Submitted:
15 March 2024
Posted:
18 March 2024
You are already at the latest version
Abstract
PCSK9 is implicated in familial hypercholesterolemia via targeting the cell surface PCSK9-LDLR complex toward lysosomal degradation. The M2 repeat in the PCSK9's C-terminal domain is essential for its extracellular function, potentially through its interaction with an unidentified “protein X”. The M2 repeat was recently shown to bind an R-x-E motif in MHC-class-I proteins (implicated in the immune system), like HLA-C, and causing their lysosomal degradation. These findings suggested a new role of PCSK9 in the immune system and that HLA-like proteins could be “protein X” candidates. However, the participation of each member of the MHC-I protein family in this process and their regulation of PCSK9's function have yet to be determined. Herein, we compared the implication of MHC-I-like proteins such as HFE (involved in iron homeostasis) and HLA-C on the extracellular function of PCSK9. Our data revealed that the M2 domain regulates the intracellular sorting of the PCSK9-LDLR complex to lysosomes, and that HFE is a new target of PCSK9 that inhibits its activity on the LDLR, whereas HLA-C enhances its function. This work suggests the potential modulation of the PCSK9 functions through interactions of HFE and HLA-C.
Keywords:
PCSK9
; LDLR
; HFE
; HLA-C
; MHC-I
; lipid metabolism
1. Introduction
The proprotein convertase subtilisin/kexin type 9 (PCSK9), discovered in 2003 by Seidah et al [1,2], is the 9th member of the proprotein convertases (PCs) family [3] and is primarily expressed in hepatocytes [1]. PCSK9 is the third gene implicated in familial hypercholesterolemia (FH3) [4] because of its ability to target the low-density lipoprotein receptor (LDLR) to lysosomes for degradation [5,6] in a non-enzymatic fashion [7,8], thereby increasing the circulating levels of LDL-cholesterol (LDLc) [9,10,11]. Accordingly, a number of strategies were proposed to silence PCSK9 activity in circulation and/or in hepatocytes, such as inhibitory monoclonal antibodies, siRNA, and CRISPR-editing, resulting in 50-60% reduction in LDLc and significantly decreased cardiovascular events [10,12,13,14,15].
Structurally, PCSK9 comprises five distinct domains, including a signal peptide, a prodomain, a catalytic domain that interacts with the EGF-A domain of LDLR, a hinge domain, and a C-terminal domain known as the Cys-His-rich domain (CHRD), composed of three repeat structures termed M1, M2 and M3 [16,17]. Although the CHRD does not affect the binding of PCSK9 to the LDLR, it is required for the extracellular activity of PCSK9 to induce LDLR degradation [10,18,19,20].
The degradation of the LDLR by PCSK9 proceeds via two distinct pathways, occurring extracellularly [3,18,19,20] or intracellularly [20,21]. In the extracellular pathway, secreted PCSK9 binds the EGF-A domain of the LDLR on the cell surface, and the PCSK9-LDLR complex is then internalized in heavy chain clathrin-coated vesicles and sorted to endosomes/lysosomes for degradation [18,20]. Thus, in presence of PCSK9, LDLR is no longer able to recycle back to the cell surface to uptake more LDLc, leading to reduced levels of LDLR at the plasma membrane, and consequently to increased levels of LDLc in circulation. In contrast, the intracellular pathway shunts the PCSK9-LDLR complex from the trans-Golgi network (TGN) to lysosomes directly via light chain clathrin-coated vesicles [21], before PCSK9 secretion and LDLR surface localization. These two sorting pathways collectively contribute to the regulation of LDLR levels and ultimately impact LDLc homeostasis. Liver hepatocytes are the main source of circulating PCSK9 [22] and the extracellular pathway is the primary route of the PCSK9-LDLR degradation [3,10].
The mechanism by which the extracellular PCSK9-LDLR complex is sorted to lysosomal compartments is not fully understood. However, the CHRD domain appears to play a critical role in this process by facilitating the interaction between PCSK9 and an unidentified partner protein referred to as "protein X" [20,23]. This interaction is essential for directing the PCSK9-LDLR heterodimer to endosomes/lysosomes for degradation. So far, several secretory proteins have been proposed as potential candidates for “protein X”, including APLP2 [24] and Sortilin [25]. However, none of them could be validated [26]. Interestingly, the cytosolic adenylyl cyclase-associated protein 1 (CAP1) was reported to bind the M1 and M3 domains of the CHRD of PCSK9 and somehow enhance its extracellular activity on the LDLR [27]. The rationale for this binding became clear after we showed that CAP1 is secreted, and then binds the M1-, M3- and pro- domains of PCSK9 allowing optimal exposure of the M2 domain, thereby enhancing its extracellular activity, but it is not crucial [23].
The M2 domain of PCSK9 was reported to be key for its extracellular activity on the LDLR, suggesting that it interacts with a hypothesized “protein X” [20,23]. Befittingly, a number of natural variants in the M2 domain including the gain-of-function (GOF) H553R and loss-of-function (LOF) Q554E led to higher and lower circulating LDLc levels, respectively [28]. Recently, it was suggested that the M2 domain of PCSK9 binds an R-x-E motif in some MHC-class I proteins (e.g., HLA-A), sending them to lysosomal degradation [29]. Among the 9-membered family of human MHC-I proteins [30], two specific members, including leukocyte antigen C (HLA-C: implicated in the immune system) and homeostatic iron regulator protein (HFE: involved in iron signaling) have captured our interest because of their likely involvement in lipid metabolism [31,32]. Recently, the 3D structure of HLA-C and its interaction with PCSK9 was modeled [23]. This work confirmed the importance of the R-x-E motif (Arg68 and Glu70) of HLA-C for its interaction with (Glu567 and Arg549) in the M2 domain of PCSK9, respectively. Removal of the interacting Arg and Glu in PCSK9 or HLA-C led to the complete LOF in the activity of PCSK9 on the LDLR. This compelling observation proposed HLA-C (and/or another MHC-I member) as a potential “protein X” that is necessary for the extracellular function of PCSK9 on the LDLR [23].
In 2020, an interesting study conducted by Demetz et al. uncovered a novel role for HFE that extends beyond its established function in iron signaling. In this work, the authors demonstrated that siRNA silencing of HFE expression in HepG2 cells resulted in elevated levels of LDLR [32]. Notably, they observed that mice carrying the HFE C282Y mutation that abrogates β2-microglobulin binding, leading to HFE retention in the endoplasmic reticulum (ER), displayed higher LDLR levels compared to wild-type (WT) mice [32]. Furthermore, a meta-analysis conducted in 2009 revealed a significant association between the HFE C282Y variation and lower levels of LDLc (-15 mg/dl) [33]. While these findings have indeed uncovered a new and important role for HFE in lipid metabolism, the specific mechanism through which HFE is implicated in the intracellular or extracellular LDLR regulation remains an open question.
In this work, our research focused on examining how HFE may regulate the extracellular PCSK9 activity on the LDLR. We also confirmed the involvement of HLA-C as a potential "protein X" and compared the trafficking pathways of HFE and HLA-C as possible opposing regulators of PCSK9 in this process. Our study indicated that HLA-C and HFE exert opposite effects on PCSK9, possibly through two distinct regulatory pathways.
2. Materials and Methods
2.1. Generation of Constructs
Human complementary DNAs (cDNAs) encoding wild-type and mutant forms of LDLR, PCSK9, HFE, and HLA-C (HFE and HLA-C WT cDNA purchased from Genscript) were generated through site-directed mutagenesis. These cDNAs were incorporated into vectors such as pIRES2-EGFP or pcDNA3.1+/C-(K)-DYK for expression. Additionally, both negative and positive control constructs were included in the experimental setup. To distinguish and track the expressed proteins, various tags like V5 and FlagM2 were introduced to the constructs. Before further analyses, the sequence integrity of each mutant construct was rigorously confirmed using Sanger DNA sequencing. Point mutations or deletion mutants were generated through a 2-step polymerase chain reaction (PCR) technique as previously described and verified by DNA sequencing [1].
The following primers were used for mutagenesis:
PCSK9-ΔM2
FP- CTACCCCAGCCAGGTCTGGAATGC
RP- TCCAGACCTGGCTGGGGTAGCAGGCAG
PCSK9-H553R
FP- CCACTGCCGCCAACAGGGCC
RP- CTGTTGGCGGCAGTGGACAC
PCSK9-Q554E
FP- CTGCCACGAACAGGGCCAC
RP- CCTGTTCGTGGCAGTGGAC
PCSK9-R549A-E567A
FP- CATGGGGACCGCTGTCCACTGCC
RP- GGCAGTGGACAGCGGTCCCCATG
FP- GCAGCTCCCACTGGGCGGTGGAGGACCTTGGC
RP- GCC AAG GTC CTC CAC CGC CCA GTG GGA GCT GC
PCSK9-R549A-Q554E-E567A
FP- CTGCCACGAACAGGGCCAC
RP- CCTGTTCGTGGCAGTGGAC
FP- CATGGGGACCGCTGTCCACTGCC
RP- GGCAGTGGACAGCGGTCCCCATG
FP- GCAGCTCCCACTGGGCGGTGGAGGACCTTGGC
RP- GCC AAG GTC CTC CAC CGC CCA GTG GGA GCT GC
LDLR-ΔCT
FP- CTATGGACCGGTAAGCCTATCCCTAAC
RP- GATAGGCTTACCGGTCCATAGAAGGAAGACCCCC
HFE-C282Y
FP- GAGCAGAGATATACGTACCAGGTGGAGCACCCAGGCCTGG
RP- CCAGGCCTGGGTGCTCCACCTGGTACGTATATCTCTGCTC
HFE-R67A-E69A
FP- CATGAGAGTCGCGCTGTGGCGCCCCGAACTCC
RP- GGAGTTCGGGGCGCCACAGCGCGACTCTCATG
HLA-C-R68A-E70A
FP- GCGACGCCGCGAGTCCGGCAGGGGCGCCGCGGGCGCCGTG
RP-CACGGCGC CGCGGCGCCCCTGCCGGACTCGCGGCGTCGC
2.2. qPCR and Sequence of Primers
Quantitative RT-PCR was performed as published before [34]. In summary: A monolayer of cells grown on a 35mm plate was lysed and homogenized using a QIAshredder spin column (Qiagen). Total RNA was isolated with a RNeasy mini kit (Qiagen). Synthesis of cDNA was done as per manufacturer’s protocol using SuperscriptTM II RT (Invitrogen) from 250 ng of total RNA. Quantitative PCR was performed with PowerUp™ SYBR™ Green Master Mix (Applied Biosystems™) using the VIIA 7 Real-Time PCR system (Applied Biosystems™). Gene expression was normalized to that of the Tata-binding protein (TBP).
The following primers from Kruse et al. [35] were used for qPCR:
TBP-FP CGAATATAATCCCAAGCGGTTT
TBP-RP GTGGTTCGTGGCTCTCTTATCC
PCSK9-FP ATCCACGCTTCCTGCTGC
PCSK9-RP CACGGTCACCTGCTCCTG
HLA-A-FP CGACGCCGCGAGCCAGA
HLA-A-RP GCGATGTAATCCTTGCCGTCGTAG
HLA-B-FP GACGGCAAGGATTACATCGCCCTGAA
HLA-B-RP CACGGGCCGCCTCCCACT
HLA-C-FP GGAGACACAGAAGTACAAGCG
HLA-C-RP CGTCGTAGGCGTACTGGTCATA
HLA-E-FP CCTACGACGGCAAGGA
HLA-E-RP CCCTTCTCCAGGTATTTGTG
HLA-F-FP GGCAGAGGAATATGCAGAGGAGTT
HLA-F-RP TCTGTGTCCTGGGTCTGTT
HLA-G-FP TTGGGAAGAGGAGACACGGAACA
HLA-G-RP AGGTCGCAGCCAATCATCCAC
HFE-FP (Origene #HP200390)
HFE-RP (Origene #HP200390)
β2M-FP CTGGGTTTCATCCATCCGACA
β2M -RP TTCACACGGCAGGCATACTCATC
2.3. Inhibition of Protein Expression by Small-Interfering RNAs (siRNAs)
siRNA analysis was performed using INTERFERin® (PolyPlus) transfection reagent according to the manufacturer’s instructions. The following siRNAs with a final concentration of 60 nM have been used: CTL siGENOME non-Targeting siRNA Pool #2 (# D-001206-14-05), ON-TARGETplus Human CLTC (1213) siRNA-SMARTpool (# L-004001-01-0005), ON-TARGETplus Human CAV1 (857) siRNA -SMARTpool, (# L-003467- 00005), siGENOME Human LDLR siRNA – SMARTpool (#M-011073-01-0005).All siRNAs were purchased from Dharmacon (Horizon Discovery). Gene silencing efficiency was assessed by Western blotting.
2.4. Cell Culture and Transfection
Various cell lines were utilized, HEK293 (human embryonic kidney-derived epithelial cells), HepG2 naïve (human hepatocellular carcinoma cells), sub-clone CHO-K1 cell line from the original Chinese hamster overran cells (CHO), CRISPR HepG2 HLA-C-/- cells (Ubigene. inc #YC-C001), and CRISPR HepG2 PCSK9-/- cells [36]. These cells were cultured in specific growth media: Dulbecco's Modified Eagle Medium (DMEM) or Eagle's Minimum Essential Medium (EMEM) supplemented with 10% fetal bovine serum (FBS; GIBCO BRL). The cells were maintained at a temperature of 37°C in an environment with 5% CO2 to simulate physiological conditions. Transfection was employed to introduce the desired genetic constructs (PCSK9, LDLR, HFE, HLA-C, and their variants) into the cells. Depending on the cell line, different transfection reagents were used: JetPEI (PolyPlus), FuGENE®HD (Promega), and jetPRIME (PolyPlus) transfection reagents for CHO-K1, HepG2, and HEK293 cells, respectively. Cells were allowed to express the introduced genes for 48h post-transfection. For HEK293 cells, a specialized protocol was followed: cells were coated with poly-L-lysine, and then seeded in large flasks (T175) to produce PCSK9 enriched media. jetPRIME transfection reagent was used for this process. After 48h, the conditioned media containing the secreted protein were collected, measured by Elisa and stored at a temperature of -80°C for subsequent analysis. Similar production has been used for all experiments. For the media swap experiment, different cells were seeded in 12-well cell culture plates, and after 24h, they were incubated with serum-free media overnight. Subsequently, cells were exposed to conditioned media produced from HEK293 cells overexpressing human PCSK9.
2.5. In-House ELISA Measurement of Human PCSK9 Levels in Media
The secreted concentrations of PCSK9 in the media were determined using an in-house luminescence-based human PCSK9 ELISA assay [37], which was conducted as follows: LumiNunc Maxisorp white assay plates were used and coated with 0.5 μg/well of anti-human PCSK9 antibody (hPCSK9-Ab). The coating was carried out by incubating the plates at 37°C for 3h and then at 4°C overnight. After the coating, the plates were subjected to washing steps to remove any unbound components. The plates were then blocked using a blocking buffer composed of PBS (Phosphate-Buffered Saline), casein at 0.1% concentration, and Merthiolate at 0.01% concentration. Calibrators were prepared by creating serial dilutions of known concentrations of a standard PCSK9 solution. Samples, which contained secreted PCSK9 from the cell culture media, were prepared by diluting them at two different dilution ratios: 1:50 and 1:100, using a dilution buffer with BSA (Bovine Serum Albumin). The calibrators and samples were added to the coated and blocked plates and allowed to incubate for 30 minutes at a temperature of 46°C. After the incubation, plates were washed again to remove any unbound materials. Subsequently, a secondary antibody known as hPCSK9-Ab-HRP (Horseradish Peroxidase) was added to the plates. The plates were then incubated for 3h at a temperature of 37°C while shaking at 300rpm. After the secondary antibody incubation, plates were washed once more. A substrate solution, specifically SuperSignal™ ELISA Femto Substrate from Pierce, was added to each well of the plate. The generated chemiluminescence was quantitated using a Pherastar luminometer from BMG Labtech. The concentrations of the secreted PCSK9 in the samples were calculated and adjusted accordingly for each experimental construct, allowing for a comparative analysis of PCSK9 secretion across different conditions or treatments.
