Submitted:
19 March 2024
Posted:
20 March 2024
You are already at the latest version
Abstract
Familial Mediterranean fever (FMF) is a systemic autoinflammatory disorder caused by inherited mutations in the MEFV (Mediterranean FeVer) gene located on chromosome 16 (16p13.3) and coding pyrin protein. Despite the existing data on MEFV mutations, the exact mechanism of their effect on the development of pathological processes leading to autoinflammation observed in FMF, remains unclear. Induced pluripotent stem cells (iPSCs) are considered an important tool for studying the molecular genetic mechanisms of various diseases due to their ability to differentiate into any cell type, including macrophages, which contribute to the development of FMF. In this study, we developed iPSCs of an Armenian patient with FMF having M694V, p.(Met694Val) (c.2080A>G, rs61752717) pathogenic mutation in exon 10 of the MEFV gene. As a result of direct differentiation, macrophages expressing CD14 and CD45 surface markers were obtained. In addition, we found that the morphology of patient macrophages derived from iPSCs with MEFV mutation was significantly different from that of differentiated from iPSCs of a healthy donor carrying wild-type MEFV gene.
Keywords:
Familial Mediterranean fever
; macrophages
; patient-specific induced pluripotent stem cells
; differentiation
; MEFV gene
1. Introduction
Familial Mediterranean fever (FMF) is a systemic autoinflammatory disorder characterized by recurrent episodes of fever and polyserositis (e.g. peritonitis, pleuritis, synovitis) symptoms. The FMF carrier frequencies are high in several eastern Mediterranean populations, ranging from 37–39% in Armenians and Iraqi Jews, to 20% in Turks, North African and Ashkenazi Jews, and Arabs which leads to a significant economic burden [1,2]. The disease is mostly caused by recessively inherited mutations in MEFV, which encodes pyrin protein playing an important role in inflammatory processes [3]. There are two “mutation hot-spots” located in the 2nd (E148Q) and 10th (M694V, M694I, M680I и V726A) exons. These mutations account for over 90% of all FMF cases [4]. Mutated pyrin causes an exaggerated inflammatory response by uncontrolled interleukin-1 (IL-1) secretion [5]. Besides the advances in molecular genetics of FMF, the molecular mechanisms underlying the disease are not fully understood. These questions have been studied using a battery of experimental and in silico methods. Thus, molecular dynamic simulations gain insight into the role of mutations on pyrin structure, function, and interactions [6,7]. Another study of polymorphonuclear neutrophils of FMF patients suggests increased sensitivity of mutated pyrin inflammasome towards cytoskeletal modifications in the absence of pathogens [8]. A recent study using different cell types (synovial fibroblasts, monocytes, macrophages) showed that inflammation-related functional assays have an anti-inflammatory effect of miR-197-3p [9]. Various cell-line-based models have been developed for a more comprehensive understanding of the etiology and pathogenesis of FMF [10]. Also, gene editing with CRISPR/Cas9 has been used to understand the effect of the MEFV E583A mutation on IL-1β secretion [11]. However, immortalized cell lines have their limitations in mimicking the disease of interest since they do not account for patient genetic variability [12], may accumulate mutations and lack genetic and cellular diversity, and mostly represent cancer-derived cells [13]. On the other side, patient primary cells have limited potential for cultivation and maintenance, posing limitations for experiments.
Induced pluripotent stem cells (iPSCs) are considered a unique tool for studying molecular genetic mechanisms of this disease, disease modeling, and screening of potential drugs [14,15,16]. The main advantage of iPSCs is the almost unlimited ability of cultivation and differentiation serving as a proper source of pluripotent stem cells and any type of cells in the living organism. IPSCs were successfully used for studying autoinflammatory [17,18], neurodegenerative [17,18,19,20,21,22,23,24], and other diseases. There are few attempts to create iPSCs for FMF patients, for example, a Turkish patient with a homozygous missense mutation (p.Met694Val) in the MEFV gene [25].
In this paper, we generated iPSCs from an Armenian FMF patient carrying a homozygous c.2080A>G (M694V) mutation in the MEFV gene. Molecular-genetic characterization proved their stemness characteristics. We further differentiated these cells into macrophage-like cells. The morphology analysis showed considerable differences between derived macrophages with mutated MEFV gene compared with macrophages derived from iPSCs with wild-type MEFV [26].
2. Results
2.1. Generation and Characteristics of iPSCs Associated with the MEFV Gene Mutation
A 20-year-old patient was admitted to the Rheumatology Department of Mikayelyan University Hospital with symptoms relevant to mixed thoracoabdominal form of FMF, pain in joints, arthritis, erysipeloid erythema, and fever. Genetic analysis of the patient revealed pathogenic homozygous missense mutation c.2080A>G (p.M694V, rs61752717) in exon 10 of the MEFV gene. We isolated peripheral blood mononuclear cells (PBMCs) in a Ficoll gradient and reprogrammed them using episomal vectors OCT4, KLF4, L-MYC, SOX2, LIN28, and Trp53 [27]. As a result, 10 independent cell lines were obtained, one of which was characterized in detail. All resulting cell lines have a large nuclear-cytoplasmic ratio, grow in densely packed iPSC-like single-layer colonies (Figure 1A), and express the early stem cell marker endogenous alkaline phosphatase (Figure 1B). Cultivation of the resulting cells was carried out on a substrate of mitotically inactivated mouse embryonic fibroblasts (MEF).
One cell line was selected for detailed characterization and was registered in the Human Pluripotent Stem Cell Registry (hPSCreg, https://hpscreg.eu, accessed on 14 March 2024) under the name RAUi002-A. We carried out qualitative (immunofluorescence) and quantitative (RT-qPCR) analyzes of this line for markers of pluripotent cells. Both analyzes demonstrate the presence in cells obtained from a patient with FMF, associated with the genetic variant c.2080A>G (M694V) in the MEFV gene, expression of the transcription factors OCT4, SOX2, and NANOG (Figure 1C,D), as well as immunofluorescence showing the expression of surface marker SSEA-4 (Figure 1C). Cytogenetic analysis (G-banding) of the obtained cells showed the presence of a normal karyotype (46,XY) (Figure 1E).
One of the main tests of pluripotency is the ability of cells to give rise to all three germ layers (ectoderm, mesoderm, and endoderm). We performed spontaneous differentiation in embryoid bodies (Figure 1F) and used immunofluorescence analysis of differentiated cells to show the expression of mesoderm markers (α-smooth muscle actin (αSMA) and the surface marker CD29), ectoderm (tubulin β 3 (TUBB3/TUJ1) and mature neural cell markers methionine aminopeptidase 2 (MAP2)), and endoderm (alpha-fetoprotein (AFP) and cytokeratin 18 (CK18)) (Figure 1G). Thus, we have demonstrated that the resulting cells are pluripotent, and we will refer to them as iPSCs.
