
Submitted:
18 March 2024
Posted:
20 March 2024
You are already at the latest version
Abstract
Atherosclerosis, a complex metabolic-immune disease characterized by a chronic inflammation driven by the buildup of lipid-rich plaques within arterial walls, has emerged as a pivotal factor in the intricate interplay between cancer and cardiovascular disease. This bidirectional relationship, marked by shared risk factors and pathophysiological mechanisms, underscores the need for a comprehensive understanding of how these two formidable health challenges intersect and influence each other. Cancer and its treatments can contribute to the progression of atherosclerosis while atherosclerosis with its inflammatory microenvironment can exert profound effects on cancer development and outcomes. Both cancer and cardiovascular disease involve intricate interactions between general and personal exposomes. In this review we want to summarize the state-of-the-art of translational data and try to show how oncologic studies on cardiotoxicity can broaden our knowledge of crucial pathways in cardiovascular biology with a positive impact on precision cardiology and cardioncology.
Keywords:
atherosclerosis
; cardiovascular disease (CVD)
; cancer
; inflammation
; immune system
; endothelium
; exposome
1. Introduction
Cardiovascular disease (CVD) and cancer are the leading causes of morbidity and mortality worldwide [1]. CVD, the most common noncommunicable condition, is responsible of almost one third of all deaths globally [2]. In Europe CVD is the most common cause of death with more than 60 million potential years of life lost annually. Mortality is higher in women compared to men while age-standardized rates of morbidity and death from CVD are higher in men than in women [3]. In 2020 a global estimation of 19.3 million new cancer cases and of almost 10.0 million cancer deaths has been made. Female breast cancer (BC) is the most commonly diagnosed cancer, with an estimated 2.3 million new cases, lung cancer is the leading cause of cancer death, with an estimated 1.8 million deaths [4]. The global cancer burden is increasing, it is expected to be 28.4 million cases in 2040, that is a 47% rise from 2020. Recent epidemiological evidence underscores that CVD and cancer are supported by shared risk factors and that 40% of cancer cases are indeed favored by environmental factors, the most important of which are well-known manageable cardiovascular risk factors (CVRFs) such as smoking, alcohol, and an unhealthy lifestyle linked to obesity [5]. Furthermore, a high-risk score for atherosclerotic cardiovascular disease (ASCVD) predicts the future development of neoplasms [6], as well as the presence of coronary calcifications at CT scan [7]. The sharing of the same risk factors between cancer and ASCVD suggests the existence of common pathogenetic mechanisms in which chronic inflammation plays a major role [8]. Among patients receiving contemporary statins therapy, inflammation assessed by high-sensitivity C-reactive protein (hs-CRP), marker of activated innate immune system, is a stronger predictor for risk of future cardiovascular events and death than cholesterol assessed by LDL-C [9,10]. These observations on a large number of patients support the results of clinical trials with anti-inflammatory agents such as monoclonal antibodies against interleukin (IL)-1β and IL-6 and colchicine in patients with ASCVD [11,12,13,14,15]. An unexpected data for canakinumab was the lower incidence of lung cancer [16]. This preliminary observation is coupled with the reduced incidence of cancer in gout patients treated with colchicine indicating the importance of inflammation and innate immunity in the development of tumors [17]. In addition, recent data indicate a potentially significant role of the inflammatory state in predicting prognosis and response to immunotherapy in cancer patients [18,19]. Increasing evidence from real world indicates that vascular damage and related ASCVD can pose a significant problem for the outcome of current cancer treatments.
Aim of this review is to summarize translational and clinical data on ASCVD and cancer and the impact of these data on precision cardiology and cardioncology, with a focus on preventive strategies.
2. The Changing Paradigm of Atherosclerosis and Related Cardiovascular Disease
Atherosclerosis is a chronic vascular disease characterized by the presence of fibroinflammatory lipid plaques in large and medium-sized arteries. Although clinical ASCVD usually affects the elderly, it is well known that the process begins in utero and evolves silently throughout life, depending on the genetic predisposition and, to a greater extent, by acquired factors due to certain behaviors and exposure to environmental factors. Over decades a lot of research in clinical medicine, pathology, epidemiology, and public health has led to the identification and characterization of environmental factors, biological agents, disease conditions, and genetic states that increase the likelihood of atherosclerosis, the main cause of CVD. To encourage an inclusive treatment paradigm of promoting and preserving cardiovascular health throughout life in populations and individuals, in 2010 the American Heart Association (AHA) defined “healthy diet, exercise, nicotine avoidance, healthy weight, healthy values of blood pressure, blood lipids, blood glucose” as “Life’s simple 7”: 7 parameters to maintain CV health [20]. In 2022 the AHA “Life’s Simple 7” was upgraded to “Life’s Essential 8” adding “healthy sleep” as an essential component of CV health, and emphasizing the crucial role of psychological and sociological issues [21]. There is indeed an increasing interest in the link between psychological health/well-being/social determinants of health and CV health due to a favorable effect of factors such as optimism, purpose in life and resilient coping and a negative effect of psychosocial stress and depression [22,23]. Moreover, some chronic stress-related pathways such as inflammatory response, glucose and lipid homeostasis, and coagulation have been identified [24]. At the same time, a growing interest in the concept of "exposome" is arising in the field of preventive cardiology to underline the importance of non-traditional risk factors in the development/progression of atherosclerosis [25,26] and in connected clinical chronic condition (e.g. diabetes) [27]. The term “exposome” was coined in 2005 by Wild [28] to weigh the impact of exposure to environmental agents on molecular pathways (genetic configuration) that, by changing the internal homeostasis, lead to chronic diseases. The exposome is an aggregation of general enviromental exposome (air, soil, light, radiation, chemical and noise pollution) and personal exposome related to individual/lifestyle factors (exercise, diet, infection, drug, stress, sleep deprivation, intestinal flora) [29]. The reasons for such interest are certified by 2021 Global Disease Burden data where unhealthy “dietary risk” and air pollution have passed cigarette smoking in the ranking of CV risk factors [30] and by the newest evidence of clinical impact of microplastics and nano-plastics contaminant in atherosclerotic plaques [31,32]. Almost all the components of the exposome raise the endocrine-sympathetic response to stress, alter the metabolic state in the obesogenic sense, increase oxidative stress and inflammation [33,34,35], furthermore they affect haematopoiesis, the circulating leukocytes, and the recruitment of monocytes in the atherosclerotic plaque [36]. In the last analysis, the trajectory of CVD is increasingly recognized as being shaped by intricate interactions between polygenic factors and environmental influences. However, unlike blood pressure, blood sugar, or blood lipid levels, practical and universally recognized tools are currently lacking to incorporate the impacts of environmental (such as pollution), socio-economic (including social deprivation, limited access to a healthy diet, opportunities for regular physical activity, and suboptimal healthcare), and psychological factors (such as stress, anxiety, and depression) into individual patient management [21,37]. As precision biomedicine advances and genomic science expands our understanding of biological systems and molecular networks underlying CVD, it is likely that we will develop tools to address these complexities. Novel methods such as immune phenotyping and "multi-omic" characterization of the epigenome, transcriptome, metabolome, and microbiome hold promise for accelerating the discovery of biomarkers and identifying individuals at higher risk. Now we can only consider their presence as "modifiers" of the risk ascertained by the classic parameters and modulate accordingly, if possible, the treatment plan for the individual patient. What we observe clinically, indeed, is that over time the landscape of atherosclerosis has changed [38]. A key factor is the considerable increase in obesity, usually associated with hypertension, and related metabolic changes such as insulin resistance, metabolic syndrome, type 2 diabetes and change in lipid pattern characterized by low high-density lipoprotein (HDL) cholesterol and elevated triglycerides-rich lipoprotein [39,40,41,42,43,44]. Nowadays traditional CVRFs may fail to predict CVD in the individual patient. In PESA (Progression of Early Subclinical Atherosclerosis) study, an ongoing longitudinal cohort study integrating serial imaging, biological and behavioral parameters associated with the progression of subclinical atherosclerosis, 63% of the 4184 enrolled asymptomatic subjects (average age 46 years) had, at the basal examination, atherosclerosis in one or more vascular districts (iliofemoral, carotid, coronary) and a third of them had a low risk according to traditional risk estimates (Framingham Heart Score < 10%) [45]. Besides confirming the concept that "lower is better" for cholesterol, the PESA study highlighted the role of triglycerides (TG) in atherosclerosis and vascular inflammation in low and moderate risk patients [46] and this result was recently confirmed in other registries [47,48]. In clinical practice, one of the most frequent causes of hypertriglyceridemia is associated with insulin resistance in type 2 diabetes mellitus. High levels of TG are a component of metabolic syndrome and are associated with the development of atherosclerosis in non-alcoholic fatty liver disease (NAFLD), where TG and remnants play a significant role. The growth of an atherosclerotic plaque is a chronic dynamic inflammatory process with many different players. Early in the atherogenesis, high levels of oxidized low-density lipoproteins (ox-LDL) and remnants of triglyceride-rich lipoproteins gain access to the subendothelial space eliciting a danger signal that activate the NOD [nucleotide oligomerization domain]-,LRR[leucine-rich repeat]-,and PYD [pyrin domain]-containing protein3 (NLRP3) inflammasome [49,50] in innate immune cells. The activated inflammasome starts a cascade of inflammatory cytokines (IL-1β, IL-6) and upregulates high-sensitivity C-reactive protein (hs-CRP) in the liver that eventually leads to atherosclerotic lesions. These processes disrupt integrity of ECs and their atheroprotective attitude that includes secretion of many vasoactive substances affecting vasodilatation, platelet function and monocyte infiltration. As a consequence, the activated “inflamed” ECs steer the recruited immune cells towards the adoption of their proinflammatory phenotypes expanding the process of tissue inflammation [51,52]. Prolonged stimulation of ECs may also contribute to the endothelial-to-mesenchymal transition that leads to fibrosis [53]. The recruitment of the immune cells has a pivotal role in atherosclerosis and seems to be the target to treat residual CV risk (beyond the lipid lowering therapies). For this reason monocytes, macrophages, dendritic cells, neutrophils, T cells and B cells are all deeply involved in the inflammatory scenario [54]. Their behaviour (how they move and change shape) comes from genetic and signalling networks [55]. In the last decades a growing interest in microbial stimuli- and endogenous ligand- induced upgrading of innate immune cells that allows an increased response against a secondary stimulation (the so-called “trained immunity”) has been widely documented. Unfortunately, “trained immunity” may have a negative effect, too. When activated in an inappropriate way it can lead to inflammatory and autoimmune diseases; . trained immunity, for example, confers a proatherogenic phenotype to monocytes and macrophages [56,57,58]. In conclusion, there has been a paradigm shift in atherogenesis: the historical view of a single culprit agent inducing atherosclerosis (either lipoproteins [59] or inflammation [60]) has been upgraded to a complex metabolic-immune process in which a metabolic switch of ECs starts the inflammatory process that involves many immune and vascular cells. Moreover, the complexity of the challenges to the ECs has also expanded in these last decades and noxious substances or abnormal hemodynamic stresses are assembled in the exposome concept [25]. The intriguing chameleonic activity of vascular cells (ECs [51,52,58,61], pericytes [62,63,64,65], smooth muscle cells [66,67,68,69]) and immune cells (dendritic cells [70], monocytes [71,72], macrophages [73,74,75,76,77], T cells [78,79,80], B cells [81,82,83], polymorphonuclear neutrophils (PMNs) [84,85,86]) that adapt their phenotypes to the multifaceted scenario of atherosclerosis is summarized in Table 1.
The remarkable inflammatory features of atherosclerosis explain the beneficial effect of drugs that target the inflammasome pathway, but a “focused” cytokine inhibition is necessary, as Ridker states, to yield an effective atheroprotective effect [87,88]. Some of the trials on the atheroprotective effects of anti-inflammatory agents are summarized in Table 2.
In the conundrum of atherosclerosis there are risk-enhancing factors such as Lp(a) [89,90] and the microbiome [91,92]. Lp(a) seems to be the strongest independent genetic risk factor for myocardial infarction and aortic stenosis [93] and is associated to increased mortality [94]. There are also somatic mutations in haematopoietic stem cells with subsequent clonal expansion of haematopoietic cells. Clonal hematopoiesis of indeterminate potential (CHIP) is associated with an expected 0.5-1.0% risk per year of leukemia but with an unexpected two-fold increase in cardiovascular risk independent of traditional risk factors. CHIP has to be considered a new CVRF [95,96,97] and plays an important role in increasing CV complication in cancer patients [98]. Another interesting component of the atherosclerotic cardiometabolic derangements is the epicardial adipose tissue (EAT), that can be both useful for the myocardium (offering a thermogenic function) and dangerous through its paracrine or vasocrine secretion of pro-inflammatory and profibrotic cytokines. EAT changes with age and in pathologic conditions and is considered pro-atherogenic for coronary arteries [99,100].
