Submitted:
29 March 2024
Posted:
29 March 2024
You are already at the latest version
Abstract
Abstract: The study used whole-genome sequencing (WGS) and bioinformatics analysis for the genomic characterization of 60 isolates of Listeria monocytogenes isolated from cattle farms, cattle abattoirs, and retail outlets in Gauteng province, South Africa. The isolates' sequence types (STs), clonal complexes (CCs), and lineages were determined using in silico multilocus sequence typing (MLST). We used BLAST-based analyses to identify virulence and antimicrobial genes, plasmids, proviruses, and the CRISPR-Cas system. The study investigated any association of the detected genes to the origin in the beef production chain of the L. monocytogenes isolates. Overall, in 60 isolates of Listeria monocytogenes, there were 7 STs, 6 CCs, 44 putative virulence factors, 2 resistance genes, 1 plasmid with AMR genes and 3 with conjugative genes, 1 CRISPR gene, and all 60 isolates were positive for proviruses. Among the 7 STs detected, ST204 (46.7%) and ST2 (21.7%) were the most prominent, with ST frequency varying significantly (p<0.001). The predominant CC detected were CC2 (21.7%) and CC204 (46.7%) in lineages I and II, respectively. Of the 44 virulence factors detected, 26 (across Listeria Pathogenicity Islands, LIPIs) were present in all the isolates. The difference in the detection frequency varied significantly (P<0.001). The two AMR genes (fosX and vga(G)) detected were present in all 60 (100%) isolates of L. monocytogenes. The only plasmid, NF033156, was present in 3 (5%) isolates. A CRISPR-Cas system was detected in 6 (10%), and all the isolates carried proviruses. Significant differences were detected in the frequencies of STs and virulence factors regarding the source and sample type of the L. monocytogenes isolates. The presence of both fosX and vga(G) genes in all the isolates from the three industries (cattle farms, abattoirs, and retail outlets) can potentially cause therapeutic implications. Our study, which characterized L. monocytogenes recovered from the three levels in the beef production chain in the country, provides the first evidence of the distribution of the pathogen with potential food safety and therapeutic implications.
Keywords:
beef production chain
; Listeria monocytogenes
; whole-genome sequencing
; sequence type
; clonal complexes
; virulence factor
; antimicrobial genes
; plasmids
; South Africa
1. Introduction
Listeria monocytogenes is the primary cause of human cases and listeriosis outbreaks and has a considerable negative economic impact on society and the food industry [1]. Although L. monocytogenes is the only recognized human pathogen among Listeria species, it is also pathogenic for animals [2,3]. L. ivanovii is the only other pathogen responsible primarily for listeriosis in animals [4], but it has been reported to cause listeriosis in humans [5].
Human listeriosis outbreaks have been documented globally, including the world’s largest outbreak reported in South Africa [3,6]. L. monocytogenes causes sporadic cases, protracted outbreaks, and even multi-country outbreaks, and the specific source may not be known [7]. The European Food Safety Authority [8] reported 2,183 confirmed invasive human cases of L. monocytogenes in 2021. In Europe, the case fatality rate is high (13.7%), similar to 2020 [8], confirming listeriosis as one of the most severe foodborne diseases.
L. monocytogenes is an important foodborne zoonotic agent, and it has been demonstrated to be present in several food types and, therefore, poses a food safety risk [9]. Meat and meat products constitute a daily human diet due to the high nutritional value of their components, such as proteins, important amino acids, vitamins, and minerals [10]. However, the nutritional components in meat function as ‘natural media’ for microorganisms such as L. monocytogenes [11]. The consumption of ready-to-eat (RTE) meat products has been described as a vehicle for approximately 30% of human listeriosis outbreaks between 2008 and 2015 [12]. Contaminated RTE meat products are the main concern for public health [13]. The ability of L. monocytogenes to survive common food processing conditions like low pH levels, a high salt concentration, low water activity, and refrigeration temperatures facilitates its proliferation in the food environment [14]. Due to the pathogen’s ubiquity, contamination of meat and meat products occurs at various processes, including RTE products [15] and distribution stages [16].
For decades, the traditional serotyping of L. monocytogenes has been used to characterize isolates recovered from several sources for investigative purposes [17]. However, researchers and diagnosticians now rely on more sensitive, specific, and accurate molecular methods to diagnose, confirm, and characterize L. monocytogenes isolates. Some of these methods include the polymerase chain reaction (PCR), multi-virulence-locus sequence typing (MVLST), multi-locus sequence typing (MLST), multilocus variable number tandem repeat analysis (MLVA), pulse-field gel electrophoresis (PFGE) and whole-genome sequencing (WGS), which are now being used [3,18,19].
The sequence types (STs) and the clonal complexes (CC) of L. monocytogenes have been used to characterize the pathogen [20,21], and numerous STs have been identified in L. monocytogenes isolates worldwide [22]. Of significance is the frequent association of some STs and CCs with isolates of L. monocytogenes that are implicated with human listeriosis, thus making the detection of these STs and CCs critical in epidemiological investigations [21,23,24,25,26].
The pathogenicity of L. monocytogenes has been associated with the possession of virulence factors, especially those present in the Listeria Pathogenicity Islands (LIPIs) [3,27,28]. The virulence factors in the LIPIs play vital roles in the pathogenicity of L. monocytogenes. For example, the LIPI-1 and LIPI-3 clusters contain genes related to the infectious life cycle and survival in the food processing environment [28]. The presence of several virulence factors, such as surface-associated internalins, listeriolysin O, and listeriolysin S (LLS) in L. monocytogenes significantly regulate its pathogenicity [29,30].
Antimicrobial resistance (AMR) genes have been documented in L. monocytogenes isolates are produced to facilitate the development of phenotypic resistance to antimicrobial agents [31]. Variable frequencies of AMR genes have been reported for L. monocytogenes isolated from cattle farms, abattoirs, and retail outlets [32,33,34]. The abuse and overuse of antimicrobial agents in human and animal populations result in developing resistance to antimicrobial agents, which is facilitated by the production of appropriate resistance genes as an adaptive response by the pathogen [35,36]. It has also been documented that the leading cause of resistance of L. monocytogenes to antimicrobials is horizontal gene transfer (HGT) of mobile genetic elements such as plasmids and transposons carrying resistant genes and the activation of efflux pump systems [36].
Plasmids are found in several bacterial pathogens, including Listeria spp. [37], and of significance is their ability to carry genetic materials with the potential to encode AMR [38]. In addition, some plasmids provide other benefits to the host cells with potential contribution to stress survival [39]. Some of the plasmids detected in L. monocytogenes include plasmid profiles (N1-011A, J1776, and pLM5578), which were detected in L. monocytogenes isolates recovered from food processing environments in South Africa [40].
Proviruses, prophages in bacterial organisms like L. monocytogenes commonly found in the Listeria genome, have been reported to play an essential role in bacterial evolution, survival, and persistence [41]. Prophages/proviruses are known to mediate defense against phage infection through diverse mechanisms in bacteria [42]. Several frequencies and types of prophages have been reported in L. monocytogenes from many sources [40,43,44,45].
The Clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas) CRISPR-cas system exists in several bacteria, including Listeria spp., which acts as an adaptive immune system of bacteria, is known to help invade the host immune system [46]. Several types of CRISPR-Cas have been reported in Listeria spp., which include Cas-type IA, Cas-type IB, and Cas-type IIA [47]. In L. monocytogenes, it has been found that 41.4% of some isolates contain putative cas genes [48,49]. Various CRISPR-Cas systems in L. monocytogenes isolates recovered from cattle farms, abattoirs, foods, food processing environments, and retail outlets have been found [46,50,51,52].
South Africa experienced a large outbreak of human listeriosis in 2017-2018 [6] caused by a strain of L. monocytogenes, ST6, due to the consumption of ‘polony,’ an RTE pork product [53]. Earlier reports in the country have documented the occurrence of listeriosis in livestock [54]. Reports exist using WGS to characterize L. monocytogenes recovered from the large human listeriosis outbreak [24], isolates of Listeria spp. obtained from beef processing environments [40] and the red meat and poultry value chain [55]. Most recently, Gana et al. [56] used WGS to characterize L. innocua isolates from cattle farms, abattoirs, and retail outlets. To date, there is a dearth of comprehensive information on the WGS analysis of L. monocytogenes circulating in the beef production chain's three levels (production, processing, and retailing) in Gauteng province, South Africa.
The specific objectives of the current study were, therefore, to apply WGS and bioinformatics analyses to characterize isolates of L. monocytogenes recovered from cattle farms, cattle abattoirs, and retail outlets in Gauteng province to unravel the diversity in the profiles of their sequence types, virulence factors, resistance genes, plasmids, CRISPR-Cas systems, and proviruses. We also investigated the potential effects of the origin of L. monocytogenes isolates (sources and sample/food types) on their profiles.
2. Materials and Methods
2.1. Origin of the Isolates of L. monocytogenes Used in Our Study
Sixty isolates of L. monocytogenes on which WGS and bioinformatics analyses were performed in the current study were recovered from cattle farms, cattle abattoirs, and retail outlets in Gauteng province. A detailed description of the isolates regarding the sources and types of samples processed has been provided in an earlier study [57].
2.2. Study Design and Sources of Samples
The study design was to conduct three cross-sectional studies, one each on cattle farms (production), cattle abattoirs (processing), and retail outlets (retailing), which constitute the three industries in the beef production system in Gauteng province. The sample size used in each industry was determined using the formula recommended by Thrusfield [58]. Our earlier report on L. innocua recovered from the same samples used in the current study has provided a flow chart for the relevant sampling to each industry [56].
2.3 Investigation of the Variables or Factors Associated with the Distribution of Genomic Characteristics of L. monocytosis Isolates
Details of the variables investigated in the current study were provided in our earlier study [56]. Briefly, investigated include the type of farm (communal, cow-calf, and feedlot) and feed (grass, grain, and silage). The size (butcheries, high throughput, and low throughput) and practices (pre- and post-evisceration) were investigated as variables at abattoirs. The effects of the size (chain, large, medium, and small) and the types of beef and beef products retailed (raw and processed beef products, including RTE products) were the variables investigated at the retail outlets.
2.4 Isolation and Identification of L. monocytogenes Isolates
The isolates of L. monocytogenes stored at -80oC were confirmed using standard bacteriological and molecular (multiplex PCR) techniques [56,57]. There were 60 isolates of L. monocytogenes comprising 11, 12, and 37 isolates originating from cattle farms, cattle abattoirs, and retail outlets, respectively studied.
2.5 DNA Extraction from L. monocytogenes Isolates
DNA was extracted from the 60 isolates of L. monocytogenes using the Qiagen DNAEasy Blood & Tissue kit, manual, Gram-positive protocol, as per the manufacturer’s instructions.
2.6 Whole-Genome Sequencing, Genomic Analysis, Assembly, and Annotation
All isolates were sequenced on an Illumina MiSeq platform (250-bp paired-end reads; Illumina, Inc., San Diego, CA, USA) using the Nextera XT library preparation kit per the manufacturer’s instructions.
Quality control, including adapter removal, was conducted using BBDuk (v.38.91; https://jgi.doe.gov/data-and-tools/bbtools/bb-tools-user-guide/bbduk-uide/; sourceforge.net/projects/bbmap/) (accessed on 6 September 2022). SPAdes v.3.15.3 [59] created a de novo assembly of each isolate with only contigs longer than 500 bp retained for further analysis. Completeness and contamination of the assemblies were assessed with CheckM v.1.1.3 [60], and taxonomic classification was performed using GTDB-Tk v.1.7.0 [61]. The details have been provided in Supplementary Data, Table S1.
2.7. In Silico MLST
Sequence Types were determined using the MLST tool [62], which makes use of the pubMLST website (https://pubmlst.org/) developed by Jolley & Maiden [63] and sited at the University of Oxford. The Wellcome Trust funded the development of that website. The latest Listeria ST scheme was obtained from BIGSdb-Lm (accessed 21 July 2023 [64] and incorporated into the MLST tool.
2.8. Resistance and Virulence Profiles
ABRicate [65] detected antimicrobial resistance genes and virulence factors. The application was run with default parameters, and the NCBI database was selected for AMR detection. This database was locally updated on 2 November 2022 and, at the time of usage, included 6,334 AMR genes (doi: 10.1128/AAC.00483-19). The “vfdb” database, updated on 2 November 2022, was used for virulence factors and contained 4,332 virulence factors (doi: 10.1093/nar/gkv1239). The virulence profile is based on the presence/absence of a virulence gene in an isolate. In essence, it is a binary matrix consisting of 0’s and 1’s, with each row representing an isolate and each column a putative virulence gene. Isolates with a similar profile based on the presence or absence of virulence factors will cluster together.
The minimum spanning trees for the virulence factors according to the different industries and sample/food types were constructed based on the presence/absence of a virulence factor. No weight was assigned to virulence factors when constructing a minimum spanning tree using a binary matrix as input. The genes from an island are considered different virulence factors.
2.9. Construction of the Phylogenetic Tree for L. monocytogenes Isolates and Correlation with Source and Type of Samples
A core SNP phylogeny was constructed using Snippy v.4.6.0 (https://github.com/tseemann/snippy) and the reference L. monocytogenes EGD-e genome (AL591824). FastTree v.2.1.11 [66] was used to infer a phylogenetic tree, which was visualized in R with ggtree [67].
2.10. Provirus Detection
GeNomad v.1.5.1 [68] facilitated virus detection by enabling aggressive filtering (“--conservative”) and score calibration (“--enable-sc-calibration”) flags in the “end-to-end” execution mode.
2.11. Detection of CRISPR-Cas System
2.12. Data Analysis
All data analyses were performed using R v.4.3.2 [72], implemented in RStudio v.2023.06.0.421 [73]. Distance matrices were calculated using the “daisy” function with the “gower” parameter specified to determine Gower distances with the R package “cluster” [74]. Minimum spanning trees were calculated using the “ape” package [75], with the “mst” function, and visualized using “igraph” [76] and “ggnetwork” [77] R packages ggstatsplot [78], ggsci [79], and ggpubr [75] were further used for data analysis and visualization. Bar charts were produced using the ggstatsplot function ggbarstats, and a Chi-squared test for given probabilities was used to test for significant differences.
3. Results
3.1. Overall Frequency of Detection of STs and Genetic Materials
For the 60 isolates of L. monocytogenes, the overall frequency of STs and genetic elements whose profiles were investigated was as follows: 7 STs were detected at a frequency from 1.7% (ST224) to 46.7% (ST204); 44 putative virulence factors across the 60 (100%) isolates from 1.7% (EcbA/fss3) to 100% (26 putative virulence factors); AMR, two genes fosX and lin found in the 60 (100%) isolates; plasmids, carrying AMR genes 3 (5%) and conjugative genes (80%); provirus, 60 (100%) and CRISPR-Cas system 6 (10%) (Class1-Subtype-I-B).
3.1.1. Influence of the Three Beef Industries (Cattle Farms, Abattoirs, and Retail) on the Frequency of STs, Virulence and AMR Genes, Plasmids, Provirus, and the CRISPR-Cas System
The frequency and distribution of STs and AMR genes in L. monocytogenes isolates in the industries are shown in Table 1. The frequency of L. monocytogenes varied significantly (P=0.002), with the lowest number detected in samples collected from cattle farms (3.4%) and the highest in retail outlet samples (9.3%). For the seven STs detected, 3 STs (12.9%), 5 STs (71.4%), and 6 STs (85.7%) were found in isolates recovered from cattle farms, abattoirs, and retail outlets, respectively, with no statistically significant difference (P=0.223). Regardless of the industry, the frequency of STs was comparatively high for ST204 (46.7%) and 21.7% (ST2) but low for ST14 (3.3%) and ST224 (1.7%), and the difference was statistically significant (P<0.001). The frequency distribution of STs across the three industries was high in cattle farm isolates for two STs, ST31 (18.2%) and ST 876 (27.3%), in cattle abattoirs for two STs, ST204 (58.3%) and ST 224 (8.3%), and in retail outlets for three STs, ST 1 (10.8%), ST2 (32.4%), and ST14 (5.4%). However, the industry had a significant (P=0.044) effect on the detection of only ST2, with a range from 0% (cattle farm) to 32.4% (retail outlet). Supplementary data, Table S2, shows the details of the sources, sample types, and STs of the 60 isolates of L. monocytogenes across the industries and sample/food types. The classification and distribution of the CCs and lineages of L. monocytogenes, according to the sources (industries and sample/food types), are shown in Supplementary data, Table S3.
