Submitted:
07 April 2024
Posted:
08 April 2024
You are already at the latest version
Abstract
Antibodies are important tools for the study of membrane proteins in biological, biophysics and structural analyses. However, the production of purified membrane proteins is still a formidable challenge due to the amphipathic character and inherent instability of these proteins. In this work, we tested a strategy for producing antibodies against an ATP-Binding Cassette (ABC) transporter without the need to produce the full membrane complex. Instead, just the extra cytoplasmic regions of the ABC-type transporter Rv1819c from Mycobacterium tuberculosis were presented in the RAD-display, a scaffold system based on the engineered Pyrococcus furiosus RadA protein developed for exposition of peptides. The RadA scaffold protein is highly stable, can tolerate long insertions and easy to produce in Escherichia coli. Based on the three-dimensional structure of Rv1891c, we selected two versions of the main extracellular loops of the permease to use as test cases for this approach. Rv1819c-derived peptides displayed on RAD display system were used for immunization of mice and production of polyclonal antibodies. These antibodies were able to recognize the transporter in M. tuberculosis cell extracts, on intact cells by flow cytometry and as purified protein. The results showed the approach can be a useful tool for in vitro analyses, screening, and evaluation of different epitopes in membrane proteins.
Keywords:
ABC transporters
; Mycobacterium tuberculosis
; antibody-mediated recognition
; membrane proteins
; RAD display
1. Introduction
Antibodies are an interesting resource for studies of membrane proteins once they can be used for different purposes. Antibodies provide high specificity for their target antigens, allowing for the selective labeling and visualization of specific proteins or domains within complex biological samples. In cell biology assays and biophysical studies, antibodies can be useful for exploring conformational changes, localization, inhibition, and other applications [1,2]. In cryo-electron microscopy (Cryo-EM), the addition of a fragment-antigen binding region (Fab region) directed to the protein of interest or to a protein belonging to a complex have many advantages. It can stabilize the protein in particular conformation, increase the size of the target and, consequently, enhance the contrast and signal-to-noise ratio, making it easier to analyze images, facilitate alignment of particles and improve three-dimensional reconstruction [3]. In X-ray crystallography, the use of antibodies, and Fab fragments, can be used to stabilize targets, drive novel crystal forms, and solve the problem of phase resolution in structure determination [4].
The development of antibodies against membrane proteins, however, is hard due the difficulties to obtain these proteins in purified form. The RAD-Display system was developed by Rossman and collaborators [5] for the presentation of small peptides in a protein to enable the study of interactions between proteins. The system was based on the thermostable recombinase RadA from Pyrococcus furiosus, engineered to contain only the ATPase domain with a replacement of so-called L2 loop with a cloning site to facilitate insertion of displayed epitopes. In this system, the coding sequences for peptides of interest are inserted with a ligation-independent cloning (LIC) into pRAD plasmid. By expressing peptides of interest in a highly stable and easily folded anchor protein, tethered at both ends, the aim is to maintain at least partially their structure (folded state) and reduce risks of proteolysis. Affinity tags on the scaffold itself facilitate large scale production and purification of the proteins.
In previous studies, we showed that this system was suitable for production of antibodies against extracellular loops of permeases from ABC transporters of Escherichia coli and Staphylococcus aureus without the need for expression and purification of complete membrane proteins [6]. The system also was applied to evaluate the immunogenicity of sequences from SARS-CoV-2 Spike protein, production of antibodies and selection of antibodies with ability to neutralize the virus infection in human cells [7].
In this study, we have evaluated the use of the RAD display system to produce antibodies against the Mycobacterium tuberculosis Rv1819c ATP-Binding Cassette (ABC) transporter. The ABC transporter Rv1819c has been characterized as a pivotal component in the transport of essential molecules, including vitamin B12 and hydrophilic compounds [8,9]. As a member of the ABC transporter superfamily, Rv1819c facilitates the translocation of these substrates across cellular membranes, utilizing energy derived from ATP hydrolysis.
Rv1819c functions as a homodimeric transport complex composed of two identical half-transporters. The TMDs are responsible for recognizing and binding substrates while the NBDs act as the energy-transducing units, utilizing ATP to power the transport process. Based on the three-dimensional structure of the transporter, we selected most prominent extracellular loop of the permease to be presented by the RAD-display system. We designed two versions of this loop, different in their length, based on their predicted antigenicity. Rv1819c-derived peptides expressed in the RAD display system were used as antigens for immunization of C57BL/6 mice and production of polyclonal antibodies. We tested the ability of the antibodies for recognition of the Rv1819c transporter using different approaches. We found that the antibodies identified the purified Rv1819c transporter in vitro, in M. tuberculosis cell extracts and on the surface of the bacterium.
2. Material and Methods
Bioinformatics analyses. The published structures of Rv1819c were used in the analysis (PDB codes: 6TQF and 6TQE). The sequence was obtained from NCBI (https://www.ncbi.nlm.nih.gov/). The structure was used to identify the extracellular loops of transmembrane helices that extend to the membrane and are exposed to the periplasm (ECLs), identification of residues, cloning design, and experimental strategy. The transporter and the ECLs were analyzed in the PyMOL viewer (Schrondinger, LLC. The PyMOL Molecular Graphics System, Version 1.2.7). After the choice of the sequences, they were submitted to the IEDB server [10] for prediction of linear B cell epitopes. All results were analyzed together with data from the literature to design cloning strategies.
Cloning, protein expression and purification. The nucleotide sequences corresponding to the short and long ECLs of Rv1819c transporter were cloned into a phRAD vector to present the peptides of interest in the RAD-Display system, using the Ligase Independent Cloning (LIC) methodology [11] following the instructions described in Rossmann and collaborators [5]. The constructs were incubated on ice for 30 minutes and then transformed into chemocompetent E. coli DH5α cells. Cloning was confirmed by digestion of the vectors with HindIII and sequencing. The vectors were then transformed into chemocompetent E. coli BL21 (DE3) cells containing the pUBS24 plasmid that encodes a tRNA with a rare anticodon for Arginine present in the RadA sequence, allowing the expression of chimeric proteins. BL21 (DE3)/pUBS520 cells transformed with the vector constructed for the expression of RAD-peps proteins grown overnight were inoculated at a ratio of 1/100 in 500 mL of 2xYT medium supplemented with 100 μg/mL of ampicillin and 25 μg/mL of kanamycin at 37ºC under stirring at 200 rpm until reaching an optical density (OD 600 nm) of 0.8, when they were induced with IPTG (isopropyl β-D-1-thiogalactopyranoside) at a final concentration of 0.4 mM. Expression occurred for 3 hours at 37ºC under shaking at 200 rpm. Aliquots of the culture were collected before induction (T0) and after 3 hours of expression (T2), for analysis of expression on SDS-polyacrylamide gel. After expression, cells were recovered by centrifugation at 5520 g for 15 minutes. The pellet was resuspended in 25 mL of equilibrium buffer with 1mM PMSF, 1 mg/mL DNAse and 1 mg/mL lysozyme at the pH appropriate to the pI of each protein, followed by incubation at 4ºC in a homogenizer for 30 minutes. Cell lysis was performed by sonication for 10 minutes (15 second pulse, 20 second pause) and the soluble fraction of the culture was obtained by centrifugation of the cell lysate at 30,500 g for 40 min at 4ºC. Purification of the proteins was carried out in 50 mM Tris HCl, 150 NaCl using the nickel affinity chromatography technique with Ni Sepharose 6 Fast Flow resin (Cytiva Life Sciences). The elution was obtained after two washes of the column with 20 mM and 50 mM and then elution with 400 mM imidazole. After IMAC, samples were purified with size exclusion chromatography (SEC) using the Superdex 200 HiLoad 16/60 column in AKTA start system (GE Healthcare) in 50 mM Tris HCl, 150 NaCl. Proteins were concentrated and quantified by Bradford.
Immunization and antibody production. Polyclonal antibodies against the RAD-peps proteins and against the RAD protein without peptide insertion were produced in female C57BL/6 mice immunized subcutaneously with 3 doses and 15-day intervals between doses. The immunization was performed combining 10 μg of the target protein with 10 μg of Aluminum hydroxide (Alum) as an adjuvant in 100 µL of saline. 15 days after the third immunization, approximately 200 µL of blood was collected from each animal via submandibular vein puncture. Serum was obtained after incubating the collected blood for 30 minutes at room temperature and 30 minutes at 4ºC, followed by centrifugation at 425 g for 30 minutes. Serum was then collected, aliquoted and stored at -20ºC.
Enzyme-linked immunosorbent assay analysis of the sera samples. Nunc high-protein binding polystyrene plates (Thermo Fisher Scientific) were sensitized with RAD, RAD_Rv1819c_S and RAD_Rv1819c_L proteins (400 ng/well) diluted in carbonate buffer pH 9.6 (100 µL/well) for 14 hours at 4ºC. A cycle of 3 washes was then carried out with PBS buffer containing 0.05% Tween-20. Blocking was carried out for 3 hours at 37ºC with 300 µL/well of buffer (5% skimmed milk, 0.5% BSA in PBS containing 0.05% Tween-20) and after a washing cycle, sera containing antibodies relating to each RAD-pep protein in dilutions starting from 1:400 in 1X PBS buffer, 5% BSA. The plate was incubated for 1 h at 37ºC and then washed. Next, the secondary antibody Anti-Mouse IgG conjugated to peroxidase (Sigma-Aldrich) was added at a dilution of 1:5000. The plate was incubated for 1 h at 37ºC and after washing, 50 µL of TMB (3.3’, 5.5’-Tetramethylbenzidine) was added per well to react with peroxidase and reveal the binding of primary antibodies to their respective target antigen. The reaction was stopped by adding 50 µL/well of 0.2 N sulfuric acid. The optical density reading was performed at a wavelength of 450 nm. The last dilution value in which the OD450nm was greater than twice the value obtained in the blank was considered as the titer value.
