of PaO2 during pregnancy found control values of PaO2 around 93 mmHg raising to 105-106 mmHg during the first trimester, and 101-106 mmHg near term [75]. Despite this increase in oxygen tension, the oxygen reserves of pregnant women are lower, making them more susceptible to hypoxic episodes during periods of apnea, and consequently to the developing fetus.
As for the PaCO2 levels, they went from 37 mmHg in the non-pregnant state to 28-29 mmHg in the first trimester and 26-30 mmHg near term. These low PaCO2 values lead to chronic respiratory alkalosis, with compensatory renal mechanisms acting to excrete bicarbonate, whose endpoint is stimulating the 2,3-diphosphoglycerate (2,3-DPG) synthesis. The increase in 2,3-DPG levels reduces the affinity of maternal hemoglobin for O2, allowing more O2 to be delivered to the fetus, whose major hemoglobin, called fetal hemoglobin or HBf, has a higher affinity for O2 than the adult hemoglobin or HbA.
The transition from fetal to postnatal breathing includes, among others, the absorption of lung fluid, gaseous expansion of the lungs, activation of the Hering-Breuer reflexes and the appearance of continuous respiration. Birth itself is a delicate process that has hindered more detailed research. Thus, many of the factors involved in the birth process are not yet fully understood. Asphyxia, umbilical cord occlusion or the rise in PaO2 with the first postnatal breath may be involved [76,77], although neither the afferent input from the carotid sinus nerve [78] nor the interruption of umbilical circulation [79] seem to play a role in this process.
5. Offspring Respiratory Disorders
Although key components of the respiratory system begin to develop in utero, such as phrenic motoneurons and diaphragm muscle, there is also an important postnatal maturation process. After birth there is a transition from fetal breathing movements to functional gas exchange. With a less efficient respiratory system, infants develop a number of counteractive mechanisms to compensate for the increased vulnerability of their respiratory system.
Looking at the evolution of some respiratory values in humans it is known that respiratory rates fall progressively with age, going from 40 breaths/min in neonates to around 12-18 breaths/min in adults, with tidal volume values (adjusted by weight) remaining virtually identical throughout life.
In addition to these numbers, chemoreceptor pCO2 sensitivity is almost mature at term, but minute ventilation is higher at any level of CO2 in neonates and infants compared to adults [80]. However, preterm infants showed an attenuated sensitivity (and slower response) to CO2 [81].
As for the peripheral pO2 receptors, they need a few weeks of readjustment [82] to gain sensitivity to the higher levels of oxygen of the environment compared to the uterus. This adaptation results in a depression of ventilatory responses to hypoxia that can last up to 6 months after birth [83]. Immediately after birth, rats showed a weak ventilatory response to a hypoxic challenge [84], even though their ventilatory rate was relatively high, probably to compensate for their immature gas exchange machinery.
Regarding the airways, they are a component of the respiratory system that also showed differences between neonates and adults. Thus, the pediatric upper and lower airways are characterized by a higher resistance to flow, mainly due to a smaller diameter, higher collapsibility, and lower pharyngeal muscle tone [85]. Focusing on the upper airways, they have an elliptical shape in neonates. From the subglottic to the cricoid ring, the airways are narrower in the transverse dimension than in the anteroposterior dimension [86]. Observations of airways in children over 1 year of age showed a circular shape. Smaller and softer, they are more prone to inspiratory collapse. The closure of newborn upper airways is behind central apneas, with values of 3 obstructive events per hour during the first 3 months of life [87]. This closure may be due to either passive pharyngeal collapse or active laryngeal closure [88]. Nevertheless, the cartilaginous structure lengthens and stiffens with maturation during childhood.
Respiratory immaturity is associated with breathing instability, while immature breathing control manifests as central apneas. Both events have been linked, although controversially, to the sudden infant death syndrome [89]. There is an improvement with progressive maturation, with the frequency and duration of central apneas decreasing during the first 12 months of life, with values of 15 apneas longer than 3 seconds per hour reported at 12 months [90].
Chest wall properties are also affected. Thus, newborns have up to 3 times higher compliance levels than adults to allow for greater expansion during breathing. The ribs also contribute to this end, as they are more flexible in the newborn, while they are positioned more horizontally than in the adults, moving only upwards, and not upwards and outwards, as in the adult rib cage. This difference is the main limitation to increase tidal volumes and the reason why infants under 12 months of age rely mainly on diaphragmatic breathing [91].
The values of the functional reserve capacity (FRC) are also subjected to significant changes, with values that increase rapidly after birth [92]. FRC is directly related to the number of alveoli; fewer alveoli in immature lungs provide less support and can lead to peripheral airways collapse. During lungs growth, they undergo a process of alveolarization which also contributes to increase the surface area available for gas exchange.
Exposure to adverse conditions in utero, such as those caused by GIH, may lead to epigenetic changes that affect the postnatal development of the respiratory control. Animal studies have shown that prenatal exposure to intermittent hypoxia can alter respiratory control mechanisms and increase the susceptibility to respiratory diseases later in life [93]. However, the effects of intermittent hypoxia protocols during pregnancy on the developing respiratory system are not yet fully understood.
