
Submitted:
11 March 2024
Posted:
12 March 2024
You are already at the latest version
Abstract
Entamoeba histolytica, throughout its lifecycle, encounters a variety of stressful conditions. This parasite possesses Heat Shock Response Elements (HSEs) which are crucial for regulating the ex-pression of various genes, aiding in its adaptation and survival. These HSEs are regulated by Heat Shock Transcription Factors (EhHSTFs). Our research has identified seven such factors in this parasite, designated as EhHSTF1 through EhHSTF7. Significantly, under heat shock condi-tions and in the presence of the antiamoebic compound emetine, EhHSTF5, EhHSTF6, and EhHSTF7 show overexpression, highlighting their essential role in gene response to these stressors. Currently, only EhHSTF7 has been confirmed to recognize the HSE in the promoter of the EhPgp5 gene (HSE_EhPgp5), leaving the binding potential of the other EhHSTFs to HSE yet to be explored. Consequently, our study aimed to examine both in vitro and in silico the oligomeri-zation and binding capabilities of the recombinant EhHSTF5 protein (rEhHSTF5) to HSE_EhPgp5. The in vitro results indicate that the oligomerization of rEhHSTF5 is concentration-dependent, with its dimeric conformation showing a higher affinity for HSE_EhPgp5 than its monomeric state. In silico analysis suggests that the alpha 3 α-helix (α3-helix) of the DNA-binding domain (DBD5) of EhHSTF5 is crucial in binding to the major groove of HSE, primarily through hydro-gen bonding and salt-bridge interactions. In summary, our results highlight the importance of oligomerization in enhancing the affinity of rEhHSTF5 for HSE_EhPgp5 and demonstrate its abil-ity to specifically recognize structural motifs within HSE_EhPgp5. These insights significantly contribute to our understanding of one of the potential molecular mechanisms employed by the parasite to efficiently respond to various stressors, thereby enabling successful adaptation and survival within its host environment.
Keywords:
Entamoeba histolytica
; rEhHSTF5
; oligomerization
; monomer
; dimer
; trimer
; HSE_ EhPgp5
; EhDBD5
; molecular docking
1. Introduction
Entamoeba histolytica (E. histolytica), the causative agent of human amoebiasis, primarily infects the intestinal tract. However, it can also spread to other organs, giving rise to severe complications such as hepatic abscesses, pneumonia, purulent pericarditis, and in rare cases, cerebral amebiasis [1,2]. It is estimated that this pathogen is responsible for approximately 50 million infections worldwide annually, with a resultant morbidity that exceeds 100,000 deaths each year [3].
Throughout the course of host infection, the parasite encounters a myriad of stressful conditions, including acidic pH levels, the presence of noxious agents, engagement with the host immune defenses, drug exposure, heavy metal toxicity, hypoxia, nutrient scarcity, and thermal fluctuations, among others. Despite these hostile environments, the parasite demonstrates remarkable adaptive capabilities, enabling it to persist and induce amoebiasis [4,5,6,7]. Therefore, it is important to understand the molecular mechanisms that trophozoites employ to overcome stressful situations and ensure their survival.
In various organisms, including Homo sapiens (H. sapiens) [8], Saccharomyces cerevisiae (S. cerevisiae) [9], Drosophila melanogaster (D. melanogaster) [10], and Caenorhabditis elegans (C. elegans) [11], highly conserved Heat Shock Transcription Factors (HSTFs) have been identified. These HSTFs are crucial in regulating stress responses, thereby safeguarding cell survival under challenging conditions. These discoveries emphasize the evolutionary conservation of stress response mechanisms and underline the significance of HSTFs in the biology of various species.
HSTFs have a structurally conserved DNA-Binding Domain (DBD) featuring three alpha helices and four folded β-sheets, an Oligomerization Domain (OD) with a coiled-coil motif for dimer and trimer formation, and a Carboxyl-terminal Transactivation Domain (CTD). The transcriptional activity of HSTFs is intricately controlled by both intra- and intermolecular interactions, as well as by a variety of Post-translational Modifications (PTMs) [12,13].
In their inactive state, HSTFs are present as monomers, but they become active when they transition into trimers, a change facilitated by specific PTMs such as phosphorylation. These modifications allow HSTFs to bind to specific sequences known as Heat Shock Elements (HSE) in the promoters of their target genes during stressful conditions. This complex regulatory mechanism underscores the critical role of HSTFs in modulating cellular responses to stress and highlights their multifaceted function in maintaining cellular homeostasis [12,14].
According to the AmoebaDB database (https://amoebadb.org/amoeba/app, accessed on 15 December 2023) [15], the E. histolytica HSFs, referred to as EhHSTFs (Heat Shock Transcription Factors), until now, are considered the second largest family after those in plants. This family consists of 7 members, identified as Ehhstf1 (EHI_008230), Ehhstf2 (EHI_049510), Ehhstf3 (EHI_087630), Ehhstf4 (EHI_142120), Ehhstf5 (EHI_137000), Ehhstf6 (EHI_008660), and Ehhstf7 (EHI_200020). Currently, it has only been reported that the EhHSTF7 protein can oligomerize and recognize the HSE within the promoter region of the EhPgp5 gene (HSE_EhPgp5) [16]. The binding capabilities of the other EhHSTFs to HSE have not yet been analyzed in vitro or in silico. Therefore, the objective of this study was to investigate both the potential oligomerization and the DNA-binding affinity of the recombinant EhHSTF5 protein (rEhHSTF5) to the HSE_EhPgp5.
2. Results
2.1. The Oligomeric Conformations of rEhHSTF5 Protein Are Recognized by α6xHis and αEhHSTF5 Antibodies
The Open Reading Frame (ORF) of the Ehhstf5 gene was successfully inserted into the pET-28a(+) plasmid, confirmed through DNA sequencing and enzymatic restriction (Supplementary Figure S1). We then proceeded to induce the expression of the recombinant EhHSTF5 protein (rEhHSTF5) in Escherichia coli (E. coli) C41, as illustrated in Figure 1a. Induction assays revealed overexpression of different proteins with approximate molecular weights of 38, 47, 76 and 114 kDa at different induction times using isopropyl-β-D-thiogalactopyranoside (IPTG). Western blot (WB) assays with the α6xHis antibody recognized the 38 and 76 kDa proteins, while the αEhHSTF5 antibody detected all four proteins (Figure 1b).
These results indicate that the 38, 76 and 114 kDa proteins probably represent different oligomerization states of the rEhHSTF5 protein, corresponding to monomer, dimer, and trimer, respectively. The 47 kDa band may represent the protein in a transition state toward oligomerization. The monomeric conformation of recombinant EhHSTF5 protein (mrEhHSF5) was consistently observed at all induction times evaluated. It peaked at 3 h after induction and gradually decreased. In contrast, detection of the dimeric conformation of rEhHSTF5 (drEhHSTF5) was minimal at 3 h but increased markedly at 6, 9 and 24 h, whereas the trimeric conformation of rEhHSTF5 (trEhHSTF5) was uniquely identified by the αEhHSTF5 antibody after 24 h of induction with IPTG. This result suggests that increasing the protein induction time leads to a higher concentration in the cellular environment, consequently inducing oligomerization.
2.2. Purification of rEhHSTF5 Protein in Its Monomeric and Dimeric Conformations
The nickel affinity chromatography was employed to purify the rEhHSTF5 protein. When the total protein extract was processed through the nickel column, a significant decrease in the concentration of mrEhHSTF5 was observed in the column filtrate, suggesting a strong affinity for the column. Conversely, the dimeric and trimeric conformation of rEhHSTF5 showed only a slight reduction in the filtrate, indicating either a lower affinity for the column or possible saturation of the column by mrEhHSTF5, preventing their retention. During the elution process, a total of 60 fractions were collected. The dimeric conformation primarily eluted in fractions 7 to 12, while the monomeric conformation was found in fractions 24 to 26. However, trEhHSTF5 was not detected in these fractions (Figure 2a–d). To verify the identities of the purified proteins corresponding to the monomeric and dimeric conformations of rEhHSTF5, Western blot assays were conducted using α6xHis and αEhHSTF5 antibodies. These assays successfully demonstrated the immunodetection of both protein conformations, as depicted in Figure 2e,f.
2.3. The rEhHSTF5 Protein Undergoes Oligomerization
Crosslinking assays were performed using the protein predominantly in its monomeric conformation (fraction 25) to demonstrate the oligomerization capability of mrEhHSTF5. In the absence of glutaraldehyde, the mrEhHSTF5 protein was the most frequently observed, while drEhHSTF5 and trEhHSTF5 were detected with less intensity. However, with the addition of glutaraldehyde, a concentration-dependent decrease in the intensity of mrEhHSTF5 was noted. At higher concentrations of glutaraldehyde, drEhHSTF5 disappeared, and the intensity of trEhHSTF5 increased (Figure 3). The formation of a 47 kDa protein was observed only in samples with glutaraldehyde at various concentrations, supporting the hypothesis that rEhHSTF5 undergoes transitional states that enable it to oligomerize into dimeric and trimeric conformations, with approximate molecular weights of 76 and 114 kDa, respectively. These molecular weights are consistent with those of proteins detected by α6xHis and αEhHSTF5 antibodies in IPTG-induced bacteria.
2.4. The EhPgp5 Gene Promoter HSE Is Recognized by the rEhHSTF5 Protein
Electrophoretic Mobility Shift Assay (EMSA) experiments were conducted to assess whether the rEhHSTF5 can recognize the HSE_EhPgp5. For this, biotinylated synthetic double-stranded oligonucleotides containing HSE_EhPgp5 were incubated with the mrEhHSTF5 (fraction 25) and the drEhHSTF5 (fraction 10).
The experiments revealed an interesting outcome: drEhHSTF5, but not mrEhHSTF5, formed a DNA-protein complex under the same experimental conditions. To determine the specificity of the drEhHSTF5-HSE interaction, both specific and nonspecific competitors were used. The formation of the drEhHSTF5-HSE complex was reduced with the specific competitor, whereas it remained evident with Poly (dI-dC), as shown in Figure 4a. This indicates that the dimeric conformation of rEhHSTF5 has a higher and more specific affinity for the HSE sequence compared to its monomeric state.
Additionally, the use of the αEhHSTF5 antibody resulted in the formation of αEhHSTF5-drEhHSTF5-HSE_EhPgp5 supercomplexes, which migrated more slowly compared to the drEhHSTF5-HSE_EhPgp5 complex in the absence of the αEhHSTF5 antibody. Interestingly, by pre-incubating mrEhHSTF5 with the specific antibody αEhHSTF5 and subsequently with biotinylated HSE, the formation of the supercomplex αEhHSTF5-mrEhHSTF5-HSE_EhPgp5 was observed. It is noteworthy that no changes in the mobility of the complex (supershifts) were detected when nonspecific antibodies were used (Figure 4b). Furthermore, increasing the concentration of the drEhHSTF5 protein led to an enhancement in the formation of both αEhHSTF5-drEhHSTF5-HSE_EhPgp5 and drEhHSTF5-HSE_EhPgp5 complexes, as illustrated in Figure 4c. These results confirm the specific binding capability of the drEhHSTF5 factor to HSE_EhPgp5.
2.5. The DNA-Binding Domain of EhHSTF5 Has a Highly Conserved Three-Dimensional Structure
To predict the behavior and binding mechanisms of the rEhHSTF5 protein in its monomeric and dimeric conformations to HSE_EhPgp5, three-dimensional (3D) models were constructed for HSE_EhPgp5 (Figure 5a), and for the DBD of the monomeric (mEhDBD5, Figure 5b) and dimeric (dEhDBD5, Figure 5c) conformations of EhHSTF5. These models revealed that both mEhDBD5 and dEhDBD5 possess the typical and highly conserved structure of HSTFs, featuring three α-helices and four β-strands. Additionally, seven β-turns, one γ-turn, and two β-hairpins were identified in the structures.
Upon assessing the stability of the primary sequence of mEhDBD5, it was noted that the three α-helices and the four strands had normalized B-factor values less than or equal to 0. This suggests that these secondary structures are rigid and stable. Conversely, regions of the amino acid sequence that do not form well-defined secondary structures (such as coils) showed B-factor values greater than or equal to 0. This indicates that these areas are less stable due to their higher flexibility or mobility, which could influence the behavior of the protein and its binding properties.
2.6. The 3D Models of mEhDBD5 and dEhDBD5 Exhibit Satisfactory Structural Quality
The 3D models of the mEhDBD5 and dEhDBD5 demonstrated excellent stereochemical quality. This quality was confirmed by analyzing the Phi/Psi torsion angles of the peptide bonds through Ramachandran analysis. In this analysis, all amino acids were found in favored regions (100%), with none in disallowed regions (0%). Additionally, on the ProSA-web server, the Z-scores for mEhDBD5 and dEhDBD5 were −6.09 and −6.67, respectively. These scores fall within the typical range for similar structures determined through crystallography, indicating the reliability of the models. The energy-based evaluation of the models further reinforced their high quality, with values being ≤0. Further validation was conducted using ERRAT, which yielded an overall quality factor of 100 for mEhDBD5 and 87.08 for dEhDBD5. This suggests a high level of accuracy and reliability in the model structures. The 3D VERIFY analysis also supported these findings, indicating that 100% of the residues in mEhDBD5 and 84.59% in dEhDBD5 had a 3D−1D score of ≥ 0.1 (Figure 6).
In addition, a comprehensive structural quality assessment of the mEhDBD5 and dEhDBD5 models was conducted, focusing on key physicochemical and geometric characteristics. This evaluation included several quality indices, including Fractional Accessible Surface Area (FASA), Fractional Residual Volume (FRV), Stereo/Packed Quality Index (SPQI), and 3D Profile Quality Index (3DPQI). These indices were crucial in providing a detailed analysis of various aspects of the models, such as surface exposure, internal packing, stereochemical configuration and overall 3D conformation. It is important to highlight that both mEhDBD5 and dEhDBD5 obtained scores that are within the range suggested by the VADAR server, indicating an excellent structural quality of the 3D models obtained (Supplementary Figure S2).
2.7. dEhDBD5 Exhibits Higher Affinity for the HSE of the EhPgp5 Gene Promoter than mEhDBD5
Through blind molecular docking, both mEhDBD5 and dEhDBD5 successfully recognized HSE_EhPgp5 (Figure 7). The dEhDBD5 exhibited a higher binding energy compared to mEhDBD5, as indicated by more negative values. Docking scores for the mEhDBD5-HSE_EhPgp5 and dEhDBD5-HSE_EhPgp5 complexes were −203.29 and −305.79, respectively, with corresponding confidence scores of 0.74 and 0.95. The first dockings were selected due to the quality of their parameters.