Western blot assays.Growth conditions. M. bovis BCG Moreau, M. tuberculosis H27Ra cells (avirulent strains) and M. tuberculosis H37Rv were grown in 100 ml of 7H9 medium with 43 mM sodium pyruvate, supplemented with 10% OADC until they reached an OD600 of 0.4. To induce the Rv1819c production, cultures were treated with ½ MIC of Isoniazid (0.015 µg /ml) for 24 hours [12]. Cellular extract preparation. M. bovis BCG and M. tuberculosis H37Ra 50 mL cultures were centrifuged, resuspended in 5 mL of PBS, PMSF 1 mM and lysozyme 1 mg/mL [13,14], and homogenized for 15 minutes at 4ºC. Then, the cultures were sonicated for 10 minutes (10 second pulse, 20 second pause). The total cell lysates were centrifuged for 1 hour, 27.000 g before applying it to a 12%, 1 mm polyacrylamide gel. M. tuberculosis H37Rv culture was centrifuged and resuspended in 10 ml of PBS, 1% PMSF and 1 mg/mL lysozyme and fractionated into 10 2 ml tubes containing ice-cold 0.1 mm silica beads. The tubes were positioned in the cell disruptor Tissue Lyser LT (QIAGEN) and cell lysis was performed with 2 sets of 2.5 min with an oscillation of 40. The lysate was transferred to clean tubes and centrifuged at 15,000 rpm to obtain the soluble fraction, which was filtered through a 0.22 µm syringe filter. Membrane fractions were obtained according to Mot and Vanderleyden [15]. After centrifugation at 30.500 g for 1 hour at 4°C to eliminate the cell wall fraction, the supernatant was centrifuged at 100.000 g for 1 h. The supernatant was gently removed and the pellet, containing the membrane protein fraction, was taken with 1X PBS and centrifuged again at 100,000 g for 1 h. The supernatant was removed, and the pellet was homogenized in buffer containing 1% (w/v) sodium deoxycholate detergent for 1 hour at 15ºC. The solution was centrifuged again at 100.000 g for 1 h and the supernatant containing the solubilized proteins was gently collected and stored at -20ºC. Western blot. For analysis of antibody binding to mycobacterial protein extracts, 35 µg/well of soluble and membrane fractions were transferred from the SDS-PAGE gel to the PVDF membrane. The transfer was carried out on a PVDF membrane at 15 V for 30 minutes in a Trans-Blot SD semi-dry transfer cell (BioRad). They were then incubated for 1 hour at room temperature under agitation in blocking buffer (TBST, Gelatin 3%). After a cycle of three 5-minute washes (TBST), the membranes were incubated with serum containing specific primary antibodies at a 1:400 dilution in buffer (TBST, Gelatin 1%) for 4 hours at room temperature. Then, the membranes were submitted to a new washing cycle and incubated with the anti-mouse IgG secondary antibody conjugated to alkaline phosphatase (A4312 – Sigma-Aldrich) at a dilution of 1:10,000 for 1 h in buffer (TBST, Gelatin 1%). Three 5-minute washes were performed again (TBST) and the membranes were developed with BCIP/NBT. For analysis of antibody binding to the purified Rv1819c transporter, purified proteins and membrane fractions were transferred from the SDS-PAGE gel to nitrocellulose membranes at 25 V, 7 minutes in a Trans-blot Turbo (BioRad). The membranes were incubated overnight in blocking buffer (I-Block 0.2%, Thermo Fisher Scientific) at 4ºC. Each of the three membranes was incubated with a different primary antibody diluted in TBST, I-Block (0.1%): α-RAD_Rv1819c_L (I), dilution 1:375 (concentration 1.515 mg/mL) and α-RAD (II) (Negative control for Rv1819c recognition), dilution 1:400 (concentration 2.230 mg/mL) both using anti-mouse alkaline phosphatase as secondary antibody (A4312) at a dilution of 1:5000 and Anti-poly-his peroxidase antibody (III) (A7058), dilution 1:5000.
Purification of α-RAD_RV1819c_L and α-RAD antibodies. To purify InG antibodies from the sera obtained after immunization of mice with RAD and RAD_Rv1819c_L, the Serum Antibody Purification Kit (Protein G) (ABCAM) was used. 500 µL of animal serum was centrifuged for 10 min at 20.000 g at 4ºC. The sample was diluted to 5 ml in wash buffer with the addition of 500 µL of 10x binding buffer. The sample was decanted 4 times in a column containing 1 ml of protein G resin, which was then washed with 6 ml of washing buffer. Elutions were made with 6 fractions of 500 µL of elution buffer. Each elution was done in tubes containing 100 µL of neutralization buffer. Samples were analyzed on SDS-PAGE gel and on Nanodrop (ThermoScientific).
Expression and purification of the M. tuberculosis Rv1819c transporter.Protein expression. E. coli MC1061 cells containing the pBAD24_Rv1819c_cHis8 vector were inoculated into 8 L of LB medium at a ratio of 1:100 containing ampicillin. Expression was performed in 5 L finned Erlenmeyer flasks containing 2 L of LB each. The culture was incubated at 37ºC, 200 rpm until reaching an OD600nm of 0.8 and expression was induced with 0.02% (w/v) L-arabinose. Expression occurred for 3 h under the same conditions and cells were recovered by centrifugation at 6000 g, 15 min, 4ºC. The pellet was resuspended in buffer with 50 mM Kpi pH 7.5, 4ºC to wash the cells, centrifuged again and resuspended in the same buffer added with 20% glycerol in a total volume of 100 ml. The suspension was quickly frozen in liquid nitrogen and stored at -80ºC. Preparation of membrane vesicles of Rv1819c transporter. The cell suspension containing the Rv1819c transporter was thawed on ice and added with a protease inhibitor tablet (Protease Inhibitor Cocktail, Roche), 0.2 mM PMSF, 1 mM MgSO4 and 20 µg/mL DNAseI. For cell lysis, the cells were subjected twice to the cell disruptor (HPL6, Maximator) previously cooled to 4ºC and equilibrated with the same buffer. The soluble fraction was obtained from centrifugation of the lysate at 16,000 g (JA-17 Fixed-Angle Rotor), 40 min, 4oC. To obtain the membrane fraction, the supernatant was subjected to ultracentrifugation at 40.000 rpm for 2.5 hours at 4ºC with the 45 TI rotor. The membrane fraction was resuspended in 50 mM Kpi buffer pH 7.5, 10% glycerol, 1 mM DTT with a “potter” homogenizer. The membranes were frozen in liquid nitrogen and stored at -80ºC. Purification of the ABC transporter Rv1819c. Purification of the transporter in 50 mM Tris pH 8.0. 300 mM NaCl, 15 mM Imidazole, 1% DDM/CHS, 10% Glycerol, 1 mM DTT was carried out using the nickel affinity chromatography technique with Ni Sepharose 6 Fast Flow resin (Cytiva Life Sciences) using 50 mM Tris pH 8.0. 300 mM NaCl, 15 mM Imidazole, 10% Glycerol, 1 mM DTT as equilibrium buffer, 50 mM Tris pH 8.0. 300 mM NaCl, 60 mM Imidazole, 0.05% DDM/CHS, 1 mM DTT as wash buffer and 50 mM Tris pH 8.0. 300 mM NaCl, 350 mM Imidazole, 0.05% DDM/CHS, 1 mM DTT as elution buffer. After IMAC, a SEC using the Superdex 200 Increase 10/300 column (Cytiva Life Sciences) in a Bio-Rad system was performed in buffer 25 mM Tris pH 8.0. 200 mM NaCl, 0.05% DDM/CHS, 1 mM DTT. A tube (500 µl) of membrane preparation was thawed on ice, solubilized in 10 ml of solubilization buffer and incubated for 1 hour at 4ºC under gentle agitation. The membrane solubilized in detergent was subjected to ultracentrifugation at 80,000 rpm for 25 min at 4ºC in the MLA80 rotor. Then, the supernatant was incubated with 1 ml of Ni-Sepharose 6 Fast Flow resin (50% v/v) in equilibration buffer for 1 hour at 4ºC under gentle agitation. Due to the DTT contained in the equilibration buffer, the resin must be pre-equilibrated with wash buffer. After incubation, the sample was decanted into a chromatography column. The resin is then washed with 20 CV of wash buffer. Elution is carried out first with 450 µl of buffer and then with 800 µl. The fractions were analyzed in Nanodrop (Thermo Fisher Scientific), centrifuged at 20.000 g, 5 min at 4ºC to remove aggregated proteins and then subjected to SEC on the column Superdex 200 increase 10/300 GL column equilibrated with buffer. The fractions referring to the chromatogram peaks were analyzed using the Nanodrop (Themo Fisher Scientific).
Statistical analysis. Statistical analyses were performed wih Graphpad Prism version 6. Specific statistical tests employed are addressed in figure legends.
