Submitted:
05 March 2024
Posted:
06 March 2024
You are already at the latest version
Abstract
52 years have passed since President Nixon launched the "War on Cancer". The goals outlined by the President were not achieved because cancer treatment applied such as chemotherapy, radiotherapy, and targeted therapy have not fully met expectations. We suggest a new chemotherapeutic strategy: disrupting the communication between cancer cells and their microenvironment by chemical means. Immunological synapses that form between cancer cells and immune or other stromal cells provide an attractive target for this approach. Synapses form ligand-receptor clusters within interface of the interacting cells. Despite their differences, synapses share common properties: intercellular protein clusters; the proximity of these proteins; and their cooperative interaction making a synapse an unified functional unit. Synapses provide the limited space for the focused intercellular exchange of signaling molecules and particles. Therefore, destruction of synapses is expected to cause collapse of various tumor types. Additionally, the clustered arrangement of synapse components offers opportunities to increase treatment safety by reducing the concentration of cell impermeable agents used, to enhance its specificity applying cross-linking reagents, thus restricting modifications of surface-exposed molecules. By attaching a cleavable cell permeable toxic agent to a crosslinker should further enhance a killing potential in treating cancer. The proposed approach promises to be simple, universal, and less expensive than existing cancer therapy methods.
Keywords:
cancer
; tumor microenvironment
; cell interaction
; synapse
; crosslinking agents
1. Introduction. Chemotherapy: Fame and Failure
The year 2023 marks 52 years since December 23, 1971, when U.S. President Nixon declared what was later called the “War on Cancer” [1,2,3]. Large allocations were made. The President promised that in five years a cure for cancer would be available, and by the year 2000, the incidence of cancer would be halved [4]. This enthusiasm was based on successful experimental results in the treatment of children with acute leukemia. Since then, progress in this “war” is still being discussed, and the results on cancer mortality are presented in different ways—either as progress or as failure. As Howy Jacobs wrote, “Nixon’s war on cancer outlasted his presidency and his life. However, after forty years we still hope for the future progress of modern therapy, even though the number of publications on this topic during this period is so large that it will soon reach the moon” [5]. There is no doubt that the war against cancer, as defined by Nixon, was lost, and its combat targets were never defeated. All efforts to significantly reduce cancer mortality have been in vain. A recent review stated: “Despite over a century of intensive efforts, the great gains promised by the War on Cancer nearly 50 years ago have not materialized” [6]. Donald Kennedy, who headed the US FDA and Stanford University, later described the war on cancer as “A Medical Vietnam” [3].
These failures have led to the fact that, despite the great merits of chemotherapy in the treatment of cancer and, especially, in the progress in our understanding of disease mechanisms, there has been a pronounced pessimism about its future prospects as a medical strategy. Thus, in 2016, Agarwal ended his brief but extremely capacious review [7] (“Is cancer chemotherapy dying?”) with the words “Today, we are moving towards non-chemotherapeutic drug therapy of malignancy. Also, today we are moving from protocol-based treatment to personalized therapy based on prognostic markers, markers predictive of drug sensitivity/resistance, markers predictive of adverse events and molecular profiling. I have no hesitation in predicting that very soon, chemotherapy may play a very small role in the cure of cancer”. Also, in his very skeptical commentary, Bizzarri notes that chemotherapy has many defects, and its effectiveness causes skepticism [8]. And even such a recognized authority in the field of oncology as one of the authors of the “Cancer hallmarks” concept, Douglas Hanahan [2], in his Editorial “Rethinking the war on cancer” (which we will discuss in more detail later) notes: “The war on cancer has largely focused on mutant cancer cells, via chemotherapy and radiotherapy and, more recently, targeted therapy... Most of the time, however, cancers eventually find ways to circumvent such targeted strikes, adapting and then reemerging as expansive and often more aggressive growths… The reality, however, is that targeted therapies are generally not curative or even enduringly effective, because of the adaptive and evasive resistance strategies developed by cancers under attack”.
However, there still remain battlegrounds where chemotherapy maintains its dominance. For example, unlike in other cancers where the substitution of chemotherapy is possible, in germ cell tumors it is very difficult—the use of platinum drugs is necessary [9]. Also, at least in some cases, chemotherapies can augment tumor immunity, for example, by inducing immunogenic cell death [10,11,12] and this hints at the prospects of searching for optimal combinations of chemo and immunotherapy.
The use of the term “targeted” requires some explanation. Traditionally, cancer is treated by surgery, radiotherapy, and chemotherapy. Chemotherapy originated from poisonous gases that were used during World War II. It has been successfully used to cure many otherwise fatal cancers, such as childhood acute lymphoblastic leukemia, Hodgkin’s lymphoma, testicular cancer, and so on. Also, a significant increase in life expectancy in many other diseases such as high-grade non-Hodgkin’s lymphomas and multiple myeloma was encouraging. However, the problem with chemotherapy is that in addition to destroying the cancer cell, it also causes widespread damage to other tissues, especially bone marrow, resulting in morbidity and even mortality. Therefore, enormous efforts of oncologists have been and continue to be aimed at increasing the specificity of the drug by directing its action to certain cancer-specific properties—targets. Regardless of whether targeting is carried out using chemical agents or genetic therapy, as we noticed as early as 2011 [13], the term “targeting” can be divided into two broad strategies: (1) the targeted destruction of tumors as a whole exploiting the features shared by all cancers, for example, relatively fast mitotic cell division, and (2) using the ideology of molecular targeted therapy, targeted at certain molecular component(s) or pathways presumably crucial for maintenance of a certain cancer type [13,14]. Previously, we analyzed in detail the reasons why molecular-targeted medicine is objectively doomed to failure [13,15]. The above quote [2] provides even more reasons why this failure is inevitable. There, Hanahan expresses an extremely important idea, which he then details in his article: “I suggest that, much like in modern warfare, the war on cancer needs to have a battlespace vision”. It is “Space” not “target” that must be taken into account for effective therapy of such a multifaceted disease as cancer. We expressed the same idea in our recent review “From the Catastrophic Objective Irreproducibility of Cancer Research and Unavoidable Failures of Molecular Targeted Therapies to the Sparkling Hope of Supramolecular Targeted Strategies” [15], pointing out the need for a multidimensional strategy.
And, nevertheless, as the authors of [16] quite rightly noted in their review, the efforts expended were not wasted: the “war” led to the accumulation of enormous (albeit still far from complete) information about the mechanisms of the occurrence and evolution of cancer. With a new understanding of the molecular mechanism(s) of disease progression, our knowledge of the disease is growing rapidly and giving rise to many new therapeutic approaches. Over the past few decades, various combinations of treatments [17] have been proposed that are currently being used to treat various types of cancer and were not common a few years ago (see, for example, [16]). At the same time, these authors, in our opinion, quite rightly point out that “chemotherapy remains a largely opted therapeutic option despite its known side effects on the patient’s physical and psychological health. Chemotherapeutic agents/pharmaceuticals served a great purpose over the past few decades and have remained the frontline choice for advanced-stage malignancies where surgery and/or radiation therapy cannot be prescribed due to specific reasons”.
Hanahan also indicates an extremely important issue, which is not always taken into account by researchers involved in fundamental aspects of cancer treatment: “Another sobering issue is the reality that many exciting new cancer treatments are very expensive... despite in many cases producing only transitory clinical benefit, posing serious cost–benefit dilemmas for patients, health insurers, and governments” [2]. We think that in the case of cancer, the modern medical “sacred cow”, personalized medicine, due to the extreme heterogeneity of cancers, faces serious difficulties in its application and formulas; this is contrary to possible generalized principles of cancer medicine. Such approaches seem especially relevant against the backdrop of a rapidly growing population. In such conditions, big medicine should focus on universal therapies. Chemotherapy can be just such an inexpensive and universal procedure, provided that a battlefield is found that ensures its supramolecular and universal action, i.e., applicable to different types of cancer.
In this short review, we would like to present arguments that not all possible potentials of cancer chemotherapy have been exhausted. Currently, almost the vast majority of procedures are aimed at intracellular processes, whether they involve the use of a higher rate of DNA replication or a cancer-specific increase or decrease in some product content. As we discussed earlier, this route of targeted therapy is doomed to fail. One of these seemingly very promising strategies may involve chemotherapeutic interventions aimed at disrupting the communication of cancer cells with their microenvironment.