The first study of the effects of GIH on respiratory responses in neonatal rats found that normoxic ventilation was increased in the GIH offspring compared to their respective controls (1- and 4-months of age), although hypoxic responses were similar [94]. They postulated that this maladaptive response to postnatal hypoxia could contribute to events such as the sudden death syndrome. Nevertheless, this study had several limitations because it didn´t distinguish between male and female pups. Our group found similar differences in basal normoxic ventilation between GIH and control rats at 1 month of age, in both, males and females (Valverde-Pérez et al., unpublished data). However, following the same rats at 3 months of age, minute ventilation values were higher only in the female GIH animals, being this ventilation decreased in the male GIH compared to their respective controls.
A study of inflammatory status in neonatal GIH-treated rats recorded respiratory motor burst in brainstem-spinal cord preparations [93]. In newborn rats (0-3 days old) burst frequency was unchanged in GIH animals, although the rhythm was more regular and the rise time tended to be faster, with no sex effects on these variables in any of the groups. When testing the respiratory motor output elicited by an inflammatory challenge, males showed a significantly reduced burst frequency compared to normal conditions, with no differences between control and GIH animals. However, in preparations from female animals, only the control preparations showed a decrease in burst frequency with the inflammatory challenge, while the GIH group showed no change. Although GIH didn´t alter the expression of pro-inflammatory factors tested in the brainstem or spinal cord (IL-1β, TNFα and COX-2), it did affect the offspring response to a later postnatal inflammatory challenge, in a sex-dependent manner. Their hypothesis is that GIH reprogrammed the neuroinflammatory response pathways in the female animals so that there is less cytokines production during inflammatory challenges. Cytokines are linked to the sensitivity of the brainstem respiratory centers, which modulate the respiratory frequency in a complex way, ranging from hyperventilation and even respiratory stress syndrome (as in the worst cases of Covid-19, associated with “storm” of cytokines) [95], to respiratory depression. If this were the case, female GIH offspring would have an impaired ability to trigger an immune response against infection, from birth, and perhaps even into adulthood, especially at the level of respiratory system pathologies.
At birth, the newborn comes into an oxygen-rich environment, and oxygenation through the lungs instead of the placenta raises tissue oxygenation within minutes. Same of oxygen forms reactive oxygen species (ROS), which are normally neutralized by antioxidant defense systems. In preterm infants, these defenses are immature, making them more prone to oxidative tissue injury. Thus, more than 65% of preterm infants suffer at least one infection during their hospitalization [96]. In addition to this, preterm infants with inflammation also have an increased incidence of apnea and are at a greater risk for adverse neurodevelopmental outcomes and mortality rates (see [97]).
The frequent episodes of intermittent hypoxia associated with preterm birth have been linked to adverse respiratory outcomes, with a recent article [98] linking them to high levels of oxidative stress. By measuring noninvasively lipids, proteins, and DNA oxidative stress biomarkers in urine samples from 1 week and 1 month old premature babies (<31 weeks of gestation), they found a direct association between intermittent hypoxia and the various oxidative products quantified. In this sense, it could be possible that most of the effects of GIH on the offspring are indirectly caused by the maternal stress. Thus, OSA events in pregnant women were associated with higher levels of systemic inflammation, which was related to the number of obstructive events, especially during the REM phases of sleep [99].
It is known that gestational stress can induce permanent epigenetic changes in the offspring, as shown by an increase in the levels of some chemokines and altered expression of their receptors in a rat model [100]. In this context, sex-specific effects of stress during gestation and development of respiratory control have been described (Fournier S et al., 2013), with a higher rate of pathological apneas in newborn male rats, which was associated with a deficit of medullar-derived serotonin (5-HT). A recent study [101] has found that GIH impairs endothelial NO synthesis in rat offspring vessels, highlighting the fact that some detrimental effects of gestational hypoxia may persist well into adulthood.
Underscoring the importance of inflammation in the neonatal respiratory system, even low levels of inflammation have been proved to impair respiratory control networks in neonatal rats [102]. Using brainstem spinal cord preparations, these authors found differential sensitivities of the respiratory control areas to inflammation. Lower levels of inflammation were more disruptive, affecting amplitude and frequency, while more severe inflammation affected frequency only.
Neonatal inflammation also affected carotid body chemosensitivity, induced spontaneous apneas in vivo and caused significant O2 desaturation [103]. Dopamine, an inhibitor of CB activity, increased its levels in the CB due to neonatal inflammation. Neonatal inflammatory responses may mirror those of adults, although it could be that neonatal signaling pathways are different (see [104]). Nevertheless, the paucity of scientific studies on GIH exposure and respiratory control makes it difficult to draw firm conclusions.
Not many studies are available either on the role of the respiratory muscles, the final effectors in the respiratory control system. A study of the diaphragm function in rats exposed to GIH found no effect on the force-generating capacity of the diaphragm [105], in either male or female animals. Similar results were observed in the same study when the animals were subjected to intermittent hypoxia shortly after birth (postnatal days 22 and 42), suggesting a relative resilience of the diaphragm muscle to hypoxic stress during the perinatal period. However, when examining the sternohyoid, one of several pharyngeal dilator muscles, the same authors found evidence of weakness in the postnatally exposed animals [106]. This effect persisted into adulthood (16 weeks of age). They suggested that this mismatch between the diaphragm and sternohyoid muscles could contribute to a higher risk of airway collapse.