3. Results
Choice of the extracellular loops of the ABC transporter and production of the RAD-Display proteins
The ABC transporter Rv1819c is a homodimer, and its three-dimensional structure was resolved in the absence and the presence of AMP-PNP (PDB codes 6TQE and 6TQF, respectively) [9]. We carefully evaluated the TDM extracellular regions of the published models and discovered that in both conformations of the transporter, the TMDs show prominent extracellular regions that might be used as antigens for production of antibodies against the transporter. A short variant of this region, consisting of residues Gly105 to Lys114 (Rv1819c_S), covers the very tip of the loop while the longer variant, residues Gln100 to Arg119 (Rv1819c_L), includes all the non-membrane embedded part of the loop and part of two helices (Figure 1).
According to the B cell epitope prediction with IEDB server [16], despite the long peptide is just an extension of the short one, the presence of residues downstream and upstream of the shorter sequence has a modulatory effect on immunogenicity since Rv1819c_L is more immunogenic than Rv1819c_S (Figure 1C, yellow area in the IEDB plot). After selection, the corresponding DNA oligonucleotides were designed (Table S1, Supplementary material) and cloned into the phRAD vector using the ligase-independent cloning (LIC) as described before [5]. Sequence-verified constructs were used to transform E. coli BL21(DE3)/pUBS520 cells for heterologous expression. Both proteins, named RAD_Rv1819c_S and RAD_Rv1819c_L were identified in the induced extracts from E. coli (Figure S1B, Supplementary material) with the corresponding expected molecular mass of 28.1 and 28.6 kDa, respectively. The empty RAD scaffold was used as positive control, resulting in expected protein of 26 kDa. The proteins were purified by immobilized nickel affinity chromatography (Ni-IMAC) followed by size-exclusion chromatography (SEC) and then, concentrated to 1 mg/mL and aliquoted in tubes of 1 mg/ml (Figure S1C and S1D, Supplementary material).
RAD-display proteins were capable to induce the antibodies’ production in C57BL/6 mice
After immunization of C57BL/6 mice with three inoculations of RAD, RAD_Rv1819c_S and RAD_Rv1819c_L proteins, the titer of the antibodies was evaluated by ELISA assays using the purified proteins immobilized on the plate. Figure 2 shows the OD450 of the antibodies in a serial dilution. The curves show the signal of the antibodies against RAD (dotted lines) and against the constructs RAD_Rv1819c_S and RAD_Rv1819c_L (solid lines) for each mouse. RAD-display with the short and long peptides induced higher level of antibodies than RAD, the results are more significant for RAD-Rv1819c_L, confirming the B cell immunogenicity predicted by the IEDB server. In addition, it is possible to see that a-RAD_Rv1819c_L responds differently for RAD and RAD_Rv1819c (Figure 2D).
Recognition of Rv1819c transporter in extract cells of M. bovis BCG Moreau and M.tuberculosis H37Ra by the produced antibodies
To evaluate if the antibodies produced in mice against the short and long peptides of the M. tuberculosis Rv1819c were able to identify the transporter in cellular extracts, we performed Western blotting assays (Figure 3). Two different strains, M. bovis BCG Moreau and M. tuberculosis H37Ra, were used in the first analysis. They share identical sequences for the selected M. tuberculosis Rv1819c peptides. Cultures of the cells were performed in 7H9 medium with 0.05% Tween 80 and 10% OADC in the absence and the presence of isoniazid, a drug that induces the positive regulation of the Rv1819c transporter [12].
α-RAD antibodies recognize RAD-display protein in extracts of induced E. coli expressing RAD_Rv1819c_S and RAD_Rv1819c_L and all the purified proteins. The antibodies generated against RAD_Rv1819c_S recognized a protein with molecular mass around 70 kDa (the transporter has 71.2 kDa) in the soluble fractions of M. bovis BCG and M. tuberculosis obtained with and without isoniazid. A weak band is also observed in the membrane fraction of H37Ra treated with isoniazid (Figure 3D). Similar results were obtained with the α-RAD_Rv1819c_L (Figure 3E) that recognized a band with the same molecular weight. Again, we note that in the membrane fractions, a weak band with expected molecular mass was recognized. The pre-immune serum did not recognize any of the samples (Figure S2, Supplementary material).
Recognition of the Rv1819c transporter by antibodies α-RAD_Rv1819c_L in extracts and cells of M. tuberculosis H37Rv
The evaluation of the recognition of the Rv1819c transporter in extracts of M. tuberculosis H37Rv by α-RAD_Rv1819c_2L antibodies was first performed using western blotting assays (Figure 4). M. tuberculosis H37Rv cells were cultivated in 7H9 medium with 0.05% Tween 80 and 10% OADC in the presence and absence of isoniazid (INH). The α-RAD_Rv1819c_L serum was effective in recognizing the purified RAD and RAD_Rv1819c_L proteins, as expected (Figure 4A, about 25 kDa). Also in this strain, the α-RAD_Rv1819c_L antibodies recognized a faint band with a molecular mass corresponding to that of the transporter in the samples collected from cultures with INH (Figure 4A). Additionally, we evaluated the interaction of α-RAD_Rv1819c_L by flow cytometry using M. tuberculosis H37Rv cells exposed to INH and grown in the presence of Tween 80 (Figure 5). The analysis was based, firstly, on the identification of populations in terms of physical properties, that is, size (FSC-A) and granularity (SSC-A), and determination of the gates (Figure S3, Supplementary material). After sample preparation and reading on the instrument, the median fluorescence intensity (MFI) measured for the binding of α-RAD_Rv1819c_L antibodies to cells was significantly higher than the MFI of α-RADs (Figure 5A) in the three dilutions tested (1 :20, 1:40 and 1:80). Additionally, the fluorescence of the FITC (fluorescein isothiocyanate) fluorophore corresponding to the binding activity of mouse sera to cells is shown in Figure 5B. Histograms show the results of mice inoculated with RAD_Rv1819c_L (gray), RAD (purple), as well as serum from pre-immunized mice (black).
Recognition of the purified M. tuberculosis Rv1819c by the α-RAD_Rv1819c_L
In addition to the western blotting and the flow cytometry analysis that suggested the recognition of Rv1819c by α-RAD_Rv1819c_L, we tested the antibodies against the purified Rv1819c transporter. For expression of the transporter, E. coli MC1061 cells containing the vector pBAD24_Rv1819c_cHis were cultivated in LB medium, and the transporter was purified by Ni-IMAC and SEC, according to Nijland and collaborators [9] (Figure S4, Supplementary material).
Three eluted fractions of the transporter were obtained after Ni-IMAC with yields of 0.12 mg/mL, 0.64 mg/mL and 0.08 mg/mL, respectively. Fraction 2 was used in SEC resulting in a single peak eluting with 10.5 mL column volume (CV), which probably indicates elution of the transporter as a dimer (147.4 kDa) (Figure S4A, Supplementary material). Fractions eluted from SEC (Figure S4A, Supplementary material, red trace) were collected and quantified as 0.19 mg/mL of Rv1819c-his. Western blotting analysis using a commercial anti-poly-His antibody with fractions from all purification steps confirmed the presence of the purified Rv1819c_His construct (Figure S4B, Supplementary material).