2.6. Western Blotting
Cultured cells underwent the following process for protein extraction and analysis: First, the cultured cells were washed to remove any residual media or contaminants. Then, a non-denaturing cell lysis buffer was used for protein extraction. The composition of the lysis buffer was as follows: 20 mM Tris-HCl (pH 8), 137 mM NaCl, 2 mM Na2EDTA, 1% NP-40 (Nonidet P-40), 10% glycerol, and 4% protease inhibitor cocktail (PIC) without EDTA. Then, Lowry assay [38] was employed to determine protein concentrations in the extracted samples. In the next step, the extracted proteins were separated by size using polyacrylamide gel electrophoresis (SDS-PAGE). Two types of gels were used: 6.5% and 8% tris-glycine gels. The separated proteins were then transferred from the gel onto PVDF (Polyvinylidene Fluoride) membranes and were incubated with specific primary antibodies that bind to the target proteins of interest. After the primary antibody incubation, secondary antibodies conjugated with Horseradish peroxidase (HRP) were applied. The membranes were analyzed and quantified using a ChemiDoc imaging system from Biorad. The following antibodies were used in this work: α-tubulin (ProteinTech #11224-1-AP [1:10,000]), HFE (Santa Cruz # sc-514405 [1:100]), HLA-C (Santa Cruz # sc-166134 [1:500]), Clathrin Heavy Chain (Abcam #ab21679 [1:1000]), Caveolin-1(D46G3) (NEB-cell signaling #3267T [1:1,000]), hPCSK9 ([39][1:2000], LDLR (R&D system #AF2148 [1:1000]), V5 (Invitrogen # R960-25 [1:5000]), monoclonal ANTI-FLAG® M2-Peroxidase (HRP) antibody produced in mouse (Sigma # A8592-1MG [1:10000]), β2M (ThermoFisher #701250 [1:1000], anti-mouse HRP(VWR #CA95017-332L [1:10000]), anti-rabbit HRP (VWR #CA95017-556L [1:10000]), and anti-goat-HRP #A5420 [1:10000]).
2.7. Immunofluorescence Assay (IF)
For the IF experiment, CRISPR HepG2 PCSK9 KO cells were cultured, and their media was replaced with a media containing 0.3 ng/ml of human PCSK9 (hPCSK9). After the media swap, cells were incubated for 24h. After 48h of incubation with PCSK9 following with serum free media, these cells were washed twice with PBS (Phosphate-Buffered Saline) to remove any residual substances. Subsequently, they were fixed using 4% paraformaldehyde. To prevent nonspecific binding of antibodies, the fixed cells were blocked with a solution of PBS containing 2% BSA (Bovine Serum Albumin) for 1h. Then, they were incubated with proper primary antibodies including LDLR (R&D system #AF2148 [1:200]), and EEA1 (Abcam #2900 [1:500]) at a temperature of 4°C overnight. The next day, plates were washed with PBS to remove unbound primary antibodies and were then incubated with an appropriate fluorescent secondary antibody including goat-Alexa 488 (Molecular probes #A-11078 [1:500]), and rabbit-Alexa 555 (Molecular probes #A-31572 [1:500]). To visualize cell nuclei, samples were stained with Hoechst dye at a concentration of 1 μg/ml. The coverslips containing the stained cells were mounted onto glass slides using Mowiol, a mounting medium. These prepared samples were then visualized using a confocal laser-scanning microscope with a high-powered objective lens (Plan-Apochromat 63 × 1.4 oil) from Carl Zeiss.
2.8. PCSK9 – LDLR (EGF-AB peptide) Binding Assay
The CircuLex human PCSK9 functional assay kit (MBL, Cat # CY8153) was used to measure the binding affinity of wild-type (WT) PCSK9 to LDLR. Media from HEK293 cells containing WT PCSK9 were incubated on HepG2 PCSK9 KO cells that transfected either with HFE or empty vector. Then, samples were collected and serially diluted. These diluted samples were then used for the binding assay. LumiNunc Maxisorp white assay plates were coated with the recombinant LDLR EGF-AB domain. Serially diluted samples of PCSK9 were added to the coated plates containing the LDLR EGF-AB domain. For each concentration of PCSK9, the absorbance at 450 nm (OD) was measured using a SpectraMax i3 plate reader. The obtained OD values were corrected for nonspecific binding and normalized to the maximum absorbance value (OD/ODmax). A binding curve was generated for each PCSK9 variant using a 4-parameter logistic (4-PL) equation. The EC50 value, which represents the concentration of PCSK9 needed for half-maximal binding to the LDL receptor EGF-AB domain, was extracted from the binding curve.
2.9. Modeling of PCSK9/HFE Complex
GlobalRAngeMolecularMatching(GRAMM*, see https://gramm.compbio.ku.edu/) webserver was used for molecular docking between HFE complexed with β-2-microglobulin (PDB: 1A6Z; chains: A and B; assumed as a receptor) and PCSK9’s CHRD (PDB: 2P4E; assumed as a ligand). HFE residues R67 and E69 of the RVE motif (UNIPROT: Q30201; residues 45 and 47 in the crystallographic structure) were taken as interface constraints for filtering 10 top models. The comparison of the structural models of the PCSK9/HLAC and PCSK9/HFE complexes was carried out using the PCSK9/HFE model described in this work and the PCSK9/HLAC model published in 2023 [23] using PyMOL.
2.10. Modeling of the Interaction between PCSK9’s N-Terminus with HLA-C and Other HLA Memnbers
The ternary complex comprising PCSK9 structured N-terminal peptide (uniprot: Q8NBP7; residues 31 to 59), extracellular region of HLA-C’s ⍺-chain (uniprot: P10321-1; residues 26 to 300) and β2-microglobulin (uniprot: P61769; residues 22 to 119) was modeled using Alphafold 2.3.1 at IDRIS HPC using NVIDIA V100 nodes (options : model_preset=multimer;use_gpu_relax;max_template_date=2022-0101; num_multimer_predictions_per_model=3). To verify if the best ranked model was compatible with previous observations of interactions between ⍺-chain of HLA-C and CHRD of PCSK-9 full length [23] a merged model was constructed by superposing both models using HLA-C/β2-microglobulin main-chain as reference. Then, HLA-C/β2-microglobulin and PSCK9 peptide 31-59 of the previous model were removed. The PSCK9 peptide 31-59 (not interacting with ⍺-chain of HLA-C was manually remodeled using Pymol (The PyMOL Molecular Graphics System, Version 2.5, Schrödinger, LLC) and energy minimization using Amber ff14SB (200 steps steepest descent + 10 gc) implemented in Chimera [40].
Alternatively, the helical N-terminal peptide PCSK9 comprising residues 32-50 (PDB: 6MV5, complexed with anti PCSK9 fab) to HLA-C ⍺-1 domain using GRAMM [41] and defining the peptide residues E34, D37 and E40 as interface constraints. Before peptide-protein docking, the side chains of HLA-C ⍺-1 domain were relaxed using fixbb application of Rosetta package [42]. A similar methodology has been used to predict the interaction of PCSK9 N-terminal peptide with other HLA members including HLA-A, HLA-B, HLA-F, HLA-G, and HFE.
3. Results
3.1. In search for “Protein X”
Analyses of the reported PCSK9's structures indicated that its M2 domain may interact with an unidentified protein, referred to as "protein X", which is essential for PCSK9's extracellular function. Prior studies proposed “protein X” as a transmembrane protein with a cytosolic tail capable of internalizing the PCSK9-LDLR [43,44] and PCSK9-LRP1 [45] complexes in the absence of the cytosolic tail (CT) of the LDLR (LDLR-ΔCT). Herein, we investigated the role of the M2 domain-“protein X” interaction on the PCSK9-LDLR trafficking by transfecting HEK293 cells with cDNAs encoding PCSK9-WT-V5, PCSK9-ΔM2-V5, and a control empty pIRES-EGFP vector to produce PCSK9-enriched media [23]. HepG2 PCSK9 KO CRISPR cells [36] were then incubated with these media (~300 ng/ml of each PCSK9 construct, estimated by ELISA) and their effects on LDLR levels were analyzed using immunofluorescence microscopy under non-permeabilized (cell surface LDLR) and permeabilized (total LDLR) cellular conditions. The results showed that WT PCSK9 significantly reduced LDLR levels both at the cell surface and intracellularly, while PCSK9-ΔM2 had a minimal impact on intracellular LDLR, but greatly reduced cell surface LDLR (Figure 1A). These data suggest that the M2 domain is not essential for the initial cell surface binding of PCSK9 to the LDLR and cell surface internalization into early endosomes, as reported previously [20], but is rather critical for the ensuing endosomal trafficking of PCSK9-LDLR complex to late endosomes and/or lysosomes for degradation.
Additionally, the impact on overall LDLR levels of PCSK9-ΔM2 was compared with PCSK9 LOF variants reported to interfere with PCSK9's interaction with the R-x-E motif of MHC-I-like proteins, namely PCSK9 R549A-E567A (PCSK9-RE) and R549A-Q554E-E567A (PCSK9-RQE) obtained from the conditioned media of HEK293 cells [23]. As expected, these M2 variants displayed the same LOF phenotype as PCSK9-ΔM2, (Figure 1B), emphasizing the importance of the PCSK9's interaction with an R-x-E motif in “protein X”, e.g., in HLA-C [23]. We next evaluated their effects on the subcellular localization of the LDLR in early endosomes (EEA1 marker). Notably, incubation of cells with either control media or those containing LOF PCSK9 derivatives (including PCSK9-ΔM2, PCSK9-RE, and PCSK9-RQE), resulted in an EEA1 punctate signal that was significantly stronger compared to WT PCSK9 (Figure 1B, see red arrows). These findings suggest that different from WT PCSK9, all PCSK9 LOF variants analyzed led to an accumulation of LDLR in early endosomes (see white arrows pointing to merged orange/yellow signals).
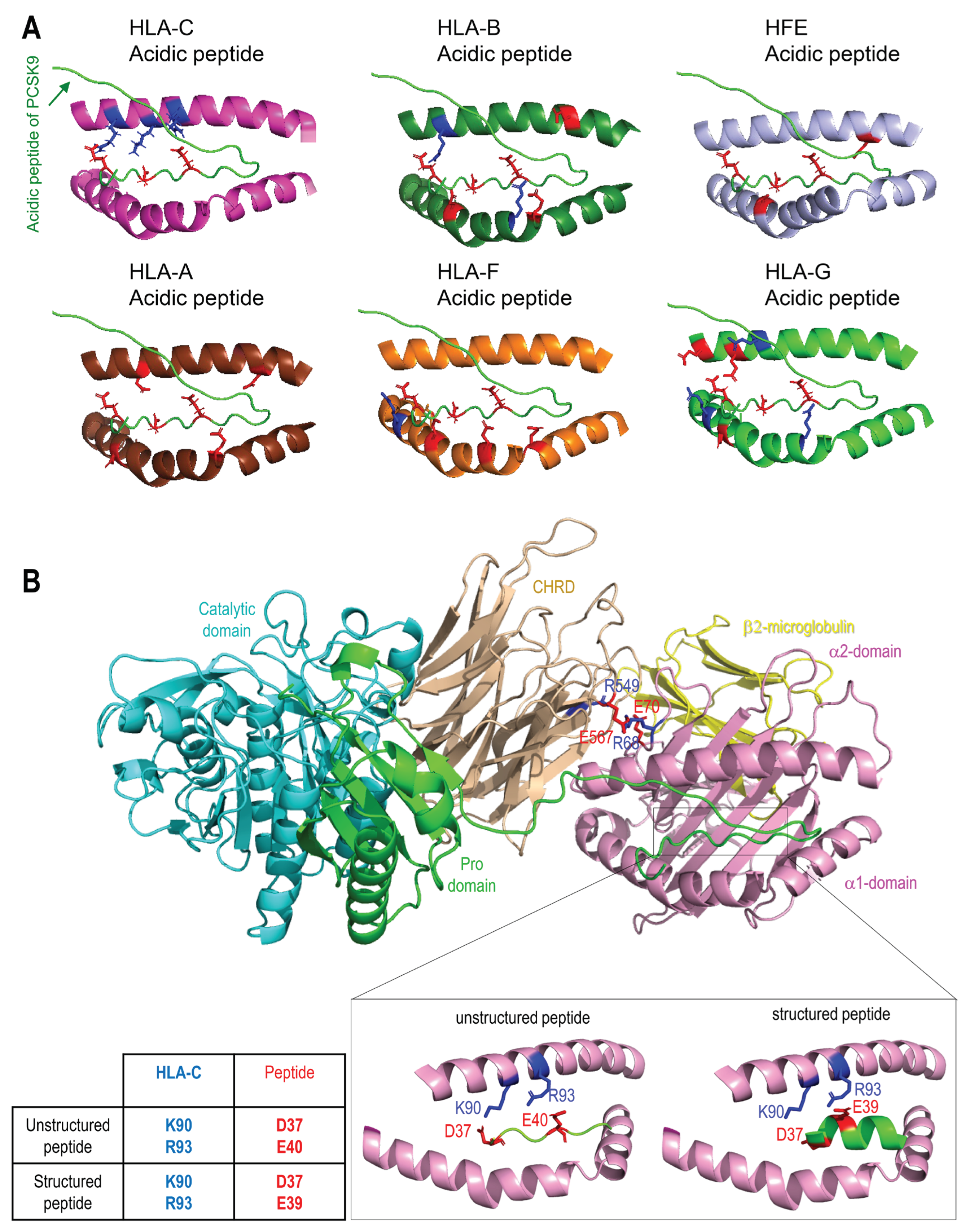
The above data demonstrated that PCSK9-ΔM2 variants that do not interact with the putative R-x-E motif in MHC-I-like proteins, result in a LOF effect on the extracellular PCSK9-induced LDLR degradation, consistent with recent findings on MHC-I-like molecules in lipid metabolism [23,29]. Accordingly, we suggested that one or more HLA member proteins [30] may serve as "protein X" in regulating PCSK9's extracellular function. Sequence alignment of MHC-I members showed that six of them (except for HLA-E) contain similarly localized R-x-E motifs in their ⍺1-chain (HLA-A, -B, -C, -F, -G and HFE) (Figure 1C). Accordingly, we measured their mRNA expression by qPCR in different hepatocyte cell lines. These data revealed that HLA-A, HLA-B, and HLA-C are the most prevalent members in naive HepG2 cells (Figure 1D), which are also rich in HepG2-PCSK9 KO cells (Figure 1E) and IHH cells (Figure 1F). In all these cells, HLA-A and HLA-B have higher mRNA expression than HLA-C (Figure 1D-F), as previously reported in other cells of the immune system [46]. The high expression of HLA-A and HLA-B remains in HepG2-HLA-C CRISPR cells lacking endogenous HLA-C (Figure 1G). Concerning HFE, even though its mRNA levels are lower than those of HLA-A, -B, and -C, they are like those of endogenous PCSK9 in naïve HepG2 cells (Figure 1D). Interestingly, our previous RNAseq data in adult mouse liver also revealed that in males and females PCSK9 and HFE levels are similar [47]. Given the recent findings of the involvement HFE and HLA-C in lipid metabolism, we have opted to concentrate on these two proteins for further studies.
3.2. HFE and HLA-C as New Targets and Regulators of Extracellular PCSK9
To investigate the potential interaction between HFE, HLA-C, and PCSK9, we expressed either HFE (WT or the common HFE-C282Y LOF variant) or HLA-C (WT), along with their common chaperone β2M, into HepG2 PCSK9 KO CRISPR and HepG2 HLA-C KO CRISPR cells, respectively. Subsequently, these cells were incubated with conditioned media of HEK293 cells containing ~300 ng of WT PCSK9 or no PCSK9 (control, empty vector) for 18h, after which they were collected for Western blot analysis.
Unexpectedly, the data from HepG2 PCSK9 KO cells revealed that in the absence of overexpressed HFE (dark bars), extracellular PCSK9 reduces endogenous LDLR levels by ~50%, but in the presence of HFE/β2M the LDLR reduction is only ~30% (Figure 2A). These data suggested that HFE could act as a negative regulator of the function of extracellular PCSK9 on the LDLR. Interestingly, extracellular PCSK9 also seems to target WT HFE to degradation, as its presence led to a ~60 % reduction in its levels (Figure 2A). In contrast, PCSK9 does not significantly enhance the degradation of the HFE-C282Y variant likely due to its retention in the ER and hence absence from the cell surface [32]. Applying different inhibitors of proteasome degradation (MG132), lysosomal degradation (NH4Cl: ammonium chloride), and autophagy (3-MA: 3-methyladenine) suggested that the degradation of HFE occurs in acidic compartments (like LDLR and HLA-C) (Figure 2B, C). These data revealed that extracellular PCSK9 enhances the degradation of both HFE and LDLR in acidic compartments, but that HFE inhibits the function of PCSK9 on LDLR.