Forty-four different putative virulence factors were detected in the 60 isolates of L. monocytogenes. From these, 26 virulence factors, including LIPI-1 genes (prfA, plcA, hly, mpl, plcB, and actA) were present in 100% of the 60 isolates. For the remaining 18 virulence factors, the carriage varied significantly from 1 isolate (1.7%) to 59 isolates (98.3%). The differences were statistically significant (P<0.001). However, the three industries had no statistically significant (P>0.05) effect on the frequency of virulence factors. Details are provided in Supplementary data, Table S4.
Only two AMR genes (fosX and lin) were detected in the study, and they were found in all 60 (100%) isolates (Table 1, Supplementary data, Table S5).
One AMR plasmid, NF033156, was detected. It was carried by three (5%) isolates, CFSAN1174456 (Retail outlet, ST204), CFSAN119117 (Abattoir, ST204) and CFSAN119138 (Retail outlet, ST204). In addition, 36 (60%) of the isolates were carriers of conjugation plasmids consisting of the following, FA_orf13; FA_orf17b, 3 (5%), MOBV, 10 (16.7%), and MOBP2, 23 (38.3%) (P<0.001). Details of both types of plasmids are shown in Supplementary data, Table S6
All 60 isolates of L. monocytogenes were carriers of proviruses in the Caudoviricetes class as shown in Supplementary data, Table S7.
A CRISPR-Cas system (Class1-Subtype-I-B_1) was present in 6 (10%) isolates, namely, CFSAN117472 (Retail outlet), CFSAN117492 (Retail outlet), CFSAN117559 (Cattle farm), CFSAN117577 (Cattle farm), CFSAN119122 (Abattoir), and CFSAN119123 (Abattoir). (Supplementary data, Table S8). The samples were predominantly ST31 (5/6) with 1 ST14, and these isolates were spread evenly across the Farm, Abattoir, and Retail industries, with 2 in each.
3.1.2. Detection of STs in L. monocytogenes According to Industries
The frequency distribution of the STs in the isolates of L. monocytogenes according to the three industries (cattle farms, cattle abattoirs, and retail outlets is shown in Figure 1. Across the 11 cattle farm isolates, there were three STs detected, ST204 was predominantly observed and statistically significant (p=2.7 x 10-3) with a higher frequency (54.5%) when compared with ST876 (27.3%) and ST31 (18.2%).
Among the five STs found in the 12 isolates recovered from the abattoirs, ST204 was predominantly detected at a statistically significant (p=2.11 x 10- 3) higher frequency (58.3%) compared to the frequency range of 8.3% (ST2, ST224, and ST876) to 16.7% (ST31) in the other four STs.
The 37 isolates of L. monocytogenes from the retail outlets yielded the highest number of different STs (n=6), with ST204 detected at the highest frequency (40.5%) and ST31 least detected (2.7%). The STs' frequency distribution differences were statistically significant (p=9.02 x 10-7).
Overall, there was a significant over-representation of ST204 in all three industries, with ST2, additionally found more often than expected in the retail industry. ST224 was found exclusively in Abattoirs, ST1, and ST14 only in Retail samples with ST2 uniquely shared between the Abattoir and Retail industries. ST31, ST204 and ST876 were distributed across all the industries.
3.1.3. Detection of STs in L. monocytogenes According to Sample/Food Types
For the eight sample/food types analyzed, the number of STs detected ranged from 2 (ST3 and ST204) in communal farm isolates to 6 (ST1, ST14, ST2, ST204, ST31, and ST876) in small retail samples. Within each sample/food type, the frequency distribution of STs varied significantly (P<0.05). (Figure 2). ST204 was the most predominantly detected, with the highest frequency in all. However, there are unique distributions of some STs. Of relevance is the fact that ST2 was detected in five sample/food types (HT abattoirs and the four types of retail outlets: small, medium, large, and chain); ST224 was found only in HT abattoirs; ST1 was present only in three sample/food types (small, medium and large retail outlets), and ST14 was found only in sample types (small and chain retailers).
3.1.4. Minimum Spanning Tree Based on Sequence Types for L. monocytogenes Detected Across the Different Industries
ST1 and ST14 were only detected in the retail industry (Figure 3). ST2 was predominant in the retail industry, with only one occurrence in abattoirs. ST31, ST204 and ST876 were spread across all three industries, with ST224 unique to the abattoirs.
3.1.5. Minimum Spanning Tree Based on Sequence Types for L. monocytogenes Detected Across the Sample/Food Types
The respective sample/food type for each isolate is displayed in Figure 4. Clustering based on ST was evident, and the spread across various sample/food types for each ST was interesting. A cluster for ST1 was found for small, medium, and large retail industries, with the ST204 cluster representing all the sample/food types in this study. The ST2 grouping represented sample/food types from all the retail sectors, i.e., small, medium, large, and chain, with one occurrence in the high throughput processing environment.
3.1.6. Phylogenies of L. monocytogenes According to STs, Industry, and Sample/Food Type
The phylogenetic tree depicted in Figure 5 indicates relatedness based on ST. The isolates were grouped according to ST, and the affinity for certain STs in the different industries was evident. The Retail industry displayed a propensity for ST1, ST2 and ST14. The promiscuous nature of ST204 was further highlighted as it was found across all three industries and all sample/food types.
3.2. Distribution of Clonal Complexes (CC) among L. monocytogenes Isolates
The 60 isolates of L. monocytogenes belonged to six CCs, three in lineage 1 (CC1: ST1 and ST876), CC2: ST2, and CC224: ST224) and three in lineage II (CC14: ST14, CC31: ST31, and CC204: ST204). A total of 25 (41.7%) and 35 (58.3%) L. monocytogenes were allocated to lineage I and lineage II, respectively (Supplementary data, Table S3). Overall, the frequency of the CCs across the sample types from cattle farms, abattoirs, and retail outlets was as follows: CC1 (18.3%), CC2 (21.7%), CC224 (1.7%), CC14 (3.3%), CC31 (8.3%), C204 (46.7%). The differences were statistically significant (P<0.05).
For the 11 L. monocytogenes isolates recovered from cattle farms (communal, cow-calf, and feedlot), only 3 CCs were detected: CC1 (27.3%, lineage I), CC31 (18.2%), and CC204 (54.5%), both classified in lineage II. The frequency of CCs across the sample types (environment, faeces, and feeds) did not vary significantly (P>0.05), but CC1 (27.3%) and CC204 (54.5%) were predominant.
Among the 12 isolates from the abattoirs (HT), five CCs were detected, namely, CC1 (8.3%), CC2 (8.3%), and CC224 (8.3%), in lineage I, and CC31 (16.7%) and CC204 (58.3%) in lineage II. The CC 204 was detected at a statistically significant (P=0.0068) higher frequency than other CCs, but the differences in the frequency of CCs across sample types (environment, faeces, and carcass) were not statistically significant (P>0.05).
Five CCs were found among the 37 isolates of L. monocytogenes recovered from retail outlets: CC1 (18.9%) and CC2 (32.4%) in lineage I, and CC14 (5.4%), CC31 (2.7%), and CC204 (40.5%) in lineage II. CC2 and CC204 were detected at statistically significant (P<0.0001) high frequencies compared to the others. However, the sample types (RTEs, milled beef, raw beef, and offal and organs) had no significant (P>0.05) effect on the frequency of CCs.
The frequency and distribution of allocated CCs of 60 L. monocytogenes isolates across the industries and sample/food types are shown in Supplementary data, Table S3.
3.3. Occurrence of Virulence Factors in L. monocytogenes Isolates According to the Industries and Sample/Food Samples
The occurrence of virulence factors detected in the L. monocytogenes isolates according to their class, industries, and sample/food type are shown in Supplementary Data, Table S9. In the 60 isolates of L. monocytogenes, a total of 44 virulence factors, of which six (prfA, plcA, Hhly, mlp, actA, and plcB) were LIPI-1 genes, were found. Additionally, 8 virulence factors in the LIPI-3 cluster were detected. Also, 6 internalin family members and other virulence factors that perform important roles in the pathogenesis of listeriosis were also detected, such as those responsible for surface protein adherence (n=1), adherence (n=4), invasion (n=6), intracellular survival (n=3), stress-related (n=3), and immune modulation (n=2), among others.
Twenty-six virulence factors were detected in all of the 60 (100%) isolates of L. monocytogenes, while 18 virulence factors were detected at a frequency range of 1.7% (1/60) for EcbA/fss3 to 98.3% (59/60) for inlA and inlP. The differences were statistically significant (P<0.001). However, the differences in the frequency of the virulence genes across the three industries (cattle farms, abattoirs, and retail outlets) were not statistically significant (P>0.05).
Shared and unique virulence factors (VF) across the industries are shown in Figure 6 for the 44 virulence factors in all the 60 isolates of L. monocytogenes. Twenty-six core VF genes were found to be shared across all the industries (bsh, clpC, clpE, clpP, fbpA, gtcA, hbp1/svpA, hbp2, hly, hpt, iap/cwhA, inlB, inlC, inlK, lap, lntA, lpeA, lplA1, lspA, mpl, oatA, pdgA, plcA, plcB, prfA, prsA2). The Abattoir and Retail industries also uniquely shared 2 VF (inlA, inlP) genes in all the samples from those industries. One VF (inlF) was present in all the abattoir samples but only in some retail and farm samples.
3.3.1. Frequency of virulence factors according to the industries
In Figure 7, the clustering of the samples based on virulence gene profiles indicated distinct groupings. These groups were aligned with the designated ST and found to be homogeneous according to the ST assignment, except ST224, which was found in a cluster otherwise populated by ST876. ST1 and ST14 were only detected in the retail industry. ST2 was further predominant in the retail industry, with only one occurrence in abattoirs. ST31, ST204 and ST876 were spread across all three industries, with ST224 unique to the abattoirs.
Overall, the main cluster consisted of a total of 28 isolates, all belonging to ST204, and was represented by all the industries, specifically farms, farms (n=6), abattoirs (n=7), and retail (n=15). The finding that the isolates belonging to the same ST have similar virulence profiles explains why this causes the main cluster.
3.3.2. Frequency of Virulence Factors According to the Sample/Food Types
In Figure 8, the tree is similar to one based on ST, but the cluster on the left is interesting. Previously, ST1 and ST876 clustered independently, whereas with the virulence factor tree, they clustered together. In the ST tree, ST224 was found within the ST876 cluster, but now it groups individually. From this, the virulence profiles for ST876 and ST1 seem very similar.
Overall, the main cluster comprised all 28 ST204 isolates, which originated from all eight sample/food types and a smaller cluster consisting of only ST2 (n=13) was distributed across five sample types: HT abattoir (n=1), large retail outlet (n=2), medium retail outlet (n=3), small retail outlet (n=3), and chain retail outlet (n=4).
3.4. Frequency of Resistance genes in L. monocytogenes Isolates
All the isolates of L. monocytogenes contained the AMR genes fosX (product - fosfomycin resistance hydrolase FosX; phenotype - Fosfomycin) and vga(G) (product - ABC-F type ribosomal protection protein Vga(G); phenotype – Lincosamide). Additional information is provided in Supplementary data, Table S5.
3.5. Occurrence of AMR Plasmids in L. monocytogenes Isolates
Only one AMR plasmid, NF033156, was detected in our study at a frequency of 5% in three isolates of L. monocytogenes (2 from retail outlets and 1 from an abattoir).
In addition, 48 (80%) isolates were carriers of conjugative plasmids comprising 36 single and 12 mixed plasmids in isolates. Three conjugative plasmids were detected with the following statistically significant (P<0.001) different distribution frequencies: MOBP2, 23 (38.3%), MOBV, 10 (16.7%), and FA_orf13; FA_orf17b, 3 (5%). The frequency of detection of the three conjugative plasmids by ST and indust- ry was as follows: MOBP2, ST876 (n=6, retail outlets:3, farms:3), ST204 (n=1, retail outlet), ST2 (n=11, retail outlets), ST31 (n=4, farm: 2, abattoir: 2) and ST1 (n=1, retail outlet); MOBV, ST2 (n=10, retail outlets); FA_orf13; FA_orf17b, ST876 (n=1, retail), ST1 (n=1, retail) and ST2 (n=1, abattoir). ST2 from retail outlets was predominant in MOBP2, 47.8% (11/23), and MOBV, 100% (10/10). Details of both types of plasmids are shown in Supplementary data, Table S6.
3.6. Frequency of Proviruses/Prophages in the Isolates of L. monocytogenes
Proviruses of the class Caudoviricetes were detected in all 60 isolates of L. monocytogenes (Supplementary data, Table S7).
3.7. Frequency of Detection of the CRISPR-Cas System in L. monocytogenes Isolates
The detection frequency of CRISPR-Cas system in L. monocytogenes was 10% (6/60) with an even distribution of positive isolate by the industry being 18.2% (2/11), 16.7% (2/12) and 5.4% (2/37) for isolates from cattle farms, abattoirs, and retail outlets (P=0.320). In the 6 samples, a CRISPR-cas subsystem (Class1-Subtype-I- B_1 was detected. The details are shown in Supplementary data, Table S8.
3.8. Provirus/Phage and AMR Co-Location (Provirus or Phage as Classification)
Proviruses of the class Caudoviricetes (Viruses; Duplodnaviria; Heunggongvirae; Uroviricota; Caudoviricetes) were detected in all 60 isolates of L. monocytogenes. In 30 of the isolates (Figure 9), the provirus was detected on the same contig as the AMR gene fosX (FOSFOMYCIN) and always within 2,500 bp of each other, with first the provirus and then fosX (strand-specific). This was found across all three industries (Farm=7, Abattoir=7, Retail=16) and only in ST31 (4 out of the 5 total in data) and ST204 (26 out of the 28 total in data). From this, it appears that the provirus is the vector for the fosX gene in certain STs, particularly ST31 and ST204 in this case. The gene space around the fosX gene was conserved in all 30 isolates with engB (GTP-binding protein EngB) always between the phage and fosX, followed by bdlA (Biofilm dispersion protein BdlA), rimJ ([Ribosomal protein S5]- alanine N-acetyltransferase), zitB (Zinc transporter ZitB), mprF (Phosphatidylglycerol lysyltransferase), Epimerase family protein and recX (Regulatory protein RecX).
3.9. Characteristics of L. monocytogenes Recovered from RTE Beef Products
A total of seven isolates of L. monocytogenes were recovered from RTE products comprising Vienna (n=1), ‘biltong’ (n=1), and beef ‘polony’ sampled from the four categories of retailers (chain, large, medium, and small) (Table 2). Isolates of L. monocytogenes of serogroup 11a and ST204 we detected only in beef polony, while ST876 and ST2 were detected in Vienna and ‘biltong’, respectively. Carriage of virulence factors was high, ranging from 32 for Biltong and polony isolates to 39 for Beef polony isolates of the 44 virulence factors detected in the study. Clonal complexes 1 and 2 were detected in 4 of the 7 RTE products. The six RTE isolates were positive for AMR genes [ga (G)] and proviruses of the class Duplodnaviria but were all negative for AMR plasmids and the CRISPR-Cas system.
4. Discussion
In the most recent outbreak of L. monocytogenes in South Africa, considered the largest in the world, L. monocytogenes ST6 was determined to be responsible. It was due to consuming contaminated ‘polony,’ an RTE product [6]. The epidemiology, WGS analysis, and the comparison of South Africa’s outbreak with reports from other countries have been documented [24,80,81]. Beef and beef-based products have been reported to be responsible for listeriosis in other countries [3,82]. As a result of the outbreak in the country, WGS analyses have been used to investigate the population structure of L. monocytogenes isolated in the meat value chain in South Africa [40,55]. However, the current study is the first to document the use of WGS and bioinformatics analyses to characterize L. monocytogenes recovered from the three levels or industries (cattle farms, cattle abattoirs, and retail outlets) of the country's beef production chain.
In our study, L. monocytogenes was detected at an overall frequency of 6.1% (60/990), comprising significantly different detection frequencies of 3.4%, 4.6%, and 9.3% for cattle farms, cattle abattoirs, and retail outlets, respectively, across Gauteng province. Variable frequencies of L. monocytogenes have been reported for samples collected elsewhere from cattle farms, cattle abattoirs, and retail outlets [12,33,83,84,85]. The differences in the frequencies of L. monocytogenes across countries may be due to different management practices in the three industries and the prevalence of the the pathogen in these countries.
