Submitted:
25 April 2024
Posted:
26 April 2024
You are already at the latest version
Abstract
Fragile X syndrome (FXS) is a genetic disorder caused by a mutation in the fragile X messenger ribonucleoprotein 1 (FMR1) gene and known to be a leading cause of inherited intellectual disability globally. It results in a range of intellectual, developmental, and behavioral problems. Fragile X premutation associated conditions (FXPAC), also caused by a smaller CGG expansion (55 to 200 CGG repeats) in the FMR1 gene, is linked to other conditions that increase morbidity and mortality for affected persons. Limited research has been conducted on the burden, characteristics, diagnosis, and management of these conditions in Africa. This comprehensive review provides an overview of the current literature on FXS and FXPAC in Africa, epidemiology, clinical features and challenges faced, such as discrimination against affected persons, limited awareness and research, poor access to resources and genetic services and treatment. It further provides an in-depth analysis of the existing worldwide data for the diagnosis and treatment of these disorders. This review will improve understanding of FXS and FXPAC in Africa by incorporating existing knowledge, identifying research gaps, and potential topics for future research to enhance the well-being of individuals and families affected by FXS and FXPAC in the region.
Keywords:
Fragile X syndrome
; Fragile X premutation associated conditions
; Africa
1. Introduction
Fragile X syndrome (FXS) first reported by Martin and Bell [1] in 1943, is an X-linked dominant genetic disorder known to be the most common cause of hereditary intellectual and developmental disability globally [2]. It is caused by a mutation that causes an abnormal expansion of a polymorphic CGG repeat in the 5’ region of the fragile X messenger ribonucleoprotein 1 (FMR1) gene leading to the silencing of the gene and a deficiency in the production of the FMRP protein which regulates numerous processes in the human body [2,3,4,5,6]. This mutation leads to a varied form of disorders depending on the CGG repeat numbers. FXS is caused by the full mutation (> 200 CGG repeats) in the 5’ region of the gene, which leads to a lack of production of the FMR1 protein (FMRP) and is typically characterized by cognitive deficits, behavioral and emotional problems including autism, and physical features which vary among different patients but may include a long face, large ears, and a prominent jaw [7,8,9]. Normal alleles have less than 45 CGG repeats, the intermediate zone also known as the ‘gray zone’ is between 45 and 54 CGG repeats and may expand in future generations, while the premutation disorders, which are caused by a small expansion at the 5’ region of FMR1 gene anywhere from 55 to 200 CGG repeats [7,10]. Usually individuals with the premutation have normal intellectual abilities although shyness, social anxiety and sometimes depression are common. A carrier male with the premutation will pass on the premutation to all his daughters who receive his only X chromosome, but all his sons will not have the mutation because they receive his Y chromosome. A carrier female with the premutation, however, will pass on the gene mutation to 50% of her children and expansion to a full mutation occurs with increasing frequency depending on the size of the premutation. Females with over 90 to 100 CGG repeats will expand to a full mutation every time that X is passed on.7
The molecular basis of FXS is the expansion of the CGG repeat sequence at the 5’ UTR of a highly conserved FMR1 gene, with 17 exons and a length of approximately 38 kb, at Xq27.3 [7,11]. This expansion leads to hypermethylation of repeat sequences and adjacent promoter regions, which decreases gene transcription so little, or no mRNA is produced, and this is called a full mutation (FM) causing FXS [12]. FXS females demonstrate widely variable symptoms and are not as severely affected as FXS males because they have two X chromosomes, and the normal one is producing FMRP. There is an inverse relationship between disease severity in FXS females and the activation ratio (AR) means the percentage of cells that have the normal X as the active X). The higher the AR the more FMRP is produced and the higher the IQ.7 Some individuals with a FM may have some cells unmethylated or some cells with the premutation and this is termed mosaicism, either from size or methylation mosaicism. If there is a lack of complete methylation or several cells with the premutation, then more FMRP is produced, and cognitive abilities are higher especially in the boys with mosaicism [13].
Fragile X premutation associated conditions (FXPAC) also known as the premutation disorders include the Fragile X- associated Tremor/Ataxia syndrome (FXTAS) which is a neurodegenerative disorder seen in older carriers, Fragile X-associated Primary Ovarian Insufficiency (FXPOI) meaning menopause before age 40, and Fragile X-associated neuropsychiatric disorders (FXAND) which includes anxiety, depression, insomnia, and other neuropsychiatric disorders [14].
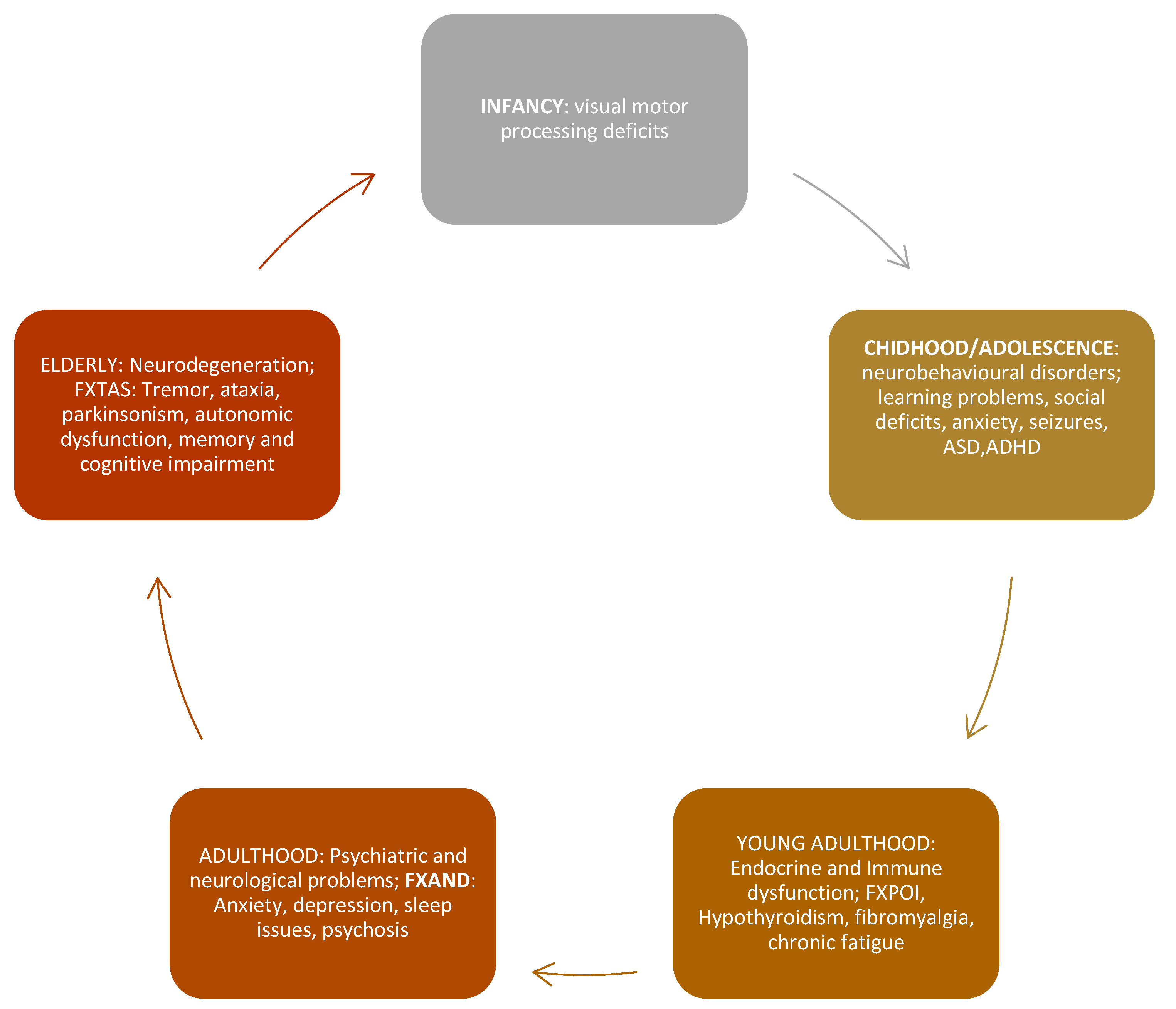
Each disorder in the premutation range is distinct and has its associated clinical characteristics (Figure 1).