2. Basic Definitions, Terms, and a Brief Presentation of Available Information
2.1. Tumor Tissue Compartments
As a rule, tumor tissue is considered to consist of three parts: 1) the tumor core (TC, in other words, a tumor nest or tumor cluster), comprising the majority of tumor cells in a certain limited area; 2) the tumor stroma (TS) with abundant stromal components surrounding the TC; and 3) the invasive margin (IM), a ~1 mm transition zone between the TC and the TS. Cancer cells in the IM may have a partial epithelial–mesenchymal transition (EMT) phenotype, expressing some EMT-determining genes that are not expressed in the central core of tumors [18]. IM plays a central role in orchestrating tumor–host interactions [19,20,21] (for a recent review, see [22]). The IM forms the front line of the tumor’s fight against the immune system, and the activity and density of immune cells in this zone may be higher than in the TC or the TC [19]. There is ambiguity in the use of the terms “stroma” and “microenvironment”. For simplicity, we will use the terminology used in Kalluri’s classic article [23]: “tumor stroma or tumor microenvironment. There are all components of a tumor except cancer cells. These are typically components of the host response to cancer cells, including the immune response. These terms are used interchangeably”.
2.2. Basement Membrane and Extracellular Matrix
The extracellular matrix (ECM) is an important component of the tumor microenvironment and is formed by a network of macromolecules in the extracellular space of tissues that provide a molecular scaffold for cell growth, survival, differentiation, and migration. The ECM provides structural support to cells and tissues and plays a key role in the functional properties of cells. The ECM is divided into two groups: the basement membrane and the interstitial matrix. The basement membrane (BM) is formed by a thin layer of ECM at the interface between the epithelial or endothelial layer and connective tissue or surrounding nerves, adipocytes, and muscle cells. The BM is primarily composed of laminin and collagen IV and serves as a structural barrier to cancer cell invasion, intravasation, and extravasation. Cancer invasion through the BM is the initial stage of tumor dissemination and metastasis [24,25,26]. In epithelial cancers, cells must penetrate the BM to metastasize. When malignant epithelial cells have broken through the basement membrane and penetrated into the adjacent stroma to a depth of 1 mm or less, microinvasion (MI) is said to be present [27].
2.3. Tumor Microenvironment (TME)
The TME mainly includes the extracellular matrix of tumor immune cells (TICs), fibroblasts, and secreted factors. TICs are often associated with sensitivity to immunotherapy and the prognosis of multiple cancers, but the prognostic role of individual cells in tumor prognosis is limited.
The mechanism of the breaching of human cancer cells into BM remains little investigated. Until recently, it was believed that this required degradation by proteases, since the cells (~10 microns) were too large to penetrate the BM pores (several nanometers). An active role in this process belongs to the tumor epithelium, immune cells, and cancer-associated fibroblasts (CAFs, see below) [28]. CAFs are thought to be capable of digesting BM components via matrix metalloproteinase (MMP) [29,30]. Recent studies indicate protease-independent BM invasion through physical forces generated by cancer cells, involving collective cellular interactions, proliferation, CAFs, and myoepithelial and immune cells. In this hypothesis, disruption of the BM, like metastasis, involves the cells pushing through the basement membrane into the stoma [26,31]. The ability of tumor cells to form various types of actin-rich protrusions, including invasive protrusions (invadopodia) and locomotor protrusions (lamellipodia [2D] or pseudopodia [3D]), may play an important role in BM disruption and tumor cell dissemination [26,32] (see below).
CAFs have also been reported to induce cellular membrane permeability through an MMP-independent mechanism; they remodeled the matrix by applying contractile forces, creating gaps between the BM fibers that allowed cancer cells to pass through [33]. However, all this information is preliminary and requires additional research [34]. It should also be noted that most tumors arise from adhesive epithelial cancer cells that have strong intercellular contacts. Metastasizing from the original tissue, epithelial tumor cells change their adhesive properties through the EMT process and synthesize proteins characteristic of mesenchymal cells, which leads to a decrease in the level of epithelial proteins and a decrease in the adhesiveness of intercellular contacts [35]
2.4. Extracellular Matrix
The interstitial matrix, which is mainly produced by stromal cells, fills the interstitial space between cells. The ECM is the noncellular stroma of the TME in which tumor cells, endothelial cells, mesenchymal cells, and various immune cells are located. It is rich in collagen types I, III, V, VI, VII, and XII, as well as proteoglycans and various glycoproteins such as TNC and fibronectin [24].
Growing research data suggest that ECM remodeling plays an important role in shaping the inflammatory and immune environment of tumors. ECM cytoskeletal remodeling, structural plasticity, and mechanical forces are critical factors for the transport, activation, and formation of immunological synapses (ISs). The ability to send and receive information through the dynamic ECM is achieved through cell–matrix adhesions. The adhesive structures of the cell matrix are mainly composed of integrins (the main receptors of the ECM), which have the unique ability to mediate bidirectional biomechanical signaling into and out of the cell. The types of contacts that these cells make with the ECM are among the most unique and have critical implications for TME biology. For example, CAF-ECM adhesions have been shown to have significant effects on CAF-regulated functions and represent stromal signatures predictive of overall patient survival [26,36].
Cells bind to the ECM with varying affinities through various specific receptors and cell membrane molecules. Through these interactions, various signals are transmitted between and within cells that regulate gene expression and cellular responses. The ECM also provides various docking sites for regulatory molecules such as cytokines, growth factors, and enzymes. Enzymatic breakdown of ECM molecules leads to remodeling of the extracellular environment and the release of various bioactive mediators. Several proteases are involved in ECM remodeling, such as plasminogen activators, matrix metalloproteinases (MMPs), and cysteine proteases [24,37]. Cancer development and progression is associated with increased extracellular matrix deposition and cross-linking. Chemical and physical signals received from the ECM are essential for cancer cell proliferation and invasion. The ECM is generally considered to be a product of stromal cells, predominantly CAFs [37]. However, it has recently become clear that cancer cells also play an important role in the production and remodeling of the ECM. Cancer cells synthesize significant amounts of collagen, fibronectin, and tenascin C (TNC). Cancer cells produce fewer extracellular matrix proteins (<10% in pancreatic cancer) than stromal cells, but a number of cancer cell-derived matrix proteins may contribute to tumorigenesis and metastasis [24,37,38,39,40].
Interaction between a cell and its microenvironment takes place by means of integrin receptors binding to extracellular matrix proteins. Cancer cell invasion and metastasis possibly depend on this interaction [41]. Recent studies highlight the ECM as an active regulator of immunity in cancer [37,42,43] in addition to its known role in regulating the behavior of the CAFs in surrounding stroma [44]. In particular, macrophages, an important component of the TME of cancer tumors, are actively involved in the remodeling of cancer ECM. It was suggested that ECM and ECM-associated molecules could play a regulatory role in antitumor immunity. This makes it possible to use tumor–ECM interactions as a target to enhance immunotherapy [45,46,47].
2.5. Paracrine and Juxtacrine: Two Types of Cell Signaling
Communication between cells describes the way a cell can provide information to other cells. There are chemical and physical communications [48]. Extracellular pathways are used first, for example, in the secretion of signaling molecules (e.g., exosomes, cytokines, and metabolites). Physical communication describes direct interactions between cells that change the behavior of neighboring cells [36]. Intercellular contacts are crucial for the existence of the organism, in particular, for the emergence and evolution of tumors. However, the complexity of interaction networks of cancer and host cells is still far from being well understood [49,50]. A fundamental analysis of the patterns of information exchange between cells was published in 2012 by Perrimon et al. [51]. See also [15,52,53].
Existing classifications of biochemical signaling processes define autocrine signals if the cell sends a signal to itself. Paracrine signaling occurs between cells situated near one another. Juxtacrine signaling takes place if cells are situated in close contact with each other. Finally, endocrine signaling represents the exchange of information, e.g., by hormones between distant cells or organs through the bloodstream [54].
In all cases, the ligands bind to the receptors of the cell receiving the signal. It is important to note that paracrine signaling molecules are released into the extracellular space and then diffuse to reach the receptor of the receiving cell. On the other hand, juxtacrine signaling requires only direct contact between cells and does not release signaling molecules into the extracellular space. Perrimon et al. distinguish three types of juxtacrine signaling: (a) a protein from one cell binds to its receptor on the surface of an adjacent cell, as occurs at synapses (see below); (b) ECM glycoprotein and the membrane protein interact, for example, a receptor on one cell binds to its ligand on the ECM secreted by another cell; and (c) the signal is transmitted directly from the cytoplasm of one cell to the cytoplasm of an adjacent cell through a small tube [51,55] (see also [55]).