Of the seven STs (ST1, ST2, ST14, ST31, ST204, ST224, and ST876) detected in our study, three were found at comparatively high frequencies for ST876 (11.7%), ST2 (21.7%), and ST204 (46.7%). Our findings are different from those reported for 217 L. monocytogenes isolates recovered from red meat and poultry value chain in South Africa [55], where a total of 20 STs were detected, comprising ST204 (14.7%), ST2 (13.8%), ST1 (11.5%), ST9 (11.1%), and ST321 (9.7%). It is pertinent to mention that the current study and two other studies, one conducted on the food chain [40] and the other on meat and meat products [55], all of which were after the large human listeriosis outbreak of 2017-2018 [6] failed to detect ST6 of L. monocytogenes which was responsible for the outbreak. However, it cannot be over-emphasized that ST204, the most frequently detected ST in the three studies, may pose a potential food safety concern regarding human listeriosis in the country since it has been associated with cases of human listeriosis elsewhere [21,86,87]. It has been documented that ST204 is the most common ST in meat products in Australia and France [88,89] and in food processing plants [90]. Furthermore, other STs observed in our study have been detected in meat and meat products and other foods implicated in cases and outbreaks of listeriosis by others [25,89,91].
It has been documented that L. monocytogenes strains are delineated into sequence types (STs) based on conventional multilocus sequence typing (MLST), which utilizes seven alleles. STs are then grouped into clonal complexes (CCs) with strains in the same sharing at least six of the seven MLST alleles [21]. The clonal complexes (CC) and lineages to which isolates of L. monocytogenes have been determined to predict the vir-ulence or pathogenicity potential of the microorganism recovered from human cases or foods [26,92,93]. This association of CCs and lineages of L. monocytogenes with virulence is linked to the type and number of virulence factors they carry. It is, therefore, it is significant that L. monocytogenes isolates allocated to CC1 and CC2in lineage, I were detected in 24 (40%) of the isolates, which have been reported to be frequently associated with human listeriosis [26,94,95,96], and CC204 in lineage II constituted 46.7% of our isolates and has been documented to be predominantly documented in foods [40,55] in South Africa and elsewhere [97,98]. It cannot be over-emphasized that in our study, all 60 isolates of L. monocytogenes were carriers of 5 LIPI-1 virulence factors (prfA, plcA, hly, mpl, and plcB) and LIPI-3 cluster, which are known to play a significant role in the virulence/pathogenicity of CC1 and CC2 L. monocytogenes [26,99,100] were carriers of 8 virulence factors (IlsA, IlsB, IlsD, IlsG, IlsH, IlsP, IlsX, and IlsY) were detected in 13.3-20% of our isolates. This is relevant to food safety, considering that 31.7% (19/60) of the CC1 and CC2 L. monocytogenes in the The current study was based on beef and beef products, including RTE products. Unsurprisingly, CC204 was detected at an overwhelmingly high frequency (40.5%-58.3%) across samples from the three industries (cattle farms, abattoirs, and retail outlets) and more importantly, 40.5% for beef and beef products.
The distribution of STs of L. monocytogenes within and across the three industries was significantly different, demonstrating that the industries were significantly associated with the STs detected. However, it is pertinent to mention that our findings that the industries were significantly associated with the STs detected may be limited to the current sampling scope, including the locations and sampling span. This is because the STs/CCs of L. monocytogenes are known to be frequently introduced and transmitted; therefore, some are expected to be found in only one location in one sampling effort [21]. A cross-sectional or ‘snapshot’ study, like ours, will be unable to infer the persistence of the CCs in that location over multiple years, thus limiting our study. It was also important to observe that some STs (ST31, ST204, and ST876) were distributed across all the industries. At the same time, ST224 was found exclusively in abattoir isolates, ST1 and ST14 were detected only in the isolate from the retail industry, and ST2 was uniquely shared between the L. monocytogenes isolates obtained from the Abattoir and Retail industries. The differences in the number and types of STs recovered in the three industries (cattle farm: 3 types, abattoirs: 5 types, retail industry: 6 types) may be explained in part by the number of isolates tested per industry, 11, 12, and 37, respectively. Furthermore, the variation in the number of sources could have contributed to the recovery of isolates of L. monocytogenes (cattle farms versus abattoirs versus retail outlets). It was also significant that ST204 was predominantly detected in three industries. Other reports have documented differences in the number and frequency of STs in L. monocytogenes from these industries by others [101,102]. Just as found with the effects of industries on the distribution of L. monocytogenes STs, the frequency of STs varied significantly according to the eight sample/food types tested. Interestingly, ST204 was detected at the highest frequency across all the sample/food types tested. Therefore, there is a possibility that ST204 is widespread among L. monocytogenes isolates in Gauteng province, as earlier documented by others [40,55]. It is also noteworthy that some STs, such as ST2, were found in as many as five sample/food types (HT abattoirs and the four types of retail outlets), whereas ST224 was found only in HT abattoirs. It cannot be over-emphasized that the distribution of the STs of L. monocytogenes, according to the sample/food types, can influence human exposure to some of these STs [47,103].
It is noteworthy that the LIPI-3 genes were detected at frequencies ranging from 13.3% (IlsP) to 20% (IlsA, IlsB, IlsG, IlsH, IlsX, and IlsY) of our isolates. This is because the LIPI-3 gene cluster is known to be involved in the infectious life cycle and survival in the food processing environment [27,28]. It has been documented that these virulence factors perform different roles and functions, such as being responsible for surface protein anchoring, adherence, invasion, immune modulation, and intracellular survival, among others; some virulence factors have been implicated in human listeriosis [3,29,103,104,105]. Equally relevant is the finding that among our 60 isolates of L. monocytogenes, 26 (59.1%) shared unique virulence factors, including virulence factors belonging to LIPI-1 and LIPI-3 genes. Matle et al. [106] similarly reported the presence of 47 similar virulence factors in six sequenced isolates of L. monocytogenes from RTE products in the country.
The current study's detection of 44 virulence factors is considerably lower than the 68 putative virulence factors earlier reported in the country [40]. The differences in the critical virulence factors detected in both studies conducted in South Africa may be accounted for partly by the origins and types of the samples from where the L. monocytogenes were isolated, and different L. monocytogenes populations resident in these locations. In our study, the L. monocytogenes isolates originated from cattle farms (faeces, feeds: grain, grass, and silage), cattle abattoirs (pre-evisceration and post-evisceration carcass swabs, chilled carcass swabs, and effluent), and from retail outlets (raw beef, offal & organs, milled beef, and RTE) in Gauteng province. On the contrary, the L. monocytogenes isolates analyzed by Mafuna et al. [40] were initially recovered from raw meat, processed meat, RTE meat products, and environmental samples collected from a commercial pig farm environment during a listeriosis outbreak [55]. Studies on L. monocytogenes recovered from meat, meat products, and other foods by others have similarly reported variable types and frequencies of virulence factors[93,107].
The clustering of virulence factors within and across the three beef industries and sample/food type, as well as the MST based on their STs, is not surprising but indicates that these variables affect the consumer’s exposure potential to virulent isolates of L. monocytogenes in agreement with published reports [108,109]. Our finding of food safety and therapeutic importance is that RTE beef products were carriers of virulent L. monocytogenes STs were also resistant genes. RTE products have been documented to be implicated in most human listeriosis cases or outbreaks [3,12]. Relevant to South Africa is that one of the three RTE beef products (Vienna, ‘polony,’ ‘biltong’) are two popular delicacies in the country. ‘Polony’ is a beef RTE product that was implicated in the recent large outbreak of human listeriosis in the country [6]. Biltong is essentially raw meat spiced and dried and has been reported to be contaminated with Salmonella [110] and Shiga toxin Escherichia coli [111] in the country. It is, therefore, a source of concern that ‘polony’ constituted 5 of the 7 RTE products where isolates of L. monocytogenes were carriers of 32-39 virulence factors, of which ST204 occurred at the highest frequency (3/5), which has been implicated in human listeriosis [86], the detection of CCs 1 and 2 (2/5), 6 LIPI-1 cluster genes (5/5), and were all positive for the fosX and vga(G) AMR genes. Our findings further confirm the potential food safety and therapeutic importance of the overwhelming detection of ST204 across all our samples, considering that the isolates found in the RTE products carried several virulence genes. Matle et al. [106] detected 142 virulence genes across the sequences of six RTE isolates, which are considerably higher than found in the RTE products in the current study. Other reports have documented the contamination of RTE meat products with L. monocytogenes and characterized the isolates regarding their virulence factors and resistance genes [3,93,107,112].
It is interesting that only two AMR genes, fosX, and vga(G), which encode phenotypic resistance to fosfomycin and lincosamide, respectively, were detected in our study, having each been found in all the 60 isolates of L. monocytogenes studied, regardless of the industries and sample/food types. This is not a surprise because, in South Africa, some antimicrobial agents, including fosfomycin, tetracycline, and sulphonamides, are legally allowed to be sold over the counter for use in the livestock industry. This has been made possible through the Fertilizers, Farm Feeds, Agricultural Remedies and Stock Remedies Act 36 of 1947. Therefore, these farmers use antimicrobial agents to treat livestock and as growth promoters in the [113,114,115] without veterinary oversight. Thus, our study's exceptionally high detection frequency of the fosX gene (100%) may have therapeutic implications. In agreement with the current study was the detection of the fosX gene in all (100%) L. monocytogenes isolates recovered RTE products of animal origin (100%) [99] and from the food chain [40] in South Africa. However, unlike our current study, where only two AMR genes, fosX and vga(G), were detected, Mafuna et al. [40] found four resistance genes (fosX, lin, norB, and mprF) in all isolates of L. monocytogenes and tetS obtained from the country's meat food chain. The origin of the samples and isolate genotypes in both studies may account for the differences in their findings. Additionally, it is known that different genotypes can have different AMR gene profiles [116]. The detection of the AMR resistance gene, fosX, in all (100%) L. monocytogenes isolates in the current study may be due to a report based on the analysis of 1,696 isolates of L. monocytogenes revealed the fosX gene to be part of the Listeria core genome, where all isolates harbored this gene [117]. Mota et al. [118] have also indicated that L.monocytogenes is currently considered to be intrinsically resistant to fosfomycin because of the lack of expression in the membrane transport systems and natural resistance to lincomycin due to the ribosomal protection of an ATP-binding cassette F (ABC-F) protein. It is, therefore, no surprise because Parra-Flores et al. [52] detected the fosX gene in 100% of L. monocytogenes recovered from RTE foods, and Hanes et al. [119] reported that 97.8% of L. monocytogenes isolated from 2010-2021 in the USA were carriers of the gene. Regardless of whether the high frequency of the fosX gene resulted from the overuse of antimicrobial agents in the cattle industry or the genes being part of the genome of Listeria spp., it is important to note that the findings may pose therapeutic complications should the gene be expressed. However, not all genes, including resistance genes, are expressed because they may be lost, limiting their application to assessing their therapeutic implications and significance [120]. The potential therapeutic effect of the high frequency of vga(G), which encodes lincosamide resistance, cannot be assessed because the antimicrobial agent is not routinely used on cattle in South Africa.
In our study of 60 isolates of L. monocytogenes, only one AMR plasmid, NF033156 was found at a low frequency of 5% (3/60), and all were from ST204 isolates recovered from an abattoir and two from retail outlets. Matle et al. [106] found no plasmid in a study on six isolates of L. monocytogenes recovered from RTE meat products. Of relevance is that Mafuna et al. [40] detected plasmids in 71% of the 143 isolates of L. monocytogenes studied, and their detection was ST-specific. Although there were differences in the types and frequencies of AMR plasmids identified in both studies, the over-representation of the plasmids in some STs is common. The differences in the types and frequencies of plasmids recovered from L. monocytogenes in both studies may be due to the origins of the samples that yielded the pathogen and the losses of plasmids. Plasmids are essential in the carriage of AMR genes and other genetic materials in L. monocytogenes and other bacteria [121,122]. Furthermore, different types of AMR plasmids have been identified in L. monocytogenes in meat products in studies by others, and it has been demonstrated that L monocytogenes may gain or lose plasmids [123].
Three conjugative plasmids were detected in 80% of the 60 isolates of L. monocytogenes tested in our study, with their detection at statistically significant different frequencies for MOBP2 (38.3%), MOBV (16.7%), and FA_orf13; FA_orf17b (5%). Notably, the occurrence of the plasmids was associated with the STs and the in- dustry, with our observation that both MOBV and MOBP2 were associated with the STs and the beef retail industries. In agreement with our findings, Mao et al. [124] demonstrated that a conjugative plasmid, pLM1686, was associated with four STs (ST87, ST59, ST9 and ST120) in China. The authors also reported that the plasmid had the self-transmissible ability and existed in various L. monocytogenes isolates, providing L. monocytogenes advantages of surviving in adverse environments. Others have described the types and roles of conjugative plasmids in L. monocytogenes [124,125].
In our study, all (100%) 60 isolates of L. monocytogenes were carriers of proviruses/ prophages. This is higher than the 90.9% (30/143) found in L. monocytogenes isolated from the food chain in the country [40]. A considerably lower likelihood of prophage- carrying isolates of L. monocytogenes was 14.4%, as suggested by Vu et al. [45], who also indicated that the prophages in L. monocytogenes are highly diverse and could be at least 16. The detection that all L. monocytogenes isolates in our study were positive for provirus/prophages is important, considering that they are known to play critical roles in L. monocytogenes including mediating defense against phage infection, bacterial survival, and persistence in stressful environments [41,42]. Interestingly, proviruses (prophages) in the class Caudoviricetes were detected in all 60 isolates of L. monocytogenes isolates in our study, their co-location with AMR gene (fosX), and being ST-specific (ST31 and ST204) indicate that the provirus /prophages may serve as the vector for the fosX gene. This finding requires further investigation. It is pertinent to mention that it has been suggested that the fosX gene is harboured on the Listeria core genome [126].
We detected the CRISPR-Cas subsystem (Class1-Subtype-I-B_1) in 10% (6/60) of the L. monocytogenes isolates, which were evenly distributed across the three industries but were highly represented in ST31, 83.3% (5/6). Parra-Flores et al. [52] recovered the CRISPR-Cas system from 71% of RTE foods sampled in Chile. Although, for comparison, no prior reports have been published on the occurrence of the CRISPR- Cas system in L. monocytogenes isolated from any source in the country, Mafuna et al. [47] detected three CRISPR-Cas system types (CAS-Type IIA system, CAS-Type IB system, and CAS-Type IIC system) in 41 non-pathogenic Listeria spp. (L. innocua and L. welshimeri) recovered from meat and food processing facilities (FPF). Regardless of the Listeria spp., the CRISPR-Cas system is known to degrade foreign genetic elements and has been documented in L. monocytogenes isolates [127,128,129]. It has been suggested that some of the CRISPR-Cas systems detected in L. monocytogenes are functional with spacers matching sequences of known Listeria phages and plasmids [129]. The CRISPR-Cas system, which exists in L. monocytogenes, acts as an adaptive immune system and has been documented to help invade the host immune system [46].
The data from our study demonstrate the occurrence of virulence factors (n=44) and STs, which were widely distributed across the three industries and eight sample/food types investigated. Others have documented similar findings [40,55,93,107]. The significantly higher distribution of certain STs and virulence factors in some food types, particularly RTE products, increases the risk of listeriosis in humans. The AMR gene, fosX, was present in all 60 isolates of L. monocytogenes from the country's beef production chain may pose a therapeutic threat since Fosfomycin, encoded by the gene, is commonly used in the country’s livestock industry. Isolates of L. monocytogenes may pose a therapeutic threat since Fosfomycin, encoded by the gene, is widely used in the country's livestock industry. The AMR gene, the CRISPR-Cas system, and proviruses/prophages are known to play some roles in the survival of L. monocytogenes in the food system [38,39,46].