The complexity of the inflammation/immunity process in acute myocardial infarction (AMI) has been recently translated in a western movie play with “Good”, “Bad” and “Ugly” characters. The “Good” players are T-cells, natural killer cells (NKs) and macrophages that protect and heal the myocardium along with the good cytokines (IL-10 and IL-2) that decrease pro-inflammatory signals [Tumour Necrosis Factor α (TNFα), monocyte chemoattractant protein-1 (MCP-1), IL-8], reduce extracellular matrix remodelling and promote the activation of regulatory T cells (T reg), type 2 helper T-cells (Th2), and NKs that favors the protective M2 phenotype of macrophages. The “Bad” players are the M1 macrophages, the foam cells and PMNs, all together they maintain a low-grade inflammation status in the late phase after AMI inducing the NLRP3 inflammasome and increasing the production of the bad IL-1α, IL-1β. The “Ugly” players are the activated PMNs, the recruited monocytes, IL-1 and IL-6; they act in the early phase after AMI soon after plaque rupture and thrombosis, this process may be amplified by NET formation [101]. Some of these characters may be applied to the process of atherosclerosis, too, as shown in Table 1 and in the Central illustration.
3. Atherosclerosis and Cancer: The Unexpected Link
Over the years, community studies have documented that adherence to the 7 ideal health metrics defined in the AHA goals [20] is associated with lower cancer incidence [6,102,103,104,105]. Cancer and CVD are intertwined [106,107,108,109] by the sharing of the same risk factors (see Koene [110] for more details) and of the fundamental physio-pathological mechanism that is represented by chronic inflammation.
4. Atherosclerosis and Cardio-Oncology: The Bidirectional Relationship Has Highlighted the Novel Issues That Need to Be Addressed
4.1. The Shared Risk Factors
(obesity, hypertension, diabetes, smoking, dyslipidemia, physical inactivity and sedentary behaviour, unhealthy diets, alcohol abuse, impaired immune response, metabolic remodeling, CHIP)
4.2. The Shared Pathways
The hallmarks of cancer (sustained proliferation, resistance to cell death, neurohormonal stress, genomic instability and inflammation) [111] have a pivotal role in CVD, too. The following biological processes shared by cancer and CVD and the candidate biomarkers that represent these shared biological processes are illustrated according to a seminal review by Narayan et coll. [112]
- Inflammation, an epiphenomenon of immune dysregulation and cell senescence, is associated to increased levels of hsCRP, IL-6 and suppression of tumorigenicity 2 (ST2). These biomarkers are linked to tissue invasion and metastasis associated with cancer, but also to tissue damage that underlies the atherosclerotic process [113,114].
- Resistance to cell death is linked to cellular stress response and apoptosis, its biomarker (the Growth differentiation factor) has a prognostic role in cancer mortality, but also in CVD (myocardial infarction, thromboembolic stroke, heart failure and stroke) [117,118]; cardiac Troponin T (cTnT), a well known marker of myocardial cell death, is also a useful marker in light chain amyloidosis [119,120].
- Neurohormonal stress leads to increased levels of cardiovascular neurohormones such as N-terminal pro-B-type natriuretic peptide, mid-regional pro-atrial natriuretic peptide and other neurohormones and diuretic hormones. These cardiovascular neurohormones have a relevant role in patients with acute or chronic heart failure (HF), but they might play a role in cancer, too, since they may be produced by some malignant cells in the vascular bed of the tumour [121,122].
- Angiogenesis with a role in EC survival and in tumorigenesis, invasion and metastasis may be measured by angiogenic biomarkers such as soluble fms-like tyrosine kinase 1, a variant of Flt-1 known also as Vascular Endothelial Growth Factor (VEGF) receptor and placental growth factor (PlGF). VEGF biology has a relevant impact on tumorigenesis and on normal cardiovascular function [123,124].
- Genomic instability such as CHIP, has an impact on both CVD and cancer. CHIP [125] is a risk factors for CVD [95], but it may be caused by atherosclerosis due to the continuous stimulation of stem cell proliferation [126]. As a of facts this reverse CHIP effect has uncovered an unexpected link between oncogenesis and atherogenesis [38] The biological explanation of the impact of a potentially pre-cancerous lesion on CVD involves an interaction between clonally-derived monocytes and macrophages and the vascular endothelium that leads to vascular inflammation and accelerated atherogenesis [95,125].
The shared pathways are illustrated in the Central illustration
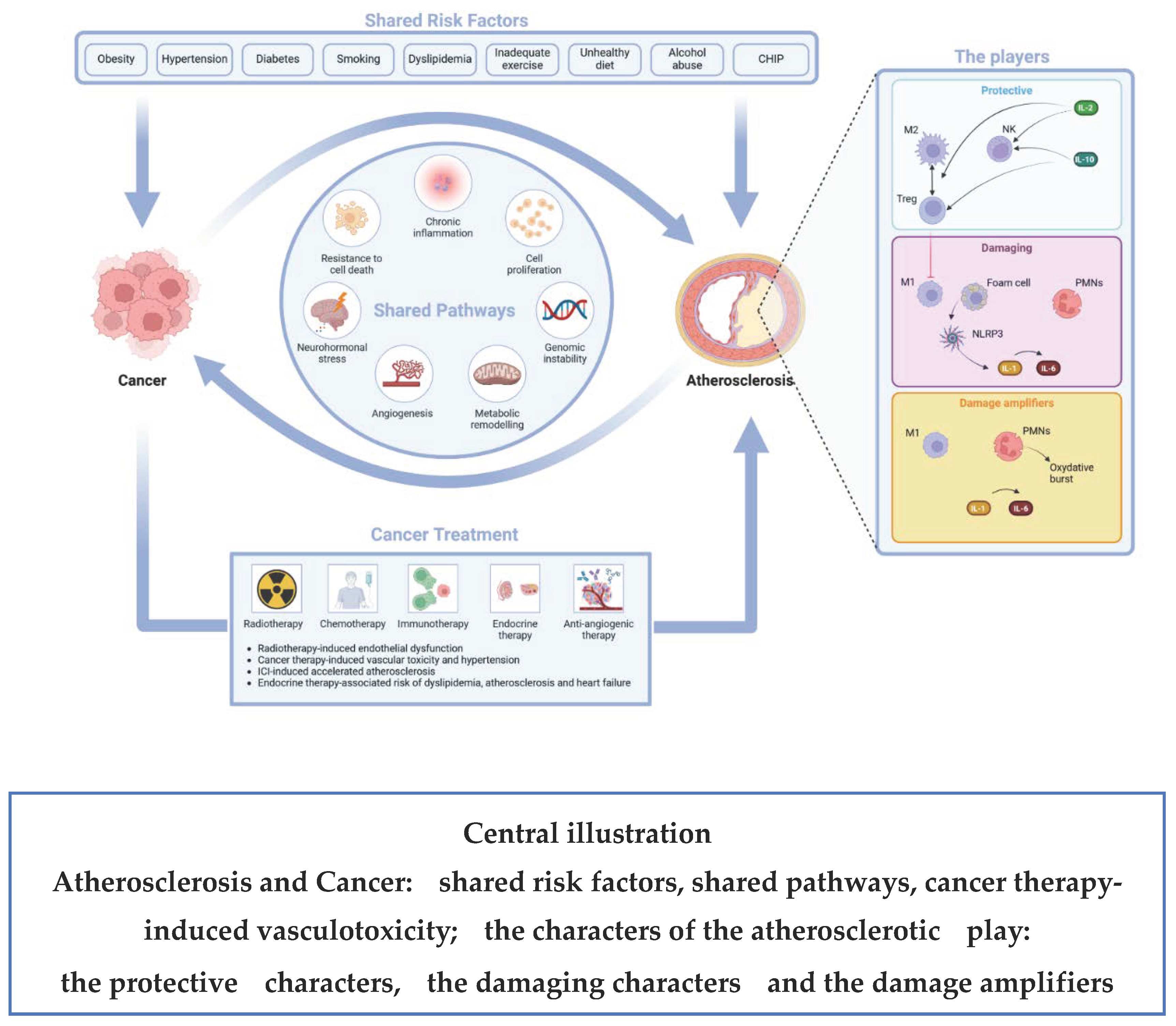
4.3. The Atherogenic Effect of Some Oncologic Treatments
- Radiotherapy-induced endothelial dysfunction. Radiation-associated damage induces the secretion of pro-inflammatory cytokines, increases the release of reactive oxygen species (ROS), causes a dysregulation of glycolysis, lipid metabolic pathways and angiogenesis and may also have a negative impact on telomere function and immunity homeostasis. The final effect is EC death (either acutely via apoptosis or chronically via EC senescence) and a disrupted EC environment [127]. Clinical phenotypes of Radiation-associated vascular damage are: accelerated CAD; cerebral events due to carotid artery disease; calcification of the ascending aorta and aortic arch and lesions of other vascular segments in the radiation field [128]. The recent BACCARAT study evaluated the association between cardiac exposure and the risk of developing calcified and non-calcified atheromatous plaques within 2 years of RT. As both calcified and non-calcified plaques were found it may be hypothesized that cardiac radiation exposure accelerates the process of atherosclerosis in already existing plaques with an increase of their calcium content (calcified plaques) and starts new non-calcified plaques [129].
- Cancer therapy-induced vasculotoxicity It is associated to traditional chemotherapies (alkylating agents, microtubule inhibitors and antimetabolites), to targeted therapies such as VEGF-Inhibitors, to Breakpoint cluster region–Abelson oncogene locus tyrosine kinase inhibitors and to multiple myeloma drugs. The majority of these drugs induce hypertension that may eventually drive atherosclerosis, but there is also a possible CV injury due to damage-associated molecular patterns (DAMPs) that sustain inflammation. There are many clinical phenotypes of vasculotoxicity that include CAD, stroke, systemic and pulmonary hypertension, vasospasm and thrombosis [130,131].
- Accelerated atherosclerosis induced by immune checkpoint inhibitor (ICI) treatment. Oncologic studies of ICI-induced cardiotoxicity have indeed shed light on the complex relationship between the immune system, inflammation and atherosclerosis. Preclinical studies have shown that the targets of ICIs [PD-1 (programmed cell death protein 1), PD-L1 (programmed death ligand 1), CTLA-4 (cytotoxic T-lymphocyte–associated protein4) are proteins with a negative regulatory role in atherosclerosis [132]. The blockage of the checkpoints may predictably lead to accelerated atherosclerosis through enhanced T cell responses, limited Treg function and infiltration of the vascular endothelium [133,134,135,136]. Pre-clinical studies have also shown that short-term ICI treatment promotes DAMPs and pro-inflammatory cytokine production [137]. In the clinical setting, a seminal study in 2842 patients and 2842 controls matched by age, a history of cardiovascular events, and cancer type has shown a 3-fold increase in atherosclerotic cardiovascular events (myocardial infarction, coronary revascularization and ischemic stroke ) after starting an ICI treatment. Moreover, in a case-crossover analysis performed by the same authors comparing an at-risk period (defined as the 2-year period after ICIs) and a control period (defined as the 2-year period before ICIs), atherosclerotic cardiovascular events significantly increased from 1.37 to 6.55 per 100 person-years at 2 years; in a subgroup of 40 patients a 3-fold higher rate of aortic plaque progression was also documented [138]. In a more recent retrospective cohort study on 1458 patients diagnosed with stages III or IV non small cell lung cancer (NSCLC) treated with (487 patients) and without (971 patients) ICI therapy and followed-up for a median time of 23.1 months, ICI therapy was associated with a 3.6-fold increase in the total risk of ASCVD events before Propensity score matching [139].
- hormone therapy-associated risk of dyslipidemia, atherosclerosis and heart failure. This effect has been proven in BC women treated with aromatase inhibitors that increase the risk of atherosclerosis, HF and dyslipidemia [140] and in prostate cancer patients treated with androgen deprivation therapy (ADT) in which the increased risk of CV events is linked to indirect modifications of CVRFs. More specifically Gonadotropin releasing hormone (GnRH) agonists increase LDL-cholesterol and triglyceride levels, visceral fat and insulin resistance, decrease lean body mass and glucose tolerance leading to accelerated atherosclerosis and coronary artery disease (CAD) events, HF and arrhythmias [141,142]. In preclinical studies orchiectomy and GnRH agonists, but not GnRH antagonists, induced long- or short-term follicular stimulating hormone elevation that, acting synergistically with TNF-α induced an amplified endothelial inflammation through elevation of vascular cell adhesion protein-1 expression thus accelerating atherosclerosis [143].
5. How Do We Measure and Quantify Atherosclerosis?
ASCVD can be imagined as a kind of continuum that originates from the earliest onset of vascular atherosclerosis at the cellular level, passes through a lasting, clinically silent, histological evolution, and eventually ends with the overt clinical complications. The time frame within which these processes are included can reach some decades, with long phases of quiescence alternating with more or less prolonged phases of instability. The ability to intercept in the individual patient the evolution of atherosclerosis before it becomes clinically manifest, and thus, given the wide time frame, ultimately to prevent its most dreaded complications, has always been and still is, one of the holy grails of CV medicine. This long path, which started from the epidemiological studies that since the 1950s have enabled the identification of the best-known CVRFs [144,145,146], has recently been enriched with more refined tools capable of identifying the factors that at the pathophysiological level initiate the atherosclerotic process and promote the transition from stable phases to acute events; increasingly sensitive and specific biomarkers of atherosclerosis; and, at the individual level, the patients who are at higher risk of acute clinical events. This goal is achieved thanks to diagnostic methods capable of investigating plaque composition and atherosclerotic lesions at higher risk of acute complications.