To confirm the presence of a pathogenic mutation in the resulting iPSC line, Sanger sequencing of DNA isolated from the patient's PBMCs and from the RAUi002-A iPSC line was performed and compared to DNA from a conditionally healthy patient. We found a substitution at position 2080 A to G in exon 10 of the MEFV gene in both samples compared to control DNA (Figure 1H, location of substitution indicated by arrow). In addition, to confirm the origin of the iPSCs derived from the patient's PBMCs, STR analysis was performed on the DNA of the PBMC sample and the RAUi002-A line. 25 loci from both samples were analyzed and their identity was shown (data available on request from the authors). This suggests that iPSCs data obtained from an FMF patient can serve as a tool to study the contribution of the p.M694V mutation in the MEFV gene to the pathogenesis of FMF disease.
RAUi002-A iPSCs were also analyzed for the presence/absence of residual episomes and culture contamination with mycoplasma. Both PCR analyses showed their complete absence (Figure 1I).
Summary characteristics of the RAUi002-A iPSC line are shown in Table 1.
2.2. Generation and Characteristic of Macrophages from RAUi002-A iPSCs
The resulting iPSC line was specifically differentiated into macrophages to obtain a relevant cell type for further studies on the pathogenesis of FMF. The previously obtained iPSC line K7-4Lf/ICGi022-A was used as a control cell line in the experiment [26]. Differentiation of iPSCs into macrophages was achieved by adding cytokines such as interleukin-3 (IL-3) and macrophage colony-stimulating factor (M-CSF) to the differentiating embryoid bodies (Figure 2A,B) to differentiation along the myeloid pathway and form a homogeneous population of monocytes. As a result, from day 14 of differentiation and over 3 weeks, monocytes were produced in the culture medium that adhered to the plastic and terminally differentiated into macrophage-like cells in the presence of M-CSF. Immunofluorescence for markers specific for mature macrophages, CD14 and CD45, confirmed that the resulting cells were macrophages (Figure 2). IPSC-derived macrophages from a healthy donor were found to have a classic cloaked, spreading morphology (Figure 2C), whereas macrophages with the pathogenic p.M694V mutation in the MEFV gene had an elongated morphology with many rounded cells (Figure 2D, middle photo, white arrows).
3. Discussion
In this study, we used the technology of reprogramming PBMCs into a pluripotent state to obtain patient-specific iPSCs from a patient with FMF associated with the pathogenic mutation p.M694V in the MEFV gene. The resulting cell line meets all the requirements of pluripotent cells, has a stem cell-like morphology, a normal karyotype, and is capable of producing derivatives of three germ layers. These cells demonstrated their ability to differentiate into macrophages which are one of the key cells involved in the disease pathogenesis [28].
Research related to the establishment of patient-derived iPSCs is expected to be a promising avenue for elucidating the pathogenesis of the diseases, disease therapy, and for drug discovery [29]. They became attractive tools for studying neurodegeneration [21,22,23,24,30], cardiac dysfunction [31,32,33], and genetic disorders, such as Duchenne’s muscular dystrophy [34]. Recently these approaches have been actively used for modeling immune-related diseases, such as systemic lupus erythematosus, systemic sclerosis, rheumatoid arthritis, and autoinflammatory syndromes (for review see in [35]). It has been shown that various cell types differentiated from patient-derived iPSCs can be further used for research into the pathogenesis of these diseases.
To our knowledge, a few attempts to generate iPSCs from FMF patients have been made [14,25]. Fidan et al. (2015) reported a cell line derived from fibroblasts of an FMF patient carrying homozygous p.Met694Val mutation in the MEFV gene [25]. In our study, we successfully reprogrammed PBMCs of FMF patient with homozygous MEFV gene mutation (M694V). This method is less invasive for the patients. Moreover, we performed the differentiation of stem cells into macrophages and analyzed morphological differences between healthy and diseased macrophages. The morphology of macrophage-like cells derived from control iPSCs was significantly different from that of cells derived from iPSCs with a mutation in the MEFV gene. Control macrophage-like cells had a flattened morphology, whereas patient-derived cells had an elongated morphology with a large number of rounded, dying cells. We saw that under the same culture conditions, macrophages with a mutation in the MEFV gene are less viable, most likely due to a pathogenic mutation. These results are well aligned with the previous observations about the structural and functional features of FMF patients’ primary immune cells. Thus, studies indicated characteristics of aged/activated cells (small cell size and granularity, up-regulated CXCR4) for polymorphic neutrophils from the patients in acute flares, while in remission mixed morphology (normal cell size and granularity, up-regulated CD11b, CD49d, CXCR4, and CD62L) has been described [8].
One of the advantages of iPSC-derived macrophages is the preservation of the initial phenotype of the cells. In previous research, it has been shown that iPSC-derived cells at different stages of differentiation demonstrate a complete switch of iPSCs to cells expressing a monocyte, macrophage, or dendritic cell-specific gene profile. Moreover, iPSC-derived LPS-induced macrophages induce the expression of classic macrophage pro-inflammatory response markers [36]. Moreover, the ability to grow an unlimited number of iPSCs and differentiate them into various cells opens multiple avenues for studying FMF pathogenesis, performing drug candidate screening, and developing gene-based therapies. Using patient-specific iPSCs from FMF patients and the CRISPR/Cas9 genome editing system, it will be possible to generate modified isogenic iPSC lines with the corrected mutation in the future. Such cell platforms will be valuable in understanding the effects of the mutations on pyrin inflammasome dysfunction in FMF.
4. Materials and Methods
4.1. Ethics Statement
The study was approved by the Ethics Committee of the Institute of Molecular Biology NAS RA (IRB 00004079, Protocol N3 from 23.08.2021). A patient provided informed consent about the use of the blood sample for planned analysis. ICGi022-A iPSC cell line obtained from a healthy donor [26] was used as a control in the experiments of macrophage differentiation and analysis of the morphological features of mutant and wild-type MEFV-carrying cells.
4.2. Detection of MEFV Mutation
Mutations in the MEFV gene in the FMF patient was determined by commercially available qPCR assay for 26 most common mutations (FMF Multiplex real-time CPR kit, SNP Biotechnology RnD Ltd, Turkey).