5. Conclusions
Our study, which used WGS and bioinformatic analyses of L. monocytogenes isolated from the beef production chain comprising three industries (cattle farms, beef abattoirs, and retail outlets) and different types of samples and foods across Gauteng province provided vital data on the pathogen. The study characterizes the L. monocytogenes regarding their STs, carriage of virulence factors, resistance genes, AMR plasmids, proviruses, and CRISPR-Cas systems, all factors that play some roles in the organism's pathogenicity. Based on the high frequency of CC1 and CC2 L. monocytogenes with most carrying LIPI-1 (73.3%-100%) and LIPI-3 (18.3%-20%) virulence factors provide evidence that they could pose a food safety risk to consumers. The predominance of these factors (STs, CCs, and virulence factors) in RTE beef products further support the risk posed by ‘polony,’ which was responsible for the recent large listeriosis outbreak in South Africa. The use of MST and phylogenies revealed clustering of the putative virulence factors according to the isolates' source and sample/food types, thus providing their relative risk of exposure. FosX was detected in all 60 isolates of L. monocytogenes, and the high frequency may be due to the over the counter availability and unsupervised use of fosfomycin encoded by the gene in the country's livestock industry. This AMR gene could, therefore, have therapeutic implications if expressed. Finally, the detection of pathogenic STs, CC1 and CC2, and putative virulence factors that have been associated with human listeriosis, the occurrence of AMR genes, plasmids, proviruses/prophages, and CRISPR-Cas system in our isolates recorded from the three industries for the first time in the country has public health, food safety, and therapeutic implications for consumers of beef and beef products in the country.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org
Author Contributions
Conceptualization, A.A.A. and N.G.; methodology, J.G., Y.C., R.E.P., A.A.A., and N.G.; software, R.E.P., Y.C., and N.G.; validation, R.E.P., and Y.C.; formal analysis, R.E.P., Y.C., A.A.A., and J.G.; resources, A.A.A., N.G., and R.M.; data curation, A.A.A.,J.G., and R.E.P.; writing— original draft preparation, A.A.A., J.G., N.G., and R.E.P.; writing—review and editing, A.A.A., J.G., N.G., R.E.P., Y.C., and R.M.; visualization, R.E.P., Y.C., N.G.,J.G., and A.A.A.; supervision, A.A.A., N.G., and R.M.; project administration, A.A.A., N.G.,and J.G.; funding acquisition, A.A.A., N.G., and R.M. All authors have read and agreed to the published version of the manuscript.
Funding
The Red Meat Research and Development, South Africa (RMRD-SA), funded the research project through a grant (REC138-19J awarded on January 01, 2019.
Institutional Review Board Statement
The study was approved and conducted under terms approved by the University Pretoria, South Africa: Animal Ethic Committee (#REC 138-19; 3 May 2020), and the Research Ethic Committee (4 November 2019); Section 20 (#12/11/1/1/8 (1131); 22 October 2019).
Informed Consent Statement
Not Applicable.
Data Availability Statement
All samples have been deposited under NCBI BioProject PRJNA215355 and can be searched based on the isolated CFSAN identifier.
Acknowledgments
We thank the ARC-OVR technicians (Kuda Jwamba, Makhado Lavhesani, Carol Matau, and Lesego Mashiane) for their assistance. We gratefully appreciate the sequencing team at the Public Health Laboratory in the U.S. South Carolina Department of Health & Environmental Control for generating the genome sequencing data for the Listeria isolates. We thank Maria Balkey for data management and submissions to NCBI.
Conflicts of Interest
The authors declare no conflict of interest. The funders did not play any role in the study’s design, in the collection, analysis, or interpretation of data, in writing the manuscript, or in the decision to publish the results. The views expressed in this article are those of the authors and do not necessarily reflect the official policy of the Department of Health and Human Services, the U.S. Food and Drug Administration, or the U.S. Governme.
References
- Olanya, O.M.; Hoshide, A.K.; Ijabadeniyi, O.A.; Ukuku, D.O.; Mukhopadhyay, S.; Niemira, B.A.; Ayeni, O. Cost estimation of listeriosis (Listeria monocytogenes) occurrence in South Africa in 2017 and its food safety implications. Food contr 2019, 102, 231–239. [Google Scholar] [CrossRef]
- Hilliard, A.; Leong, D.; O’Callaghan, A.; Culligan, E.P.; Morgan, C.A.; DeLappe, N.; Hill, C.; Jordan, K.; Cormican, M.; Gahan, C.G. Genomic characterization of Listeria monocytogenes isolates associated with clinical listeriosis and the food production environment in Ireland. Genes 2018, 9, 171. [Google Scholar] [CrossRef]
- Koopmans, M.M.; Brouwer, M.C.; Vázquez-Boland, J.A. and van de Beek, D.Human listeriosis. Clin Microbio Rev 2023, 36, e00060–19. [Google Scholar] [CrossRef]
- Chand, P.; Sadana, J.R. Outbreak of Listeria ivanovii abortion in sheep in India. Vet Record 1999, 145, 83–84. [Google Scholar] [CrossRef]
- Guillet, C.; Join-Lambert, O.; Le Monnier, A.; Leclercq, A.; Mechaï, F.; Mamzer-Bruneel, M.F.; Bielecka, M.K.; Scortti, M.; Disson, O.; Berche, P.; Vazquez-Boland, J. Human listeriosis caused by Listeria ivanovii. Emerg Infect Dis 2010, 16, 136. [Google Scholar] [CrossRef] [PubMed]
- Allam, M.; Tau, N.; Smouse, S.L.; Mtshali, P.S.; Mnyameni, F.; Khumalo, Z.T.; Ismail, A.; Govender, N.; Thomas, J.; Smith, A.M. Whole-genome sequences of Listeria monocytogenes sequence type 6 isolates associated with a large foodborne outbreak in South Africa, 2017 to 2018. Geno Anno 2018, 6, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Halbedel, S.; Prager, R.; Fuchs, S.; Trost, E.; Werner, G.; Flieger, A. Whole-genome sequencing of recent Listeria monocytogenes isolates from Germany reveals population structure and disease clusters. J.Clin Microbio 2018, 56, 10–1128. [Google Scholar] [CrossRef]
- European Food Safety Authority and European Centre for Disease Prevention and Control. The European Union One Health 2021 Zoonoses Report. EFSA J 2022, 2, e07666. [Google Scholar]
- Cufaoglu, G.; Ambarcioglu, P.; Ayaz, N.D. Meta-analysis of the prevalence of Listeria spp. and antibiotic resistant Listeria monocytogenes isolates from foods in Turkey. Lwt 2021, 144, 111210. [Google Scholar] [CrossRef]
- İnci̇li̇, G.K.; Aydemi̇r, M.E.; Akgöl, M.; Kaya, B.; Kanmaz, H.; Öksüztepe, G.; Hayaloğlu, A.A. Effect of Rheum ribes L. juice on the survival of Listeria monocytogenes, Escherichia coli O157: H7 and Salmonella Typhimurium and chemical quality on vacuum packaged raw beef. LWT 2021, 150, 112016. [Google Scholar] [CrossRef]
- Zhang, L.; Moosekian, S.R.; Todd, E.C.; Ryser, E.T. Growth of Listeria monocytogenes in different retail delicatessen meats during simulated home storage. J. food Prot 2012, 75, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Demaître, N.; De Reu, K.; Haegeman, A.; Schaumont, D.; De Zutter, L.; Geeraerd, A.; Rasschaert, G. Study of the transfer of Listeria monocytogenes during the slaughter of cattle using molecular typing. Meat sci 2021, 175, 108450. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, W.; Sun, T.; Gorris, L.G.; Wang, X.; Liu, B.; Dong, Q. The prevalence of Listeria monocytogenes in meat products in China: A systematic literature review and novel meta-analysis approach. Inte J. food Microbio 2020, 312, 108358. [Google Scholar] [CrossRef]
- Sibanda, T.; Buys, E.M. (2022). Listeria monocytogenes pathogenesis: The role of stress adaptation. Microorganisms 2022, 10, 1522. [Google Scholar] [CrossRef]
- Tsaloumi, S.; Aspridou, Z.; Tsigarida, E.; Gaitis, F.; Garofalakis, G.; Barberis, K.; Koutsoumanis, K. Quantitative risk assessment of Listeria monocytogenes in ready-to-eat (RTE) cooked meat products sliced at retail stores in Greece. Food Microbio 2021, 99, 103800. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Sun, T.; Wang, X.; Liu, Q.; Dong, Q. Probabilistic model for estimating Listeria monocytogenes concentration in cooked meat products from presence/absence data. Food Res Inte 2020, 131, 109040. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, J.D.; Borucki, M.K.; Mandrell, R.E.; Gorski, L. Serotyping of Listeria monocytogenes by enzyme-linked immunosorbent assay and identification of mixed-serotype cultures by colony immunoblotting. J. Clin Microbio 2003, 41, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Hunt, K.; Jordan, K. Pulsed-Field Gel Electrophoresis (PFGE) Analysis of Listeria monocytogenes. Listeria Monocytogenes: Meth Proto, 2021; 79–88. [Google Scholar] [CrossRef]
- Hurley, D.; Luque-Sastre, L.; Parker, C.T.; Huynh, S.; Eshwar, A.K.; Nguyen, S.V.; Fanning, S. Whole-genome sequencing-based characterization of 100 Listeria monocytogenes isolates collected from food processing environments over a four-year period. MSphere 2019, 4, e00252-19. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Chang, X.; Qin, S.; Song, Y.; Tian, J.; Ma, A. Analysis of 90 Listeria monocytogenes contaminated in poultry and livestock meat through whole-genome sequencing. Food Res Inte 2022, 159, 111641. [Google Scholar] [CrossRef]
- Lee, S.; Chen, Y.; Gorski, L.; Ward, T.J.; Osborne, J.; Kathariou, S. Listeria monocytogenes source distribution analysis indicates regional heterogeneity and ecological niche preference among serotype 4b clones. mBio. 2018, 9, e00396–18. [Google Scholar] [CrossRef]
- Haase, J.K.; Didelot, X.; Lecuit, M.; Korkeala, H.L. monocytogenes MLST Study Group.; Achtman, M. The ubiquitous nature of Listeria monocytogenes clones: a large-scale M ultilocus S equence T yping study. Enviro Microbio 2014, 16, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Papić, B.; Kušar, D.; Zdovc, I.; Golob, M.; Pate, M. Retrospective investigation of listeriosis outbreaks in small ruminants using different analytical approaches for whole genome sequencing-based typing of Listeria monocytogenes. Infec, Gen Evolu 2020, 77, 104047. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Tau, N.P.; Smouse, S.L.; Allam, M.; Ismail, A.; Ramalwa, N.R.; Thomas, J. Outbreak of Listeria monocytogenes in South Africa, 2017–2018: Laboratory activities and experiences associated with whole-genome sequencing analysis of isolates. Foodborne Path Dis 2019, 16, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Wu, Q.; Zhang, J.; Chen, M.; Guo, W. Analysis of multilocus sequence typing and virulence characterization of Listeria monocytogenes isolates from Chinese retail ready-to-eat food. Front Microbio 2016, 7, 168. [Google Scholar] [CrossRef] [PubMed]
- Moura, A.; Lefrancq, N.; Wirth, T.; Leclercq, A.; Borges, V.; Gilpin, B.; Dallman, T.J.; Frey, J.; Franz, E.; Nielsen, E.M.; Thomas, J.; Pightling, A.; Howden, B.P.; Tarr, C.L.; Gerner-Smidt, P.; Cauchemez, S.; Salje, H.; Brisse, S.; Lecuit, M. Listeria CC1 Study Group. Emergence and global spread of Listeria monocytogenes main clinical clonal complex. Sci Adv 2021, 7, 49. [Google Scholar] [CrossRef]
- Lopez-Valladares, G.; Danielsson-Tham, M.L.; Tham, W. Implicated food products for listeriosis and changes in serovars of Listeria monocytogenes affecting humans in recent decades. Foodborne Path Dis 2018, 15, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Wiktorczyk-Kapischke, N.; Skowron, K.; Wałecka-Zacharska, E. Genomic and pathogenicity islands of Listeria monocytogenes—overview of selected aspects. Front Mole Biosci 2023, 10, 1161486. [Google Scholar] [CrossRef]
- Cotter, P.D.; Draper, L.A.; Lawton, E.M.; Daly, K.M.; Groeger, D.S.; Casey, P.G.; Hill, C. Listeriolysin S, a novel peptide haemolysin associated with a subset of lineage I Listeria monocytogenes. PLoS pathogens 2008, 4, e1000144. [Google Scholar] [CrossRef]
- Shen, J.; Rump, L.; Zhang, Y.; Chen, Y.; Wang, X.; Meng, J. Molecular subtyping and virulence gene analysis of Listeria monocytogenes isolates from food. Food Microbio 2013, 35, 58–64. [Google Scholar] [CrossRef]
- Aminov, R.I. A brief history of the antibiotic era: lessons learned and challenges for the future. Front Microbio 2010, 1, 134. [Google Scholar] [CrossRef]
- Calvo-Arrieta, K.; Matamoros-Montoya, K.; Arias-Echandi, M.L.; Huete-Soto, A.; Redondo-Solano, M. (2021). Presence of Listeria monocytogenes in ready-to-eat meat products sold at retail stores in Costa Rica and analysis of contributing factors. J. Food Prot 2021, 84, 1729–1740. [Google Scholar] [CrossRef] [PubMed]
- Obaidat, M.M.; Kiryluk, H.; Rivera, A.; Stringer, A.P. Molecular serogrouping and virulence of Listeria monocytogenes from local dairy cattle farms and imported beef in Jordan. LWT 2020, 127, 109419. [Google Scholar] [CrossRef]
- Shourav, A.H.; Hasan, M.; Ahmed, S. Antibiotic susceptibility pattern of Listeria spp. isolated from cattle farm environment in Bangladesh. J. Agri Food Res 2020, 2, 100082. [Google Scholar] [CrossRef]
- Luque-Sastre, L.; Arroyo, C.; Fox, E.M.; McMahon, B.J.; Bai, L.I.; Li, F.; Fanning, S. Antimicrobial resistance in Listeria species. Microbio Spect 2018, 6, 6–4. [Google Scholar] [CrossRef]
- Wiktorczyk-Kapischke, N.; Skowron, K.; Grudlewska-Buda, K.; Wałecka-Zacharska, E.; Korkus, J.; Gospodarek-Komkowska, E. Adaptive response of Listeria monocytogenes to the stress factors in the food processing environment. Front Microbio 2021, 12, 710085. [Google Scholar] [CrossRef]
- Chmielowska, C.; Korsak, D.; Chapkauskaitse, E.; Decewicz, P.; Lasek, R.; Szuplewska, M.; Bartosik, D. Plasmidome of Listeria spp.—The repA-Family Business. Inte J. Mole Sci 2021, 22, 10320. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.S.; Call, D.R.; Broschat, S.L. PARGT: A software tool for predicting antimicrobial resistance in bacteria. Sci rep 2020, 10, 11033. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Esser, S.; Anast, J.M.; Cortes, B.W. A large-scale sequencing-based survey of plasmids in Listeria monocytogenes reveals global dissemination of plasmids. Front Microbio 2021, 12, 653155. [Google Scholar] [CrossRef] [PubMed]
- Mafuna, T.; Matle, I.; Magwedere, K.; Pierneef, R.E.; Reva, O.N. Whole genome-based characterization of Listeria monocytogenes isolates recovered from the food chain in South Africa. Front Microbio 2021, 12, 669287. [Google Scholar] [CrossRef]
- Vu, H.T.K.; Stasiewicz, M.J.; Benjakul, S.; Vongkamjan, K. Genomic analysis of prophages recovered from Listeria monocytogenes lysogens found in seafood and seafood-related environment. Microorganisms 2021, 9, 1354. [Google Scholar] [CrossRef]
- Bondy-Denomy, J.; Qian, J.; Westra, E.R.; Buckling, A.; Guttman, D.S.; Davidson, A.R.; Maxwell, K.L. Prophages mediate defense against phage infection through diverse mechanisms. ISME J. 2016, 10, 2854–2866. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Knabel, S.J. Prophages in Listeria monocytogenes contain single-nucleotide polymorphisms that differentiate outbreak clones within epidemic clones. J. Clin Microbio 2008, 46, 1478–1484. [Google Scholar] [CrossRef] [PubMed]
- Vu, H.T.K.; Benjakul, S.; Vuddhakul, V.; Vongkamjan, K. Host Range of Listeria Prophages Induced from Lysogenic Listeria Isolates from Foods and Food-related Environments in Thailand. Food App Biosci Inno Techn 2019, 18, 2. [Google Scholar] [CrossRef]
- Vu, H.T.K.; Benjakul, S.; Vongkamjan, K. Characterization of Listeria prophages in lysogenic isolates from foods and food processing environments. PLoS One 2019, 14, e0214641. [Google Scholar] [CrossRef] [PubMed]
- Espinoza-Mellado, M.D.R.; Vilchis-Rangel, R.E. Review of CRISPR-Cas systems in Listeria species: current knowledge and perspectives. Inte J.Microbio 2022. [Google Scholar] [CrossRef]
- Mafuna, T.; Matle, I.; Magwedere, K.; Pierneef, R.E.; Reva, O.N. Comparative Genomics of Listeria Species Recovered from Meat and Food Processing Facilities. Microbio Spect 2022, 10, e01189–22. [Google Scholar] [CrossRef] [PubMed]
- Hupfeld, M.; Trasanidou, D.; Ramazzini, L.; Klumpp, J.; Loessner, M.J.; Kilcher, S. A functional type II-A CRISPR–Cas system from Listeria enables efficient genome editing of large non-integrating bacteriophage. Nucleic acids res 2018, 46, 6920–6933. [Google Scholar] [CrossRef]
- Deveau, H.; Garneau, J.E.; Moineau, S. CRISPR/Cas system and its role in phage-bacteria interactions. Ann Rev Microbio 2010, 64, 475–493. [Google Scholar] [CrossRef]
- Menchaca, A.; Dos Santos-Neto, P.C.; Mulet, A.P.; Crispo, M. CRISPR in livestock: From editing to printing. Theriogenology 2020, 150, 247–254. [Google Scholar] [CrossRef]
- Ohadi, E.; Azarnezhad, A.; Lotfollahi, L.; Asadollahi, P.; Kaviar, V.H.; Razavi, S.; Sadeghi Kalani, B. Evaluation of Genetic Content of the CRISPR Locus in Listeria monocytogenes Isolated From Clinical, Food, Seafood and Animal Samples in Iran. Cur Microbio 2023, 80, 388. [Google Scholar] [CrossRef]
- Parra-Flores, J.; Holý, O.; Bustamante, F.; Lepuschitz, S.; Pietzka, A.; Contreras-Fernández, A.; Castillo, C.; Ovalle, C.; Alarcón-Lavín, M.P.; Cruz-Córdova, A.; Xicohtencatl-Cortes, J. Virulence and antibiotic resistance genes in Listeria monocytogenes strains isolated from ready-to-eat foods in Chile. Front Microbio 2022, 12, 796040. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Govender, N.; McCarthy, K.M.; Erasmus, L.K.; Doyle, T.J.; Allam, M.; Ismail, A.; Ramalwa, N.; Sekwadi, P.; Ntshoe, G.; Shonhiwa, A. Outbreak of listeriosis in South Africa associated with processed meat. New Eng J.Med 2020, 382, 632–643. [Google Scholar] [CrossRef]
- Du Toit, I.F. An outbreak of caprine listeriosis in the Western Cape. J. South Afri Vet Asso 1977, 48, 39–40. [Google Scholar]
- Matle, I.; Mafuna, T.; Madoroba, E.; Mbatha, K.R.; Magwedere, K.; Pierneef, R. Population structure of non-ST6 Listeria monocytogenes isolated in the red meat and poultry value chain in South Africa. Microorganisms 2020, 8, 1152. [Google Scholar] [CrossRef] [PubMed]
- Gana, J.; Gcebe, N.; Pierneef, R.E.; Chen, Y.; Moerane, R.; Adesiyun, A.A. Genomic Characterization of Listeria innocua Isolates Recovered from Cattle Farms, Beef Abattoirs, and Retail Outlets in Gauteng Province, South Africa. Pathogens 2023, 12, 1062. [Google Scholar] [CrossRef] [PubMed]
- Gana, J. Prevalence, risk factors, and molecular characterization of Listeria species from cattle farms, beef abattoirs and retail outlets in Gauteng, South Africa. Ph.D. Thesis, University of Pretoria, South Africa, 2022. [Google Scholar]
- Thrusfield, M. Sample size determination. Vet. Epidemiol. 2007, 3, 185–189. [Google Scholar]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. Available online: http://www.genome.org/cgi/doi/10.1101/gr.186072.114. [CrossRef] [PubMed]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef]
- Seemann, T. mlst Github. Available online: https://github.com/tseemann/mlst (accessed on 7 July 2023).