At the cell surface, HFE usually binds transferrin receptor 1 (TfR1). Under high levels of circulating iron, HFE dissociates from TfR1 to activate the ERK/MAP signaling pathway for hepcidin expression [48,49,50,51]. This dissociation may increase the availability of HFE at the cell surface for its interaction with extracellular PCSK9 and subsequently may result in a higher inhibitory effect on PCSK9’s function on the LDLR. To test this possibility, HepG2 CRISPR PCSK9 KO cells were transfected with WT HFE and then incubated with PCSK9-enriched media containing either 200 μg/ml ferric ammonium citrate (FAC) or 200 μM iron chelating factors (DFA: deferoxamine) [52] for 18h. The addition of DFA dramatically increases the total LDLR levels regardless of the presence of WT PCSK9 due to the presence of an iron regulatory element (IRE) at 3’UTR of LDLR that stabilizes and increases LDLR expression [52]. Intriguingly, in the presence of FAC, PCSK9’s activity on LDLR is completely blocked by HFE (Figure 2D). Therefore, disrupting the interaction of HFE with TfR1 likely results in higher levels of available HFE at the cell surface and hence greater inhibition of PCSK9’s function on the LDLR.
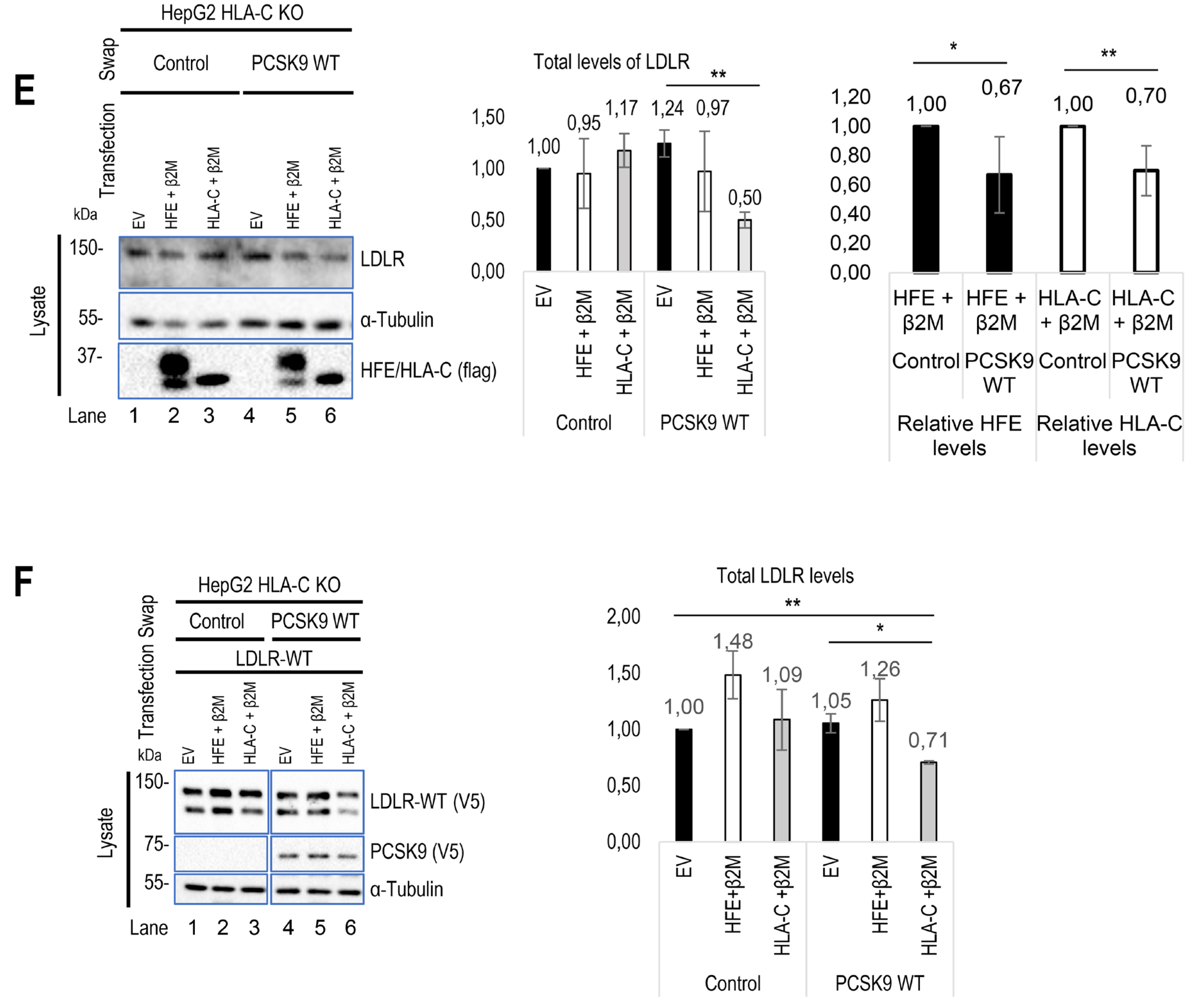
To further confirm the critical impact of HLA-C on the extracellular PCSK9 ability to enhance the degradation of the LDLR [23], we initially attempted to silence HLA-C expression by siRNA in HepG2 cells. However, we were unsuccessful (data not shown)[32], possibly due to the high endogenous expression levels of HLA-C (Figure 1D). Previously, we found that extracellular PCSK9 didn't impact LDLR in CHO-K1 cells [39], but the reason was unclear. Since CHO-K1 cells were reported to lack endogenous HLA-C expression [53], we compared the effect of extracellular PCSK9 on LDLR in CHO-K1 cells overexpressing HLA-C-WT-FlagM2 or its R68-x-E70 mutant HLA-C-R68A-E70A-FlagM2, which no longer binds PCSK9 [23]. Interestingly, HLA-C significantly enhanced PCSK9's function compared to control and to the LOF mutant HLA-C-R68A-E70A-FlagM2 (data not shown). In the presence of overexpressed HLA-C in CHO-K1 cells, exogenous PCSK9 significantly reduced LDLR levels by ~40%, supporting HLA-C's ability to enhance the function of PCSK9. Since these ovarian cells are distinct from hepatocytes, and because of our inability to significantly reduce HLA-C levels from HepG2 cells by siRNA, we completely silenced HLA-C mRNA expression in HepG2 cells using the CRISPR-Cas9 technology, thereby generating HepG2 CRISPR HLA-C KO cells (Figure 1G). In the latter, our data confirmed that, in the absence of HLA-C, extracellular PCSK9 has no significant effect on total LDLR levels (either endogenous or overexpressed) (Figure 2E, F). In contrast, overexpression of HLA-C in these cells recaptured PCSK9’s function towards LDLR degradation and significantly reduced endogenous LDLR levels by ~50% (Figure 2E). Similarly, in these cells, overexpression of HLA-C and LDLR revealed that extracellular PCSK9 also reduced the levels of overexpressed LDLR by ~30% (Figure 2F). These results support the notion that HLA-C is a potential “protein X” candidate implicated in the sorting of the PCSK9-LDLR complex to lysosomes for degradation [23]. Overall, our data further revealed that although HLA-C is critical for PCSK9-induced degradation of the LDLR, it is dispensable for the observed ~30% PCSK9-induced HFE degradation (Figure 2E).
Figure 2.
Regulatory effect of HLA-C and HFE on PCSK9 and vice versa. (A) The effect of cell surface HFE on extracellular PCSK9. HepG2 lacking endogenous PCSK9 were transiently transfected with an empty vector (EV), HFE-WT-flagM2, HFE-C282Y-flagM2, β2M-flagM2 and were incubated with conditioned media enriched with extracellular PCSK9 or empty vector (control). The effect of HFE on the extracellular activity of PCSK9 has been analyzed by WB (SDS/PAGE on 8% Tris-glycine gel) analysis. The quantification of total LDLR and HFE levels are shown in respective charts. (B) and (C) Cellular inhibitors were used to inhibit lysosomal degradation (NH4CL: ammonium chloride), proteasome degradation (MG132), or autophagy (3-MA: 3-methyladenine). The data suggests the possible lysosomal degradation of HFE. (D) Effect of iron on HFE function. HepG2 PCSK9 KO cells were transfected with WT HFE and β2M, then incubated with conditioned media from HEK293 cells expressing an empty vector (control) or WT PCSK9. Following the incubation with conditioned media, cells were treated with either ferric ammonium citrate (FAC) or deferoxamine (DFA) to analyze the function of HFE on PCSK9 in different iron conditions.
Figure 2.
Regulatory effect of HLA-C and HFE on PCSK9 and vice versa. (A) The effect of cell surface HFE on extracellular PCSK9. HepG2 lacking endogenous PCSK9 were transiently transfected with an empty vector (EV), HFE-WT-flagM2, HFE-C282Y-flagM2, β2M-flagM2 and were incubated with conditioned media enriched with extracellular PCSK9 or empty vector (control). The effect of HFE on the extracellular activity of PCSK9 has been analyzed by WB (SDS/PAGE on 8% Tris-glycine gel) analysis. The quantification of total LDLR and HFE levels are shown in respective charts. (B) and (C) Cellular inhibitors were used to inhibit lysosomal degradation (NH4CL: ammonium chloride), proteasome degradation (MG132), or autophagy (3-MA: 3-methyladenine). The data suggests the possible lysosomal degradation of HFE. (D) Effect of iron on HFE function. HepG2 PCSK9 KO cells were transfected with WT HFE and β2M, then incubated with conditioned media from HEK293 cells expressing an empty vector (control) or WT PCSK9. Following the incubation with conditioned media, cells were treated with either ferric ammonium citrate (FAC) or deferoxamine (DFA) to analyze the function of HFE on PCSK9 in different iron conditions.
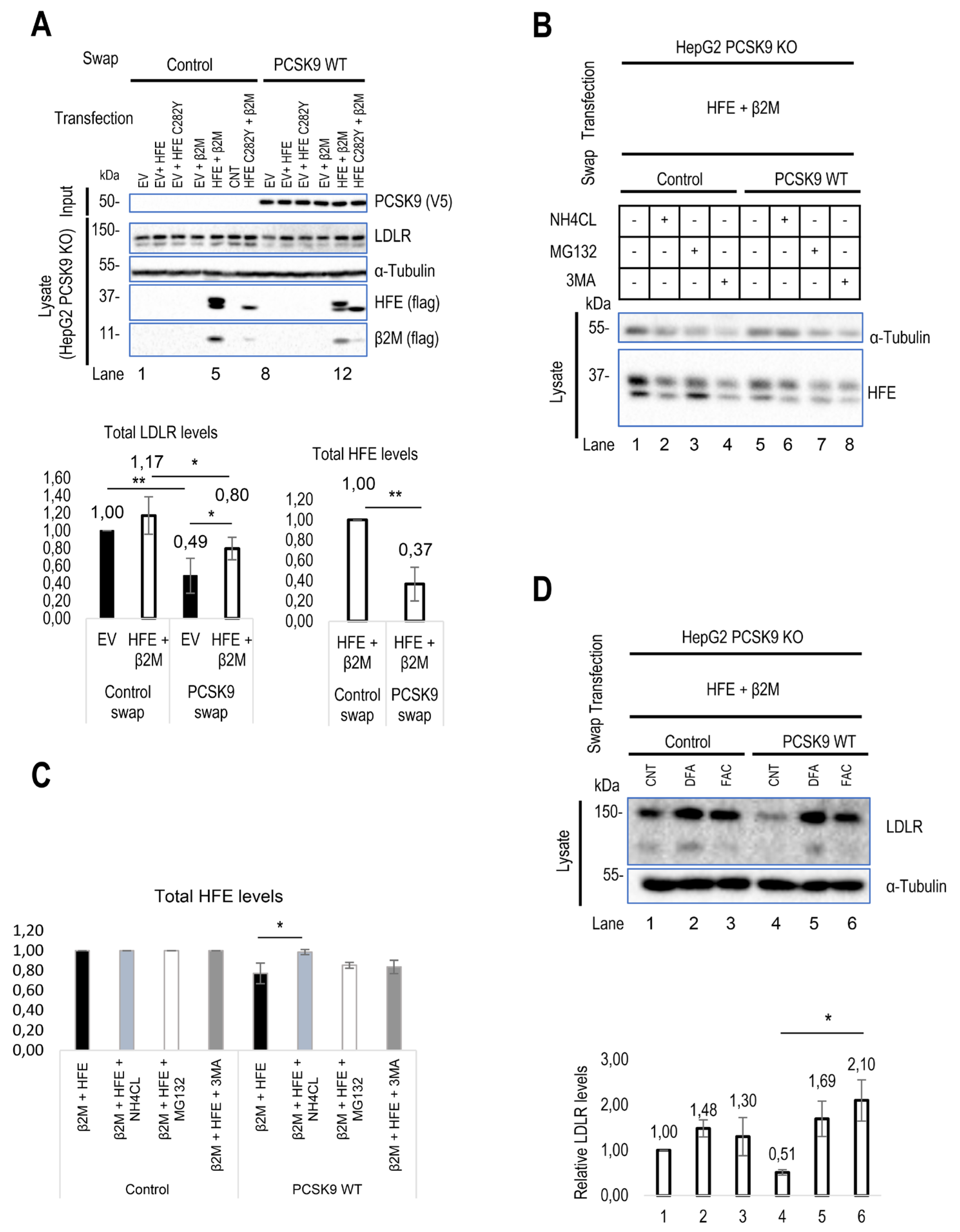
Figure 2.
Continued. (E) HLA-C impact on extracellular PCSK9 in HepG2 HLA-C KO cells. These cells were transfected with an empty vector (EV), WT HLA-C, or WT HFE and then incubated with conditioned media from HEK293 cells expressing an empty vector (control) or WT PCSK9. The total levels of endogenous LDLR were quantified. (F) HepG2 HLA-C KO cells were co-transfected with (empty vector (EV) + WT LDLR), (WT HLA-C + WT LDLR), or (WT HFE + WT LDLR) and then incubated with conditioned media from HEK293 cells expressing an empty vector (control) or WT PCSK9. The total levels of overexpressed LDLR were quantified. All cell Lysates were extracted to be analyzed by WB (SDS/PAGE on 8% or 6.5% Tris-glycine gel). Data are representative of three independent experiments. Protein levels were normalized to the control protein, α-tubulin. Quantifications are averages ± standard deviation (SD). * p<0.05; ** p<0.01; ***p<0.001 (two-sided t-test). NS: non-significant.
Figure 2.
Continued. (E) HLA-C impact on extracellular PCSK9 in HepG2 HLA-C KO cells. These cells were transfected with an empty vector (EV), WT HLA-C, or WT HFE and then incubated with conditioned media from HEK293 cells expressing an empty vector (control) or WT PCSK9. The total levels of endogenous LDLR were quantified. (F) HepG2 HLA-C KO cells were co-transfected with (empty vector (EV) + WT LDLR), (WT HLA-C + WT LDLR), or (WT HFE + WT LDLR) and then incubated with conditioned media from HEK293 cells expressing an empty vector (control) or WT PCSK9. The total levels of overexpressed LDLR were quantified. All cell Lysates were extracted to be analyzed by WB (SDS/PAGE on 8% or 6.5% Tris-glycine gel). Data are representative of three independent experiments. Protein levels were normalized to the control protein, α-tubulin. Quantifications are averages ± standard deviation (SD). * p<0.05; ** p<0.01; ***p<0.001 (two-sided t-test). NS: non-significant.