The latter case is characteristic of gap junctions, which, for the transport of ions and small soluble molecules, connect the cytoplasm of two neighboring cells through the clustering of tens to thousands of intercellular channels. Gap junctions are involved in a wide range of biological processes and consist of bonds that include six proteins—connexins [56]. Immunological synapse (IS), predominantly involving membrane ligands and receptors, is formed at the interface between a T cell and an antigen-presenting cell (APC) during the adaptive immune response, or a cancer cell. Virological synapses are similar to ISs [57].
Recently, an unexpected juxtacrine communication mechanism was identified that utilizes characteristic protrusions of plasma membrane cells to facilitate sensing of the extracellular environment, intercellular information exchange, and materials processes for chemical signaling through direct contact over long distances. Cells can use filopodia for this purpose—exploratory cytoplasmic projections consisting of F-actin [58]. Filopodia can extend over 800 µm and have a diameter of 100–500 nm [59]. Cell-connecting tunnel nanotubes (TNTs) are F-actin-based structures that form direct cytoplasmic connections between distant cells. TNT has been observed in a variety of cell types, including neuronal, immune, cancer, and stromal cells. Two types of intercellular connections can be formed: cytoneme and TNT [59]. In the case of a cytonema, there is no cytoplasmic connection between two cells; this type of connection is considered non-tubular. To transmit signals, they use a specific ligand–receptor interaction between the tip of the cytoneme and the target cell. TNTs are F-actin-based tubular junctions that are distinct from filopodia and cytonemes because they allow the exchange of cell surface molecules and cytoplasmic contents between distant cells [58,60]. TNTs are long-range cytoplasmic intercellular channels for direct intercellular communication, independent of soluble factors. Uniquely, these structures enable the rapid exchange of cellular cargo between connected, non-contiguous cells, including organelles, vesicles, molecules, ions, and pathogens [60]. Cytonemes can reach lengths of several hundred microns (most mammalian cells have a diameter of 10 to 100 microns). The length of the filopodia is consistent with the radius of distribution of signal molecules secreted [51,61,62]. These protrusions can deliver signals in both ways: from the sender to the recipient and back [48,54,60,63,64,65,66,67]. The protrusions are heterogeneous [57] and can form, for example, TNTs or tumor microtubes (MTs), which consist of actin and function as intercellular bridges connecting a variety of cell types. TMs are longer and have a larger diameter compared with TNTs observed in vitro [58,60,62,68,69]. TNTs play an important role in signaling through various types of immune cells [70]. They can transport DNA, mitochondrial DNA organelles, vehicles, exosomes, proteins, genetic material, ions and small molecules, viral RNA, and non-coding RNA from cell to cell under physiological and pathological conditions [71,72,73]. The connections of multiple cells by TNTs form functional cellular networks. TNTs are minimally present in adult healthy tissues (may be except the immune system) in physiological conditions (see Figure 1). It is suggested that TNTs are stimulated by different diseases and inflammatory stimuli and may be involved in disease transmission [74]. The existence of the TNTs between cancer cells and stromal cells ex vivo and in vivo was reported [75]. This difference between cancer and healthy cells allows us to think about its therapeutic applications.
It has been suggested [60] that intercellular communication via TNTs and the TME may promote tumor survival and progression. These membrane tubes can connect cells over considerable distances. Cancer cells form TNTs with other cancer cells as well as with microenvironmental cells [65,66,80]. The protrusions provide membrane continuity between cells located at distances exceeding 100 μm. These tubes can be open-ended to maintain cytoplasmic continuity or closed-ended cytonemes, which allow intercellular ligand–receptor signaling [69]. In a solid tumor, cancer cells can be spread, making communication through gap junctions or exosomes not possible. But TNTs and the TME can allow direct distant physical contact. Intercellular communication between tumor cells and the tumor microenvironment is involved in metastasis [60]. The existence of TNTs in many types of cells was confirmed in numerous recent studies [77]. TNTs can also form a membrane junction between the donor cell and the target cell, which may promote IS formation [81] or gap junction signaling [63,69]. TNTs can be used by pathogens, especially viruses, to facilitate their spread.
Of all the methods of direct communication, synapses appear to be the best supramolecular target. This kind of contact, common to all types of immune cells, use for cell–cell signaling direct ligand–receptor adhesion [56,82,83,84,85,86]. IS has also been used for intercellular communication mediated by vesicular traffic, as well as a site for the active release of soluble molecules [87].
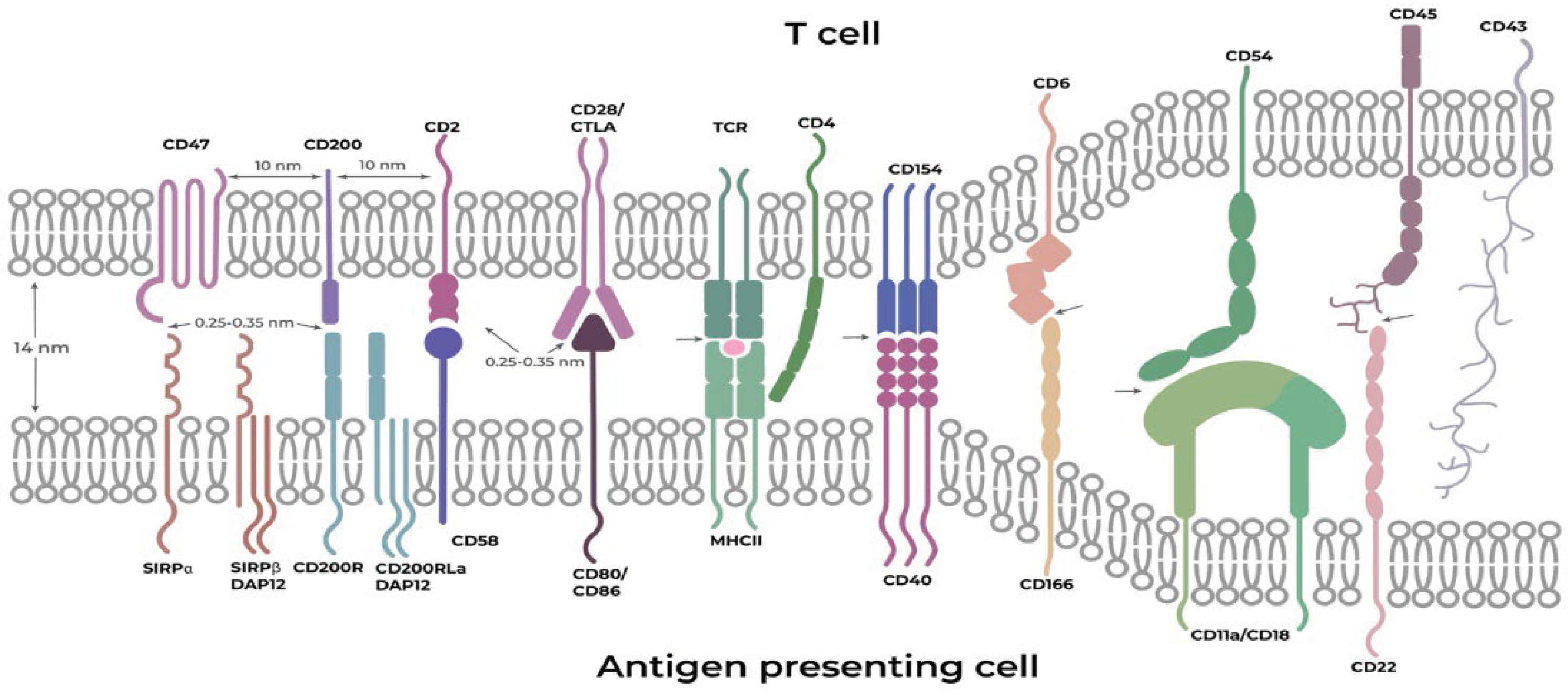
Synapses are specific intercellular interfaces that have a unique membrane organization and provide highly informative signal transmission between contacting cells. Cell–cell interfaces represent unique compartments that can serve as supramolecular targets for medical intervention [54,88]. They are heterogeneous, starting with a variable width of cleft formed between T cells and antigen-presenting cells, whose average interfacial gap is ~14 nm (Figure 2), continuing with a variable length of the synapse itself from short interfaces (hundreds of nanometers) to very long ones, extending over several micrometers and ending with a variable duration of the synapse, starting from short transient contacts and continuing to quite long, permanent neuronal contacts. The lateral organization of proteins within junctions plays an important role in the integration and regulation of signals from multiple sources. During the formation of IS, TCR molecules quickly form submicron-sized microclusters, which work as sites of active signaling [88].