FXS and FXPAC have not been adequately studied in Africa, and there is sparse data on the burden, diagnosis, and management of these conditions in the continent.
In this comprehensive review, we aim to explore the prevalence of FXS and FXPAC in Africa, highlighting the challenges and gaps in research, as well as potential strategies for improving diagnosis and management of these conditions in the region.
2. Epidemiology of FXS in Africa
The burden of FXS and FXPAC in Africa has received relatively little attention in scientific literature. Research on FXS in Africa is sparse, due to limited awareness, diagnostic services, and personnel, with most studies focusing on other continents such as North America and Europe [15,16,17]. However, there is some data that suggests that the prevalence of FXS in Africa may be underreported due to a lack of awareness and diagnostic services. In addition, the genetic diversity of African populations may affect the prevalence and presentation of FXS in this region[18,19].
It is estimated that FXS affects a similar proportion of the population in Africa as in other parts of the world, with a prevalence of approximately 1 in 5,000 males and 1 in 4,000 -8,000 females [20]. A large, 20-year retrospective population review conducted by Essop et al [21] among over 2000 individuals with intellectual disability in urban South Africa found a 5.2% prevalence rate of FXS among African participants. Another population study on intellectually disabled institutionalized adolescent and adult males in rural South Africa reported a slightly higher prevalence rate of 6.1% for FXS among participants studied [22]. Although both studies were conducted in the same country, the population studies were different. The former was conducted on those with intellectual disability in the general population, while the latter was among institutionalized males. This could have accounted for the mild disparity in rates.
In Egypt, North Africa, Meguid et al [23] found a rate of 6.4% (16/250) among intellectually disabled in-school male children and adolescents with FXS. This was almost like a study by Goldman and Jenkins [22] among intellectually disabled institutionalized males in South Africa.
In contrast, Kamga et al [24] in Yaounde, Cameroon, central Africa after cascade testing, reported a 4 in 19 (21%) prevalence rate among boys with intellectual disability in a large extended family. This high prevalence rate could be explained by the founder effect (participants tested were descendants of the founder).
Latunji [25] in Ibadan, South-west, Nigeria reported a 6.83% (4/69) prevalence rate of FXS among intellectually disabled children and adults who were enrolled in special schools or living in homes. This rate was like the findings by Meguid et al [23] in Egypt despite the variations in the population studied, sample size and diagnostic method. Latunji’s [25] study used both cytogenetic and molecular methods in diagnosis, whereas Meguid et al [23] used a molecular method (PCR). However, these studies are limited in scope and may not accurately reflect the true prevalence of FXS in Africa.
2.1. Epidemiology of FXPAC in Africa
FXPAC includes a group of disorders caused by the premutation which makes a higher level of mRNA than normal causing RNA toxicity and leading to clinical involvement, so this is very different from those with a FM and FXS [3,7]. These disorders are in the premutation range, implying that affected persons are unmethylated, CGG repeats in the range of 55 to 200 and the level of FMRP is usually in the normal range [14,20,26]. They were previously thought to be carriers who were largely unaffected but progressive research over the years has revealed that in fact premutation carriers may have a plethora of symptoms and signs that largely cause morbidity and mortality if left unattended [14]. The premutation (PM) carriers with greater than 120 CGG repeats have a mild reduction of FMRP levels which can lead to mildly prominent ears and hyperextensibility of finger joints similar to those with FXS [7,14,27]. The pathogenesis of PM disorders in male and female carriers can be explained by markedly elevated FMR1 mRNA, directly related to the length of the repeats, leading to activation of cellular stress pathways, sequestration of proteins important for neuronal function, mitochondrial dysfunction and other RNA-mediated toxicity [14,28,29,30].
Alleles in the ‘gray zone’ also known as intermediate alleles with 45 to 54 CGG repeats, may occasionally expand to the premutation range or even the full mutation after the second generation [31].
2.2. Studies on Fragile X Premutation Associated Conditions in Africa
While there is limited research on FXS in Africa, even less is known about FXPAC, such as FXTAS, FXPOI and the most recently named Fragile X- associated neuropsychiatric disorders (FXAND). These conditions do not cause intellectual disability but can still have significant impact on affected individuals.
In a large study of unrelated participants in Johannesburg, South Africa, the prevalence of the premutation was noted to be 0.54% (12/2239) among participants [21]. This prevalence rate is slightly higher than rate of about 0.4% documented in the systematic review by Hunter et al [32], which did not include African studies and other global estimates of PM; 0.1-0.3% in males and 0.3-0.4% in females [33,34]. Another study among 148 unrelated, institutionalized South African males with intellectual disability found the prevalence of premutation to be zero (0) among participants.22 This is not an unexpected finding considering the fact that participants in this study all had intellectual disability which is common in the FM but not in those with the PM [3,35,36,37,38]. A study in Egypt reported a prevalence of FXPOI of 40% in carriers, which is also higher than global estimates [17]. Although there is limited data on the prevalence of these conditions in Africa, these studies suggest that FXPAC may indeed be more common in certain African populations than previously thought, highlighting the need for further research.
2.3. FXPOI
Premature ovarian insufficiency is a diverse condition typified by a premature decline in the function of the ovaries before 40 years old [39]. It affects approximately 3.5% of women globally [40]. It is also known as hypergonadotropic hypogonadism and characteristic features include low estrogen levels, elevated gonadotropins (FSH > 30 U/I) and either primary or secondary amenorrhea of 4 to 6 months [41].
POI remains idiopathic as the cause is still unknown, however some factors have been implicated as possible mechanisms including genetic, metabolic, environmental toxins, autoimmune disorders, pelvic surgery, cancer treatment with chemotherapy and or, radiotherapy [39]. Genetics has been implicated to play a major role in the pathogenesis of POI [42,43]. POI has been associated mainly with the X chromosome and autosomal mutations. Turner syndrome (45, X), with a complete loss of one X chromosome, caused by haploinsufficiency of inactivated X-linked genes is known to be a predominant genetic cause of syndromic POF [44,45]. A notable report in the history of FXPAC, is the report of premature menopause (FXPOI) by conference attendees who were women in their early thirties and mothers of boys with FXS. This event happened in 1986 before the discovery of the FMR1 gene [14]. Now the FMR1 premutation is recognized to be the most common genetic etiologic factor for POI [46], with FXPOI occurring in about 1 in every 5 women with the premutation [7,14,26,47]. The prevalence of the PM in women is estimated at 1 in 150, with over 1 million women in the United States known to have the premutation allele [48]. These data on how prevalent FXPOI is, remain to be seen particularly in settings that have diagnostic challenges [49]. Reports have shown that delays in the diagnosis of FXPOI occur in women who knew their premutation status and presented with symptoms of POI. This delay was even more pronounced in women who did not [50]. These women usually present with either primary or secondary infertility, hot flashes, osteoporosis, and other associated features of low estrogen levels [7,47].