Intermolecular analysis indicated that both mEhDBD5 and dEhDBD5 primarily bind to the major groove of HSE_EhPgp5 through the α3-helix. On the sense strand of HSE_EhPgp5, mEhDBD5 formed five hydrogen bond interactions and one salt bridge, while on the complementary strand, it engaged in seven hydrogen bond interactions and one salt bridge. In contrast, dEhDBD5 showed more extensive interactions: eight hydrogen bonds and four salt bridges on the sense strand, and nine hydrogen bonds and six salt bridges on the complementary strand (Supplementary Table S1).
In the mEhDBD5-HSE_EhPgp5 complex, the amino acids Arg 55 and Asn 58 interact with the adenine of the “GAA” motif in the sense strand, while Lys 92 forms a salt bridge with the phosphate group located between two adenines in “GAAA”. In the complementary strand, Ile 1 and Tyr 60 interact with an adenine of the “AAG” motif. In contrast, in the dEhDBD5-HSE_EhPgp5 complex, we identified that monomer 1 (in red) interacted in the sense strand with Gln 56 with the adenine “GAA”, Ser 48, Asn 49, Ser 52 with “GAA”, and His 47 formed a salt bridge with the phosphate group located between the adenines in “GAA”. Monomer 2 (in yellow) interacted in the complementary strand with Glu 66 with the cytosine of the “CTT”motif, Arg 55 with “AAG”, Ser 52 with “AAG”, and Gln 56 with “AAG”. We identified 3 salt bridges, Lys 64 between two adenines in “GAAA” in the sense strand, and in the complementary strand, Lys 64 between 2 adenines of the “AAG” motif, and His 47 with the phosphate group located between the adenine and guanine in “AAG” in the complementary strand (Figure 7a–c).
Interestingly, in the crystallized complexes of KlHSTF (PDB ID: 3HTS), HsHSTF1 (PDB ID: 5D5X), HsHSTF2 (PDB ID: 5D8L), and in the in silico predictions for the EhHSTF7-HSE_Ehpgp5 complex [16], the amino acids Arg, Ser, and Gln from α3-helix play a crucial role in mediating interactions with the “GAA” and “TTC” motifs (Figure 8c). Complementarily, upon aligning and generating a logo of the amino acids present in the α2-helix and α3-helix of EhDBD5 with 28 sequences of HSTF factors from diverse species, including the EhHSTF family, we found that α3-helix exhibits a high degree of conservation. Notably, amino acids such as Ser, Phe, Arg, Gln, Leu, Asn, Tyr, and Gly are prominently conserved (Figure 7d).
The analysis of the physicochemical properties of the complexes revealed that the dEhDBD5-HSE_EhPgp5, containing an additional monomer compared to the mEhDBD5-HSE_EhPgp5, demonstrated more significant aromatic interactions, hydrogen bonds, and charges. This complex also exhibited increased hydrophobicity and a higher ionization capacity. Furthermore, it possessed a more extensive solvent-exposed surface area than its monomeric counterpart, mEhDBD5-HSE_EhPgp5 (Supplementary Figure S3).
2.8. The Intermolecular Interaction Mechanism of the dEhDBD5-HSE Complex Is Highly Conserved
To provide a detailed comparison between the dEhDBD5-HSE_EhPgp5 complex and similar crystallographic complexes, the RaptorX server was utilized. This server offers two key parameters for comparison: the Structural Similarity Percentage (TMscore) and the Root Mean Square Deviation (RMSD). A TMscore value of 60% or higher indicates a significant similarity in conformation and structure between compared complexes, while RMSD measures the proximity of superimposed structures in three-dimensional space.
Our in silico analysis revealed that the dEhDBD5-HSE_EhPgp5 complex displayed TMscore values of 81% or greater and RMSDs of 1.4 or lower when compared with crystallographic complexes of K. lactis, S. cerevisiae-C. thermophilum (chimera), H. sapiens-C. thermophilum (chimera), synthetic H. sapiens, and native H. sapiens, as shown in Figure 8b–f. Upon overlaying all these structures, an average TMscore of 89.8% and an RMSD of 1.17 were observed. This suggests a high degree of structural conservation and indicates functional conservation, as evidenced by the similar spatial conformation when binding to HSEs (Figure 8g). A particularly noteworthy finding emerged when only the α2-helix and α3-helix of the DBDs were superimposed, resulting in an average TMscore exceeding 85.5% (Figure 8h). Additionally, upon comparing the primary amino acid sequences of these 3D structures, the α2-helix and α3-helix of EhDBD5 demonstrated identity and homology percentages above 73% in comparison with the evaluated crystallographic structures. This further emphasizes the significant structural and functional conservation of these domains, particularly in their roles related to DNA binding and transcriptional regulation.
Analysis of helix α2 and helix α3 in EhDBD5 at the primary sequence level showed that their identity and homology was 73% and 76% or higher, respectively, compared to the same helices in the five evaluated crystallographic structures. Additionally, at the 3D structural level, these α-helices in EhDBD5 exhibited TM scores of 73.5 or higher, suggesting a conserved folding pattern. This result also underscores the high conservation of amino acids such as Asn, Ser, Phe, Val, Arg, Gln, Leu, and Tyr in α3-helix (Figure 8i).
3. Discussion
In this study, we demonstrated that the rEhHSTF5 protein undergoes oligomerization, forming dimers and trimers in protein extracts from bacteria harboring the recombinant protein. Induction with IPTG for 3 h enhances the formation of mrEhHSTF5, while induction at 24 h favors the formation of drEhHSTF5 and trEhHSTF5, suggesting that oligomerization is concentration-dependent. Protein oligomerization has been documented in HSTFs from various species. HSTF1[8], HSTF2 [17], and HSTF4 [18] from H. sapiens; HSTF1 from A. thaliana [19], HSTF1 from D. melanogaster [20], and EhHSTF7 from E. histolytica [16], among others. These transcription factors tend to oligomerize in cellular environments with elevated protein concentrations, while remaining as monomers at lower concentrations.
On the other hand, the 47 kDa protein expressed during IPTG induction, which was recognized by the αEhHSTF5 antibody, was also observed upon addition of glutaraldehyde to the purified mrEhHSTF5 protein (fraction 25), suggesting that the 47 kDa protein may represent a stable conformation. This conformation could be an important transient state for dimer and trimer formation, which allows the interaction and coordinated assembly of the subunits, maintaining the stability of the oligomeric structure as occurs in different proteins [21,22], including Superoxide Dismutase 1 (SOD1)[22], or Heat Shock Protein 90 (HSP90) [23], among others.
In WB assays, we found that the oligomerization of the mrEhHSTF5 protein can hide the polyhistidine tag, which makes it difficult to detect using the α6xHis antibody in induced bacteria extracts. Furthermore, after conducting nickel-affinity chromatography, we were unsuccessful in purifying the 114 kDa protein. This reinforces the idea that the polyhistidine region is hidden, making it difficult to bind to the nickel column. This observation is consistent with the behavior of the rEhHSTF7 protein reported by Bello et al. [16], where, under similar experimental conditions, they were unable neither to detect the trimeric conformation of the protein using the α6xHis antibody nor to purify it through nickel affinity purification. Moreover, it has been previously demonstrated that the oligomerization of recombinant proteins can alter the accessibility of polyhistidine tags, potentially affecting their binding capacity to affinity matrices[24,25,26].
HSTFs are recognized for their role in the heat stress response and their ability to bind to HSEs to regulate gene expression [27,28,29]. The oligomerization and stability of these factors are of vital importance for their proper function, as their ability to bind to HSE and activate transcription depends on the formation of stable oligomeric structures. HSTF1 and HSTF2 from H. sapiens form homotrimers or heterotrimers with other HSTFs. This oligomer formation increases the structural stability and their affinity for HSE, resulting in an increased ability to regulate gene expression. Therefore, oligomerization is a critical factor to achieve the necessary stability to recognize HSE [30,31,32]. This fact is in concordance with our EMSA assays demonstrating that the dimeric conformation of the rEhHSTF5 protein exhibits a higher affinity for binding to the HSE_EhPgp5. In contrast, the monomeric conformation under the same experimental conditions did not bind with the HSE_EhPgp5, confirming the necessity of oligomerization to achieve greater stability and, consequently, higher affinity to the HSE_EhPgp5. Supershift assays validated the EhHSTF5-HSE interaction, forming the αEhHSTF5-drEhHSTF5-HSE supercomplex.
The binding of mrEhHSTF5 to the HSE_EhPgp5 was not detectable by EMSA assays. However, when the protein was preincubated with the αEhHSTF5 antibody, the formation of a highly specific supercomplex was observed. These results suggest that the antibody binding to mrEhHSTF5 induces a conformational change that exposes the DBD and, consequently, enables the binding to the HSE_EhPgp5. A similar finding was previously reported by Zimarino et al. [33], who found that the binding activity to the HSE by the inactive form of the HSTF from D. melanogaster could be induced in vitro by the addition of a polyclonal antibody against the purified factor; proposing that binding of an anti-HSTF antibody led to a conformational change, exposing its DBD to activate its transcriptional capability. Moreover, the studies performed with HsHSTF1 showed that its DBD is hidden when it is in a monomeric conformation, but is exposed when it is oligomerized, which allows it to bind more easily to HSE [28,30,34]. Similarly, we suggest the possibility that EhDBD5 is hidden in the monomeric conformation of EhHSTF5 and is exposed upon oligomerization or antibody binding, which would more effectively facilitate interaction with HSE_EhPgp5.
In silico assays is an important tool for understanding protein activity and molecular mechanisms involved [35].
Through our in silico blind molecular docking studies, we identified that mEhDBD5 and dEhDBD5 exhibit binding to HSE_EhPgp5. Notably, dEhDBD5 shows significantly higher binding affinity (−305.79) compared to mEhDBD5 (−203.29).
We previously performed molecular docking under the same conditions as in this study, focusing on the trimeric conformation (tEhDBD5) of the EhHSTF5 protein and discovered its binding to the same HSE with a Docking score of −459 [36]. This clearly indicates that oligomerization of EhHSTF5 into dimers and trimers progressively improves its stability and affinity towards HSE_EhPgp5. This behavior has also been demonstrated in other factors such as KlHSTF [37], HsHSTF1 [38], and HsHSTF2 [39], among others.
We identified that α3-helix plays a crucial role in the interaction of the DBD domain with the major groove of the HSE, especially in the structural motifs “GAA” and “TTC”, through hydrogen bonds and salt bridges. These results are in agreement with various crystallized complexes (HSTF-HSE) where it has been observed that α3-helix is responsible for direct contact with the major groove of the HSE. In the KlHSTF-HSE complex, for example, amino acids involved in recognizing the structural motifs 5´-TTC-3´, 3´-AAG-5´, and 3´-CTT-5´ are Arg 250, Ser 247, His 242, Gln 252, and Lys 259 [37]. In the HsHSTF1-HSE crystallographic complex, it has been observed that Arg 71, Ser 68, and Gln 72 recognize the structural motifs 5´-GAA-3´ and 3´-CTT-5´ [38], while in the HsHSTF2-HSE complex, Arg 109, Arg 63, Ser 60, and Lys 72 participate in recognizing the motifs 5´-TTC-3´, 5´-GAA-3´, and 3´-CTT-5´[39].
Notably, during the analysis of sequences comprising α3-helix, including EhDBD5, high conservation of amino acids such as Ser, Arg, Gln, Asn, Tyr, and Gly were observed in several species, suggesting their significant relevance in HSE binding. In fact, it has been reported that mutation of amino acids Ser 247, Arg 250, and Asn 253 in α3-helix of K. lactis leads to a lethal phenotype, further underlining the importance of these amino acids in HSTFs [37].
An important aspect in the interaction between HSTFs and HSEs is that certain amino acids can interact with the phosphate group of the HSE backbone through salt bridges. This is the case for amino acids Ser 68, Arg 71, and Gln 72 in the HsHSTF1-HSE complex, and Lys 72, Asn 57, Ser 60, and Lys 53 in the HsHSTF2-HSE complex [38]. This interaction enhances the stability of the DBD, facilitating correct binding of α3-helix to HSE. In our study, we observed a similar interaction mechanism mediated by salt bridges. We identified the presence of two salt bridges in the mEhDBD5_HSE_EhPgp5 complex, and ten in the dEhDBD5_HSE_EhPgp5 complex. Hydrogen bonds are also crucial for the stability of the complexes. Interestingly, we observed 12 intermolecular interactions in mEhDBD5_HSE_EhPgp5 and 17 in dEhDBD5_HSE_EhPgp5 (Supplementary Table S1). This suggests that the dimeric conformation, by forming a higher number of salt bridges and hydrogen bonds during its binding to HSE, might be promoting greater stability compared to the monomeric conformation.
Consistently, upon evaluating the physicochemical properties, we observed that the dEhDBD5-HSE_EhPgp5 complex, compared to the mEhDBD5-HSE_EhPgp5 complex, exhibits more aromatic interactions, indicating a higher affinity at the binding interface. It also displays a stronger electric charge, suggesting more robust ionic interactions. Additionally, the complex is more hydrophobic, which enhances its structural stability. Notably, the ionization capacity implies diverse electrostatic interactions. The increased exposure to solvent suggests a more intricate structure and higher solubility. These features indicate a complex network of intramolecular and intermolecular interactions in the dimeric complex, potentially contributing to its greater stability compared to the monomeric conformation, as has been documented in numerous studies for different proteins [40,41,42,43]
A study that highlights a similar functional and structural behavior to rEhHSTF5 is the report by Bello et al. [16] which revealed that the rEhHSTF7 factor can oligomerize in a concentration-dependent manner within total extracts of C41 bacteria, and this oligomerization mechanism was verified by αEhHSTF7 and α6xHis antibodies. In addition, successful purification of the protein in its oligomeric states has been achieved through nickel affinity chromatography. Analogously to the current study, EMSA and supershift assays have demonstrated that the oligomeric form of E. histolytica rEhHSTF7 can recognize the HSE_EhPgp5 gene. It is interesting to observe that through in silico tools, it was confirmed that α3-helix also recognizes the 3´-AAG-5´ motif, primarily through the amino acids Arg 72 and Ser 69. Moreover, it has been shown that the expression of the EhPgp5 gene decreases when a specific siRNA is used to knockdown the expression of the Ehhstf7 gene in E. histolytica trophozoites growth with 8 µM of Emetine, underlining the significance of transcriptional modulation by this factor under stress conditions. It is worth highlighting that the EhPGP5 protein of E. histolytica is recognized as a multidrug resistance protein, acting as a pump to expel drugs, and enhancing amoebic survival when exposed to amoebicidal drugs. In this work, we observed that oligomeric rEhHSTF5 can recognize the HSE_EhPgp5 gene similarly to rEhHSTF7, suggesting that both factors could collaborate in regulating the gene transcription of EhPgp5 and, consequently, help the survival of the amoeba by responding to stressors present in its environment.