Another physical feature of cell–cell boundaries is a consequence of the different sizes of cell surface molecules and the limited size of the gap between membranes at the interfaces. The height of proteins can determine whether they can be accommodated within an interface and participate in cell–cell interaction enhancement [88,89].
Figure 2.
Schematic illustration of interactions between an antigen-presenting cell and a T cell. Approximate distances between neighboring synapse components on the same cell–cell interface surface were approximated from the data of [90]. Approximate distances between interacting ligands and receptors were estimated based on the approximate hydrogen bond sizes (hydrogen bond 0.276 nm and typical ~0.45 nm distance of hydrophobic contacts).
Figure 2.
Schematic illustration of interactions between an antigen-presenting cell and a T cell. Approximate distances between neighboring synapse components on the same cell–cell interface surface were approximated from the data of [90]. Approximate distances between interacting ligands and receptors were estimated based on the approximate hydrogen bond sizes (hydrogen bond 0.276 nm and typical ~0.45 nm distance of hydrophobic contacts).
3. General Physicochemical Principles of Direct Intercellular Contacts
3.1. Two Postulate Principle
The extracellular protein–protein interactions vary from very strong to very weak and observed between membrane-embedded receptor proteins. Intercellular recognition between two opposing membranes, therefore, requires large multivalent arrays (clusters) consisting of hundreds, or even thousands, of receptors that increase the total avidity of the interaction with the ligands to a point high enough to prompt a signaling event [88,91]. For example, T cells are capable of detecting a single ligand on antigen-presenting cells; however, nearly ten of them are necessary for T cell activation [92]. As for signaling by secreted ligands, the range of signaling elicited by the ligand-producing cell can span tens of cell diameters. The distribution of ligands over such a distance generates a graded signaling profile, which leads in many cases to distinct responses at different distances from the source of signals, rather than a single on/off switch. Various factors affect the final distribution of the ligand and hence the resulting signaling profile [15,51]. The interactions are highly diverse; however, many of them require large clusters to be sufficiently strong, as was mentioned above. A common feature of the clusters is the close location of their components on the surface (about 10 nm), and despite the different quantitative compositions, they have similar qualitative features. It should be noted that an extremely important feature of clusters is that their components interact cooperatively [15,88].
In the following discussion, we will use the postulate principle, which is defined as “statements or claims assumed to be true especially as the basis of a process of reasoning” (Merriam-Webster Dictionary).
Postulate 1: Extracellular vesicles—highly problematic mean of intercellular communication.
All cells release extracellular vesicles (EVs) that can be broadly divided into two categories, ectosomes and exosomes. Ectosomes pinch off the surface of the plasma membrane and embrace microvesicles, microparticles, and large vesicles, ~50 nm to 1 μm in diameter. Exosomes are particles with a size of ~40 to 160 nm (average ~100 nm) in diameter [93]. Fibroblasts according to a popular point of view can be “educated” by cancer cells and also secrete EVs and establish intercellular communication that possibly benefits cancer progression [94].
It is widely believed that EVs mediate intercellular communications by functioning as messengers (for a recent review see [94,95]). EVs contain as cargo various biomolecules, in particular, nucleic acids and proteins. It has been hypothesized that the cargo being transferred from cell to cell may cause phenotypic changes in the recipient cells. The hypothesis that EVs could transfer RNA-encoded information from cell to cell was put forward by Valadi et al. [96] in 2007. The title of this publication, “Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells”, speaks for itself. Actually, the authors demonstrated that EVs purified from murine cell lines do contain small RNAs, and these RNAs may be found in human cells after exposition to EVs. Due to limits in the sensibility and specificity of the experimental techniques used in their research, the weak signal observed might have been a false positive. Also, this transfer may be a consequence of the use in vitro of non-physiologically high concentrations of EVs not corresponding to the in vivo conditions. However, since then, quite a lot of articles have described how EVs may participate in cross-talk among cancer and stromal cells. This information was obtained mainly from in vitro experiments, and adequate confirmatory in vivo studies have not been reported [97]. There are numerous difficulties in purification, standardization of materials and methods, and inadequate bioassays for determining the functionality of EVs for obtaining reliable evidence of intercellular trafficking [98].
Let us continue our reasoning with the quantitative data that we managed to find in the literature [99].
In order for a cancer cell bunch to be called a tumor, it must contain about 200 cells (0.2 mm diameter). Each cell is about 20 μm in diameter. A 1 cm3 cancer has about 100 million cells, a 0.5 cm3 cancer has about 10 million cells, and a 1 mm3 cancer has about 100 thousand cells. It is reasonable to assume that exosomes are secreted into the stromal environment from the IM cancer compartment (see above). Let us try to make a semi-quantitative assessment of the scale of this secretion.
First of all, let us try to estimate the secretion rate (SR) of exosomes from the cells. Although the data are very sparse, they allow calculating a first approximation. Breast cancer cells reportedly shed ~60 to 65 exosomes per cell per hour [93]. SR 240–107 EVs per cell per min was reported for six cancerous cell lines [100]. It should be mentioned that the authors acknowledge that the determined SR of EVs in previous studies may differ by orders of magnitude! In any case, these figures are sharply different from other publications and we will not use them in our further calculation. Medulloblastoma cells, glioblastoma and melanoma cells, and normal human fibroblasts release about 100–300 EVs per cell per hour [7]. The cell-specific EV secretion rate from six cancerous cell lines in human blood plasma estimates were highest for monocytes (2700 EVs/cell/hour) [101].
With a tumor volume of 1 cm3, it contains 108 cells [99] and its radius is ~0.6 cm. The surface of such a tumor is 4πR2 = 4.32 cm2 = 4.32 × 1014 nm2 (1cm2 = 1014 nm2). It contains 4.32 × 1014/1.2 × 109 = 3.6 × 105 cells on the surface monolayer of the IM.
Let us assume for simplicity that each cell secretes 100 exosomes. This means that every hour a tumor with a volume of 1 cm3 releases about 3.6 × 107 exosomes per hour into the medium from the surface monolayer. If we assume that the IM width is 1 mm and the cell diameter is 100 microns, then the IM contains approximately 10 cell monolayers. If they all secrete exosomes, then the number of exosomes secreted into the stroma increases by an order of magnitude. These are diverse exosomes with different sets of information. In addition, to interact with stromal cells, exosomes must penetrate the BM barrier [102]ю Apparently, the probability of encountering an exosome with the information required for the “desirable” information for a cancer cell and subsequent productive adaptation of the exosome by the target cell is not so high, although it seems difficult to calculate this probability due to many uncertainties. This supports the hypothesis that simple uptake of exosomes emitted by cancer tumors by stromal cells is unlikely to be used as a source of instructions for reprogramming stromal cells, in particular, to convert them into CAFs.
As early as 2012, one of the authors of this review (ES) published a critical paper [103] questioning: (1) whether data obtained in vitro on RNA transfer are physiologically relevant in terms of really existing in in vivo molar concentrations, and (2) whether the potential of exosomes and other EVs to deliver information (e.g., through nucleic acids) is physiologically relevant. The conclusion was that this is extremely unlikely. Despite a sufficient number of objective analytical reviews that in general agree with this point of view (for most recent analytical reviews see [97,98,104,105]), the flow of articles beginning with words like the following: “Extracellular vesicles are key contributors to cancer where they play an integral role in cell-cell communication and transfer pro-oncogenic molecules to recipient cells thereby conferring a cancerous phenotype” does not subside. Apparently, in our science the effect still exists, which could be called the “William Faulkner effect”, which is described as follows: “They all talked at once, their voices insistent and contradictory and impatient, making of unreality a possibility, then a probability, then an incontrovertible fact, as people will when their desires become words.” (William Faulkner “Sound and Fury”). However, the transportation of these particles through TNTs, the MT, and synapses cannot be excluded [60,106].
Postulate 2: During the evolution of a cancer tumor, the transfer of information between cells occurs mainly through direct physical interactions between cells.