3.3. Interaction of PCSK9 with HFE and HLA-C
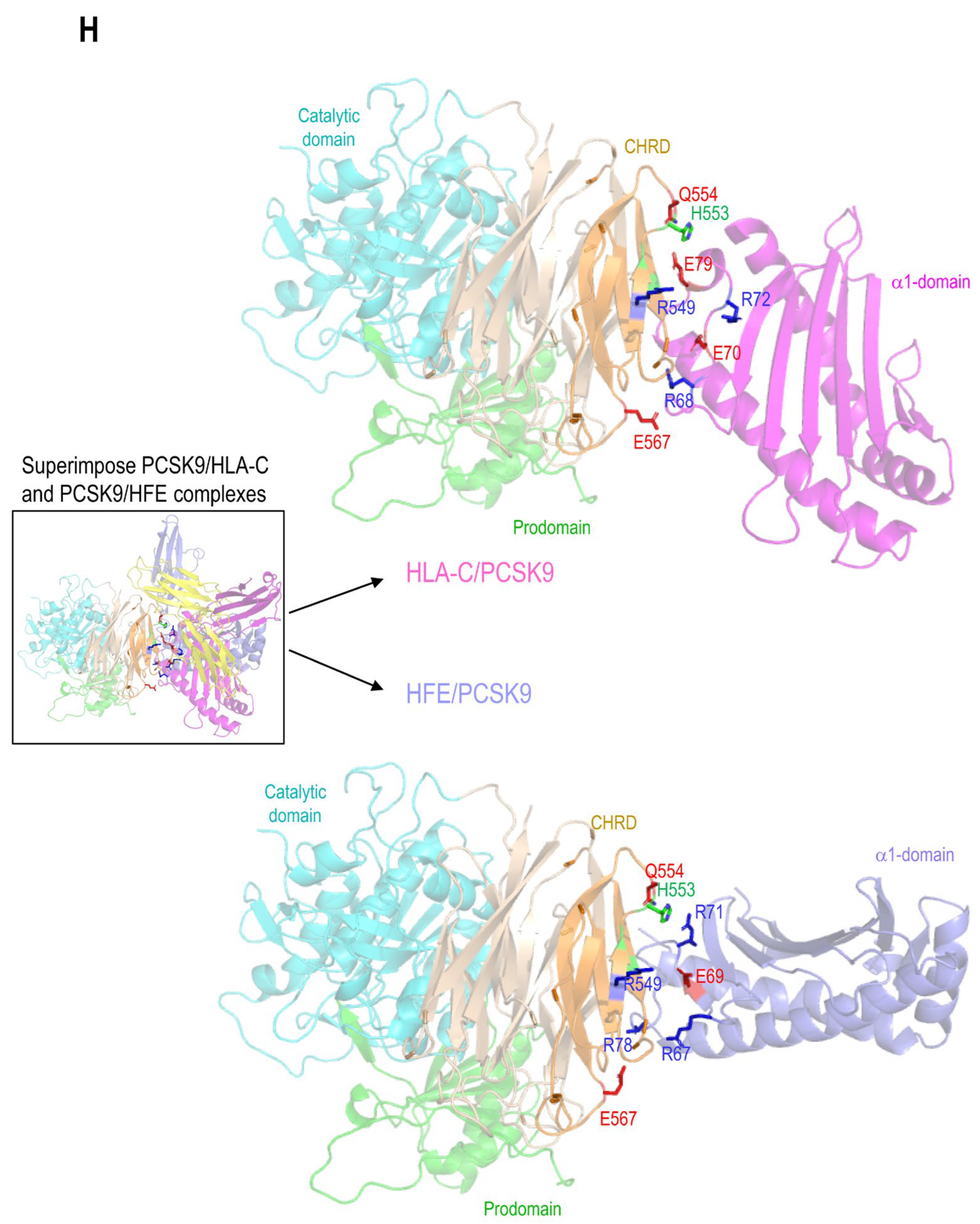
Recently, the 3D structure of HLA-C and PCSK9 interaction was modeled and confirmed the importance of the R68-G-E70 motif on HLA-C for the interaction with the M2 domain of PCSK9 [23]. This model predicted that R68 and E70 on HLA-C bind to E567 and R549 on PCSK9, respectively [23]. HFE's structural similarity to HLA-C, along with its similar R67-V-E69 motif, led us to hypothesize that it could also bind the M2 domain of PCSK9 via its R-x-E motif. Hence, we modeled the M2 domain interaction with HFE using the GRAMM-X web server. The resulting structure proposes that PCSK9 uses a similar R-x-E motif as HLA-C to interact with HFE (Figure 3A). For 5 out of 10 best obtained models, the M2 of PCSK9’s CHRD is implicated in contacts with HFE and 2 of them are clearly in competition with HLA-C. Of these two models, one model exhibits similar contacts compared to HLA-C. Specifically, R67 and E69 on HFE are predicted to interact with E567 and R549 on PCSK9 (Figure 3A). Interestingly, we noticed that the R67-V-E69 motif in HFE is preceded by a reverse motif E64-S-R66 giving the palindromic motif E64-x-R66-R67-x-E69 (Figure 1C). In this model, likely key contacts predicted include R549 in the M2 domain of PCSK9 with E69 in the α1 domain of HFE, E567 in the PCSK9’s M2 domain with R67 and/or R78 in the HFE α1 domain, and Q554 in the PCSK9’s M2 domain with R71 in the α1 domain of HFE (Figure 3A, B). The interaction of HFE with natural PCSK9 variants including Q554E (LOF for LDLR and HLA-C) and H553R (GOF for LDLR and HLA-C) were also modeled (Figure 3B) [23,28,54]. Such 3D structure modeling suggests that due to the positive charge of R71 on HFE, PCSK9 Q554E could enhance HFE's function, while PCSK9 H553R may hinder it (Figure 3B).
To support the importance of the predicted sites implicated in the HFE-PCSK9 interaction, we mutated/deleted WT PCSK9 (R549A-E567A, R549A-Q554E-E567A, H553R and ΔM2) and WT HFE (R67A-E69A) to remove the modeled binding sites mentioned above. As predicted, PCSK9 variants lacking the HFE interaction sites were all functionally impaired (LOF) and were not able to degrade HFE (Figure 3C). Interestingly, our data also showed that PCSK9 WT can still interact with HFE R67A-E69A, although it lacks the R-x-E motif. We postulate that this could be because of the presence of palindromic sequence of E64-x-R66-R67-x-E69 on HFE. Alternatively, our refined 3D structure analysis further suggested the presence of another Arg on HFE (R78) that may be closer to PCSK9 E567 compared to R67 (Figure 3D).
Furthermore, in HepG2 PCSK9 KO CRISPR cells overexpression of WT HLA-C or its inactive R68A-E70A mutant did not significantly enhance the effect on WT PCSK9 compared to control (Figure 3E). We presume that the reason for this observation is that the endogenous WT HLA-C is highly expressed in HepG2 cells (Figure 1E) and already reached its maximum effect on PCSK9. Thus, with low transfection efficiency of HepG2 cells (~20-30%) it would be hard to see an additional or inhibitory effect of over-expressed WT HLA-C or its LOF mutant on endogenous LDLR. Interestingly, HLA-C increased the activity of the extracellular PCSK9 H553R GOF variant on LDLR (Figure 3E). This may suggest that the overexpressed HLA-C isoform used (a highly polymorphic gene) may be different than that of the endogenous HLA-C in HepG2 cells and that it may interact better with PCSK9 H553R than WT PCSK9.
As HLA-C and HFE interact with the same residues of PCSK9, we hypothesize that these proteins might be potential competitors. To test this and to eliminate the dominant expression of endogenous HLA-C, we co-expressed HFE and HLA-C in HepG2 HLA-C KO cells. Surprisingly, a similar activity of PCSK9 on both HFE and HLA-C was observed in the absence or presence of the other homologue (Figure 3 F, G). These data suggest possible distinct trafficking pathways for HFE compared to HLA-C, and that their localization might be different, and subsequently they may meet PCSK9 in different places/compartments. The superposition of the 2 models of the HFE/PCSK9 and HLA-C/PCSK9 complexes confirmed that HFE and HLA-C could interact with a similar motif of PCSK9 (R549 and E567 in M2 domain) via their respective R-x-E motifs (HFE: R67 (or R78) and E69; HLA-C: R68 and E70) (Figure 3H). Therefore, HFE and HLA-C could potentially interact with PCSK9 depending on their availability, and subsequently either activate (Figure 2E, F) or inhibit (Figure 2A, D) the activity of PCSK9 on LDLR.
Figure 3.
PCSK9’s direct interaction with HLA-C compared to HFE. (A) Molecular modeling of the interaction of the M2 subdomain of PCSK9 with the α1 domain of HFE suggests the interaction of the R549-x-E567 motif of PCSK9 with the R67-x-E69 motif of HFE. (B) Further analysis of the 3D modeling of PCSK9 interaction with HFE (like what has been published before for HLA-C [23]), suggests the presence of another interaction site on HFE (R71) with PCSK9 that could be sensitive to PCSK9 natural mutations Q554E (GOF for HFE) and H553R (LOF for HFE). The HLA-C interaction with Q554E and H553R model is adopted from [23].
Figure 3.
PCSK9’s direct interaction with HLA-C compared to HFE. (A) Molecular modeling of the interaction of the M2 subdomain of PCSK9 with the α1 domain of HFE suggests the interaction of the R549-x-E567 motif of PCSK9 with the R67-x-E69 motif of HFE. (B) Further analysis of the 3D modeling of PCSK9 interaction with HFE (like what has been published before for HLA-C [23]), suggests the presence of another interaction site on HFE (R71) with PCSK9 that could be sensitive to PCSK9 natural mutations Q554E (GOF for HFE) and H553R (LOF for HFE). The HLA-C interaction with Q554E and H553R model is adopted from [23].
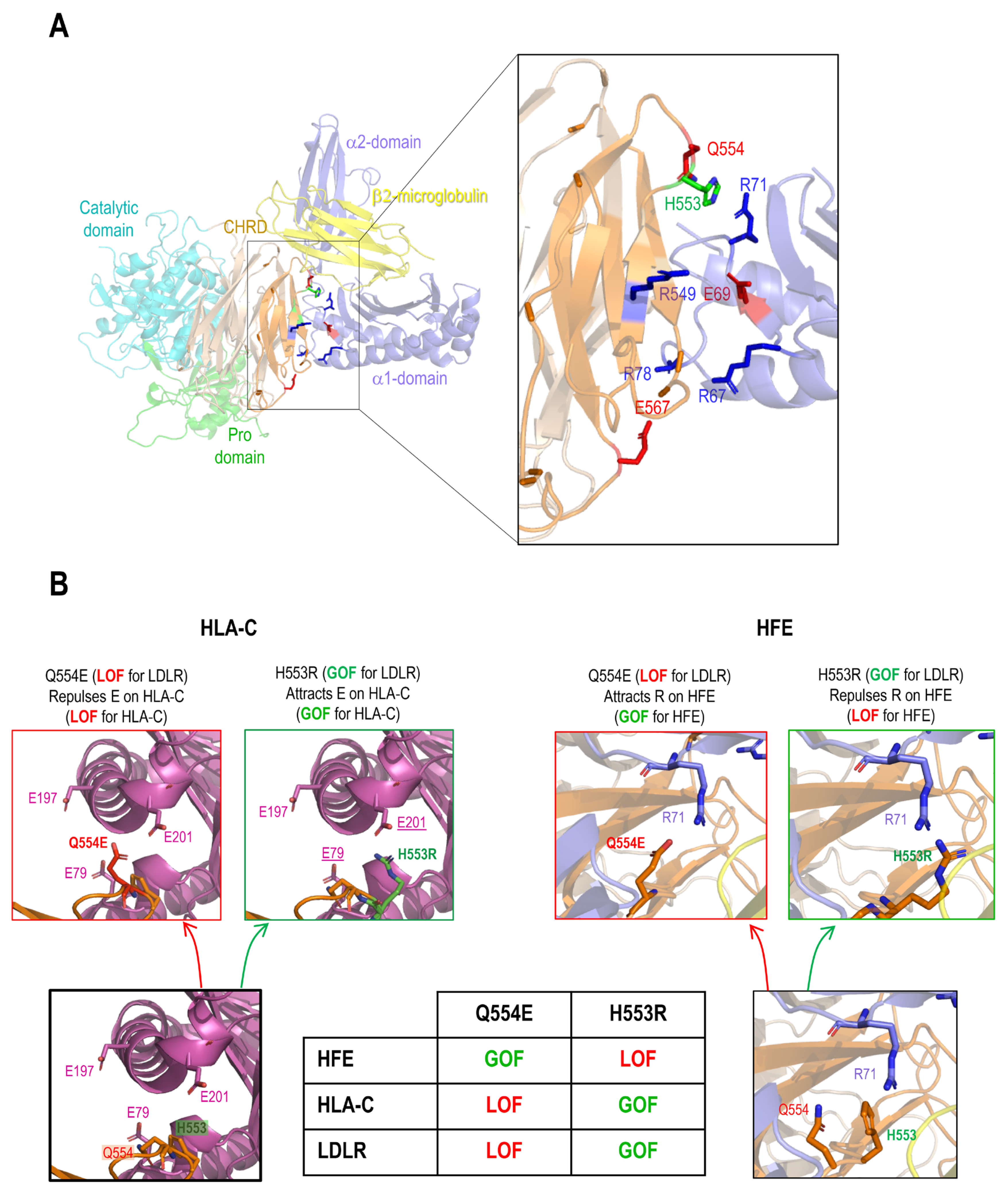
Figure 3.
Continued. (C) HepG2 PCSK9 KO cells were transfected with either, empty vector (EV), WT HFE or HFE R67A-E69A variant and then incubated with conditioned media from HEK293 cells expressing an empty vector (control), WT PCSK9, and proposed LOF variants on PCKS9 (ΔM2, R549A-E567A, R549A-Q554E-E567A, and H553R). Cell Lysates were extracted to be analyzed by WB (SDS/PAGE on 8% Tris-glycine gel). The cell-based data confirmed the predicted interaction sites by 3D structure modeling. (D) Further 3D structure modeling revealed the second arginine at position 78 that could interact better with PCSK9 E567. (E) HLA-C interaction with PCSK9. HepG2 PCSK9 KO cells were transfected with either, empty vector (EV), WT HLA-C or HLA-C R68A-E70A variant and then incubated with conditioned media from HEK293 cells expressing an empty vector (control), WT PCSK9, and proposed LOF variants on PCKS9 (ΔM2, R549A-E567A, R549A-Q554E-E567A, and H553R). Cell Lysates were extracted to be analyzed by WB (SDS/PAGE on 8% Tris-glycine gel). The cell-based data confirmed the predicted interaction sites by 3D structure modeling [23].
Figure 3.
Continued. (C) HepG2 PCSK9 KO cells were transfected with either, empty vector (EV), WT HFE or HFE R67A-E69A variant and then incubated with conditioned media from HEK293 cells expressing an empty vector (control), WT PCSK9, and proposed LOF variants on PCKS9 (ΔM2, R549A-E567A, R549A-Q554E-E567A, and H553R). Cell Lysates were extracted to be analyzed by WB (SDS/PAGE on 8% Tris-glycine gel). The cell-based data confirmed the predicted interaction sites by 3D structure modeling. (D) Further 3D structure modeling revealed the second arginine at position 78 that could interact better with PCSK9 E567. (E) HLA-C interaction with PCSK9. HepG2 PCSK9 KO cells were transfected with either, empty vector (EV), WT HLA-C or HLA-C R68A-E70A variant and then incubated with conditioned media from HEK293 cells expressing an empty vector (control), WT PCSK9, and proposed LOF variants on PCKS9 (ΔM2, R549A-E567A, R549A-Q554E-E567A, and H553R). Cell Lysates were extracted to be analyzed by WB (SDS/PAGE on 8% Tris-glycine gel). The cell-based data confirmed the predicted interaction sites by 3D structure modeling [23].