It is important to emphasize the capability of the rEhHSTF5 factor to recognize the structural motifs “GAA” and “TTC” within the HSE, which expands the potential for recognition not only of the HSE_EhPgp5 gene [16,44], but also, under specific conditions, the HSEs present in other promoters regulated by this element, such as the seven HSEs identified in EhrabB [45], three in Ehhsp100 [46], and four in the Ehmlbp gene promoter [47]. These proteins are of great significance for the parasite, as EhRABB plays a pivotal role in cytoskeleton regulation, cellular motility, potential cyst formation, invasion, and parasitic pathogenicity [45,48,49,50,51]. EhHSP100 is involved in amoeba response to heat and oxidative stress, protection against cell damage and modulation of pathogenicity [46]. Furthermore, EhMLBP is a mRNA-binding protein known to interact specifically with the 3' untranslated region of mRNAs of various genes of the parasite, suggesting its potential role in posttranscriptional regulation and messenger RNA stability [47,52,53,54].
Recently, Dorantes et al. [36] conducted an in silico analysis of HSEs located in the promoter regions (ranging from −500 to +50 bp) of the 8,343 genes present in the E. histolytica genome. This analysis revealed the presence of 2578 HSEs in total, of which 1412 HSEs were in the promoter regions of 1010 hypothetical genes, and 1,166 HSEs in the promoter regions of 957 coding genes. Remarkably, 24% of the genes could potentially be regulated by HSEs. Furthermore, it was observed that these HSEs are situated in promoters of genes associated with various functions, including ATP-dependent activity, binding, catalytic activity, cytoskeletal motor activity, molecular adaptor activity, molecular function regulator, molecular transducer activity, structural molecule activity, transcription regulator activity, translation regulator activity, and transporter activity, among others.
These findings further underscore the functional importance of the EhHSTF5 factor as a potential regulator of various genes that harbor HSEs in their promoters in E. histolytica. This ability could allow the parasite to dynamically adapt to its environment, thereby ensuring its survival across diverse physiological conditions.
Current in vitro experiments are underway to evaluate the significance of the “GAA” and “TTC” motifs. Our approach involves targeted manipulation and mutation of these nucleotides within the HSE_EhPgp5 sequence to understand their impact on binding affinity with rEhHSTF5. Furthermore, conducting assays on E. histolytica trophozoites will deepen our comprehension of how oligomerization affects the affinity of EhHSTF5 for various HSEs, particularly under stress conditions. Simultaneously, we will proceed to evaluate the relevance of EhHSTF5 in the parasite by specifically blocking its expression through siRNAs.
In summary, as illustrated in Figure 9, our findings indicate that the mrEhHSTF5 protein, with a molecular weight of 38 kDa, can form dimeric (76 kDa) and trimeric (114 kDa) structures. This oligomerization is crucial, as it increases the stability of the protein, which translates into a higher affinity for HSE_EhPgp5.
4. Materials and Methods
4.1. Cloning of the Ehhstf5 Gene and Expression of the rEhHSTF5 Protein
The Ehhstf5 gene was cloned into the pET-28a(+) plasmid by GenScript (Piscataway, NJ, USA). The gene was flanked by the restriction sites of the BamHI and XhoI enzymes at the 5' and 3' ends, respectively. Additionally, a 6-histidine tag was incorporated at the 5' end of the plasmid. The insertion of the Ehhstf5 gene was confirmed through gene amplification. The forward primer (5'-ATCCGATCGGATGAGTGAACCACACAAACA-3') and reverse primer (5'-CGACTCGACTTTAAAACTTCCATGGAATTT-3') oligonucleotides were employed. The amplification process started with an initial denaturation step at 94°C for 5 min, followed by 30 cycles. Each cycle involved denaturation at 94°C for 1 min, primer annealing at 49.4°C for 1 min and 30 s, and extension of the new strand at 72°C for 1 min. Following the cycles, there was an incubation at 72°C for 7 min to conclude the amplification process. The resulting PCR product was sequenced using the ABI PRISM 3130 sequencer (Thermo Fisher Scientific, Waltham, MA, USA).
The constructed plasmid pET-28a(+)-Ehhstf5 was used to transform competent cells of E. coli C41 strain (Sigma-Aldrich, San Luis, MO, USA). Expression of the recombinant rEhHSTF5 protein was induced using 1 mM Isopropyl-beta-D-thiogalactopyranoside (IPTG) (Thermo Fisher Scientific, Waltham, MA, USA) with agitation at 220 rpm in SOC medium for 24 h at 37°C. The progress of recombinant protein induction was observed at 3, 6, 9, and 24 h. Resulting bacterial cells were resuspended in lysis buffer (20 mM Tris-HCl, pH 8, 1 mg/ml lysozyme, and 1% Triton X-100) and then lysed at 4°C by sonication using an Ultrasonic Processor equipment (MRC, laboratory instruments, Harlow, ESX, UK) at 60% amplitude. Five cycles of 15 s were applied with 15 s rest intervals. The obtained soluble proteins were quantified using the Bradford method [55]. Integrity and expression of the rEhHSTF5 protein were assessed through 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) using 20 μg of protein.
4.2. Immunodetection of rEhHSTF5 Protein by Western Blotting
The proteins separated by electrophoresis were transferred to PVDF membranes with 0.45 μm pores (Sigma-Aldrich, Basel, Switzerland). The membranes were incubated for 30 min with the primary antibody anti-6His (α6xHis) (Sigma-Aldrich, Basel, Switzerland) at a dilution of 1:3000, or with a polyclonal antibody anti-EhHSTF5 (αEhHSTF5) at a dilution of 1:1000, generated in Balb/c mice by SPIDDSNNVELP (GLBiochem, Shanghai, China) EhHSTF5 peptide immunization. The membranes were subsequently incubated for 30 min with the secondary antibody goat anti-mouse IgG (H+L) conjugated to horseradish peroxidase (Thermo Fisher Scientific, Waltham, MA, USA) at a dilution of 1:20,000. Detection was performed through chemiluminescence using Immobilon™ Western chemiluminescent HRP substrate (Millipore, Burlington, MA, USA).
4.3. Purification of Recombinant Protein rEhHSTF5
Bacteria containing the overexpressed rEhHSTF5 protein were suspended in buffer I (20 mM Tris-HCl, 500 mM NaCl, and 25 mM Imidazole, pH 8.5) and lysed by sonication. The soluble fraction was obtained through centrifugation at 20,000 xg for 45 min at 4°C, followed by filtration using 0.22 μm pore size filters. The purification of the recombinant protein was conducted through nickel affinity chromatography using the ÄKTA pure™ chromatography system (Cytiva, Marlborough, MA, USA). Briefly, the protein lysate was loaded onto a 5 mL HisTrap FF column (Cytiva, Marlborough, MA, USA), which was subsequently washed with 5 column volumes (25 ml) of buffer I, and the protein was eluted from the column with buffer II (20 mM Tris-HCl, 500 mM NaCl, 500 mM Imidazole, pH 8.5). The purification of the rEhHSTF5 protein was evaluated by Western Blot following the methodology described previously.
4.4. Oligomerization States of the mrEhHSTF5 Protein
The oligomerization of the monomeric rEhHSTF5 protein (mrEhHSTF5) was assessed by incubating 20 µg of purified protein at 25°C in the presence of glutaraldehyde (Sigma-Aldrich, St. Louis, MO, USA) at concentrations of 0.5%, 1.5%, and 2.5% for 15 min. The oligomerization reaction was stopped by adding 10 µl of a loading buffer (2X) containing 100 mM Tris-Cl at pH 6.8, 4% SDS (sodium dodecyl sulfate; electrophoresis-grade), 0.2% bromophenol blue, 20% glycerol, 200 mM DTT (dithiothreitol), and 200 mM β-mercaptoethanol. Oligomerization states were evaluated through 12% SDS-PAGE gel electrophoresis.
4.5. Biotinylation and Hybridization of the HSE_EhPgp5 Sequence
The sense (5'-ATAGAAATTTTTCATA-3') and antisense (3'-TATCTTTAAAAAGTAT-5') oligonucleotides of the HSE_EhPgp5 were synthesized at Integrated DNA Technologies (IDT) (Coralville, IA, USA). The 3' end of the single-stranded oligonucleotides was biotinylated using the Biotin 3' End DNA Labeling Kit (Thermo Fisher Scientific, Waltham, MA, USA), following the manufacturer's instructions. Subsequently, the oligonucleotides were hybridized in equimolar ratios in an Axygen-Maxigene thermal cycler (Thermo Fisher Scientific, Waltham, MA, USA) at 95°C for 5 min, followed by sequential descending incubations from 10°C to 25°C, holding each temperature for one min.
4.6. Electrophoretic Mobility Shift Assays (EMSA)
The interaction between the rEhHSTF5 factor and HSE_EhPgp5 was evaluated using 0.7 μM of monomeric protein (mrEhHSTF5) or dimeric protein (drEhHSTF5). The proteins were incubated for 30 min at room temperature with the components of the Lightshift EMSA kit (Thermo Fisher Scientific, Waltham, MA, USA), including 2 µl of binding buffer, 1 µl of Poly (dI-dC) (50 ng/µl), 1 µl of glycerol (50%), 1 µl of MgCl2 (100 mM), and 300 µg of BSA. For competition assays, a non-biotinylated probe (specific competitor) or Poly (dI-dC) was used as an unspecific competitor at molar excesses of 150 and 350 times, respectively. Prior to incubation with the binding mixture and the biotinylated probe, competitors were preincubated for 30 min at 4°C with mrEhHSTF5 and drEhHSTF5. The samples were subjected to electrophoresis on non-denaturing 0.5X-TBE polyacrylamide gels and then transferred to PVDF membranes (Sigma-Aldrich, Basel, Switzerland). The formation of the rEhHSTF5-HSE complex was detected through chemiluminescence using the provided kit solution.
4.7. Electrophoretic Mobility Supershift Assay
The αEhHSTF5 antibody was preincubated at a 1:3000 dilution with mrEhHSTF5 or drEhHSTF5 protein for 30 min. This was followed by another 30 min incubation with the binding mixture and biotinylated probe under the same conditions. Preimmune serum (serum from mice not immunized with the immunogenic peptide of EhHSTF5) or an unrelated commercial antibody (αGAPDH, 1:3000) were used as controls. The procedure for detecting complexes was carried out following the EMSA conditions described previously. Furthermore, a kinetics study involving an increase in drEhHSTF5 concentration (from 0.7 to 2.8 µM) was conducted to confirm the consistency and correspondence of the formed complexes with the drEhHSTF5-HSE_EhPgp5 complex.
4.8. Three-Dimensional Modeling of the HSE_EhPgp5
We utilized UCSF Chimera version 1.15 software (San Francisco, CA, USA) to construct the three-dimensional model of HSE_EhPgp5, using the sense sequence 5'-ATAGAAATTTTTCATA-3' and antisense sequence 3'-TATCTTTAAAAAGTAT-5'. The resulting double-stranded structure in conformation b was saved in .pdb format for subsequent bioinformatics analysis.
4.9. 3D Modeling, Structural Validation, and Stability Analysis of mEhDBD5 and dEhDBD5
The amino acid sequence 46-137, corresponding to the DNA-binding domain (DBD) of the EhHSTF5 factor (EhDBD5), was obtained from the AmoebaDB platform (https://amoebadb.org/amoeba/app, accessed on 2 November 2023) with the ID EHI_137000 to generate the 3D models of the DNA-binding domain in monomeric (mEhDBD5) and dimeric (dEhDBD5) conformations. The SWISS-MODEL server (https://swissmodel.expasy.org/, accessed on 2 November 2023) was utilized to model the 3D structures of mEhDBD5 and dEhDBD5, using the DBD of human HSTF2 in complex with HSE as a template, deposited with the ID 5D8L in the Protein Data Bank (PDB) (https://www.rcsb.org, accessed on 2 November 2023). The obtained models were structurally validated using various servers, including PDBsum for Ramachandran analysis (http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/GetPage.pl?pdbcode=index.html, accessed on 5 November 2023), ProSA-web (https://prosa.services.came.sbg.ac.at/prosa.php, accessed on 10 November 2023), ERRAT and VERIFY 3D contained in UCLA-DOE LAB-SAVES v6.0 (https://saves.mbi.ucla.edu, accessed on 8 December 2023), and VADAR (http://vadar.wishartlab.com/index.html?, accessed on 10 December 2023). Additionally, the structural stability of mEhDBD5 was predicted using the normalized B-factor or temperature factor calculated by the I-Tasser server (https://zhanggroup.org/I-TASSER/, accessed on 15 December 2023).
4.10. In Silico Molecular Docking
Blind molecular docking simulations were conducted using the HDOCK server (http://hdock.phys.hust.edu.cn/, accessed on 20 December 2023) to assess the intermolecular interactions between HSE_EhPgp5 and the mEhDBD5 and dEhDBD5 domains of the EhHSTF5 factor. During this process, the .pdb file corresponding to HSE_EhPgp5 was used as ligand, while the mEhDBD5 and dEhDBD5 domains were used as receptors. The selection of dockings was based on the most negative values obtained in the Docking Score, representing binding affinity. Additionally, selections were made based on the Confidence score.
4.11. Analysis of the Physicochemical Properties of the mEhDBD5-HSE_EhPgp5 and dEhDBD5-HSE_EhPgp5 Complexes
The obtained complexes, mEhDBD5-HSE_EhPgp5 and dEhDBD5-HSE_EhPgp5, were subjected to a comprehensive analysis of physicochemical properties. This analysis covered Aromatics, H-bonds, Charge, Hydrophobicity, Ionizability, and Solvent Accessible Surface (SAS) and was conducted using BIOVIA Discovery Studio, version v.21 (San Diego, CA, USA).