Another study demonstrated increased prevalence of premature menopause among premutation carrier women who had either an average or normal IQ, which is expected in those with the PM [51]. Women who carry the premutation alleles have increased risk of developing FXPOI [7,14,47,52]. There is an increased risk for women with these highly unstable, usually unmethylated premutation alleles to give birth to offspring in the FM if they conceive. There is a bell-shaped relationship between FXPOI in PM carriers and the CGG repeat number, with the greatest incidence of FXPOI found among those with CGG repeats between 85 and 100 [46,53,54]. Premutation carriers occur in 7 out of every 100 of sporadic cases of POI while the prevalence increases with a familial history and about 21 in 100 women have the premutation with familial POI [39]. In contrast, a lower prevalence rate of approximately 1% has been reported for POI in the general population [54,55]. However, there is still a slim chance of fertility in women with FXPOI, thus, the name was changed from premature ovarian failure (POF) to POI a few years ago [44].
Both premutation carriers and healthcare personnel have demonstrated a great need to identify risk and predictive markers for FXPOI, particularly because of the increased prevalence of women who are premutation carriers from several screening studies [56,57,58,59,60,61,62,63].
There is limited research on the prevalence of FXPOI in Africa. Essop and Krause [21] reported an estimated 33% prevalence of POI (1 in 3 females tested who had a PM) in POI referrals in Johannesburg, South Africa in their 20-year review. This is significantly higher than the rates reported by previous authors, suggesting that FXPOI and other FXPAC may indeed be more prevalent than presumed [14].
The treatment of FXPOI includes hormonal replacement and psychotherapy if needed and this is discussed in detail elsewhere [64].
2.4. FXTAS
FXTAS as its name implies is a neurodegenerative syndrome associated with the premutation and characterized by tremor and ataxia; specific clinical and radiological criteria make one qualify for a diagnosis of this disorder [14,65,66].
FXTAS like other conditions in the FXPAC spectrum is caused by trinucleotide expansion of 55 to 200 CGG repeats in the FMR1 gene [67]. The pathogenesis revolves around markedly elevated FMR1 mRNA, cellular stress, increase in reactive oxygen species (ROI), mitochondrial dysfunction and further RNA toxicity, leading to CGG-binding protein sequestration with the excess mRNA and repeat-associated non-AUG-initiated (RAN) translation leading to production of a toxic protein, FMRpolyG [14,29,30].
The characteristic pathological findings consist of intranuclear inclusions that are positive for ubiquitin. These inclusions are seen in neurons, astrocytes, and Purkinje cells in the brain, spinal cord, and autonomic ganglia throughout the body [68,69,70].
This neurodegenerative disorder usually starts in older carriers above 50 years, is more common and more severe in males than females [71,72]. Women are less susceptible to FXTAS since about half of their neurons and glial cells produce the normal X chromosome instead of the X chromosome with the premutation. This results in reduced toxicity [73].
FXTAS is so diverse that it may present with a variety of features beyond the tremor and ataxia including parkinsonism, executive function deficits, memory loss, cognitive deficits with eventual dementia, brain atrophy, white matter disease characterized by hyperintensities on MRI involving the middle cerebellar peduncles (MCP sign), the splenium and periventricular areas, cerebellar, motor, autonomic dysfunction, neuropsychiatric symptoms like anxiety, irritability, apathy and depression, and peripheral neuropathy [67,74,75,76,77,78,79,80,81,82,83].
The conclusive diagnostic criteria for this medical condition require the presence of one major clinical symptom (either gait ataxia or intention tremor) and one major radiological finding (white matter lesions in the MCP on MRI) in an affected individual [65,66,84]. Other secondary indicators such as neuropathy, executive function impairments, Parkinsonism, overall brain atrophy, and damage to the splenium of the Corpus Callosum can be utilized to establish a Probably or Possible diagnosis of FXTAS when combined with an intention tremor or ataxia [65,84].
This prevalence increases with increasing age and is said to affect almost 75% of male carriers by 80 years of age [66].
Some medications can improve tremors such as primidone or a beta blocker, but ataxia is more difficult to treat. Psychiatric problems such as depression or anxiety or irritability can be treated with a selective serotonin reuptake inhibitor (SSRI). There is some evidence that memantine can improve memory or focus [65], but studies have not been carried out regarding treatment of dementia. Newer medications that can improve mitochondrial function, such as oral allopregnanolone or Anavex 2-73 will hopefully lead to greater improvement in this disorder [14].
There is limited research on FXTAS in Africa. In fact, from our literature search, no study was found on FXTAS, thus highlighting the need for awareness, testing, resources, and research in this area since FXTAS mimics various neurological disorders.
2.5. FXAND
A more recent concept introduced an additional categorization called FXAND to specifically address the higher occurrence of mental health issues, immunological illnesses, and several other medical conditions in women with a premutation [73].
The conditions under the umbrella of FXAND can begin in childhood including anxiety presenting as severe shyness and ADHD in both males and females with the premutation. Often anxiety can worsen in adulthood and can be accompanied by insomnia and depression. Chronic fatigue, chronic pain from migraines or muscle aches or fibromyalgia are common. Autoimmune problems such as autoimmune thyroiditis can occur, but more severe problems, such as lupus or multiple sclerosis have also been reported in 1 to 3% of carriers of the premutation [66].
Treatment of FXAND includes SSRIs as noted above, particularly for anxiety, depression, or obsessive-compulsive thinking. Avoidance of CNS toxins including alcohol, pesticides, illegal drugs, smoking, and excessive opioids can help alleviate symptoms and slow progression to FXTAS. In addition, exercise can be very helpful for anxiety and depression and chronic pain or fatigue. Therapy is always indicated for psychiatric problems and antioxidants may also help to relieve oxidative stress [85].
2.6. Clinical and Behavioral Phenotypes of FXS
The cognitive abilities in those with FXS depend largely on FMRP levels. The level of FMRP is related to the presence of mosaicism in males and to the AR in females [86]. Once the CGG repeat is above 273 in the FM whether methylated or unmethylated there is no FMRP produced so it makes no difference if the CGG repeat number is 400 or 1000 [87].
Mildly low FMRP levels are associated with less severe symptoms, normal or borderline intelligence and less severe learning disabilities as in many females with the FM. In contrast, very low or absent FMRP levels leads to moderate to severe cognitive impairment and intellectual disability [11].
Production of FMRP also determines physical characteristics in affected individuals [11]. Characteristic facial features are seen in 8 out of 10 post pubertal men and these include a long face, prominent ears, and large testicles (macroorchidism) [86].
However, in young children approximately 30 to 40% may not demonstrate typical facial features. A prominent forehead and hyperextensible finger joints are seen in most children. Enlarged testicles or macroorchidism do not occur until puberty. In 1991, Hagerman et al [35] developed a checklist to aid clinicians easily identify patients with FXS. This checklist contained 13 items (intellectual disability, hyperactivity, short attention span, tactile defensiveness, hand-flapping, hand-biting, poor eye contact, perseverative speech, hyperextensible metacarpophalangeal joints, large or prominent ears, large testicles, Simian crease or Sydney line, and family history of intellectual disability). Five or more clinical criteria were associated with a much higher risk for FXS (Table 1).
A few years after the Hagerman’s checklist was developed [35], Maes et al [38] developed a 28-item checklist comprising 7 physical and 21 behavioral characteristics for the identification of patients in need for further referral for gene testing. Physical features listed include narrow and elongated face, high forehead, prominent lower jaw, large protruding ears, macroorchidism, hyperextensible finger joints and other hyperextensible joints. Behavioral features mentioned in the checklist are hyperactivity, sensory oversensitivity, impulsivity, being chaotic, shyness, gaze avoidance, being too helpful, approach-avoidance conflict, fearfulness, gaiety/cheerfulness, hypersensitivity for changes, hand-biting, stereotypic hand movement, flapping hands, and arms, turning away the face, tactile defensiveness, rapid speed of language, being talkative, perseveration, echolalia, and imitation of language. It is pertinent to note that this checklist, while containing a lot of items, may be repetitive, and vary across cultural lines and co-morbidities like autism spectrum disorders.