6. Conclusions
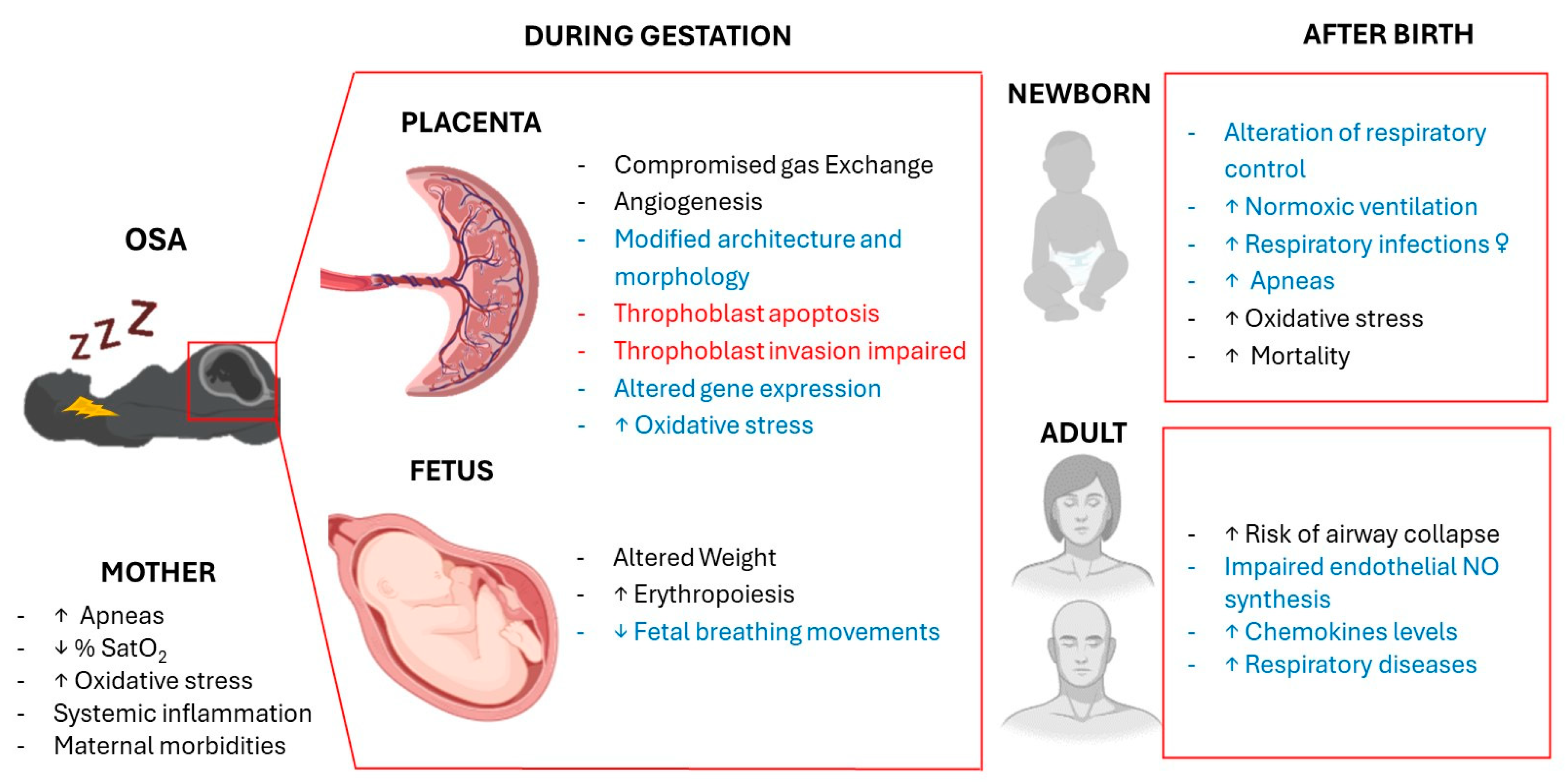
Overall, gestational intermittent hypoxia can have remarkable effects on both maternal and fetal health, with a concomitant risk of adverse pregnancy outcomes (Figure 1).
Multidisciplinary care involving obstetricians, sleep specialists, and respiratory therapists should be the approach adopted by national health services for the comprehensive management of sleep apnea during pregnancy. Early detection of OSA during pregnancy should be an essential tool for timely intervention. Questionnaires and polysomnography should be first line tools to help clinicians identify women at higher risk for OSA and respiratory disturbances.
Once identified, Continuous Positive Airway Pressure (CPAP) therapy is usually the treatment of choice. It helps improve upper airway patency and maternal oxygenation. Other interventions include lifestyle changes, such as weight management, positional therapy, and avoidance of sedatives and alcohol.
