
Submitted:
18 April 2024
Posted:
19 April 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The COVID-19 pandemic remains a serious public health problem globally. During winter flu seasons, more aggressive SARS-CoV-2 infections with fatalities have been documented, indicating that influenza co-infections may significantly impact the disease outcome of COVID-19. Both influenza and SARS-CoV-2 viruses share many similarities in their transmission and cellular tropism for their replication in the human respiratory tract. However, it is still unclear how the two pathogens interplay to ensure their survival in the same lung microenvironment. In addition, clinical studies on influenza co-infections in COVID-19 patients do not provide conclusive evidence on how influenza co-infection mechanistically modifies the disease outcome in COVID-19. This mini-review discusses various viral as well as host factors that potentially influence the survival or synergism of these two respiratory pathogens in the infected lung microenvironment.
Keywords:
influenza
; SARS-CoV-2
; co-infection
; inflammation
; lung pathology
Introduction
Since its emergence in December 2019, the devastating COVID-19 pandemic has culminated in over 7.0 million fatalities globally according to a World Health Organization report. [1] Although SARS-CoV-2 continued to cause disease throughout the year from 2020 to 2022, significant increases in hospitalizations and mortalities have been reported during the winter flu seasons globally. This could be due to climatic conditions, which are more favorable for virus growth transmission, and/or the possibility for co-infections with circulating other respiratory pathogens. Among the various respiratory pathogens that could cause co-infections, influenza viruses have been identified as a significant risk in exacerbating lung disease in COVID-19. SARS-CoV-2 belongs to the coronavirus family and is related to 2003-SARS-CoV and Middle East Respiratory Syndrome (MERS) viruses. Structurally, both SARS-CoV-2 and influenza viruses are enveloped, single-stranded RNA viruses, and the RNA is encapsidated with nucleoprotein. While SARS-CoV-2 contains positive sense single large RNA, influenza viruses contain negative sense segmented RNA as their genome. Both viruses share similar modes of transmission and infection sites in the upper and lower respiratory tract. [2] Although both viruses exhibit different entry mechanisms, simultaneous infections, and the continuous presence of these two pathogens in the same lung environment could result in serious complications and clinical illness. It is believed that SARS-CoV-2 will likely cause recurring infections and based on the literature on seasonal and pandemic influenza outbreaks, future viral epidemics are unavoidable. Thus, it is highly essential to understand the interplay between SARS-CoV-2 and influenza viruses, especially during winter flu seasons to minimize morbidities and mortalities. This review provides an overview of influenza co-infections in SARS-CoV-2 and describes how influenza, and SARS-CoV-2 viruses compete for survival within the lung microenvironment.
Impact of Influenza Epidemic Season on SARS-CoV-2 Infection and Disease Outcome
Patients with COVID-19 develop early symptoms such as high fever, cough, myalgia, and dyspnea, but most infected patients recover from mild or subclinical infections. However, less than 10 % of COVID-19 patients, especially older adult patients, suffer from severe lung disease, which necessitates admission into intensive care units, assisted ventilation, and other supportive therapies. A significant number of these severely ill patients eventually develop progressive clinical manifestations of acute respiratory distress syndrome (ARDS) and succumb due to impaired oxygen intake capacity and multi-organ dysfunction. [3,4] Pre-existing chronic illnesses, co-morbidities, other health conditions, and microbial co-infections have been associated with increased fatalities among critically ill COVID-19 patients. [5,6,7,8]
The phenomenon of co-infection involves different viral, fungal, or bacterial species, with varying combinations. [9,10,11,12] There was a marked increase in SARS-CoV-2 infections and fatalities during the winter flu seasons of 2020 and 2021, indicating that influenza co-infections may have a significant exacerbating impact on the disease outcome of COVID-19. [9] Further, a sudden hike in COVID-19 cases reported during the flu season of 2022 in China indicates the need for effective surveillance to monitor co-circulation of SARS-CoV-2 and influenza viruses in future. Influenza and SARS-COV-2 share similar clinical and epidemiologic features – hence, understanding the co-infections is imperative to ensure optimal management during flu epidemic seasons. The presence of another virus may give rise to the phenomenon of viral interference. Influenza interference during SARS-CoV-2 infection may significantly impact survival, virus spread, virulence, and the pattern of immune responses. Numerous clinical reports from various countries document aggravated pathophysiology among COVID-19 patients with influenza co-infection. [13,14,15,16] Early reports from China and Iran noted over 50% of severely ill hospitalized COVID-19 patients were positive for influenza co-infections. [13,14]
Influenza co-infections significantly increase in-hospital mortalities compared to SARS-CoV-2 mono-infection, or other viral or bacterial co-infections. [15] A clinical study revealed increased death rates among hospitalized patients with co-infections, compared to either of these pathogens alone indicating a lethal synergism between the two respiratory viruses. [16] Interestingly, a study on the prevalence of influenza and SARS-CoV-2 co-infections reported to the Center for Disease Control (CDC) surveillance system between 2020-2022, described more severe disease and mortalities among young patients (<18 years) with influenza co-infections. [17] This raises a concern about the underuse of antivirals or influenza vaccines among the young population. A recent clinical report identified avian influenza (H5N1) coinfection with SARS-CoV-2 and despite severe pneumonia, the patient recovered by use of antiviral drugs together with the standard care. [18] Numerous clinical reports also documented no differences in clinical signatures, and unaltered disease severities between patients with influenza co-infections compared to SARS-CoV-2 infection alone. [19,20] These findings raise questions on how these two viruses interact and interplay for survival and indicate dichotomous effects in influenza co-infection. There is a need to understand how these pathogens interact and modulate lung pathophysiology and immune signatures while sharing the same pulmonary microenvironment. Furthermore, due to their high similarities in clinical presentations and immunopathologic mechanisms between influenza and COVID-19, the detection of these viral pathogens is essential for the assessment of disease progression and for effective treatment to prevent hospital-associated deaths.