- Jolley, K.A.; Maiden, M.C. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 2010, 11, 1–11. [Google Scholar] [CrossRef]
- Moura, A.; Criscuolo, A.; Pouseele, H.; Maury, M.M.; Leclercq, A.; Tarr, C.; Björkman, J.T.; Dallman, T.; Reimer, A.; Enouf, V. Whole genome-based population biology and epidemiological surveillance of Listeria monocytogenes. Nat. Microbio. 2016, 2, 16185. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Abricate, Github. Available online: https://github.com/tseemann/abricate (accessed on 28 July 2023).
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–approximately maximum-likelihood trees for large alignments. PloS one 2010, 5, e9490. [Google Scholar] [CrossRef]
- Yu, G. Using ggtree to visualize data on tree-like structures. Curr. Proto. Bioinform. 2020, 69, e96. [Google Scholar] [CrossRef]
- Camargo, A.P.; Roux, S.; Schulz, F.; Babinski, M.; Xu, Y.; Hu, B.; Chain, P.S.; Nayfach, S.; Kyrpides, N.C. Identification of mobile genetic elements with geNomad. Nat Biotechno 2023, 1–10. [Google Scholar] [CrossRef]
- Abby, S.S.; Néron, B.; Ménager, H.; Touchon, M.; Rocha, E.P. MacSyFinder: a program to mine genomes for molecular systems with an application to CRISPR-Cas systems. PloS one 2014. 9, e110726. [CrossRef]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res 2018, 46, W246–W251. [Google Scholar] [CrossRef] [PubMed]
- Grissa, I.; Vergnaud, G.; Pourcel, C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic acids res 2007, 35, W52–W57. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Team, R. RStudio: Integrated Development Environment for R; RStudio, PBC: Boston, MA, USA, 2022. [Google Scholar]
- Maechler, M. Finding groups in data: Cluster analysis extended Rousseeuw et al. R Package Version 2019, 2, 242–248. [Google Scholar]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. Int. J. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Tyner, R.C.; Briatte, F.; Hofmann, H. Network Visualization with ggplot2. R J. 2017, 9, 26–59. [Google Scholar] [CrossRef]
- Ratil, I. Visualizations with statistical details: The ‘ggstatsplot’ approach. J. Open. Source Soft. 2021, 61, 3167. [Google Scholar]
- Xiao, N. ggsci: Scientific journal and Sci-Fi themed color palettes for ‘ggplot2’. In R Package Version; R Core Team: Vienna, Austria, 2018. [Google Scholar]
- Kaptchouang Tchatchouang, C.D.; Fri, J.; De Santi, M.; Brandi, G.; Schiavano, G.F.; Amagliani, G.; Ateba, C.N. Listeriosis outbreak in South Africa: a comparative analysis with previously reported cases worldwide. Microorganisms 2020, 8, 135. [Google Scholar] [CrossRef] [PubMed]
- Kayode, A.J.; Semerjian, L.; Osaili, T.; Olapade, O.; Okoh, A.I. Occurrence of multidrug-resistant Listeria monocytogenes in environmental waters: a menace of environmental and public health concern. Front Envir Sci 2021, 9, 737435. [Google Scholar] [CrossRef]
- Matle, I.; Mbatha, K.R.; Madoroba, E. A review of Listeria monocytogenes from meat and meat products: Epidemiology, virulence factors, antimicrobial resistance and diagnosis. Onderstepoort J. Vet Res 2020, 1–20. Available online: https://hdl.handle.net/10520/ejc-opvet-v87-n1-a9. [CrossRef] [PubMed]
- Hurtado, A.; Ocejo, M.; Oporto, B. Salmonella spp. and Listeria monocytogenes shedding in domestic ruminants and characterization of potentially pathogenic strains. Vet Microbio 2017, 210, 71–76. [Google Scholar] [CrossRef]
- Zhao, Q.; Hu, P.; Li, Q.; Zhang, S.; Li, H.; Chang, J.; Lu, S. Prevalence and transmission characteristics of Listeria species from ruminants in farm and slaughtering environments in China. Emer Micr Infect 2021, 10, 356–364. [Google Scholar] [CrossRef]
- Rossi, F.; Amadoro, C.; Conficoni, D.; Giaccone, V.; Colavita, G. Occurrence, diversity of Listeria spp. isolates from food and food-contact surfaces and the presence of virulence genes. Microorganisms 2020, 8, 294. [Google Scholar] [CrossRef]
- Fox, E.M.; Allnutt, T.; Bradbury, M.I.; Fanning, S.; Chandry, P.S. (2016). Comparative genomics of the Listeria monocytogenes ST204 subgroup. Front Microbio 2016, 7, 2057. [Google Scholar] [CrossRef] [PubMed]
- Jennison, A.V.; Masson, J.J.; Fang, N.X.; Graham, R.M.; Bradbury, M.I.; Fegan, N.; Fox, E.M. Analysis of the Listeria monocytogenes population structure among isolates from 1931 to 2015 in Australia. Front Microbio 2017, 8, 603. [Google Scholar] [CrossRef]
- Ebner, R.; Stephan, R.; Althaus, D.; Brisse, S.; Maury, M.; Tasara, T. (2015). Phenotypic and genotypic characteristics of Listeria monocytogenes strains isolated during 2011–2014 from different food matrices in Switzerland. Food Contr 2015, 57, 321–326. [Google Scholar] [CrossRef]
- Kwong, J.C.; Mercoulia, K.; Tomita, T.; Easton, M.; Li, H.Y.; Bulach, D.M.; Stinear, T.P.; Seemann, T.; Howden, B.P. 2016. Prospective whole-genome sequencing enhances national surveillance of Listeria monocytogenes. J. Clin Microbio 2016, 54, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Martín, B.; Perich, A.; Gómez, D.; Yangüela, J.; Rodríguez, A.; Garriga, M.; Aymerich, T. Diversity and distribution of Listeria monocytogenes in meat processing plants. Food Microbio 2014, 44, 119–127. [Google Scholar] [CrossRef]
- Bespalova, T.Y.; Mikhaleva, T.V.; Meshcheryakova, N.Y.; Kustikova, O.V.; Matovic, K.; Dmitrić, M.; Zaitsev, S.S.; Khizhnyakova, M.A.; Feodorova, V.A. Novel sequence types of Listeria monocytogenes of different origin obtained in the Republic of Serbia. Microorganisms 2021, 9, 1289. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Doijad, S.; Wang, W.; Lian, K.; Pan, X.; Koryciński, I.; Hu, Y.; Tan, W.; Ye, S.; Wang, Z.; Pan, Z.; Chakraborty, T.; Jiao, X. Genetic Diversity of Listeria monocytogenes Isolates from Invasive Listeriosis in China. Foodborn Path Dis 2020, 17, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Centorotola, G.; Ziba, M.W.; Cornacchia, A.; Chiaverini, A.; Torresi, M.; Guidi, F.; Cammà, C.; Bowa, B.; Mtonga, S.; Magambwa, P.; D'Alterio, N.; Scacchia, M.; Pomilio, F.; Muuka, G. Listeria monocytogenes in ready to eat meat products from Zambia: phenotypical and genomic characterization of isolates. Front Microbio, 2023; 14, 1228726. [Google Scholar] [CrossRef]
- Myintzaw, P.; Pennone, V.; McAuliffe, O.; Begley, M.; Callanan, M. Association of Virulence, Biofilm, and Antimicrobial Resistance Genes with Specific Clonal Complex Types of Listeria monocytogenes. Microorganisms 2023, 11, 1603. [Google Scholar] [CrossRef] [PubMed]
- Schiavano, G.F.; Ateba, C.N.; Petruzzelli, A.; Mele, V.; Amagliani, G.; Guidi, F.; De Santi, M.; Pomilio, F.; Blasi, G.; Gattuso, A.; Di Lullo, S.; Rocchegiani, E.; Brandi, G. 2021. Whole-Genome Sequencing Characterization of Virulence Profiles of Listeria monocytogenes Food and Human Isolates and In Vitro Adhesion/Invasion Assessment. Microorganisms 2021, 10, 62. [Google Scholar] [CrossRef]
- Wang, Y.; Meng, F.; Deng, X.; Yang, Y.; Li, S.; Jiao, X.; Li, S.; Liu, M. Genomic epidemiology of hypervirulent Listeria monocytogenes CC619: Population structure, phylodynamics and virulence. Microbio Res 2024, 280, 127591. [Google Scholar] [CrossRef] [PubMed]
- Madden, R.H.; Hutchison, M.; Jordan, K.; Pennone, V.; Gundogdu, O.; Corcionivoschi, N. 2018. Prevalence and persistence of Listeria monocytogenes in premises and products of small food business operators in Northern Ireland. Food Contr 2018, 87, 70–78. [Google Scholar] [CrossRef]
- Melero, B.; Stessl, B.; Manso, B.; Wagner, M.; Esteban-Carbonero, O.J.; Hernandez, M.; Rovira, J.; Rodriguez-Lazaro, D. Listeria monocytogenes colonization in a newly established dairy processing facility. Inter J. Food Microbio 2019, 289, 64–71. [Google Scholar] [CrossRef]
- Gorski, L.; Cooley, M.B.; Oryang, D.; Carychao, D.; Nguyen, K.; Luo, Y.; Weinstein, L.; Brown, E.; Allard, M.; Mandrell, R.E.; Chen, Y. Prevalence and Clonal Diversity of over 1,200 Listeria monocytogenes Isolates Collected from Public Access Waters near Produce Production Areas on the Central California Coast during 2011 to 2016. Appl Envi Microbio 2022, 88, e00357-22. [Google Scholar] [CrossRef] [PubMed]
- Chenal-Francisque, V.; Lopez, J.; Cantinelli, T.; Caro, V.; Tran, C.; Leclercq, A.; Lecuit, M.; Brisse, S. Worldwide distribution of major clones of Listeria monocytogenes. Emer Inf Dis 2011, 17, 1110–2. [Google Scholar] [CrossRef]
- Matle, I.; Mbatha, K.R.; Lentsoane, O.; Magwedere, K.; Morey, L.; Madoroba, E. Occurrence, serotypes, and characteristics of Listeria monocytogenes in meat and meat products in South Africa between 2014 and 2016. J. Food Saf. 2019, 39, e12629. [Google Scholar] [CrossRef]
- Tomáštíková, Z.; Gelbíčová, T.; Karpíšková, R. Population structure of Listeria monocytogenes isolated from human listeriosis cases and from ready-to-eat foods in the Czech Republic. J.Food Nutr Res 2019, 58, 2. [Google Scholar]
- Segerman, B. The most frequently used sequencing technologies and assembly methods in different time segments of the bacterial surveillance and RefSeq genome databases. Front. Cell. Infect. Microbio. 2020, 10, 527102. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Boland, J.A.; Kuhn, M.; Berche, P.; Chakraborty, T.; Domı́nguez-Bernal, G.; Goebel, W.; González-Zorn, B.; Wehland, J.; Kreft, J. Listeria pathogenesis and molecular virulence determinants. Clin Microbio Rev 2001, 14, 584–640. [Google Scholar] [CrossRef] [PubMed]
- Gilmour, M.W.; Graham, M.; Van Domselaar, G.; Tyler, S.; Kent, H.; Trout-Yakel, K.M.; Larios, O.; Allen, V.; Lee, B.; Nadon, C. High-throughput genome sequencing of two Listeria monocytogenes clinical isolates during a large foodborne outbreak. BMC Geno 2010, 11, 1–15. [Google Scholar] [CrossRef]
- Matle, I.; Pierneef, R.; Mbatha, K.R.; Magwedere, K.; Madoroba, E. Genomic diversity of common sequence types of Listeria monocytogenes isolated from ready-to-eat products of animal origin in South Africa. Genes 2019, 10, 1007. [Google Scholar] [CrossRef]
- Centorotola, G.; Ziba, M.W.; Cornacchia, A.; Chiaverini, A.; Torresi, M.; Guidi, F.; Cammà, C.; Bowa, B.; Mtonga, S.; Magambwa, P.; D’Alterio, N. Listeria monocytogenes in ready to eat meat products from Zambia: phenotypical and genomic characterization of isolates. Front Microbio 2023, 14. [Google Scholar] [CrossRef]
- Kayode, A.J.; Okoh, A.I. Assessment of the molecular epidemiology and genetic multiplicity of Listeria monocytogenes recovered from ready-to-eat foods following the South African listeriosis outbreak. Sci Rep 2022, 12, 20129. [Google Scholar] [CrossRef]
- Wagner, E.; Zaiser, A.; Leitner, R.; Quijada, N.M.; Pracser, N.; Pietzka, A.; Ruppitsch, W.; Schmitz-Esser, S.; Wagner, M.; Rychli, K. Virulence characterization and comparative genomics of Listeria monocytogenes sequence type 155 strains. BMC Geno 2020, 21, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Ji, X.; Wu, Y.; Xu, W.; WANG, F.; Huang, X. Molecular Characterization of Listeria monocytogenes Strains Isolated from imported Food in China from 14 countries/regions, 2003-2018. Front Cell Infect Microbio 2023, 13, 1287564. [Google Scholar] [CrossRef]
- Manqele, A. Prevalence and characterization of Salmonella spp. in slaughter cattle, abattoir environment, and meat sold at retail outlets in Gauteng, South Africa. University of Pretoria, South Africa. 2018.