4.12. In Silico Analysis of Intermolecular Interactions in the EhDBD5-HSE_EhPgp5 Complex
To identify the intermolecular interactions within the mEhDBD5-HSE_EhPgp5 and dEhDBD5-HSE_EhPgp5 complexes, the Protein-Ligand Interaction Profiler (PLIP) server (https://plip-tool.biotec.tu-dresden.de/plip-web/plip/index, accessed on 29 December 2023) was used. Interactions were assessed with a bond length cutoff of 4 Å, following the recommendations of the server.
4.13. Structural Similarity of 3D Proteins
The RaptorX server (http://raptorx6.uchicago.edu/, accessed on 2 January 2024) was used to perform 3D structure superposition and obtain structural similarity and root mean square error (RMSD). Among the structures deposited on the PDB platform used for the superposition, the DBD of HSTF in complex with an HSE was used, including that of Kluyveromyces lactis (K. lactis) (ID; 3HTS), Saccharomyces cerevisiae-Chaetomium thermophilum (S. cerevisiae-C. thermophilum, chimera) (ID; 5D5X), Homo sapiens-Chaetomium thermophilum (H. sapiens-C. thermophilum, chimera) (ID; 5D5W), H. sapiens (ID; 7DCU) and the synthetic H. sapiens construct (ID; 5D8L). We were also able to obtain only DBD α2-helix and α3-helix in .pdb format from these structures, which were used to calculate the percentage of structural similarity.
4.14. Conservation of DBD α2-Helix and α3-Helix
To evaluate the conservation degree of α-helices 2 and 3 within the DBDs of HSTFs, an amino acid sequence analysis was conducted using the Clustal Omega platform (https://www.ebi.ac.uk/jdispatcher/msa/clustalo, accessed on 6 November 2023). This analysis included the sequences of the seven EhHSTFs of E. histolytica previously mentioned, as well as sequences obtained from the National Center for Biotechnology Information platform (NCBI) (https://www.ncbi.nlm.nih.gov/, accessed on 6 November 2023) of HSTF1, HSTF2 and HSTF4 from H. sapiens (HsHSTF1, 2 and 4 , IDs: 3297, 3298 and 3299), as well as sequences from Mus musculus (MmHSTF1, ID: 15499), S. cerevisiae (ScHSTF1, ID: 1322586), K. lactis (KlHSTF1, ID: 1751387658), Drosophila melanogaster (DmHSTF1, ID: 37068), Caenorhabditis elegans (CeHSTF1, ID: 45643032), Gallus gallus (GgHSTF1, ID: 76523668686), Arabidopsis thaliana (AtHSTF1, ID: 827496), Danio rerio (DrHSTF1, ID: 134026298), Xenopus laevis (XlHSTF1, ID: 148222464), Pteropus vampyrus (PvHSTF1, ID: 759162997), Sus scrofa (SsHSTF1, ID: 100511321), Brugia malayi (BmHSTF1, ID: CRZ24125. 1), Dasypus novemcinctus (DnHSTF1, ID: 101424131), Cavia porcellus (CpHSTF1, ID: 101787582), Phascolarctos cinereus (PcHSTF1, ID: 1190400766), Pan troglodytes (PtHSTF1, ID: 741396), Bos taurus (BtHSTF1, ID: 506235) and Capra hircus (ChHSTF1, ID: 102178552).
Simultaneously, amino acid sequences were extracted from the crystals deposited in the PDB database for K. lactis (ID: 3HTS), S. cerevisiae-C. thermophilum (ID: 5D5X), H. sapiens-C. thermophilum (ID: 5D5W), synthetic H. sapiens construct (ID: 5D8L), and H. sapiens (ID: 7DCU) to perform a sequence alignment and determine the percentage of identity and homology.
Complementarily, logos were constructed using the amino acids from the aforementioned sequences through the WebLogo platform (https://weblogo.berkeley.edu/logo.cgi, accessed on 7 November 2023).
4.15. 3D Structure Viewer
PyMol, version 2.5.0, (Nueva York, NY, USA) and BIOVIA Discovery Studio were used to visualize and generate images for different bioinformatics analyses.
4.16. Relative Expression and Graphics
The ImageJ software, version 1.54, (Bethesda, MD, USA) was used to quantify the relative protein expression through pixel analysis. For the statistical analysis, a two-way ANOVA (Analysis of Variance) was utilized, followed by Tukey's multiple comparison test, in accordance with the American Psychological Association (APA) format for adjusted p-value analysis, detailed as follows: *p ≤ 0.033, **p ≤ 0.002, and ***p ≤ 0.001. The data are expressed as the mean ± standard deviation, derived from a minimum of three independent experiments. For graph creation, the GraphPad Prism software, version 8.0.1 (Boston, MA, USA), was used, allowing for a precise and detailed graphical representation of the results.
5. Conclusion
The oligomerization of the rEhHSTF5 factor is a crucial process that enhances its affinity for recognizing the HSE of the E. histolytica EhPgp5 gene. The specific ability of drEhHSTF5 to identify the structural motifs “GAA” and “TTC” within the HSE suggests its potential role in regulating the expression not only of this gene, but also of others that have HSEs in their promoters and that participate in fundamental processes for the parasite, including developmental processes, growth, metabolism, infection, and response to different types of stress, among others. Therefore, EhHSTF5 may play a fundamental role in the adaptation of the parasite to its environment and contribute to its survival.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, S.P.-M., M.d.C.G.-G, and D.G.P.-I.; methodology, S.P.-M., M.d.C.G.-G., D.G.P.-I., S.V.S-H., M.O.M-F., C.A.R-L., M.A. R., and V. S-M.; software, S.P.-M.; S.V.S-H., and C.A.R-L.; validation, D.G.P.-I., and M.d.C.G.-G.; formal analysis, S.P.-M., D.G.P.-I., M.d.C.G.-G., C.A.R-L., and M.A. R.; investigation, S.P.-M., M.d.C.G.-G., and D.G.P.-I.; resources, M.d.C.G.-G., and D.G.P.-I.; writing—original draft preparation, S.P.-M.; writing—review and editing, S.P.-M., M.d.C.G.-G., D.G.P.-I., C.A.R-L., and M.A. R.; visualization, S.P.-M., S.V.S-H., M.d.C.G.-G., D.G.P.-I., C.A.R-L., and M.A. R.; supervision, M.d.C.G.-G., and D.G.P.-I.; project administration, M.d.C.G.-G., D.G.P.-I., and S.P.-M.; funding acquisition, M.d.C.G.-G., and D.G.P.-I. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Instituto Politécnico Nacional, with project grants provided by the Secretaría de Investigación y Posgrado (SIP) to M.d.C.G.-G in 2021 (20211325), 2022 (20220709), and 2023 (20230880) and to D.G.P.-I in 2023 (20230881).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Acknowledgments
To CONAHCYT for their invaluable contribution through the postgraduate scholar-ship granted to S.P.-M, and S.V.S-H.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Salles, J.M.; Moraes, L.A.; Salles, M.C. Hepatic Amebiasis. Braz. J. Infect. Dis. Off. Publ. Braz. Soc. Infect. Dis. 2003, 7, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Varghese, V.; Kansal, A.; Bhardwaj, S.; Sharma, A. Cerebral Amebiasis: An Uncommon Cerebral Abscess. Ann. Indian Acad. Neurol. 2021, 24, 445–447. [Google Scholar] [CrossRef]
- Chou, A.; Austin, R.L. Entamoeba Histolytica Infection. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Pineda, E.; Perdomo, D. Entamoeba Histolytica under Oxidative Stress: What Countermeasure Mechanisms Are in Place? Cells 2017, 6, 44. [Google Scholar] [CrossRef] [PubMed]
- Uribe, E.; Rosales, C. Immune Response to the Enteric Parasite Entamoeba histolytica. Physiology (Bethesda) 2020, 35, 244–260. [Google Scholar] [CrossRef]
- Guillén, N. Pathogenicity and Virulence of Entamoeba Histolytica, the Agent of Amoebiasis. Virulence 2023, 14, 2158656. [Google Scholar] [CrossRef] [PubMed]
- Ankri, S. Entamoeba Histolytica—Gut Microbiota Interaction: More Than Meets the Eye. Microorganisms 2021, 9, 581. [Google Scholar] [CrossRef] [PubMed]
- Hentze, N.; Le Breton, L.; Wiesner, J.; Kempf, G.; Mayer, M.P. Molecular Mechanism of Thermosensory Function of Human Heat Shock Transcription Factor Hsf1. eLife 2016, 5, e11576. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Liu, L.; Sun, L.; Lin, X.; Lin, L.; Zhang, C. Genome-Wide Analysis Reveals Hsf1 Maintains High Transcript Abundance of Target Genes Controlled by Strong Constitutive Promoter in Saccharomyces Cerevisiae. Biotechnol. Biofuels Bioprod. 2023, 16, 72. [Google Scholar] [CrossRef]
- Jedlicka, P.; Mortin, M.A.; Wu, C. Multiple Functions of Drosophila Heat Shock Transcription Factor in Vivo. EMBO J. 1997, 16, 2452–2462. [Google Scholar] [CrossRef]
- Morton, E.A.; Lamitina, T. C. Elegans HSF-1 Is an Essential Nuclear Protein That Forms Stress Granule-like Structures Following Heat Shock. Aging Cell 2013, 12, 112–120. [Google Scholar] [CrossRef]
- Ota, A.; Enoki, Y.; Yamamoto, N.; Sawai, M.; Sakurai, H. Evolutionarily Conserved Domain of Heat Shock Transcription Factor Negatively Regulates Oligomerization and DNA Binding. Biochim. Biophys. Acta 2013, 1829, 930–936. [Google Scholar] [CrossRef]
- Xu, Y.-M.; Huang, D.-Y.; Chiu, J.-F.; Lau, A.T.Y. Post-Translational Modification of Human Heat Shock Factors and Their Functions: A Recent Update by Proteomic Approach. J. Proteome Res. 2012, 11, 2625–2634. [Google Scholar] [CrossRef]
- Sakurai, H.; Enoki, Y. Novel Aspects of Heat Shock Factors: DNA Recognition, Chromatin Modulation and Gene Expression. FEBS J. 2010, 277, 4140–4149. [Google Scholar] [CrossRef]
- Aurrecoechea, C.; Barreto, A.; Brestelli, J.; Brunk, B. P.; Caler, E. V.; Fischer, S.; Gajria, B.; Gao, X.; Gingle, A.; Grant, G.; Harb, O. S.; Heiges, M.; Iodice, J.; Kissinger, J. C.; Kraemer, E. T.; Li, W.; Nayak, V.; Pennington, C.; Pinney, D. F.; Pitts, B.; Roos, D. S.; Srinivasamoorthy, G.; Stoeckert, C. J., Jr.; Treatman, C.; Wang, H. AmoebaDB and MicrosporidiaDB: Functional Genomic Resources for Amoebozoa and Microsporidia Species. Nucleic Acids Res. 2011, 39, D612–D619. [Google Scholar] [CrossRef] [PubMed]
- Bello, F.; Orozco, E.; Benítez-Cardoza, C.G.; Zamorano-Carrillo, A.; Reyes-López, C.A.; Pérez-Ishiwara, D.G.; Gómez-García, C. The Novel EhHSTF7 Transcription Factor Displays an Oligomer State and Recognizes a Heat Shock Element in the Entamoeba Histolytica Parasite. Microb. Pathog. 2022, 162, 105349. [Google Scholar] [CrossRef] [PubMed]
- Alastalo, T.-P.; Hellesuo, M.; Sandqvist, A.; Hietakangas, V.; Kallio, M.; Sistonen, L. Formation of Nuclear Stress Granules Involves HSF2 and Coincides with the Nucleolar Localization of Hsp70. J. Cell Sci. 2003, 116, 3557–3570. [Google Scholar] [CrossRef]
- Enoki, Y.; Mukoda, Y.; Furutani, C.; Sakurai, H. DNA-Binding and Transcriptional Activities of Human HSF4 Containing Mutations That Associate with Congenital and Age-Related Cataracts. Biochim. Biophys. Acta BBA - Mol. Basis Dis. 2010, 1802, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Ohama, N.; Nakajima, J.; Kidokoro, S.; Mizoi, J.; Nakashima, K.; Maruyama, K.; Kim, J.-M.; Seki, M.; Todaka, D.; Osakabe, Y.; Sakuma, Y.; Schöffl, F.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Arabidopsis HsfA1 Transcription Factors Function as the Main Positive Regulators in Heat Shock-Responsive Gene Expression. Mol. Genet. Genomics MGG 2011, 286, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Gonsalves, S. E.; Moses, A. M.; Razak, Z.; Robert, F.; Westwood, J. T. Whole-genome Analysis Reveals That Active Heat Shock Factor Binding Sites Are Mostly Associated with Non-heat Shock Genes in Drosophila melanogaster. PLoS One 2011, 6, e15934. [Google Scholar] [CrossRef] [PubMed]