A 15 item checklist adapted and modified from Hagerman’s [35] original 13-item checklist was developed by Indian researchers more than a decade later, to reflect the cultural variations in dysmorphology in FXS [37]. Items included physical (large ears, elongated face, broad forehead, large testicles, hyperextensible joints and simian crease) and neurological characteristics (intellectual disability, hyperactivity, gaze avoidance, positive family history of intellectual disability, hand biting, hand flapping, short attention span, tactile defensiveness and perseverative speech).
A meta-analysis by Lubala et al [88] developed a concise 7-item universal clinical checklist for FXS (soft and velvety skin with redundancy of skin on the dorsum of hand, flat feet, large and prominent ears, plantar crease, large testes in post pubertal boys, family history of intellectual disability and autistic-like behavior), based on findings from Europe and Asia because of insufficient data from Africa. This lack of data from Africa prompted the recent development of another checklist by Lubala et al [36] to reflect their findings in African subjects from three families in Congo, central Africa. This is likely the first attempt to phenotypically differentiate FXS in Africa from other cultures, bearing in mind cultural variations in clinical and behavioral phenotypes in different dysmorphologies like FXS. Items mentioned in the physical features include elongated face, soft and velvety skin, and plantar crease (which were seen in all cases), large testicles, hyperextensible joints, simian crease and prominent ears were seen in over two-third of cases, while about 14 of cases had microcephaly which is an unusual finding in FXS. The behavioral characteristics include severe intellectual disability, hyperactivity, short attention span and autistic-like features which were seen in all cases. This checklist may be utilized albeit cautiously in resource-limited settings as a diagnostic aid for FXS.
2.7. Challenges in Diagnosing FXS and FXPAC in Africa
One of the major challenges in diagnosing FXS and FXPAC in Africa is the lack of awareness among healthcare professionals and the general population about the condition. This has also been reported in other developing nations[57]. Many individuals with FXS may go undiagnosed or misdiagnosed due to lack of clinical expertise, leading to delays in receiving appropriate care and support.
Another challenge is the limited access to genetic testing, resources, inadequate healthcare infrastructure and specialized healthcare services in many African countries [16,17,21,89].
Genetic testing for FXS and associated disorders now employs more advanced molecular technologies like Southern blot and newer PCR-based methods compared to older methods of testing [90]. These are often not widely available, and healthcare facilities may not have the necessary expertise to diagnose and manage the condition effectively [16,21]. Many countries are advocating for newborn screening for FXS and there has been significant strides in this regard [10,63,91]. Attempts have been made by a few researchers on the possibility of using clinical and behavioral phenotypes as a means of diagnosis in resource poor settings with conflicting findings [36,89]. Lubala et al [36] in Congo found clinical and behavioral phenotypes like checklists reported by Hagerman et al [35], Maes et al [38] and Guruju et al [37]. In contrast, Lumaka et al [89] in Congo, central Africa, noted that none of the 105 individuals assessed with checklists reported by previous authors had FXS after molecular testing, further highlighting the need for gene testing for suspected cases. The paucity of genetic services in Africa may result in a lack of accurate data on the prevalence of FXS in Africa and hinders efforts to improve diagnosis and treatment of the condition.
An additional challenge is stigma and discrimination against individuals and families with fragile X syndrome or other intellectual disabilities [92]. This stigma from cultural beliefs may hinder early identification and testing because families and or communities may conceal affected individuals to prevent alienation by unaffected community members. Kamga et al [93] in their qualitative study in Cameroon, reported the experience of a mother who had 2 children with FXS. She was a descendant of the founder of a powerful royal family and despite this, they clearly experienced stigma, isolation, and name-calling. The disorder was said to have been a curse on the founder of the family which then transmitted to his descendants. Cultural beliefs about intellectual disabilities contribute to under-diagnosis and under-treatment of these conditions [92].
2.8. Genetic Testing for FXS and FXPAC in Africa
Genetic testing for FXS and FXPAC is essential for accurate diagnosis, genetic counseling, and management of these conditions. However, access to genetic testing in Africa is limited, with most centers located in urban regions [21], and there is a lack of awareness among some healthcare providers about the importance of testing for FXS and its associated conditions. Efforts to improve access to genetic testing and training for healthcare providers are needed to ensure that individuals with FXS and FXPAC receive the appropriate care and support.
2.9. Clinical Management of FXS and FXPAC in Africa
The clinical management of FXS and FXPAC should start from a detailed history taking, which should consider all important aspects of the history, including the developmental, nutritional, social history, drug history, allergy history and family history. After this, a thorough physical examination should be conducted on the individual; in the case of FXS, look for phenotypical features; in FXPAC, check for tell-tale signs relevant to the condition and other co-morbidities. For example, FXTAS patients may have ataxia and tremor, which can be detected by a neurological examination. Next step will be to get genetic testing if the patient has not had testing, and if it is available. Subsequently, treatment proper should be done.
The treatment of FXS/FXPAC can be pharmacological and non-pharmacological.
3. Clinical Management of FXS
Individuals with FXS typically enjoy a normal state of health, apart from frequently occurring ear infections across early life, seizures that may affect around 12-15% of cases, and connective tissue issues, such as joint dislocations, hernias and collapsable Eustachian tubes predisposing to otitis media resulting from the absence or insufficiency of FMRP [20].
Recurrent ear infections are prevalent in early childhood among individuals with FXS, affecting more than 50% of males with FXS [94]. This issue is likely attributed to a combination of factors including the shape of the face, collapsible Eustachian tubes, enlarged tonsils or adenoids which may block the Eustachian tube causing recurrent infections. The treatment of recurrent otitis media infections in childhood includes the use of antibiotics for the acute infection in addition to insertion of pressure equalizing (PE) tubes. Language is boosted and significantly improves after the insertion of PE tubes.7
3.1. Seizures
The prevalence of seizures in FXS is higher compared to the general population [94,95]. A wide variation exists in the results of prevalence from initial studies on FXS patients in genetics, epilepsy and neurology clinics compared with the community. The prevalence rate found in the genetics clinics was between 9% and 27%, epilepsy and neurology clinics-14% to 44%, while the community was 12% to 18%. This could have occurred because of a selection bias in the population studied.95 FXS males are at a higher risk for seizures than females with seizures occurring in 12 % of males versus 3% in females [94,95]. However, the information at hand indicates the actual population prevalence of seizure incidence at 10% to 15% [7].
Many types of seizures do occur in FXS although partial seizures are presumed to be the most frequently occurring type. However, generalized seizures are quite common; others are simple, complex partial and status seizures. An electroencephalogram (EEG) should be carried out in patients with possible seizures to help differentiate seizure type e.g. benign Rolandic seizures vs others.
Risk of Autism spectrum disorder (ASD) increases in people with FXS and seizures. Similarly, the chances of seizure occurrence were higher in those who had FXS and co-occurring ASD [95]. Monotherapy with non-sedating and non-behaviorally aggravating anti-seizure medications are preferred and achieve seizure control, with most patients being seizure free. Medications are usually discontinued after 2 years without any seizure episode [7].
Preferred medications with minimal side effects are Keppra (levetiracetam) and oxcarbazepine (Trileptal), and Lamictal (lamotrigine) or Depakote (valproic acid) for refractory seizures[7].