4.3. Reprogramming of PBMCs into iPSCs
PBMCs of a patient with FMF were isolated as described here [22]. iPSCs were obtained by overexpression of reprogramming factors OCT4, KLF4, L-MYC, SOX2, LIN28, and Trp53 using a set of episomal vectors (ID Addgene #41855–58, #41813–14) as previously described [22].
IPSCs were propagated onto feeder layer of mitotically inactivated mouse embryonic fibroblasts (MEF) in iPSC-medium: 82% KnockOut DMEM medium, 15% KoSR, 2 mM Gluta-MAX, 100 U/ml penicillin-streptomycin, 0.1 mM MEM NEAA (all Thermo Fisher Scientific, Waltham, MA, USA), 0.1 mM β-Mercaptoethanol (Sigma-Aldrich, Darmstadt, Germany), 10 ng/ml basic FGF (SCI Store, Moscow, Russia).
IPSCs were passaged using TrypLE Express (Thermo Fisher Scientific, Waltham, MA, USA), splitting 1:10 in the iPSC medium with the addition of 2 µM Thiazovivin (Sigma-Aldrich, Darmstadt, Germany) for the first 24 hours.
4.4. In Vitro Spontaneous Differentiation of RAUi002-A into Three Germ Layers
Differentiation capacity of the iPSCs was estimated by spontaneous differentiation in embryoid bodies as described earlier [37].
4.5. Immunofluorescent Staining of RAUi002-A iPSC Line
For immunofluorescence staining, cells growing on Chambered Coverglass 8-well plates (Thermo Fisher Scientific, Waltham, MA, USA) were fixed with 4% PFA (Sigma-Aldrich, Darmstadt, Germany), permeabilized with 0.5% Triton-X (Thermo Fisher Scientific, Waltham, MA, USA) in PBS for 30 minutes, and incubated in blocking buffer containing 1% BSA (Sigma-Aldrich, Darmstadt, Germany) in PBS at room temperature. Primary antibodies were diluted in a blocking buffer according to Table 2. Cell preparations were incubated with primary antibodies overnight at +4°C. Preparations were washed with PBS twice for 15 min, and secondary antibodies were added for 1.5 hours at room temperature. After incubation, cell preparations were washed twice with PBS and stained with DAPI. Manufacturers, catalog numbers and dilutions of all used antibodies are listed in Table 2. The preparations were analyzed using Nikon Eclipse Ti-E microscope and NIS Elements software.
4.6. qPCR Analysis of Expression of Pluripotency Markers in RAUi002-A iPSC Line
For RNA isolation, 2*106 cells were lysed in 1 ml TRIzol reagent (Ambion by Life technologies, Carlsbad CA, USA), and processed according to the manufacturer's protocols. The cDNA was synthesized by reverse transcription of 1 μg RNA using M-MuLV reverse transcriptase (Biolabmix, Novosibirsk, Russia).
Quantitative PCR (qPCR) was performed on a LightCycler 480 II system (Roche, Basel, Switzerland) using BioMaster HS-qPCR SYBR Blue 2 × (Biolabmix, Novosibirsk, Russia) with the following program: 95 °C 5 min; 40 cycles: 95 °C 10 s, 60 °C 1 min. The primers used are listed in Table 2. qPCR reactions for each sample were run in triplicate. CT values of the samples for NANOG, OCT4, and SOX2 expression were normalized to beta-2-microglobulin (B2M), and the results were processed using the ΔΔCT method.
4.7. Karyotyping of RAUi002-A iPSC Line
Karyotype analysis was performed as described earlier [22]. For chromosome banding, samples were stained with DAPI (4,6-diamino-2-phenylindole) solution (200 ng/mL, in 2xSSC) for 5 minutes, rinsed in 2xSSC buffer and water. Air-dried slides were covered with 7-10 μL antifade (Vector, United States) under a coverslip. Analysis of preparations was performed using an Axioplan 2 microscope (Zeiss, Germany) equipped with a CV-M300 CCD camera (JAI Corp., Japan) at the Center for Collective Use of Microscopic Analysis of Biological Objects at the Institute of Cytology and Genetics, Siberian Branch of the Russian Academy of Sciences. ISIS 5 software (MetaSystems Group, Inc., United States) was used for metaphase processing and chromosome folding.
4.8. Genotyping of RAUi002-A iPSC Line
Sanger sequencing was used to confirm the mutation in the MEFV gene in the RAUi002-A iPSC line. To confirm the absence of MEFV mutations, Sanger sequencing was performed also for the line K7-4Lf/ICGi022-A used as a control sample. The list of primers used is shown in Table 2. Genome DNA was isolated using Quick-DNA Miniprep Kit (Zymo Research, Irvine, CA, USA). PCR reactions were run on a T100 thermal cycler (Bio-Rad) using BioMaster HS-Taq PCR-Color (2×) (Biolabmix, Novosibirsk, Russia) with the program: 95 ◦C, 3 min; further 35 cycles: 95 ◦C, 30 s; 60 ◦С, 30 s; 72 ◦C, 30 s; and 72 ◦C, 5 min. For Sanger sequencing, we used BigDye Terminator V. 3.1. Cycle Sequencing Kit (Applied Biosystems, Austin, TX, USA). Sequencing reactions were analyzed on an ABI 3130XL genetic analyzer at the Genomics Center of the SB RAS (http://www.niboch.nsc.ru/doku.php/corefacility, accessed on 14 March 2024).
STR profiling was performed using AmpFlSTR Identifiler (Applied Biosystems) and Investigator HDplex (QIAGEN) by Genoanalytica company (https://www.genoanalytica.ru, accessed on 14 March 2024).
4.9. Detection of Mycoplasma and Reprogramming Vectors in RAUi002-A iPSC Line
4.10. Differentiation of RAUi002-A iPSC Line into Macrophages
Differentiation of iPSCs into macrophages was performed according to a previously published protocol [39] with modifications. IPSCs were placed on a Petri dish (D60 mm) coated with mitotically inactivated MEFs. Dense iPSC colonies were detached with 0.15% collagenase type IV (Thermo Fisher Scientific, Waltham, MA, USA), washed with medium, and transferred to a Petri dish (D60 mm) coated with 1% agarose (Sigma-Aldrich, Darmstadt, Germany) in iPSC medium without the addition of bFGF. On day 4 of culture, the formed embryoid bodies were transferred to 3 wells of a 6-well plate coated with 0.1% gelatin (Sigma-Aldrich, Darmstadt, Germany) for spreading and differentiation into monocyte-like cells in RPMI medium supplemented with 10% fetal bovine serum, 2 mM GlutaMax, 100 U/ml penicillin-streptomycin, 0. 1 mM MEM NEAA, 1 mM sodium pyruvate (all Thermo Fisher Scientific, Waltham, MA, USA), 0.1 mM 2-mercaptoethanol (2-mce, Sigma-Aldrich, Darmstadt, Germany), 25 ng/ml IL-3 and 100 ng/ml M-CSF (both SCI Store, Moscow, Russia). From day 14-19 of culture, the cell suspension was collected from embryoid bodies containing monocyte-like cells, centrifuged at 300 g for 5 min, and seeded onto Chambered Coverglass 8-well plates pretreated with 0.1% gelatin for immunofluorescence staining.