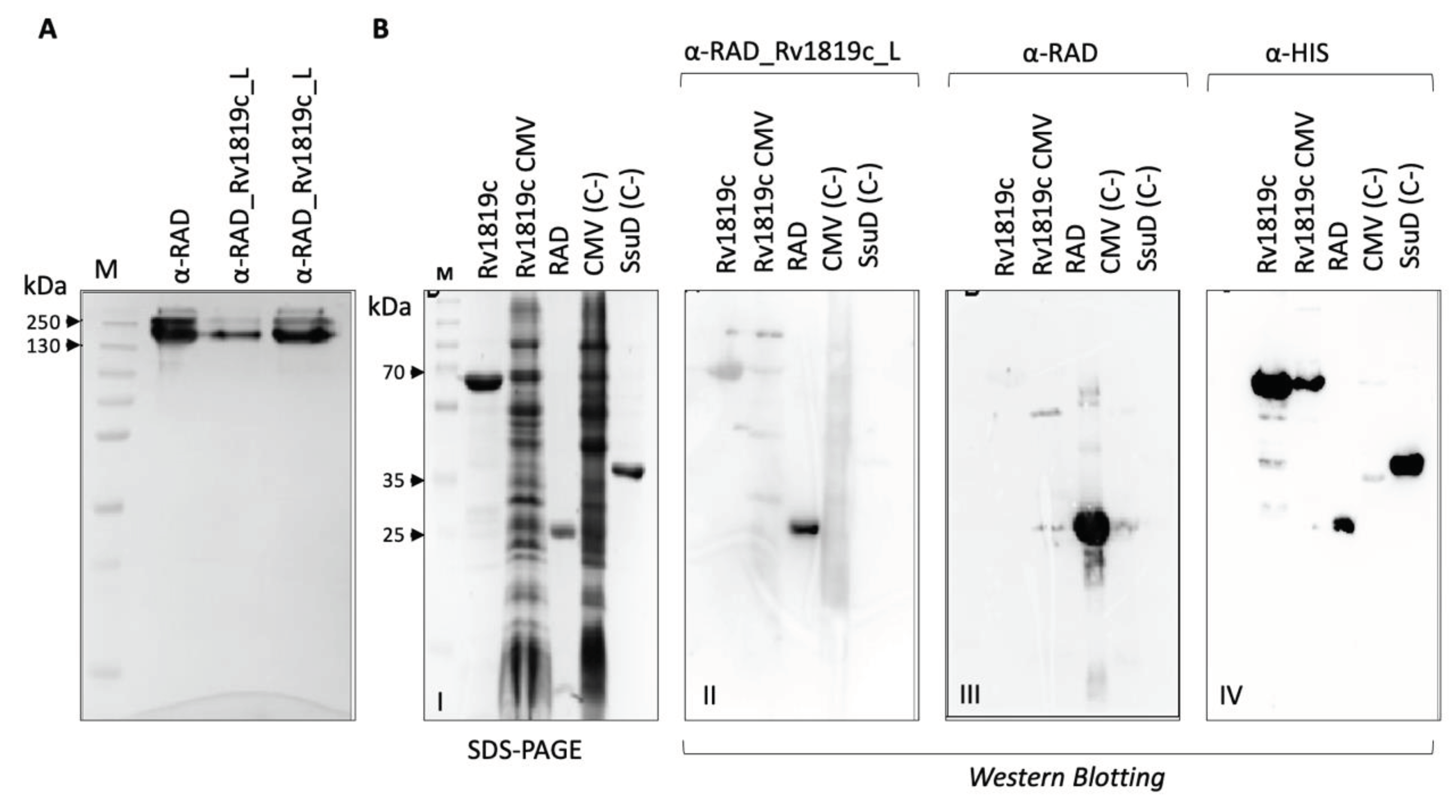
To carry out the assays with the ABC transporter Rv1819c, the IgG fraction of the sera was purified using a commercial kit with a protein G column. After optimizing the purification, pure samples of IgG antibodies (150 kDa) were obtained from the α-RAD (one aliquot of 2.230 mg/ml) and α-RAD_Rv1819c_L (two aliquots of 0.425 mg/ml and 1.515mg/ml, respectively) (Figure 6A). To evaluate the recognition of the Rv1819c protein by the α-RAD_Rv1819c-L, western blotting assays were performed with α-RAD_Rv1819c_L, α-RAD and α-His. Antibodies were tested for recognition of the purified full-length transporter Rv1819c_His, the membrane fraction from E. coli MC1061 cells used to express the transporter, the epitope-free RAD-display protein, the membrane fraction from E. coli MC1061 cells not induced (negative control for α-RAD-Rv1819c_2L) and an unrelated purified protein (SsuD_his) containing a 8-His tag (negative control for α-RAD_Rv1819c_L) (Figure 6B-I to IV).
The α-RAD_Rv1819c_L recognizes a band of size corresponding to the Rv1819c transporter (approximately 73.7 kDa) in the purified and membrane fraction, indicating recognition of the target protein (Figure 6B-II). Antibodies raised against RAD-display strongly recognized the purified RAD protein (approximately 26 kDa) (Figure 6B-III). The results obtained with the α-His antibody show that all proteins are identified (Figure 6B-IV), as expected, and highlight the recognition of the transporter in the purified fraction. The results so far suggest that the α-RAD_Rv1819c_L antibodies recognize the transporter, which validates the strategy proposed in the work.
4. Discussion
Antibodies are an interesting resource for studies of membrane proteins such as identification, exploring conformational changes, transport inhibition and localization [2,17]. In structural biology, antibodies can be used to increase the size of the targets, facilitating the reconstruction of the 3D structure in cryo-EM studies [18,19,20], expanding the surface area available for the formation of crystal contacts in protein crystallization or still ensuring less flexibility and heterogeneity of samples for the formation of crystals [1,21,22].
In clinical practice, detecting membrane proteins, is crucial as it helps identify mechanisms potentially linked to distinct diseases and increased drug resistance [23,24]. Efflux pumps are one of the most important drug resistance mechanisms involved in cancer treatment and have been considered important targets for modulation [25]. Similarly, efflux pumps are upregulated in bacteria during treatment with antibiotics and could become candidate tools to treat infectious diseases [26,27]. However, the use of other targets like the transmembrane permeases for these means is sparsely explored, primarily due to the complications in their production, high hydrophobicity of the TM segments necessitating the use of detergents and uncertain immunogenicity of these proteins [28,29].
In this work, aiming to avoid the problems of membrane protein expression and purification, we present the RAD display as a quick and effective alternative for expressing small epitopes from membrane proteins as antigens for antibody production. As a model, we used a well characterized ABC transporter for vitamin B12 from M. tuberculosis, Rv1819c. Rv1819c is needed for the maintenance of chronic infection in mice, mediates the uptake of antimicrobial peptides and other toxins, and is also essential for the import of vitamin B12 (cobalamin) in M. tuberculosis [8,9,30]. Once its three-dimensional structure is available in different conformations [9], we could explore extracellular regions of permeases exposed to the periplasm. The choice of these specific regions was based on their localization (outside the membrane) and in the possibility that antibodies that target those regions could inhibit the transport. The constructs with the two potential immunogenic epitopes (short and long) were expressed in a soluble form and hence easily purified using Ni-IMAC followed by SEC.
Via in silico analysis the long epitope of Rv1819 TMD was predicted by IEDB to be more immunogenic than the short one and the ELISA results confirmed the superiority of a longer epitope. Fortunately, the RAD-display system easily tolerates the insertion of long peptides, without much restriction on their folding due to the flexibility of the system. In addition, our results confirm that the IEDB can be reliable tool for preliminary analysis of peptides, with greater agility and speed in the screening process for possible antigens of membrane proteins.
Interestingly, although the antibody titers produced against the short peptide were not as abundant as for the long peptide, when we take into account the recognition of the RAD scaffold, these antibodies also recognized the transporter in soluble fractions of M. bovis BCG and M. tuberculosis H37Ra suggesting both epitopes were able to induce antibodies’ production. Rv1819c shares 99.7% and 100% of sequence identity with its orthologs from M. bovis and M. tuberculosis H37Ra, respectively, and the sequences chose were identical. The Rv1819c corresponding gene in these lineages is assigned as probable drug transporter. The induction of the transporter from M. bovis has been shown in isolates from different animals that show resistance not only to isoniazid but also to other first and second line antibiotics [31]. However, it was not detected in the membrane fractions, where we anticipated stronger recognition. It is possible that the protocol used and treatment of cells to obtain membrane fractions was more harmful. Otherwise, in M. tuberculosis H37Rv, the results show that INH has an effect in western blot assays and in flow cytometry. In flow cytometry, we observed that α-RAD antibodies have nonspecific interactions with the M. tuberculosis cells, but not with the Xanthomonas citri cells. Even so, the MFI observed for the α-RAD_Rv1819c_L antibodies in comparison with the control cells and exposed to α-RAD was significantly higher. Importantly, we must consider that we worked with a sample of polyclonal antibodies and that the target peptide corresponds to a small portion of the RAD display. In fact, it is not possible to quantify how much of peptide-specific antibodies are present in the sample. Therefore, the recognition of the purified Rv1819c corroborated the previous results and showed the strategy applied with RAD display can be a useful tool for screening of efflux pumps epitopes, with different perspectives.
5. Conclusions
The Rad display system was a useful, simple, and cheap tool for production of M. tuberculosis Rv1819c membrane protein epitopes and induction of specific antibodies. The system eliminated the need for expression and purification of full membrane proteins complex and can be used with different targets in parallel, which could open perspectives of applications in vaccine development, protein-protein interaction studies and diagnosis.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
SCT: Conceptualization, methodology, writing original draft preparation; MFCA: immunization, Flow cytometry experiments; AG: Experimental design, writing review and editing; LCSF: writing review and editing; AB: Conceptualization, supervision, writing review and editing All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Fundação de Amparo à Pesquisa de São Paulo (FAPESP), grant number 2018/20162-9, Conselho Nacional de Desenvolvimento Científico (CNPq), grant number 401505/2016-2. SCT received fellowship from Fundação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), grant number 88887.497909/2020-00 and Erasmus + International Credit Mobility scholarship.
Acknowledgments
We thank the members of the Applied Structural Biology Laboratory and the Laboratory of Development of Vaccines from the Institute of Biomedical Sciences for the technical support. We also thank Mark Nijland from the Groningen Biomolecular Sciences and Biotechnology Institute (GBB), University of Groningen for the help with the purification of the Rv1819c transporter.
Conflicts of Interest statement
The authors declare that they have no conflict of interest.
Ethics statement
To obtain the polyclonal antibodies to the proteins of interest, wild type (WT) C57BL/6 mice were purchased from the Faculty of Veterinary Medicine of the University of São Paulo. All procedures for maintenance and manipulation of mice were approved by the ethics committee for animal experimentation (protocol number CEUA 050/2014), following rules from the National Council for Control of Animal Experimentation (CONCEA).