3.2. Pharmacotherapy
There has been great advancement in the treatment of FXS. FXS currently has no cure and as such, treatment aims to manage symptoms the individual presents with. Stimulants work well for ADHD symptoms after 5years of age and guanfacine helps with hyperactivity in children younger than 5 years [96]. Anxiety is pervasive in both males and females with FXS and an SSRI such as sertraline can be helpful. A controlled trial of low dose sertraline (2.5 to 5mg per day) was carried out in children with FXS ages 2 to 6 years old and sertraline was superior to placebo in many developmental subtests on the Mullen Scales for Early Learning [97]. If aggression is problematic then low doses of an atypical antipsychotic such as aripiprazole or risperidone have been helpful in children with FXS [96]. Currently, there are targeted treatments for FXS. These include metformin which lowers the mTOR pathway that is elevated in FXS. Open label studies of metformin have shown improvement in IQ in 2 adults with FXS treated with metformin for a year [98]. In an open-label study of young children with FXS ages 2 to 7 years old, metformin improved developmental testing on the Mullen Scales compared to previously published cognitive testing in children with FXS [99].
A controlled trial of metformin is currently under way in children and adults ages 6 to 40 years old and results will be available in the summer of 2024.
Cannabidiol (CBD) is a GABA agonist that also impacts the serotonin system, and it is anti-inflammatory. In a controlled trial in children ages 3 to 18 with FXS it was efficacious in the primary outcome measure of social avoidance from the Aberrant Behavior Checklist but only in those who were > 90% methylated [100]. CBD was manufactured so there was no tetrahydrocannabinol (THC) and it was applied to the skin as a gel twice a day. It was very well tolerated, and caregiver reports demonstrated an improvement in aggression, anxiety and tantrums so that the families were able to take their children out in public, for instance to restaurants without an outburst so this improved their quality of life [100]. However, the FDA was concerned that those with less than 90% methylation or those who were mosaic did not demonstrate significant efficacy, so a second phase 3 trial is now being carried out at multiple international sites.
Perhaps the most exciting, targeted treatment for FXS is the Tetra drug called BPN14770 (Zitolamast) which is a phosphodiesterase 4D (PDE4D) inhibitor that inhibits the breakdown of cyclic adenosine monophosphate (cAMP) which is too low in FXS. So, this PDE4D allows cAMP to rise to normal levels which enhances neuronal connectivity. The results of a controlled trial in adults with FXS demonstrated efficacy by improving subtests on the National Institute of Health (NIH) toolbox including oral reading, picture vocabulary and overall Crystallized Intelligence Composite score [101]. This is an exciting result seeing cognition improve in adults so currently both adolescent and adult studies are being carried out at multiple centers in a phase 3 trial.
3.3. Non- Pharmacological Therapy
Behavioral therapy is beneficial in patients with FXS.
Additionally, there is a need for research on the efficacy of various treatment options for FXS in African populations, as well as the development of culturally appropriate interventions for affected individuals and their families. Guidance for behavioral interventions can be found in the literature and, also on the National Fragile X Foundation website at www.fragileX.org.
3.4. Conclusion
The burden of FXS and FXPAC in Africa, though important, is still understudied and warrants further research and attention. Challenges in diagnosing and managing FXS in Africa include limited awareness, research, access to genetic testing and specialized healthcare services, and discrimination against affected persons.
These challenges need to be addressed to improve diagnosis, treatment, and support for individuals and families with FXS and FXPAC in the African continent.
3.5. Future Line of Study
Future research on FXS/FXPAC in Africa should focus on conducting large-scale epidemiological studies, developing culturally appropriate and acceptable screening tools, improving access to genetic testing and diagnostic services, raising awareness and education about the disorders even among health workers, and addressing stigma and discrimination against individuals with intellectual disabilities. Furthermore, there is a need for more research on genetic variability across different ethnicities and subregions in Africa and how this influences the burden and clinical picture of FXS/FXPAC. This will contribute to a deeper understanding of these disorders in the African region.
Author Contributions
Conceptualization, all authors.; writing original draft preparation, M.C.N.P and M.I.I., figure preparation M.C.N.P and H.R., manuscript review and editing: all authors. All authors have read and agreed to the published version of the manuscript.
Funding
This work was partially supported by NICHD grant HD036071, the MIND IDDRC grant from NICHD P50 HD103526, the Azrieli Foundation, the Tides Foundation, and the Victor LaFave III Fund
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
This work was partially supported by NICHD grant HD036071, the MIND IDDRC grant from NICHD P50 HD103526, the Azrieli Foundation, the Tides Foundation, and the Victor LaFave III Fund
Conflicts of Interest
RH has received funding from Zynerba and Tetra Pharma companies to carry out treatment studies in those with Fragile X Syndrome. The other authors declare no conflict of interest.
References
- Purdon Martin J, Bell J. A pedigree of mental defect showing sex-linkage. J Neurol Psychiatry. 1943, 6, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Verkerk AJ, Pieretti Y, Sutcliffe JS, Fu YH, A Kuhl DP, Pixxuti A, et al. Identification of a Gene (HIM?-1) Containing a CGG Repeat Coincident with a Breakpoint Cluster Region Exhibiting Length Variation in Fragile X Syndrome. Vol. 65, Cell. 1991.
- Chonchaiya W, Schneider A, Hagerman RJ. Fragile X: A Family of Disorders. Vol. 56, Advances in Pediatrics. 2009. p. 165–86.
- Bagni C, Tassone F, Neri G, Hagerman R. Fragile X syndrome: Causes, diagnosis, mechanisms, and therapeutics. Vol. 122, Journal of Clinical Investigation. 2012. p. 4314–22.
- Berry-Kravis E, Knox A, Hervey C. Targeted treatments for fragile X syndrome. J Neurodev Disord. 2011, 3, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Levenga J, de Vrij FMS, Oostra BA, Willemsen R. Potential therapeutic interventions for fragile X syndrome. Vol. 16, Trends in Molecular Medicine. 2010. p. 516–27.
- Hagerman, RJ. Overview of FXS and Fragile X Spectrum Disorders. In: Hagerman Randi J, Hagerman Paul J, editors. Fragile X Syndrome and Premutation Disorders Clinics in Developmental Medicine Fragile X Syndrome and Premutation Disorders New Developments and Treatments. London: Mac Keith Press; 2020. p. 1–4.
- Gross C, Berry-Kravis EM, Bassell GJ. Therapeutic strategies in fragile X syndrome: Dysregulated mGluR signaling and beyond. Vol. 37, Neuropsychopharmacology. 2012. p. 178–95.
- Mclennan Y, Polussa J, Tassone F, Hagerman R. Fragile X Syndrome. Vol. 12, Current Genomics. 2011.
- Fernandez-Carvajal I, Walichiewicz P, Xiaosen X, Pan R, Hagerman PJ, Tassone F. Screening for expanded alleles of the FMR1 gene in blood spots from newborn males in a Spanish population. Journal of Molecular Diagnostics. 2009, 11, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Hagerman RJ, Hagerman PJ. Fragile X syndrome. In: Outcomes in Neurodevelopmental and Genetic Disorders [Internet]. Cambridge University Press; 2002. p. 198–219. Available from: https://www.cambridge.org/core/product/identifier/CBO9780511543876A015/type/book_part.
- Hornstra IK, Nelson DL, Warren ST, Yang TP. High resolution methylation analysis of the FMR1 gene trinucleotide repeat region in fragile X syndrome [Internet]. Vol. 2, Human Molecular Genetics. 1993. Available from: http://hmg.oxfordjournals.org/.
- Berlin CI, Hood LJ, Morlet T, Rose K, Brashears S. Auditory Neuropathy/Dys-Synchrony: Diagnosis and Management. Vol. 9, Mental Retardation and Developmental Disabilities Research Reviews. 2003. p. 225–31.