5.1. Risk Scores and Mendelian Randomization
Randomized controlled trials (RCTs) are considered the gold standard design to infer causality and this is true even for CVRF role in atherosclerosis development. However RCTs are expensive and time consuming, and often difficult to conduct particularly if, as is the case for CVRF identification, a long follow-up is needed. Other limits are represented by possible poor long-term compliance and ethical issues about random treatment allocation. Therefore, the relationship of modifiable CVRFs with CV events has mostly been investigated using observational study design, such as case-control studies and cohort studies. The limitations of this type of approach are the presence of confounding factors and the so-called reverse causation bias, that make any causal relationship that may be demonstrated less reliable, given that confounding factors and reverse causation bias can distort the findings. Observational and cohort studies conducted over the past 75 years, starting with the Framingham study, have enabled the construction of a number of risk scores that can be applied in different populations [147,148].
The most recent European guidelines on cardiovascular prevention in clinical practice [37] recommend the use of the Systematic Coronary Risk Evaluation (SCORE2) and of Systematic Coronary Risk Evaluation Older Persons (SCORE2-OP) systems. The risk cards in this system allow estimation of the 10-year risk of CVD in four European geographical risk regions. In Europe, maps have been developed for low, moderate, high and very high-risk regions. This rating system takes into account risk factors such as non-HDL cholesterol, systolic blood pressure, smoking, sex and age, drawing different risk categories (low, moderate, high, and very high) according to possibly associated additional CVRFs [149]. In recent years, in addition to the traditional CVRFs identified since the 1950s, several new risk factors for atherosclerosis have been identified, including many diseases that increase systemic inflammation, (such as gout, inflammatory bowel disease, autoimmune collagen vascular diseases, and psoriasis), some maternal (pre-eclampsia, delivering a child of low birth weight, preterm delivery, and premature or surgical menopause) and early childhood occurring factors (early-life trauma in young and middle-aged individuals with a history of AMI), and some lifestyle factors other than unbalanced diet and sedentary lifestyle, such as low socioeconomic status and air pollution.
Since 2003, with the pioneering works of Iacobellis et al., the presence of epicardial fat has also been increasingly proposed and better defined as a risk factor not only for coronary atherosclerosis but also for atrial fibrillation and HF in relation to its localization at the atrial or pericoronary level, respectively [150,151]. Adipocyte aggregates between the myocardium and the visceral layer of the epicardium have unique anatomic and functional characteristics, given the close proximity and interactions with the heart owing to the shared circulation and the absence of muscle fascia separating the two organs. EAT tissue can be clinically measured with cardiac imaging techniques that can help to predict and stratify cardiovascular risk [100].
Finally, to overcome the limitations of observational and cohort studies, genetics was used to better define the cause-and-effect relationships between CVRFs and CV events. Large scale genome wide association studies (GWAS) conducted over the last decades have identified numerous genetic variants associated with different cardiometabolic profiles and risk factors. From these discoveries came the so-called Mendelian randomization studies that use the genetic differences present in the study population as a "natural experiment" to improve inferences about the cause-and-effect relationship between hypothetical CVRFs and CV events derived from prior observational studies. Mendelian randomization studies have several advantages over RCTs, given that they are usually cheaper and faster to create since they are derived from existing, large scale, GWAS database.
5.2. Biomarkers
An ideal clinical biomarker should contain the following characteristics [152]: (1) clinical relevance, (2) sensitivity and specificity to treatment effects, (3) reliability, (4) practicality, and (5) simplicity. Based on these general principles, it is possible to profile the ideal biomarker for the atherosclerotic process (Table 3).
The atherosclerotic process involves multiple pathophysiological mechanisms. Inflammation of the vessel wall has long been recognized as central to the initiation of the atherosclerotic process, as well as to its progression and transition to stages of clinical instability. Not surprisingly, many biomarkers directly or indirectly related to the inflammatory process (Table 4) have been proposed as useful in monitoring atherosclerotic pathology, first in experimental models and then clinically. The endothelium can be considered the target organ of the pathophysiological processes that initiate atherosclerosis. A recent review points out the differences between an activated endothelium and a dysfunctional endothelium, the former being a very early stage of dysfunction [153]. Biomarkers of endothelial activation are: endothelial adhesion molecules, cytokines, C-reactive protein, CD62E+/E-selectin activated endothelial microparticles, oxidation of LDL, asymmetric dimethylarginine and endocan. Biomarkers of endothelial dysfunction are matrix metalloproteinases such as MMP-7, MMP-9, ANGPTL2, endoglin, annexin V, endothelial apoptotic microparticles and serum homocysteine. The recent discovery of exosomes both as diagnostic and therapeutic tools has boosted research on their use. Exosomes that derive from ECs may be responsible for the changing phenotype of vascular smooth muscle cells [154,155].
5.3. Imaging Techniques
Since coronary angiography in particular is an invasive, expensive, not widely available, and not risk-free procedure, clinicians and researchers have focused on two alternative strategies: the first has been the aforementioned study of biomarkers and genetic variants that can predict future atherosclerosis-related clinical events, while the second has been the study of atherosclerosis in more accessible vascular districts, such as the carotid and lower extremity arteries, using noninvasive methods, mainly ultrasound.
A unique feature is the ability to study calcifications in the coronary arteries.
- i.
- Coronary artery calcium (CAC) has become a useful tool to detect and quantify calcified subclinical atherosclerotic burden. The most widely used method to quantify CAC is the Agatston method expressed as the product of total calcium area and a quantized peak calcium density weighting factor defined by the calcification attenuation in Hounsfied units on non-contrast computed tomography [156], in addition CAC may be identified on scans scheduled for other reasons [157]. In the ITALUNG trial CAC was assessed in a baseline low-dose computed tomography performed in 1364 participants aged 59-69 years and a smoking history ≥20 pack-years in a lung cancer screening program with a follow up of 22 years. CAC score was graded as absent, mild, moderate and severe. In the study moderate or severe CAC was significantly associated with CV mortality after adjustment for traditional CVRFs [158]. CAC progression may be also a marker of accelerated atherosclerosis, as shown in a recent study regarding ICI therapy in cancer [139]. CAC score is unreliable with statin or PCSK9 inhibitor treatment [159,160].
- ii.
- Computed Tomography Angiography: the “actionable lump” concept (the detection of early subclinical atherosclerosis where intervention is still possible) and the importance of a proactive monitoring of silent atherosclerosis [161].
There is indeed a large body of literature examining the promise of imaging markers for early detection of subclinical CVD to ameliorate primary prevention approach. Available data are in favour of CAC as a strong marker of ASCVD risk. On the other hand, the absence of CAC or zero CAC has an even greater value as a negative predictive tool, with a remarkably favorable prognosis in older adults with guideline recommendations to consider deferral of lipid lowering therapies in subjects with zero CAC [162,163]. Another intriguing finding is breast arterial calcification (BAC), a form of medial artery calcification that can be detected on routine mammography, which has been proposed in recent years as a sex-specific imaging marker for the early detection of subclinical CVD. However, the pathophysiology of CAC, BAC, and calcium deposits at other cardiovascular sites (valve and aorta calcification) differs, as do their prognostic implications. For example, BAC is a form of medial artery calcification whose formation is regulated by the expression of osteogenic genes in a manner similar to bone tissue. Unlike the intimal calcifications expressed by CAC, BAC is formed independently of lipid deposition and macrophage activation, processes typical of classic atherosclerosis. However, these intimal calcifications are thought to contribute to endothelial dysfunction and reduced vascular compliance. Similarly, valvular calcifications are likely to result from mechanisms that combine classic atherosclerotic and more strictly osteogenic processes. Even if BAC, CAC, aortic calcification and valvular calcification are correlated with each other and all have been associated with CVD, we have to take into account the aforementioned heterogenous pathophysiologic processes involved in their origin, which could make a difference in their clinical utility as an early marker of atherosclerosis. In conclusion, we can say that BAC, as well as other areas of extracoronary calcifications, can be useful as a general cardiovascular risk marker, and in this sense its identification should lead to a vigorous implementation of Life Simple 7, having a positive impact on both CV risk and oncology risk. However, at present, the absence of BAC (zero BAC) does not have the same negative predictive value as zero CAC and is not equivalent to low CV risk [164].
In a 2024 published paper, Parveen Grag et al. [165] reported that carotid plaque burden, CAC, and low ankle/brachial pressure index were the only 3 tests that robustly predicted future atherosclerotic events in people of middle age or older. The authors conducted a critical review of measurements used to infer the presence of subclinical atherosclerosis in the major conduit vessels and focused on the predictive capability of these tests for future CV events (defined as stroke, myocardial infarction, and chronic ischemic limb disease) independent of the presence of conventional CVRFs. The authors preferred studies with > 10,000 person-years of follow-up, limited to carotid, coronary, aorta and lower limb arteries, and performed a meta-analysis of the results reporting adjusted hazard ratios (HRs) with 95% confidence intervals. In the carotid artery (8 studies), the presence of plaques was independently associated with future strokes (HR 1.89, 95% CI 1.04-3.44) and cardiac events (7 studies, HR 1.77, 95% CI 1.19-2.62). Coronary calcifications (5 studies) were found to be associated with acute coronary events (HR 1.54, 95% CI 1.07-2.07), while an increase in their severity, as expressed by the Agatston score, was associated with a significant increase in risk in 13 studies. Seven studies showed that an ankle-brachial index (ABI) < 0.9 was associated with an increased risk of cardiovascular death (HR 2.01, 95% CI 1.43-2.81).
What is new on the horizon for early atherosclerosis imaging?
Magnetic resonance imaging (MRI)
The ability of MRI to accurately detect the presence of lipid deposits, the sine qua non of atherosclerosis, in various vascular territories, has been known for more than 20 years [166]. Technological advances that will soon make more "portable" MRIs available, suggest a possible future use of this method also in large epidemiologic studies [167]. More recently, the combination of MRI and positron emission tomography (MRI-PET) has proven to be a promising imaging modality for studying the inflammation that underlies the various stages of the atherosclerotic process, with the possibility of whole-body studies [168].
The use of near-infrared spectroscopy, which chemically characterizes the plaque, in conjunction with various intravascular imaging techniques (most commonly intravascular ultrasound or optical coherence tomography) allows accurate study of the composition and characteristics of atherosclerotic plaques, although this method requires an invasive approach [169].
Limitations and unresolved questions
However, it should be remembered that there is currently no solid evidence to support specific therapeutic interventions in the presence of subclinical atherosclerosis. It is certainly very suggestive to think that aggressive interventions on CVRFs such as dyslipidemia or high inflammatory burden may be able to alter the natural history of preclinical atherosclerosis, but this needs to be demonstrated with ad hoc designed clinical trials, which will not be easy to conduct because they require adequate sample sizes and long follow-ups. In addition, to date, studies of subclinical atherosclerosis have typically focused on middle-aged or older populations (range 40 to 70 years). It is possible that the identification of subclinical atherosclerosis may be of even greater value in younger individuals, in whom it is likely that currently used tests may not be sensitive enough to identify the earliest atherosclerotic lesions. This, of course, also underscores the need to develop new diagnostic methods capable of identifying the earliest stages of the disease.
Funding
“This study was partially funded by Italian Ministry of Health – Ricerca Corrente Annual Program 2025”.
Conflicts of Interest
“The authors declare no conflicts of interest.” the results”.
References
- World Health Organization, WHO Department of Data and Analytics. WHO Methods and Data Sources for Global Burden of Disease Estimates, 2000-2019. W: Geneva, Switzerland, 2020.