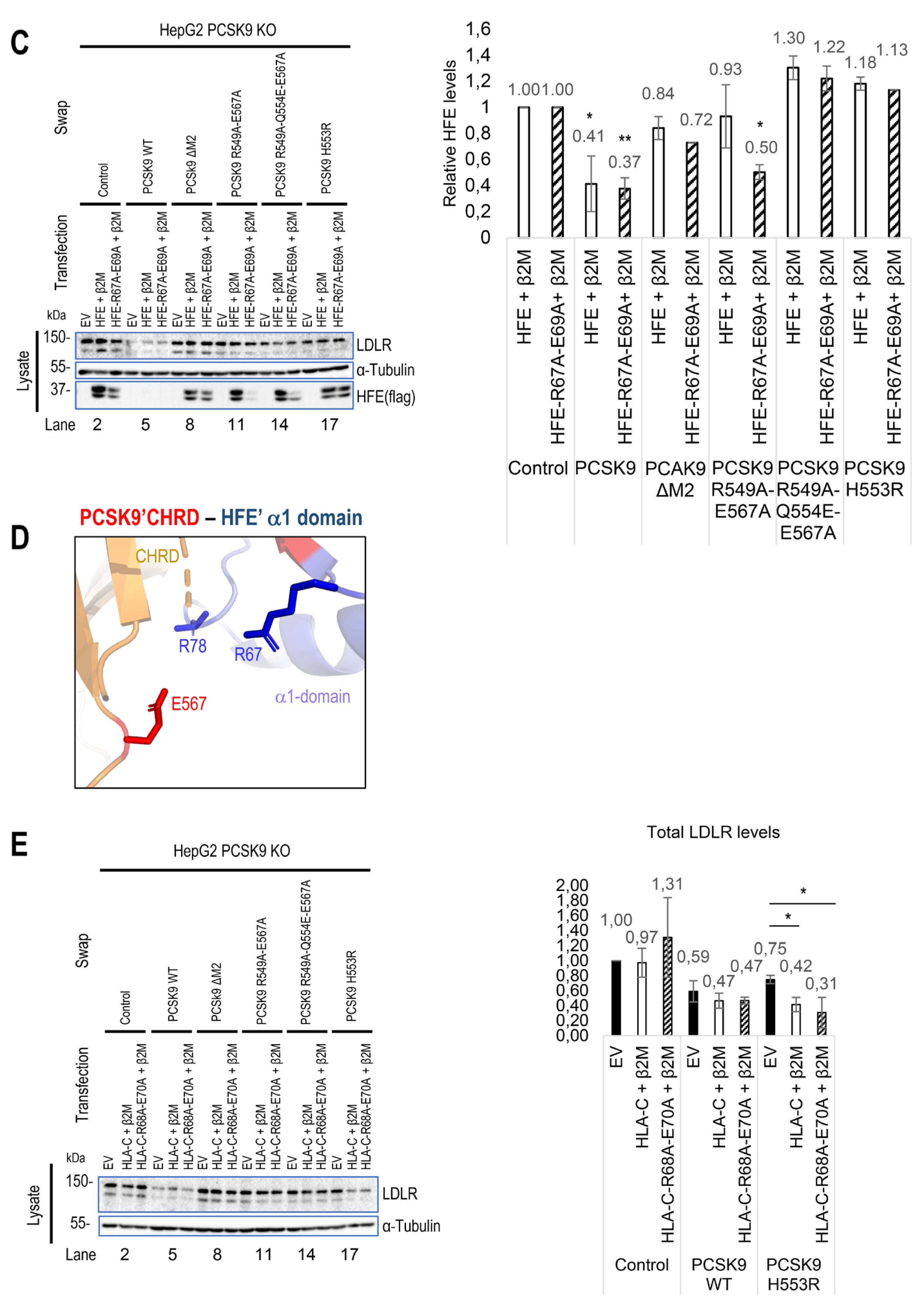
Figure 3.
Continued. (F) and (G) Studying the potential competition of HFE with HLA-C to interact with PCSK9 and get degraded. HepG2 HLA-C KO cells were co-transfected with empty vector (EV), WT HLA-C, WT HFE, or WT HLA-C+WT HFE and then incubated with conditioned media from HEK293 cells expressing an empty vector (control) or WT PCSK9. Total levels of HFE and HLA-C were quantified in respective charts.
Figure 3.
Continued. (F) and (G) Studying the potential competition of HFE with HLA-C to interact with PCSK9 and get degraded. HepG2 HLA-C KO cells were co-transfected with empty vector (EV), WT HLA-C, WT HFE, or WT HLA-C+WT HFE and then incubated with conditioned media from HEK293 cells expressing an empty vector (control) or WT PCSK9. Total levels of HFE and HLA-C were quantified in respective charts.
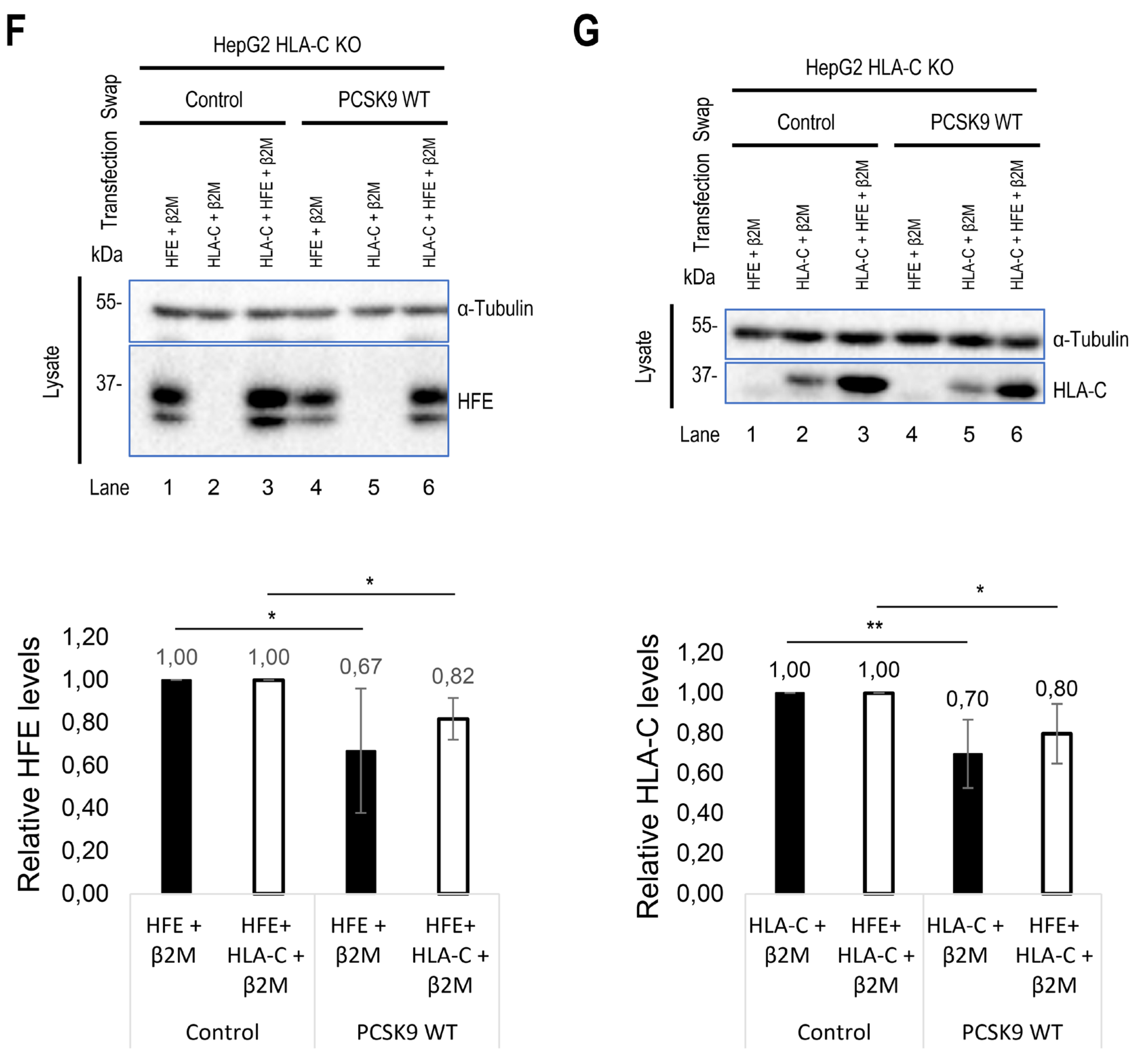
Figure 3.
Continued. (H) 3D modeling of possible similarities of HFE with HLA-C and their interaction with PCSK9. All protein levels were normalized to the control protein, α-tubulin. Data are representative of three independent experiments. Quantifications are averages ± standard deviation (SD). * p<0.05; ** p<0.01; ***p<0.001 (two-sided t-test). NS: non-significant.
Figure 3.
Continued. (H) 3D modeling of possible similarities of HFE with HLA-C and their interaction with PCSK9. All protein levels were normalized to the control protein, α-tubulin. Data are representative of three independent experiments. Quantifications are averages ± standard deviation (SD). * p<0.05; ** p<0.01; ***p<0.001 (two-sided t-test). NS: non-significant.
3.4. HFE and HLA-C Trafficking
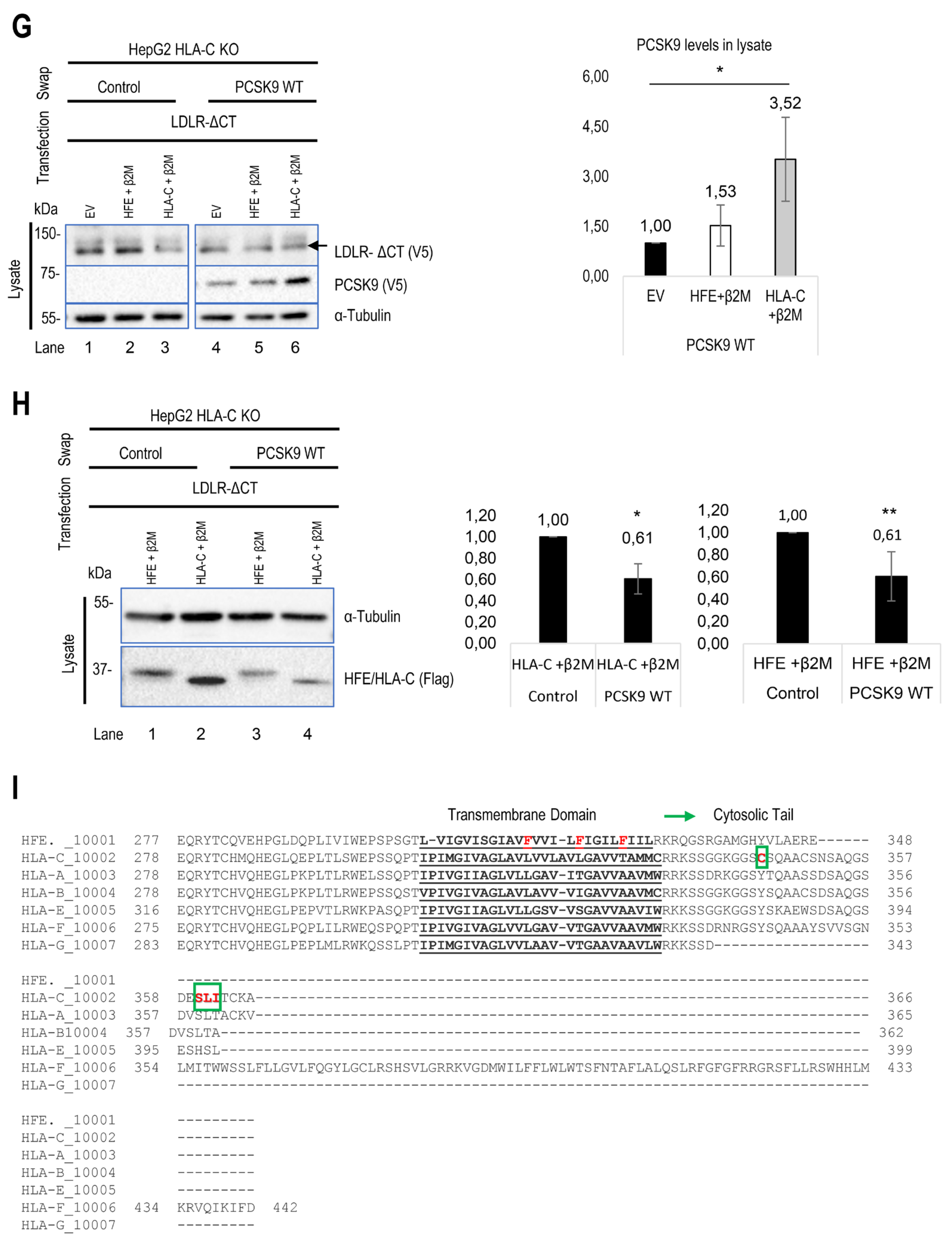
Prior studies indicated that LDLR internalization occurs via clathrin-coated vesicles [18,23,39,55]. Given HLA-C's involvement in LDLR degradation by PCSK9, we suggest that HLA-C also uses clathrin-coated vesicles for internalization. Additionally, HFE coupled with TfR1 is also known to undergo internalization through clathrin-coated vesicles [49]. However, an analysis of HFE's transmembrane domain reveals 3 unique aromatic Phe separated by four residues (F316xxxxF321xxxxF326) that might interact with caveolae [56] (Figure 4I). To determine which endocytosis pathway is dominant for each protein, we knocked down either caveolin 1 (siCav1) or clathrin heavy chain (siCHC) [23] and tested the effect of their absence on the degradation of HLA-C and HFE by PCSK9. As a result, while HLA-C degradation was inhibited by silencing of CHC only, HFE degradation was affected by knocking down either CHC or Cav1 (Figure 4A-C). Since the presence of HFE seems to inhibit PCSK9’s function on LDLR (Figure 2A, D), we postulated that HFE might compete with LDLR to interact with PCSK9. To test this hypothesis, we estimated the binding affinity of LDLR to PCSK9 in the presence or absence of HFE using the CircuLex human PCSK9 functional assay kit (MBL, #CY8153) [57]. The data showed that PCSK9 has the same binding affinity to the EGF-A domain of LDLR either in the presence or absence of HFE/β2M (Figure 4D). Indeed, the absence of LDLR (using an siRNA approach) inhibits PCSK9 function on HFE (Figure 4E). Therefore, HFE not only fails to compete with LDLR for interaction with PCSK9 but also depends on LDLR for its degradation by PCSK9, probably because of the short cytosolic tail of HFE (Figure 4I). This is consistent with prior research indicating that HFE alone cannot internalize into cells and needs to be coupled with another transmembrane protein such as TfR1 [58]. In contrast to HFE and in agreement with a previous study [29] HLA-C degradation by PCSK9 does not require LDLR (Figure 4F). Indeed, co-expression of LDLR-ΔCT-V5 [45] and HLA-C-FlagM2 in HepG2 HLA-C KO cells showed that in the presence of HLA-C, PCSK9 is better internalized and still active on LDLR (Figure 4G; compare lanes 6 and 4). However, the absence of LDLR cytosolic tail did not affect the activity of PCSK9 on either HFE or HLA-C (Figure 4H). We suggest that this observation could be due to the presence of endogenous LDLR in those cell lines that could help for internalization of HFE. This result agrees with the notion that the cytosolic tail of the LDLR is dispensable for the enhanced degradation of LDLR by PCSK9 [38,39], and further emphasized the crucial role of “protein X”, e.g., HLA-C, for the sorting of the PCSK9-LDLR complex to degradation compartments.
Figure 4.
Distinct trafficking of HFE compared to HLA-C. (A-C) HepG2 HLA-C KO cells were transfected with siRNA against clathrin heavy chain (siCHC), siRNA against caveolin 1 (siCav1), or non-targeting siRNA. 24h later, these cells were transfected with either an empty vector (EV), WT HFE, or WT HLA-C and then incubated with conditioned media from HEK293 cells expressing an empty vector (control) or WT PCSK9. (D) The binding affinity of WT PCSK9 to the LDLR was measured using the CircuLex human PCSK9 functional assay kit. The results of the affinity curve suggest that the presence of HFE/β2M does not affect the interaction of PCSK9 with the EGF-AB domain of LDLR. (E) and (F) HepG2 PCSK9 KO cells were transfected with siRNA against LDLR or non-targeting siRNA (Scramble). 24h later, these cells were transfected with either an empty vector (EV), or WT HLA-C/HFE and then incubated with conditioned media from HEK293 cells expressing an empty vector (control) or WT PCSK9. The effect of LDLR on either HFE or HLA-C was measured.
Figure 4.
Distinct trafficking of HFE compared to HLA-C. (A-C) HepG2 HLA-C KO cells were transfected with siRNA against clathrin heavy chain (siCHC), siRNA against caveolin 1 (siCav1), or non-targeting siRNA. 24h later, these cells were transfected with either an empty vector (EV), WT HFE, or WT HLA-C and then incubated with conditioned media from HEK293 cells expressing an empty vector (control) or WT PCSK9. (D) The binding affinity of WT PCSK9 to the LDLR was measured using the CircuLex human PCSK9 functional assay kit. The results of the affinity curve suggest that the presence of HFE/β2M does not affect the interaction of PCSK9 with the EGF-AB domain of LDLR. (E) and (F) HepG2 PCSK9 KO cells were transfected with siRNA against LDLR or non-targeting siRNA (Scramble). 24h later, these cells were transfected with either an empty vector (EV), or WT HLA-C/HFE and then incubated with conditioned media from HEK293 cells expressing an empty vector (control) or WT PCSK9. The effect of LDLR on either HFE or HLA-C was measured.