References
- Zimmermann, I.; Egloff, P.; Hutter, C.A.; Arnold, F.M.; Stohler, P.; Bocquet, N.; Hug, M.N.; Huber, S.; Siegrist, M.; Hetemann, L.; et al. Synthetic Single Domain Antibodies for the Conformational Trapping of Membrane Proteins. Elife 2018, 7, e34317. [Google Scholar] [CrossRef]
- Thul, P.J.; Åkesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. A Subcellular Map of the Human Proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef]
- Zhang, H.; Huang, C.-S.; Yu, X.; Lee, J.; Vaish, A.; Chen, Q.; Zhou, M.; Wang, Z.; Min, X. Cryo-EM Structure of ABCG5/G8 in Complex with Modulating Antibodies. Commun Biol 2021, 4, 526. [Google Scholar] [CrossRef]
- Holcomb, J.; Spellmon, N.; Zhang, Y.; Doughan, M.; Li, C.; Yang, Z. Protein Crystallization: Eluding the Bottleneck of X-Ray Crystallography. AIMS Biophys 2017, 4, 557–575. [Google Scholar] [CrossRef]
- Rossmann, M.; J Greive, S.; Moschetti, T.; Dinan, M.; Hyvönen, M. Development of a Multipurpose Scaffold for the Display of Peptide Loops. Protein Eng Des Sel 2017, 30, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Guerra, G.S. Desenvolvimento de uma estratégia de inibição de transportadores ABC utlizando anticorpos; University of Sao Paulo: Sao Paulo.
- Cioffi, V.B.; De Castro-Amarante, M.F.; Lulla, A.; Andreata-Santos, R.; Cruz, M.C.; Moreno, A.C.R.; De Oliveira Silva, M.; De Miranda Peres, B.; De Freitas Junior, L.H.G.; Moraes, C.B.; et al. SARS-CoV-2 Spike Protein Peptides Displayed in the Pyrococcus Furiosus RAD System Preserve Epitopes Antigenicity, Immunogenicity, and Virus-Neutralizing Activity of Antibodies. Sci Rep 2023, 13, 16821. [Google Scholar] [CrossRef] [PubMed]
- Gopinath, K.; Venclovas, C.; Ioerger, T.R.; Sacchettini, J.C.; McKinney, J.D.; Mizrahi, V.; Warner, D.F. A Vitamin B₁₂ Transporter in Mycobacterium Tuberculosis. Open Biol 2013, 3, 120175. [Google Scholar] [CrossRef] [PubMed]
- Rempel, S.; Gati, C.; Nijland, M.; Thangaratnarajah, C.; Karyolaimos, A.; de Gier, J.W.; Guskov, A.; Slotboom, D.J. A Mycobacterial ABC Transporter Mediates the Uptake of Hydrophilic Compounds. Nature 2020, 580, 409–412. [Google Scholar] [CrossRef]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 Update. Nucleic Acids Res 2019, 47, D339–D343. [Google Scholar] [CrossRef]
- Aslanidis, C.; de Jong, P.J. Ligation-Independent Cloning of PCR Products (LIC-PCR). Nucleic Acids Res 1990, 18, 6069–6074. [Google Scholar] [CrossRef]
- Garima, K.; Pathak, R.; Tandon, R.; Rathor, N.; Sinha, R.; Bose, M.; Varma-Basil, M. Differential Expression of Efflux Pump Genes of Mycobacterium Tuberculosis in Response to Varied Subinhibitory Concentrations of Antituberculosis Agents. Tuberculosis (Edinb) 2015, 95, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Rezwan, M.; Lanéelle, M.-A.; Sander, P.; Daffé, M. Breaking down the Wall: Fractionation of Mycobacteria. J Microbiol Methods 2007, 68, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.; Li, H.; Ma, B.; Weiss, R.; Bendayan, D.; Abramovitz, L.; Ben-Shalom, N.; Mor, M.; Pinko, E.; Bar Oz, M.; et al. Human Antibodies Targeting a Mycobacterium Transporter Protein Mediate Protection against Tuberculosis. Nat Commun 2021, 12, 602. [Google Scholar] [CrossRef]
- Mot, R.D.; Vanderleyden, J. Application of Two-Dimensional Protein Analysis for Strain Fingerprinting and Mutant Analysis of Azospirillum Species. Can. J. Microbiol. 1989, 35, 960–967. [Google Scholar] [CrossRef]
- Ponomarenko, J.V.; Bourne, P.E. Antibody-Protein Interactions: Benchmark Datasets and Prediction Tools Evaluation. BMC Struct Biol 2007, 7, 64. [Google Scholar] [CrossRef] [PubMed]
- Bordignon, E.; Seeger, M.A.; Galazzo, L.; Meier, G. From in Vitro towards in Situ: Structure-Based Investigation of ABC Exporters by Electron Paramagnetic Resonance Spectroscopy. FEBS Lett 2020, 594, 3839–3856. [Google Scholar] [CrossRef]
- Wu, S.; Avila-Sakar, A.; Kim, J.; Booth, D.S.; Greenberg, C.H.; Rossi, A.; Liao, M.; Li, X.; Alian, A.; Griner, S.L.; et al. Fabs Enable Single Particle cryoEM Studies of Small Proteins. Structure 2012, 20, 582–592. [Google Scholar] [CrossRef]
- Bailey, L.J.; Sheehy, K.M.; Dominik, P.K.; Liang, W.G.; Rui, H.; Clark, M.; Jaskolowski, M.; Kim, Y.; Deneka, D.; Tang, W.-J.; et al. Locking the Elbow: Improved Antibody Fab Fragments as Chaperones for Structure Determination. Journal of Molecular Biology 2018, 430, 337–347. [Google Scholar] [CrossRef]
- Rizk, S.S.; Kouadio, J.-L.K.; Szymborska, A.; Duguid, E.M.; Mukherjee, S.; Zheng, J.; Clevenger, C.V.; Kossiakoff, A.A. Engineering Synthetic Antibody Binders for Allosteric Inhibition of Prolactin Receptor Signaling. Cell Commun Signal 2015, 13, 1. [Google Scholar] [CrossRef]
- Kermani, A.A. A Guide to Membrane Protein X-Ray Crystallography. FEBS J 2021, 288, 5788–5804. [Google Scholar] [CrossRef]
- Hutchings, C.J.; Colussi, P.; Clark, T.G. Ion Channels as Therapeutic Antibody Targets. MAbs 2018, 11, 265–296. [Google Scholar] [CrossRef]
- Zamek-Gliszczynski, M.J.; Sangha, V.; Shen, H.; Feng, B.; Wittwer, M.B.; Varma, M.V.S.; Liang, X.; Sugiyama, Y.; Zhang, L.; Bendayan, R.; et al. Transporters in Drug Development: International Transporter Consortium Update on Emerging Transporters of Clinical Importance. Clin Pharmacol Ther 2022, 112, 485–500. [Google Scholar] [CrossRef]
- Liu, X. Overview: Role of Drug Transporters in Drug Disposition and Its Clinical Significance. Adv Exp Med Biol 2019, 1141, 1–12. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A View on Drug Resistance in Cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef]
- Choi, Y.H.; Yu, A.-M. ABC Transporters in Multidrug Resistance and Pharmacokinetics, and Strategies for Drug Development. Curr Pharm Des 2014, 20, 793–807. [Google Scholar] [CrossRef]
- Yadav, D.; Rao, G.S.N.K.; Paliwal, D.; Singh, A.; Shadab, S. Insight into the Basic Mechanisms and Various Modulation Strategies Involved in Cancer Drug Resistance. Curr Cancer Drug Targets 2023, 23, 778–791. [Google Scholar] [CrossRef]
- Pandey, A.; Shin, K.; Patterson, R.E.; Liu, X.-Q.; Rainey, J.K. Current Strategies for Protein Production and Purification Enabling Membrane Protein Structural Biology. Biochem Cell Biol 2016, 94, 507–527. [Google Scholar] [CrossRef]
- Birch, J.; Quigley, A. The High-Throughput Production of Membrane Proteins. Emerg Top Life Sci 2021, 5, 655–663. [Google Scholar] [CrossRef]
- Domenech, P.; Kobayashi, H.; LeVier, K.; Walker, G.C.; Barry, C.E. BacA, an ABC Transporter Involved in Maintenance of Chronic Murine Infections with Mycobacterium Tuberculosis. J Bacteriol 2009, 191, 477–485. [Google Scholar] [CrossRef]
- De Rossi, E.; Aínsa, J.A.; Riccardi, G. Role of Mycobacterial Efflux Transporters in Drug Resistance: An Unresolved Question. FEMS Microbiol Rev 2006, 30, 36–52. [Google Scholar] [CrossRef]
Figure 1.
Rv1819c extracellular loops chosen for display in RAD system. A Homodimeric three-dimensional structure of Rv1819c (Rempel et al, 2020) in gray cartoon with the highlighted extracellular regions in the TMDs (magenta). P: periplasmic region; IM: inner membrane. Two sequences of amino acids, short (B) and long (C) were chosen and submitted to the Bepipred Linear Epitope Prediction 2.0 program for prediction of B cell epitopes. The yellow peak corresponds to antigenic residues in each sequence according to Kolaskar and Tongaonkar (1990) antigenicity scale. The AlphaFold models of RAD_Rv1819c_S and RAD_Rv1819c_L are shown in yellow cartoon with the epitopes in magenta.
Figure 1.
Rv1819c extracellular loops chosen for display in RAD system. A Homodimeric three-dimensional structure of Rv1819c (Rempel et al, 2020) in gray cartoon with the highlighted extracellular regions in the TMDs (magenta). P: periplasmic region; IM: inner membrane. Two sequences of amino acids, short (B) and long (C) were chosen and submitted to the Bepipred Linear Epitope Prediction 2.0 program for prediction of B cell epitopes. The yellow peak corresponds to antigenic residues in each sequence according to Kolaskar and Tongaonkar (1990) antigenicity scale. The AlphaFold models of RAD_Rv1819c_S and RAD_Rv1819c_L are shown in yellow cartoon with the epitopes in magenta.