Author Contributions
Conceptualization, J.P-L and A.R; writing original draft preparation, J.P-L and A.R.; writing—review and editing, E.O. and E.V-P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Canever JB, Zurman G, Vogel F, Sutil DV, Diz JBM, Danielewicz AL, Moreira BS, Cimarosti HI, de Avelar NCP. Worldwide prevalence of sleep problems in community-dwelling older adults: A systematic review and meta-analysis. Sleep Med. 2024, 119, 118–134. [CrossRef] [PubMed]
- Heinzer R, Vat S, Marques-Vidal P, Marti-Soler H, Andries D, Tobback N, Mooser V, Preisig M, Malhotra A, Waeber G, Vollenweider P, Tafti M, Haba-Rubio J. Prevalence of sleep-disordered breathing in the general population: The HypnoLaus study. Lancet Respir Med. 2015, 3, 310–318. [CrossRef] [PubMed]
- Mediano O, González Mangado N, Montserrat JM, Alonso-Álvarez ML, Almendros I, Alonso-Fernández A, et al. Documento internacional de consenso sobre apnea obstructiva del sueno. Arch Bronconeumol. 2022, 58, 52–68. [CrossRef] [PubMed]
- Chaudhary B, Dasti S, Park Y, Brown T, Davis H, Akhtar B. Hour-to-hour variability of oxygen saturation in sleep apnea. Chest 1998, 113, 719–722. [CrossRef] [PubMed]
- Gonzalez C, Almaraz L, Obeso A, Rigual R. Carotid body chemoreceptors: From natural stimuli to sensory discharges. Physiol Rev. 1994, 74, 829–898. [CrossRef] [PubMed]
- Song, R.; Baker, T.L.; Watters, J.J.; Kumar, S. Obstructive Sleep Apnea-Associated Intermittent Hypoxia-Induced Immune Responses in Males, Pregnancies, and Offspring. Int. J. Mol. Sci. 2024, 25, 1852. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar NR, Peng YJ, Nanduri J. Carotid body hypersensitivity in intermittent hypoxia and obtructive sleep apnoea. J Physiol. 2023, 601, 5481–5494. [CrossRef] [PubMed]
- Young, T. Rationale, design and findings from the Wisconsin Sleep Cohort Study: Toward understanding the total societal burden of sleep disordered breathing. Sleep Med Clin. 2009, 4, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Pien, G.W.; Pack, A.I.; Jackson, N.; Maislin, G.; Macones, G.A.; Schwab, R.J. Risk factors for sleep-disordered breathing in pregnancy. Thorax 2014, 69, 371–377. [Google Scholar] [CrossRef]
- Zhu B, Bronas UG, Carley DW, Lee K, Steffen A, Kapella MC, Izci-Balserak B. Relationships between objective sleep parameters and inflammatory biomarkers in pregnancy. Ann N Y Acad Sci. 2020, 1473, 62–73. [CrossRef]
- Louis JM, Koch MA, Reddy UM, Silver RM, Parker CB, Facco FL, Redline S, Nhan-Chang CL, Chung JH, Pien GW, Basner RC, Grobman WA, Wing DA, Simhan HN, Haas DM, Mercer BM, Parry S, Mobley D, Carper B, Saade GR, Schubert FP, Zee PC. Predictors of sleep-disordered breathing in pregnancy. Am J Obstet Gynecol. 2018, 218, e1–e521. [CrossRef]
- Eleftheriou D, Athanasiadou KI, Sifnaios E, Vagiakis E, Katsaounou P, Psaltopoulou T, Paschou SA, Trakada G. Sleep disorders during pregnancy: An underestimated risk factor for gestational diabetes mellitus. Endocrine 2024, 83, 41–50. [CrossRef] [PubMed]
- Louis JM, Mogos MF, Salemi JL, Redline S, Salihu HM. Obstructive sleep apnea and severe maternal-infant morbidity/mortality in the United States, 1998-2009. Sleep 2014, 37, 843–849. [CrossRef] [PubMed]
- Khalyfa A, Cortese R, Qiao Z, Ye H, Bao R, Andrade J, Gozal, D. Late gestational intermittent hypoxia induces metabolic and epigenetic changes in male adult offspring mice. J Physiol 2017, 595, 2551–2568. [CrossRef] [PubMed]
- Dahlgren J, Samuelsson AM, Jansson T, Holmäng A. Interleukin-6 in the maternal circulation reaches the rat fetus in mid-gestation. Pediatr Res. 2006, 60, 47–51. [CrossRef] [PubMed]
- Contreras G, Gutiérrez M, Beroíza T, Fantín A, Oddó H, Villarroel L, Cruz E, Lisboa C. Ventilatory drive and respiratory muscle function in pregnancy. Am Rev Respir Dis. 1991, 144, 837–841. [CrossRef]
- Lyons HA, Antonio R. The sensitivity of the respiratory center in pregnancy and after administration of progesterone. Trans Assoc Am Physicians 1959, 72, 173–180.
- Weinberger SE, Weiss ST, Cohen WR, Johnson, TS. Pregnancy and the lung. Am Rev Respir Dis 1980, 121, 559–581. [CrossRef] [PubMed]
- McAuliffe F, Kametas N, Costello J, Rafferty GF, Greenough A, Nicolaides K. Respiratory function in singleton and twin pregnancy. BJOG 2002, 109, 765–769. [CrossRef]
- Izci B, Vennelle M, Liston WA, Dundas KC, Calder AA, Douglas NJ. Sleep-disordered breathing and upper airway size in pregnancy and post-partum. Eur Respir J. 2006, 27, 321–327. [CrossRef]
- Izci B, Riha RL, Martin SE, Vennelle M, Liston WA, Dundas KC, Calder AA, Douglas NJ. The upper airway in pregnancy and pre-eclampsia. Am J Respir Crit Care Med. 2003, 167, 137–140. [CrossRef] [PubMed]
- Bende M, Gredmark T. Nasal stuffiness during pregnancy. Laryngoscope 1999, 109, 1108–1110. [CrossRef] [PubMed]
- Williams PE, Goldspink G. Changes in sarcomere length and physiological properties in immobilized muscle. J Anat. 1978, 127 Pt 3, 459–468.