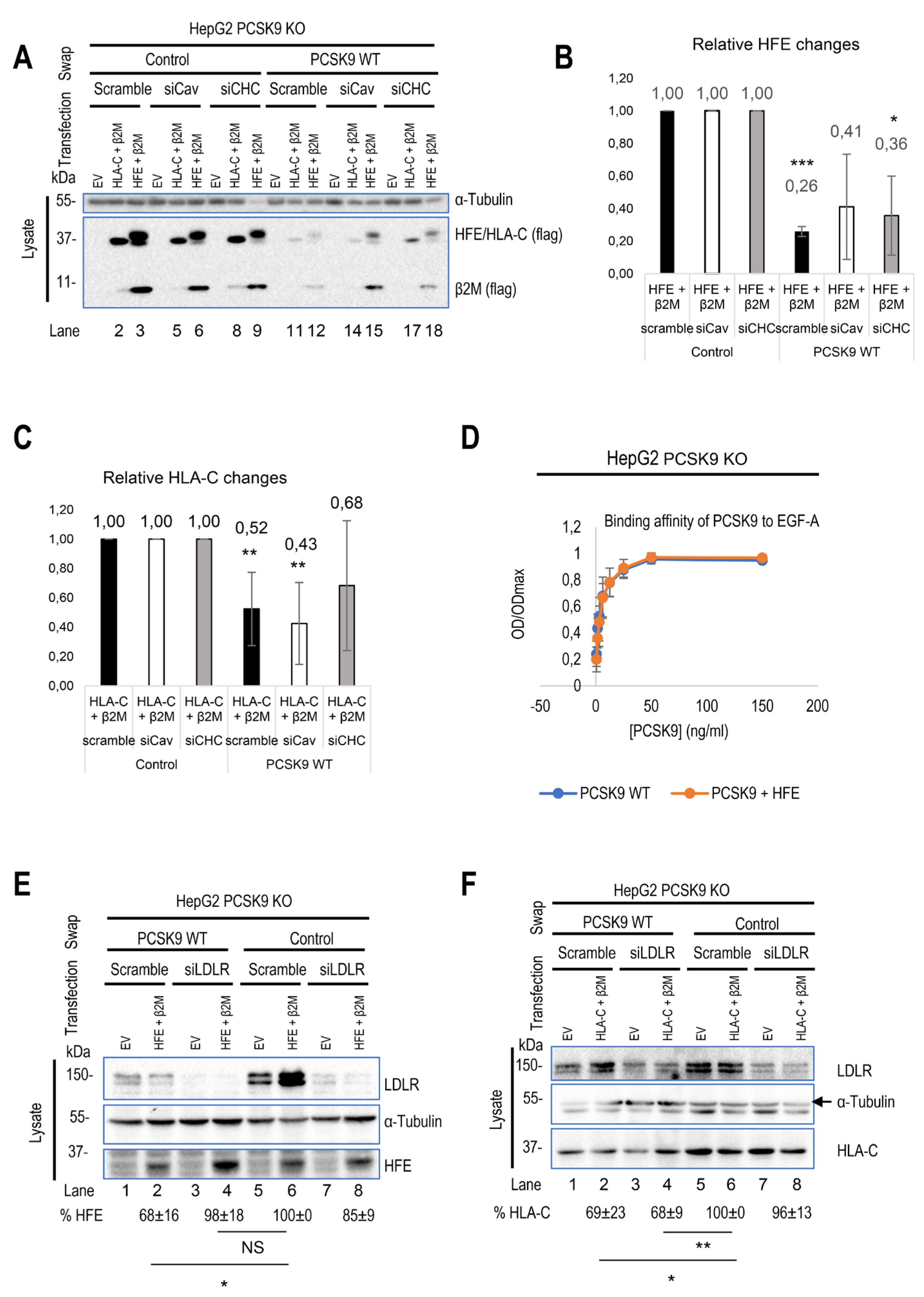
Figure 4.
Continued. (G) and (H) HepG2 HLA-C KO cells were co-transfected with (empty vector (EV)+ ΔCT LDLR), (WT HLA-C+ ΔCT LDLR), or (WT HFE+ΔCT LDLR) and then incubated with conditioned media from HEK293 cells expressing an empty vector (control) or WT PCSK9. The effect of ΔCT LDLR on either (G) PCSK9 internalization or (H) HFE/HLA-C degradation was measured in prospective charts. (I) Comparison of cytosolic and transmembrane domains of HFE with other major HLA family members. Notice the unique residues (in red) in the transmembrane domain of HFE and in the cytosolic tail of HLA-C (in red surrounded by green boxes). All cell Lysates were extracted to be analyzed by WB (SDS/PAGE on 8% Tris-glycine gel). All protein levels were normalized to the control protein, α-tubulin. Data are representative of three independent experiments. Quantifications are averages ± standard deviation (SD). * p<0.05; ** p<0.01; ***p<0.001 (two-sided t-test). NS: non-significant.
Figure 4.
Continued. (G) and (H) HepG2 HLA-C KO cells were co-transfected with (empty vector (EV)+ ΔCT LDLR), (WT HLA-C+ ΔCT LDLR), or (WT HFE+ΔCT LDLR) and then incubated with conditioned media from HEK293 cells expressing an empty vector (control) or WT PCSK9. The effect of ΔCT LDLR on either (G) PCSK9 internalization or (H) HFE/HLA-C degradation was measured in prospective charts. (I) Comparison of cytosolic and transmembrane domains of HFE with other major HLA family members. Notice the unique residues (in red) in the transmembrane domain of HFE and in the cytosolic tail of HLA-C (in red surrounded by green boxes). All cell Lysates were extracted to be analyzed by WB (SDS/PAGE on 8% Tris-glycine gel). All protein levels were normalized to the control protein, α-tubulin. Data are representative of three independent experiments. Quantifications are averages ± standard deviation (SD). * p<0.05; ** p<0.01; ***p<0.001 (two-sided t-test). NS: non-significant.
3.5. Modeling of the Interaction between PCSK9’s N-Terminus and HLA-C
Superimposition of the models of the PCSK9/HLA-C and PCSK9/HFE complexes (Figure 3H) shows that HLA-C and HFE both bind the CHRD of PCSK9 via R549 and E567, however, the orientation of the ⍺1-chain of both HLA- C and HFE in the two complexes are slightly different. The N-terminal end of the PCSK9 prodomain appears closer to the ⍺1-chain of HLA-C than that of HFE (Figure 3H). The possible interaction of PCSK9 N-terminal peptide with ⍺1-chain of each HLA member was next tested. This prediction suggested that among all members, only HLA-C’s ⍺1-chain could optimally interact with the N-terminal acidic peptide (aa 31-59) of PCSK9 (Figure 5A). To further support the possible interaction of the above acidic peptide with the antigenic pocket of HLA-C, we evaluated whether this unstructured part of PCSK9 (unstructured in PDB:2P4E) presented a length compatible to reach and possibly bind the antigenic pocket of HLA-C. Thus, modeling of the interaction of the PCSK9 residues 31-59 and the antigenic pocket of HLA-C was carried out. All the 15 models generated by Alphafold suggested an interaction between the PCSK9’s unstructured N-terminal residues 38-44 (peptide: YEELVLA) with HLA-C peptide binding pocket (Figure 5B). The mean pLDDT values (used to estimate prediction confidence) for the hotspot interaction region of the best ranked model (residues 37-43, DYEELVL) was 85, suggesting it was modeled with high confidence (Figure 5B). The positioning of PCSK9’ N-terminal structured peptide in an α-helix (PBD:6MV5, complexed with anti PCSK9 fab, [59]) was also positioned in this antigenic pocket of HLA-C ⍺-1 domain, using GRAMM. This allowed us to evaluate the possible influence of the structure of the acidic peptide on its binding to the HLA-C pocket (Figure 5B). The positioning of the PCSK9 prodomain peptide in the HLA-C pocket suggests possible contacts between Lys90 and Arg93 of HLA-C with, respectively, Asp37 and Glu40 for the unstructured peptide and Asp37 and Glu39 for the structured peptide.
The full PCSK9/HLA-C ⍺1-chain/β2-microglobulin ternary model argue in favor of a plausible interaction of PCSK9/HLA-C simultaneously by PCSK9’s CHRD and N-terminal peptide. The former implicating residues E567 and E549 of the CHRD’s M2 module with residues R68 and E70 of HLA-C α1-chain. The CHRD/⍺1-chain of HLA-C interaction is also reinforced by the proximity between the three CHRD’s M2 domain Histidines (537, 553 and 551) and Glu79 of HLA-C (Figure S1), which may be enhanced in acidic pHs of endosomes whereupon these His in the M2 domain would be positively charged.
All MHC-I molecules have a similar structure (Figure S2A). The presence of positively or negatively charged residues at the HLA-C antigenic pocket (Figure S2B) as well as the electrostatic potential (Figure S2C) were evaluated and compared to other MHC-I antigenic pockets. The prevalence of positively charged residues in the HLA-C antigenic pocket supports possible binding of an acidic peptide such as that of the PCSK9 prodomain.
Figure 5.
Modeling of interaction between PCSK9’s N-terminus with HLA members. Molecular modeling of the interaction of PCSK9’ 31-59 propeptide with all HLA proteins. (B) Molecular modeling of the interaction of PCSK9’ 31-59 unstructured propeptide (green) with HLA-C antigenic pocket of HLA-C (magenta). A zoom view shows possible proximities between basic amino acids of HLA-C pocket (blue) and acid residues of unstructured (left) or structured (right) peptide (red).
Figure 5.
Modeling of interaction between PCSK9’s N-terminus with HLA members. Molecular modeling of the interaction of PCSK9’ 31-59 propeptide with all HLA proteins. (B) Molecular modeling of the interaction of PCSK9’ 31-59 unstructured propeptide (green) with HLA-C antigenic pocket of HLA-C (magenta). A zoom view shows possible proximities between basic amino acids of HLA-C pocket (blue) and acid residues of unstructured (left) or structured (right) peptide (red).
4. Discussion
The detailed understanding of the trafficking and regulation of PCSK9 and LDLR in the liver and other tissues remains incomplete. Since the inhibition of PCSK9 presents a potent strategy for treating cardiovascular disorders (CVDs), understanding the detailed trafficking of this protein and its possible implications in other cellular processes holds the potential to extend the advantages of this established treatment beyond CVD [10,60]. The discovery of HLA-A2 as a new target of PCSK9 led to the combination of PCSK9 inhibitors [29] or antibodies [61], with PD-1 antibodies in cancer therapy. This combination has shown a potential in enhancing responses in breast and colorectal cancers compared to PD-1 antibody treatment alone [29,61].
Recently, we introduced a novel model for the clathrin-coated sorting of the PCSK9-LDLR complex that requires at least two partner proteins including CAP1 and an unidentified "protein X". CAP1 interacts with PCSK9's M1 and M3 domains, as well as acidic residues in the N-terminal segment of PCSK9’s prodomain [23], thereby exposing the M2 domain of PCSK9 for efficient interaction with “protein X”. In this work, HLA-C is proposed as a bona fide “protein X” candidate for PCSK9’s function on LDLR [23,27]. Immunofluorescence microscopy [62] revealed that lack of the M2 domain (more specifically residues R549, Q554, E567) leading to loss of “protein X” interaction with PCSK9 [23], results in a complete LOF of PCSK9 on LDLR degradation, but has no effect on the endocytosis of the PCSK9-LDLR complex (Figure 1A, B). This suggests that “protein X” becomes critical following endocytosis of this complex, likely to sort it to lysosomes for degradation. Additionally, we showed that the presence or absence of these residues is critical for PCSK9’s binding to either HFE or HLA-C (Figure 3A-H). In addition, our cell-based assays in CHO-K1 and HepG2 CRISPR HLA-C KO cells confirmed the crucial role of HLA-C for PCSK9’s function, since in the absence of this protein PCSK9 no longer reduces LDLR levels (Figure 2E, F). Notably, HLA-C still enhances the internalization of the PCSK9-LDLR complex in the absence of the LDLR’s C-terminal cytosolic domain (Figure 4G, I). We hypothesized that this may be due to the presence of a di-Leu motif (Leu-Ile362; Figure 4I) reported to be critical for the lysosomal sorting of HLA-C [63]. Preliminary data in HepG2 CRISPR HLA-C KO cells revealed that both Leu361 and the unique Cys345 (Figure S2B) are needed for HLA-C activation of extracellular PCSK9 function on LDLR (not shown). These data point to the uniqueness of HLA-C to act as “protein X” via cytosolic tail sequences regulating lysosomal targeting (Leu-Ile362) and membrane association (possibly palmitoylation of Cys345). Notably, HLA-C also significantly increased the PCSK9’s activity of the supposedly GOF PCSK9 H553R variant [23,28] on LDLR (Figure 3E), supporting the proposed model of PCSK9-HLA-C interaction where PCSK9’s Arg553 interacts better than the native His553 with a negatively charged cluster consisting of Glu79, Glu197 and Glu201 in HLA-C (Figure 3B) [23].
The resemblance of HFE’s crystal structure to HLA-C, along with its prior connection to LDLR regulation, motivated us to study its potential regulatory effect on PCSK9’s function. Our 3D modeling and cellular analysis revealed that PCSK9's R-x-E motif could interact with HFE's Arg67 (or Arg78) and Glu69, like HLA-C interactions. However, modeling of PCSK9 natural variants (Q554E and H553R) suggested that they may exert opposite effects on HFE compared to HLA-C, likely due to the positive charge of Arg71 in HFE (Figure 3A, B). Furthermore, our data uncovered a negative regulatory effect of HFE on PCSK9’s function on LDLR, which could be stimulated under certain physiological conditions such as elevated iron levels (Figure 2A, D). TfR1 binds the HFE’s α1 and α2 domains [64]. Our 3D modeling also suggests the involvement of the α1 domain (Arg67/78, Glu69, and Arg71) of HFE in its interaction with PCSK9 (Figure 3A), suggesting a potential competition of TfR1 with PCSK9 to interact with HFE. Elevated iron levels lead to the dissociation of HFE from TfR1, increasing its potential availability for PCSK9 at the cell surface. Apart from the regulatory effect of HFE on extracellular PCSK9, we discovered that this protein is a new target for extracellular PCSK9 for lysosomal degradation, requiring LDLR (Figure 2A-C, 4E), suggesting the possible implication of PCSK9 in iron metabolism.
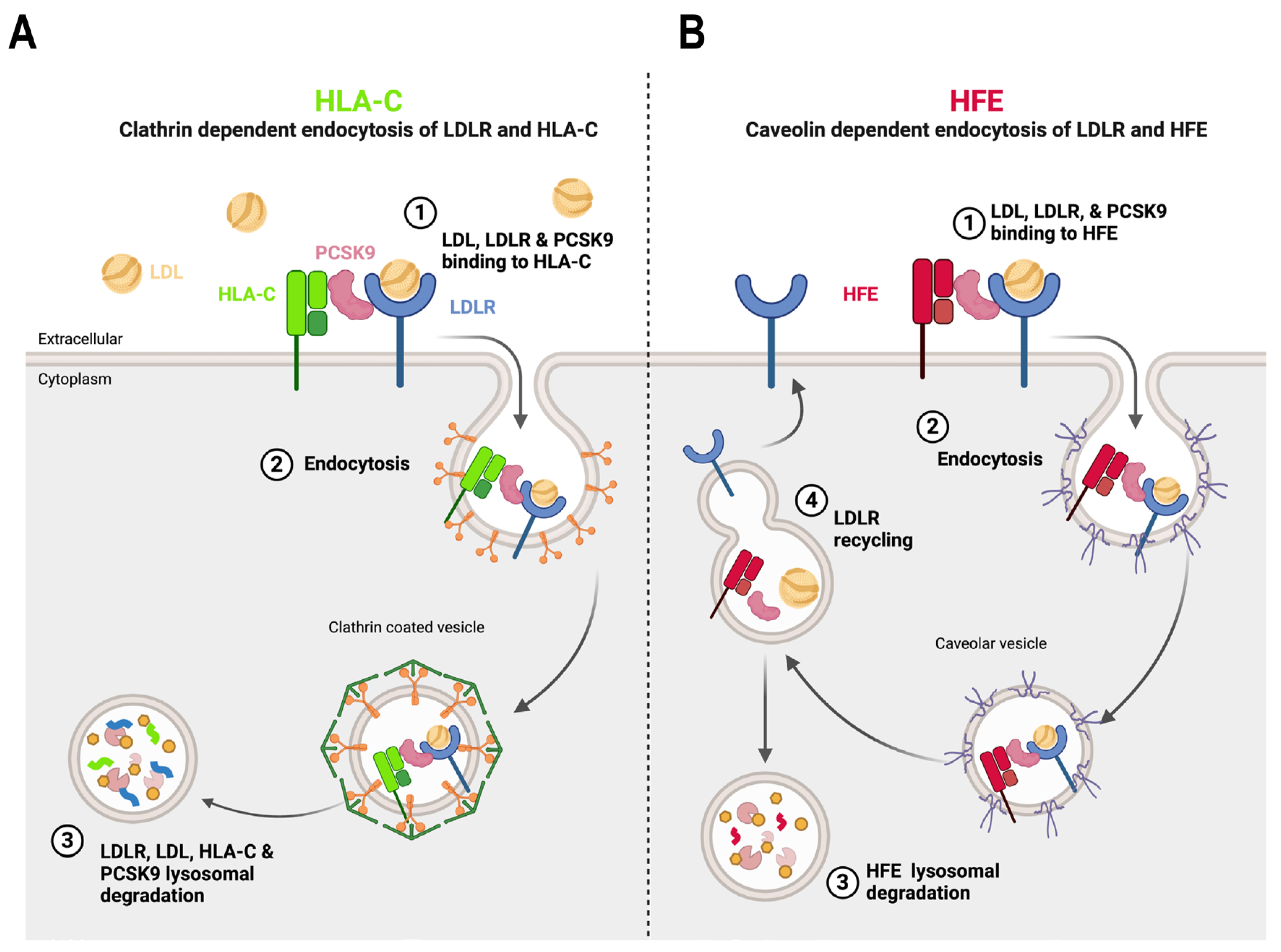
While HLA-C positively regulates the extracellular activity of PCSK9 on the LDLR, PCSK9 doesn't rely on HLA-C for HFE degradation, suggesting distinct regulatory pathways of PCSK9 by HFE compared to HLA-C. Previous studies have established that PCSK9 and LDLR internalization occurs through clathrin-coated vesicles [18,23,39,55]. The present investigation revealed that in HepG2 cells distinct endocytosis pathways HFE and HLA-C exist. While the degradation of HLA-C was reduced by the removal of the clathrin heavy chain (CHC), the degradation of HFE was decreased by the absence of either CHC or caveolin (Cav1) suggesting that PCSK9-HFE sorts to lysosomes via clathrin-coated and caveolin-dependent vesicles (Figure 4A-C).