Alveolar Epithelium: A Common Target for Influenza and SARS-CoV-2 Replication
Both influenza and SARS-COV-2 viruses can target different epithelial cells from the upper and lower respiratory tract. The infection of the alveolar epithelial lining directly correlates with the disease severity in both viral infections. Structurally, the alveolar lining is covered by two types of epithelial cells: alveolar type I (AT-I) and type II (AT-II) pneumocytes. AT-I cells are squamous and thin membranous cells, important for gas exchange and fluid homeostasis; while AT-II cells are small cuboidal-shaped cells that produce surfactant proteins and lipids. Both AT-I and AT-II cells are permissive for influenza and SARS-CoV-2 infection, which induces cytopathic damage, [21,22,23] leading to alveolar disintegration, loss of surfactant, and impairment of gas exchange. The loss of alveolar integrity subsequently injures the structural integrity of the extracellular matrix present in the underneath interstitium and endothelial damage in the capillary network, ultimately collapsing the alveolar architecture. The loss of gas exchange due to alveolar damage contributes to massive inflammation and cytokine storm, which eventually progresses into pathologic manifestations of ARDS. [3]
Despite their similar cell tropism, these viruses utilize different routes of entry into the target cells. The influenza virus undergoes endocytosis via the interaction of its hemagglutinin with sialic acid receptors on the epithelial membrane. SARS-CoV-2 gains cell entry via its spike binding to angiotensin-converting enzyme-2 (ACE2) receptor. The spike-AT-II binding triggers protease-mediated cleavage of the spike protein by the transmembrane Serine Protease 2 (TMPRSS2) causing the fusion of the viral membrane with the host cell membrane and subsequent endocytosis of the virus. The initial infection and replication of both influenza and SARS-CoV-2 in AT-II cells trigger the induction of the interferon and other cytokines by surrounding cells as part of the innate immune response in clearing the virus at the early phase of infection. These early inflammatory responses damage adjacent AT-I cells as well as capillary endothelial cells within the walls of the thin alveolar epithelial interstitium, which further mobilizes more inflammatory immune cells to the infection site. The damage and disruption of the alveolar epithelial lining subsequently develop pathological alterations of hyaline membrane formation, which is a major characteristic feature identified in both severe influenza SARS-CoV-2 infected patients. Interestingly, the replication of SARS-CoV-2 occurs at a slower rate compared to the influenza virus in tracheobronchial and alveolar type II cells. [24] The influenza virus infection displayed high viral antigen staining at 48 hours post-infection and in contrast, SARS-CoV-2 growth was found at a slower rate and of less magnitude. The differences in the growth kinetics could significantly impact infectivity and pathogenesis when both viruses share the same microenvironment. [24]
Influenza and SARS-CoV-2 Co-Infection: Competition or Sinister Alliance?
The synergism that occurs during influenza and SARS-CoV-2 co-infection is not yet completely understood. Two possible scenarios of this co-infection can be envisaged, i.e., (i) secondary SARS-CoV-2 infection following primary influenza exposure; and (ii) secondary influenza infection following primary SARS-CoV-2 infection. In both scenarios, the co-existence of both pathogens in the same lung compartment is possible. This may allow both viruses to either compete for their survival in the phenomenon of competitive viral suppression or to exhibit synergism, which may cause exacerbated lung pathology (Figure 1).
The competition for survival of these viruses may depend on the time of infection, virus inoculum size, and interactions between virus and host-related entities. Typically, the replication of one virus may inhibit the growth of another virus through competitive suppression. Interestingly, analyses through mathematical modeling of viral co-infections hypothesize that viruses with faster growth rates will likely suppress those viruses with slow growth rates when competing for survival. SARS-CoV-2 has slow growth compared to influenza virus thus the influenza co-infection could reduce SARS-CoV-2 growth. [24] Several mechanisms have been proposed on how influenza can inhibit the growth of the SARS-CoV-2. The type I IFN genes (IL-1α and IL-1β) are activated upon recognition of viral nucleotides by pattern recognition receptors (PRRs) such as toll-like receptors (TLRs) or retinoic acid-inducible gene I (RIG-I). This triggers antiviral effects via JAK-STAT pathway-mediated expression of IFN-stimulated-genes (ISGs) followed by synthesis and release of protein kinase R (PKR), Mx proteins, and 2′,5′-oligoadenylate synthetase (OAS) which degrade viral RNA. [25] In vitro infection of Vero cells with influenza was shown to block SARS-CoV-2 replication kinetics via induction of IFN-I/-III. [25] In support of these findings, hamsters co-infected with SARS-CoV-2 and influenza displayed decreased SARS-COV-2 loads by 5 days post-infection, while influenza viral replication remained high, thus indicating suppression of SARS-COV-2 in a co-infection scenario. Influenza-induced type I IFN also induces apoptosis in surrounding epithelial cells via Fas ligand signaling, [26,27] thus limiting the access of ACE2 receptors for invading SARS-CoV-2. [28] Similarly, prior SARS-CoV-2 infection may restrict influenza superinfection via induction of AT-II apoptosis or NOD-like receptor-mediated pyroptosis. [29] Indeed cells undergoing pyroptotic death aid in eliminating infected cells and thus restrict the replicative niche and survival of the opportunistic intracellular pathogens. [29] In another instance, competition may exist within the same target cell. If both viruses infect the same epithelial cell, they may compete at different stages of virus replication, including endocytic entry, viral genome replication, packing, and release of progeny viruses. One study showed that influenza infection downregulates the expression of ACE2, thereby preventing SARS-CoV-2 entry and replication. [30] In addition, non-structural genes expressed by influenza and SARS-CoV-2 play a critical role in evading the host's innate immune response to facilitate their infection of susceptible cells. [31,32] The SARS-CoV-2 NSP1 gene suppresses expression of IFN-α signaling by attenuating STAT1 and STAT2 phosphorylation via downregulating IFN-β protein, IFN-λ1, and IL-8 through binding to 18s rRNA of the 40S ribosome in the host cell. [32,33]