3.2. Immune and Other Cells within Tumor Compartments: A Quick Glance
Tumor-infiltrating immune cells and stromal cells are thought to play a crucial role in tumor progression and response to therapy. Cytotoxic T lymphocytes (CTLs), myeloid antigen-presentation cells, and CAFs have various preferable locations within tumors [19,107].
There was a higher enrichment of immune infiltrate in the stroma compared with the other tumor cell compartment, but it was approximately equal in the tumor core and the IM, at least in colorectal cancer [108] and metastatic pleural mesothelioma [109]. The cancer ECM excludes some types of immune cells (for example, infiltrating CD8+ T cells) but may increase the content of others, such as macrophages and neutrophils [110]. The content of tumor-infiltrating lymphocytes (TILs) may be essential for prognostic purposes [111]. CD8+ T cells are the main effectors of anticancer immunity. They recognize and destroy cancer cells containing cancer neoantigens. On the other hand, Treg cells, M2 macrophages, and myeloid-derived suppressor cells can cause immunosuppression and enhance tumor growth [112].
CAFs, one of the major components of the TME, originate from different predecessors and are functionally heterogeneous [113,114,115]. Most of the CAFs are thought to appear from normal fibroblasts due to activation by cancer cell stimuli. The most recent information concerning the function of CAFs in tumor progression can be found in [34,116,117]. We would think that the most reliable way for cancer cells to transform various stromal cells into CAFs is direct contact between the two cells [118], with subsequent transfer of either signal molecules or vesicles (see below).
CAFs possibly participate in cancer invasion and metastasis using signaling between CAFs, cancer cells, and the ECM by direct contact, secretion of cytokines, or EVs. Myofibroblastic CAFs (myCAFs) may directly interact with malignant cells; on the other hand, inflammatory CAFs are more distant from the cancer cells and, possibly, used for this goal of secretion of IL-6 and other cytokines [119]. In 2017, Öhlund et al. demonstrated that some myCAFs were mainly positioned close to tumor foci and required juxtacrine interactions with cancer cells for their formation [78,79]. CAFs participate in the tumor–immune cell crosstalk. Normal fibroblasts retard tumor growth, but some subsets of CAFs may accelerate cancer cell proliferation, invasion, enhance drug resistance, and reduce anti-tumor immunity [112].
To better understand the pathways of information exchange between cells, it is important to evaluate the distances separating cells from each other in tumor compartments. To date, the distance between immune cells and stromal cells in these fields is not well understood [19]. The distance between CD8+ T cells and tumor cells and the distance between Tregs and tumor cells were estimated by [120]. Generally, total T-cell infiltration was comparable between the CT and the IM: 349.2 cells/mm2 in the CT vs. 359.6 cells/mm2 in the IM. CD8+ T cells in the CT were more clustered than in the IM (average distance between cells: 24.8 μm vs. 61.2 μm) and located closer to tumor cells (39.4 μm vs 104.6 μm). Tregs were found to be located farther away from tumor cells in the CT than in the IM: (221.0 μm vs 164.8 μm). There were no differences regarding the spatial analysis in the IM. In general, immune cell quantity was maximal at a distance of approximately 15–30 μm from tumor cells (typical eukaryotic cell size is 10 to 30 microns) [19,120]. Also, FoxP3+ TILs must be located within a distance between 30 and 110 μm of CD8+ T cells [121,122]. The distribution of distances between Ki67+ (monoclonal antibody Ki67 positive) carcinoma cells and nearest CAFs in breast carcinoma tumors on average was about 25 μm [123].
Summarizing this incomplete and scattered information, we can, however, conclude that both immune cells from each other and immune cells from cancer cells in tumor compartments are located at a distance of 30–100 microns. This distance is quite surmountable with the help of TNT and TMT, but the question arises of how they, in an environment filled with extracellular matrix, reach each other in order to form close contact, 2–20 nanometers, for the formation of gap junctions or synapses. This will be discussed in the following sections. Perhaps the search systems of cells, for example filopodia, are involved in this process, and help cells come together at such a close distance. One can make a cautious assumption that rapprochement can occur after the formation of nanotubes due to their contraction, although it is usually believed that, on the contrary, tubes arise after the synapse (see, for example, [82]).
3.3. Fibroblasts, Immune and Cancer Cells Interactions
Epithelial cancer cells (ECCs) and other components of the TME do not physically interact before BM degradation [36]. At this point, the cancer cells turn invasive, and physical communication between them and stromal cells participates in the metastatic process [48]. When it starts, ECCs possibly expose on the surface adhesion molecules, such as cadherins, integrins, connexins, and others [36] and their contact with neighboring cells and immune cells and CAFs form, possibly, synapse-like connections. Physical cell–cell communication between fibroblasts and other cell types can influence their chemical communication with immune cells [36,124]. There is only indirect evidence for the formation of synapse-like structures between CAFs and cancer and immune cells. The main one is associated with the expression of an inhibitory receptor expressed by T cells as a synapse component, Programmed Cell Death Protein 1 (PD-1), whose main ligand PD-L1 is expressed in cancer cells and surrounding stromal cells [125,126,127,128]. Until recently, PD-1 was considered only expressed on the surface of immune cells, whereas its ligands, PD-L1 and PD-L2, are only expressed in tumor cells. However, recent studies revealed the intrinsic expression of PD-1 in melanoma and some other cancers. Tumor-intrinsic PD-1 expression seems to be widespread in many tumor types [126]. It was indicated that close proximity between PD-1–PD-L1 and ISs is required for PD-L1 function to disturb the T-cell receptor (TCR)–major histocompatibility complex (MHC) interaction [129]. The cis-interaction between PD-1 and PD-L1 was reported [130] to occur if both were expressed on the same cell surface, which indicates their close positions on the cell membrane [66]. T cells also interact with a number of cell surface ligands displayed by CAFs, including JAM2, OX40L, and PD-L2, thus significantly increasing the duration of contact time between T cells and CAFs [131].
All these data clearly indicate that, at least where the PD1–PD-L1 interaction is detected, the formation of a synapse-like structure is inevitable, including the formation of clusters of ligand–receptor pairs. Taking into account the general laws of direct cell–cell interaction, which require the formation of synapse-like cluster structures to form a strong contact (see above), it is quite reasonable to assume that during the interaction of CAFs with immune and tumor cells, in addition to TNT and TMT, synapses are also formed that can serve as supramolecular chemotherapeutic targets. Considering that in tumors PD-1 is co-expressed with PD-L1 on many types of TME cells, in particular, on tumor-infiltrating macrophages, MDSCs, dendritic cells, etc., one can suggest that synapse-like interactions may be rather general in the tumor microenvironment. Naturally, many authors suggest the existence of other types of direct contact between TME cells and cancer cells, for example, gap or adherence junctions [65].
3.4. Communication of the Cancer and Microenvironmental cells—a Supramolecular Target for Chemotherapy
We are witnessing the failure of molecular targeting in medicine [2]. Some authors (see, for example, [132]) consider this as a crisis of the paradigm of the “magic bullet” theory. The extreme heterogeneity of the cancer tumor, the extreme complexity of the process of its evolution, and the unimaginably complex metastatic finale themselves create an intractable complexity. But this complexity is not only caused by the involvement of a huge number of processes and components but also the insurmountable unpredictability of the emergent properties of the system that arise as a result of their countless interactions form principally unsolvable problems [133]. This is why chemotherapy, despite its side effects on the patient’s health, still remains an unavoidable therapeutic option and remains the choice for advanced-stage malignancies where surgery and/or radiation therapy cannot be prescribed [16]. There also exist some new ways of using chemotherapy; for example, the use of epigenetics as the chemotherapeutical target [134,135]. Also, combination chemotherapy combining independently active cancer therapies enables us to overcome, to some extent, tumor heterogeneity [17,136]. However, the most universal and reliable way to overcome tumor resistance to treatments may be to redirect them to the tumor microenvironment [15,132]. In recent years, immunotherapy has become highly popular in the treatment of tumors. However, most patients do not respond to treatment or develop resistance. Therefore, in order to achieve a better therapeutic effect, the combination of immune checkpoint inhibition and other therapies is one of the fruitful strategies [14].