- Froeliger A, Deneux-Tharaux C, Madar H, Bouchghoul H, Le Ray C, Sentilhes L. TRAAP study group. Closed- or open-glottis pushing for vaginal delivery: A planned secondary analysis of the TRAnexamic Acid for Preventing postpartum hemorrhage after vaginal delivery study. Am J Obstet Gynecol. 2024, 230, S879–S889.e4. [CrossRef] [PubMed]
- Lui B, Burey L, Ma X, Kjaer K, Abramovitz SE, White RS. Obstructive sleep apnea is associated with adverse maternal outcomes using a United States multistate database cohort, 2007-2014. Int J Obstet Anesth. 2021, 45, 74–82. [CrossRef] [PubMed]
- Maltepe E, Fisher SJ. Placenta: The forgotten organ. Annu Rev Cell Dev Biol. 2015, 31, 523–552. [CrossRef]
- Zamudio, S. The placenta at high altitude. High Alt. Med. Biol. 2003, 4, 171–191. [Google Scholar] [CrossRef]
- Hutter, D. , Kingdom, J., Jaeggi, E. Causes and mechanisms of intrauterine hypoxia and its impact on the fetal cardiovascular system: A review. Int. J. Pediatr. 2010, 2010, 401323. [Google Scholar] [CrossRef]
- Sferruzzi-Perri AN, Camm EJ. The Programming Power of the Placenta. Front Physiol. 2016, 7, 33. [CrossRef]
- Vaughan, O.R. , Sferruzzi-Perri, A. N., Coan, P.M., Fowden, A. L. Environmental regulation of placental phenotype: Implications for fetal growth. Reprod. Fertil. Dev. 2012, 24, 80–96. [Google Scholar] [CrossRef]
- Kalisch-Smith JI, Simmons DG, Dickinson H, Moritz KM. Review: Sexual dimorphism in the formation, function and adaptation of the placenta. Placenta 2017, 54, 10–16. [CrossRef]
- Hung TH, Burton GJ. Hypoxia and reoxygenation: A possible mechanism for placental oxidative stress in preeclampsia. Taiwan J Obstet Gynecol. 2006, 45, 189–200. [CrossRef] [PubMed]
- Caruso M, Evangelista M, Parolini O. Human term placental cells: Phenotype, properties and new avenues in regenerative medicine. Int J Mol Cell Med. 2012, 1, 64–74.
- Goplerud JM, Delivoria-Papadopoulos M. Physiology of the placenta—Gas exchange. Ann Clin Lab Sci 1985, 15, 270–278.
- Browne VA, Julian CG, Toledo-Jaldin L, Cioffi-Ragan D, Vargas E, Moore LG. Uterine artery blood flow, fetal hypoxia and fetal growth. Philos Trans R Soc Lond B Biol Sci 2015, 370, 20140068. [CrossRef] [PubMed]
- Ravishankar S, Bourjeily G, Lambert-Messerlian G, He M, De Paepe ME, Gündoğan F. Evidence of Placental Hypoxia in Maternal Sleep Disordered Breathing. Pediatr Dev Pathol. 2015, 18, 380–386. [CrossRef] [PubMed]
- Al-Gubory KH, Fowler PA, Garrel C. The roles of cellular reactive oxygen species, oxidative stress and antioxidants in pregnancy outcomes. Int J Biochem Cell Biol. 2010, 42, 1634–1650. [CrossRef]
- Hung TH, Skepper JN, Burton GJ. In vitro ischemia-reperfusion injury in term human placenta as a model for oxidative stress in pathological pregnancies. Am J Pathol. 2001, 159, 1031–1043. [CrossRef] [PubMed]
- Lavie, L. Obstructive sleep apnoea syndrome--an oxidative stress disorder. Sleep Med Rev. 2003, 7, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Cuffe JS, Walton SL, Singh RR, Spiers JG, Bielefeldt-Ohmann H, Wilkinson L, Little MH, Moritz KM. Mid- to late term hypoxia in the mouse alters placental morphology, glucocorticoid regulatory pathways and nutrient transporters in a sex-specific manner. J Physiol. 2014, 592, 3127–3141. [CrossRef]
- Higgins JS, Vaughan OR, Fernandez de Liger E, Fowden AL, Sferruzzi-Perri AN. Placental phenotype and resource allocation to fetal growth are modified by the timing and degree of hypoxia during mouse pregnancy. J Physiol. 2016, 594, 1341–1356. [CrossRef]
- Rosario GX, Konno T, Soares MJ. Maternal hypoxia activates endovascular trophoblast cell invasion. Dev Biol. 2008, 314, 362–375. [CrossRef] [PubMed]
- Valverde-Pérez E, Prieto-Lloret J, Gonzalez-Obeso E, Cabero MI, Nieto ML, Pablos MI, Obeso A, Gomez-Niño A, Cárdaba-García RM, Rocher A, Olea E. Effects of Gestational Intermittent Hypoxia on Placental Morphology and Fetal Development in a Murine Model of Sleep Apnea. Adv Exp Med Biol. 2023, 1427, 73–81. [CrossRef] [PubMed]
- Wakefield SL, Lane M, Mitchell M. Impaired mitochondrial function in the preimplantation embryo perturbs fetal and placental development in the mouse. Biol Reprod. 2011, 84, 572–580. [CrossRef]
- Adelman DM, Gertsenstein M, Nagy A, Simon MC, Maltepe E. Placental cell fates are regulated in vivo by HIF-mediated hypoxia responses. Genes Dev. 2000, 14, 3191–3203. [CrossRef] [PubMed]