- Karamanos, T. K.; Jackson, M. P.; Calabrese, A. N.; Goodchild, S. C.; Cawood, E. E.; Thompson, G. S.; Kalverda, A. P.; Hewitt, E. W.; Radford, S. E. Structural Mapping of Oligomeric Intermediates in an Amyloid Assembly Pathway. eLife 2019, 8, e46574. [Google Scholar] [CrossRef]
- Teilum, K.; Smith, M. H.; Schulz, E.; Christensen, L. C.; Solomentsev, G.; Oliveberg, M.; Akke, M. Transient Structural Distortion of Metal-Free Cu/Zn Superoxide Dismutase Triggers Aberrant Oligomerization. Proc Natl Acad Sci U S A 2009, 106, 18273–18278. [Google Scholar] [CrossRef]
- Lee, C.-C.; Lin, T.-W.; Ko, T.-P.; Wang, A. H.-J. The Hexameric Structures of Human Heat Shock Protein 90. PLoS One 2011, 6, e19961. [Google Scholar] [CrossRef]
- Kuo, W.-H.K.; Chase, H.A. Exploiting the Interactions between Poly-Histidine Fusion Tags and Immobilized Metal Ions. Biotechnol. Lett. 2011, 1075–1084. [Google Scholar] [CrossRef]
- Kenig, M.; Peternel, S.; Gaberc-Porekar, V.; Menart, V. Influence of the Protein Oligomericity on Final Yield after Affinity Tag Removal in Purification of Recombinant Proteins. J. Chromatogr. A 2006, 1101, 293–306. [Google Scholar] [CrossRef]
- Young, C. L.; Britton, Z. T.; Robinson, A. S. Recombinant Protein Expression and Purification: A Comprehensive Review of Affinity Tags and Microbial Applications. Biotechnol J. 2012, 7, 620–634. [Google Scholar] [CrossRef] [PubMed]
- Scharf, K.-D.; Berberich, T.; Ebersberger, I.; Nover, L. The Plant Heat Stress Transcription Factor (Hsf) Family: Structure, Function and Evolution. Biochim. Biophys. 2012, 1819, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Feng, N.; Feng, H.; Wang, S.; Punekar, A.S.; Ladenstein, R.; Wang, D.-C.; Zhang, Q.; Ding, J.; Liu, W. Structures of Heat Shock Factor Trimers Bound to DNA. iScience 2021, 24, 102951. [Google Scholar] [CrossRef]
- Mittal, D.; Enoki, Y.; Lavania, D.; Singh, A.; Sakurai, H.; Grover, A. Binding Affinities and Interactions among Different Heat Shock Element Types and Heat Shock Factors in Rice (Oryza Sativa L.). FEBS J. 2011, 278, 3076–3085. [Google Scholar] [CrossRef] [PubMed]
- Sarge, K.D.; Murphy, S.P.; Morimoto, R.I. Activation of Heat Shock Gene Transcription by Heat Shock Factor 1 Involves Oligomerization, Acquisition of DNA-Binding Activity, and Nuclear Localization and Can Occur in the Absence of Stress. Mol. Cell. Biol. 1993, 13, 1392–1407. [Google Scholar] [CrossRef]
- Enoki, Y.; Sakurai, H. Diversity in DNA Recognition by Heat Shock Transcription Factors (HSFs) from Model Organisms. FEBS Lett. 2011, 585, 1293–1298. [Google Scholar] [CrossRef]
- Zandi, E.; Tran, T.N.; Chamberlain, W.; Parker, C.S. Nuclear Entry, Oligomerization, and DNA Binding of the Drosophila Heat Shock Transcription Factor Are Regulated by a Unique Nuclear Localization Sequence. Genes Dev. 1997, 11, 1299–1314. [Google Scholar] [CrossRef]
- Zimarino, V.; Wilson, S.; Wu, C. Antibody-Mediated Activation of Drosophila Heat Shock Factor in Vitro. Science 1990, 249, 546–549. [Google Scholar] [CrossRef]
- Gomez-Pastor, R.; Burchfiel, E.T.; Thiele, D.J. Regulation of Heat Shock Transcription Factors and Their Roles in Physiology and Disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 4–19. [Google Scholar] [CrossRef]
- Bhat, G. R.; Sethi, I.; Rah, B.; Kumar, R.; Afroze, D. Innovative in Silico Approaches for Characterization of Genes and Proteins. Front Genet. 2022, 13, 865182. [Google Scholar] [CrossRef]
- Dorantes-Palma, D.; Pérez-Mora, S.; Azuara-Liceaga, E.; Pérez-Rueda, E.; Pérez-Ishiwara, D. G.; Coca-González, M.; Medel-Flores, M. O.; Gómez-García, C. Screening and Structural Characterization of Heat Shock Response Elements (HSEs) in Entamoeba histolytica Promoters. Int. J. Mol. Sci. 2024, 25, 1319. [Google Scholar] [CrossRef]
- Littlefield, O.; Nelson, H. C. A New Use for the 'Wing' of the 'Winged' Helix-Turn-Helix Motif in the HSF-DNA Cocrystal. Nat Struct Biol. 1999, 6, 464–470. [Google Scholar] [CrossRef]
- Neudegger, T.; Verghese, J.; Hayer-Hartl, M.; Hartl, F.U.; Bracher, A. Structure of Human Heat-Shock Transcription Factor 1 in Complex with DNA. Nat. Struct. Mol. Biol. 2016, 23, 140–146. [Google Scholar] [CrossRef]
- Jaeger, A.M.; Pemble, C.W.; Sistonen, L.; Thiele, D.J. Structures of HSF2 Reveal Mechanisms for Differential Regulation of Human Heat-Shock Factors. Nat. Struct. Mol. Biol. 2016, 23, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Nishi, H.; Hashimoto, K.; Madej, T.; Panchenko, A.R. Evolutionary, Physicochemical, and Functional Mechanisms of Protein Homooligomerization. Prog. Mol. Biol. Transl. Sci. 2013, 117, 3–24. [Google Scholar] [CrossRef] [PubMed]
- Reisjalali, M.; Burgos-Mármol, J.J.; Manurung, R.; Troisi, A. Local Structuring of Diketopyrrolopyrrole (DPP)-Based Oligomers from Molecular Dynamics Simulations. Phys. Chem. Chem. Phys. 2021, 23, 19693–19707. [Google Scholar] [CrossRef] [PubMed]
- Ponstingl, H.; Kabir, T.; Gorse, D.; Thornton, J.M. Morphological Aspects of Oligomeric Protein Structures. Prog. Biophys. Mol. Biol. 2005, 89, 9–35. [Google Scholar] [CrossRef]
- Rinauro, D.J.; Chiti, F.; Vendruscolo, M.; Limbocker, R. Misfolded Protein Oligomers: Mechanisms of Formation, Cytotoxic Effects, and Pharmacological Approaches against Protein Misfolding Diseases. Mol. Neurodegener. 2024, 19, 20. [Google Scholar] [CrossRef]
- Nieto, A.; Pérez Ishiwara, D.G.; Orozco, E.; Sánchez Monroy, V.; Gómez García, C. A Novel Heat Shock Element (HSE) in Entamoeba Histolytica That Regulates the Transcriptional Activation of the EhPgp5 Gene in the Presence of Emetine Drug. Front. Cell. Infect. Microbiol. 2017, 7. [Google Scholar] [CrossRef]
- Romero-Díaz, M.; Gómez, C.; López-Reyes, I.; Martínez, M.B.; Orozco, E.; Rodríguez, M.A. Structural and Functional Analysis of the Entamoeba Histolytica EhrabB Gene Promoter. BMC Mol. Biol. 2007, 8, 82. [Google Scholar] [CrossRef] [PubMed]
- Bernes, S.; Siman-Tov, R.; Ankri, S. Epigenetic and Classical Activation of Entamoeba Histolytica Heat Shock Protein 100 (EHsp100) Expression. FEBS Lett. 2005, 579, 6395–6402. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.; Kushnir, O.; Tovy, A.; Siman Tov, R.; Ankri, S. The Entamoeba Histolytica Methylated LINE-Binding Protein EhMLBP Provides Protection against Heat Shock. Cell. Microbiol. 2012, 14, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Nakada-Tsukui, K.; Saito-Nakano, Y.; Ali, V.; Nozaki, T. A Retromerlike Complex Is a Novel Rab7 Effector That Is Involved in the Transport of the Virulence Factor Cysteine Protease in the Enteric Protozoan Parasite Entamoeba Histolytica. Mol. Biol. Cell 2005, 16, 5294–5303. [Google Scholar] [CrossRef] [PubMed]
- Juárez-Hernández, L.J.; García-Pérez, R.M.; Salas-Casas, A.; García-Rivera, G.; Orozco, E.; Rodríguez, M.A. Entamoeba Histolytica: The over Expression of a Mutated EhRabB Protein Produces a Decrease of in Vitro and in Vivo Virulence. Exp. Parasitol. 2013, 133, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Hernandes-Alejandro, M.; Calixto-Gálvez, M.; López-Reyes, I.; Salas-Casas, A.; Cázares-Ápatiga, J.; Orozco, E.; Rodríguez, M.A. The Small GTPase EhRabB of Entamoeba Histolytica Is Differentially Expressed during Phagocytosis. Parasitol. Res. 2013, 112, 1631–1640. [Google Scholar] [CrossRef] [PubMed]
- Javier-Reyna, R.; Montaño, S.; García-Rivera, G.; Rodríguez, M. A.; González-Robles, A.; Orozco, E. EhRabB Mobilises the EhCPADH Complex Through the Actin Cytoskeleton During Phagocytosis of Entamoeba histolytica. Cell Microbiol. 2019, 21, e13071. [Google Scholar] [CrossRef] [PubMed]
- Lavi, T.; Siman-Tov, R.; Ankri, S. EhMLBP Is an Essential Constituent of the Entamoeba Histolytica Epigenetic Machinery and a Potential Drug Target. Mol. Microbiol. 2008, 69, 55–66. [Google Scholar] [CrossRef]
- Katz, S.; Trebicz-Geffen, M.; Ankri, S. Stress Granule Formation in Entamoeba Histolytica: Cross-Talk between EhMLBP, EhRLE3 Reverse Transcriptase and Polyubiquitinated Proteins. Cell. Microbiol. 2014, 16, 1211–1223. [Google Scholar] [CrossRef] [PubMed]
- Lavi, T.; Siman-Tov, R.; Ankri, S. Insights into the Mechanism of DNA Recognition by the Methylated LINE Binding Protein EhMLBP of Entamoeba Histolytica. Mol. Biochem. Parasitol. 2009, 166, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Induction and immunodetection of the oligomeric states of the rEhHSTF5 protein. The integrity of total proteins extracted from transformed and IPTG-induced bacteria was evaluated by SDS-PAGE. A Molecular Weight Marker (MWM) and IPTG induction times ranging from 0 to 24 h were included. The gel was stained with Coomassie Brilliant Blue G-250 to visualize protein bands (a). Subsequently, immunodetection was performed by Western blot using α6xHis and αEhHSTF5 specific antibodies to detect the distinct conformations of rEhHSTF5 (b). The graphs display the relative protein expression, calculated as the average of pixel values obtained from three independent samples. Expression was normalized using the endogenous control, GAPDH. The statistical analysis was performed using a two-way ANOVA, followed by the Tukey's multiple comparison test. Significant differences between experimental groups are denoted with (*). The significance levels used were **p≤ 0.002, and ***p≤ 0.001.
Figure 1.
Induction and immunodetection of the oligomeric states of the rEhHSTF5 protein. The integrity of total proteins extracted from transformed and IPTG-induced bacteria was evaluated by SDS-PAGE. A Molecular Weight Marker (MWM) and IPTG induction times ranging from 0 to 24 h were included. The gel was stained with Coomassie Brilliant Blue G-250 to visualize protein bands (a). Subsequently, immunodetection was performed by Western blot using α6xHis and αEhHSTF5 specific antibodies to detect the distinct conformations of rEhHSTF5 (b). The graphs display the relative protein expression, calculated as the average of pixel values obtained from three independent samples. Expression was normalized using the endogenous control, GAPDH. The statistical analysis was performed using a two-way ANOVA, followed by the Tukey's multiple comparison test. Significant differences between experimental groups are denoted with (*). The significance levels used were **p≤ 0.002, and ***p≤ 0.001.
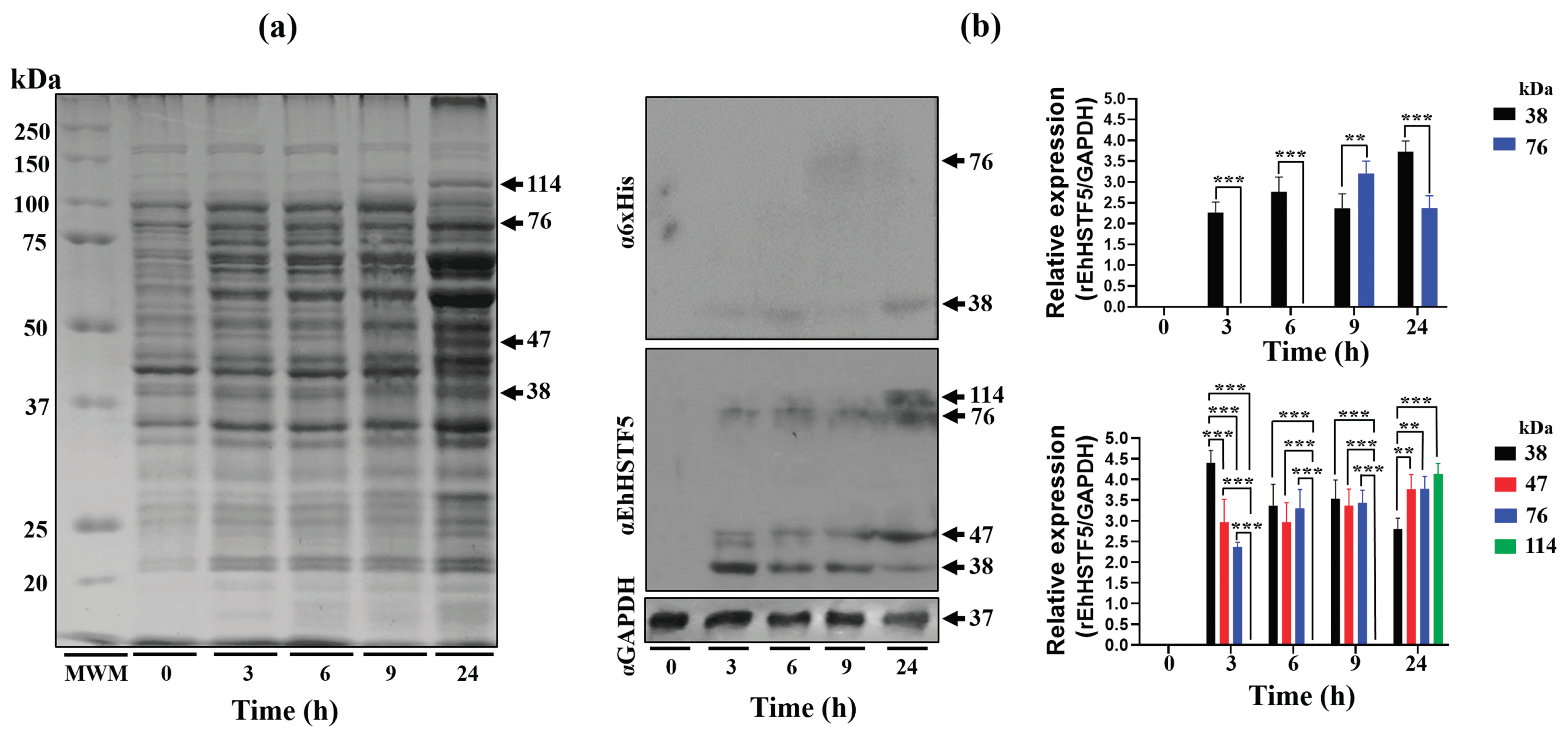
Figure 2.