Author Contributions
Conceptualization, S.M.Z., R.Z., A.A. and V.S.V.; methodology, E.V.G.; validation, E.V.G. and S.P.M.; formal analysis, A.A.M.; investigation, E.V.G., J.M.M., S.P.M., L.V.K., V.H.H.; resources, S.M.Z.; data curation, R.Z., A.A., S.M.Z. and E.V.G.; interpretation of data, S.M.Z., R.Z., A.A., V.S.V.; writing—original draft preparation, E.V.G. and A.A.M.; writing—review and editing, R.Z. and A.A.; visualization, E.V.G., R.Z. and A.A.; supervision, S.M.Z., R.Z. and A.A.; project administration, R.Z. and A.A.; funding acquisition, S.M.Z. and R.Z. All authors have read and agreed to the published version of the manuscript.
Funding
The cell reprogramming and characterization was funded by the Ministry of Science and Higher Education of the Russian Federation, Agreement No. 075-15-2021-1063 of 09/28/2021. PBMCs isolation and molecular-genetic characterisation work was supported by the Higher Science and Education Committee of the Ministry of Health RA, in the frames of the research project N 21SCG-1F010 and research grant provided by the Armenian Engineers and Scientists of America (PI: Dr. Roksana Zakharyan). The immunofluorescent imaging was performed using resources of the Common Facilities Center of Microscopic Analysis of Biological Objects, ICG SB RAS (https://ckp.icgen.ru/ckpmabo/, accessed on 14 March 2024), supported by the Budget project of the Institute of Cytology and Genetics (FWNR-2022-0015).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of the Institute of Molecular Biology NAS RA (IRB 00004079, Protocol N3 from 23.08.2021).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are openly available in the Human Pluripotent Stem Cell Registry (https://hpscreg.eu/cell-line/RAUi002-A and https://hpscreg.eu/cell-line/ICGi022-A, all accessed on 14 March 2024).
Acknowledgments
The equipment of the Meshalkin National Medical Research Center, Ministry of Health of the Russian Federation (Novosibirsk, Russia) was used for routine cell imaging.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Chae, J.J.; Aksentijevich, I.; Kastner, D.L. Advances in the understanding of familial Mediterranean fever and possibilities for targeted therapy. Br J Haematol. 2009, 146, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Booty, M.G.; Chae, J.J.; Masters, S.L.; Remmers, E.F.; Barham, B.; Le, J.M.; Barron, K.S.; Holland, S.M.; Kastner, D.L.; Aksentijevich, I. Familial Mediterranean fever with a single MEFV mutation: where is the second hit? Arthritis Rheum. 2009, 60, 1851–1861. [Google Scholar] [CrossRef] [PubMed]
- Cantarini, L.; Rigante, D.; Brizi, M.G.; Lucherini, O.M.; Sebastiani, G.D.; Vitale, A.; Gianneramo, V.; Galeazzi, M. Clinical and biochemical landmarks in systemic autoinflammatory diseases. Ann Med. 2012, 44, 664–673. [Google Scholar] [CrossRef] [PubMed]
- French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet. 1997, 17, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Özen, S.; Batu, E.D.; Demir, S. Familial Mediterranean Fever: recent developments in pathogenesis and new recommendations for management. Front. Immunol. 2017, 8, 253. [Google Scholar] [CrossRef] [PubMed]
- Sargsyan, A.; Sahakyan, H.; Nazaryan, K. Effect of colchicine binding site inhibitors on the tubulin intersubunit interaction. ACS Omega 2023, 8, 29448–29454. [Google Scholar] [CrossRef] [PubMed]
- Arakelov, G.; Arakelov, V.; Nazaryan, K. Complex formation dynamics of native and mutated pyrin's B30.2 domain with caspase-1. Proteins 2018, 86, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Martirosyan, A.; Poghosyan, D.; Ghonyan, S.; Mkrtchyan, N.; Amaryan, G.; Manukyan, G. Transmigration of neutrophils from patients with Familial Mediterranean fever causes increased cell activation. Front. Immunol. 2021, 12, 672728. [Google Scholar] [CrossRef]
- Akkaya-Ulum, Y.Z.; Akbaba, T.H.; Tavukcuoglu, Z.; Chae, J.J.; Yilmaz, E.; Ozen, S.; Balci-Peynircioglu, B. Familial Mediterranean fever-related miR-197-3p targets IL1R1 gene and modulates inflammation in monocytes and synovial fibroblasts. Sci. Rep. 2021, 11, 685. [Google Scholar] [CrossRef]
- Mezher, N.; Mroweh, O.; Karam, L.; Ibrahim, J.N.; Kobeissy, P.H. Experimental models in Familial Mediterranean Fever (FMF): Insights into pathophysiology and therapeutic strategies. Exp. Mol. Pathol. 2024, 135, 104883. [Google Scholar] [CrossRef]
- Wang, Q.; Jin, T.; Jian, Sh.; Han, X.; Song, H.; Zhou, Q.; Yu, X. A dominant pathogenic MEFV mutation causes atypical pyrin-associated periodic syndromes. JCI Insight. 2023, 8, e172975. [Google Scholar] [CrossRef] [PubMed]
- Volpato, V.; Webber, C. Addressing variability in iPSC-derived models of human disease: guidelines to promote reproducibility. Dis. Model. Mech. 2020, 13, dmm042317. [Google Scholar] [CrossRef] [PubMed]
- Gorringe, K.L.; Chin, S.F.; Pharoah, P.; Staines, J.M.; Oliveira, C.; Edwards, P.A.W.; Caldas, C. Evidence that both genetic instability and selection contribute to the accumulation of chromosome alterations in cancer. Carcinogenesis 2005, 26, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Shiba, T.; Tanaka, T.; Ida, H.; Watanabe, M.; Nakaseko, H.; Osawa, M.; Shibata, H.; Izawa, K.; Yasumi, T.; Kawasaki, Y.; Saito, M.K.; Takita, J.; Heike, T.; Nishikomori, R. Functional evaluation of the pathological significance of MEFV variants using induced pluripotent stem cell-derived macrophages. J. Allergy Clin. Immunol. 2019, 144, 1438–1441.e12. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.K. Elucidation of the pathogenesis of autoinflammatory diseases using iPS cells. Children (Basel) 2021, 8, 94. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.G. & Daley, G.Q. Induced pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Genet. 2019, 20, 377–388. [Google Scholar] [CrossRef]
- Hew, M.; O’Connor, K.; Edel, M.J.; Lucas, M. The possible future roles for iPSC-derived therapy for autoimmune diseases. J. Clin. Med. 2015, 4, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Shiba, T.; Honda, Y.; Izawa, K.; Yasumi, T.; Saito, M.K.; Nishikomori, R. Induced pluripotent stem cell-derived monocytes/macrophages in autoinflammatory diseases. Front. Immunol. 2022, 13, 870535. [Google Scholar] [CrossRef]