- Tassone F, Protic D, Allen EG, Archibald AD, Baud A, Brown TW, et al. Insight and Recommendations for Fragile X-Premutation-Associated Conditions from the Fifth International Conference on FMR1 Premutation. Vol. 12, Cells. Multidisciplinary Digital Publishing Institute (MDPI); 2023.
- Essop FB, Krause A. Diagnostic, carrier and prenatal genetic testing for fragile X syndrome and other FMR-1-related disorders in Johannesburg, South Africa: A 20-year review. South African Medical Journal. 2013, 103, 994–998. [Google Scholar] [CrossRef] [PubMed]
- Wonkam A, Tekendo CN, Sama DJ, Zambo H, Dahoun S, Béna F, et al. Initiation of a medical genetics service in sub-Saharan Africa: Experience of prenatal diagnosis in Cameroon. Eur J Med Genet. 2011, 54, 4. [Google Scholar]
- Meguid NA, Ismail MF, El-Mahdy RS, Barakat MA, El-Awady MK. Simple molecular diagnostic method for Fragile X syndrome in Egyptian patients: Pilot study [Internet]. Available from: www.actabp.pl.
- Peprah EK, Allen EG, Williams SM, Woodard LM, Sherman SL. Genetic diversity of the Fragile X syndrome Gene (FMR1) in a large sub-saharan West African population. Ann Hum Genet. 2010, 74, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Chiurazzi P, Destro-Bisol G, Genuardi M, Oostra BA, Spedini G, Neri G. Extended Gene Diversity at the FMRl Locus and Neighbouring CA Repeats in a Sub-Saharan Population. Vol. 64, American Journal of Medical Genetics. 1996.
- Hagerman RJ, Berry-Kravis E, Hazlett HC, Bailey DB, Moine H, Kooy RF, et al. Fragile X syndrome. Nat Rev Dis Primers. 2017, 3, 1. [Google Scholar]
- Essop FB, Krause A. Diagnostic, carrier and prenatal genetic testing for fragile X syndrome and other FMR-1-related disorders in Johannesburg, South Africa: A 20-year review. South African Medical Journal. 2013, 103, 994–998. [Google Scholar] [CrossRef]
- Goldman A, Jenkins T, Krause A. Molecular evidence that fragile X syndrome occurs in the South African black population [1]. Vol. 35, Journal of Medical Genetics. 1998. p. 878.
- Abdel Meguid N, El Awady MK. Prevalence of fragile X syndrome among school-age Egyptian males [Internet]. 2007.Available from: https://www.researchgate.net/publication/285380905.
- Kengne Kamga K, De Vries J, Nguefack S, Munung NS, Wonkam A. Explanatory models for the cause of Fragile X Syndrome in rural Cameroon. J Genet Couns. 2021, 30, 1727–1736. [Google Scholar] [CrossRef]
- Latunji, CA. Fragile X allelemorphism among the mentally retarded and in affected families. Scientific Research and Essay [Internet]. Available from: http://www.academicjournals.org/sre [cited 2024 Mar 12]. 2009, 4, 1123–1131. [Google Scholar]
- Wittenberger MD, Hagerman RJ, Sherman SL, McConkie-Rosell A, Welt CK, Rebar RW, et al. The FMR1 premutation and reproduction. Vol. 87, Fertility and Sterility. 2007. p. 456–65.
- Hunter JE, Epstein MP, Tinker SW, Charen KH, Sherman SL. Fragile X-associated primary ovarian insufficiency: Evidence for additional genetic contributions to severity. Genet Epidemiol. 2008, 32, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Kenneson A, Zhang F, Hagedorn CH, Warren ST. Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Vol. 10, Human Molecular Genetics. 2001.
- Allen EG, He W, Yadav-Shah M, Sherman SL. A study of the distributional characteristics of FMR1 transcript levels in 238 individuals. Hum Genet. 2004, 114, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Tassone F, Hagerman RJ, Taylor AK, Gane LW, Godfrey TE, Hagerman PJ. Elevated Levels of FMR1 mRNA in Carrier Males: A New Mechanism of Involvement in the Fragile-X Syndrome. Vol. 66, Am. J. Hum. Genet. 2000.
- Biancalana V, Glaeser D, McQuaid S, Steinbach P. EMQN best practice guidelines for the molecular genetic testing and reporting of fragile X syndrome and other fragile X-associated disorders. European Journal of Human Genetics. 2015, 23, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Hunter J, Rivero-Arias O, Angelov A, Kim E, Fotheringham I, Leal J. Epidemiology of fragile X syndrome: A systematic review and meta-analysis. Am J Med Genet A. 2014, 164, 1648–1658. [Google Scholar] [CrossRef] [PubMed]
- Hagerman RJ, Hagerman PJ. The fragile X premutation: into the phenotypic fold. Curr Opin Genet Dev. 2002, 12, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, PJ. The fragile X prevalence paradox. Vol. 45, Journal of Medical Genetics. 2008. p. 498–9.
- Hagerman RJ, Amiri K, Cronister A. Fragile X Checklist. Vol. 38, Journal of Medical Genetics. 1991.
- Kasole Lubala T, Kayembe-Kitenge T, Lubala N, Kanteng G, Luboya O, Hagerman R, et al. Fragile X syndrome in Democratic Republic of Congo: dysmorphic, cognitive and behavioral findings in 14 subjects from three families. Clin Dysmorphol. 2024, 33, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Guruju MR, Lavanya K, Thelma BK, Sujatha M, OmSai VR, Nagarathna V, et al. Assessment of a clinical checklist in the diagnosis of fragile X syndrome in India. Journal of Clinical Neuroscience. 2009, 16, 1305–1310. [Google Scholar] [CrossRef] [PubMed]
- Maes B, Fryns J, Ghesquiere P, Borghgraef M. Phenotypic checklist to screen for Fragile X syndrome in people with mental retardation. Ment retard. 2000, 38, 207–215. [Google Scholar] [CrossRef]
- Bouali N, Hmida D, Mougou S, Bouligand J, Lakhal B, Dimessi S, et al. Analysis of FMR1 gene premutation and X chromosome cytogenetic abnormalities in 100 Tunisian patients presenting premature ovarian failure. Ann Endocrinol (Paris). 2015, 76, 671–678. [Google Scholar] [CrossRef]
- Li M, Zhu Y, Wei J, Chen L, Chen S, Lai D. The global prevalence of premature ovarian insufficiency: a systematic review and meta-analysis. Climacteric [Internet]. 2023 [cited 2024 Mar 24];26, 95–102. Available from: https://www.tandfonline.com/doi/abs/10.1080/13697137.2022.2153033.
- Sopiarz N, Sparzak PB. Primary Ovarian Insufficiency. StatPearls [Internet]. 2023 Mar 6 [cited 2024 Mar 24]; Available from: https://www.ncbi.nlm.nih.gov/books/NBK589674/.
- Goswami D, Conway GS. Premature ovarian failure. Vol. 68, Hormone Research. 2007. p. 196–202.
- Simpson JL, Rajkovic A. Ovarian Differentiation and Gonadal Failure. Vol. 89, J. Med. Genet. (Semin. Med. Genet.). 1999.
- Persani L, Rossetti R, Cacciatore C. Genes involved in human premature ovarian failure. Vol. 45, Journal of Molecular Endocrinology. 2010. p. 257–79.
- Zinn AR, Ross JL. Turner syndrome and haploinsufficiency [Internet]. Available from: http://biomednet.com/elecref/0959437X00800322.
- Sherman SL. Premature Ovarian Failure in the Fragile X Syndrome. Vol. 97, J. Med. Genet. (Semin. Med. Genet.). 2000.