Purification and immunodetection of oligomeric conformations of the rEhHSTF5 protein. The oligomeric conformations of the rEhHSTF5 protein were monitored before and after loading onto the HisTrap FF column. The total proteins obtained from bacteria transformed with the plasmid construct and induced with IPTG (24 h) were passed through filters with a pore size of 0.22 μm (flowthrough, FT), and the proteins that did not bind to the column (waste, W) are shown (a). The chromatogram and SDS-PAGE assays revealed that the drEhHSTF5 protein was eluted in fractions 7 to 12, while the mrEhHSTF5 protein was eluted in fractions 24 to 26 (b-d). These proteins were detected using α6xHis and αEhHSTF5 antibodies in WB assays (e, f). In the chromatogram (b), the stepped black lines represent the absorbance reading at 280 nm during the elution time, indicating the presence of proteins being eluted from the affinity column. Panel (c) to (f), “F” denotes the fraction number obtained from the chromatography, and “MWM” indicates molecular weight marker.
Figure 2.
Purification and immunodetection of oligomeric conformations of the rEhHSTF5 protein. The oligomeric conformations of the rEhHSTF5 protein were monitored before and after loading onto the HisTrap FF column. The total proteins obtained from bacteria transformed with the plasmid construct and induced with IPTG (24 h) were passed through filters with a pore size of 0.22 μm (flowthrough, FT), and the proteins that did not bind to the column (waste, W) are shown (a). The chromatogram and SDS-PAGE assays revealed that the drEhHSTF5 protein was eluted in fractions 7 to 12, while the mrEhHSTF5 protein was eluted in fractions 24 to 26 (b-d). These proteins were detected using α6xHis and αEhHSTF5 antibodies in WB assays (e, f). In the chromatogram (b), the stepped black lines represent the absorbance reading at 280 nm during the elution time, indicating the presence of proteins being eluted from the affinity column. Panel (c) to (f), “F” denotes the fraction number obtained from the chromatography, and “MWM” indicates molecular weight marker.
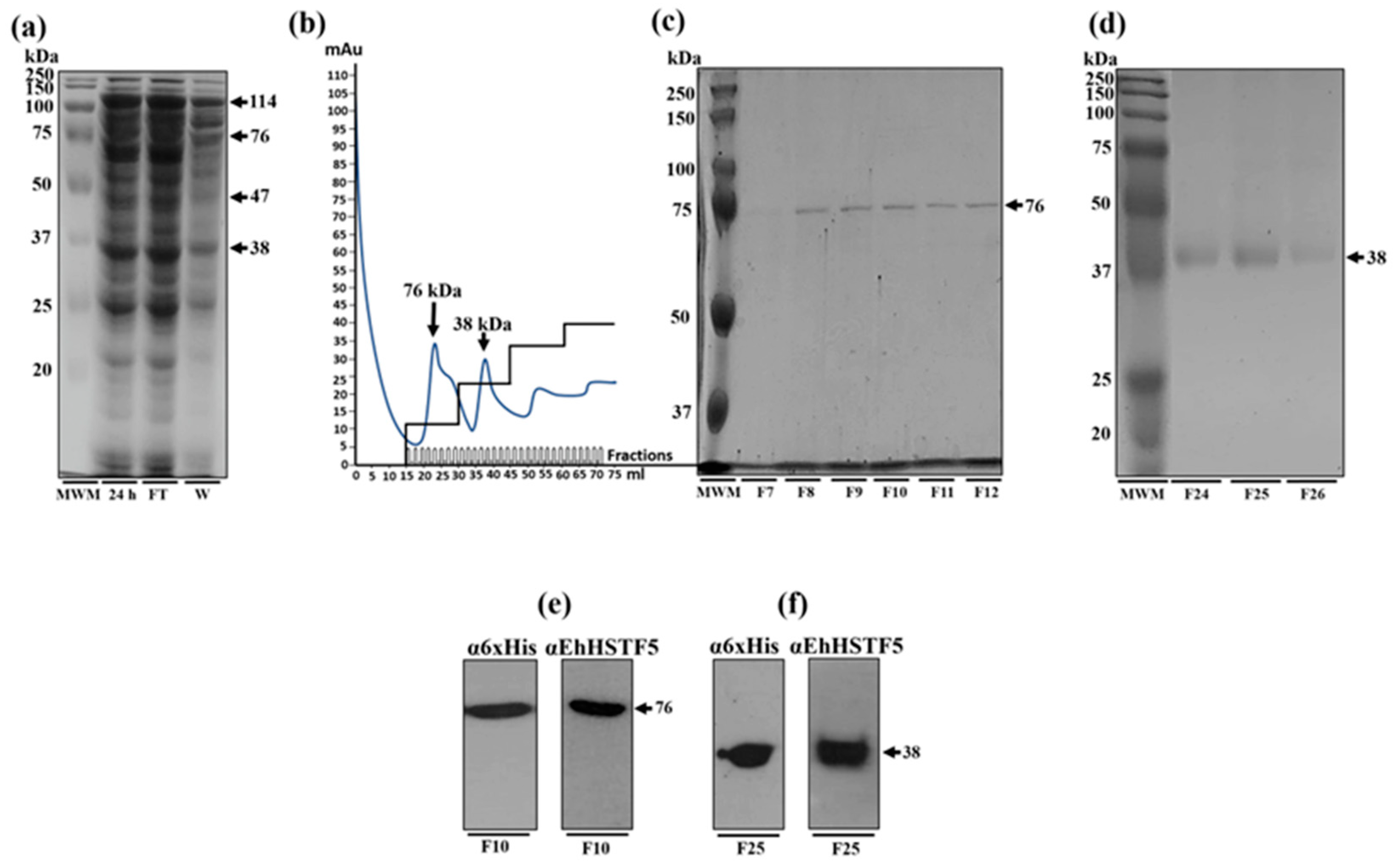
Figure 3.
mrEhHSTF5 oligomerization induction with increasing concentrations of glutaraldehyde. Oligomerization states were evaluated by 12% SDS-PAGE gel electrophoresis stained with Coomassie Brilliant Blue G-250. The statistical analysis was performed using a two-way ANOVA, followed by the Tukey's multiple comparison test. Significant differences between experimental groups are denoted with (*). The significance levels used were *p≤ 0.033, **p≤ 0.002, and ***p≤ 0.001.
Figure 3.
mrEhHSTF5 oligomerization induction with increasing concentrations of glutaraldehyde. Oligomerization states were evaluated by 12% SDS-PAGE gel electrophoresis stained with Coomassie Brilliant Blue G-250. The statistical analysis was performed using a two-way ANOVA, followed by the Tukey's multiple comparison test. Significant differences between experimental groups are denoted with (*). The significance levels used were *p≤ 0.033, **p≤ 0.002, and ***p≤ 0.001.
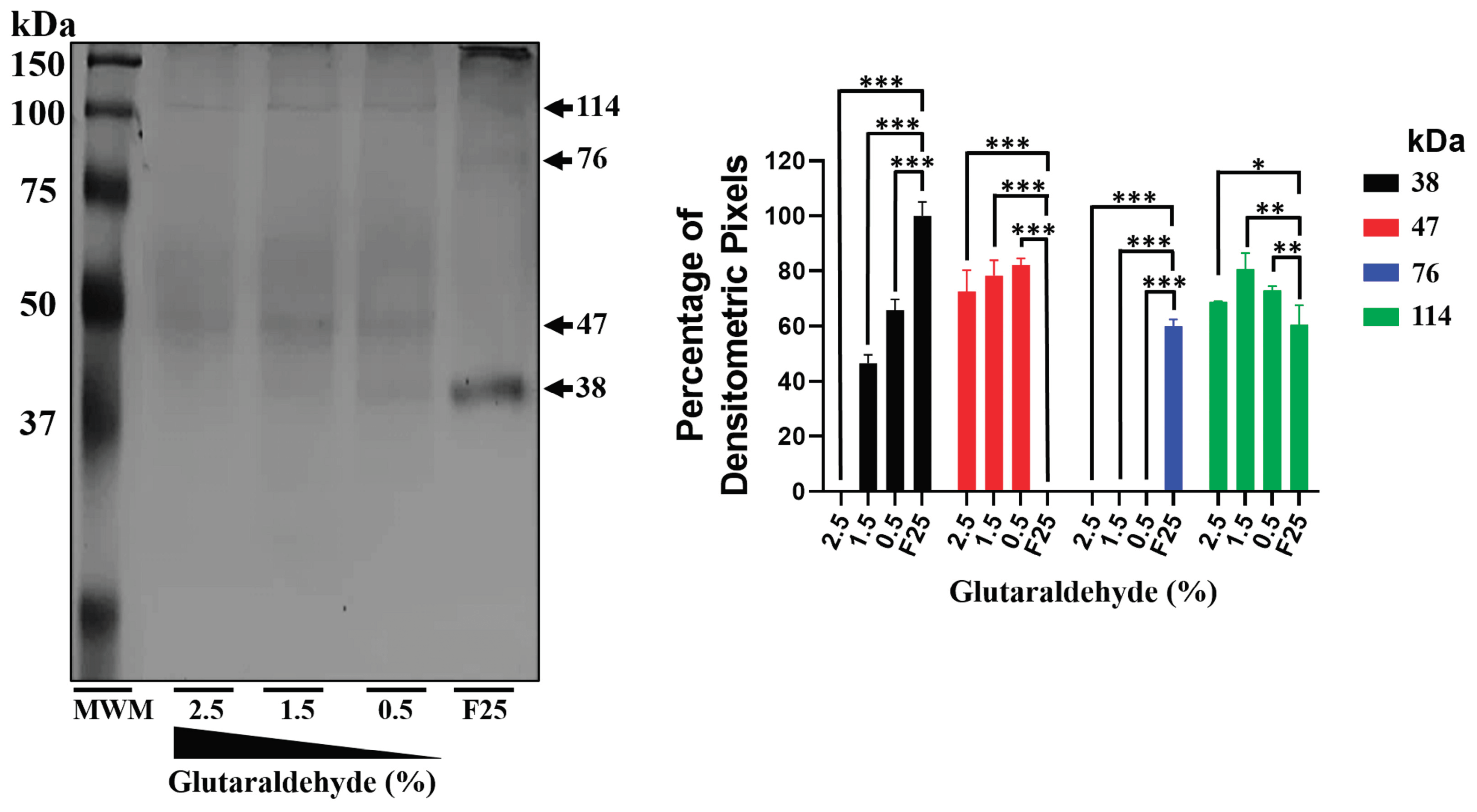
Figure 4.
HSE recognition of the EhPgp5 gene by the rEhHSTF5 protein. EMSA assays was performed using the monomeric and dimeric conformations of the rEhHSTF5 protein. A specific competitor (non-biotinylated HSE_EhPgp5) and a nonspecific competitor (Poly(dI-dC)) were used (a). For the supershift assays, the specific antibody αEhHSTF5 was utilized for both conformations of the rEhHSTF5 protein. Unrelated antibodies, including mouse preimmune serum (PS) and αGAPDH antibody, were also used as negative controls (b). Likewise, a kinetic study with increasing drEhHSTF5 concentration (from 0.7 to 2.8 µM) drEhHSTF5 was performed in supershift assays (c). In the image legend, the plus sign (+) indicates the presence of a component in the reaction, while the minus sign (−) denotes its absence. A graphical illustration of each condition used in the EMSA and supershift tests is provided at the bottom. .
Figure 4.
HSE recognition of the EhPgp5 gene by the rEhHSTF5 protein. EMSA assays was performed using the monomeric and dimeric conformations of the rEhHSTF5 protein. A specific competitor (non-biotinylated HSE_EhPgp5) and a nonspecific competitor (Poly(dI-dC)) were used (a). For the supershift assays, the specific antibody αEhHSTF5 was utilized for both conformations of the rEhHSTF5 protein. Unrelated antibodies, including mouse preimmune serum (PS) and αGAPDH antibody, were also used as negative controls (b). Likewise, a kinetic study with increasing drEhHSTF5 concentration (from 0.7 to 2.8 µM) drEhHSTF5 was performed in supershift assays (c). In the image legend, the plus sign (+) indicates the presence of a component in the reaction, while the minus sign (−) denotes its absence. A graphical illustration of each condition used in the EMSA and supershift tests is provided at the bottom. .