- Song W, Chang WL, Shan D, Gu Y, Gao L, Liang S, Guo H, Yu J, Liu X. Intermittent Hypoxia Impairs Trophoblast Cell Viability by Triggering the Endoplasmic Reticulum Stress Pathway. Reprod Sci. 2020, 27, 477–487. [CrossRef] [PubMed]
- Giannubilo SR, Cecati M, Marzioni D, Ciavattini A. Circulating miRNAs and Preeclampsia: From Implantation to Epigenetics. Int J Mol Sci. 2024, 25, 1418. [CrossRef] [PubMed]
- Myatt L, Webster RP. Vascular biology of preeclampsia. J Thromb Haemost. 2009, 7, 375–384. [CrossRef] [PubMed]
- Morton JS, Care AS, Davidge ST. Mechanisms of Uterine Artery Dysfunction in Pregnancy Complications. J Cardiovasc Pharmacol. 2017, 69, 343–359. [CrossRef] [PubMed]
- Lang U, Baker RS, Braems G, Zygmunt M, Künzel W, Clark KE. Uterine blood flow--a determinant of fetal growth. Eur J Obstet Gynecol Reprod Biol. 2003, 110 Suppl. 1, S55–S61. [CrossRef]
- Reynolds LP, Redmer DA. Utero-placental vascular development and placental function. J Anim Sci 1995, 73, 1839–1851. [CrossRef]
- Genbacev O, Zhou Y, Ludlow JW, Fisher SJ. Regulation of human placental development by oxygen tension. Science 1997, 277, 1669–1672. [CrossRef]
- Jauniaux E, Poston L, Burton GJ. Placental-related diseases of pregnancy: Involvement of oxidative stress and implications in human evolution. Hum Reprod Update 2006, 12, 747–755. [CrossRef] [PubMed]
- Zygmunt M, Herr F, Münstedt K, Lang U, Liang OD. Angiogenesis and vasculogenesis in pregnancy. Eur J Obstet Gynecol Reprod Biol. 2003, 110, S10–S18. [CrossRef] [PubMed]
- Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003, 9, 669–676. [CrossRef] [PubMed]
- Ramakrishnan S, Anand V, Roy S. Vascular endothelial growth factor signaling in hypoxia and inflammation. J Neuroimmune Pharmacol. 2014, 9, 142–160. [CrossRef] [PubMed]
- Dailey L, Ambrosetti D, Mansukhani A, Basilico C. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev 2005, 16, 233–247. [CrossRef] [PubMed]
- Wang K, Jiang YZ, Chen DB, Zheng J. Hypoxia enhances FGF2- and VEGF-stimulated human placental artery endothelial cell proliferation: Roles of MEK1/2/ERK1/2 and PI3K/AKT1 pathways. Placenta 2009, 30, 1045–1051. [CrossRef] [PubMed]
- Almendros I, Martínez-Ros P, Farré N, Rubio-Zaragoza M, Torres M, Gutiérrez-Bautista ÁJ, Carrillo-Poveda JM, Sopena-Juncosa JJ, Gozal D, Gonzalez-Bulnes A, Farré R. Placental oxygen transfer reduces hypoxia-reoxygenation swings in fetal blood in a sheep model of gestational sleep apnea. J Appl Physiol 2019, 127, 745–752. [CrossRef] [PubMed]
- Almendros I, Farré R, Planas AM, Torres M, Bonsignore MR, Navajas D, Montserrat JM. Tissue oxygenation in brain, muscle, and fat in a rat model of sleep apnea: Differential effect of obstructive apneas and intermittent hypoxia. Sleep 2011, 34, 1127–1133. [CrossRef]
- Cahill LS, Zhou YQ, Seed M, Macgowan CK, Sled JG. Brain sparing in fetal mice: BOLD MRI and Doppler ultrasound show blood redistribution during hypoxia. J Cereb Blood Flow Metab 2014, 34, 1082–1088. [CrossRef]
- Carter, A.M. Placental gas exchange and the oxygen supply to the fetus. Compr Physiol 2015, 5, 1381–1403. [Google Scholar] [CrossRef]
- Giussani, D.A. The fetal brain sparing response to hypoxia: Physiological mechanisms. J Physiol 2016, 594, 1215–1230. [Google Scholar] [CrossRef]
- Iqbal W, Ciriello J. Effect of maternal chronic intermittent hypoxia during gestation on offspring growth in the rat. Am J Obstet Gynecol, 2013, 209, 564.e1–564.e9. [CrossRef] [PubMed]
- Cortese R, Khalyfa A, Bao R, Andrade J, Gozal D. Epigenomic profiling in visceral white adipose tissue of offspring of mice exposed to late gestational sleep fragmentation. Int J Obes 2015, 39, 1135–1142. [CrossRef] [PubMed]
- Cortese R, Gileles-Hillel A, Khalyfa A, Almendros I, Akbarpour M, Khalyfa AA, Qiao Z, Garcia T, Andrade J, Gozal D. Aorta macrophage inflammatory and epigenetic changes in a murine model of obstructive sleep apnea: Potential role of CD36. Sci Rep 2017, 7, 43648. [CrossRef] [PubMed]
- Koos, B.J. Breathing and sleep states in the fetus and at birth. In: Marcus CL, Carroll JL, Donnelly DF, Loughlin GM, editors. Sleep and breathing in children: Developmental changes in breathing during sleep. 2nd ed. New York, NY: Informa Healthcare; 2008. p. 1–17.