- Okano, H. & Morimoto, S. iPSC-based disease modeling and drug discovery in cardinal neurodegenerative disorders. Cell Stem Cell 2022, 29, 189–208. [Google Scholar] [CrossRef]
- Pandey, Sh.; Jirásko, M.; Lochman, J.; Chvátal, A.; Dvorakova, M.C.; Kučera, R. iPSCs in neurodegenerative disorders: a unique platform for clinical research and personalized medicine. J. Pers. Med. 2022, 12, 1485. [Google Scholar] [CrossRef]
- Grigor’eva, E.V.; Malankhanova, T.B.; Surumbayeva, A.; Pavlova, S.V.; Minina, J.M.; Kizilova, E.A.; Suldina, L.A.; Morozova, K.N.; Kiseleva, E.; Sorokoumov, E.D.; et al. Generation of GABAergic striatal neurons by a novel iPSC differentiation protocol enabling scalability and cryopreservation of progenitor cells. Cytotechnology. 2020, 72, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Grigor’eva, E.V.; Kopytova, A.E.; Yarkova, E.S.; Pavlova, S.V.; Sorogina, D.A.; Malakhova, A.A.; Malankhanova, T.B.; Baydakova, G.V.; Zakharova, E.Y.; Medvedev, S.P.; Pchelina, S.N.; et al. Biochemical Characteristics of iPSC-Derived Dopaminergic Neurons from N370S GBA Variant Carriers with and without Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 4437. [Google Scholar] [CrossRef] [PubMed]
- Malankhanova, T.B.; Suldina, L.A.; Grigor’eva, E.V.; Medvedev, S.P.; Minina, J.M.; Morozova, K.N.; Kiseleva, E.; Zakian, S.M.; Malakhova, A.A. A human induced pluripotent stem cell–derived isogenic model of Huntington’s disease based on neuronal cells has several relevant phenotypic abnormalities. J. Pers. Med. 2020, 10, 215. [Google Scholar] [CrossRef] [PubMed]
- Ustyantseva, E.; Pavlova, S.V.; Malakhova, A.A.; Ustyantsev, K.; Zakian, S.M.; Medvedev, S.P. Oxidative stress monitoring in iPSC-derived motor neurons using genetically encoded biosensors of H2O2. Sci Rep. 2022, 12, 8928. [Google Scholar] [CrossRef] [PubMed]
- Fidan, K.; Kavaklıoğlu, G.; Ebrahimi, A.; Özlü, C.; Ay, N.Z.; Ruacan, A.; Gül, A.; Önder, T.T. Generation of integration-free induced pluripotent stem cells from a patient with Familial Mediterranean Fever (FMF). Stem Cell Res. 2015, 15, 694–696. [Google Scholar] [CrossRef] [PubMed]
- Malakhova, A.A.; Grigor’eva, E. V.; Pavlova, S. V.; Malankhanova, T.B.; Valetdinova, K.R.; Vyatkin, Y. V.; Khabarova, E.A.; Rzaev, J.A.; Zakian, S.M.; Medvedev, S.P. Generation of induced pluripotent stem cell lines ICGi021-A and ICGi022-A from peripheral blood mononuclear cells of two healthy individuals from Siberian population. Stem Cell Res. 2020, 48. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Yamakawa, T.; Matsumura, Y.; Sato, Y.; Amano, N.; Watanabe, A.; Goshima, N.; Yamanaka, S. An efficient nonviral method to generate integration-free human-induced pluripotent stem cells from cord blood and peripheral blood cells. Stem Cells. 2013, 31, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Savran, Y.; Sari, I.; Kozaci, D.L.; Gunay, N.; Onen, F.; Akar, S. Increased levels of macrophage migration inhibitory factor in patients with Familial Mediterranean Fever. Int. J. Med. Sci. 2013, 10, 836–839. [Google Scholar] [CrossRef]
- Nicholson, M.W.; Ting, C.Y.; Chan, D.Z.H.; Cheng, Y.; Lee, Y.; Hsu, C.; Huang, C.; Hsieh, P.C.H. Utility of iPSC-derived cells for disease modeling, drug development, and cell therapy. Cells 2022, 11, 1853. [Google Scholar] [CrossRef]
- Chamberlain, S.J. Disease modelling using human iPSCs. Hum. Mol. Genet. 2016, 25, R173–R181. [Google Scholar] [CrossRef]
- Csöbönyeiová, M.; Polák, S.; Danišovič, L. Perspectives of induced pluripotent stem cells for cardiovascular system regeneration. Exp. Biol. Med (Maywood). 2015, 240, 549–556. [Google Scholar] [CrossRef]
- Rikhtegar, R.; Pezeshkian, M.; Dolati, S.; Safaie, N.; Rad, A.A.; Mahdipour, M.; Nouri, M.; Jodati, A.R.; Yousefi, M. Stem cells as therapy for heart disease: iPSCs, ESCs, CSCs, and skeletal myoblasts. Biomed. Pharmacother. 2019, 109, 304–313. [Google Scholar] [CrossRef]
- Parrotta, E.I.; Lucchino, V.; Scaramuzzino, L.; Scalise, S.; Cuda, G. Modeling cardiac disease mechanisms using induced pluripotent stem cell-derived cardiomyocytes: progress, promises and challenges. Int. J. Mol. Sci. 2020, 21, 4354. [Google Scholar] [CrossRef] [PubMed]
- Danisovic, L.; Culenova, M.; Csobonyeiova, M. Induced pluripotent stem cells for Duchenne muscular dystrophy modeling and therapy. Cells 2018, 7, 253. [Google Scholar] [CrossRef]
- Shoda, H.; Natsumoto, B.; Fujio, K. Investigation of immune-related diseases using patient-derived induced pluripotent stem cells. Inflamm. Regen. 2023, 43, 51. [Google Scholar] [CrossRef]
- Monkley, S.; Krishnaswamy, J.K.; Göransson, M.; Clausen, M.; Meuller, J.; Thörn, K.; Hicks, R.; Delaney, S.; Stjernborg, L. Optimised generation of iPSC-derived macrophages and dendritic cells that are functionally and transcriptionally similar to their primary counterparts. PLoS One 2020, 15, e0243807. [Google Scholar] [CrossRef]
- Grigor’eva, E.V.; Malakhova, A.A.; Ghukasyan, L.; Hayrapetyan, V.; Atshemyan, S.; Vardanyan, V.; Zakian, S.M.; Zakharyan, R.; Arakelyan, A. Generation of three induced pluripotent stem cell lines (RAUi001-A, RAUi001-B and RAUi001-C) from peripheral blood mononuclear cells of a healthy Armenian individual. Stem Cell Research. 2023, 71, 103147. [Google Scholar] [CrossRef] [PubMed]
- Choppa, P.C.; Vojdani, A.; Tagle, C.; Andrin, R.; Magtoto, L. Multiplex PCR for the detection of mycoplasma fermentans, M. hominis and M. penetrans in cell cultures and blood samples of patients with chronic fatigue syndrome. Mol. Cell. Probes. 1998, 12, 301–308. [Google Scholar] [CrossRef]
- Wilgenburg, B.; Browne, C.; Vowles, J.; Cowley, S.A. Efficient, long term production of monocyte-derived macrophages from human pluripotent stem cells under partly-defined and fully-defined conditions. PLoS One. 2013, 8, e71098. [Google Scholar] [CrossRef]
Figure 1.