- Onyeka, L.O.; Adesiyun, A.A.; Keddy, K.H.; Madoroba, E.; Manqele, A.; Thompson, P.N. Shiga toxin–producing Escherichia coli contamination of raw beef and beef-based ready-to-eat products at retail outlets in Pretoria, South Africa. J. Food Prot 2020, 83, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Phung, T.; Tran, T.; Pham, D.; To, A.; Le, H. Occurrence and molecular characteristics of Listeria monocytogenes isolated from ready-to-eat meats in Hanoi, Vietnam. Italian J. Food Saf 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Durso, L.M.; Cook, K.L. Impacts of antibiotic use in agriculture: what are the benefits and risks? Curr 0pin Microbio 2014, 19, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Henton, M.M.; Eagar, H.A.; Swan, G.E.; Van Vuuren, M. Part VI. Antibiotic management and resistance in livestock production. SAMJ: South Afric Med J 2011, 101, 583–586. [Google Scholar]
- Van, T.T.H.; Yidana, Z.; Smooker, P.M.; Coloe, P.J. Antibiotic use in food animals worldwide, with a focus on Africa: Pluses and minuses. J. Glob Antimicro Resis 2020, 20, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Scortti, M.; Han, L.; Alvarez, S.; Leclercq, A.; Moura, A.; Lecuit, M.; Vazquez-Boland, J. Epistatic control of intrinsic resistance by virulence genes in Listeria. PLoS Genetics 2018, 14, e1007525. [Google Scholar] [CrossRef] [PubMed]
- Mota, M.I.; Vázquez, S.; Cornejo, C.; D'Alessandro, B.; Braga, V.; Caetano, A.; Betancor, L.; Varela, G. Does Shiga toxin-producing Escherichia coli and Listeria monocytogenes contribute significantly to the burden of antimicrobial resistance in Uruguay? Front Vet Sci 2020, 7, 583930. [Google Scholar] [CrossRef]
- Hanes, R.M.; Huang, Z. Investigation of Antimicrobial Resistance Genes in Listeria monocytogenes from 2010 through to 2021. Inte J. Envir Res Pub Health 2022, 19, 5506. [Google Scholar] [CrossRef]
- Salyers, A.A.; Amabile-Cuevas, C.F. Why are antibiotic resistance genes so resistant to elimination? Antimicro Agents Chemo 1997, 41, 2321–2325. [Google Scholar] [CrossRef] [PubMed]
- Hingston, P.; Brenner, T.; Truelstrup Hansen, L.; Wang, S. Comparative analysis of Listeria monocytogenes plasmids and expression levels of plasmid-encoded genes during growth under salt and acid stress conditions. Toxins 2019, 11, 426. [Google Scholar] [CrossRef] [PubMed]
- Lebrun, M.; Loulergue, J.; Chaslus-Dancla, E.; Audurier, A. Plasmids in Listeria monocytogenes in relation to cadmium resistance. Appl Enviro Microbio 1992, 58, 3183–3186. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Hoffmann, M.; Allard, M.W.; Brown, E.W.; Chen, Y. Microevolution and gain or loss of mobile genetic elements of outbreak-related Listeria monocytogenes in food processing environments identified by whole genome sequencing analysis. Front Microbio 2020, 11, 866. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Wang, Y.; Gan, L.; Sun, H.; Wang, Y.; Li, L.; Ji, S.; Song, Z.; Jiang, H.; Ye, C. Function and distribution of the conjugative plasmid pLM1686 in foodborne Listeria monocytogenes in China. Inte J. Food Microbio 2021, 352, 109261. [Google Scholar] [CrossRef]
- Charpentier, E.; Gerbaud, G.; Courvalin, P. Conjugative mobilization of the rolling-circle plasmid pIP823 from Listeria monocytogenes BM4293 among gram-positive and gram-negative bacteria. J. Bacterio 1999, 181, 3368–3374. [Google Scholar] [CrossRef]
- Scortti, M.; Han, L.; Alvarez, S.; Leclercq, A.; Moura, A.; Lecuit, M.; Vazquez-Boland, J. Epistatic control of intrinsic resistance by virulence genes in Listeria. PLoS Genet. 2018, 14, e1007525, Erratum in: PLoS Genet. 2018 Oct 15;14(10):e1007727. [Google Scholar] [CrossRef]
- Di, H.; Ye, L.; Yan, H.; Meng, H.; Yamasak, S.; Shi, L. Comparative analysis of CRISPR loci in different Listeria monocytogenes lineages. Biochem Biophy Res Comm 2014, 454, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Kuenne, C.; Billion, A.; Mraheil, M.A.; Strittmatter, A.; Daniel, R.; Goesmann, A.; Barbuddhe, S.; Hain, T.; Chakraborty, T. Reassessment of the Listeria monocytogenes pan-genome reveals dynamic integration hotspots and mobile genetic elements as major components of the accessory genome. BMC Geno 2013, 14, 1–19. [Google Scholar] [CrossRef]
- Parsons, C.; Brown, P.; Kathariou, S. Use of bacteriophage amended with CRISPR-Cas systems to combat antimicrobial resistance in the bacterial foodborne pathogen Listeria monocytogenes. Antibiotics 2021, 10, 308. [Google Scholar] [CrossRef]
Figure 1.
Frequency of L. monocytogenes sequence types by industry. Significant over-representation of ST204 was found in all three industries, with ST2 additionally found more often than expected in retail. Farms only displayed 3 of the 7 STs detected, followed by Abattoirs with 5 STs and the Retail industry with 6-7 STs. ST224 was found exclusively in Abattoirs, ST1, and ST14, in retail samples, with ST2 uniquely shared between the Abattoir and retail industries. ST31, ST204, and ST876 were distributed across all the industries.
Figure 1.
Frequency of L. monocytogenes sequence types by industry. Significant over-representation of ST204 was found in all three industries, with ST2 additionally found more often than expected in retail. Farms only displayed 3 of the 7 STs detected, followed by Abattoirs with 5 STs and the Retail industry with 6-7 STs. ST224 was found exclusively in Abattoirs, ST1, and ST14, in retail samples, with ST2 uniquely shared between the Abattoir and retail industries. ST31, ST204, and ST876 were distributed across all the industries.
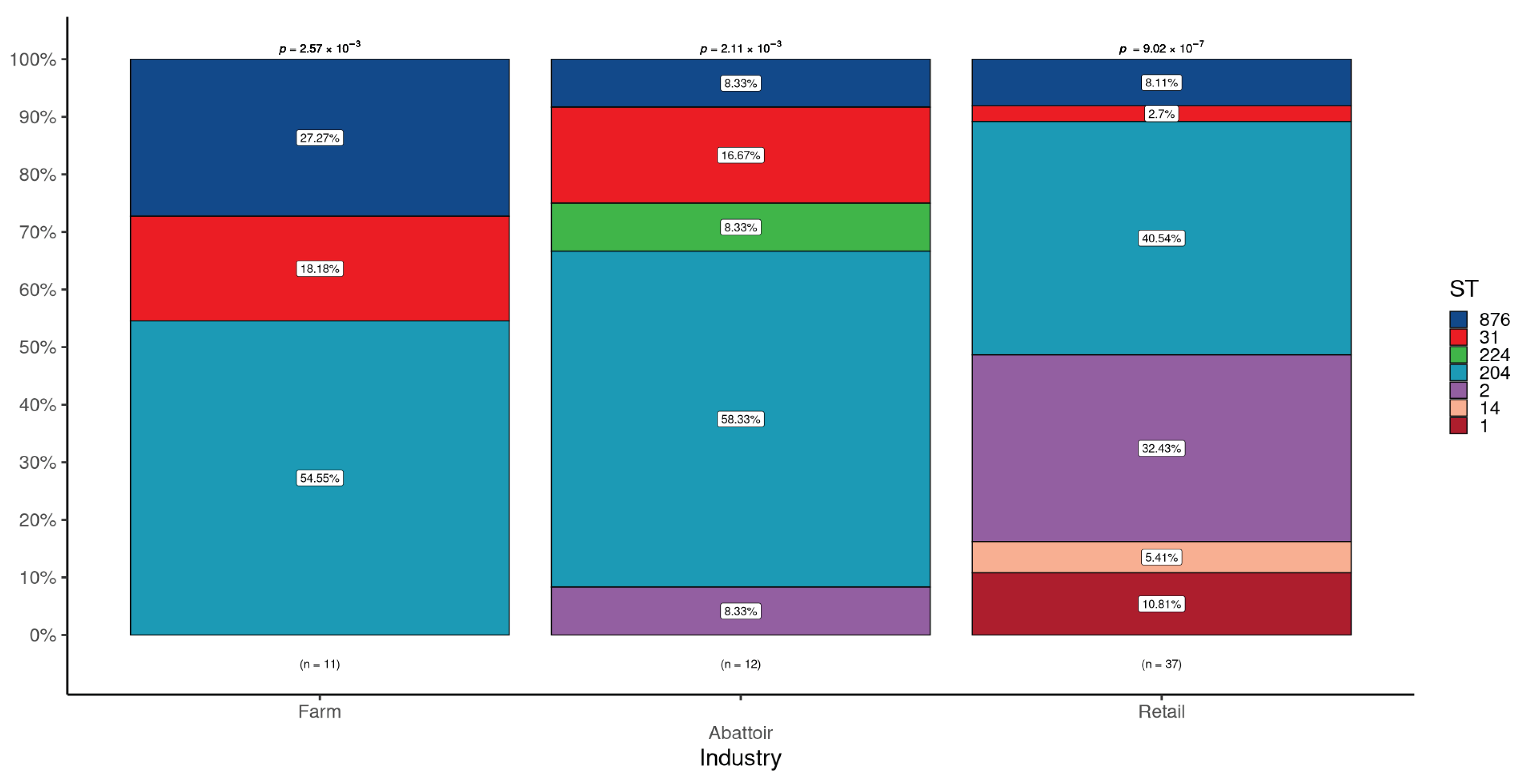
Figure 2.
Frequency of L. monocytogenes sequence types by sample/food type. Significant over- representation of ST204 was found in High Throughput (HT), Large Retail, and Chain Retail.
Figure 2.
Frequency of L. monocytogenes sequence types by sample/food type. Significant over- representation of ST204 was found in High Throughput (HT), Large Retail, and Chain Retail.
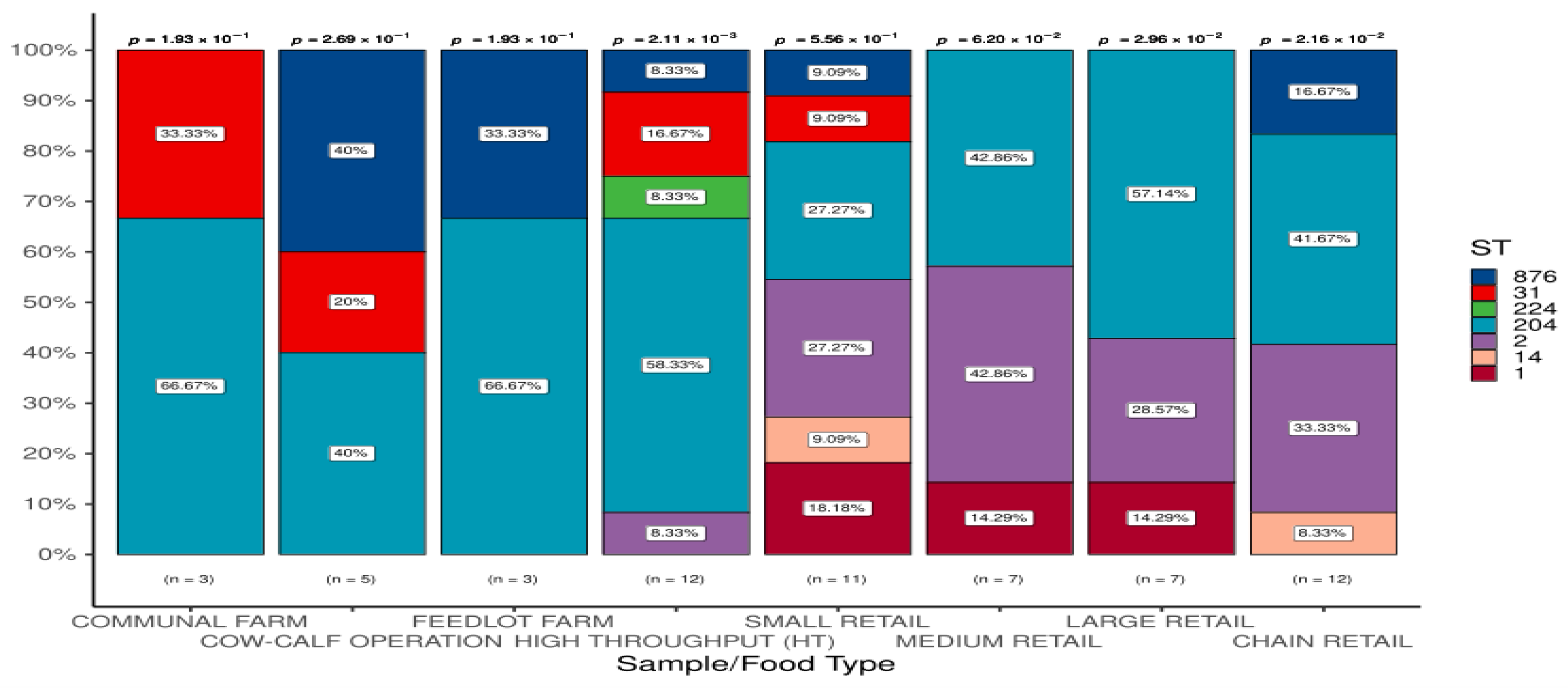
Figure 3.
Minimum spanning tree based on sequence types for L. monocytogenes detected across the different industries. Shared STs across the industries are visible in the multicoloured clusters with STs unique to particular industries evident based on more homogeneous coloured clusters.
Figure 3.
Minimum spanning tree based on sequence types for L. monocytogenes detected across the different industries. Shared STs across the industries are visible in the multicoloured clusters with STs unique to particular industries evident based on more homogeneous coloured clusters.