- The Global Cardiovascular Risk Consortium; Magnussen, C. ; Ojeda, F.M.; Leong, D.P.; Alegre-Diaz, J.; Amouyel, P.; Aviles-Santa, L.; De Bacquer, D.; Ballantyne, C.M.; Bernabé-Ortiz, A.; et al. Global Effect of Modifiable Risk Factors on Cardiovascular Disease and Mortality. New Engl. J. Med. 2023, 389, 1273–1285. [Google Scholar] [CrossRef]
- Townsend, N.; Kazakiewicz, D.; Wright, F.L.; Timmis, A.; Huculeci, R.; Torbica, A.; Gale, C.P.; Achenbach, S.; Weidinger, F.; Vardas, P. Epidemiology of cardiovascular disease in Europe. Nat. Rev. Cardiol. 2021, 19, 133–143. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Tran, K.B.; Lang, J.J.; Compton, K.; Xu, R.; Acheson, A.R.; Henrikson, H.J.; Kocarnik, J.M.; Penberthy, L.; Aali, A.; Abbas, Q.; et al. The global burden of cancer attributable to risk factors, 2010–19: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 2022, 400, 563–591. [Google Scholar] [CrossRef]
- Lau, E.S.; Paniagua, S.M.; Liu, E.; Jovani, M.; Li, S.X.; Takvorian, K.; Suthahar, N.; Cheng, S.; Splansky, G.L.; Januzzi, J.L.; et al. Cardiovascular Risk Factors Are Associated With Future Cancer. JACC: CardioOncology 2021, 3, 48–58. [Google Scholar] [CrossRef]
- Dzaye, O.; Berning, P.; A Dardari, Z.; Mortensen, M.B.; Marshall, C.H.; Nasir, K.; Budoff, M.J.; Blumenthal, R.S.; Whelton, S.P.; Blaha, M.J. Coronary artery calcium is associated with increased risk for lung and colorectal cancer in men and women: the Multi-Ethnic Study of Atherosclerosis (MESA). Eur. Hear. J. - Cardiovasc. Imaging 2021, 23, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Kobold, S. Inflammation: a common contributor to cancer, aging, and cardiovascular diseases—expanding the concept of cardio-oncology. Cardiovasc. Res. 2019, 115, 824–829. [Google Scholar] [CrossRef]
- Ridker, P.M.; Bhatt, D.L.; Pradhan, A.D.; Glynn, R.J.; MacFadyen, J.G.; E Nissen, S.; PROMINENT, REDUCE-IT, and STRENGTH Investigators. Inflammation and cholesterol as predictors of cardiovascular events among patients receiving statin therapy: a collaborative analysis of three randomised trials. Lancet 2023, 401, 1293–1301. [Google Scholar] [CrossRef]
- Ridker, P.M.; Lei, L.; Louie, M.J.; Haddad, T.; Nicholls, S.J.; Lincoff, A.M.; Libby, P.; Nissen, S.E. ; on behalf of the CLEAR Outcomes Investigators Inflammation and Cholesterol as Predictors of Cardiovascular Events Among 13 970 Contemporary High-Risk Patients With Statin Intolerance. Circ. 2024, 149, 28–35. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Ridker, P.M.; Devalaraja, M.; Baeres, F.M.M.; Engelmann, M.D.M.; Hovingh, G.K.; Ivkovic, M.; Lo, L.; Kling, D.; Pergola, P.; Raj, D.; et al. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021, 397, 2060–2069. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.-C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.-F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef]
- Soehnlein, O.; Libby, P. Targeting inflammation in atherosclerosis — from experimental insights to the clinic. Nat. Rev. Drug Discov. 2021, 20, 589–610. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J.; Ridker, P.; Lorenzatti, A.; Krum, H.; Varigos, J.; et al. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.-C.; Chang, S.-J.; Hsieh, M.-C. Colchicine Significantly Reduces Incident Cancer in Gout Male Patients. Medicine 2015, 94, e1570. [Google Scholar] [CrossRef]
- Yang, T.; Hao, L.; Yang, X.; Luo, C.; Wang, G.; Cai, C.L.; Qi, S.; Li, Z. Prognostic value of derived neutrophil-to-lymphocyte ratio (dNLR) in patients with non-small cell lung cancer receiving immune checkpoint inhibitors: a meta-analysis. BMJ Open 2021, 11, e049123. [Google Scholar] [CrossRef]
- Han, C.-L.; Meng, G.-X.; Ding, Z.-N.; Dong, Z.-R.; Chen, Z.-Q.; Hong, J.-G.; Yan, L.-J.; Liu, H.; Tian, B.-W.; Yang, L.-S.; et al. The Predictive Potential of the Baseline C-Reactive Protein Levels for the Efficiency of Immune Checkpoint Inhibitors in Cancer Patients: A Systematic Review and Meta-Analysis. Front. Immunol. 2022, 13, 827788. [Google Scholar] [CrossRef]
- Lloyd-Jones, D.M.; Hong, Y.; Labarthe, D.; Mozaffarian, D.; Appel, L.J.; Van Horn, L.; Greenlund, K.; Daniels, S.; Nichol, G.; Tomaselli, G.F.; et al. Defining and setting national goals for cardiovascular health promotion and disease reduction: The American heart association’s strategic impact goal through 2020 and beyond. Circulation 2010, 121, 586–613. [Google Scholar] [CrossRef]
- Lloyd-Jones, D.M.; Allen, N.B.; Anderson, C.A.; Black, T.; Brewer, L.C.; Foraker, R.E.; Grandner, M.A.; Lavretsky, H.; Perak, A.M.; Sharma, G.; et al. Life’s Essential 8: Updating and Enhancing the American Heart Association’s Construct of Cardiovascular Health: A Presidential Advisory From the American Heart Association. Circ. 2022, 146, E18–E43. [Google Scholar] [CrossRef]
- Boehm, J.K.; Soo, J.; Chen, Y.; Zevon, E.S.; Hernandez, R.; Lloyd-Jones, D.; Kubzansky, L.D. Psychological Well-being’s Link with Cardiovascular Health in Older Adults. Am. J. Prev. Med. 2017, 53, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Ogunmoroti, O.; Osibogun, O.; Spatz, E.S.; Okunrintemi, V.; Mathews, L.; Ndumele, C.E.; Michos, E.D. A systematic review of the bidirectional relationship between depressive symptoms and cardiovascular health. Prev. Med. 2021, 154, 106891. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Z.; Wang, Y.-X.; Jiang, C.-L. Inflammation: The Common Pathway of Stress-Related Diseases. Front. Hum. Neurosci. 2017, 11, 316–316. [Google Scholar] [CrossRef] [PubMed]
- Münzel, T.; Sørensen, M.; Hahad, O.; Nieuwenhuijsen, M.; Daiber, A. The contribution of the exposome to the burden of cardiovascular disease. Nat. Rev. Cardiol. 2023, 20, 651–669. [Google Scholar] [CrossRef] [PubMed]
- A Montone, R.; Camilli, M.; Calvieri, C.; Magnani, G.; Bonanni, A.; Bhatt, D.L.; Rajagopalan, S.; Crea, F.; Niccoli, G. Exposome in ischaemic heart disease: beyond traditional risk factors. Eur. Hear. J. 2024, 45, 419–438. [Google Scholar] [CrossRef] [PubMed]
- Beulens, J.W.J.; Pinho, M.G.M.; Abreu, T.C.; Braver, N.R.D.; Lam, T.M.; Huss, A.; Vlaanderen, J.; Sonnenschein, T.; Siddiqui, N.Z.; Yuan, Z.; et al. Environmental risk factors of type 2 diabetes—an exposome approach. Diabetologia 2021, 65, 263–274. [Google Scholar] [CrossRef]
- Wild, C.P. Complementing the Genome with an “Exposome”: The Outstanding Challenge of Environmental Exposure Measurement in Molecular Epidemiology. Cancer Epidemiology Biomarkers Prev. 2005, 14, 1847–1850. [Google Scholar] [CrossRef]
- Rappaport, S.M.; Smith, M.T. Environment and Disease Risks. Science 2010, 330, 460–461. [Google Scholar] [CrossRef]
- Vaduganathan, M.; Mensah, G.A.; Turco, J.V.; Fuster, V.; Roth, G.A. The Global Burden of Cardiovascular Diseases and Risk. J. Am. Coll. Cardiol. 2022, 80, 2361–2371. [Google Scholar] [CrossRef]
- Marfella, R.; Prattichizzo, F.; Sardu, C.; Fulgenzi, G.; Graciotti, L.; Spadoni, T.; D’onofrio, N.; Scisciola, L.; La Grotta, R.; Frigé, C.; et al. Microplastics and Nanoplastics in Atheromas and Cardiovascular Events. New Engl. J. Med. 2024, 390, 900–910. [Google Scholar] [CrossRef]
- Lee, S.E.; Yoon, H.K.; Kim, D.Y.; Jeong, T.S.; Park, Y.S. An Emerging Role of Micro- and Nanoplastics in Vascular Diseases. Life 2024, 14, 255. [Google Scholar] [CrossRef]
- Bhatnagar, A. Environmental Determinants of Cardiovascular Disease. Circ. Res. 2017, 121, 162–180. [Google Scholar] [CrossRef]
- Sheikh, A.M.; Yano, S.; Tabassum, S.; Nagai, A. The Role of the Vascular System in Degenerative Diseases: Mechanisms and Implications. Int. J. Mol. Sci. 2024, 25, 2169. [Google Scholar] [CrossRef] [PubMed]
- Bagby, S.P.; Martin, D.; Chung, S.T.; Rajapakse, N. From the Outside In: Biological Mechanisms Linking Social and Environmental Exposures to Chronic Disease and to Health Disparities. Am. J. Public Heal. 2019, 109, S56–S63. [Google Scholar] [CrossRef]
- Janssen, H.; Koekkoek, L.L.; Swirski, F.K. Effects of lifestyle factors on leukocytes in cardiovascular health and disease. Nat. Rev. Cardiol. 2023, 21, 157–169. [Google Scholar] [CrossRef]
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.; Bäck, M.; Benetos, A.; Biffi, A.; Boavida, J.-M.; Capodanno, D.; et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur Heart J. 2021, 42, 3227–3337. [Google Scholar] [CrossRef]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef]
- Zhu, Z.; Bundy, J.D.; Mills, K.T.; Bazzano, L.A.; Kelly, T.N.; Zhang, Y.; Chen, J.; He, J. Secular Trends in Cardiovascular Health in US Adults (from NHANES 2007 to 2018). Am. J. Cardiol. 2021, 159, 121–128. [Google Scholar] [CrossRef]
- Elías-López, D.; for the Metabolic Syndrome Study Group; Vargas-Vázquez, A. ; Mehta, R.; Bautista, I.C.; Olvera, F.D.R.; Gómez-Velasco, D.; Valdes, P.A.; Aguilar-Salinas, C.A. Natural course of metabolically healthy phenotype and risk of developing Cardiometabolic diseases: a three years follow-up study. BMC Endocr. Disord. 2021, 21, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lhoste, V.P.F.; Zhou, B.; Mishra, A.; Bennett, J.E.; Filippi, S.; Asaria, P.; Gregg, E.W.; Danaei, G.; Ezzati, M. Cardiometabolic and renal phenotypes and transitions in the United States population. Nat. Cardiovasc. Res. 2023, 3, 46–59. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhu, Z.; Bundy, J.D.; Dorans, K.S.; Chen, J.; Hamm, L.L. Trends in Cardiovascular Risk Factors in US Adults by Race and Ethnicity and Socioeconomic Status, 1999-2018. JAMA 2021, 326, 1286–1298. [Google Scholar] [CrossRef] [PubMed]
- Kouvari, M.; Panagiotakos, D.B.; Yannakoulia, M.; Georgousopoulou, E.; Critselis, E.; Chrysohoou, C.; Tousoulis, D.; Pitsavos, C.; The ATTICA Study Investigators. Transition from metabolically benign to metabolically unhealthy obesity and 10-year cardiovascular disease incidence: The ATTICA cohort study. Metabolism 2019, 93, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.X.; Chaudhary, N.; Akinyemiju, T. Metabolic Syndrome Prevalence by Race/Ethnicity and Sex in the United States, National Health and Nutrition Examination Survey, 1988–2012. Prev. Chronic Dis. 2017, 14, E24. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Friera, L.; Peñalvo, J.L.; Fernández-Ortiz, A.; Ibañez, B.; López-Melgar, B.; Laclaustra, M.; Oliva, B.; Mocoroa, A.; Mendiguren, J.; de Vega, V.M.; et al. Prevalence, Vascular Distribution, and Multiterritorial Extent of Subclinical Atherosclerosis in a Middle-Aged Cohort. Circ. 2015, 131, 2104–2113. [Google Scholar] [CrossRef] [PubMed]
- Raposeiras-Roubin, S.; Rosselló, X.; Oliva, B.; Fernández-Friera, L.; Mendiguren, J.M.; Andrés, V.; Bueno, H.; Sanz, J.; de Vega, V.M.; Abu-Assi, E.; et al. Triglycerides and Residual Atherosclerotic Risk. J. Am. Coll. Cardiol. 2021, 77, 3031–3041. [Google Scholar] [CrossRef] [PubMed]
- Wadström, B.N.; Pedersen, K.M.; Wulff, A.B.; Nordestgaard, B.G. Elevated remnant cholesterol, plasma triglycerides, and cardiovascular and non-cardiovascular mortality. Eur. Hear. J. 2023, 44, 1432–1445. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-B.; Arsanjani, R.; Hong, S.-J.; Yi, J.-J.; Yi, S.-W. Impact of hypertriglyceridaemia on cardiovascular mortality according to low-density lipoprotein cholesterol in a 15.6-million population. Eur. J. Prev. Cardiol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Sterling, K.; Wang, Z.; Zhang, Y.; Song, W. The role of inflammasomes in human diseases and their potential as therapeutic targets. Signal Transduct. Target. Ther. 2024, 9, 1–30. [Google Scholar] [CrossRef]
- Tall, A.R.; Bornfeldt, K.E. Inflammasomes and Atherosclerosis: a Mixed Picture. Circ. Res. 2023, 132, 1505–1520. [Google Scholar] [CrossRef]
- Gimbrone, M.A. Jr.; García-Cardeña, G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118:620–636. [CrossRef]
- Eelen, G.; de Zeeuw, P.; Simons, M.; Carmeliet, P. Endothelial Cell Metabolism in Normal and Diseased Vasculature. Circ. Res. 2015, 116, 1231–1244. [Google Scholar] [CrossRef]
- Hsu, T.; Nguyen-Tran, H.-H.; Trojanowska, M. Active roles of dysfunctional vascular endothelium in fibrosis and cancer. J. Biomed. Sci. 2019, 26, 86. [Google Scholar] [CrossRef] [PubMed]
- Engelen, S.E.; Robinson, A.J.B.; Zurke, Y.-X.; Monaco, C. Therapeutic strategies targeting inflammation and immunity in atherosclerosis: how to proceed? Nat. Rev. Cardiol. 2022, 19, 522–542. [Google Scholar] [CrossRef] [PubMed]
- Crainiciuc, G.; Palomino-Segura, M.; Molina-Moreno, M.; Sicilia, J.; Aragones, D.G.; Li, J.L.Y.; Madurga, R.; Adrover, J.M.; Aroca-Crevillén, A.; Martin-Salamanca, S.; et al. Behavioural immune landscapes of inflammation. Nature 2022, 601, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Quintin, J.; van der Meer, J.W. Trained Immunity: A Memory for Innate Host Defense. Cell Host Microbe 2011, 9, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Domínguez-Andrés, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Andrés, J.; dos Santos, J.C.; Bekkering, S.; Mulder, W.J.M.; van der Meer, J.W.M.; Riksen, N.P.; Joosten, L.A.B.; Netea, M.G. Trained immunity: adaptation within innate immune mechanisms. Physiol. Rev. 2023, 103, 313–346. [Google Scholar] [CrossRef] [PubMed]
- Anitschkow, N.; Chalatow, S. On experimental cholesterin steatosis and its significance in the origin of some pathological processes (1913). Arteriosclerosis 1983;3: 178–82.