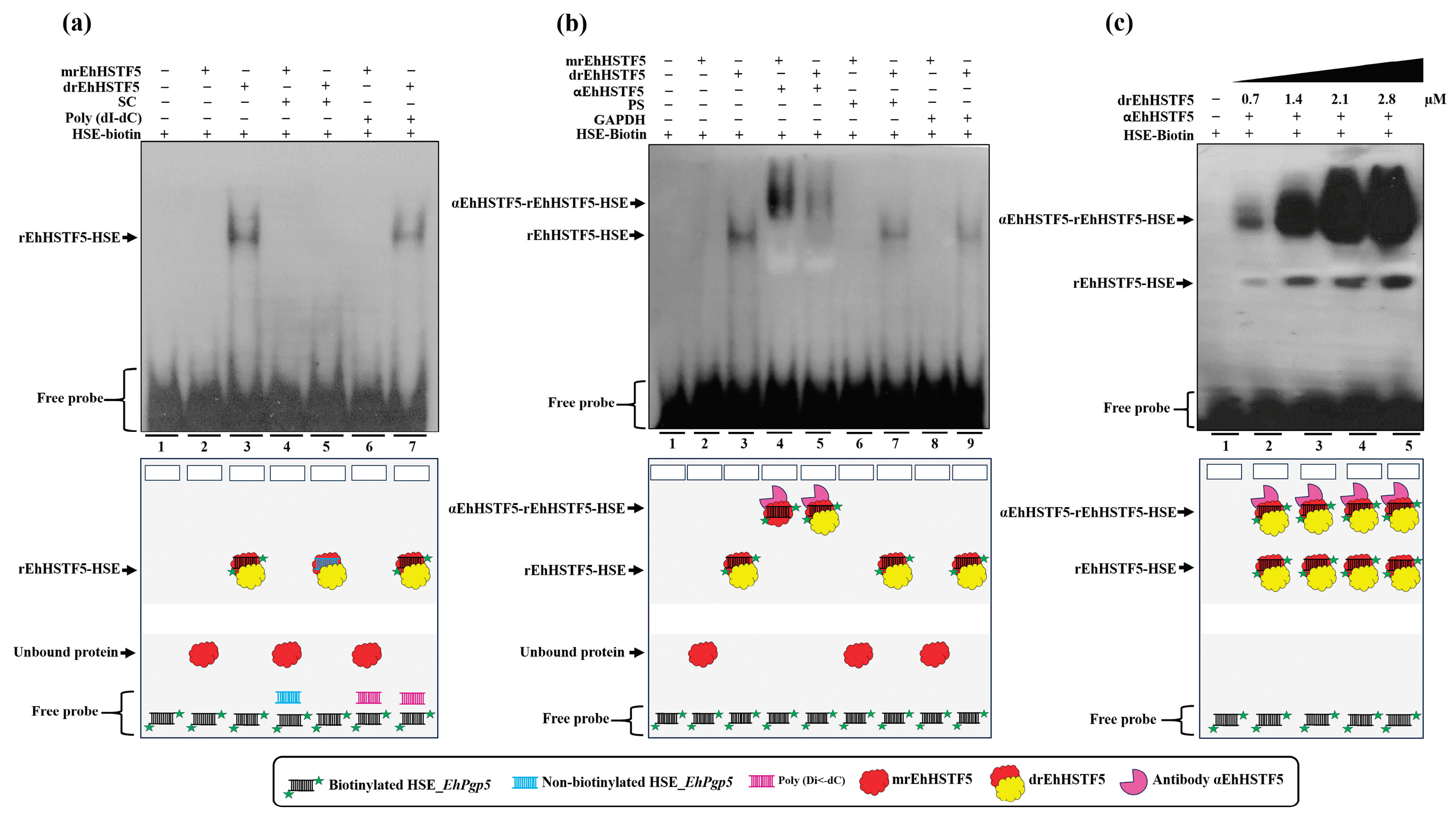
Figure 5.
Construction of 3D models. The 3D structures of HSE_EhPgp5 (a), mEhDBD5 (b), and dEhDBD5 (c) were obtained. At the bottom of the EhDBDs, the secondary structure of a monomer is displayed using a color code, while the graph illustrates its stability, assessed in silico through the normalized B-factor.
Figure 5.
Construction of 3D models. The 3D structures of HSE_EhPgp5 (a), mEhDBD5 (b), and dEhDBD5 (c) were obtained. At the bottom of the EhDBDs, the secondary structure of a monomer is displayed using a color code, while the graph illustrates its stability, assessed in silico through the normalized B-factor.
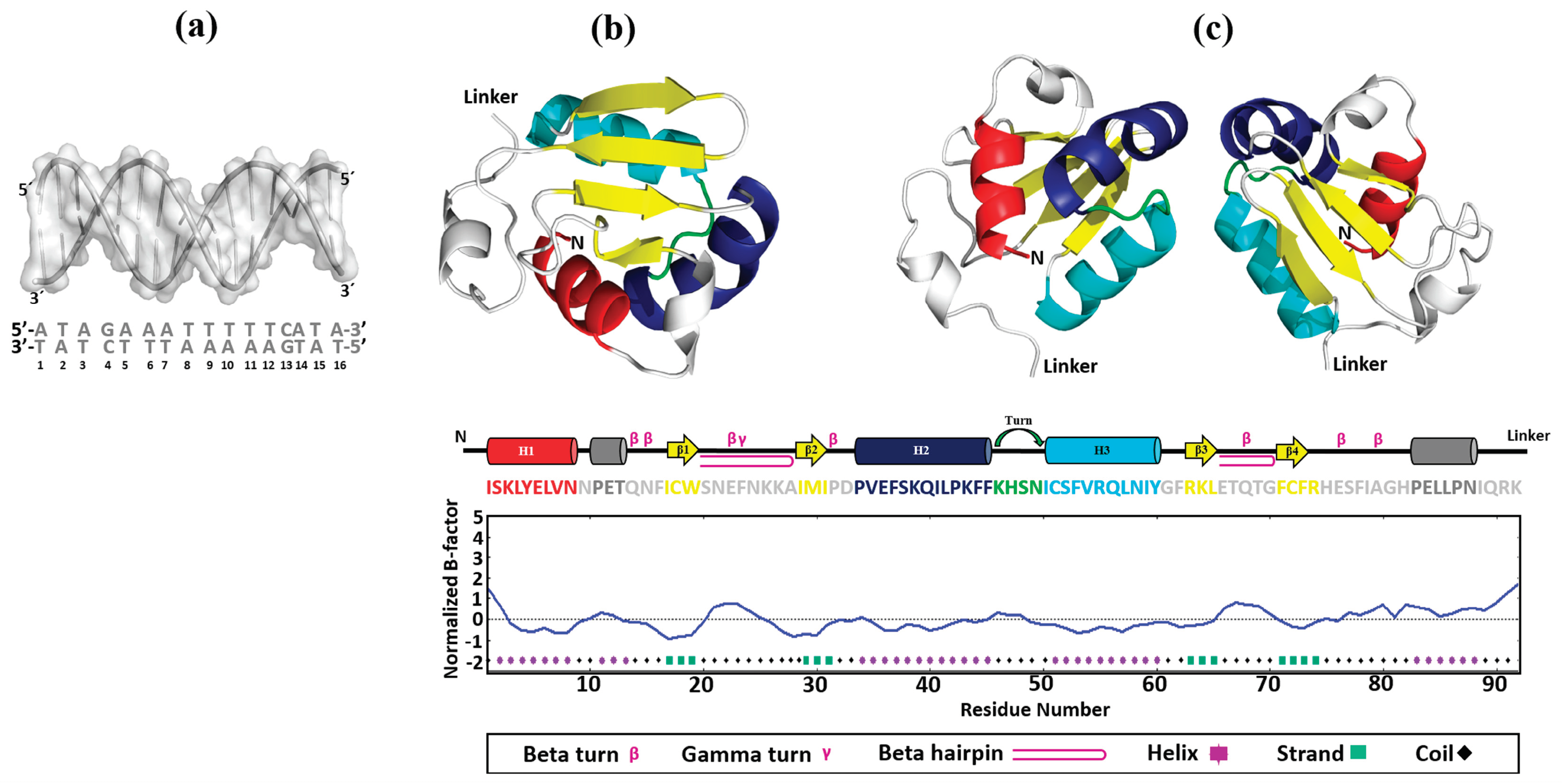
Figure 6.
Validation of 3D structures. The 3D models mEhDBD5 (a) and dEhDBD5 (b) were validated using Ramachandran analysis, which provides detailed information about the distribution of dihedral angles of amino acids in favorable and unfavorable regions. The capital letters A, B, and L are used to designate the Most Favoured Regions, while the lowercase letters a, b, l, and p represent the Additional Allowed Regions. The tilde (~a, ~b, ~l, ~p) is utilized to indicate the Generously Allowed Regions. The label 'XX' is employed for the Disallowed Regions. Furthermore, a value of −1 is assigned to a torsion angle located within a non-permissible region. The models were subjected to the ProSA-web server for a validation based on Z-score and global energy, where values ≤0 indicate absence of structural errors. ERRAT was used for error quantification, where an asterisk (*) indicates the error value. White bars in the graph represent instances without structural errors, while yellow bars indicate amino acids with structural errors falling within the 95-99% error range, and red bars identify errors exceeding 99%. Verify 3D validates protein structures by evaluating their 3D conformation through comparison with a set of experimental structures. It assigns a score, suggesting an acceptable average 3D−1D score of ≥ 0.1 for validation.
Figure 6.
Validation of 3D structures. The 3D models mEhDBD5 (a) and dEhDBD5 (b) were validated using Ramachandran analysis, which provides detailed information about the distribution of dihedral angles of amino acids in favorable and unfavorable regions. The capital letters A, B, and L are used to designate the Most Favoured Regions, while the lowercase letters a, b, l, and p represent the Additional Allowed Regions. The tilde (~a, ~b, ~l, ~p) is utilized to indicate the Generously Allowed Regions. The label 'XX' is employed for the Disallowed Regions. Furthermore, a value of −1 is assigned to a torsion angle located within a non-permissible region. The models were subjected to the ProSA-web server for a validation based on Z-score and global energy, where values ≤0 indicate absence of structural errors. ERRAT was used for error quantification, where an asterisk (*) indicates the error value. White bars in the graph represent instances without structural errors, while yellow bars indicate amino acids with structural errors falling within the 95-99% error range, and red bars identify errors exceeding 99%. Verify 3D validates protein structures by evaluating their 3D conformation through comparison with a set of experimental structures. It assigns a score, suggesting an acceptable average 3D−1D score of ≥ 0.1 for validation.
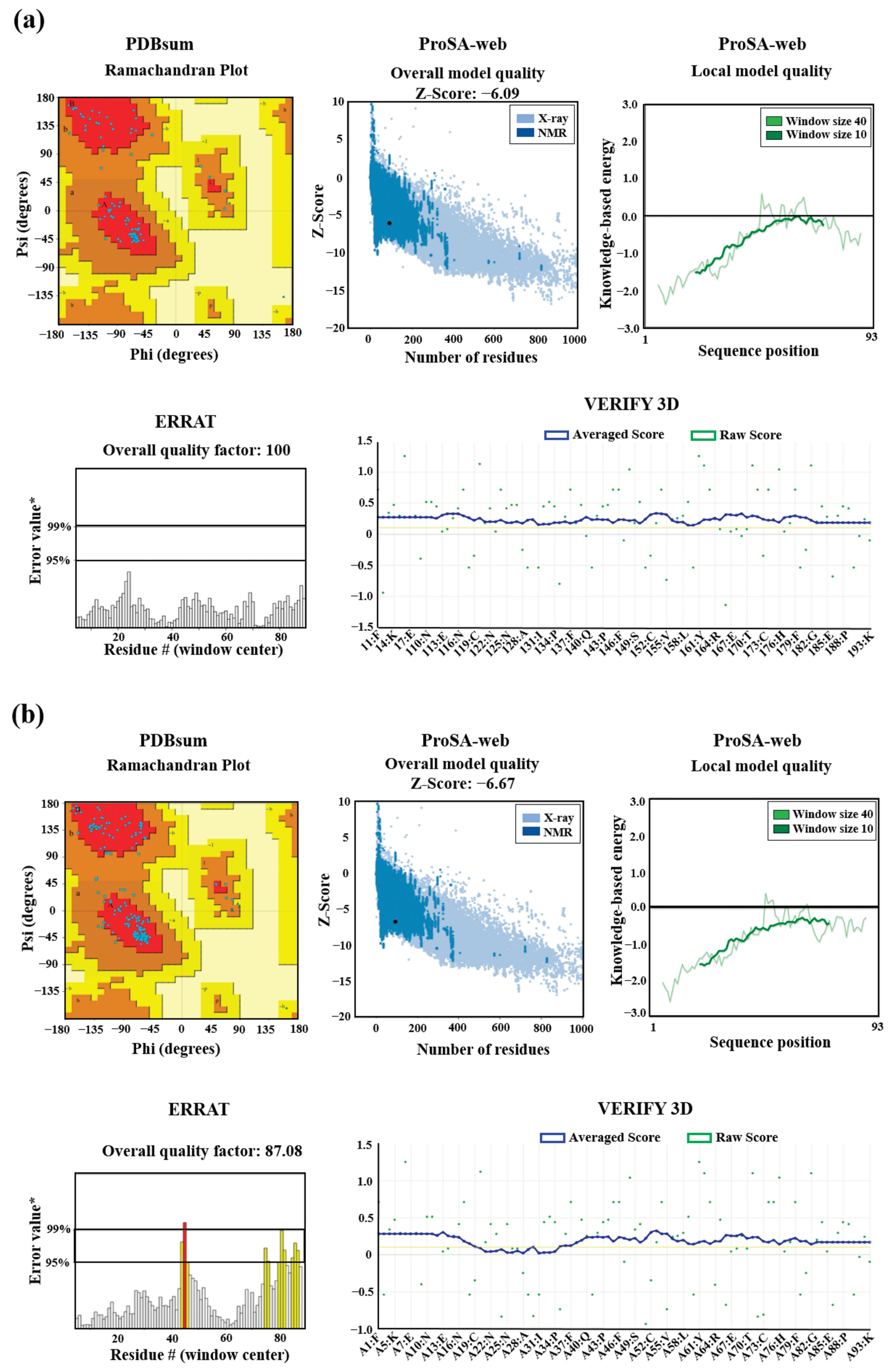
Figure 7.
Molecular docking and intermolecular evaluation of the mEhDBD5-HSE_EhPgp5 and dEhDBD5-HSE_ EhPgp5 complexes. Blind dockings were performed with the domains mEhDBD5 or dEhDBD5 and the HSE_EhPgp5. Using the PLIP server, intermolecular interactions of amino acids involved in the interaction with the sense and antisense strands of HSE were estimated. A close-up of the area of interaction is shown in the boxes, where amino acids from mEhDBD5 (EhDBD5(1)) are illustrated in red, while the analogue of the second EhDBD5 domain from dEhDBD5 (EhDBD5(2)) is shown in yellow (a) and (b). The amino acids involved in the interaction with the amino acids of the mEhDBD7-HSE, KlHSTF-HSE, HsHSTF1-HSE, and HsHSTF2-HSE complexes were compared (c). The high conservation degree of α2-helix and α3-helix of DBDs from 28 species, including the EhHSTFs family, is shown (d). .
Figure 7.
Molecular docking and intermolecular evaluation of the mEhDBD5-HSE_EhPgp5 and dEhDBD5-HSE_ EhPgp5 complexes. Blind dockings were performed with the domains mEhDBD5 or dEhDBD5 and the HSE_EhPgp5. Using the PLIP server, intermolecular interactions of amino acids involved in the interaction with the sense and antisense strands of HSE were estimated. A close-up of the area of interaction is shown in the boxes, where amino acids from mEhDBD5 (EhDBD5(1)) are illustrated in red, while the analogue of the second EhDBD5 domain from dEhDBD5 (EhDBD5(2)) is shown in yellow (a) and (b). The amino acids involved in the interaction with the amino acids of the mEhDBD7-HSE, KlHSTF-HSE, HsHSTF1-HSE, and HsHSTF2-HSE complexes were compared (c). The high conservation degree of α2-helix and α3-helix of DBDs from 28 species, including the EhHSTFs family, is shown (d). .