Accordingly, we propose two internalization pathways for the PCSK9-LDLR complex depending on its interaction with either HLA-C or HFE. Under normal conditions due to the high expression levels of HLA-C, it interacts with the PCSK9-LDLR complex and sorts it to lysosomes for degradation via clathrin-coated vesicles. However, under elevated iron levels, HFE’s may bind the PCSK9-LDLR, and the complex may then be internalized into caveolin-positive endosomes. Such a competitive pathway may prevent the binding of HLA-C to PCSK9 and hence the degradation of the LDLR (Figure 6). It is still unclear why different from HFE, the caveolin-dependent pathway requires the LDLR for degradation, but the latter is not degraded. Similar pathways have been observed for the TGF-β receptor, in which its internalization can occur either through clathrin-coated pits (for signal transduction) or caveolin-positive vesicles (for its degradation), where some physiological conditions, like high potassium levels, could favor one pathway over the other [65].
Our results and molecular models advocate in favor of a more general crosstalk between PCSK9 and HLA molecules, which may have profound implications in immunology. Our current model for PCSK9/HLA-C complex proposes that the PCSK9’s acidic N-terminus could bind to peptide binding pocket of the latter (Figure 5A, B). Future works using site-directed mutagenesis on the proposed interface should provide more substantial evidence for validation or refutation of this model and the possible regulation of the levels of HLA-C at the cell surface by the acidic domain of PCSK9. Finally, the possible implication of the acidic domain of PCSK9 in the regulation of its intracellular activity and its effect on antigen presentation by HLA members and/or their cell surface localization begs for more detailed analysis of these phenomena.
Figure 6.
The proposed model of PCSK9’s regulation by either HFE or HLA-C. The model is created by using BioRender.com. (A) In normal conditions within hepatocytes, where the expression of HLA-C is significantly higher compared to HFE, it interacts with the M2 domain and prodomain of PCSK9. The resulting complex of PCSK9-LDLR-HLA-C is internalized via clathrin-coated vesicles, which transport the entire complex to lysosomal compartments for degradation. (B) However, in certain physiological conditions, such as elevated levels of circulating iron, HFE could also interact with PCSK9. In this scenario, the entire complex is likely internalized with the assistance of caveolin-dependent vesicles. This alternative endocytosis pathway leads LDLR to recycle back to the cell surface, while only HFE is directed towards degradation.
Figure 6.
The proposed model of PCSK9’s regulation by either HFE or HLA-C. The model is created by using BioRender.com. (A) In normal conditions within hepatocytes, where the expression of HLA-C is significantly higher compared to HFE, it interacts with the M2 domain and prodomain of PCSK9. The resulting complex of PCSK9-LDLR-HLA-C is internalized via clathrin-coated vesicles, which transport the entire complex to lysosomal compartments for degradation. (B) However, in certain physiological conditions, such as elevated levels of circulating iron, HFE could also interact with PCSK9. In this scenario, the entire complex is likely internalized with the assistance of caveolin-dependent vesicles. This alternative endocytosis pathway leads LDLR to recycle back to the cell surface, while only HFE is directed towards degradation.
5. Conclusions
In summary, our findings highlight the significance of PCSK9's M2 domain and HLA-C in the extracellular activity of PCSK9 for lysosomal degradation of LDLR. Furthermore, our results suggest an opposite regulatory effect HFE compared to HLA-C, which utilizes different endocytosis pathways when interacting with PCSK9. The interaction of each protein could potentially influence the fate of PCSK9-bound proteins in the cellular trafficking process.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Figure S1: Graphic representation of the 3 Histidine residues in the PCSK9’ CHRD in contact with Glu79 of HLA-C; Figure S2: Structure and interaction sites of all MHC-I molecules.
Author Contributions
Conceptualization, S.M. and N.G.S.; methodology, S.M. and A.E.; software, C.F-G. and O.H.P.R.; validation, S.M., A.B.D.O., A.E. and R.E.; formal analysis, S.M., N.G.S., C.F-G. and O.H.P.R.; data curation, S.M. and N.G.S.; writing—original draft preparation, S.M. and N.G.S.; writing— review and editing, S.M., C.F-G. and N.G.S.; visualization, S.M., C.F-G. and N.G.S; supervision, N.G.S.; project administration, N.G.S.; funding acquisition, N.G.S.. All authors have read and agreed to the published version of the manuscript.
Funding
“This research was funded by a CIHR Foundation grant (NGS: # 148363), a Canada Research Chairs in Precursor Proteolysis (N.G.S.: # 950-231335) and a Leducq Foundation grant (N.G.S: # 13 CVD 03). The salary of S.M. was supported by an IRCM CAL construction scholarship (2020-2021) and later by an FRQS fellowship (# 302040; 2021-2023).
Acknowledgments
The authors would like to acknowledge the expert secretarial help of Jisca Borgela in the preparation of this manuscript. We would also like to thank Dr. Kostas Pantopoulos, Dr. May Faraj for fruitful discussions and Mailys Le Dévéhat, and Josée Hamelin for technical assistance, as well as the IRCM confocal microscopy, Dominic Filion and the IRCM sequencing facility. This work was granted access to the HPC resources of IDRIS under the allocation 2023-AD010714267 made by GENCI.
Conflicts of Interest
“The authors declare no conflict of interest.”
References
- Seidah, N.G.; Benjannet, S.; Wickham, L.; Marcinkiewicz, J.; Jasmin, S.B.; Stifani, S.; Basak, A.; Prat, A.; Chretien, M. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proceedings of the National Academy of Sciences of the United States of America 2003, 100, 928-933. [CrossRef]
- Seidah, N.G. The PCSK9 discovery, an inactive protease with varied functions in hypercholesterolemia, viral infections, and cancer. J Lipid Res 2021, 62, 100130. [CrossRef]
- Seidah, N.G.; Prat, A. The biology and therapeutic targeting of the proprotein convertases. Nat Rev Drug Discov 2012, 11, 367-383. [CrossRef]
- Abifadel, M.; Varret, M.; Rabes, J.P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nature Genetics 2003, 34, 154-156. [CrossRef]
- Maxwell, K.N.; Breslow, J.L. Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proceedings of the National Academy of Sciences of the United States of America 2004, 101, 7100-7105.
- Benjannet, S.; Rhainds, D.; Essalmani, R.; Mayne, J.; Wickham, L.; Jin, W.; Asselin, M.C.; Hamelin, J.; Varret, M.; Allard, D.; et al. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. Journal of Biological Chemistry 2004, 279, 48865-48875. [CrossRef]
- McNutt, M.C.; Lagace, T.A.; Horton, J.D. Catalytic activity is not required for secreted PCSK9 to reduce low density lipoprotein receptors in HepG2 cells. Journal of Biological Chemistry 2007, 282, 20799-20803.
- Poirier, S.; Mayer, G.; Benjannet, S.; Bergeron, E.; Marcinkiewicz, J.; Nassoury, N.; Mayer, H.; Nimpf, J.; Prat, A.; Seidah, N.G. The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. Journal of Biological Chemistry 2008, 283, 2363-2372.
- Seidah, N.G.; Abifadel, M.; Prost, S.; Boileau, C.; Prat, A. The Proprotein Convertases in Hypercholesterolemia and Cardiovascular Diseases: Emphasis on Proprotein Convertase Subtilisin/Kexin 9. Pharmacol Rev 2017, 69, 33-52. [CrossRef]
- Seidah, N.G.; Prat, A. The Multifaceted Biology of PCSK9. Endocr Rev 2022, 43, 558-582. [CrossRef]
- Lambert, G.; Sjouke, B.; Choque, B.; Kastelein, J.J.; Hovingh, G.K. The PCSK9 decade. J Lipid Res 2012, 53, 2515-2524. [CrossRef]
- Seidah, N.G.; Prat, A.; Pirillo, A.; Catapano, A.L.; Norata, G.D. Novel strategies to target proprotein convertase subtilisin kexin 9: beyond monoclonal antibodies. Cardiovasc Res 2019, 115, 510-518. [CrossRef]
- Oostveen, R.F.; Khera, A.V.; Kathiresan, S.; Stroes, E.S.G.; Fitzgerald, K.; Harms, M.J.; Oakes, B.L.; Kastelein, J.J.P. New Approaches for Targeting PCSK9: Small-Interfering Ribonucleic Acid and Genome Editing. Arterioscler Thromb Vasc Biol 2023, 43, 1081-1092. [CrossRef]
- Lee, R.G.; Mazzola, A.M.; Braun, M.C.; Platt, C.; Vafai, S.B.; Kathiresan, S.; Rohde, E.; Bellinger, A.M.; Khera, A.V. Efficacy and Safety of an Investigational Single-Course CRISPR Base-Editing Therapy Targeting PCSK9 in Nonhuman Primate and Mouse Models. Circulation 2023, 147, 242-253. [CrossRef]
- Davis, J.R.; Wang, X.; Witte, I.P.; Huang, T.P.; Levy, J.M.; Raguram, A.; Banskota, S.; Seidah, N.G.; Musunuru, K.; Liu, D.R. Efficient in vivo base editing via single adeno-associated viruses with size-optimized genomes encoding compact adenine base editors. Nat Biomed Eng 2022, 6, 1272-1283. [CrossRef]
- Cunningham, D.; Danley, D.E.; Geoghegan, K.F.; Griffor, M.C.; Hawkins, J.L.; Subashi, T.A.; Varghese, A.H.; Ammirati, M.J.; Culp, J.S.; Hoth, L.R.; et al. Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat Struct Mol Biol. 2007, 14, 413-419. [CrossRef]
- Piper, D.E.; Jackson, S.; Liu, Q.; Romanow, W.G.; Shetterly, S.; Thibault, S.T.; Shan, B.; Walker, N.P. The Crystal Structure of PCSK9: A Regulator of Plasma LDL-Cholesterol. Structure. 2007, 15, 545-552.
- Holla, O.L.; Cameron, J.; Berge, K.E.; Ranheim, T.; Leren, T.P. Degradation of the LDL receptors by PCSK9 is not mediated by a secreted protein acted upon by PCSK9 extracellularly. BMC. Cell Biol. 2007, 8, 9-20.
- Zhang, D.W.; Garuti, R.; Tang, W.J.; Cohen, J.C.; Hobbs, H.H. Structural requirements for PCSK9-mediated degradation of the low-density lipoprotein receptor. Proceedings of the National Academy of Sciences of the United States of America 2008, 105, 13045-13050.
- Saavedra, Y.G.; Day, R.; Seidah, N.G. The M2 module of the Cys-His-rich domain (CHRD) of PCSK9 is needed for the extracellular low density lipoprotein receptor (LDLR) degradation pathway. J Biol. Chem. 2012, 287, 43492-43501.
- Poirier, S.; Mayer, G.; Poupon, V.; McPherson, P.S.; Desjardins, R.; Ly, K.; Asselin, M.C.; Day, R.; Duclos, F.J.; Witmer, M.; et al. Dissection of the endogenous cellular pathways of PCSK9-induced LDLR degradation: Evidence for an intracellular route. Journal of Biological Chemistry 2009, 284, 28856-28864. [CrossRef]
- Zaid, A.; Roubtsova, A.; Essalmani, R.; Marcinkiewicz, J.; Chamberland, A.; Hamelin, J.; Tremblay, M.; Jacques, H.; Jin, W.; Davignon, J.; et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9): Hepatocyte-specific low-density lipoprotein receptor degradation and critical role in mouse liver regeneration. Hepatology 2008, 48, 646-654. [CrossRef]
- Fruchart Gaillard, C.; Ouadda, A.B.D.; Ciccone, L.; Girard, E.; Mikaeeli, S.; Evagelidis, A.; Le Devehat, M.; Susan-Resiga, D.; Lajeunesse, E.C.; Nozach, H.; et al. Molecular interactions of PCSK9 with an inhibitory nanobody, CAP1 and HLA-C: Functional regulation of LDLR levels. Mol Metab 2023, 67, 101662. [CrossRef]
- Devay, R.M.; Shelton, D.L.; Liang, H. Characterization of Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) Trafficking Reveals a Novel Lysosomal Targeting Mechanism via Amyloid Precursor-like Protein 2 (APLP2). J Biol. Chem. 2013, 288, 10805-10818.