- Ross, R. Atherosclerosis is an inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Galis, Z.S. Exploring the Role of Endothelial Cell Resilience in Cardiovascular Health and Disease. Arter. Thromb. Vasc. Biol. 2020, 41, 179–185. [Google Scholar] [CrossRef]
- Hayes, K.L. Pericytes in Type 2 Diabetes. In Pericyte Biology in Disease; Birbrair, A., Ed.; (Advances in Experimental Medicine and Biology); Springer International Publishing: Cham, Switzerland, 2019; Volume 1147, pp. 265–278. ISBN 978-3-030-16907-7. [Google Scholar]
- Dabravolski, S.A.; Markin, A.M.; Andreeva, E.R.; Eremin, I.I.; Orekhov, A.N.; Melnichenko, A.A. Molecular Mechanisms Underlying Pathological and Therapeutic Roles of Pericytes in Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 11663. [Google Scholar] [CrossRef]
- Kumar, A.; D’souza, S.S.; Moskvin, O.V.; Toh, H.; Wang, B.; Zhang, J.; Swanson, S.; Guo, L.-W.; Thomson, J.A.; Slukvin, I.I. Specification and Diversification of Pericytes and Smooth Muscle Cells from Mesenchymoangioblasts. Cell Rep. 2017, 19, 1902–1916. [Google Scholar] [CrossRef]
- Yao, H.; Sun, Z.; Zang, G.; Zhang, L.; Hou, L.; Shao, C.; Wang, Z. Epidemiological Research Advances in Vascular Calcification in Diabetes. J. Diabetes Res. 2021, 2021, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Cui, Z.-Y.; Huang, X.-F.; Zhang, D.-D.; Guo, R.-J.; Han, M. Inflammation and atherosclerosis: signaling pathways and therapeutic intervention. Signal Transduct. Target. Ther. 2022, 7, 131. [Google Scholar] [CrossRef] [PubMed]
- Alencar, G.F.; Owsiany, K.M.; Karnewar, S.; Sukhavasi, K.; Mocci, G.; Nguyen, A.T.; Williams, C.M.; Shamsuzzaman, S.; Mokry, M.; Henderson, C.A.; et al. Stem Cell Pluripotency Genes Klf4 and Oct4 Regulate Complex SMC Phenotypic Changes Critical in Late-Stage Atherosclerotic Lesion Pathogenesis. Circulation 2020, 142, 2045–2059. [Google Scholar] [CrossRef] [PubMed]
- Wirka, R.C.; Wagh, D.; Paik, D.T.; Pjanic, M.; Nguyen, T.; Miller, C.L.; Kundu, R.; Nagao, M.; Coller, J.; Koyano, T.K.; et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 2019, 25, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Feil, S.; Fehrenbacher, B.; Lukowski, R.; Essmann, F.; Schulze-Osthoff, K.; Schaller, M.; Feil, R. Transdifferentiation of Vascular Smooth Muscle Cells to Macrophage-Like Cells During Atherogenesis. Circ. Res. 2014, 115, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Clement, M.; Raffort, J.; Lareyre, F.; Tsiantoulas, D.; Newland, S.; Lu, Y.; Masters, L.; Harrison, J.; Saveljeva, S.; Ma, M.K.; et al. Impaired Autophagy in CD11b + Dendritic Cells Expands CD4 + Regulatory T Cells and Limits Atherosclerosis in Mice. Circ. Res. 2019, 125, 1019–1034. [Google Scholar] [CrossRef]
- Tabas, I.; Lichtman, A.H. Monocyte-Macrophages and T Cells in Atherosclerosis. Immunity 2017, 47, 621–634. [Google Scholar] [CrossRef]
- Bekkering, S.; Blok, B.A.; Joosten, L.A.B.; Riksen, N.P.; van Crevel, R.; Netea, M.G. In Vitro Experimental Model of Trained Innate Immunity in Human Primary Monocytes. Clin. Vaccine Immunol. 2016, 23, 926–933. [Google Scholar] [CrossRef]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: a dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef]
- Barrett, T.J. Macrophages in Atherosclerosis Regression. Arter. Thromb. Vasc. Biol. 2020, 40, 20–33. [Google Scholar] [CrossRef]
- Fredman, G.; Tabas, I. Boosting Inflammation Resolution in Atherosclerosis. Am. J. Pathol. 2017, 187, 1211–1221. [Google Scholar] [CrossRef]
- Owsiany, K.M.; Alencar, G.F.; Owens, G.K. Revealing the Origins of Foam Cells in Atherosclerotic Lesions. Arter. Thromb. Vasc. Biol. 2019, 39, 836–838. [Google Scholar] [CrossRef]
- Yurdagul, A.; Doran, A.C.; Cai, B.; Fredman, G.; Tabas, I.A. Mechanisms and Consequences of Defective Efferocytosis in Atherosclerosis. Front. Cardiovasc. Med. 2018, 4, 86. [Google Scholar] [CrossRef] [PubMed]
- Saigusa, R.; Winkels, H.; Ley, K. T cell subsets and functions in atherosclerosis. Nat. Rev. Cardiol. 2020, 17, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Ketelhuth, D.F.; Hansson, G.K. Adaptive Response of T and B Cells in Atherosclerosis. Circ. Res. 2016, 118, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Ait-Oufella, H.; Salomon, B.L.; Potteaux, S.; Robertson, A.-K.L.; Gourdy, P.; Zoll, J.; Merval, R.; Esposito, B.; Cohen, J.L.; Fisson, S.; et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat. Med. 2006, 12, 178–180. [Google Scholar] [CrossRef] [PubMed]
- Sage, A.P.; Tsiantoulas, D.; Binder, C.J.; Mallat, Z. The role of B cells in atherosclerosis. Nat. Rev. Cardiol. 2018, 16, 180–196. [Google Scholar] [CrossRef] [PubMed]
- Nus, M.; Sage, A.P.; Lu, Y.; Masters, L.; Lam, B.Y.H.; Newland, S.; Weller, S.; Tsiantoulas, D.; Raffort, J.; Marcus, D.; et al. Marginal zone B cells control the response of follicular helper T cells to a high-cholesterol diet. Nat. Med. 2017, 23, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.; A Newland, S.; Jiang, W.; Giakomidi, D.; Zhao, X.; Clement, M.; Masters, L.; Corovic, A.; Zhang, X.; Drago, F.; et al. Marginal zone B cells produce ‘natural’ atheroprotective IgM antibodies in a T cell–dependent manner. Cardiovasc. Res. 2024, 120, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Silvestre-Roig, C.; Braster, Q.; Ortega-Gomez, A.; Soehnlein, O. Neutrophils as regulators of cardiovascular inflammation. Nat. Rev. Cardiol. 2020, 17, 327–340. [Google Scholar] [CrossRef]
- Soehnlein, O.; Wantha, S.; Simsekyilmaz, S.; Döring, Y.; Megens, R.T.A.; Mause, S.F.; Drechsler, M.; Smeets, R.; Weinandy, S.; Schreiber, F.; et al. Neutrophil-Derived Cathelicidin Protects from Neointimal Hyperplasia. Sci. Transl. Med. 2011, 3, 103ra98–103ra98. [Google Scholar] [CrossRef]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; MacFadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Rosenberg, Y.; Iturriaga, E.; et al. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. New Engl. J. Med. 2019, 380, 752–762. [Google Scholar] [CrossRef]
- Ridker, P.M. Anticytokine Agents: Targeting Interleukin Signaling Pathways for the Treatment of Atherothrombosis. Circ. Res. 2019, 124, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Wang, M.; Pirruccello, J.P.; Ellinor, P.T.; Ng, K.; Kathiresan, S.; Khera, A.V. Lp(a) (Lipoprotein[a]) Concentrations and Incident Atherosclerotic Cardiovascular Disease: New Insights From a Large National Biobank. Arterioscler Thromb Vasc Biol. 4: 2021;4, 2021. [Google Scholar]
- Virani, S.S.; Koschinsky, M.L.; Maher, L.; Mehta, A.; Orringer, C.E.; Santos, R.D.; Shapiro, M.D.; Saseen, J.J. Global think tank on the clinical considerations and management of lipoprotein(a): The top questions and answers regarding what clinicians need to know. Prog. Cardiovasc. Dis. 2022, 73, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Candelli, M.; Franza, L.; Cianci, R.; Pignataro, G.; Merra, G.; Piccioni, A.; Ojetti, V.; Gasbarrini, A.; Franceschi, F. The Interplay between Helicobacter pylori and Gut Microbiota in Non-Gastrointestinal Disorders: A Special Focus on Atherosclerosis. Int. J. Mol. Sci. 2023, 24, 17520. [Google Scholar] [CrossRef] [PubMed]
- Luqman, A.; Hassan, A.; Ullah, M.; Naseem, S.; Ullah, M.; Zhang, L.; Din, A.U.; Ullah, K.; Ahmad, W.; Wang, G. Role of the intestinal microbiome and its therapeutic intervention in cardiovascular disorder. Front. Immunol. 2024, 15, 1321395. [Google Scholar] [CrossRef] [PubMed]
- Afshar, M.; Thanassoulis, G. Lipoprotein(a): new insights from modern genomics. Curr. Opin. Infect. Dis. 2017, 28, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Arsenault, B.; Pelletier, W.; Kaiser, Y.; Perrot, N.; Couture, C. ; Khaw, K-T.; et al. Long-Term Exposure to Elevated Lipoprotein(a) Levels Influences Human Longevity. SSRN Electronic Journal. 2019: 10.2139/ssrn. 3404. [Google Scholar]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Marnell, C.S.; Bick, A.; Natarajan, P. Clonal hematopoiesis of indeterminate potential (CHIP): Linking somatic mutations, hematopoiesis, chronic inflammation and cardiovascular disease. J. Mol. Cell. Cardiol. 2021, 161, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Pasupuleti, S.K.; Ramdas, B.; Burns, S.S.; Palam, L.R.; Kanumuri, R.; Kumar, R.; Pandhiri, T.R.; Dave, U.P.; Yellapu, N.K.; Zhou, X.; et al. Obesity-induced inflammation exacerbates clonal hematopoiesis. J. Clin. Investig. 2023, 133. [Google Scholar] [CrossRef]
- Fuster, J.J. Clonal hematopoiesis and cardiovascular disease in cancer patients and survivors. Thromb. Res. 2022, 213, S107–S112. [Google Scholar] [CrossRef]
- McAninch, E.A.; Fonseca, T.L.; Poggioli, R.; Panos, A.L.; Salerno, T.A.; Deng, Y.; Li, Y.; Bianco, A.C.; Iacobellis, G. Epicardial adipose tissue has a unique transcriptome modified in severe coronary artery disease. Obesity 2015, 23, 1267–1278. [Google Scholar] [CrossRef]
- Iacobellis, G. Epicardial adipose tissue in contemporary cardiology. Nat. Rev. Cardiol. 2022, 19, 593–606. [Google Scholar] [CrossRef] [PubMed]
- A Matter, M.; Paneni, F.; Libby, P.; Frantz, S.; E Stähli, B.; Templin, C.; Mengozzi, A.; Wang, Y.-J.; Kündig, T.M.; Räber, L.; et al. Inflammation in acute myocardial infarction: the good, the bad and the ugly. Eur. Hear. J. 2023, 45, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Dam, R.M.V.; Li, T.; Spiegelman, D.; Franco, O.H.; Hu, F.B. Combined impact of lifestyle factors on mortality: prospective cohort study in US women. BMJ 2008, 337, a1440–a1440. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen-Torvik, L.J.; Shay, C.M.; Abramson, J.G.; Friedrich, C.A.; Nettleton, J.A.; Prizment, A.E.; Folsom, A.R.; J, A.; J, M.; R, d.B.; et al. Ideal Cardiovascular Health Is Inversely Associated With Incident Cancer. Circ. 2013, 127, 1270–1275. [Google Scholar] [CrossRef] [PubMed]
- van Sloten, T.T.; Climie, R.E.; Deraz, O.; Périer, M.-C.; Valentin, E.; Fayosse, A.; Sabia, S.; Weiderpass, E.; Jouven, X.; Goldberg, M.; et al. Is the number of ideal cardiovascular health metrics in midlife associated with lower risk of cancer? Evidence from 3 European prospective cohorts. CMAJ Open 2023, 11, E774–E781. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yu, H.; Huang, T.; Huang, N.; Liang, H. Importance of ideal cardiovascular health metrics in the risk of colorectal cancer among people aged 50 years or older: a UK Biobank cohort study. BMJ Open 2022, 12, e059642. [Google Scholar] [CrossRef] [PubMed]
- de Boer, R.A.; Meijers, W.C.; van der Meer, P.; van Veldhuisen, D.J. ; Cancer and heart disease: associations and relations. 1: Eur J Heart Fail 2019;21, 2019. [Google Scholar]
- Hasin, T.; Iakobishvili, Z.; Weisz, G. Associated Risk of Malignancy in Patients with Cardiovascular Disease: Evidence and Possible Mechanism. Am. J. Med. 2017, 130, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Hasin, T.; Gerber, Y.; Weston, S.A.; Jiang, R.; Killian, J.M.; Manemann, S.M.; Cerhan, J.R.; Roger, V.L. Heart Failure After Myocardial Infarction Is Associated With Increased Risk of Cancer. J. Am. Coll. Cardiol. 2016, 68, 265–271. [Google Scholar] [CrossRef]
- Koelwyn, G.J.; Aboumsallem, J.P.; Moore, K.J.; de Boer, R.A. Reverse cardio-oncology: Exploring the effects of cardiovascular disease on cancer pathogenesis. J. Mol. Cell. Cardiol. 2021, 163, 1–8. [Google Scholar] [CrossRef]
- Koene, R.J.; Prizment, A.E.; Blaes, A.; Konety, S.H.; J, S.; K, R.; H, W.; D, E.; R, S.; D, S.; et al. Shared Risk Factors in Cardiovascular Disease and Cancer. Circ. 2016, 133, 1104–1114. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. 5: Cell 2000;100, 2000. [Google Scholar]
- Narayan, V.; Thompson, E.W.; Demissei, B.; Ho, J.E.; Januzzi, J.L.; Ky, B. Mechanistic Biomarkers Informative of Both Cancer and Cardiovascular Disease. J. Am. Coll. Cardiol. 2020, 75, 2726–2737. [Google Scholar] [CrossRef]
- Larsen, K.M.; Minaya, M.K.; Vaish, V.; Pena, M.M.O. The role of IL-33/ST2 pathway in tumorigenesis. Int J Mol Sci 2018;19:2676.