Characteristics of the iPSC cell lines RAUi002-A. (A) Morphology of iPSC colonies. (B) Histochemical detection of alkaline phosphatase (AP). (C) Immunofluorescent staining for pluripotency markers OCT4 (red signal), NANOG (green signal), SSEA-4 (green signal), TRA-1-60 (red signal). (D) Quantitative analysis of the expression of pluripotency markers (OCT4, NANOG, SOX2) using RT-qPCR. Error bars show standard deviation. (E) Karyotype analysis (G-banding) (46,XY). (F) Morphology of embryoid bodies on the 18th day of differentiation. (G) Immunofluorescent staining for differentiation markers: αSMA (red signal) and CD29 (green signal) (mesoderm); TUBB3/TUJ1 (red signal) and MAP2 (green signal) (ectoderm); AFP (red signal) and CK18 (green signal) (endoderm). Nuclei are stained with DAPI (blue signal). (H) Chromatograms of MEFV gene regions of PBMCs of a patient with FMF, and iPSCs with wild-type MEFV [26]. The detected polymorphisms are marked with arrows. (I) PCR test for mycoplasma and episomes of the iPSC line (RAUi002-A). Scale bars for (A-C) and (G) - 100 μm. Scale bar for (F) - 500 μm.
Figure 1.
Characteristics of the iPSC cell lines RAUi002-A. (A) Morphology of iPSC colonies. (B) Histochemical detection of alkaline phosphatase (AP). (C) Immunofluorescent staining for pluripotency markers OCT4 (red signal), NANOG (green signal), SSEA-4 (green signal), TRA-1-60 (red signal). (D) Quantitative analysis of the expression of pluripotency markers (OCT4, NANOG, SOX2) using RT-qPCR. Error bars show standard deviation. (E) Karyotype analysis (G-banding) (46,XY). (F) Morphology of embryoid bodies on the 18th day of differentiation. (G) Immunofluorescent staining for differentiation markers: αSMA (red signal) and CD29 (green signal) (mesoderm); TUBB3/TUJ1 (red signal) and MAP2 (green signal) (ectoderm); AFP (red signal) and CK18 (green signal) (endoderm). Nuclei are stained with DAPI (blue signal). (H) Chromatograms of MEFV gene regions of PBMCs of a patient with FMF, and iPSCs with wild-type MEFV [26]. The detected polymorphisms are marked with arrows. (I) PCR test for mycoplasma and episomes of the iPSC line (RAUi002-A). Scale bars for (A-C) and (G) - 100 μm. Scale bar for (F) - 500 μm.
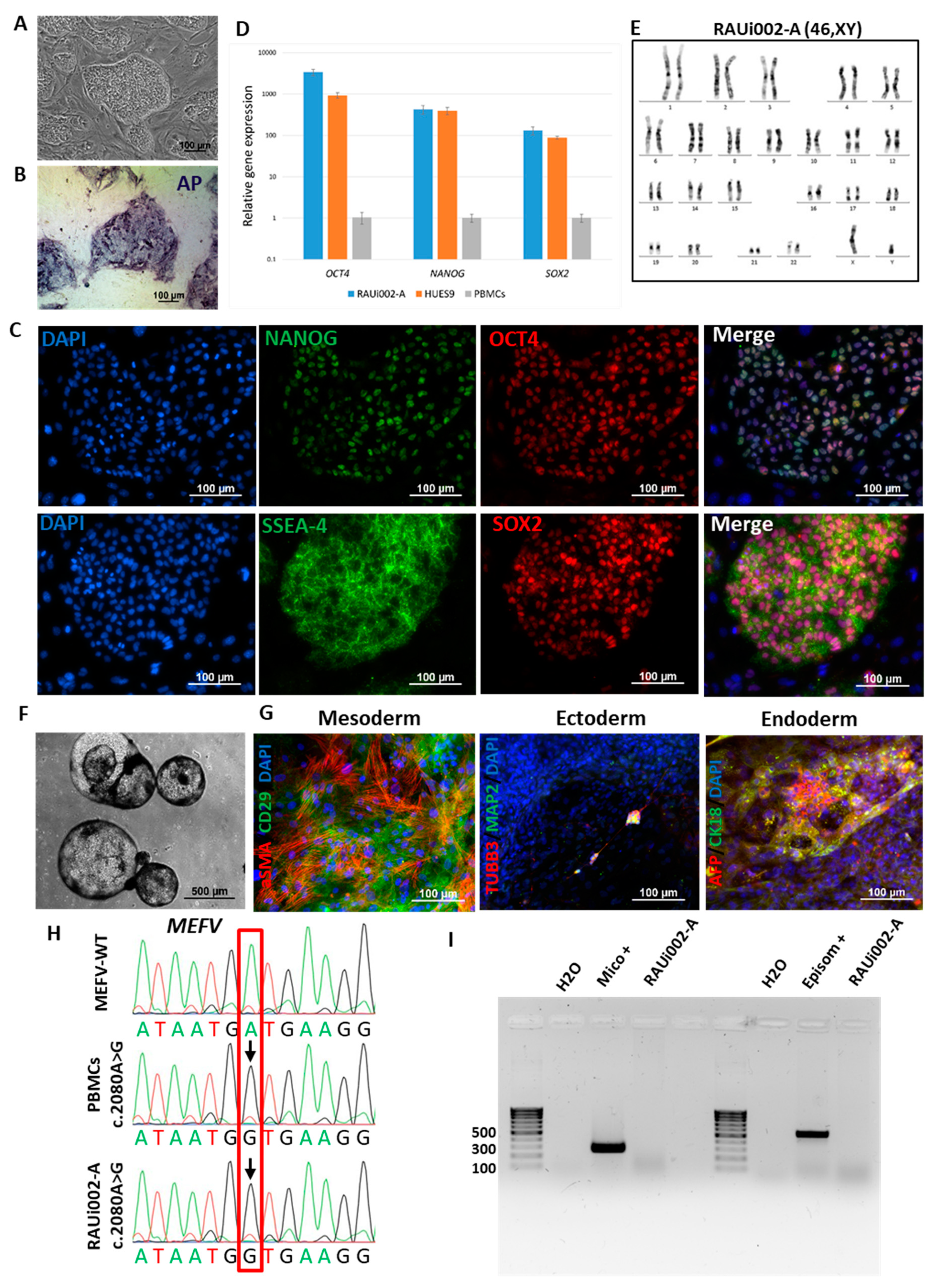
Figure 2.