On the other hand, SARS-CoV-2-influenza co-infection may aggravate the disease outcome. Prior influenza infection has been linked to upregulation of the ACE2 receptor expression and an increase in SARS-CoV-2 infectivity in A549 lung epithelial cells. [34] In support of this, >5-fold increase in SARS-CoV-2 infectivity was observed in A549, Calu-3 (human adenocarcinoma epithelial cell line), and normal human bronchial epithelial cells when pre-infected with influenza virus, thus suggesting a lethal synergism of infection during influenza co-infections in various cell types. In another study, a 2-3-fold increase in ACE2 mRNA levels was observed in influenza-infected A549 cells. However, co-infection with SARS-CoV-2 increased >28-fold increase in the expression of ACE-2 mRNA thus suggesting positive feedback of ACE-2 expression in a co-infection scenario that contributes to increased infectivity of SARS-CoV-2 in lung epithelial cells. [35] Interestingly, the increased SARS-CoV-2 infectivity observed in prior influenza-infected A549 cells was completely blocked when ACE-2 expression was knocked down, thus suggesting that induction of ACE2 is key for aggravated SARS-CoV-2 infectivity in a Co-infection. [34]
Another mechanism of lethal synergism was linked to the activation of type-1 membrane-bound protease, furin, which activates both influenza hemagglutinin protein and cleaves SARS-CoV-2 spike protein, thereby facilitating both viral entry into the host cell. [36,37] The activity of furin increases during influenza, thus prior influenza infection may augment predisposition to SARS-CoV-2 during the flu season. Likewise, SARS-CoV-2 superinfection at 48 hours after the influenza challenge of mice exacerbates lung pathology compared to SARS-CoV-2 infection alone. [34] In another study, sequential infection with influenza at 4 weeks after SARS-CoV-2 infection of ferrets leads to increased clinical signs and inflammation responses compared to influenza infection alone. [38] The increase in susceptibility was observed even among animals challenged with mild SARS-CoV-2, thus indicating the increased risk of influenza during COVID-19. Similarly, hamsters co-infected with influenza A virus plus SARS-CoV-2 exhibited increased loss of body weight, prolonged pneumonia, and severity of the disease than animals infected with either of the pathogens alone. Interestingly, immunohistochemical analysis of co-infected hamster lungs did not show co-localization of SARS-CoV-2 and influenza, thus indicating that these viruses in the infected environment may compete for survival thus inhibiting each other or both viruses are replicating in two adjacent areas in the infected-lungs. In addition, increased IL-6 levels in co-infected animals compared to either of the pathogen infected alone suggests involvement of IL-6 in aggravated lung pneumonia. [39] Immunopathologic analysis of lungs of ferrets co-infected with influenza and SARS-CoV-2 exhibited extensive inflammation in the nasal cavity and lungs, compared to either of these viruses infected alone. [40] Further, influenza, but not SARS-CoV-2 displayed high viral shedding and transmission by direct contact with cohoused animals. In a case report, influenza, and SARS-COV-2 co-infection in a 32-year-old man exhibited severe lung pneumonia. Despite standard care together with administration of anti-IL-6 monoclonal antibodies and anti-viral agent against influenza, the patient succumbed to infection with respiratory failure, thus indicating a poor prognosis in influenza co-infection. [41] Overall, the ability of the influenza virus to aggravate SARS-CoV-2-mediated pathogenesis and infectivity suggests that influenza co-infections could be a major target in reducing SARS-CoV-2-related morbidities and mortalities in future COVID-19 outbreaks.
Viruses including human rhinovirus (HRV), human parainfluenza (HPI), and respiratory syncytial virus (RSV) are the most common human respiratory viruses that cause the common cold and circulate throughout the year. Several clinical reports identified co-infection of these respiratory pathogens in severely ill COVID-19 patients. To understand the impact of these respiratory pathogens on SARS-CoV-2 infectivity, a study was conducted using lung epithelial cells co-infected with different respiratory viruses together with SARS-CoV-2. Interestingly, co-infection of alveolar epithelial cells with HRV, HPI, and RSV did not show any effect on SARS-COV-2 viral loads, although these viruses displayed their active replication during co-infection. However, a lethal synergism was observed only when SARS-CoV-2 was co-infected with influenza virus, thus suggesting that influenza co-infection could have a detrimental effect on epithelial survival causing exacerbating the epithelial injury and subsequent lung damage. [34] In a clinical study, an increase in cough and dyspnea was observed in patients co-infected with SARS-CoV-2 and HRV compared to either of the virus-alone infected patients. However, no significant number of ICU admissions or deaths were observed between mono-infected and co-infected patients. [42] A recent study revealed that co-infection with RSV enhances the severity of the disease in SARS-CoV-2 omicron infection in young adults. These findings warrant further investigations on how RSV co-infections impact the children on the disease outcome. [17,43]
Immunopathology: A Common Mediator of Acute Lung Injury in a Co-Infection
The aggravated lung injury in influenza co-infections could be a cumulative effect of virus-inflicted cytopathic effects together with immunopathology. Infection with both influenza and SARS-CoV-2 elicits massive inflammatory cellular and cytokine responses, which play a central role in the disruption of the thin alveolar epithelial-capillary barrier. Golden Syrian hamsters co-infected with influenza and SARS-CoV-2 displayed extensive inflammatory cell influx, vascular injury, and pulmonary edema compared to single virus-infected hamsters. [44] In another study increased lung pathology was observed in influenza and SARS-CoV-2 co-infected hamsters compared to either of these pathogens challenged alone. Further increased IL-6 levels were observed in co-infected animals suggesting that IL-8-mediated signaling could be involved in increased severity of the disease. [39] Further, SARS-CoV-2 infection 24 hours before the 2009 pandemic influenza virus infection displayed severe clinical signs, loss of body weight, extensive alveolitis, perivascular and peribranchial inflammatory cellular influx high inflammation, and acute lung injury, and in contrast, prior influenza infection ameliorated SARS-CoV-2 infectivity and lung injury.