- Aimo, A.; Januzzi, J.L.; Bayes-Genis, A.; Vergaro, G.; Sciarrone, P.; Passino, C.; Emdin, M. Clinical and Prognostic Significance of sST2 in Heart Failure. J. Am. Coll. Cardiol. 2019, 74, 2193–2203. [Google Scholar] [CrossRef]
- Wang, L.; Guo, X.-L. Molecular regulation of galectin-3 expression and therapeutic implication in cancer progression. Biomed. Pharmacother. 2016, 78, 165–171. [Google Scholar] [CrossRef]
- Yu, X.; Sun, Y.; Zhao, Y.; Zhang, W.; Yang, Z.; Gao, Y.; Cai, H.; Li, Y.; Wang, Q.; Bian, B.; et al. Prognostic Value of Plasma Galectin-3 Levels in Patients With Coronary Heart Disease and Chronic Heart Failure. Int. Hear. J. 2015, 56, 314–318. [Google Scholar] [CrossRef]
- Emmerson, P.J.; Duffin, K.L.; Chintharlapalli, S.; Wu, X. GDF15 and growth control. Front Physiol 2018;9:1712.
- Wollert, K.C.; Kempf, T.; Wallentin, L. Growth Differentiation Factor 15 as a Biomarker in Cardiovascular Disease. Clin. Chem. 2017, 63, 140–151. [Google Scholar] [CrossRef]
- Cardinale, D.; Sandri, M.T.; Colombo, A.; Colombo, N.; Boeri, M.; Lamantia, G.; Civelli, M.; Peccatori, F.; Martinelli, G.; Fiorentini, C.; et al. Prognostic Value of Troponin I in Cardiac Risk Stratification of Cancer Patients Undergoing High-Dose Chemotherapy. Circ. 2004, 109, 2749–2754. [Google Scholar] [CrossRef]
- Kumar, S.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Colby, C.; Laumann, K.; Zeldenrust, S.R.; Leung, N.; Dingli, D.; et al. Revised Prognostic Staging System for Light Chain Amyloidosis Incorporating Cardiac Biomarkers and Serum Free Light Chain Measurements. J. Clin. Oncol. 2012, 30, 989–995. [Google Scholar] [CrossRef]
- Pavo, N.; Raderer, M.; Hülsmann, M.; Neuhold, S.; Adlbrecht, C.; Strunk, G.; Goliasch, G.; Gisslinger, H.; Steger, G.G.; Hejna, M.; et al. Cardiovascular biomarkers in patients with cancer and their association with all-cause mortality. Heart 2015, 101, 1874–1880. [Google Scholar] [CrossRef]
- Widiapradja, A.; Chunduri, P.; Levick, S.P. The role of neuropeptides in adverse myocardial remodeling and heart failure. Cell. Mol. Life Sci. 2017, 74, 2019–2038. [Google Scholar] [CrossRef]
- Ky, B.; French, B.; Ruparel, K.; Sweitzer, N.K.; Fang, J.C.; Levy, W.C.; Sawyer, D.B.; Cappola, T.P. The Vascular Marker Soluble Fms-Like Tyrosine Kinase 1 Is Associated With Disease Severity and Adverse Outcomes in Chronic Heart Failure. J. Am. Coll. Cardiol. 2011, 58, 386–394. [Google Scholar] [CrossRef]
- Yang, F.; Jin, C.; Jiang, Y.-J.; Li, J.; Di, Y.; Fu, D.-L. Potential role of soluble VEGFR-1 in antiangiogenesis therapy for cancer. Expert Rev. Anticancer. Ther. 2011, 11, 541–549. [Google Scholar] [CrossRef]
- Bick, A.G.; Weinstock, J.S.; Nandakumar, S.K.; Fulco, C.P.; Bao, E.L.; Zekavat, S.M.; Szeto, M.D.; Liao, X.; Leventhal, M.J.; Nasser, J.; et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 2020, 586, 763–768. [Google Scholar] [CrossRef]
- Heyde, A.; Rohde, D.; McAlpine, C.S.; Zhang, S.; Hoyer, F.F.; Gerold, J.M.; Cheek, D.; Iwamoto, Y.; Schloss, M.J.; Vandoorne, K.; et al. Increased stem cell proliferation in atherosclerosis accelerates clonal hematopoiesis. Cell 2021, 184, 1348–1361. [Google Scholar] [CrossRef]
- Venkatesulu, B.P.; Mahadevan, L.S.; Aliru, M.L.; Yang, X.; Bodd, M.H.; Singh, P.K.; Yusuf, S.W.; Abe, J.-I.; Krishnan, S. Radiation-Induced Endothelial Vascular Injury. JACC: Basic Transl. Sci. 2018, 3, 563–572. [Google Scholar] [CrossRef]
- Desai, M.Y.; Windecker, S.; Lancellotti, P.; Bax, J.J.; Griffin, B.P.; Cahlon, O.; Johnston, D.R. Prevention, Diagnosis, and Management of Radiation-Associated Cardiac Disease. J. Am. Coll. Cardiol. 2019, 74, 905–927. [Google Scholar] [CrossRef]
- Honaryar, M.K.; Allodji, R.; Jimenez, G.; Lapeyre, M.; Panh, L.; Camilleri, J.; Broggio, D.; Ferrières, J.; De Vathaire, F.; Jacob, S. Early Development of Atherosclerotic Plaques in the Coronary Arteries after Radiotherapy for Breast Cancer (BACCARAT Study). J. Cardiovasc. Dev. Dis. 2023, 10, 299. [Google Scholar] [CrossRef]
- Herrmann, J. Vascular toxic effects of cancer therapies. Nat. Rev. Cardiol. 2020, 17, 503–522. [Google Scholar] [CrossRef]
- Terwoord, J.D.; Beyer, A.M.; Gutterman, D.D. Endothelial dysfunction as a complication of anti-cancer therapy. Pharmacol. Ther. 2022, 237, 108116–108116. [Google Scholar] [CrossRef]
- Strauss, L.; Mahmoud, M.A.A.; Weaver, J.D.; Tijaro-Ovalle, N.M.; Christofides, A.; Wang, Q.; Pal, R.; Yuan, M.; Asara, J.; Patsoukis, N.; et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]
- Fernandez, D.M.; Rahman, A.H.; Fernandez, N.F.; Chudnovskiy, A.; Amir, E.-A.D.; Amadori, L.; Khan, N.S.; Wong, C.K.; Shamailova, R.; Hill, C.A.; et al. Single-cell immune landscape of human atherosclerotic plaques. Nat. Med. 2019, 25, 1576–1588. [Google Scholar] [CrossRef]
- Bu, D.-X.; Tarrio, M.; Maganto-Garcia, E.; Stavrakis, G.; Tajima, G.; Lederer, J.A.; Jarolim, P.; Freeman, G.J.; Sharpe, A.H.; Lichtman, A.H. Impairment of the Programmed Cell Death-1 Pathway Increases Atherosclerotic Lesion Development and Inflammation. Arter. Thromb. Vasc. Biol. 2011, 31, 1100–1107. [Google Scholar] [CrossRef]
- Lee, J.; Zhuang, Y.; Wei, X.; Shang, F.; Wang, J.; Zhang, Y.; Liu, X.; Yang, Y.; Liu, L.; Zheng, Q. Contributions of PD-1/PD-L1 pathway to interactions of myeloid DCs with T cells in atherosclerosis. J. Mol. Cell. Cardiol. 2009, 46, 169–176. [Google Scholar] [CrossRef]
- Matsumoto, T.; Sasaki, N.; Yamashita, T.; Emoto, T.; Kasahara, K.; Mizoguchi, T.; Hayashi, T.; Yodoi, K.; Kitano, N.; Saito, T.; et al. Overexpression of Cytotoxic T-Lymphocyte–Associated Antigen-4 Prevents Atherosclerosis in Mice. Arter. Thromb. Vasc. Biol. 2016, 36, 1141–1151. [Google Scholar] [CrossRef]
- Quagliariello, V.; Passariello, M.; Di Mauro, A.; Cipullo, C.; Paccone, A.; Barbieri, A.; Palma, G.; Luciano, A.; Buccolo, S.; Bisceglia, I.; et al. Immune checkpoint inhibitor therapy increases systemic SDF-1, cardiac DAMPs Fibronectin-EDA, S100/Calgranulin, galectine-3, and NLRP3-MyD88-chemokine pathways. Front. Cardiovasc. Med. 2022, 9, 930797. [Google Scholar] [CrossRef]
- Drobni, Z.D.; Alvi, R.M.; Taron, J.; Zafar, A.; Murphy, S.P.; Rambarat, P.K.; Mosarla, R.C.; Lee, C.; Zlotoff, D.A.; Raghu, V.K.; et al. Association between immune checkpoint inhibitors with cardiovascular events and atherosclerotic plaque. Circulation 2020, 142, 2299–2311. [Google Scholar] [CrossRef]
- Gong, B.; Guo, Y.; Li, Y.; Wang, J.; Zhou, G.; Chen, Y.-H.; Nie, T.; Yang, M.; Luo, K.; Zheng, C.; et al. Immune checkpoint inhibitors in cancer: the increased risk of atherosclerotic cardiovascular disease events and progression of coronary artery calcium. BMC Med. 2024, 22, 44. [Google Scholar] [CrossRef]
- Matthews, A.; Stanway, S.; E Farmer, R.; Strongman, H.; Thomas, S.; Lyon, A.R.; Smeeth, L.; Bhaskaran, K. Long term adjuvant endocrine therapy and risk of cardiovascular disease in female breast cancer survivors: systematic review. BMJ 2018, 363, k3845. [Google Scholar] [CrossRef]
- Bhatia, N.; Santos, M.; Jones, L.W.; Beckman, J.A.; Penson, D.F.; Morgans, A.K.; Moslehi, J.; Poulsen, C.B.; Mortensen, M.B.; Koechling, W.; et al. Cardiovascular Effects of Androgen Deprivation Therapy for the Treatment of Prostate Cancer. Circ. 2016, 133, 537–541. [Google Scholar] [CrossRef]
- Hu, J.-R.; Duncan, M.S.; Morgans, A.K.; Brown, J.D.; Meijers, W.C.; Freiberg, M.S.; Salem, J.-E.; Beckman, J.A.; Moslehi, J.J. Cardiovascular Effects of Androgen Deprivation Therapy in Prostate Cancer: Contemporary Meta-Analyses. Arter. Thromb. Vasc. Biol. 2020, 40, E55–E64. [Google Scholar] [CrossRef]
- Wang, Q.; Han, J.; Liang, Z.; Geng, X.; Du, Y.; Zhou, J.; Yao, W.; Xu, T. FSH Is Responsible for Androgen Deprivation Therapy-Associated Atherosclerosis in Mice by Exaggerating Endothelial Inflammation and Monocyte Adhesion. Arter. Thromb. Vasc. Biol. 2024. [Google Scholar] [CrossRef]
- Dawber, T.R.; Meadors, G.F.; Moore, F.E., Jr. Epidemiological Approaches to Heart Disease: The Framingham Study. Am. J. Public Health 1951, 41, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Dawber, T.R.; Moore, F.E.; Mann, G.V. II. Coronary Heart Disease in the Framingham Study. Am. J. Public Heal. Nations Heal. 1957, 47, 4–24. [Google Scholar] [CrossRef] [PubMed]
- Kannel, W.B.; Castelli, W.P.; Gordon, T. Cholesterol in the Prediction of Atherosclerotic Disease. Ann. Intern. Med. 1979, 90, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, S.S.; Levy, D.; Vasan, R.S.; Wang, T.J. The Framingham Heart Study and the epidemiology of cardiovascular disease: a historical perspective. Lancet 2013, 383, 999–1008. [Google Scholar] [CrossRef]
- D'Agostino, R.B., Sr.; Vasan, R.S.; Pencina, M.J.; Wolf, P.A.; Cobain, M.; Massaro, J.M.; Kannel, W.B. General Cardiovascular Risk Profile for Use in Primary Care: The Framingham Heart Study. Circulation 2008, 117, 743–753. [Google Scholar] [CrossRef]
- SCORE2 working group and ESC Cardiovascular risk collaboration. SCORE2 risk prediction algorithms: new models to estimate 10-year risk of cardiovascular disease in Europe. Eur. Hear. J. 2021, 42, 2439–2454. [Google Scholar] [CrossRef]
- Iacobellis, G.; Assael, F.; Ribaudo, M.C.; Zappaterreno, A.; Alessi, G.; Di Mario, U.; Leonetti, F. Epicardial Fat from Echocardiography: A New Method for Visceral Adipose Tissue Prediction. Obes. Res. 2003, 11, 304–310. [Google Scholar] [CrossRef]
- Iacobellis, G.; Ribaudo, M.C.; Assael, F.; Vecci, E.; Tiberti, C.; Zappaterreno, A.; Di Mario, U.; Leonetti, F. Echocardiographic Epicardial Adipose Tissue Is Related to Anthropometric and Clinical Parameters of Metabolic Syndrome: A New Indicator of Cardiovascular Risk. J. Clin. Endocrinol. Metab. 2003, 88, 5163–5168. [Google Scholar] [CrossRef]
- Lesko, L.; Atkinson, A. Use of Biomarkers and Surrogate Endpoints in Drug Development and Regulatory Decision Making: Criteria, Validation, Strategies. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 347–366. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Biomarkers of endothelial activation and dysfunction in cardiovascular diseases. Rev. Cardiovasc. Med. 2022, 23, 73. [Google Scholar] [CrossRef]