However, it is evident that to defeat cancer a more universal strategy is highly desirable. In this connection, we will return to Hanahan’s paper, which we cited above [2]: “A military battlespace is a strategic approach that takes an integrative, holistic view of war, incorporating information about the enemy’s characteristics and armamentarium, precise topographical maps of all potential battlefields and war zones…. The metaphorical war on cancer needs to adopt an analogous cancer battlespace plan, integrating knowledge about similar variable……, including: a census of a cancer’s variously specialized cells, the basis of their corruptions (e.g., genetic mutations, reprogrammed regulatory circuitry), and their lines of communication …”.
Analysis of such a strategic plan as applied to a tumor, combined with the principle of Occam’s razor, which recommends choosing the most economical path to solve a problem, and the analogy with military operations, suggests that the destruction of the lines of communication that turns a cancerous tumor into a kind of living organism may be necessary and sufficient means of tumor destruction.
As we tried to demonstrate above, the arsenal of intercellular communications is diverse and covers all elements of the existence of the tumor “organism” [87,137]. The list of players implicated in cell–cell recognition and adhesion has grown to include the cadherin superfamily comprising classical, atypical- and proto-cadherins, nectins, CAMs, connexins, lectins, eph/Ephrin, and others. Such a rich palette of adhesion proteins has the potential to provide radically different effects upon cell–cell contact, from repulsion to adhesion and everything in between [138,139]. A total of 66% of drugs in the DrugBank target the surface proteins. A plasma membrane with its embedded proteins creates the environment for all these proteins to function cooperatively, thus achieving the optimal physiological output [140].
Although TNTs and the TM have been considered potential drug targets, however, we are still very far from translating these ideas into clinical practice [26,141,142]. We will discuss synapses and synapse-like structures as the most reliable supramolecular targets.
As mentioned above, the development of T cell-mediated immunity includes the assembly of the ISs—the complicated interface between the T cell and the APC [52,87,88,89,137,143,144,145,146,147,148,149,150,151]. The knowledge acquired to date about the mechanisms of IS assembly underscores this structure as a robust pharmacological target [137]. An outstanding example of the efficiency of this postulate is tumor immune checkpoint therapy. The suggestion formulated above—that not only does direct contact occur between the tumor and immune cells but it also occurs between the tumor and other stromal cells [53]—opens a new platform for universal chemotherapy.
4. Simple Principles of Specific Chemical Effects on Synapses
Sola dosis facit venenum (“Only the dose makes the poison”)
Theophrastus Philippus Aureolus Bombastus von Hohenheim (Paracelsus)
With all the differences in synapses, they retain common properties: 1) connections between cells are provided by the interaction of clustered protein ligands and receptors; 2) these connections are located close to each other; and 3) collective interaction forms a common functional unit and is cooperative in nature. In addition, and very importantly, synapses are not only clusters of ligand–receptor interactions but also a limited space providing a focused cell–cell exchange of signaling molecules and particles, such as cytokines and signaling vesicles. Consequently, synapse destruction should lead to multidimensional functional tumor collapses. In addition, the emergence of tumor resistance to this effect is practically impossible. This effect is universal for various tumors—”one size fits all”. IS has already been indicated as a robust pharmacological target [88,137]. The various activities of molecules involved in IS assembly and function in one local and narrow space provide the necessary platform for supramolecular blow at “one target” [88,137].
The cluster arrangement of interacting components in the cell–cell interface of the synapse creates four unique opportunities to increase specificity when the concentration of the agent is reduced:
1. From simple kinetic considerations, the probability of damage to at least one component of the cluster by a certain reagent that specifically interacts with proteins exceeds the probability of damage to an individual protein on the cell surface or located in the extracellular matrix in proportion to the number of proteins involved in the formation of the synapse. It becomes possible to sharply reduce the dose of the reagent by hundreds of times and achieve its synapse-specific action, thus implementing the concept of Paracelsus that “materials which are poisonous in large doses may be curative in small doses” [152].
2. The specificity of the reagent can be further increased by using cross-linking reagents. In this case, the reagent, having contacted one of the components of the synapsoidal cluster, almost automatically reacts with one of the neighboring ones on the same surface or on opposite surfaces. This makes the modification durable (Figure 3A).
3. Non-cell-permeable crosslinkers can be used, which further increases specificity by limiting modification to only surface-exposed molecules. Examples of crosslinking agents are presented in Figure 3B.
4. Finally, one can enhance the effect by attaching an activated reagent to the crosslinker through a cleavable connection, which, after crosslinking, can be activated and separated from the crosslinker and, upon entering the cell, disrupt its vital functions (Figure 3C).
The Figure 3 demonstrates one possible strategy for using the above features of synapses for cancer therapy.
To summarize, the proposed approach uses a chemical attack on the vulnerable communication field of the cancer tumor with the environment by blocking the synapses formed by cancer cells and cells of the immune system and the synapse-like contacts of fibroblasts with cancer cells forming with a high probability.
5. Instead of a Conclusion: Immunochemotherapy as a General Strategy Aimed at Poorly Fortified Areas of Cancer Tumors
It is now clear that the current gene-targeted cancer therapy paradigm must be replaced by a more effective one. To beat cancer, we must discard desperate attempts to influence the intracellular interactome of cancer, which is varied and complex, and twist our attention to a more accessible system of interactions between cancer cells with the surrounding microenvironment. The survival of cancer cells depends on their interaction with their TME. Disruption of these connections, which are vital for the development of tumors, is a difficult but realizable and promising task from the point of view of real therapeutic effects. A major example of the practicality of this concept is tumor checkpoint immunotherapy. However, this approach destroys only a few of the multiple contacts of cancer cells with the TME, and perhaps because of this limitation, a rather large proportion of patients do not respond to one or another immunotherapy. More tumor–TME interactions must be disrupted to allow the development of more robust therapeutic strategies.
In a sense, an ideology comparable to that described here was used with a combination of immune checkpoint therapy with so-called weaponized antibodies, and tumor-targeting antibodies coupled with toxic chemicals. Recently, a great success of such a strategy was reported in treating bladder cancer. First, the monoclonal antibody pembrolizumab, which is a repressor of synapse-embedded ligand PD1 hindering immune T-cells, was blocked (this is immune checkpoint therapy, which releases T-cells for antitumor activity). Then, the treatment with a bifunctional agent, antibody–drug conjugate (ADC), enfortumab vedotin was undertaken. Enfortumab is an antibody against a member of a family of adhesion proteins, nectins (nectin-4), exposed on the tumor surface [153]. Nectins are clustered on some tumor surfaces and take part in synapse formation with immune cells. Binding partners of the nectin protein family of adhesion molecules are widely expressed in the immune system [154,155]. The nectin-targeting antibodies were coupled through a cleavable linker with a toxic chemical (monomethyl auristatin E, MMAE) that interrupts cell division by disrupting replication spindle microtubules [156].
In this strategy, as well as in our suggestions, the first blow is struck at the synapse: the authors of the work described by [156] block repressive synapse component PD-1; in our suggestion, it is a general blow directed at synaptic protein clusters. In the second blow, the authors use a toxin, MMAE, attached with a cleavable link to separate antibodies. In our approach, a toxin, which can be the same MMAE or any other toxic intracellular agent, is a part of the same agent that destroys the synapse. However, the procedure suggested here looks considerably more simple, universal, and less expensive.
It should be noted here that the chemotherapeutic destruction of cancer cells using, for example, suicide gene therapy also leads to the destruction of metastases. This was noted in an early review [157], in our own work [158] and review, considering more recent experimental data [159]. Also, such an effect was reported in other chemotherapeutic cancer treatments, as well as in more recent work on chemotherapy by other authors. It has already been mentioned above that, at least in some cases, chemotherapies can augment tumor immunity, for example, by inducing immunogenic cell death [10,11,12]. Most likely, a similar general anticancer immune response should be expected using the proposed strategy. There is every reason to believe that the chemotherapy constructs proposed here can be delivered intratumorally. Recently, this delivery principle was discussed in detail and it was indicated that intratumoral injections with antitumor agents can be a workable strategy to reach higher local drug concentrations while at the same time reducing the risk of side effects (see, for example, [160,161]).