An excessive lung influx of neutrophils was observed in both severe influenza and SARS-CoV-2 infections. Upon recruiting to the infectious lung microenvironment, neutrophils exhibit dysregulated activity and generate neutrophil extracellular traps (NETs), which are composed of DNA decorated with toxic components such as histones, neutrophil elastase (NE), and cathepsins. [45] The released NETs function like a double-edged sword to damage the alveolar-capillary bed; trigger epithelial and endothelial necrosis and injure pulmonary vasculature. Further, histones released extracellularly aggregate platelet activation triggering microvascular thrombosis. [46,47] In a co-infection scenario, these insults may precipitously intensify causing collapse of the alveolar-capillary architecture leading to clinical development of ARDS. Further, the damaged alveolar-capillary barrier may aid in the systemic dissemination of these viruses and facilitate the adhesion and colonization of opportunistic bacterial or fungal pathogens. [48,49] Influenza co-infection can likely pose a recurring global problem during flu seasons – hence, it is imperative to elucidate the underlying mechanisms of this important co-infection to explore and design novel therapeutic interventions.
On the other hand, influenza-SARS-CoV-2 co-infection in K-18-hACE2 mice exhibited not only increased and prolonged inflammatory cellular infiltrations in infected lungs, but also lymphopenia with reduced levels of IgG, and decreased neutralizing antibody titers in the blood. Interestingly, these mice also exhibited reduced CD4+ T cells against both viruses. [50] These findings highlight that co-infection would impact the protective adaptive immunity and subsequent long-term impairment of immune response. Further studies are required to investigate whether immunopathogenesis in influenza-SARS-CoV-2 co-infection could be a cumulative effect of aggravated innate immune response together with impaired adaptive immune response.
Does Vaccination against Influenza Decrease the Risk of COVID-19?
Annual influenza vaccines drastically reduce intensive care unit (ICU) admissions among elderly and high-risk individuals during flu seasons in the USA and Europe. Influenza vaccination also decreases hospitalization and pneumonia caused by other viral or bacterial infections. [51,52] Several clinical studies indicate that prior influenza vaccination diminishes susceptibility to SARS-CoV-2 infection and reduces the burden of COVID-19 by improving the clinical outcome. In a retrospective cohort study, prior influenza vaccination demonstrated significant reductions in COVID-19-related hospitalizations, length of stay in the intensive care unit, and need for invasive respiratory support via mechanical ventilation when compared to unvaccinated individuals. [53] Notably, influenza vaccination in ferrets exhibited significant protection against influenza-alone or influenza and SARS-CoV-2 co-infected animals, thus suggesting that influenza vaccination could help in preventing disease severity of SARS-CoV-2 during flu seasons. [39] Although the precise mechanisms of this protection are yet unclear, there is a possibility for bystander protective effects through the induction of cell-mediated immune responses. One hypothesis is that restoration of Th1 cell immunity, and a decrease in the cytokine storm, could alleviate SARS-CoV-2 infections. [54] On the other hand, prior flu vaccination may also trigger non-specific innate immune responses that may aid in the early clearance of SARS-CoV-2. Although specific antibodies and T cell-mediated responses are generated against specific antigens in the classical immune response, the induction of proinflammatory responses may provide non-specific immunity and protection against non-specific and unrelated pathogens. [54] Quadrivalent inactivated influenza vaccination could elicit a strong cytokine response following stimulation of immune cells, thus supporting that seasonal influenza vaccination could mitigate SARS-CoV-2 infection. [55,56] Interestingly, intranasal vaccination with influenza vectored COVID-19 vaccine that contains NS1-deleted influenza virus carrying receptor binding domain of COVID-19 (dNS1-RBD) has shown broad spectrum protection in hamsters against challenges with different variants of SARS-CoV-2 variants. [57] The combination vaccine decreased viral loads and attenuated pro-inflammatory cytokine (IL-6, IL-1β, and IFN-γ) thus alleviating immunopathology and tissue injury after SARS-CoV-2 infection. These findings are important, as the severity of the disease COVID-19 is due to a combination of viral-inflicted tissue damage together with immunopathology mediated by overactive immune cells and inflammatory cytokines. Notably, in one study, protection by prior influenza vaccination has been shown to protect against influenza-SARS-CoV-2 co-infection. However, prior immunity against SARS-CoV-2 does not protect in a co-infection scenario. The protection of influenza vaccination has been associated with neutralizing antibodies, but not CD4+ and CD8+ T cells. [58] Further studies warrant on mechanisms of how the prior influenza immune response helps in alleviating influenza/SARS-CoV-2 comorbidity. BCG produces a heterologous immune response that was found to be effective against cancers such as bladder cancer by reducing tumor progression and recurrence of cancer. Heterogeneous effects against SARS-CoV-2 infections were also observed in influenza and Bacille Calmette-Guérin (BCG) vaccinations. [59] BCG vaccine prepared from the live attenuated strain of Mycobacterium bovis. More experimental evidence is needed to better understand the mechanisms of how heterogeneous immunity can prevent or mitigate SARS-CoV-2 pathophysiology.
Conclusions and Future Directions
The risk of influenza co-infections during the COVID-19 pandemic remains unclear. Notwithstanding this, the dramatic increase in SARS-CoV-2 infections and fatalities during influenza epidemic seasons supports the notion that influenza co-infection exacerbates lung damage. Both influenza and SARS-CoV-2 infections give rise to complex cytopathic and immunologic reactions in the lungs and other organs. These reactions may either aggravate lung and organ pathogenesis, or these viruses may compete and inhibit the other for their survival in the pulmonary micro-compartments. Given that both viruses are likely to continue to co-circulate every year, it is critical to continue effective surveillance and to characterize novel variants of SARS-CoV-2 and influenza viruses. During forthcoming winters, a double viral threat is anticipated that may require the administration of COVID-19 vaccines together with seasonal influenza vaccines to alleviate the respiratory disease burden, especially among vulnerable populations. Additionally, the protective effects of influenza vaccination against severe outcomes of SARS-CoV-2 infection suggest a promising avenue for reducing the burden of COVID-19. Future vaccination strategies should explore potential cross-protection conferred by influenza vaccines, while also investigating the heterogeneous effects of different vaccination approaches. As the world grapples with evolving respiratory virus infections, future research endeavors should unravel the complexities of co-infections, guide surveillance strategies, and inform targeted interventions, crucial for optimizing public health responses during influenza epidemic seasons and their impact on SARS-CoV-2 infection and disease outcomes.