- Wang, C.; Li, Z.; Liu, Y.; Yuan, L. Exosomes in atherosclerosis: performers, bystanders, biomarkers, and therapeutic targets. Theranostics 2021, 11, 3996–4010. [Google Scholar] [CrossRef] [PubMed]
- Lao, K.H.; Zeng, L.; Xu, Q. Endothelial and smooth muscle cell transformation in atherosclerosis. Curr. Opin. Infect. Dis. 2015, 26, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Agatston, A.S.; Janowitz, W.R.; Hildner, F.J.; Zusmer, N.R.; Viamonte, M., Jr.; Detrano, R. Quantification of coronary artery calcium using ultrafast computed tomography. J. Am. Coll. Cardiol. 1990, 15, 827–832. [Google Scholar] [CrossRef]
- Razavi, A.C.; Agatston, A.S.; Shaw, L.J.; De Cecco, C.N.; van Assen, M.; Sperling, L.S.; Bittencourt, M.S.; Daubert, M.A.; Nasir, K.; Blumenthal, R.S.; et al. Evolving Role of Calcium Density in Coronary Artery Calcium Scoring and Atherosclerotic Cardiovascular Disease Risk. JACC: Cardiovasc. Imaging 2022, 15, 1648–1662. [Google Scholar] [CrossRef]
- Mascalchi, M.; Puliti, D.; Romei, C.; Picozzi, G.; De Liperi, A.; Diciotti, S.; Bartolucci, M.; Grazzini, M.; Vannucchi, L.; Falaschi, F.; et al. Moderate-severe coronary calcification predicts long-term cardiovascular death in CT lung cancer screening: The ITALUNG trial. Eur. J. Radiol. 2021, 145, 110040. [Google Scholar] [CrossRef]
- Robinson, J.G.; Williams, K.J.; Gidding, S.; Borén, J.; Tabas, I.; Fisher, E.A.; Packard, C.; Pencina, M.; Fayad, Z.A.; Mani, V.; et al. Eradicating the Burden of Atherosclerotic Cardiovascular Disease by Lowering Apolipoprotein B Lipoproteins Earlier in Life. J. Am. Hear. Assoc. 2018, 7, e009778. [Google Scholar] [CrossRef]
- de Isla, L.P.; Díaz-Díaz, J.L.; Romero, M.J.; Muñiz-Grijalvo, O.; Mediavilla, J.D.; Argüeso, R.; Muñoz-Torrero, J.F.S.; Rubio, P.; Álvarez-Baños, P.; Ponte, P.; et al. Alirocumab and Coronary Atherosclerosis in Asymptomatic Patients with Familial Hypercholesterolemia: The ARCHITECT Study. Circ. 2023, 147, 1436–1443. [Google Scholar] [CrossRef]
- Williams, K.J. Eradicating Atherosclerotic Events by Targeting Early Subclinical Disease: It Is Time to Retire the Therapeutic Paradigm of Too Much, Too Late. Arter. Thromb. Vasc. Biol. 2024, 44, 48–64. [Google Scholar] [CrossRef]
- Selvam, P.V.; Grandhi, G.R.; Leucker, T.M.; Zadeh, A.; Gulati, M.; Blumenthal, R.S.; Whelton, S.P. Recent advances in cardiovascular risk assessment: The added value of non-invasive anatomic imaging. J. Cardiovasc. Comput. Tomogr. 2024. [Google Scholar] [CrossRef] [PubMed]
- Ansari, S.; Pourafkari, L.; Kinninger, A.; Manubolu, V.; Budoff, M.J. Risk stratifying individuals with zero, minimal, and mild coronary artery calcium for cardiovascular disease by determining coronary plaque burden. J. Cardiovasc. Comput. Tomogr. 2023. [Google Scholar] [CrossRef]
- Grant, J.K.; Orringer, C.E. Coronary and Extra-coronary Subclinical Atherosclerosis to Guide Lipid-Lowering Therapy. Curr. Atheroscler. Rep. 2023, 25, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Garg, P.K.; Bhatia, H.S.; Allen, T.S.; Grainger, T.; Pouncey, A.L.; Dichek, D.; Virmani, R.; Golledge, J.; Allison, M.A.; Powell, J.T. Assessment of Subclinical Atherosclerosis in Asymptomatic People In Vivo: Measurements Suitable for Biomarker and Mendelian Randomization Studies. Arter. Thromb. Vasc. Biol. 2024, 44, 24–47. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, R.P.; Fuster, V.; Badimon, J.J.; Fisher, E.A.; Fayad, Z.A.; A, M.; M, B.; T, K.; G, M.; J, D.; et al. MRI and Characterization of Atherosclerotic Plaque. Arter. Thromb. Vasc. Biol. 2002, 22, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Wald, L.L.; McDaniel, P.C.; Witzel, T.; Stockmann, J.P.; Cooley, C.Z. Low-cost and portable MRI. J. Magn. Reson. Imaging 2019, 52, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Senders, M.L.; Calcagno, C.; Tawakol, A.; Nahrendorf, M.; Mulder, W.J.M.; Fayad, Z.A. PET/MR imaging of inflammation in atherosclerosis. Nat. Biomed. Eng. 2022, 7, 202–220. [Google Scholar] [CrossRef] [PubMed]
- Gurgoglione, F.L.; Denegri, A.; Russo, M.; Calvieri, C.; Benatti, G.; Niccoli, G. Intracoronary Imaging of Coronary Atherosclerotic Plaque: From Assessment of Pathophysiological Mechanisms to Therapeutic Implication. Int. J. Mol. Sci. 2023, 24, 5155. [Google Scholar] [CrossRef]

Table 1.
The chameleonic vascular and immune players of atherosclerosis with their “good”, “bad” or “ugly” behaviours.
Table 1.
The chameleonic vascular and immune players of atherosclerosis with their “good”, “bad” or “ugly” behaviours.
| Players | Phenotypes | “good” behaviour | “bad” behavior | “ugly” behavior |
|---|---|---|---|---|
| Endothelial cells (EC): secrete vasoactive substances (e.g. endothelin-1, nitric oxide, etc) affecting Vascular smooth muscle cells, platelets and white blood cells. | Heterogeneous ECs to suit heterogeneous endothelium, capable of angiogenic and metabolic switch. | Healthy ECs are excellent sensors of the hemodynamic forces of blood flow. | In the early stages of atherosclerosis, high levels of ox-LDL and remnants of tryglyceride-rich lipoproteins gain access to the subendothelial space eliciting a danger signal that activate the NLRP3- inflammasome in innate immune cells and the inflammatory pathways leading to endothelial dysfunction | Inflammation begets inflammation: and this vicious circle leads to the advanced stages of atherosclerosis |
| In atherosclerosis ECs are activated by trapped lipoproteins | EC may exhibit trained immunity | ECs have a pivotal role in endothelial resilience , the ability to cope with many stressors or challenges (exposomes) | ||
| [51,52,58,61] | ||||
| Pericytes: perivascular cells derived from human pluripotent stem cells (HPSCs) and located around ECs | a) Due to their common origin from HPSCs, pericytes can differentiate into other cells of the mesenchymal lineage such as monocytes | Support vascular stability by preventing matrix degradation, play a relevant role in differentiation, angiogenesis, regeneration, immunomodulation and blood flow regulation | Dysmetabolic-driven alteration of pericytes in diabetes contributes to the plaque formation | In advanced atherosclerosis pericytes are involved in plaque neovascularization, inflammation and vascular calcification process |
| [62,63,64,65] | ||||
| Vascular smooth muscle cells exhibit a contractile phenotype in the healthy arterial wall, if stimulated by ECs through PGDF-BB and TNF-α, they can switch to a synthetic phenotype that increases the production of ECM, exosome, and proinflammatory cytokines | b) Different phenotypes with beneficial and detrimental role in atherogenesis | VSMC stabilize fibrous cap in advanced atherosclerosis and produce ECM (fibroblast-like features) | Lipid-induced transformation in macrophage-like and foam cell-like phenotypes exhibiting pro-inflammatory behavior and increasing the vulnerability of the plaque | |
| [66,67,68,69] | (macrophage-like features) | |||
| Dendritic cells (DCs): bridge the innate and adaptive immune response involved in the scenario of the evolving plaque [66,70] | c) Preclinical studies: pro-atherogenic and anti-atherogenic function | In Ldlr-/- mice fed with high fat diet, autophagy disruption in DCs limits atherogenesis. | In humans dendritic cell numbers are connected to vulnerability of the atherosclerotic plaque. | |
| Monocytes in a homeostatic state populate blood, bone marrow and spleen. | Classical vs non- classical monocytes. | Non-classical monocytes are “on patrol” to maintain vascular endothelial | Classical monocytes (CD14+ CD16-in humans, Ly6Chigh in mice) recruited to atherosclerotic plaques exhibit phenotypic heterogeneity differentiating into dendritic cells and macrophages. | Preclinical studies in mice: splenic classical monocytes (Ly6Chigh) increase plaque and its instability. |
| [71,72] | Monocytes may be rewired by metabolic stimuli (e.g. ox-LDL) to become a “trained” immune cell. | Recruited monocytes have also an impact on atherosclerosis regression. | In humans and mice monocytosis is associated with increased severity of atherosclerosis | |
| Macrophages | M1 macrophages | M2 macrophages clear lipids and secrete anti-inflammatory factors (e.g. Il-10 and collagen) | M1 macrophages favor the accumulation of intracellular lipids and increase the secretion of proinflammatory factors (e.g. TNF-α IL-1 β and IL-6). | M1 |
| [73,74,75,76,77] | M2 macrophages | Recruited monocyte-derived macrophages remove apoptotic cells (efferocytosis), eliciting the secretion of anti-inflammatory cytokines and hampering the progression of atherosclerotic plaque | Macrophages may appear as foamy cells | When reprogrammed macrophages lose their efferocytic ability, apoptotic cells undergo post-apoptotic necrosis, release pro-inflammatory mediators giving a boost to the progression of the plaque |
| Macrophages may be upgraded in “trained” immune cells. | ||||
| T cells have a role in all stages of atherosclerosis CD4+T cells are prevalent in mouse atherosclerotic | Atheroprotective phenotype (T reg) and pro-atherogenic phenotype (T helper 1) | T reg can silence inflammation through the elaboration of the immunomodulatory cytokine transforming growth factor beta and by secreting IL-10 (pre-clinical studies) | pro- inflammatory phenotype (T helper 1 | |
| plaque and exhibit a pro-inflammatory atherogenic phenotype. | cells): activated T cells have a direct role in the arterial wall or help B cells in antibody production | |||
| [78,79,80] | ||||
| B cells are classified into B1 cells (subdivided in B1a and B1b cells), mainly produced in the fetal liver, and B2 cells (subdivided in T1 and T2 marginal zone progenitor) | B1 cells atheroprotective in mice | B1 cells exhibit an atheroprotective behavior in mice due to the production of IgM antibodies that block the uptake of oxLDL by macrophages in atherosclerotic lesions. | B2 cells exhibit a pro- atherogenic behavior through antibody | |