In this connection, we would like to return to the highly important problem of the practical application of cancer therapy in the context of a rapidly growing human population, which we have already touched upon in the introduction and which is addressed, in particular, by one of the leaders of modern oncology, Douglas Hanahan [2]. His concern is understandable, as many beautiful and even brilliant technologies based on the excellent achievements of modern molecular biology and genetics have turned out to be extremely expensive. As for the cost, personalized drugs are breaking all records. In 2017, the FDA approved two CAR-T drugs, namely Kymriah and Yescarta [162]. However, the drug treatments are extremely costly. For instance, a one-time infusion of Kymriah or Yescarta costs USD 373,000 for adults with advanced lymphomas. In the case of children and young adults with acute lymphoblastic leukemia, Kymriah treatment costs USD 475,000. Moreover, apart from the drug cost, many patients encounter severe side effects that may require them to stay in a hospital intensive care unit for weeks. Consequently, the overall treatment expenses can exceed USD 1 million [163]. Recently [164], the top five most expensive FDA-approved gene therapies were published, and the least expensive one costs USD 850,000. And the most expensive one costs USD 5,800,000. With such a high upfront price, only some patients can manage to pay for the medications. Also, such prices leave low- and middle-income countries out of personalized treatment (for a recent review, see [165]). We think that the approach proposed in this review would serve as basis for a desirable, inexpensive, and universal clinical procedure.
Funding
This research was funded by grants from the Moscow Government (research project no. 0903-2).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Brawley, O.W.; Goldberg, P. The 50 years’ war: The history and outcomes of the National Cancer Act of 1971. Cancer 2021, 127, 4534–4540. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Rethinking the war on cancer. Lancet 2014, 383, 558–563. [Google Scholar] [CrossRef]
- Surh, Y.-J. The 50-year war on cancer revisited: should we continue to fight the enemy within? Journal of Cancer Prevention 2021, 26, 219. [Google Scholar] [CrossRef]
- Henson, D.E. 25th Anniversary of the Signing of the National Cancer Act, December 23, 1971. Introduction. Cancer 1996, 78, 2582–2583. [Google Scholar] [CrossRef]
- Jacobs, H. Warhead. EMBO Rep 2011, 12, 91. [Google Scholar] [CrossRef]
- Sonnenschein, C.; Soto, A.M. Over a century of cancer research: Inconvenient truths and promising leads. PLoS biology 2020, 18, e3000670. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, M. Is cancer chemotherapy dying? Asian journal of transfusion science 2016, 10, S1. [Google Scholar] [CrossRef] [PubMed]
- Bizzarri, M. Do new anticancer drugs really work? A serious concern. Organisms. Journal of Biological Sciences 2017, 1, 9–10. [Google Scholar] [CrossRef]
- Singh, R.; Fazal, Z.; Freemantle, S.J.; Spinella, M.J. Mechanisms of cisplatin sensitivity and resistance in testicular germ cell tumors. Cancer Drug Resist 2019, 2, 580–594. [Google Scholar] [CrossRef]
- Emens, L.A.; Middleton, G. The interplay of immunotherapy and chemotherapy: harnessing potential synergies. Cancer Immunol Res 2015, 3, 436–443. [Google Scholar] [CrossRef]
- Wargo, J.A.; Reuben, A.; Cooper, Z.A.; Oh, K.S.; Sullivan, R.J. Immune Effects of Chemotherapy, Radiation, and Targeted Therapy and Opportunities for Combination With Immunotherapy. Semin Oncol 2015, 42, 601–616. [Google Scholar] [CrossRef]
- Wu, J.; Waxman, D.J. Immunogenic chemotherapy: Dose and schedule dependence and combination with immunotherapy. Cancer Lett 2018, 419, 210–221. [Google Scholar] [CrossRef]
- Sverdlov, E.D. Genetic surgery—a right strategy to attack cancer. Curr Gene Ther 2011, 11, 501–531. [Google Scholar] [CrossRef]
- Liu, Z.; Ren, Y.; Weng, S.; Xu, H.; Li, L.; Han, X. A New Trend in Cancer Treatment: The Combination of Epigenetics and Immunotherapy. Front Immunol 2022, 13, 809761. [Google Scholar] [CrossRef]
- Alekseenko, I.; Kondratyeva, L.; Chernov, I.; Sverdlov, E. From the Catastrophic Objective Irreproducibility of Cancer Research and Unavoidable Failures of Molecular Targeted Therapies to the Sparkling Hope of Supramolecular Targeted Strategies. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Anand, U.; Dey, A.; Chandel, A.K.S.; Sanyal, R.; Mishra, A.; Pandey, D.K.; De Falco, V.; Upadhyay, A.; Kandimalla, R.; Chaudhary, A.; et al. Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics. Genes Dis 2023, 10, 1367–1401. [Google Scholar] [CrossRef]
- Li, Q.; Lei, X.; Zhu, J.; Zhong, Y.; Yang, J.; Wang, J.; Tan, H. Radiotherapy/Chemotherapy-Immunotherapy for Cancer Management: From Mechanisms to Clinical Implications. Oxidative Medicine and Cellular Longevity 2023, 2023, 7530794, PMID: 36778203; PMCID: PMC9911251. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.; Dai, L.J.; Wu, S.Y.; Xiao, Y.; Ma, D.; Jiang, Y.Z.; Shao, Z.M. Spatial architecture of the immune microenvironment orchestrates tumor immunity and therapeutic response. J Hematol Oncol 2021, 14, 98. [Google Scholar] [CrossRef]
- Hendry, S.; Salgado, R.; Gevaert, T.; al., e. Assessing Tumor-infiltrating Lymphocytes in Solid Tumors: A Practical Review for Pathologists and Proposal for a Standardized Method From the International Immunooncology Biomarkers Working Group: Part 1: Assessing the Host Immune Response, TILs in Invasive Breast Carcinoma and Ductal Carcinoma In Situ, Metastatic Tumor Deposits and Areas for Further Research. Adv Anat Pathol 2017, 24, 235-251. [CrossRef]
- Koelzer, V.H.; Lugli, A. The tumor border configuration of colorectal cancer as a histomorphological prognostic indicator. Front Oncol 2014, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Liu, J.; Qian, H.; Zhuang, Q. Cancer-associated fibroblasts: from basic science to anticancer therapy. Experimental & Molecular Medicine 2023, 55, 1322–1332. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat Rev Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.F.; Xu, R.H. Function of cancer cell-derived extracellular matrix in tumor progression. J. Cancer. Metastasis. Treat. 2016, 2, 357–364. [Google Scholar] [CrossRef]
- Chang, J.; Chaudhuri, O. Beyond proteases: Basement membrane mechanics and cancer invasion. J Cell Biol 2019, 218, 2456–2469. [Google Scholar] [CrossRef] [PubMed]
- Brás, M.M.; Sousa, S.R.; Carneiro, F.; Radmacher, M.; Granja, P.L. Mechanobiology of Colorectal Cancer. Cancers 2022, 14, 1945. [Google Scholar] [CrossRef] [PubMed]
- Bane, A. Ductal carcinoma in situ: what the pathologist needs to know and why. Int J Breast Cancer 2013, 2013, 914053. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.C.; Lohmer, L.L.; Hagedorn, E.J.; Sherwood, D.R. Traversing the basement membrane in vivo: a diversity of strategies. Journal of Cell Biology 2014, 204, 291–302. [Google Scholar] [CrossRef]
- Nazari, S.S.; Doyle, A.D.; Yamada, K.M. Mechanisms of Basement Membrane Micro-Perforation during Cancer Cell Invasion into a 3D Collagen Gel. Gels 2022, 8. [Google Scholar] [CrossRef]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nature Cell Biology 2007, 9, 1392–1400. [Google Scholar] [CrossRef]