Author Contributions
TN: VC, PM, VK, KV, SMG, and MR drafted the manuscript and revised the final form. All authors contributed to the article and approved the submitted version.
Funding
The authors have no conflicts of interest and funding to declare.
Conflicts of Interest
All the authors declare no conflicts of Interest.
References
- Coronavirus disease-19. World Health Organization. Last accessed 2024 April 2024. Available from http://www.Coronavirus disease (COVID-19) (who. int).
- Wiemken, T.L.; Khan, F.; Puzniak, L.; Yang, W.; Simmering, J.; Polgreen, P.; Nguyen, J.L.; Jodar, L.; McLaughlin, J.M. Seasonal trends in COVID-19 cases, hospitalizations, and mortality in the United States and Europe. Sci Rep 2013, 13, 3886. [Google Scholar] [CrossRef] [PubMed]
- Matera, M.G.; Rogliani, P.; Calzetta, L.; et al. Pharmacological management of COVID-19 patients with ARDS (CARDS): A narrative review. Respir Med. 2020, 171, 106114. [Google Scholar] [CrossRef] [PubMed]
- Kazi, A.W.; Summer, R.; Sundaram, B.; George, G. Lung recovery with prolonged ECMO following fibrotic COVID-19 acute respiratory distress syndrome. Am J Med Sci. 1016. [Google Scholar]
- Ejaz, H.; Alsrhani, A.; Zafar, A.; Javed, H.; Junaid, K.; Abdalla, A.E.; Abosalif, K.O.A.; Ahmed, Z.; Younas, S. COVID-19 and comorbidities: Deleterious impact on infected patients. J Infect Public Health. 2020, 13, 1833–1839. [Google Scholar] [CrossRef] [PubMed]
- Gremese, E.; Tolusso, B.; Bruno, D.; Paglionico, A.M.; Perniola, S.; Ferraccioli, G.; Alivernini, S. COVID-19 illness: Different comorbidities may require different immunological therapeutic targets. Eur J Clin Invest. 2023, e14096. [Google Scholar] [CrossRef] [PubMed]
- Rosano, G.; Jankowska, E.A.; Ray, R.; Metra, M.; Abdelhamid, M.; Adamopoulos, S.; Anker, S.D.; Bayes-Genis, A.; Belenkov, Y.; Gal, T.B.; et al. COVID-19 vaccination in patients with heart failure: a position paper of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. 2021, 23, 1806–1818. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Almodóvar, A.S.; Nahata, M.C. Medication Adherence in Medicare-Enrolled Older Adults with Chronic Obstructive Pulmonary Disease before and during the COVID-19 Pandemic. J Clin Med. 2022, 11, 6985. [Google Scholar] [CrossRef] [PubMed]
- Jones, N. How COVID-19 is changing the cold and flu season. Nature. 2020, 588, 388–390. [Google Scholar] [CrossRef]
- Losier, A.; Gupta, G.; Caldararo, M.; Dela Cruz, C.S. The Impact of Coronavirus Disease 2019 on Viral, Bacterial, and Fungal Respiratory Infections. Clin Chest Med. 2023, 44, 407–423. [Google Scholar] [CrossRef] [PubMed]
- Markovskaya, Y.; Gavioli, E.M.; Cusumano, J.A.; Glatt, A.E. Coronavirus disease 2019 (COVID-19): Secondary bacterial infections and the impact on antimicrobial resistance during the COVID-19 pandemic. Antimicrob Steward Health Epidemiol. 2022, 2, e114. [Google Scholar] [CrossRef]
- Chamola, V.; Mohammadi, R.; Nair, H.; Goyal, A.; Patel, A.; Hassija, V.; Bassetti, M.; Narang, P.; Paredes, R.; Santos, J.R.; et al. COVID-19-associated mucormycosis: A review of an emergent epidemic fungal infection in the era of COVID-19 pandemic. J Res Med Sci. 2022, 27, 57. [Google Scholar]
- Ma, S.; Lai, X.; Chen, Z.; Tu, S.; Qin, K. Clinical characteristics of critically ill patients co-infected with SARS-CoV-2 and the influenza virus in Wuhan, China. Int J Infect Dis. 2020, 96, 683–87. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, S.A.; Safamanesh, S.; Ghasemzadeh-Moghaddam, H.; Ghafouri, M.; Azimian, A. High prevalence of SARS-CoV-2 and influenza A virus (H1N1) coinfection in dead patients in northeastern Iran. J Med Virol. 2021, 93, 1008–1012. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Zhang, Z.; Guo, Y.; Shi, J.; Pei, G.; Yao, Y.; Liao, W.; Zeng, R. Lopinavir/ritonavir is associated with pneumonia resolution in COVID-19 patients with influenza coinfection: A retrospective matched-pair cohort study. J Med Virol. 