- Sparks, R.P.; Arango, A.S.; Jenkins, J.L.; Guida, W.C.; Tajkhorshid, E.; Sparks, C.E.; Sparks, J.D.; Fratti, R.A. An Allosteric Binding Site on Sortilin Regulates the Trafficking of VLDL, PCSK9, and LDLR in Hepatocytes. Biochemistry 2020, 59, 4321-4335. [CrossRef]
- Butkinaree, C.; Canuel, M.; Essalmani, R.; Poirier, S.; Benjannet, S.; Asselin, M.C.; Roubtsova, A.; Hamelin, J.; Marcinkiewicz, J.; Chamberland, A.; et al. Amyloid precursor-like protein 2 and sortilin do not regulate the PCSK9-mediated low density lipoprotein receptor degradation but interact with each other. Journal of Biological Chemistry 2015, 290, 18609-18620. [CrossRef]
- Jang, H.D.; Lee, S.E.; Yang, J.; Lee, H.C.; Shin, D.; Lee, H.; Lee, J.; Jin, S.; Kim, S.; Lee, S.J.; et al. Cyclase-associated protein 1 is a binding partner of proprotein convertase subtilisin/kexin type-9 and is required for the degradation of low-density lipoprotein receptors by proprotein convertase subtilisin/kexin type-9. Eur Heart J 2020, 41, 239-252. [CrossRef]
- Kotowski, I.K.; Pertsemlidis, A.; Luke, A.; Cooper, R.S.; Vega, G.L.; Cohen, J.C.; Hobbs, H.H. A Spectrum of PCSK9 Alleles Contributes to Plasma Levels of Low-Density Lipoprotein Cholesterol. American Journal of Human Genetics 2006, 78, 410-422. [CrossRef]
- Liu, X.; Bao, X.; Hu, M.; Chang, H.; Jiao, M.; Cheng, J.; Xie, L.; Huang, Q.; Li, F.; Li, C.Y. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature 2020, 588, 693-698. [CrossRef]
- Klein, J.; Sato, A. The HLA system. First of two parts. N Engl J Med 2000, 343, 702-709. [CrossRef]
- Stasiak, M.; Zawadzka-Starczewska, K.; Tymoniuk, B.; Stasiak, B.; Lewiński, A. Associations between Lipid Profiles and Graves' Orbitopathy can Be HLA-Dependent. Genes (Basel) 2023, 14. [CrossRef]
- Demetz, E.; Tymoszuk, P.; Hilbe, R.; Volani, C.; Haschka, D.; Heim, C.; Auer, K.; Lener, D.; Zeiger, L.B.; Pfeifhofer-Obermair, C.; et al. The haemochromatosis gene Hfe and Kupffer cells control LDL cholesterol homeostasis and impact on atherosclerosis development. Eur Heart J 2020, 41, 3949-3959. [CrossRef]
- Pankow, J.S.; Boerwinkle, E.; Adams, P.C.; Guallar, E.; Leiendecker-Foster, C.; Rogowski, J.; Eckfeldt, J.H. HFE C282Y homozygotes have reduced low-density lipoprotein cholesterol: the Atherosclerosis Risk in Communities (ARIC) Study. Transl Res 2008, 152, 3-10. [CrossRef]
- Susan-Resiga, D.; Girard, E.; Essalmani, R.; Roubtsova, A.; Marcinkiewicz, J.; Derbali, R.M.; Evagelidis, A.; Byun, J.H.; Lebeau, P.F.; Austin, R.C.; Seidah, N.G. Asialoglycoprotein receptor 1 is a novel PCSK9-independent ligand of liver LDLR cleaved by furin. J Biol Chem 2021, 297, 101177. [CrossRef]
- Kruse, V.; Hamann, C.; Monecke, S.; Cyganek, L.; Elsner, L.; Hübscher, D.; Walter, L.; Streckfuss-Bömeke, K.; Guan, K.; Dressel, R. Human Induced Pluripotent Stem Cells Are Targets for Allogeneic and Autologous Natural Killer (NK) Cells and Killing Is Partly Mediated by the Activating NK Receptor DNAM-1. PLoS One 2015, 10, e0125544. [CrossRef]
- Ben Djoudi Ouadda, A.; Gauthier, M.S.; Susan-Resiga, D.; Girard, E.; Essalmani, R.; Black, M.; Marcinkiewicz, J.; Forget, D.; Hamelin, J.; Evagelidis, A.; et al. Ser-Phosphorylation of PCSK9 (Proprotein Convertase Subtilisin-Kexin 9) by Fam20C (Family With Sequence Similarity 20, Member C) Kinase Enhances Its Ability to Degrade the LDLR (Low-Density Lipoprotein Receptor). Arterioscler Thromb Vasc Biol 2019, 39, 1996-2013. [CrossRef]
- Dubuc, G.; Tremblay, M.; Pare, G.; Jacques, H.; Hamelin, J.; Benjannet, S.; Boulet, L.; Genest, J.; Bernier, L.; Seidah, N.G.; Davignon, J. A new method for measurement of total plasma PCSK9: clinical applications. Journal of Lipid Research 2010, 51, 140-149.
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. Journal of Biological Chemistry 1951, 193, 265-275.
- Nassoury, N.; Blasiole, D.A.; Tebon, O.A.; Benjannet, S.; Hamelin, J.; Poupon, V.; McPherson, P.S.; Attie, A.D.; Prat, A.; Seidah, N.G. The Cellular Trafficking of the Secretory Proprotein Convertase PCSK9 and Its Dependence on the LDLR. Traffic. 2007, 8, 718-732. [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 2004, 25, 1605-1612. [CrossRef]
- Singh, A.; Copeland, M.M.; Kundrotas, P.J.; Vakser, I.A. GRAMM Web Server for Protein Docking. Methods Mol Biol 2024, 2714, 101-112. [CrossRef]
- Kuhlman, B.; Dantas, G.; Ireton, G.C.; Varani, G.; Stoddard, B.L.; Baker, D. Design of a novel globular protein fold with atomic-level accuracy. Science 2003, 302, 1364-1368. [CrossRef]
- Strom, T.B.; Holla, O.L.; Tveten, K.; Cameron, J.; Berge, K.E.; Leren, T.P. Disrupted recycling of the low density lipoprotein receptor by PCSK9 is not mediated by residues of the cytoplasmic domain. Mol. Genet. Metab 2010, 101, 76-80.
- Holla, O.L.; Strom, T.B.; Cameron, J.; Berge, K.E.; Leren, T.P. A chimeric LDL receptor containing the cytoplasmic domain of the transferrin receptor is degraded by PCSK9. Mol. Genet. Metab 2010, 99, 149-156.
- Canuel, M.; Sun, X.; Asselin, M.C.; Paramithiotis, E.; Prat, A.; Seidah, N.G. Proprotein convertase subtilisin/kexin type 9 (PCSK9) can mediate degradation of the low density lipoprotein receptor-related protein 1 (LRP-1). PLoS ONE 2013, 8, e64145.
- Johnson, J.K.; Wright, P.W.; Li, H.; Anderson, S.K. Identification of trophoblast-specific elements in the HLA-C core promoter. Hla 2018, 92, 288-297. [CrossRef]
- Roubtsova, A.; Chamberland, A.; Marcinkiewicz, J.; Essalmani, R.; Fazel, A.; Bergeron, J.J.; Seidah, N.G.; Prat, A. PCSK9 deficiency unmasks a sex/tissue-specific subcellular distribution of the LDL and VLDL receptors in mice. Journal of Lipid Research 2015, 56, 2133-2142. [CrossRef]
- Lebron, J.A.; Bennett, M.J.; Vaughn, D.E.; Chirino, A.J.; Snow, P.M.; Mintier, G.A.; Feder, J.N.; Bjorkman, P.J. Crystal structure of the hemochromatosis protein HFE and characterization of its interaction with transferrin receptor. Cell 1998, 93, 111-123. [CrossRef]
- Gross, C.N.; Irrinki, A.; Feder, J.N.; Enns, C.A. Co-trafficking of HFE, a nonclassical major histocompatibility complex class I protein, with the transferrin receptor implies a role in intracellular iron regulation. J Biol Chem 1998, 273, 22068-22074. [CrossRef]
- Ganz, T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 2003, 102, 783-788.
- Zhang, A.S.; Davies, P.S.; Carlson, H.L.; Enns, C.A. Mechanisms of HFE-induced regulation of iron homeostasis: Insights from the W81A HFE mutation. Proc Natl Acad Sci U S A 2003, 100, 9500-9505. [CrossRef]
- Guillemot, J.; Asselin, M.C.; Susan-Resiga, D.; Essalmani, R.; Seidah, N.G. Deferoxamine stimulates LDLR expression and LDL uptake in HepG2 cells. Mol Nutr. Food Res. 2016, 60, 600-608.
- Parolini, F.; Biswas, P.; Serena, M.; Sironi, F.; Muraro, V.; Guizzardi, E.; Cazzoletti, L.; Scupoli, M.T.; Gibellini, D.; Ugolotti, E.; et al. Stability and Expression Levels of HLA-C on the Cell Membrane Modulate HIV-1 Infectivity. J Virol 2018, 92. [CrossRef]
- Mayer, G.; Poirier, S.; Seidah, N.G. Annexin A2 is a C-terminal PCSK9-binding protein that regulates endogenous low density lipoprotein receptor levels. Journal of Biological Chemistry 2008, 283, 31791-31801.
- Qian, Y.W.; Schmidt, R.J.; Zhang, Y.; Chu, S.; Lin, A.; Wang, H.; Wang, X.; Beyer, T.P.; Bensch, W.R.; Li, W.; et al. Secreted PCSK9 downregulates low density lipoprotein receptor through receptor-mediated endocytosis. Journal of Lipid Research 2007, 48, 1488-1498.
- Puddu, A.; Maggi, D. Emerging Role of Caveolin-1 in GLP-1 Action. Front Endocrinol (Lausanne) 2021, 12, 668012. [CrossRef]
- Mikaeeli, S.; Susan-Resiga, D.; Girard, E.; Ben Djoudi Ouadda, A.; Day, R.; Prost, S.; Seidah, N.G. Functional analysis of natural PCSK9 mutants in modern and archaic humans. Febs j 2020, 287, 515-528. [CrossRef]
- Ramalingam, T.S.; West, A.P., Jr.; Lebron, J.A.; Nangiana, J.S.; Hogan, T.H.; Enns, C.A.; Bjorkman, P.J. Binding to the transferrin receptor is required for endocytosis of HFE and regulation of iron homeostasis. Nat. Cell Biol. 2000, 2, 953-957.
- Ultsch, M.; Li, W.; Eigenbrot, C.; Di Lello, P.; Lipari, M.T.; Gerhardy, S.; AhYoung, A.P.; Quinn, J.; Franke, Y.; Chen, Y.; et al. Identification of a Helical Segment within the Intrinsically Disordered Region of the PCSK9 Prodomain. J Mol Biol 2019, 431, 885-903. [CrossRef]
- Seidah, N.G.; Garçon, D. Expanding Biology of PCSK9: Roles in Atherosclerosis and Beyond. Curr Atheroscler Rep 2022, 1-10. [CrossRef]
- Wang, R.; Liu, H.; He, P.; An, D.; Guo, X.; Zhang, X.; Feng, M. Inhibition of PCSK9 enhances the antitumor effect of PD-1 inhibitor in colorectal cancer by promoting the infiltration of CD8(+) T cells and the exclusion of Treg cells. Front Immunol 2022, 13, 947756. [CrossRef]
- Seidah, N.G.; Garcon, D. Expanding Biology of PCSK9: Roles in Atherosclerosis and Beyond. Curr Atheroscler Rep 2022, 24, 821-830. [CrossRef]
- Tan, P.K.; Waites, C.; Liu, Y.; Krantz, D.E.; Edwards, R.H. A leucine-based motif mediates the endocytosis of vesicular monoamine and acetylcholine transporters. J Biol Chem 1998, 273, 17351-17360. [CrossRef]
- Bennett, M.J.; Lebron, J.A.; Bjorkman, P.J. Crystal structure of the hereditary haemochromatosis protein HFE complexed with transferrin receptor. Nature 2000, 403, 46-53. [CrossRef]
- Di Guglielmo, G.M.; Le Roy, C.; Goodfellow, A.F.; Wrana, J.L. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat Cell Biol 2003, 5, 410-421. [CrossRef]
Figure 1.
Extracellular importance of M2 domain of PCSK9 and mRNA expression of HLA members. (A) Immunofluorescence staining of total LDLR (permeabilized conditions) vs. cell surface LDLR (non-permeabilized conditions) levels in the presence of PCSK9-ΔM2 compared to WT PCSK9. (B) Immunostaining of total LDLR along with early endosomal (EEA1) marker in the presence of PCSK9 ΔM2, PCSK9 R549A-E567A (RE), and PCSK9 R549A-Q554E-E567A (RQE). Blue, Nuclei; green, LDLR; red, EEA1. Scale bar, 1μm. (C) location of R-x-E motif in the amino acid sequence of each HLA member. mRNA levels of HLA protein and PCSK9 in (D) HepG2 naïve, (E) HepG2 CRISPR PCSK9 KO, (F) IHH, and (G) HepG2 CRISPR HLA-C KO, cells by qPCR experiment. mRNA values were normalized to the mRNA expression of the housekeeping gene (TATA box binding protein: TBP) to calculate the relative mRNA expression of each MHC-I member and compare it to the expression of PCSK9 and β2M. Data are representative of three independent experiments. Quantifications are averages ± standard deviation (SD). * p<0.05; ** p<0.01; ***p<0.001 (two-sided t-test). NS: non-significant.
Figure 1.
Extracellular importance of M2 domain of PCSK9 and mRNA expression of HLA members. (A) Immunofluorescence staining of total LDLR (permeabilized conditions) vs. cell surface LDLR (non-permeabilized conditions) levels in the presence of PCSK9-ΔM2 compared to WT PCSK9. (B) Immunostaining of total LDLR along with early endosomal (EEA1) marker in the presence of PCSK9 ΔM2, PCSK9 R549A-E567A (RE), and PCSK9 R549A-Q554E-E567A (RQE). Blue, Nuclei; green, LDLR; red, EEA1. Scale bar, 1μm. (C) location of R-x-E motif in the amino acid sequence of each HLA member. mRNA levels of HLA protein and PCSK9 in (D) HepG2 naïve, (E) HepG2 CRISPR PCSK9 KO, (F) IHH, and (G) HepG2 CRISPR HLA-C KO, cells by qPCR experiment. mRNA values were normalized to the mRNA expression of the housekeeping gene (TATA box binding protein: TBP) to calculate the relative mRNA expression of each MHC-I member and compare it to the expression of PCSK9 and β2M. Data are representative of three independent experiments. Quantifications are averages ± standard deviation (SD). * p<0.05; ** p<0.01; ***p<0.001 (two-sided t-test). NS: non-significant.
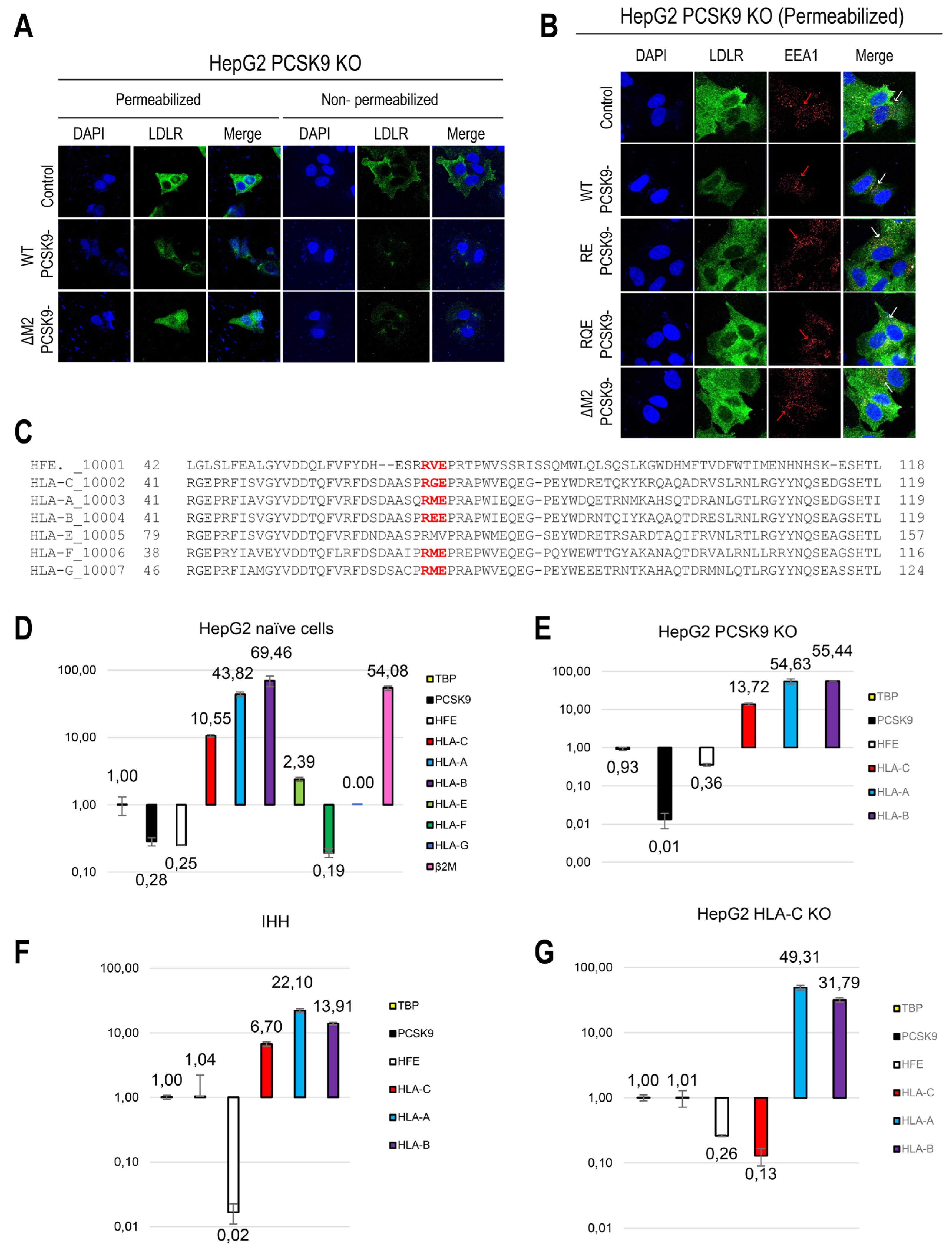
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