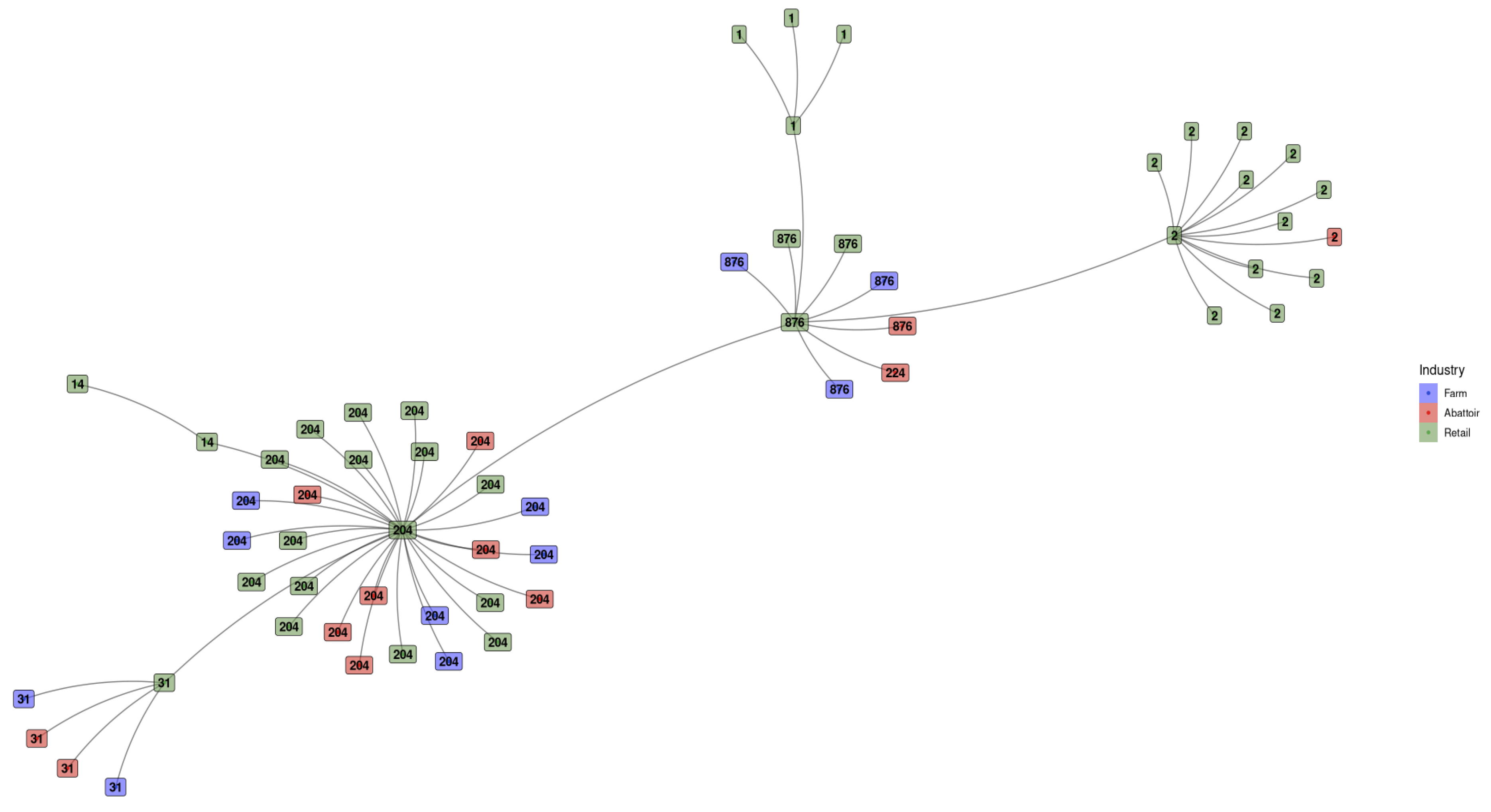
Figure 4.
Minimum spanning tree based on sequence types for L. monocytogenes detected across the different sample/food types. ST1 is spread across medium, large, and chain retail but is not detected in small retail settings, with ST14 seen in small and large retail. ST2 was detected in all the retail sectors, including high throughput. Clusters with a high diversity of colours represent STs found across numerous sample/food types.
Figure 4.
Minimum spanning tree based on sequence types for L. monocytogenes detected across the different sample/food types. ST1 is spread across medium, large, and chain retail but is not detected in small retail settings, with ST14 seen in small and large retail. ST2 was detected in all the retail sectors, including high throughput. Clusters with a high diversity of colours represent STs found across numerous sample/food types.
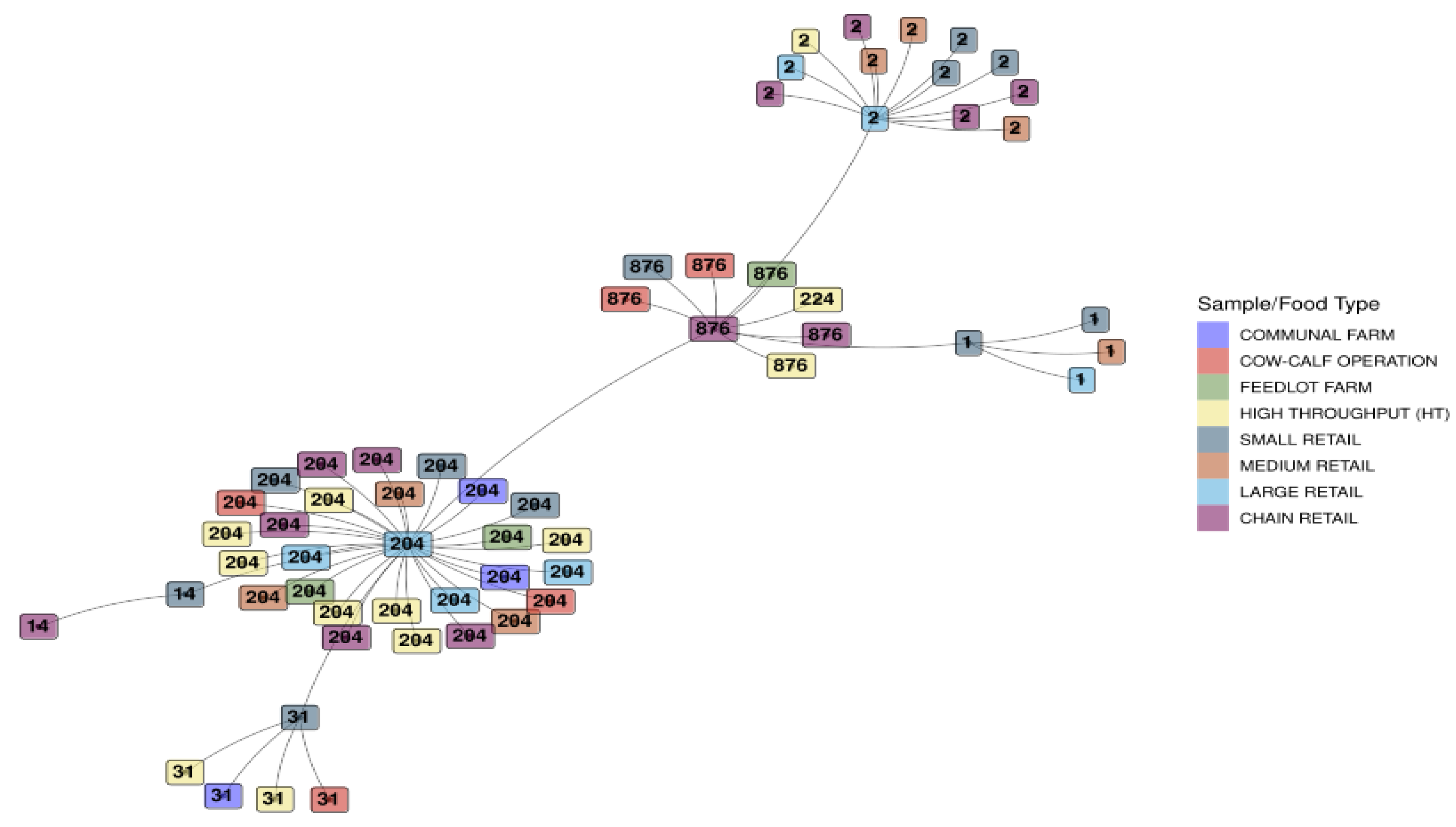
Figure 5.
Phylogeny of L. monocytogenes was detected across the industries based on SNPs present in all samples (core SNPs). The first colour legend indicates the ST, the second the industry for each isolate, and the third represents the Sample/Food Type. The tree demonstrates the grouping of isolates by ST based on the sequential use of colours in the first colour legend, with the second colour legend displaying an affinity for specific STs within industries.
Figure 5.
Phylogeny of L. monocytogenes was detected across the industries based on SNPs present in all samples (core SNPs). The first colour legend indicates the ST, the second the industry for each isolate, and the third represents the Sample/Food Type. The tree demonstrates the grouping of isolates by ST based on the sequential use of colours in the first colour legend, with the second colour legend displaying an affinity for specific STs within industries.
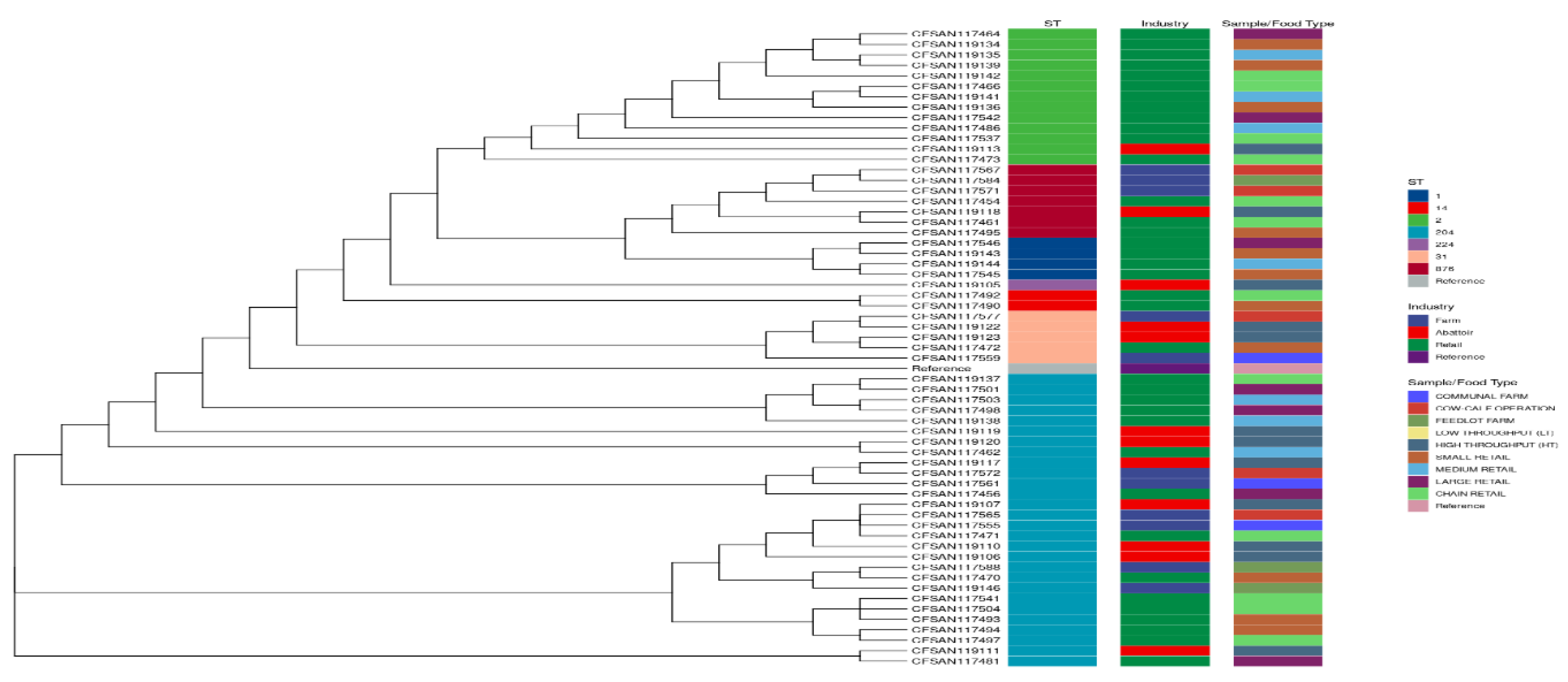
Figure 6.
Shared and unique virulence factor (VF) genes across the industries. A total of 44 different VF genes were found in the 60 isolates. Farm isolates had 26 (59.1%; 26/44) VF genes shared by all 11 samples, Abattoirs had 29 (65.9%) from 12 isolates and the Retail industry had 28 (63.6%) VF genes found in all 37 samples. Each industry's core VF gene list was inspected for shared and unique genes between the different industries.
Figure 6.
Shared and unique virulence factor (VF) genes across the industries. A total of 44 different VF genes were found in the 60 isolates. Farm isolates had 26 (59.1%; 26/44) VF genes shared by all 11 samples, Abattoirs had 29 (65.9%) from 12 isolates and the Retail industry had 28 (63.6%) VF genes found in all 37 samples. Each industry's core VF gene list was inspected for shared and unique genes between the different industries.
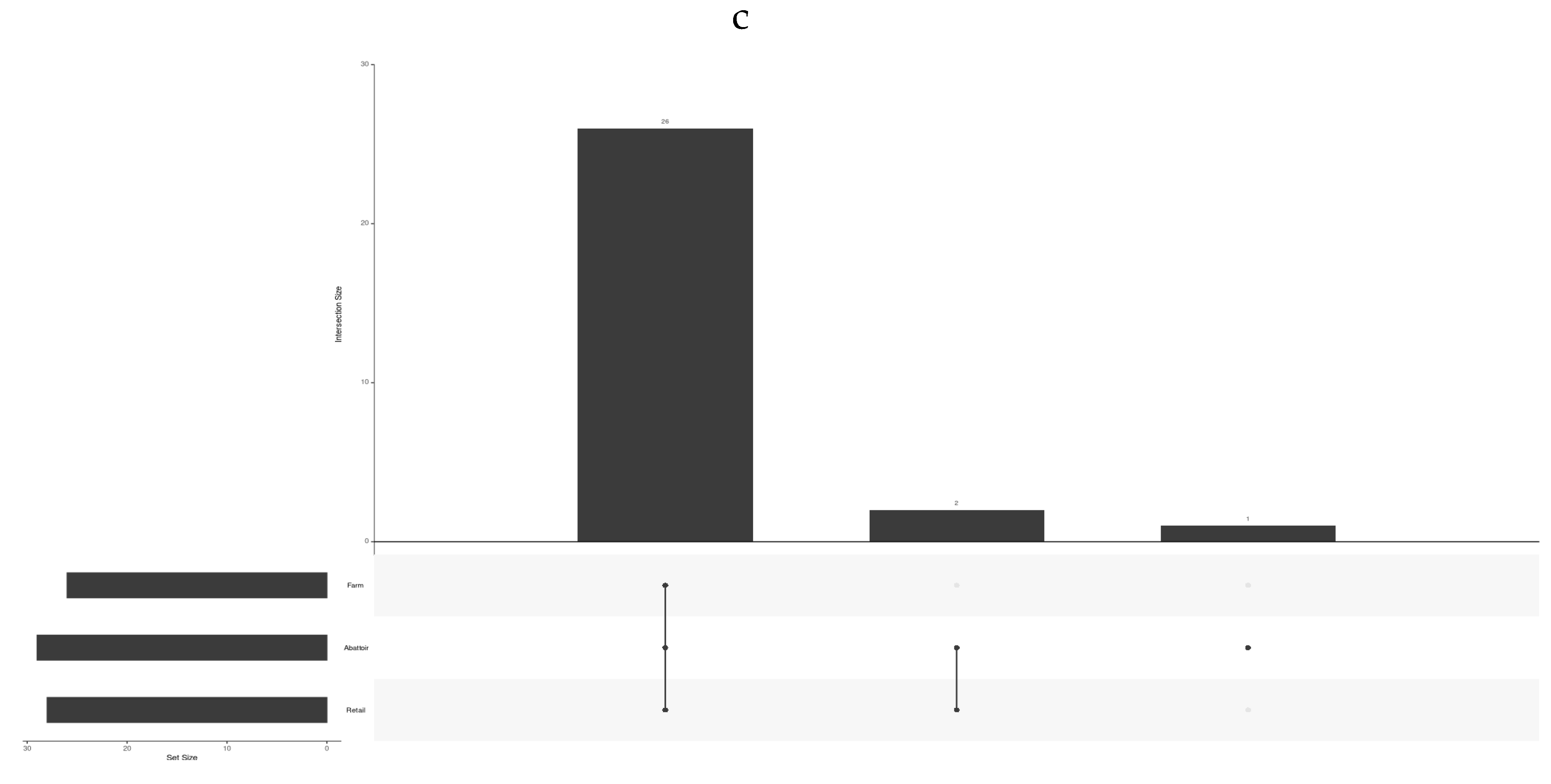
Figure 7.
Minimum spanning tree based on presence/absence of virulence factors for L. monocytogenes detected across the different industries. Each isolate is represented by sequence type as text and coloured by industry. This is similar to one based on ST, but of interest is the cluster on the left. Previously, ST1 and ST876 clustered independently, but they clustered together with the virulence factor tree. In the ST tree, ST224 is found within the ST876 cluster, but now it groups individually. From this, the virulence profiles for ST876 and ST1 are very similar.
Figure 7.
Minimum spanning tree based on presence/absence of virulence factors for L. monocytogenes detected across the different industries. Each isolate is represented by sequence type as text and coloured by industry. This is similar to one based on ST, but of interest is the cluster on the left. Previously, ST1 and ST876 clustered independently, but they clustered together with the virulence factor tree. In the ST tree, ST224 is found within the ST876 cluster, but now it groups individually. From this, the virulence profiles for ST876 and ST1 are very similar.