- Allen EG, Sullivan AK, Marcus M, Small C, Dominguez C, Epstein MP, et al. Examination of reproductive aging milestones among women who carry the FMR1 premutation. Human Reproduction. 2007, 22, 2142–2152. [Google Scholar] [CrossRef]
- Seltzer MM, Baker MW, Hong J, Maenner M, Greenberg J, Mandel D. Prevalence of CGG expansions of the FMR1 gene in a US population-based sample. American Journal of Medical Genetics, Part B: Neuropsychiatric Genetics. 2012, 159 B, 589–597.
- Allen EG, Charen K, Hipp HS, Shubeck L, Amin A, He W, et al. Refining the risk for fragile X–associated primary ovarian insufficiency (FXPOI) by FMR1 CGG repeat size. Genetics in Medicine. 2021, 23, 1648–1655. [Google Scholar] [CrossRef]
- Hipp HS, Charen KH, Spencer JB, Allen EG, Sherman SL. Reproductive and gynecologic care of women with fragile X primary ovarian insufficiency (FXPOI). Menopause. 2016, 23, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Cronister A, Schreiner R, Wittenberger M, Amiri K, Harris K, Hagerman RJ. Heterozygous Fragile X Female: Historical, Physical, Cognitive, and Cytogenetic Features.
- Murray A. Premature Ovarian Failure and the FMR1 Gene. 2000.
- Mailick MR, Hong J, Greenberg J, Smith L, Sherman S. Curvilinear association of CGG repeats and age at menopause in women with FMR1 premutation expansions. American Journal of Medical Genetics, Part B: Neuropsychiatric Genetics. 2014, 165, 705–711. [Google Scholar] [CrossRef]
- Sullivan AK, Marcus M, Epstein MP, Allen EG, Anido AE, Paquin JJ, et al. Association of FMR1 repeat size with ovarian dysfunction. Human Reproduction. 2005, 20, 402–412. [Google Scholar] [CrossRef]
- Jin M, Yu YQ, Huang HF. An update on primary ovarian insufficiency. Vol. 55, Science China Life Sciences. 2012. p. 677–86.
- Archibald AD, McClaren BJ, Caruana J, Tutty E, King EA, Halliday JL, et al. The Australian Reproductive Genetic Carrier Screening Project (Mackenzie’s Mission): Design and Implementation. J Pers Med. 2022, 12, 11. [Google Scholar]
- Tassone F, Budimirovic D, Hopkins Medicine J, Cvjetkovic S. Fragile X-Associated Disorders in Serbia: Baseline Quantitative and Qualitative Survey of Knowledge, Attitudes, and Practices Among Medical Professionals. 2018, Available from: www.frontiersin.org.
- Archibald AD, Hickerton CL, Jaques AM, Wake S, Cohen J, Metcalfe SA. “It’s about having the choice”: Stakeholder perceptions of population-based genetic carrier screening for fragile X syndrome. Am J Med Genet A. 2013, 161, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Archibald AD, Jaques AM, Wake S, Collins VR, Cohen J, Metcalfe SA. “It’s something i need to consider”: Decisions about carrier screening for fragile X syndrome in a population of non-pregnant women. Am J Med Genet A. 2009, 149, 2731–2738. [Google Scholar]
- Hantash FM, Goos DM, Crossley B, Anderson B, Zhang K, Sun W, et al. FMR1 premutation carrier frequency in patients undergoing routine population-based carrier screening: Insights into the prevalence of fragile X syndrome, fragile X-associated tremor/ataxia syndrome, and fragile X-associated primary ovarian insufficiency in the United States. Genetics in Medicine. 2011, 13, 39–45. [Google Scholar]
- Lee S, Taylor JL, Redmond C, Hadd AG, Kemppainen JA, Haynes BC, et al. Validation of Fragile X Screening in the Newborn Population Using a Fit-for-Purpose FMR1 PCR Assay System. In: Journal of Molecular Diagnostics. Elsevier B.V.; 2020. p. 346–54.
- Cronister A, Teicher J, Rohlfs EM, Donnenfeld A, Hallam S. Prevalence and Instability of Fragile X Alleles Implications for Offering Fragile X Prenatal Diagnosis. Vol. 111, Obstet Gynecol. 2008.
- Tassone F, Iong KP, Tong TH, Lo J, Gane LW, Berry-Kravis E, et al. FMR1 CGG allele size and prevalence ascertained through newborn screening in the United States [Internet]. 2013. Available from: http://genomemedicine.com/content/4/12/100.
- Hunter Jessica Ezzell, Wheeler Anne C, Allen Emily Grave, Wald Kaitlyn, Rajkovic Aleksandar, Hagerman RJ, et al. Women’s issues in Fragile X Spectrum Disorders. In: Hagerman RJ, Hagerman PJ, editors. Fragile X Syndrome and Premutation Disorders New Developments and Treatments [Internet]. London: Mac Keith Press; 2020. p. 75–82. Available from: www.mackeith.co.uk.
- Martinez-Cerdeno V, Wang Jun Yi, Grigsby Jim, Hall Deborah, Hagerman RJ. FXTAS: New Advances and Treatments In: Fragile X syndrome and Premutation Disorders New developments and treatments. In: Hagerman RJ, Hagerman PJ, editors. London: Mac Keith Press; 2020. p. 83–93.
- Hagerman RJ, Hagerman P. Fragile X-associated tremor/ataxia syndrome-features, mechanisms and management. Vol. 12, Nature Reviews Neurology. Nature Publishing Group; 2016. p. 403–12.
- Hall DA, Birch RC, Anheim M, Jønch AE, Pintado E, O’Keefe J, et al. Emerging topics in FXTAS. J Neurodev Disord. 2014, 6, 1. [Google Scholar]
- Wenzel HJ, Hunsaker MR, Greco CM, Willemsen R, Berman RF. Ubiquitin-positive intranuclear inclusions in neuronal and glial cells in a mouse model of the fragile X premutation. Brain Res. 2010, 1318, 155–66. [Google Scholar] [CrossRef]
- Greco CM, Berman RF, Martin RM, Tassone F, Schwartz PH, Chang A, et al. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS). Brain. 2006, 129, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, P. Fragile X-associated tremor/ataxia syndrome (FXTAS): Pathology and mechanisms. Vol. 126, Acta Neuropathologica. 2013. p. 1–19.
- Leehey, MA. Fragile X- Associated Tremor/Ataxia Syndrome: Clinical Phenotype, Diagnosis, and Treatment. Journal of Investigative Medicine. 2009, 57, 830–836. [Google Scholar] [CrossRef] [PubMed]
- Jacquemont S, Hagerman RJ, Leehey M, Grigsby J, Zhang L, Brunberg JA, et al. Fragile X Premutation Tremor/Ataxia Syndrome: Molecular, Clinical, and Neuroimaging Correlates. Vol. 72, Am. J. Hum. Genet. 2003.
- Hagerman RJ, Protic D, Rajaratnam A, Salcedo-Arellano MJ, Aydin EY, Schneider A. Fragile X-Associated Neuropsychiatric Disorders (FXAND). Vol. 9, Frontiers in Psychiatry. Frontiers Media S.A.; 2018.
- Robertson EE, Hall DA, McAsey AR, O’Keefe JA. Fragile X-associated tremor/ataxia syndrome: phenotypic comparisons with other movement disorders. Vol. 30, Clinical Neuropsychologist. Routledge; 2016. p. 849–900.
- Hall DA, O’keefe JA. Fragile X-Associated Tremor Ataxia Syndrome: The Expanding Clinical Picture, Pathophysiology, Epidemiology, and Update on Treatment [Internet]. 2012. Available from: http://tremorjournal.org/article/view/56.