Differentiation of iPSCs into macrophages and characteristics of the resulting cells. (A) Morphology of embryoid bodies on day 4 of differentiation of iPSCs line RAUi002-A. (B) Morphology of spread out embryoid bodies on the 5th day after plating of the RAUi002-A line. (C) Immunofluorescence analysis on macrophage-specific marker CD14 of macrophages derived from control iPSCs line ICGi022-A. (D) Immunofluorescent analysis of macrophage-specific markers CD14 and CD45 of macrophages carrying a mutation c.2080A>G (M694V) in the MEFV gene. White arrows indicate rounded cells. Nuclei are stained with DAPI (blue signal). All scale bars: 100 μm.
Figure 2.
Differentiation of iPSCs into macrophages and characteristics of the resulting cells. (A) Morphology of embryoid bodies on day 4 of differentiation of iPSCs line RAUi002-A. (B) Morphology of spread out embryoid bodies on the 5th day after plating of the RAUi002-A line. (C) Immunofluorescence analysis on macrophage-specific marker CD14 of macrophages derived from control iPSCs line ICGi022-A. (D) Immunofluorescent analysis of macrophage-specific markers CD14 and CD45 of macrophages carrying a mutation c.2080A>G (M694V) in the MEFV gene. White arrows indicate rounded cells. Nuclei are stained with DAPI (blue signal). All scale bars: 100 μm.
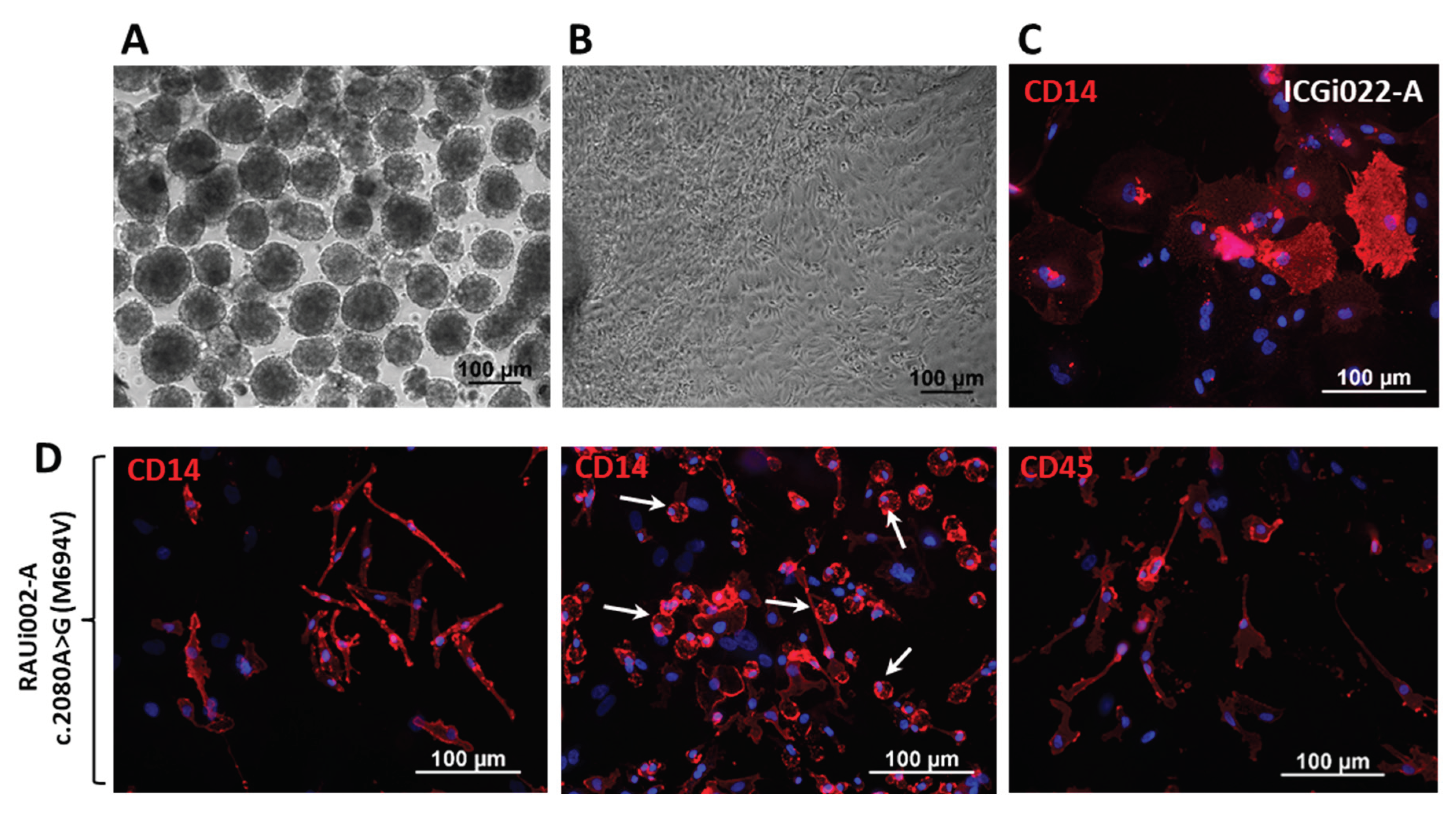

Table 1.