- Koos BJ, Rajaee A. Fetal breathing movements and changes at birth. Adv Exp Med Biol. 2014, 814, 89–101. [CrossRef] [PubMed]
- Patrick J, Campbell K, Carmichael L, Natale R, Richardson B. A definition of human fetal apnea and the distribution of fetal apneic intervals during the last ten weeks of pregnancy. Am J Obstet Gynecol. 1980, 136, 471–477. [CrossRef] [PubMed]
- Arduini D, Rizzo G, Giorlandino C, Valensise H, Dell'Acqua S, Romanini C. The development of fetal behavioural states: A longitudinal study. Prenat Diagn. 1986, 6, 117–124. [CrossRef]
- Patrick J, Campbell K, Carmichael L, Natale R, Richardson B. Patterns of human fetal breathing during the last 10 weeks of pregnancy. Obstet Gynecol. 1980, 56, 24–30.
- Koos BJ, Matsuda K, Power GG. Fetal breathing and cardiovascular responses to graded methemoglobinemia in sheep. J Appl Physiol 1990, 69, 136–140. [CrossRef] [PubMed]
- Platt LD, Manning FA, Lemay M, Sipos L. Human fetal breathing: Relationship to fetal condition. Am J Obstet Gynecol. 1978, 132, 514–518. [CrossRef] [PubMed]
- Tauman R, Many A, Deutsch V, Arvas S, Ascher-Landsberg J, Greenfeld M, Sivan Y. Maternal snoring during pregnancy is associated with enhanced fetal erythropoiesis--a preliminary study. Sleep Med. 2011, 12, 518–522. [CrossRef]
- Templeton A, Kelman GR. Maternal blood-gases, (PAo2-Pao2), physiological shunt and VD/VT in normal pregnancy. Br J Anaesth 1976, 48, 1001–1004. [CrossRef]
- Dawes, GS. Foetal and neonatal physiology; a comparative study of the changes at birth. Year Book Medical Publishers, 1968. [Google Scholar]
- Longo, L.D. The Rise of Fetal and Neonatal Physiology: Basic Science to Clinical Care, Perspectives in Physiology. Am Physiol Society 2013. [CrossRef]
- Herrington RT, Harned HS Jr, Ferreiro JI, Griffin CA 3rd. The role of the central nervous system in perinatal respiration: Studies of chemoregulatory mechanisms in the term lamb. Pediatrics 1971, 47, 857–864. [CrossRef]
- Chou PJ, Ullrich JR, Ackerman BD. Time of onset of effective ventilation at birth. Biol Neonate. 1974, 24, 74–81. [CrossRef]
- Cohen G, Katz-Salamon M. Development of chemoreceptor responses in infants. Respir Physiol Neurobiol. 2005, 149, 233–42. [CrossRef]
- Rigatto H, De La Torre Verduzco R, Gates DB. Effects of O2 on the ventilatory response to CO2 in preterm infants. J Appl Physiol. 1975, 39, 896–899. [CrossRef]
- Kumar P, Hanson MA. Re-setting of the hypoxic sensitivity of aortic chemoreceptors in the new-born lamb. J Dev Physiol. 1989, 11, 199–206.
- Richardson HL, Parslow PM, Walker AM, Harding R, Horne RS. Maturation of the initial ventilatory response to hypoxia in sleeping infants. J Sleep Res. 2007, 16, 117–127. [CrossRef]
- Liu Q, Fehring C, Lowry TF, Wong-Riley MT. Postnatal development of metabolic rate during normoxia and acute hypoxia in rats: Implication for a sensitive period. J Appl Physiol 2009, 106, 1212–1222. [CrossRef]
- Trachsel D, Erb TO, Hammer J, von Ungern-Sternberg BS. Developmental respiratory physiology. Paediatr Anaesth. 2022, 32, 108–117. [CrossRef] [PubMed]
- Wani TM, Rafiq M, Akhter N, AlGhamdi FS, Tobias JD. Upper airway in infants-a computed tomography-based analysis. Paediatr Anaesth. 2017, 27, 501–505. [CrossRef] [PubMed]
- Sanchez I, Vega-Briceño L, Muñoz C, Mobarec S, Brockman P, Mesa T, Harris P. Polysomnographic findings in 320 infants evaluated for apneic events. Pediatr Pulmonol. 2006, 41, 215–221. [CrossRef]
- Praud JP, Reix P. Upper airways and neonatal respiration. Respir Physiol Neurobiol. 2005, 149, 131–141. [CrossRef]
- Ramanathan R, Corwin MJ, Hunt CE, Lister G, Tinsley LR, Baird T, Silvestri JM, Crowell DH, Hufford D, Martin RJ, Neuman MR, Weese-Mayer DE, Cupples LA, Peucker M, Willinger M, Keens TG. Collaborative Home Infant Monitoring Evaluation (CHIME) Study Group. Cardiorespiratory events recorded on home monitors: Comparison of healthy infants with those at increased risk for SIDS. JAMA 2001, 285, 2199–2207. [CrossRef] [PubMed]
- Daftary AS, Jalou HE, Shively L, Slaven JE, Davis SD. Polysomnography Reference Values in Healthy Newborns. J Clin Sleep Med. 2019, 15, 437–443. [CrossRef]
- Hershenson MB, Colin AA, Wohl ME, Stark AR. Changes in the contribution of the rib cage to tidal breathing during infancy. Am Rev Respir Dis. 1990, 141, 922–925. [CrossRef]
- Rosen, C.L. Maturation of breathing during sleep. In: Marcus CL, Carroll JL, Donnelly DF, Loughlin GM, editors. Sleep and breathing in children: Developmental changes in breathing during sleep. 2nd ed. New York, NY: Informa Healthcare; 2008. p. 117–30.