2021, 93, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Stowe, J.; Tessier, E.; Zhao, H.; Guy, R.; Muller-Pebody, B.; Zambon, M.; Andrews, N.; Ramsay, M.; Lopez Bernal, J. Interactions between SARS-CoV-2 and influenza, and the impact of coinfection on disease severity: a test-negative design. Int J Epidemiol. 2021, 50, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Grohskopf, L.A.; Blanton, L.H.; Ferdinands, J.M.; Chung, J.R.; Broder, K.R.; Talbot, H.K.; Morgan, R.L.; Fry, A.M. Prevention and Control of Seasonal Influenza with Vaccines: Recommendations of the Advisory Committee on Immunization Practices - United States, 2022-23 Influenza Season. MMWR Recomm Rep. 2022, 71, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Dai, Z.; Shi, P.; Li, Y.; Zhu, C. Severe pneumonia with co-infection of H5N1 and SARS-CoV-2: a case report. BMC Infect Dis. 2024, 24, 31. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Zhang, M.; Xing, L.; Wang, K.; Rao, X.; Liu, H.; Tian, J.; Zhou, P.; Deng, Y.; Shang, J. The epidemiology and clinical characteristics of co-infection of SARS-CoV-2 and influenza viruses in patients during COVID-19 outbreak. J Med Virol. 2020, 92, 2870–2873. [Google Scholar] [CrossRef]
- Konala, V.M.; Adapa, S.; Naramala, S.; Chenna, A.; Lamichhane, S.; Garlapati, P.R.; Balla, M.; Gayam, V. A Case Series of Patients Coinfected With Influenza and COVID-19. J Investig Med High Impact Case Rep. 2020, 2324709620934674. [Google Scholar] [CrossRef]
- Rahman, M.; Irmler, M.; Keshavan, S.; Introna, M.; Beckers, J.; Palmberg, L.; Johanson, G.; Ganguly, K.; Upadhyay, S. Differential Effect of SARS-CoV-2 Spike Glycoprotein 1 on Human Bronchial and Alveolar Lung Mucosa Models: Implications for Pathogenicity. Viruses. 2021, 13, 2537. [Google Scholar] [CrossRef]
- Narasaraju, T.; Edwin, Y.; Ramar, P.S.; et al. Excessive Neutrophils and Neutrophil Extracellular Traps Contribute to Acute Lung Injury of Influenza Pneumonitis. Am J Pathol. 2011, 179, 199–210. [Google Scholar] [CrossRef]
- Al-Qahtani, A.A.; Pantazi, I.; Alhamlan, F.S.; Alothaid, H.; Matou-Nasri, S.; Sourvinos, G.; Vergadi, E.; Tsatsanis, C. SARS-CoV-2 modulates inflammatory responses of alveolar epithelial type II cells via PI3K/AKT pathway. Front Immunol. 2022, 13, 1020624. [Google Scholar] [CrossRef] [PubMed]
- Zarkoob, H.; Allué-Guardia, A.; Chen, Y.C.; Garcia-Vilanova, A.; Jung, O.; Coon, S.; Song, M.J.; Park, J.G.; Oladunni, F.; Miller, J.; et al. Modeling SARS-CoV-2 and influenza infections and antiviral treatments in human lung epithelial tissue equivalents. Commun Biol. 2022, 5, 810. [Google Scholar] [CrossRef]
- Oishi, K.; Horiuchi, S.; Minkoff, J.M.; tenOever, B.R. The Host Response to Influenza A Virus Interferes with SARS-CoV-2 Replication during Coinfection. J Virol. 2022, 96, e0076522. [Google Scholar] [CrossRef]
- Killip, M.J.; Fodor, E.; Randall, R.E. Influenza virus activation of the interferon system. Virus Res. 2015, 209, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.; Chen, L.; Wu, H. Enhanced Programmed Cell Death Protein 1/Programmed Cell Death Ligand 1 Expression Induced by Severe Influenza A Virus Infection Impairs Host's Antiviral Response. Viral Immunol. 2022, 35, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Bittner, Z.A.; Schrader, M.; George, S.E.; Amann, R. Pyroptosis and Its Role in SARS-CoV-2 Infection. Cells. 11, 1717. [CrossRef] [PubMed]
- Kuriakose, T.; Kanneganti, T.D. Pyroptosis in Antiviral Immunity. Curr Top Microbiol Immunol. 1007. [Google Scholar]
- Liu, X.; Yang, N.; Tang, J.; Liu, S.; Luo, D.; Duan, Q.; Wang, X. Downregulation of angiotensin-converting enzyme 2 by the neuraminidase protein of influenza A (H1N1) virus. Virus Res. 2014, 185, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Varghese, P.M.; Kishore, U.; Rajkumari, R. Innate and adaptive immune responses against Influenza A Virus: Immune evasion and vaccination strategies. Immunobiology. 2022, 227, 152279. [Google Scholar] [CrossRef] [PubMed]
- Low, Z.Y.; Zabidi, N.Z.; Yip, A.J.W.; Puniyamurti, A.; Chow, V.T.K.; Lal, S.K. SARS-CoV-2 Non-Structural Proteins and Their Roles in Host Immune Evasion. Viruses. 202, 14, 1991.