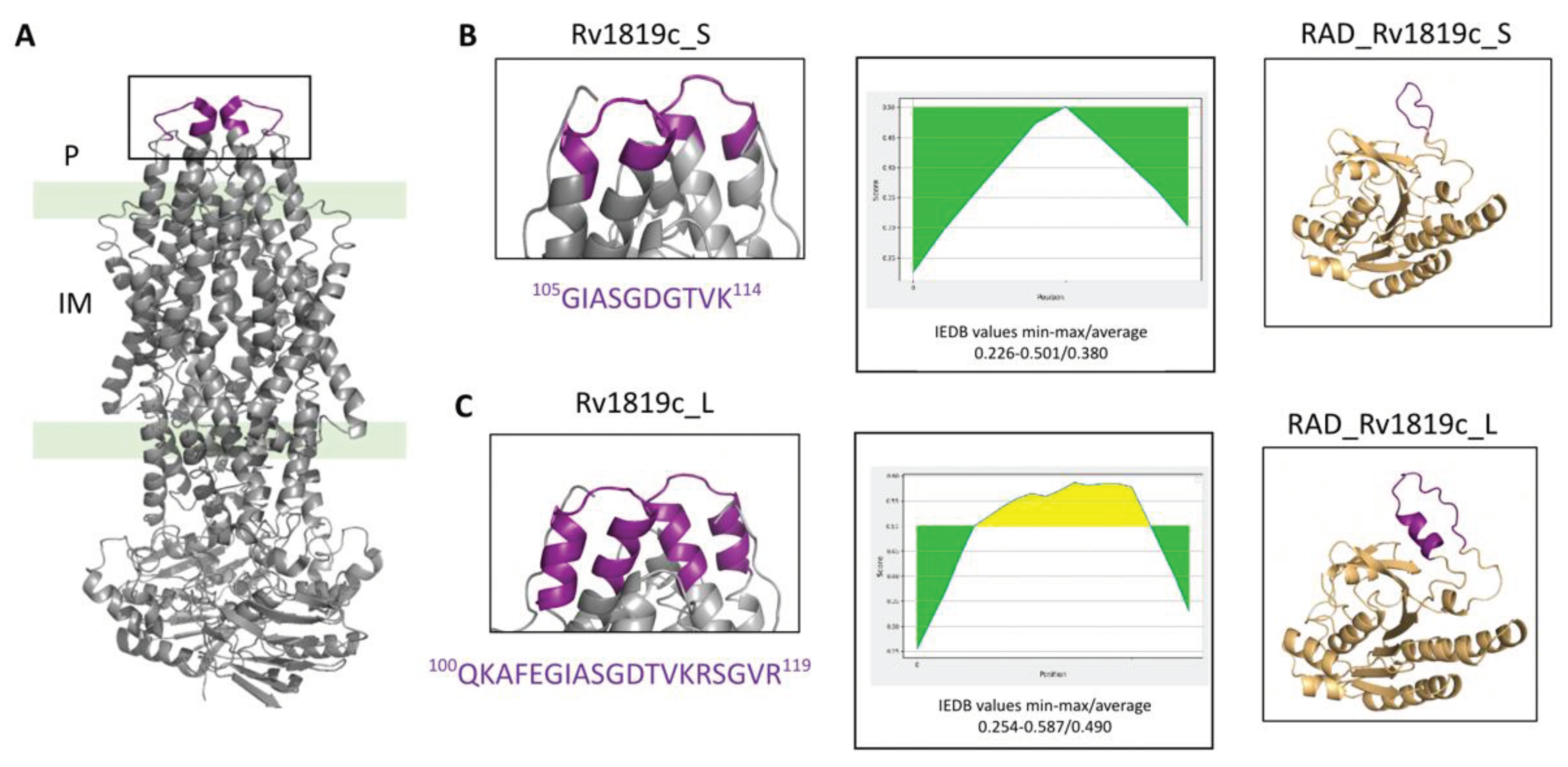
Figure 2.
Titration of the sera produced in C57BL/6 mice against RAD, RAD_ Rv1819c_S and RAD_ Rv1819c_L by ELISA. A Serum α-RAD against RAD-scaffold; B Serum α-RAD_Rv1819c_S against RAD (dotted lines) and RAD_Rv1819c_S (solid lines); C Serum α-RAD_Rv1819c_L against RAD (dotted lines) and RAD_Rv1819c_L (solid lines). D Detail of the results obtained with α-RAD_Rv1819c_L. The absorbance (450nm) of the sera produced against each protein was evaluated starting from a dilution of 1:800 to 1:51200. Plates were coated with 400 ng/well of the proteins and the sera used as primary antibodies. Anti-mouse IgG HRP antibody was used as a secondary antibody. The sera were obtained from the immunizations in three different mice. Solid line: Serum against RAD displaying the corresponding epitope.
Figure 2.
Titration of the sera produced in C57BL/6 mice against RAD, RAD_ Rv1819c_S and RAD_ Rv1819c_L by ELISA. A Serum α-RAD against RAD-scaffold; B Serum α-RAD_Rv1819c_S against RAD (dotted lines) and RAD_Rv1819c_S (solid lines); C Serum α-RAD_Rv1819c_L against RAD (dotted lines) and RAD_Rv1819c_L (solid lines). D Detail of the results obtained with α-RAD_Rv1819c_L. The absorbance (450nm) of the sera produced against each protein was evaluated starting from a dilution of 1:800 to 1:51200. Plates were coated with 400 ng/well of the proteins and the sera used as primary antibodies. Anti-mouse IgG HRP antibody was used as a secondary antibody. The sera were obtained from the immunizations in three different mice. Solid line: Serum against RAD displaying the corresponding epitope.
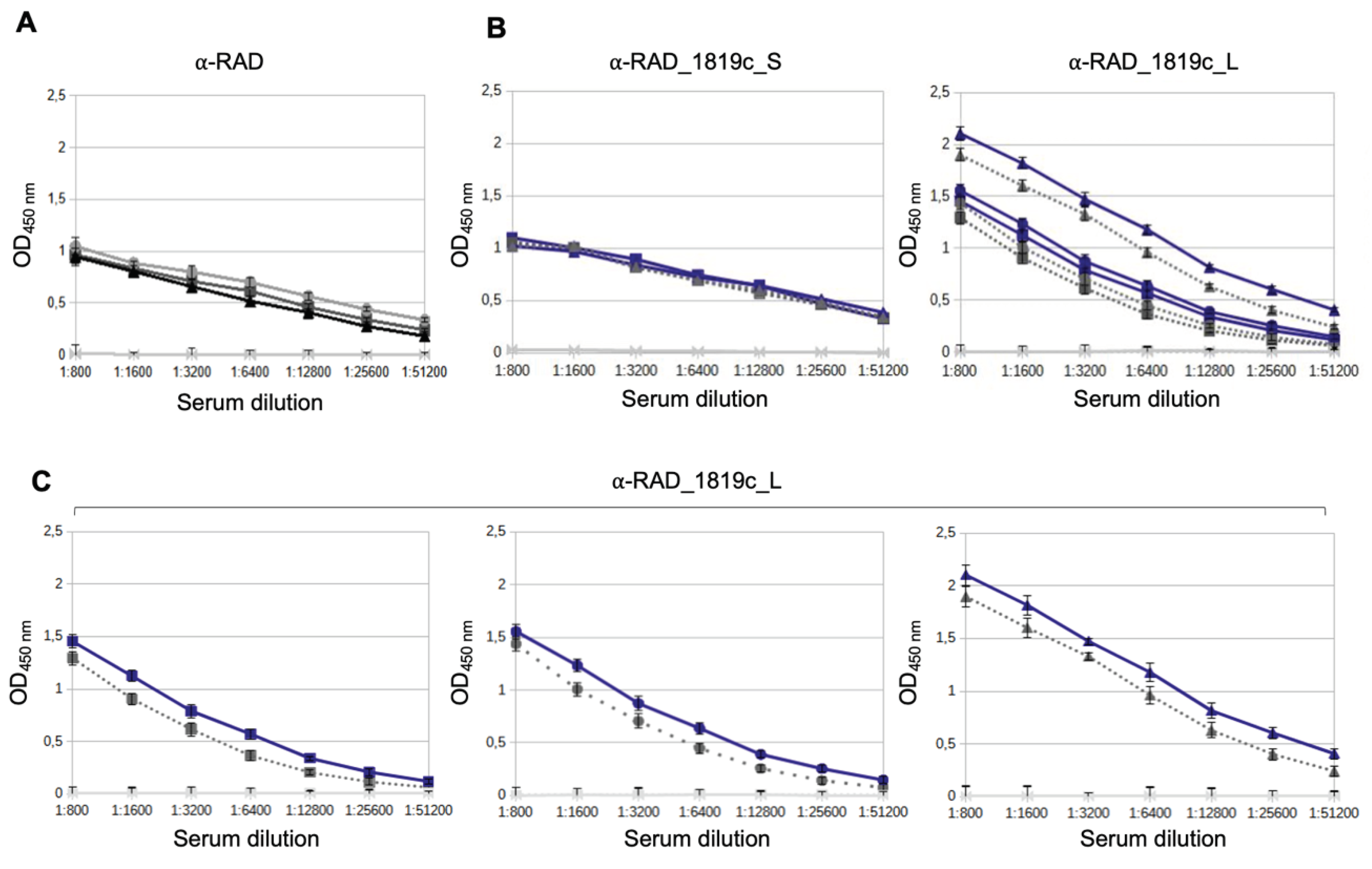
Figure 3.