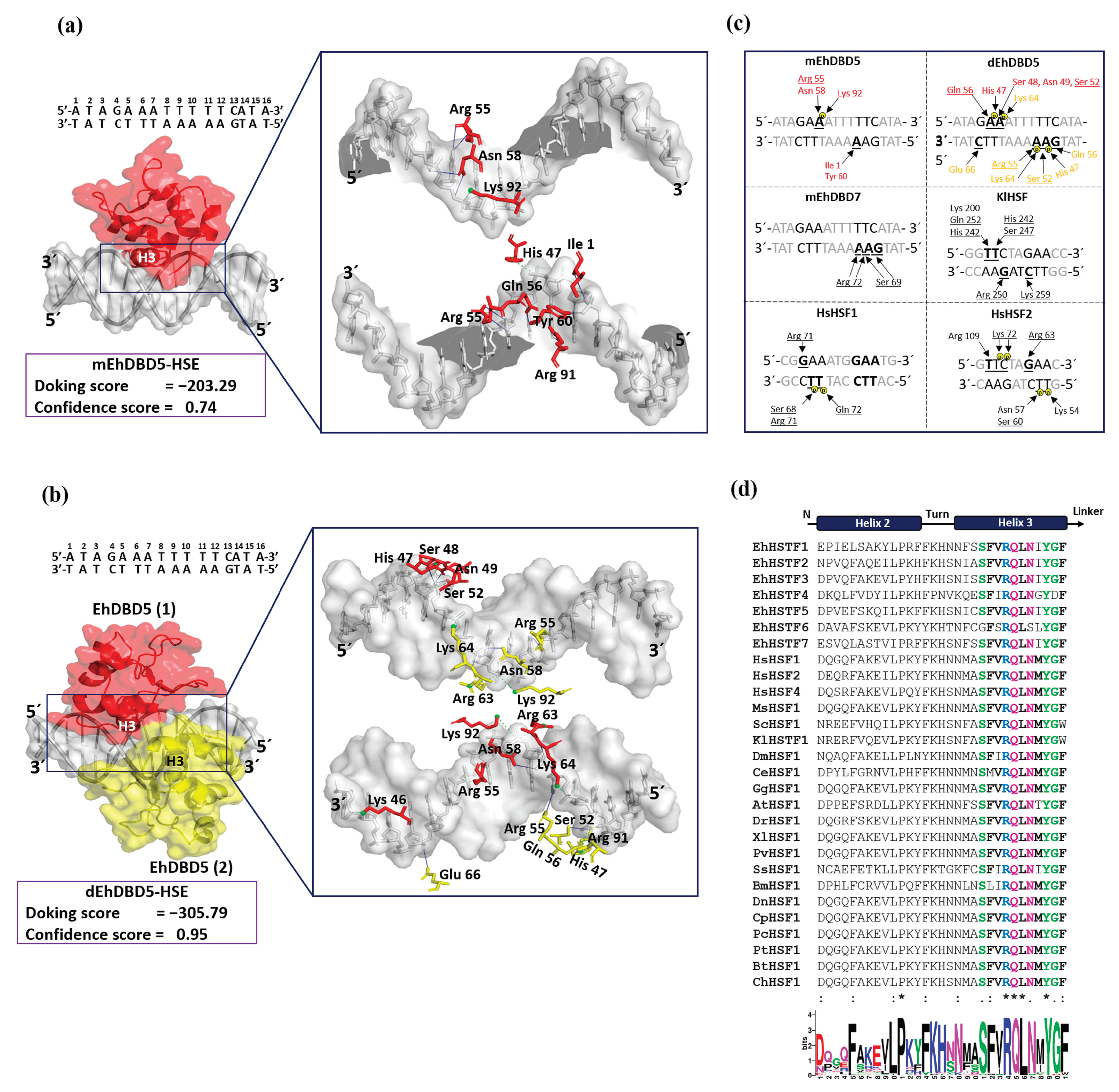
Figure 8.
3D structural similarity between DBDs-HSEs complexes. Based on the obtained dEhDBD5-HSE_EhPgp5 complex (a), overlay comparisons were performed with crystallographic structures deposited in the PDB database. These included the K. lactis DBD-HSE complex (ID: 3HTS) (b), S. cerevisiae-C. thermophilum complex (ID: 5D5X) (c), H. sapiens-C. thermophilum complex (ID: 5D5W) (d), synthetic H. sapiens construction (ID: 5D8L) (e), and H. sapiens (ID: 7DCU) (f). A joint overlay of all these structures was performed (g) and focusing solely on the wing domain (h). In addition, a significant degree of conservation was identified through the alignment of amino acid sequences of helices 2 and 3 (i). The squares illustrate the TM-scores in percentages, while the RMSD is in Angstroms (Å).
Figure 8.
3D structural similarity between DBDs-HSEs complexes. Based on the obtained dEhDBD5-HSE_EhPgp5 complex (a), overlay comparisons were performed with crystallographic structures deposited in the PDB database. These included the K. lactis DBD-HSE complex (ID: 3HTS) (b), S. cerevisiae-C. thermophilum complex (ID: 5D5X) (c), H. sapiens-C. thermophilum complex (ID: 5D5W) (d), synthetic H. sapiens construction (ID: 5D8L) (e), and H. sapiens (ID: 7DCU) (f). A joint overlay of all these structures was performed (g) and focusing solely on the wing domain (h). In addition, a significant degree of conservation was identified through the alignment of amino acid sequences of helices 2 and 3 (i). The squares illustrate the TM-scores in percentages, while the RMSD is in Angstroms (Å).
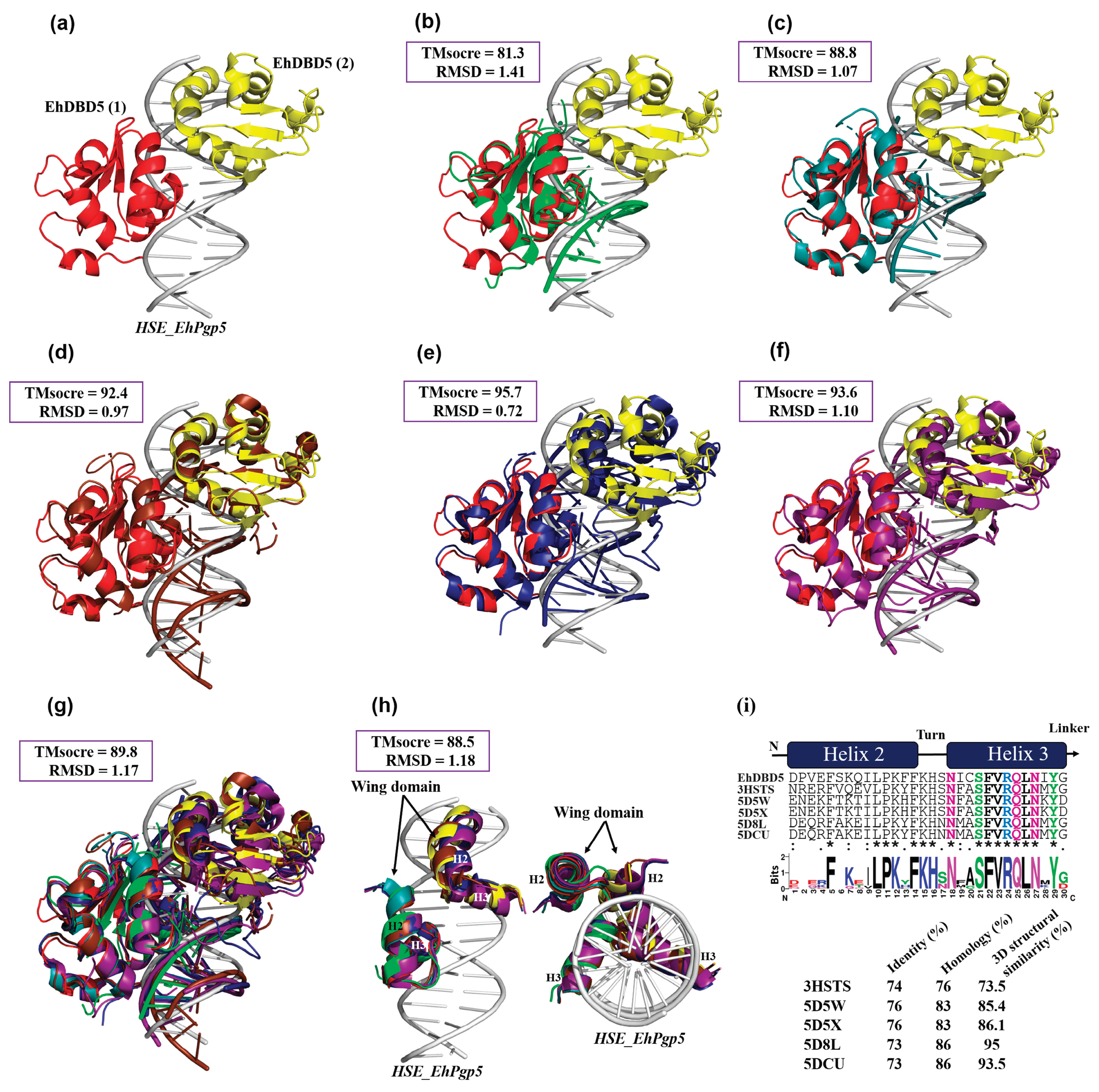
Figure 9.
Our in vitro findings demonstrate that the mrEhHSTF5 protein is capable of oligomerizing, forming dimers and trimers. Furthermore, through EMSA assays and in silico molecular docking, we discovered that the dimeric conformation exhibits greater affinity for HSE_EhPgp5 compared to the monomeric conformation. Dorantes et al. [36] reported higher affinity (-459) binding of tEhHSTF5 to the same HSE, supporting our results and indicating that the degree of oligomerization increases the affinity towards HSE_EhPgp5. OD; oligomerization domain, DBD; DNA binding domain, Ds; Docking score.
Figure 9.
Our in vitro findings demonstrate that the mrEhHSTF5 protein is capable of oligomerizing, forming dimers and trimers. Furthermore, through EMSA assays and in silico molecular docking, we discovered that the dimeric conformation exhibits greater affinity for HSE_EhPgp5 compared to the monomeric conformation. Dorantes et al. [36] reported higher affinity (-459) binding of tEhHSTF5 to the same HSE, supporting our results and indicating that the degree of oligomerization increases the affinity towards HSE_EhPgp5. OD; oligomerization domain, DBD; DNA binding domain, Ds; Docking score.
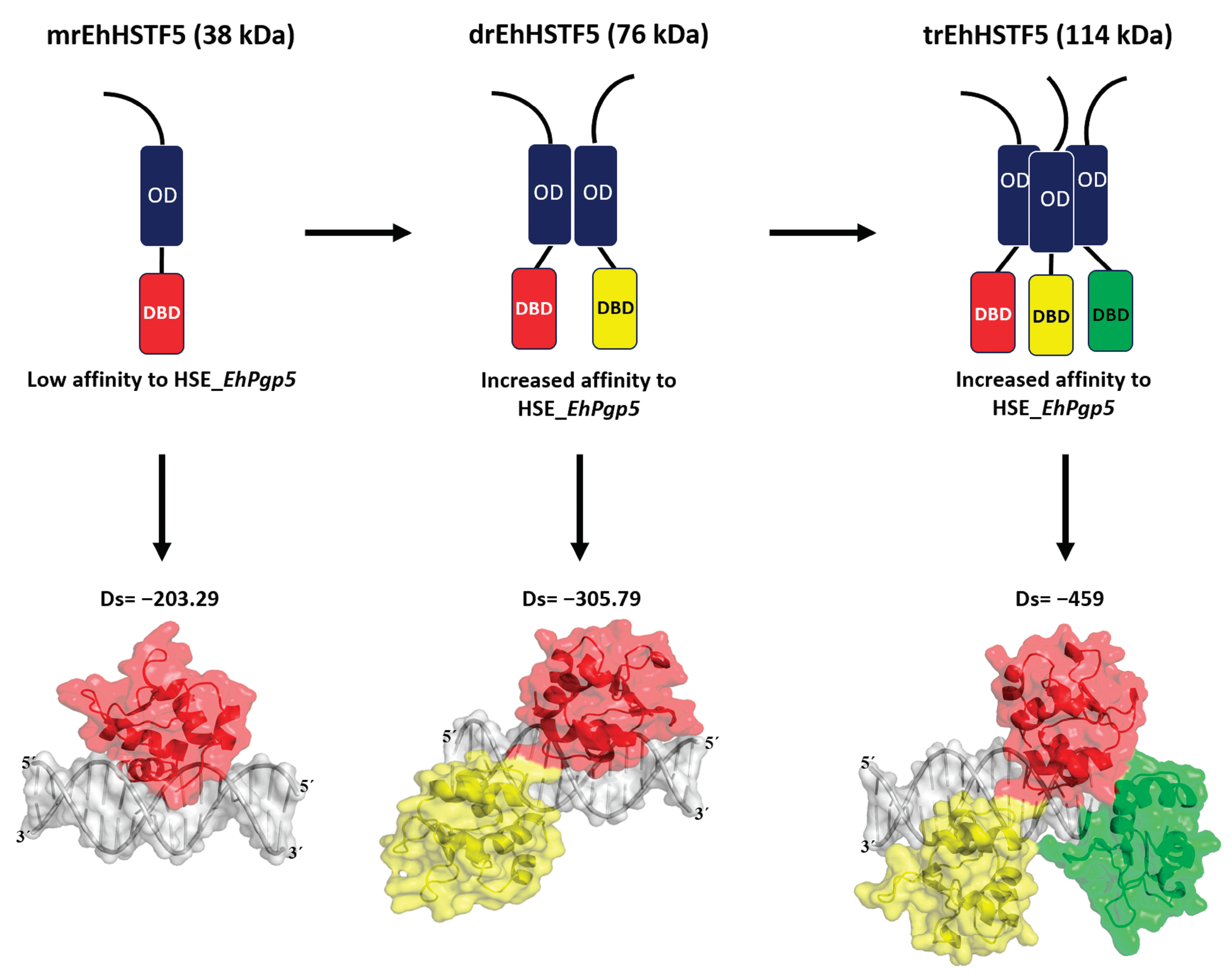
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