- Thoms, M.; Buschauer, R.; Ameismeier, M.; Koepke, L.; Denk, T.; Hirschenberger, M.; Kratzat, H.; Hayn, M.; Mackens-Kiani, T.; Cheng, J.; et al. Structural Basis for Translational Shutdown and Immune Evasion by the Nsp1 Protein of SARS-CoV-2. Science. 2020, 369, 1249–1255. [Google Scholar] [CrossRef]
- Bai, L.; Zhao, Y.; Dong, J.; Liang, S.; Guo, M.; Liu, X.; Wang, X.; Huang, Z.; Sun, X.; Zhang, Z.; et al. Coinfection with influenza A virus enhances SARS-CoV-2 infectivity. Cell Res. 2021, 31, 395–403. [Google Scholar] [CrossRef]
- Ziegler, C.G.K.; et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell. 2020, 181, 1016–1035. [Google Scholar] [CrossRef] [PubMed]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 2020, 176, 104742. [Google Scholar] [CrossRef] [PubMed]
- Kido, H.; Okumura, Y.; Takahashi, E.; Pan, H.Y.; Wang, S.; Yao, D.; Yao, M.; Chida, J.; Yano, M. Role of host cellular proteases in the pathogenesis of influenza and influenza-induced multiple organ failure. Biochim. Biophys. Acta 2012, 1824, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Vilas Boas de Melo, C.; Peters, F.; van Dijken, H.; Lenz, S.; van de Ven, K.; Wijsman, L.; Gomersbach, A.; Schouten, T.; van Kasteren, P.B.; van den Brand, J.; et al. Influenza Infection in Ferrets with SARS-CoV-2 Infection History. Microbiol Spectr. 2022, 10, e0138622. [Google Scholar] [CrossRef]
- Kinoshita, T.; Watanabe, K.; Sakurai, Y.; Nishi, K.; Yoshikawa, R.; Yasuda, J. Co-infection of SARS-CoV-2 and influenza virus causes more severe and prolonged pneumonia in hamsters. Sci Rep. 2021, 11, 21259. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Skarlupka, A.L.; Jang, H.; Blas-Machado, U.; Holladay, N.; Hogan, R.J.; Ross, T.M. SARS-CoV-2 and Influenza A Virus Coinfections in Ferrets. J Virol. 2022, 96, e0179121. [Google Scholar] [CrossRef] [PubMed]
- Lew, S.; Manes, P.; Smith, B. Coinfection with SARS-CoV-2 and influenza A virus in a 32-year-old man. Am. J. Case Rep. 2020, 21, e926092. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Fernandez, R.; González-Martínez, F.; Perez-Moreno, J.; González-Sánchez, M.I.; Toledo Del Castillo, B.; Mingueza de la Paz, I.; Diaz Pozo, L.; Mejias, A.; Ramilo, O. Clinical Relevance of RSV and SARS-CoV-2 Coinfections in Infants and Young Children. Pediatr Infect Dis J. 2023, 42, e473–e475. [Google Scholar] [CrossRef] [PubMed]
- Le Glass, E.; Hoang, V.T.; Boschi, C.; Ninove, L.; Zandotti, C.; Boutin, A.; Bremond, V.; Dubourg, G.; Ranque, S.; Lagier, J.C.; et al. Incidence and Outcome of Coinfections with SARS-CoV-2 and Rhinovirus. Viruses. 2021, 13, 2528. [Google Scholar] [CrossRef]
- Zhang, A.J.; Lee, A.C.; Chan, J.F.; Liu, F.; Li, C.; Chen, Y.; Chu, H.; Lau, S.Y.; Wang, P.; Chan, C.C.; et al. Coinfection by Severe Acute Respiratory Syndrome Coronavirus 2 and Influenza A(H1N1)pdm09 Virus Enhances the Severity of Pneumonia in Golden Syrian Hamsters. Clin Infect Dis. 2021, 72, e978–e992. [Google Scholar] [CrossRef]
- Pérez-Guerrero, P.; Illanes-Álvarez, F.; Márquez-Ruiz, D.; Campaña-Gómez, I.; Cuesta-Sancho, S.; Márquez-Coello, M.; Girón-González, J.A. Implication of Neutrophils Extracellular Traps in the Pathogenesis of SARS-CoV-2 pneumonia. Biomedicines. 2022, 10, 2638. [Google Scholar] [CrossRef] [PubMed]
- Ashar, H.K.; Mueller, N.C.; Rudd, J.M.; Snider, T.A.; Achanta, M.; Prasanthi, M.; Pulavendran, S.; Thomas, P.G.; Ramachandran, A.; Malayer, J.R.; et al. The Role of Extracellular Histones in Influenza Virus Pathogenesis. Am J Pathol. 2018, 188, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, D.V.; Shreenivas, S.S.; Hudock, K.M. Role of Acute Thrombosis in Coronavirus Disease 2019. Crit Care Clin. 2022, 38, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Langford, B.J.; So, M.; Raybardhan, S.; Leung, V.; Westwood, D.; MacFadden, D.R.; Soucy, J.R.; Daneman, N. Bacterial co-infection and secondary infection in patients with COVID-19: a living rapid review and meta-analysis. Clin Microbiol Infect. 