Western blotting for detection of the Rv1819c transporter in extracts of M. bovis BCG Moreau and M. tuberculosis H37Ra by α-RAD_Rv1819c_S and α-RAD_Rv1819c_L. Western blotting of the samples with α-RAD (A), α-RAD_Rv1819c_S (B) and α-RAD_Rv1819c_L (C). M: molecular weight marker; T0: Lysate of non-induced E. coli cells; T2: Lysate of induced E. coli cells expressing indicated proteins; RAD, RAD_Rv1819c_S or RAD_Rv1819c_L: purified proteins used as antigens; BCG: M. bovis BCG Moreau; H37Ra: M. tuberculosis H37Ra; S: soluble fraction of the cells; M: Fraction of cellular membrane; INH: isoniazid.
Figure 3.
Western blotting for detection of the Rv1819c transporter in extracts of M. bovis BCG Moreau and M. tuberculosis H37Ra by α-RAD_Rv1819c_S and α-RAD_Rv1819c_L. Western blotting of the samples with α-RAD (A), α-RAD_Rv1819c_S (B) and α-RAD_Rv1819c_L (C). M: molecular weight marker; T0: Lysate of non-induced E. coli cells; T2: Lysate of induced E. coli cells expressing indicated proteins; RAD, RAD_Rv1819c_S or RAD_Rv1819c_L: purified proteins used as antigens; BCG: M. bovis BCG Moreau; H37Ra: M. tuberculosis H37Ra; S: soluble fraction of the cells; M: Fraction of cellular membrane; INH: isoniazid.
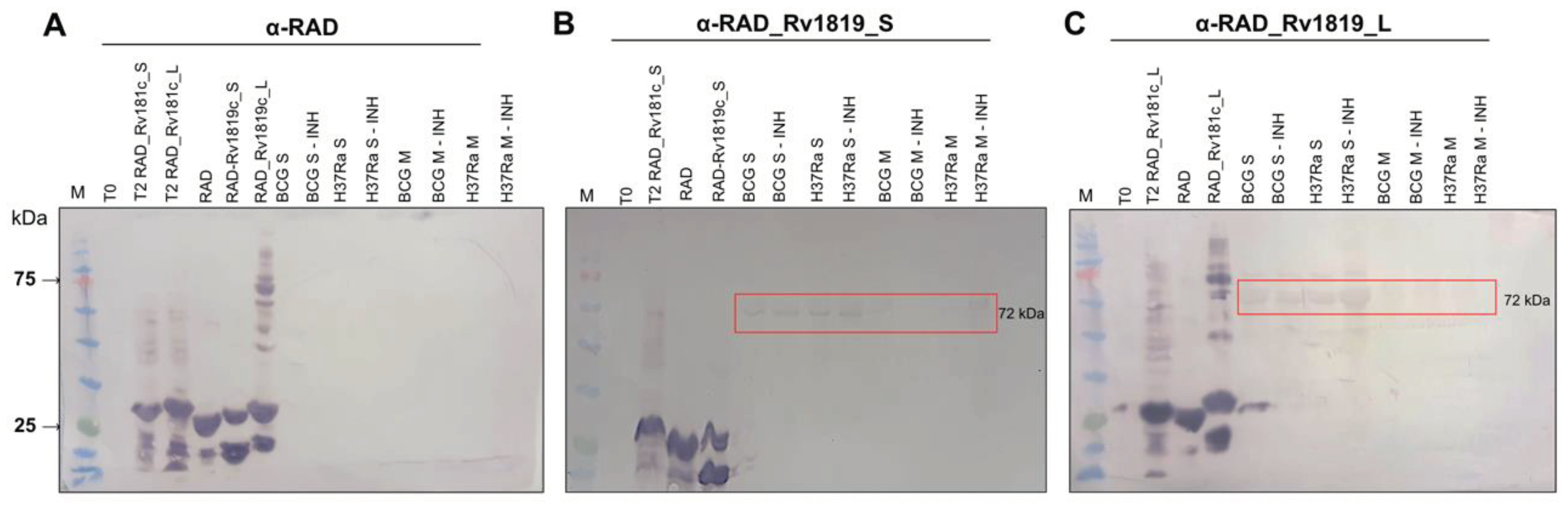
Figure 4.
Recognition of the ABC transporter Rv1819c in protein extracts of M. tuberculosis H37Rv. Western blotting assay for detection of Rv1819c in protein extracts of M. tuberculosis H37Rv. A 10% SDS-PAGE with purified RAD and RAD_Rv1819c_L proteins and extracts from M. tuberculosis cultivated in 7H9 broth with 10% OADC and 7H9 broth with 10% OADC added with 0.05 μg/ml Isoniazid (INH) for transporter induction. B PVDF membrane exposed to α-RAD_Rv1819c_L serum at a dilution of 1:200. The red box highlights the presence of a ~75 kDa band identified only in M. tuberculosis extract samples with INH.
Figure 4.
Recognition of the ABC transporter Rv1819c in protein extracts of M. tuberculosis H37Rv. Western blotting assay for detection of Rv1819c in protein extracts of M. tuberculosis H37Rv. A 10% SDS-PAGE with purified RAD and RAD_Rv1819c_L proteins and extracts from M. tuberculosis cultivated in 7H9 broth with 10% OADC and 7H9 broth with 10% OADC added with 0.05 μg/ml Isoniazid (INH) for transporter induction. B PVDF membrane exposed to α-RAD_Rv1819c_L serum at a dilution of 1:200. The red box highlights the presence of a ~75 kDa band identified only in M. tuberculosis extract samples with INH.
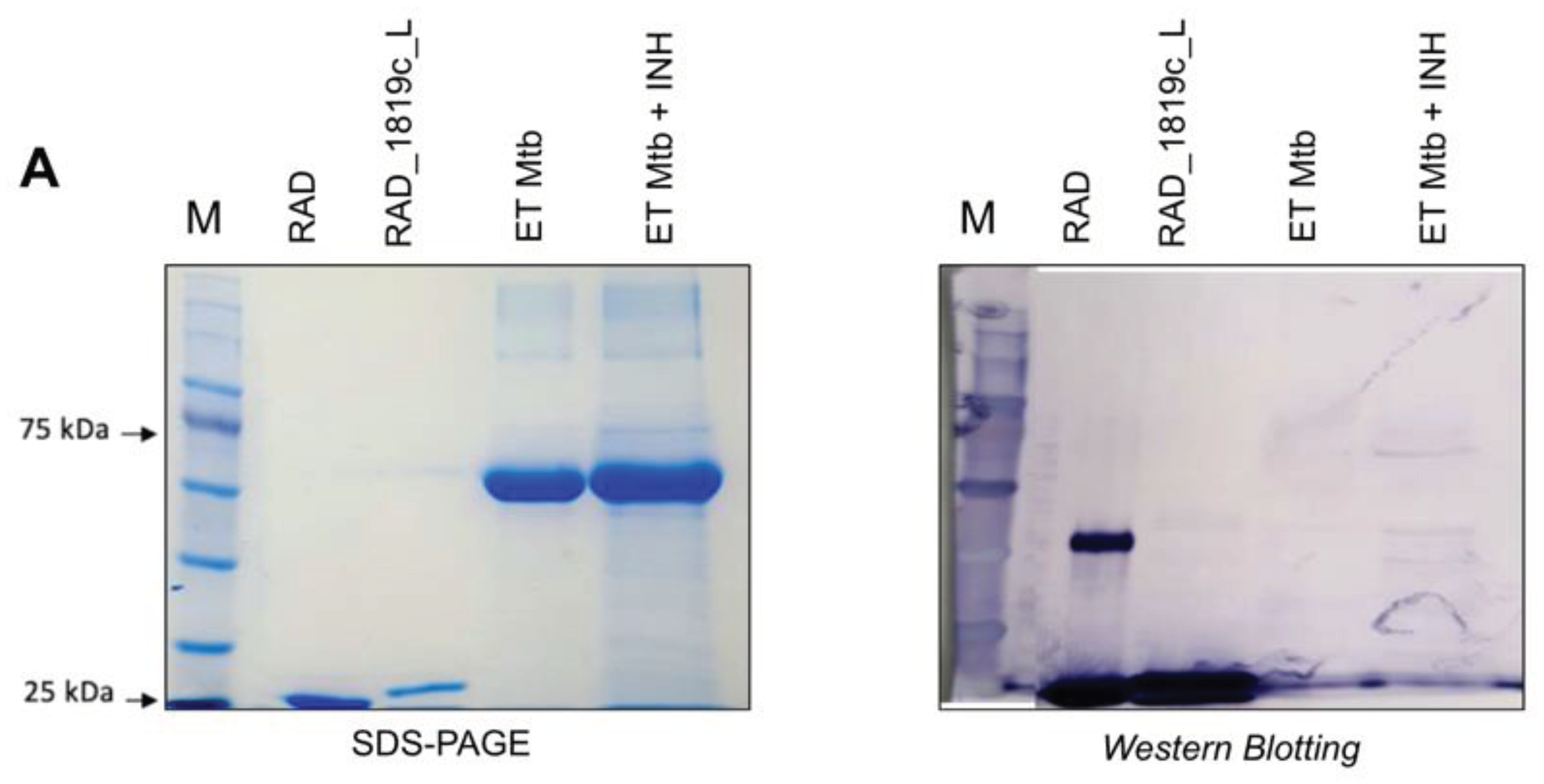
Figure 5.