Characteristics and validation of the new line iPSCs RAUi002-A.
| Classification | Test | Result | Data |
|---|---|---|---|
| Morphology | Photography Bright field | Normal | Figure 1A |
| Pluripotency status | Qualitative analysis: Alkaline phosphatase staining | Positive | Figure 1B |
| Qualitative analysis: Immunocytochemistry | Positive staining for pluripotency markers: OCT3/4, SOX2, NANOG, SSEA-4 | Figure 1C | |
| Quantitative analysis: RT-qPCR | Expression of pluripotency markers: NANOG, OCT4, SOX2 | Figure 1D | |
| Genotype | Karyotype (G-banding) | 46,XY | Figure 1E |
| Mutation analysis | Sanger sequencing of DNA from patient's PBMCs and iPSCs | Homozygous p.M694V (c.2080A>G, rs61752717) in exon 10 of the MEFV gene | Figure 1H |
| Differentiation potential | Embryoid body formation | Positive staining for germ layer markers: ɑSMA and CD29 (mesoderm); MAP2 and TUBB3/TUJ1 (ectoderm); CK18/AFP (endoderm) | Figure 1G |
| Specific pathogen-free status | Mycoplasma | Negative | Figure 1I |
Table 2.
Reagents details.
| Antibodies used for immunocytochemistry | |||
| Antibody | Dilution | Company Cat # and RRID | |
| Pluripotency Markers | Mouse IgG2b anti-OCT3/4 (C-10) | 1:200 | Santa Cruz Biotechnology, Dallas, TX, USA, Cat# sc-5279, RRID:AB_628051 |
| Mouse IgG3 anti-SSEA-4 | 1:200 | Abcam, Cambridge, UK, Cat# ab16287, RRID:AB_778073 | |
| Mouse IgG1 anti-NANOG | 1:200 | Santa Cruz Biotechnology, Dallas, TX, USA, Cat# sc-293121, RRID:AB_2665475 | |
| Rabbit IgG anti-SOX2 | 1:500 | Cell Signaling, Danvers, MA, USA, Cat# 3579, RRID:AB_2195767 | |
| Differentiation Markers | Mouse IgG2a anti- αSMA | 1:100 | Dako, Glostrup, Denmark, Cat# M0851, RRID:AB_2223500 |
| Mouse IgG1 anti-CD29 (Integrin beta 1) (TS2/16) | 1:100 | Thermo Fisher Scientific,Waltham, MA, USA, Cat # 14-0299-82, RRID:AB_1210468 | |
| Mouse IgG2a anti-AFP | 1:250 | Sigma-Aldrich, Darmstadt, Germany,Cat# A8452, RRID:AB_258392 | |
| Mouse IgG2a anti-Tubulin β 3 (TUBB3)/ Clone: TUJ1 | 1:1000 | BioLegend, San Diego, CA, USA, Cat# 801201, RRID:AB_2313773 | |
| Chicken IgG anti MAP2 | 1:1000 | Abcam, Cambridge, UK, Cat# аb5392, RRID:AB_2138153 | |
| Mouse IgG1 anti-CK18 | 1:200 | Millipore, Burlington, VT, USA Cat# MAB3234, RRID:AB_94763 | |
| Macrophage-specific Markers | Mouse IgG2b, κ anti-CD14 APC (Clone MφP9) | 1:30 | BD Biosciences, Franklin Lakes, NJ, USA, Cat# 345787, RRID:AB_2868813 |
| Mouse IgG1, κ anti-CD45 PerCP-Cy5.5 CE | 1:20 | BD Biosciences, Franklin Lakes, NJ, USA, Cat# 332784, RRID:AB_2868632 | |
| Secondary antibodies | Goat anti-Mouse IgG3 Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | 1:400 | Thermo Fisher Scientific,Waltham, MA, USA, Cat# A-21151, RRID:AB_2535784 |
| Goat anti-Mouse IgG2b Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 | 1:400 | Thermo Fisher Scientific,Waltham, MA, USA, Cat# A-21144, RRID:AB_2535780 | |
| Goat anti-Rabbit IgG (H + L) Alexa Fluor 568 | 1:400 | Thermo Fisher Scientific,Waltham, MA, USA, Cat# A-11011, RRID:AB_143157 | |
| Goat anti-Mouse IgG1 Alexa Fluor 488 | 1:400 | Thermo Fisher Scientific,Waltham, MA, USA, Cat# A-21121, RRID:AB_2535764 | |
| Goat anti-Mouse IgG1 Alexa Fluor 568 | 1:400 | Thermo Fisher Scientific, Waltham, MA, USA, Cat# A21124, RRID:AB_2535766 | |
| Goat anti-Mouse IgG2a Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 | 1:400 | Thermo Fisher Scientific,Waltham, MA, USA, Cat # A-21134, RRID:AB_2535773 | |
| Goat anti-Chicken IgY (H + L) Alexa Fluor 488 | 1:400 | Abcam, Cambridge, UK, Cat # ab150173, RRID:AB_2827653 | |
| Primers | |||
| Target | Size of band | Forward/Reverse primer (5′-3′) | |
| Episomal plasmid vectors detection | EBNA-1 | 61 bp | TTCCACGAGGGTAGTGAACC/ TCGGGGGTGTTAGAGACAAC |
| Mycoplasma detection | 16S ribosomal RNA gene | 280 bp | GGGAGCAAACAGGATTAGATACCCT/ TGCACCATCTGTCACTCTGTTAACCTC |
| House-keeping gene (RT-qPCR) | beta-2-microglobulin | 280 bp | TAGCTGTGCTCGCGCTACT/ TCTCTGCTGGATGACGTGAG |
| Pluripotency marker (RT-qPCR) | NANOG | 116 bp | TTTGTGGGCCTGAAGAAAACT/ AGGGCTGTCCTGAATAAGCAG |
| OCT4 | 94 bp | CTTCTGCTTCAGGAGCTTGG/ GAAGGAGAAGCTGGAGCAAA |
|
| SOX2 | 100 bp | GCTTAGCCTCGTCGATGAAC/ AACCCCAAGATGCACAACTC |
|
| Targeted mutation analysis | MEFV | 297 bp | TGGGATCTGGCTGTCACATTG/ CATTGTTCTGGGCTCTCCGAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