2020, 26, 1622–1629. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kaur, A.; Chowdhary, A. Fungal pathogens and COVID-19. Curr Opin Microbiol. 2023, 75, 102365. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Nguyen, T.Q.; Casel, M.A.B.; Rollon, R.; Kim, S.M.; Kim, Y.I.; Yu, K.M.; Jang, S.G.; Yang, J.; Poo, H.; et al. Coinfection with SARS-CoV-2 and Influenza A Virus Increases Disease Severity and Impairs Neutralizing Antibody and CD4+ T Cell Responses. J Virol. 2022, 96, e0187321. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.L.; El-Hennawy, D. The possible beneficial adjuvant effect of influenza vaccine to minimize the severity of COVID-19. Med Hypotheses. 2020, 140, 109752. [Google Scholar] [CrossRef] [PubMed]
- Marín-Hern_andez, D.; Schwartz, R.E.; Nixon, D.F. Epidemiological evidence for association between higher influenza vaccine uptake in the elderly and lower COVID-19 deaths in Italy. J Med Virol. 2021, 93, 64–65. [Google Scholar] [CrossRef] [PubMed]
- Fink, G.; Orlova-Fink, N.; Schindler, T.; Grisi, S.; Ferrer, A.P.S.; Daubenberger, C.; Brentani, A. Inactivated trivalent influenza vaccination is associated with lower mortality among patients with COVID-19 in Brazil. BMJ Evid Based Med. 1549. [Google Scholar]
- Sahin, U.; Muik, A.; Derhovanessian, E.; Vogler, I.; Kranz, L.M.; Vormehr, M.; Baum, A.; et al. COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses. Nature. 2020, 586, 594–599. [Google Scholar] [CrossRef]
- Benn, C.S.; Netea, M.G.; Selin, L.K.; Aaby, P. A small jab - a big effect: nonspecific immunomodulation by vaccines. Trends Immunol. 2013, 34, 431–439. [Google Scholar] [CrossRef]
- Debisarun, P.A.; Gössling, K.L.; Bulut, O.; Kilic, G.; Zoodsma, M.; Liu, Z.; Oldenburg, M.; Rüchel, N.; Zhang, B.; Xu, C.J.; et al. Induction of trained immunity by influenza vaccination - impact on COVID-19. PLoS Pathog. 2021, 17, e1009928. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jiang, Y.; He, J.; Chen, J.; Qi, R.; Yuan, L.; Shao, T.; Zhao, H.; Chen, C.; Chen, Y.; et al. Intranasal influenza-vectored COVID-19 vaccine restrains the SARS-CoV-2 inflammatory response in hamsters. Nat Commun. 2023, 14, 4117. [Google Scholar] [CrossRef] [PubMed]
- Achdout, H.; Vitner, E.B.; Politi, B.; Melamed, S.; Yahalom-Ronen, Y.; Tamir, H.; Erez, N.; Avraham, R.; Weiss, S.; Cherry, L.; et al. Increased lethality in influenza and SARS-CoV-2 coinfection is prevented by influenza immunity but not SARS-CoV-2 immunity. Nat Commun. 2021, 12, 5819. [Google Scholar] [CrossRef]
- O'Connor, E.; Teh, J.; Kamat, A.M.; Lawrentschuk, N. Bacillus Calmette Gu_erin (BCG) vaccination use in the fight against COVID-19 - what's old is new again? Future Oncol. 2020, 16, 1323–1325. [Google Scholar] [CrossRef] [PubMed]
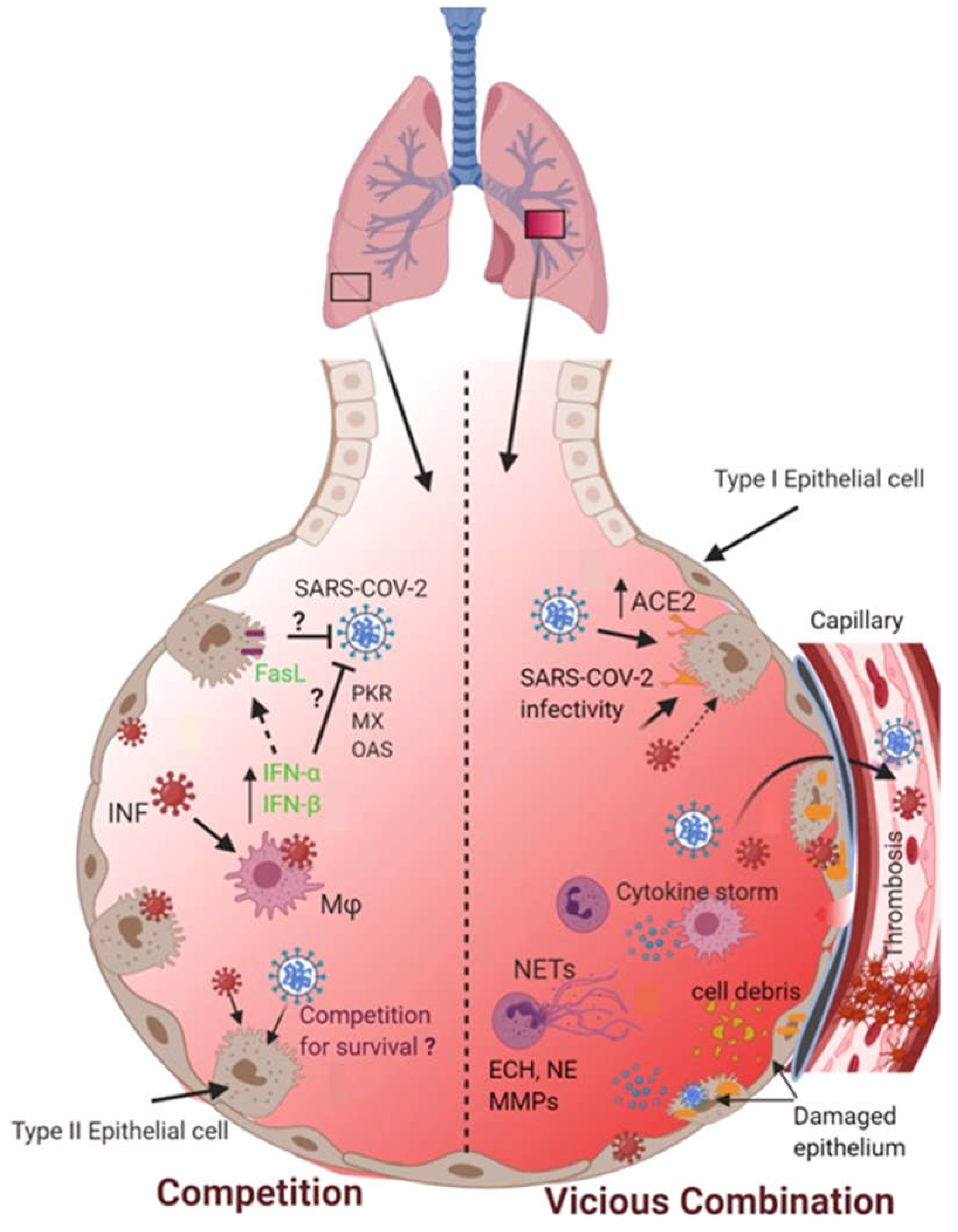
Figure 1.
The potential fate of co-infection of SARS-CoV-2 and influenza viruses in the alveolar microenvironment. Two possible scenarios may occur when both SARS-CoV-2 and influenza viruses encounter and co-infect the lungs. Firstly, co-infection may cause these two viral pathogens to compete for their survival. On the other hand, co-infection could result in a vicious combination that may exacerbate lung pathophysiology by causing alveolar-capillary damage, overwhelming inflammatory responses, NETs release, and formation of pulmonary vascular thrombosis.
Figure 1.
The potential fate of co-infection of SARS-CoV-2 and influenza viruses in the alveolar microenvironment. Two possible scenarios may occur when both SARS-CoV-2 and influenza viruses encounter and co-infect the lungs. Firstly, co-infection may cause these two viral pathogens to compete for their survival. On the other hand, co-infection could result in a vicious combination that may exacerbate lung pathophysiology by causing alveolar-capillary damage, overwhelming inflammatory responses, NETs release, and formation of pulmonary vascular thrombosis.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