- Johnson SM, Randhawa KS, Epstein JJ, Gustafson E, Hocker AD, Huxtable AG, Baker TL, Watters JJ. Gestational intermittent hypoxia increases susceptibility to neuroinflammation and alters respiratory motor control in neonatal rats. Respir Physiol Neurobiol. 2018, 256, 128–142. [CrossRef]
- Gozal D, Reeves SR, Row BW, Neville JJ, Guo SZ, Lipton AJ. Respiratory effects of gestational intermittent hypoxia in the developing rat. Am J Respir Crit Care Med. 2003, 167, 1540–1547. [CrossRef] [PubMed]
- Soy M, Keser G, Atagündüz P, Tabak F, Atagündüz I, Kayhan S. Cytokine storm in COVID-19: Pathogenesis and overview of anti-inflammatory agents used in treatment. Clin Rheumatol. 2020, 39, 2085–2094. [CrossRef] [PubMed]
- Stoll BJ, Hansen NI, Adams-Chapman I, Fanaroff AA, Hintz SR, Vohr B, Higgins RD. National Institute of Child Health and Human Development Neonatal Research Network. Neurodevelopmental and growth impairment among extremely low-birth-weight infants with neonatal infection. JAMA 2004, 292, 2357–2365. [CrossRef]
- Di Fiore JM, Martin RJ, Gauda EB. Apnea of prematurity--perfect storm. Respir Physiol Neurobiol. 2013, 189, 213–222. [CrossRef]
- Raffay TM, Di Fiore JM, Chen Z, Sánchez-Illana Á, Vento M, Piñeiro-Ramos JD, Kuligowski J, Martin RJ, Tatsuoka C, Minich NM, MacFarlane PM, Hibbs AM. Hypoxemia events in preterm neonates are associated with urine oxidative biomarkers. Pediatr Res. 2023, 94, 1444–1450. [CrossRef] [PubMed]
- Alonso-Fernández A, Ribot Quetglas C, Herranz Mochales A, Álvarez Ruiz De Larrinaga A, Sánchez Barón A, Rodríguez Rodríguez P, Gil Gómez AV, Pía Martínez C, Cubero Marín JP, Barceló Nicolau M, Cerdà Moncadas M, Codina Marcet M, De La Peña Bravo M, Barceló Bennasar A, Iglesias Coma A, Morell-Garcia D, Peña Zarza JA, Giménez Carrero MP, Durán Cantolla J, Marín Trigo JM, Piñas Cebrian MC, Soriano JB, García-Río F. Influence of Obstructive Sleep Apnea on Systemic Inflammation in Pregnancy. Front Med 2021, 8, 674997. [CrossRef] [PubMed]
- Ślusarczyk J, Trojan E, Głombik K, Budziszewska B, Kubera M, Lasoń W, Popiołek-Barczyk K, Mika J, Wędzony K, Basta-Kaim A. Prenatal stress is a vulnerability factor for altered morphology and biological activity of microglia cells. Front Cell Neurosci. 2015, 9, 82. [CrossRef] [PubMed]
- Zhao M, Lei J, Deng F, Zhao C, Xu T, Ji B, Fu M, Wang X, Sun M, Zhang M, Gao Q. Gestational Hypoxia Impaired Endothelial Nitric Oxide Synthesis Via miR-155-5p/NADPH Oxidase/Reactive Oxygen Species Axis in Male Offspring Vessels. J Am Heart Assoc. 2024, 13, e032079. [CrossRef] [PubMed]
- Morrison NR, Johnson SM, Hocker AD, Kimyon RS, Watters JJ, Huxtable AG. Time and dose-dependent impairment of neonatal respiratory motor activity after systemic inflammation. Respir Physiol Neurobiol. 2020, 272, 103314. [CrossRef] [PubMed]
- Master ZR, Porzionato A, Kesavan K, Mason A, Chavez-Valdez R, Shirahata M, Gauda EB. Lipopolysaccharide exposure during the early postnatal period adversely affects the structure and function of the developing rat carotid body. J Appl Physiol 2016, 121, 816–827. [CrossRef] [PubMed]
- Beyeler SA, Hodges MR, Huxtable AG. Impact of inflammation on developing respiratory control networks: Rhythm generation, chemoreception and plasticity. Respir Physiol Neurobiol. 2020, 274, 103357. [CrossRef] [PubMed]
- McDonald FB, Dempsey EM, O'Halloran KD. Effects of Gestational and Postnatal Exposure to Chronic Intermittent Hypoxia on Diaphragm Muscle Contractile Function in the Rat. Front Physiol. 2016, 7, 276. [CrossRef]
- McDonald FB, Dempsey EM, O'Halloran KD. Early Life Exposure to Chronic Intermittent Hypoxia Primes Increased Susceptibility to Hypoxia-Induced Weakness in Rat Sternohyoid Muscle during Adulthood. Front Physiol. 2016, 7, 69. [CrossRef]
Figure 1.
Effects of gestational intermittent hypoxia on maternal, fetal and offspring respiratory system. In black, effects in humans; in red, in vitro effects; in blue, effects in animal models.
Figure 1.
Effects of gestational intermittent hypoxia on maternal, fetal and offspring respiratory system. In black, effects in humans; in red, in vitro effects; in blue, effects in animal models.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