Flow cytometry to evaluate the interaction of serum from mice inoculated with the RAD_Rv1819c_L (α−RAD_Rv1819c_L) protein to M. tuberculosis H37Rv cells. A Mean fluorescence intensity (MFI) values were calculated on the FITC SSC-A subset using FlowJo v.10.8.1. Both serum samples were tested at 1:20, 1:40 and 1:80 dilutions against cells expressing the Rv1819c transporter. Statistically significant differences were determined using two-tailed unpaired t-test, (*p = 0.0341, **p = 0.0011, ***p = 0002) in GraphPad Prism V. B Histograms showing fluorescence (FITC) corresponding to binding activity between cells and sera from mice inoculated with RAD_Rv1819c_L (gray), RAD (purple), as well as serum from pre-immunized mice (black). The figure also shows the fluorescence detected from cells without incubation with serum or Alexa Fluor 488-conjugated secondary antibody (blue) and from cells incubated with secondary antibody alone (orange). I: serum α-RAD_Rv1819_L and α-RAD dilution 1:20; II: serum α-RAD_Rv1819c_L and α-RAD dilution 1:40; III: Binding activity of α-RAD (serum dilution 1:20) to Xanthomonas citri (negative control for cells, green) and M. tuberculosis H37Rv (purple).
Figure 5.
Flow cytometry to evaluate the interaction of serum from mice inoculated with the RAD_Rv1819c_L (α−RAD_Rv1819c_L) protein to M. tuberculosis H37Rv cells. A Mean fluorescence intensity (MFI) values were calculated on the FITC SSC-A subset using FlowJo v.10.8.1. Both serum samples were tested at 1:20, 1:40 and 1:80 dilutions against cells expressing the Rv1819c transporter. Statistically significant differences were determined using two-tailed unpaired t-test, (*p = 0.0341, **p = 0.0011, ***p = 0002) in GraphPad Prism V. B Histograms showing fluorescence (FITC) corresponding to binding activity between cells and sera from mice inoculated with RAD_Rv1819c_L (gray), RAD (purple), as well as serum from pre-immunized mice (black). The figure also shows the fluorescence detected from cells without incubation with serum or Alexa Fluor 488-conjugated secondary antibody (blue) and from cells incubated with secondary antibody alone (orange). I: serum α-RAD_Rv1819_L and α-RAD dilution 1:20; II: serum α-RAD_Rv1819c_L and α-RAD dilution 1:40; III: Binding activity of α-RAD (serum dilution 1:20) to Xanthomonas citri (negative control for cells, green) and M. tuberculosis H37Rv (purple).
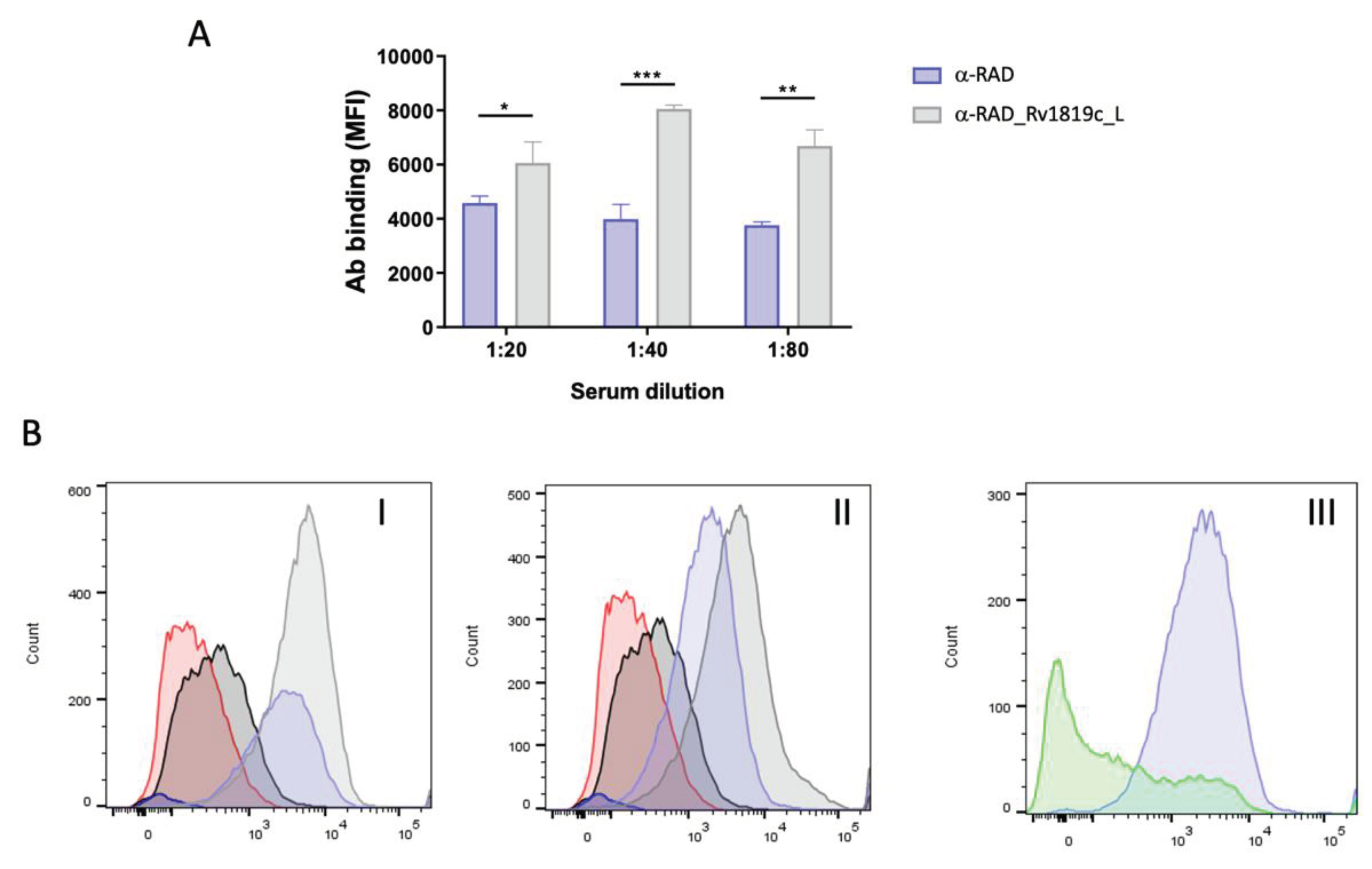
Figure 6.
Detection of the purified Rv1819c transporter by α-RAD_Rv1819c_L antibodies. A Purification of the IgG fraction of the sera produced. 12% polyacrylamide gel presenting samples of the IgG fraction (150 kDa) from purified α-RAD and α-RAD_Rv1819c_L sera. B Recognition of the Rv1819c transporter by Western blotting. I: 12% acrylamide gel electrophoresis (SDS-PAGE, mirror gel) containing samples of the purified complete Rv1819c-His transporter, membrane fraction of the E. coli MC1061 used to express the transporter, the epitope-free RAD protein, the membrane fraction from uninduced E. coli MC1061 cells (negative control for α-RAD_Rv1819c_L) and an unrelated purified protein (hSsuD) containing a tag with 8 histidine (negative control for α-RAD_Rv1819c_L). Samples of proteins and protein extracts were applied in equal quantities to three nitrocellulose membranes, each incubated with a different antibody. II: α-RAD_Rv1819c_L, dilution 1:375 (concentration 1.515 mg/mL) and III: α-RAD, dilution 1:400 (concentration 2.230 mg/mL) and anti-mouse alkaline phosphatase secondary antibody (A4312) at a dilution of 1:5000 (Negative control for Rv1819c recognition) and IV: α-His, dilution 1:5000 and anti-poly-his peroxidase secondary antibody (A7058).
Figure 6.
Detection of the purified Rv1819c transporter by α-RAD_Rv1819c_L antibodies. A Purification of the IgG fraction of the sera produced. 12% polyacrylamide gel presenting samples of the IgG fraction (150 kDa) from purified α-RAD and α-RAD_Rv1819c_L sera. B Recognition of the Rv1819c transporter by Western blotting. I: 12% acrylamide gel electrophoresis (SDS-PAGE, mirror gel) containing samples of the purified complete Rv1819c-His transporter, membrane fraction of the E. coli MC1061 used to express the transporter, the epitope-free RAD protein, the membrane fraction from uninduced E. coli MC1061 cells (negative control for α-RAD_Rv1819c_L) and an unrelated purified protein (hSsuD) containing a tag with 8 histidine (negative control for α-RAD_Rv1819c_L). Samples of proteins and protein extracts were applied in equal quantities to three nitrocellulose membranes, each incubated with a different antibody. II: α-RAD_Rv1819c_L, dilution 1:375 (concentration 1.515 mg/mL) and III: α-RAD, dilution 1:400 (concentration 2.230 mg/mL) and anti-mouse alkaline phosphatase secondary antibody (A4312) at a dilution of 1:5000 (Negative control for Rv1819c recognition) and IV: α-His, dilution 1:5000 and anti-poly-his peroxidase secondary antibody (A7058).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