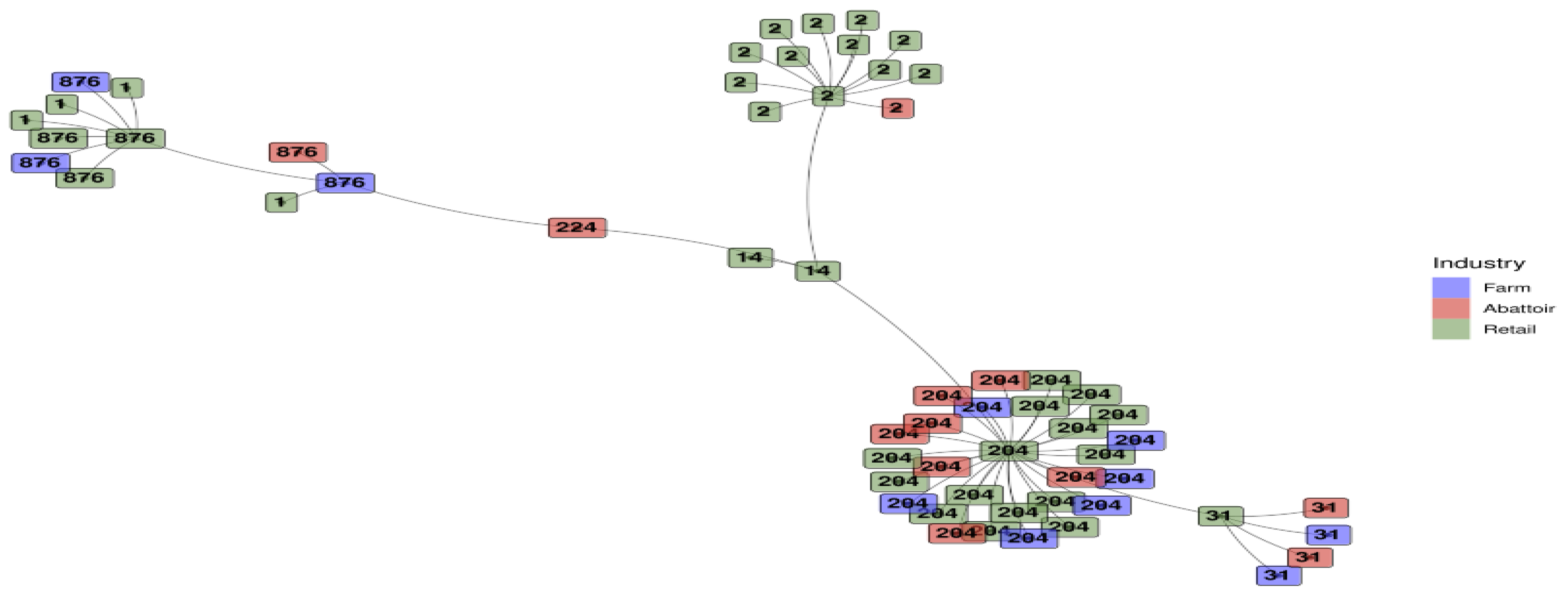
Figure 8.
Minimum spanning tree based on the presence/absence of virulence factors for L. monocytogenes detected across the Sample and food types. Each sample is represented by sequence type as text and coloured by industry. This tree is similar to one based on ST, but the cluster on the left is interesting. Previously, ST1 and ST876 clustered independently, whereas with the virulence factor tree, they clustered together. In the ST tree, ST224 was found within the ST876 cluster, but now it groups individually. From this, the virulence profiles for ST876 and ST1 are very similar.
Figure 8.
Minimum spanning tree based on the presence/absence of virulence factors for L. monocytogenes detected across the Sample and food types. Each sample is represented by sequence type as text and coloured by industry. This tree is similar to one based on ST, but the cluster on the left is interesting. Previously, ST1 and ST876 clustered independently, whereas with the virulence factor tree, they clustered together. In the ST tree, ST224 was found within the ST876 cluster, but now it groups individually. From this, the virulence profiles for ST876 and ST1 are very similar.
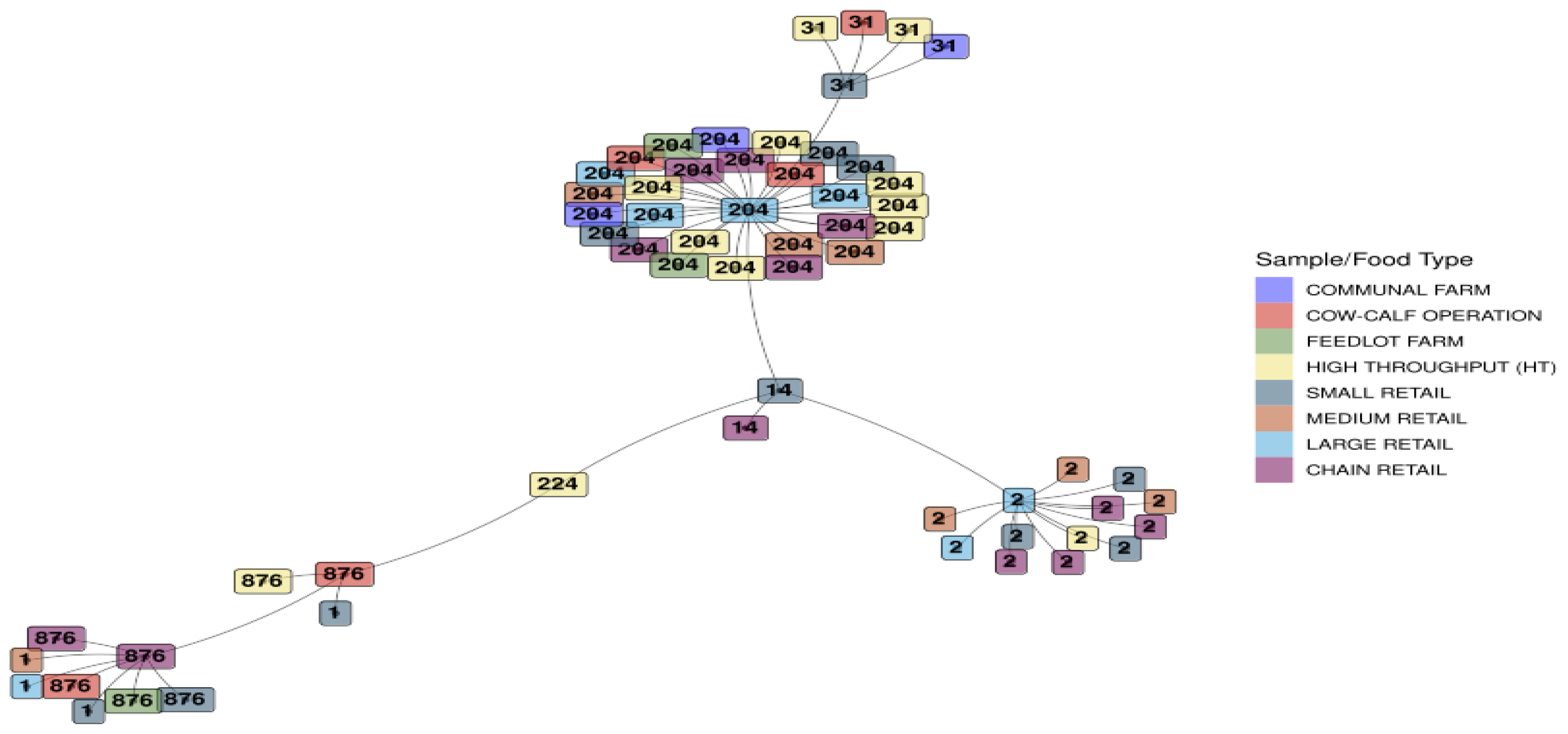
Figure 9.
Provirus/phage and AMR co-location (Provirus or phage as classification).
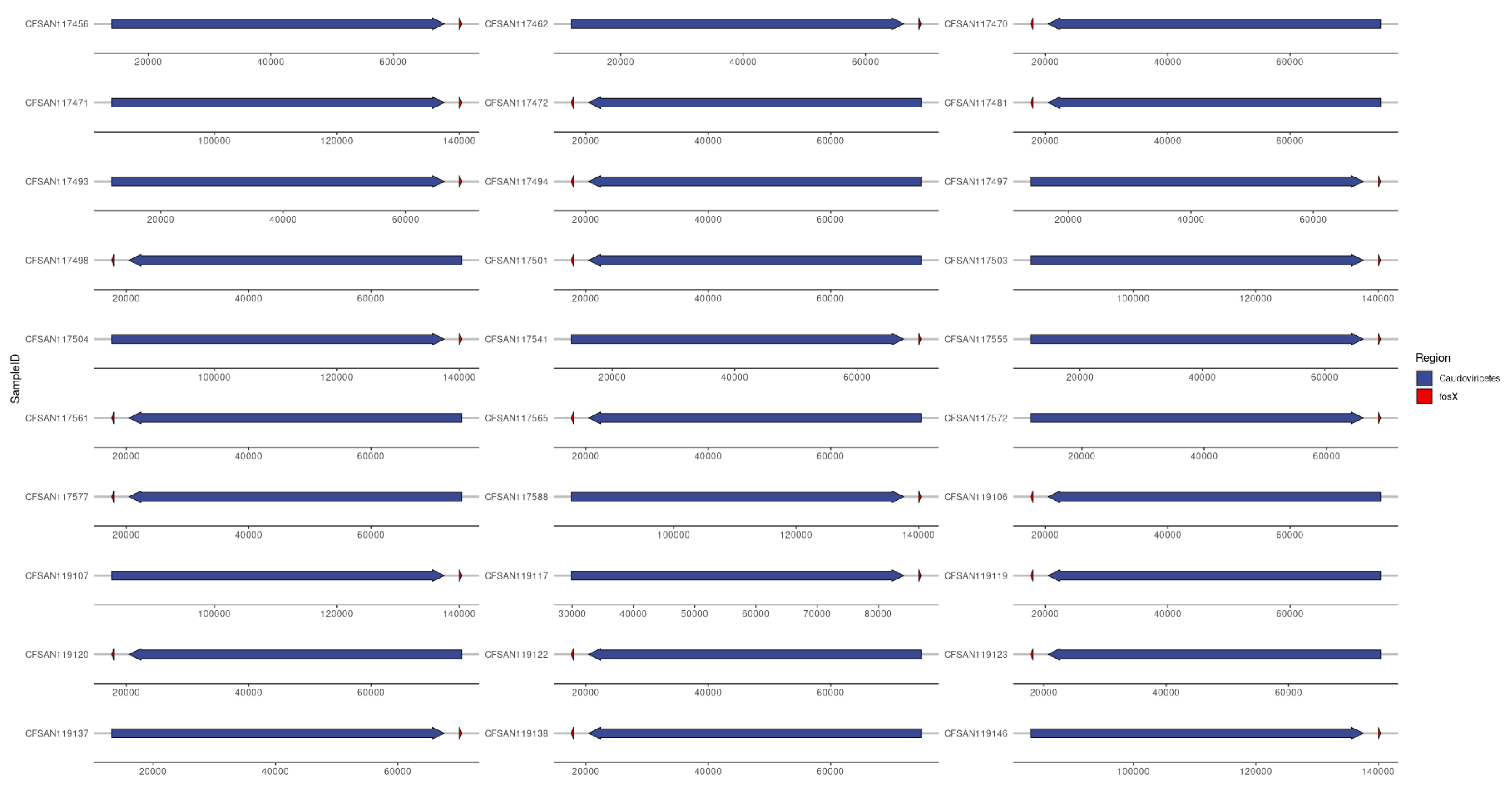

Table 1.
Characteristics of L. monocytogenes isolates according to the three industries (farm, abattoir, and retail) according to STs and AMR genes.
Table 1.
Characteristics of L. monocytogenes isolates according to the three industries (farm, abattoir, and retail) according to STs and AMR genes.
| Industry | No. of samples tested |
No. (%) positive for L. monocytogenes |
No. of isolates tested |
No. (%) of isolates L. monocytogenes that belong to ST: | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ST1 | ST2 | ST14 | ST31 | ST204 | ST224 | ST876 | ||||
| Cattle farms a | 328 | 11 (3.4) | 11 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (18.2) | 6 (54.5) | 0 (0.0) | 3 (27.3) |
| Abattoirs b | 262 | 12 (4.6) | 12 | 0 (0.0) | 1 (8.3) | 0 (0.0) | 2 (16.7) | 7 (58.3) | 1 (8.3) | 1 (8.3) |
| Retail outlets c | 400 | 37 (9.3) | 37 | 4 (10.8) | 12 (32.4) | 2 (5.4) | 1 (2.7) | 15 (40.5) | 0 (0.0) | 3 (8.1) |
| p-value | 0.002 | 0.560 | 0.044 | 1 | 0.134 | 0.475 | 1 | 0.204 | ||
| Total | 990 | 60 (6.1) d | 60 | 4 (6.7) | 13 (21.7) | 2 (3.3) | 5 (8.3) | 28 (46.7) | 1 (1.7) | 7 (11.7) |
a Comprising communal farms (n = 10; 3 isolates), cow-calf farms (n = 10; 5 isolates), and feedlots (n = 3; 3 isolates). b Abattoirs consisting of high-throughput, HT (n = 6; 12 isolates), low throughput, LT (n = 2; 0 isolates). c Retail outlets made up of chain outlets (n = 30; 12 isolates), large (n = 10; 7 isolates), medium (n = 6; 7 isolates), and small (n = 2; 11 isolate). d All 60 isolates of L. monocytogenes from the three industries were positive for AMR genes, fosX, and lin.
Table 2.
Occurrence of serogroups, STs, virulence factors, clonal complexes, AMR genes, plasmids, AMR plasmid CRISPR-Cas, and proviruses in L. monocytogenes recovered from RTE beef products.
Table 2.
Occurrence of serogroups, STs, virulence factors, clonal complexes, AMR genes, plasmids, AMR plasmid CRISPR-Cas, and proviruses in L. monocytogenes recovered from RTE beef products.
| Characteristics of seven L. monocytogenes isolates recovered from RTE products: | |||||||
|---|---|---|---|---|---|---|---|
| Isolate ID# | CFSAN117454 | CFSAN117466 | CFSAN117473 | CFSAN117497 | CFSAN117498 | CFSAN117503 | CFSAN117545 |
| Source | Chain-Retail | Chain-Retail | Chain-Retail | Chain-Retail | Large-Retail | Medium-Retail | Small-Retail |
| Sample type | Vienna | Biltonga | Beef polonyb | Beef polonyb | Beef polonyb | Beef polonyb | Beef polonyb |
| Serogroup | IVb | Ivb | IVb | 11a | 11a | 11a | IVb |
| MLST | 876 | 2 | 2 | 204 | 204 | 204 | 1 |
| Clonal complex | 1 | 2 | 2 | 204 | 204 | 204 | 1 |
| No. of virulence factors | 39 | 32 | 32 | 35 | 34 | 34 | 39 |
| LIPI-1 | Positivec | Positive | Positive | Positive | Positive | Positived | Positivee |
| Internalins A&B | Positive | Positive | Positive | Positive | Positive | Positive | Positive |
| LIPI-3 | Positive | Negative | Negative | Negative | Negative | Negative | Negative |
| Othersf | Positive | Positive | Positive | Positive | Positive | Positive | Positive |
| AMR gene: FosX | Positive | Positive | Positive | Positive | Positive | Positive | Positive |
| AMR gene: vga(G) | Positive | Positive | Positive | Positive | Positive | Positive | Positive |
| AMR Plasmid | Negative | Negative | Negative | Negative | Negative | Negative | Negative |
| CRISPR-cas | Negative | Negative | Negative | Negative | Negative | Negative | Negative |
| Proviruses | Positive | Positive | Positive | Positive | Positive | Positive | Positive |
| aBiltong: A delicacy made of spiced dried raw meat (beef and game) widely consumed in the country | |||||||
| bBeef polony: A popularly consumed product responsible for the 2018-2019 large outbreak of human listeriosis in South Africa | |||||||
| cOf the six LIPI-1 virulence, negative for the actA gene | |||||||
| dOf the six LIPI-1 virulence, negative for the hly gene | |||||||
| eOf the six LIPI-1 virulence, negative for the actA gene | |||||||
fOthers include virulence factors responsible for adherence, surface protein anchoring, invasion, stress-related, immune modulation, and intracellular survival.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