| [81,82,83] | When challenged by a high fat/high cholesterol diet marginal zone B cells switch to an atheroprotective programme mediated by Atf3, Nr4a1, Pdl1 | responses that stimulate | ||
| adaptive immunity | ||||
| Proatherogenic phenotype. | Reparative phenotype exhibited during thrombotic | Neutrophils secrete ROS increasing the permeability of the ECs and inducing NLRP3 inflammasome. Neutrophils attract | NET formation that stimulates the NLRP3 inflammasome and produce IL-1β (preclinical study). | |
| Reparative phenotype | events when neutrophils promote endothelial repair and angiogenesis (arterial healing) | monocytes and | In this scenario NLRP3 inflammasome requires a second hit to be fully activated. | |
| can activate macrophages via extrusion of their | Defective efferocytosis that leads to DAMPS accumulation. | |||
| NETs . | ||||
| Players | Phenotypes | “good” behaviour | “bad” behavior | “ugly” behavior |
|
Endothelial cells (EC): secrete vasoactive substances (e.g. endothelin-1, nitric oxide, etc) affecting Vascular smooth muscle cells, platelets and white blood cells. In atherosclerosis ECs are activated by trapped lipoproteins [51,52,58,61] |
Heterogeneous ECs to suit heterogeneous endothelium, capable of angiogenic and metabolic switch. EC may exhibit trained immunity |
Healthy ECs are excellent sensors of the hemodynamic forces of blood flow. ECs have a pivotal role in endothelial resilience , the ability to cope with many stressors or challenges (exposomes) |
In the early stages of atherosclerosis, high levels of ox-LDL and remnants of tryglyceride-rich lipoproteins gain access to the subendothelial space eliciting a danger signal that activate the NLRP3- inflammasome in innate immune cells and the inflammatory pathways leading to endothelial dysfunction | Inflammation begets inflammation: and this vicious circle leads to the advanced stages of atherosclerosis |
|
Pericytes: perivascular cells derived from human pluripotent stem cells (HPSCs) and located around ECs [62,63,64,65] |
Due to their common origin from HPSCs, pericytes can differentiate into other cells of the mesenchymal lineage such as monocytes | Support vascular stability by preventing matrix degradation, play a relevant role in differentiation, angiogenesis, regeneration, immunomodulation and blood flow regulation |
Dysmetabolic-driven alteration of pericytes in diabetes contributes to the plaque formation | In advanced atherosclerosis pericytes are involved in plaque neovascularization, inflammation and vascular calcification process |
|
Vascular smooth muscle cells exhibit a contractile phenotype in the healthy arterial wall, if stimulated by ECs through PGDF-BB and TNF-α, they can switch to a synthetic phenotype that increases the production of ECM, exosome, and proinflammatory cytokines [66,67,68,69] |
Different phenotypes with beneficial and detrimental role in atherogenesis | VSMC stabilize fibrous cap in advanced atherosclerosis and produce ECM (fibroblast-like features) |
Lipid-induced transformation in macrophage-like and foam cell-like phenotypes exhibiting pro-inflammatory behavior and increasing the vulnerability of the plaque (macrophage-like features) |
|
| Dendritic cells (DCs): bridge the innate and adaptive immune response involved in the scenario of the evolving plaque [66,70] | Preclinical studies: pro-atherogenic and anti-atherogenic function | In Ldlr-/- mice fed with high fat diet, autophagy disruption in DCs limits atherogenesis. | In humans dendritic cell numbers are connected to vulnerability of the atherosclerotic plaque. | |
|
Monocytes in a homeostatic state populate blood, bone marrow and spleen. [71,72] |
Classical vs non- classical monocytes. Monocytes may be rewired by metabolic stimuli (e.g. ox-LDL) to become a “trained” immune cell. |
Non-classical monocytes are “on patrol” to maintain vascular endothelial Recruited monocytes have also an impact on atherosclerosis regression. |
Classical monocytes (CD14+ CD16-in humans, Ly6Chigh in mice) recruited to atherosclerotic plaques exhibit phenotypic heterogeneity differentiating into dendritic cells and macrophages. |
Preclinical studies in mice: splenic classical monocytes (Ly6Chigh) increase plaque and its instability. In humans and mice monocytosis is associated with increased severity of atherosclerosis |
|
Macrophages [73,74,75,76,77] |
M1 macrophages M2 macrophages Macrophages may be upgraded in “trained” immune cells. |
M2 macrophages clear lipids and secrete anti-inflammatory factors (e.g. Il-10 and collagen) Recruited monocyte-derived macrophages remove apoptotic cells (efferocytosis), eliciting the secretion of anti-inflammatory cytokines and hampering the progression of atherosclerotic plaque |
M1 macrophages favor the accumulation of intracellular lipids and increase the secretion of proinflammatory factors (e.g. TNF-α IL-1 β and IL-6). Macrophages may appear as foamy cells |
M1 When reprogrammed macrophages lose their efferocytic ability, apoptotic cells undergo post-apoptotic necrosis, release pro-inflammatory mediators giving a boost to the progression of the plaque |
|
T cells have a role in all stages of atherosclerosis CD4+T cells are prevalent in mouse atherosclerotic plaque and exhibit a pro-inflammatory atherogenic phenotype. [78,79,80] |
Atheroprotective phenotype (T reg) and pro-atherogenic phenotype (T helper 1) | T reg can silence inflammation through the elaboration of the immunomodulatory cytokine transforming growth factor beta and by secreting IL-10 (pre-clinical studies) |
pro- inflammatory phenotype (T helper 1 cells): activated T cells have a direct role in the arterial wall or help B cells in antibody production |
|
|
B cells are classified into B1 cells (subdivided in B1a and B1b cells), mainly produced in the fetal liver, and B2 cells (subdivided in T1 and T2 marginal zone progenitor) [81,82,83] |
B1 cells atheroprotective in mice When challenged by a high fat/high cholesterol diet marginal zone B cells switch to an atheroprotective programme mediated by Atf3, Nr4a1, Pdl1 |
B1 cells exhibit an atheroprotective behavior in mice due to the production of IgM antibodies that block the uptake of oxLDL by macrophages in atherosclerotic lesions. |
B2 cells exhibit a pro- atherogenic behavior through antibody responses that stimulate adaptive immunity |
|
| Proatherogenic phenotype. Reparative phenotype |
Reparative phenotype exhibited during thrombotic events when neutrophils promote endothelial repair and angiogenesis (arterial healing) |
Neutrophils secrete ROS increasing the permeability of the ECs and inducing NLRP3 inflammasome. Neutrophils attract monocytes and can activate macrophages via extrusion of their NETs . |
NET formation that stimulates the NLRP3 inflammasome and produce IL-1β (preclinical study). In this scenario NLRP3 inflammasome requires a second hit to be fully activated. Defective efferocytosis that leads to DAMPS accumulation. |
|
Legend: CAD coronary artery disease; DAMPs danger-associated molecular patterns ; ECM extracellular matrix; NET Neutrophils Extracellular Trap; NLRP3 NOD [nucleotide oligomerization domain]-,LRR[leucine-rich repeat]-,and PYD[pyrin domain]-containing protein3 ; PDGF platelet derived growth factor: TNF-α Tumour necrosis factor- α; Atf3 Activating Transcription Factor-3; Nr4a1 nuclear receptor subfamily 4 group a1; Pdl1 Programmed death ligand-1.
Table 2.
Anti-inflammatory agents and CVD.
| Study | Anti-inflammatory agent | Target | Population | Effect on inflammation biomarker | Clinical effects | References |
|---|---|---|---|---|---|---|
| CANTOS | 3 doses of canakinumab (50 mg, 150mg and 300 mg), subcutaneously (s.c.) every 3 months vs placebo |
IL-1β | 10,061 patients with previous myocardial infarction and hsCRP≥2mg/l | Reduction of hsCRP (for all the doses) | The dose of 150 mg s.c.every 3 months was associated to a significant reduction of recurrent CV events |
Ref. 11 NEJM 2017;377:1119 |
| CIRT | Low-dose methotrexate (15-20 mg weekly) |
No specific target | 4,786 patients with known atherosclerosis and either DM orMS | No reduction of IL-1β, IL-6. | No reduction of CV event rates | Ref. 87 NEJM 2019;380:752 |
| RESCUE | Ziltivekimab 7·5 mg, 15 mg, or 30 mg every 4 weeks up to 24 weeks | IL-6 | 264 patients with high CV risk> (age≥18 years, moderate to severe CKD, hsCRP≥2mg/l) | Reduction in biomarkers of inflammation (hsCRP) and thrombosis (e.g fibrinogen) | Reduction in biomarkers of inflammation (hsCRP) and thrombosis (e.g fibrinogen) | Ref.12 Lancet 2021;397:2060 |
| COLCOT | Low dose colchicine (0.5 mg daily) | Inhibition of tubulin polymerization and alteration of leukocyte responsiveness. | Patients with recent myocardial infarction: 2366 patients assigned to colchicine and 2379 to placebo |
. | Significant reduction of ischemic CV events | Ref 13 NEJM 2019;381:2497 |
| Lo-Do-Co2 | Low dose colchicine (0.5 mg daily) | inhibition of tubulin polymerization and alteration of leukocyte responsiveness. |
Patients with chronic coronary artery disease in stable condition: 2762 patients assigned to colchicine and 2760 to placebo. |
31% lower relative risk of CV death, spontaneous myocardial infarction, ischemic stroke, or coronary revascularization in patients treated with colchicine compared to placebo |
Ref 14 NEJM 2020;383:1838 . |
CANTOS Canakinumab Anti-Inflammatory Thrombosis Outcomes Study; CIRT Cardiovascular Inflammation Reduction Trial; COLCOT Colchicine Cardiovascular Outcomes Trial; CV cardiovascular; hsCPR high sensitivity C reactive Protein.
Table 3.
Ideal Atherosclerosis Biomarker.
|
|
|
|
|
|
|
|
|
Table 4.
Pro-and anti-inflammatory cells and mediators in atherosclerosis.
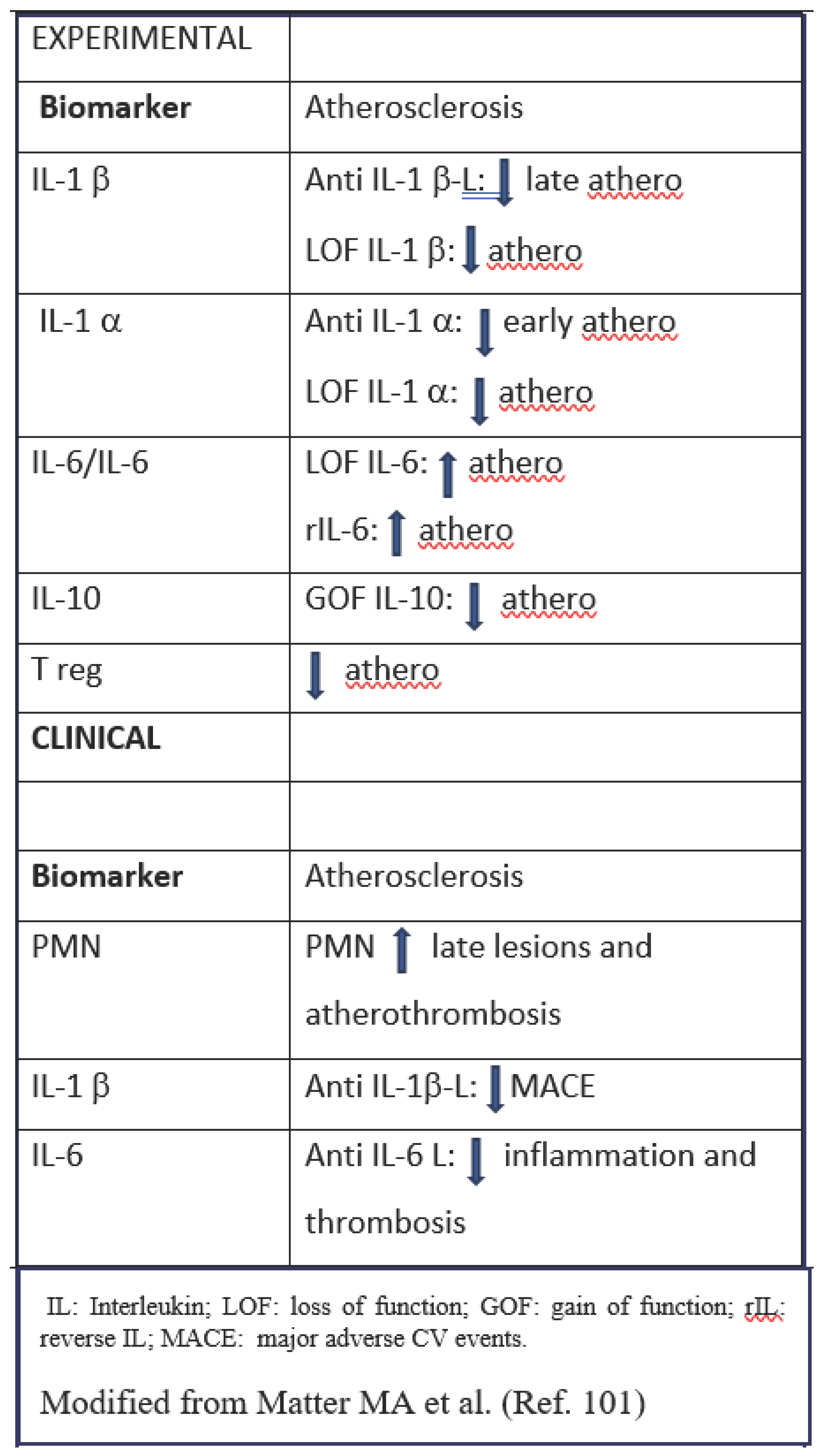
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