- Berry-Kravis E, Goetz CG, Leehey MA, Hagerman RJ, Zhang L, Li L, et al. Neuropathic features in fragile X premutation carriers. Am J Med Genet A. 2007, 143, 19–26. [Google Scholar]
- Kraff J, Tang HT, Cilia R, Canesi M, Pezzoli G, Goldwurm S, et al. Screen for Excess FMR1 Premutation Alleles Among Males With Parkinsonism [Internet]. Available from: http://www.parkinson.it.
- Hagerman RJ, Leehey ; M, Heinrichs ; W, Med ;, Tassone ; F, Wilson ; R, et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. 2001.
- Zhao C, Liu Y, Wang Y, Li H, Zhang B, Yue Y, et al. A Chinese case of fragile X-associated tremor/ataxia syndrome (FXTAS) with orthostatic tremor:case report and literature review on tremor in FXTAS. Vol. 20, BMC Neurology. BioMed Central Ltd.; 2020.
- Cabal-Herrera AM, Tassanakijpanich N, Salcedo-Arellano MJ, Hagerman RJ. Fragile X-associated tremor/ataxia syndrome (FXTAS): Pathophysiology and clinical implications. Vol. 21, International Journal of Molecular Sciences. MDPI AG; 2020. p. 1–23.
- Peters N, Kamm C, Asmus F, Holinski-Feder E, Kraft E, Dichgans M, et al. Intrafamilial variability in fragile x-associated tremor/ataxia syndrome. Movement Disorders. 2006, 21, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Fraint A, Vittal P, Szewka A, Bernard B, Berry-Kravis E, Hall DA. New observations in the fragile X-associated tremor/ataxia syndrome (FXTAS) phenotype. Front Genet. 2014, 5(SEP).
- Islam F, Lee W. A Case of Generalized Chorea Presenting as an Early Feature of Fragile-X Associated Tremor/Ataxia Syndrome. Mov Disord Clin Pract. 2020, 7, 464–466. [Google Scholar] [CrossRef] [PubMed]
- Hall DA, Birch RC, Anheim M, Jønch AE, Pintado E, O’Keefe J, et al. Emerging topics in FXTAS. J Neurodev Disord. 2014, 6, 1. [Google Scholar]
- Sodhi DK, Hagerman R. Fragile X Premutation: Medications, Therapy and Lifestyle Advice. Vol. 14, Pharmacogenomics and Personalized Medicine. Dove Medical Press Ltd; 2021. p. 1689–99.
- Saldarriaga W, Carrera G, Tassone F, Yuriko González-Teshima L, Forero-Forero JV, Ayala-Zapata S, et al. Fragile X Syndrome Síndrome de X Frágil. Vol. 45, Colombia Médica. 2014.
- Randol JL, Kim K, Ponzini MD, Tassone F, Falcon AK, Hagerman RJ, et al. Variation of FMRP Expression in Peripheral Blood Mononuclear Cells from Individuals with Fragile X Syndrome. Genes (Basel) 2024, 15. [Google Scholar]
- Lubala TK, Lumaka A, Kanteng G, Mutesa L, Mukuku O, Wembonyama S, et al. Fragile X checklists: A meta-analysis and development of a simplified universal clinical checklist. Mol Genet Genomic Med. 2018, 6, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Lumaka A, Lubala TK, Race V, Peeters H, Lukusa P, Devriendt K. Usefulness of fragile X checklist and CGG distribution in specialized institutions in Kinshasa, DR Congo. J Community Genet. 2019, 10, 153–159. [Google Scholar] [CrossRef]
- Siomi H, Siomi MC, Nussbaum RL, Dreyfuss G. The Protein Product of the Fragile X Gene, FMRI, Has Characteristics of an RNA-Binding Protein. Vol. 74, Cell. 1993.
- Bailey DB, Berry-Kravis E, Gane LW, Guarda S, Hagerman R, Powell CM, et al. Fragile X Newborn Screening: Lessons Learned From a Multisite Screening Study. Vol. 139, PEDIATRICS. 2017.
- Ali A, King M, Strydom A, Hassiotis A. Self-reported stigma and symptoms of anxiety and depression in people with intellectual disabilities: Findings from a cross sectional study in England. J Affect Disord. 2015, 187, 224–231. [Google Scholar] [CrossRef]
- Kengne Kamga K, Munung NS, Nguefack S, Wonkam A, De Vries J. Negotiating political power and stigma around fragile X Syndrome in a rural village in Cameroon: A tale of a royal family and a community. Mol Genet Genomic Med. 2021, 9, 3. [Google Scholar]
- Kidd SA, Lachiewicz A, Barbouth D, Blitz RK, Delahunty C, McBrien D, et al. Fragile X syndrome: A review of associated medical problems. Vol. 134, Pediatrics. American Academy of Pediatrics; 2014. p. 995–1005.
- Berry-Kravis E, Raspa M, Loggin-Hester L, Bishop E, Holiday D, Bailey DB. Seizures in fragile X syndrome: Characteristics and comorbid diagnoses. Vol. 115, American Journal on Intellectual and Developmental Disabilities. 2010. p. 461–72.
- Hagerman RJ, Berry-Kravis E, Kaufmann WE, Ono MY, Tartaglia N, Lachiewicz A, et al. Advances in the treatment of fragile x Syndrome. Vol. 123, Pediatrics. 2009. p. 378–90.
- Greiss Hess L, Fitzpatrick SE, Nguyen D V. , Chen Y, Gaul KN, Schneider A, et al. A randomized, double-blind, placebo-controlled trial of low-dose sertraline in young children with fragile X syndrome. Journal of Developmental and Behavioral Pediatrics. 2016, 37, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Protic D, Aydin EY, Tassone F, Tan MM, Hagerman RJ, Schneider A. Cognitive and behavioral improvement in adults with fragile X syndrome treated with metformin-two cases. Mol Genet Genomic Med. 2019, 7, 1–7. [Google Scholar]
- Biag HMB, Potter LA, Wilkins V, Afzal S, Rosvall A, Salcedo-Arellano MJ, et al. Metformin treatment in young children with fragile X syndrome. Mol Genet Genomic Med. 2019, 7, 1–13. [Google Scholar]
- Berry-Kravis E, Hagerman R, Budimirovic D, Erickson C, Heussler H, Tartaglia N, et al. A randomized, controlled trial of ZYN002 cannabidiol transdermal gel in children and adolescents with fragile X syndrome (CONNECT-FX). J Neurodev Disord. 2022, 14, 1.
- Berry-Kravis EM, Harnett MD, Reines SA, Reese MA, Ethridge LE, Outterson AH, et al. Inhibition of phosphodiesterase-4D in adults with fragile X syndrome: a randomized, placebo-controlled, phase 2 clinical trial. Nat Med. 2021, 27, 862–870. [Google Scholar] [CrossRef]
Figure 1.
Fragile X- Premutation Associated Conditions involvement throughout life (Adapted from Tassone et al [14].)
Figure 1.
Fragile X- Premutation Associated Conditions involvement throughout life (Adapted from Tassone et al [14].)

| Not present (score 0) | Borderline or present in the past (score 1) | Definitely present (score 2) | |
|---|---|---|---|
| Intellectual disability | |||
| Hyperactivity | |||
| Short attention span | |||
| Tactilely defensive | |||
| Hand-flapping | |||
| Hand biting | |||
| Poor eye contact | |||
| Perseverative speech | |||
| Hyperextensible metacarpophalangeal joints | |||
| Large or prominent ears | |||
| Large testicles | |||
| Simian crease or Sydney line | |||
| Family history of intellectual disabilities | |||
| Total score | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