- Rice, A.; Cortes, E.; Lachowski, D.; Oertle, P.; Matellan, C.; Thorpe, S.D.; Ghose, R.; Wang, H.; Lee, D.A.; Plodinec, M.; et al. GPER Activation Inhibits Cancer Cell Mechanotransduction and Basement Membrane Invasion via RhoA. Cancers (Basel) 2020, 12. [Google Scholar] [CrossRef]
- Bravo-Cordero, J.J.; Hodgson, L.; Condeelis, J.S. Spatial regulation of tumor cell protrusions by RhoC. Cell Adh Migr 2014, 8, 263–267. [Google Scholar] [CrossRef]
- Glentis, A.; Oertle, P.; Mariani, P.; Chikina, A.; El Marjou, F.; Attieh, Y.; Zaccarini, F.; Lae, M.; Loew, D.; Dingli, F.; et al. Cancer-associated fibroblasts induce metalloprotease-independent cancer cell invasion of the basement membrane. Nature Communications 2017, 8, 924. [Google Scholar] [CrossRef]
- D’Arcangelo, E.; Wu, N.C.; Cadavid, J.L.; McGuigan, A.P. The life cycle of cancer-associated fibroblasts within the tumour stroma and its importance in disease outcome. British Journal of Cancer 2020, 122, 931–942. [Google Scholar] [CrossRef]
- Garrido-Urbani, S.; Vonlaufen, A.; Stalin, J.; De Grandis, M.; Ropraz, P.; Jemelin, S.; Bardin, F.; Scheib, H.; Aurrand-Lions, M.; Imhof, B.A. Junctional adhesion molecule C (JAM-C) dimerization aids cancer cell migration and metastasis. Biochimica et Biophysica Acta (BBA)—Molecular Cell Research 2018, 1865, 638–649. [Google Scholar] [CrossRef]
- Gardiner, J.C.; Cukierman, E. Meaningful connections: Interrogating the role of physical fibroblast cell-cell communication in cancer. Adv Cancer Res 2022, 154, 141–168. [Google Scholar] [CrossRef]
- Karamanos, N.K.; Piperigkou, Z.; Passi, A.; Gotte, M.; Rousselle, P.; Vlodavsky, I. Extracellular matrix-based cancer targeting. Trends Mol Med 2021, 27, 1000–1013. [Google Scholar] [CrossRef]
- Popova, N.V.; Jücker, M. The Functional Role of Extracellular Matrix Proteins in Cancer. Cancers (Basel) 2022, 14. [Google Scholar] [CrossRef]
- Rijal, G.; Becker, S. Carcinoma Cell-Based Extracellular Matrix Modulates Cancer Cell Communication. 2022. [CrossRef]
- Tian, C.; Öhlund, D.; Rickelt, S.; Lidström, T.; Huang, Y.; Hao, L.; Zhao, R.T.; Franklin, O.; Bhatia, S.N.; Tuveson, D.A.; et al. Cancer Cell–Derived Matrisome Proteins Promote Metastasis in Pancreatic Ductal Adenocarcinoma. Cancer Research 2020, 80, 1461–1474. [Google Scholar] [CrossRef]
- Doyle, A.D.; Nazari, S.S.; Yamada, K.M. Cell-extracellular matrix dynamics. Phys Biol 2022, 19. [Google Scholar] [CrossRef]
- Vyas, M.; Demehri, S. The extracellular matrix and immunity: breaking the old barrier in cancer. Trends in Immunology 2022. [Google Scholar] [CrossRef]
- Sun, X.; Wu, B.; Chiang, H.C.; Deng, H.; Zhang, X.; Xiong, W.; Liu, J.; Rozeboom, A.M.; Harris, B.T.; Blommaert, E.; et al. Tumour DDR1 promotes collagen fibre alignment to instigate immune exclusion. Nature 2021, 599, 673–678. [Google Scholar] [CrossRef]
- Chakravarthy, A.; Khan, L.; Bensler, N.P.; Bose, P.; De Carvalho, D.D. TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nature communications 2018, 9, 4692. [Google Scholar] [CrossRef]
- Robertson, C.; Sebastian, A.; Hinckley, A.; Rios-Arce, N.D.; Hynes, W.F.; Edwards, S.A.; He, W.; Hum, N.R.; Wheeler, E.K.; Loots, G.G. Extracellular matrix modulates T cell clearance of malignant cells in vitro. Biomaterials 2022, 282, 121378. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Yang, T.-H.O.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A. The immune landscape of cancer. Immunity 2018, 48, 812–830.e814. [Google Scholar] [CrossRef]
- Sutherland, T.E.; Dyer, D.P.; Allen, J.E. The extracellular matrix and the immune system: A mutually dependent relationship. Science 2023, 379, eabp8964. [Google Scholar] [CrossRef]
- Schwager, S.C.; Taufalele, P.V.; Reinhart-King, C.A. Cell-Cell Mechanical Communication in Cancer. 2019, 12, 1-14. [CrossRef]
- Almagro, J.; Messal, H.A. Volume imaging to interrogate cancer cell-tumor microenvironment interactions in space and time. Frontiers in Immunology 2023, 14. [Google Scholar] [CrossRef]
- Nishida-Aoki, N.; Gujral, T.S. Emerging approaches to study cell-cell interactions in tumor microenvironment. Oncotarget 2019, 10, 785–797. [Google Scholar] [CrossRef]
- Perrimon, N.; Pitsouli, C.; Shilo, B.Z. Signaling mechanisms controlling cell fate and embryonic patterning. Cold Spring Harb Perspect Biol 2012, 4, a005975. [Google Scholar] [CrossRef]
- Alekseenko, I.V.; Chernov, I.P.; Kostrov, S.V.; Sverdlov, E.D. Are Synapse-Like Structures a Possible Way for Crosstalk of Cancer with Its Microenvironment? Cancers (Basel) 2020, 12. [Google Scholar] [CrossRef]
- Sverdlov, E. Missed Druggable Cancer Hallmark: Cancer-Stroma Symbiotic Crosstalk as Paradigm and Hypothesis for Cancer Therapy. Bioessays 2018, 40, e1800079. [Google Scholar] [CrossRef]
- Mattes, B.; Scholpp, S. Emerging role of contact-mediated cell communication in tissue development and diseases. Histochem Cell Biol 2018, 150, 431–442. [Google Scholar] [CrossRef]
- Gilbert, S.F. “Juxtacrine Signaling” In Developmental biology 6ed.; (ed.), N.b., Ed.; Sunderland, Mass.: Sinauer Assoc.: 2000.
- Oliveira, M.C.; Verswyvel, H.; Smits, E.; Cordeiro, R.M.; Bogaerts, A.; Lin, A. The pro- and anti-tumoral properties of gap junctions in cancer and their role in therapeutic strategies. Redox Biol 2022, 57, 102503. [Google Scholar] [CrossRef]
- Marzo, L.; Gousset, K.; Zurzolo, C. Multifaceted roles of tunneling nanotubes in intercellular communication. Front Physiol 2012, 3, 72. [Google Scholar] [CrossRef] [PubMed]
- Abounit, S.; Zurzolo, C. Wiring through tunneling nanotubes--from electrical signals to organelle transfer. J Cell Sci 2012, 125, 1089–1098. [Google Scholar] [CrossRef]
- Casas-Tintó, S.; Portela, M. Cytonemes, Their Formation, Regulation, and Roles in Signaling and Communication in Tumorigenesis. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef]
- Roehlecke, C.; Schmidt, M.H.H. Tunneling Nanotubes and Tumor Microtubes in Cancer. Cancers (Basel) 2020, 12. [Google Scholar] [CrossRef]
- Hall, E.T.; Daly, C.A.; Zhang, Y.; Dillard, M.E.; Ogden, S.K. Fixation of Embryonic Mouse Tissue for Cytoneme Analysis. JoVE (Journal of Visualized Experiments) 2022, e64100. [CrossRef]
- Yamashita, Y.M.; Inaba, M.; Buszczak, M. Specialized Intercellular Communications via Cytonemes and Nanotubes. Annu Rev Cell Dev Biol 2018, 34, 59–84. [Google Scholar] [CrossRef]
- Allegra, A.; Di Gioacchino, M.; Cancemi, G.; Casciaro, M.; Petrarca, C.; Musolino, C.; Gangemi, S. Specialized Intercellular Communications via Tunnelling Nanotubes in Acute and Chronic Leukemia. Cancers (Basel) 2022, 14. [Google Scholar] [CrossRef]
- Eugenin, E.A. Role of cell-to-cell communication in cancer: New features, insights, and directions. 2019, 2, e1228. [CrossRef]
- Sato, A.; Rahman, N.I.A.; Shimizu, A.; Ogita, H. Cell-to-cell contact-mediated regulation of tumor behavior in the tumor microenvironment. Cancer science 2021, 112, 4005–4012. [Google Scholar] [CrossRef]
- Dominiak, A.; Chelstowska, B.; Olejarz, W.; Nowicka, G. Communication in the Cancer Microenvironment as a Target for Therapeutic Interventions. Cancers (Basel) 2020, 12. [Google Scholar] [CrossRef]
- Ljubojevic, N.; Henderson, J.M.; Zurzolo, C. The Ways of Actin: Why Tunneling Nanotubes Are Unique Cell Protrusions. Trends Cell Biol 2021, 31, 130–142. [Google Scholar] [CrossRef]
- Pinto, G.; Brou, C.; Zurzolo, C. Tunneling Nanotubes: The Fuel of Tumor Progression? Trends Cancer 2020, 6, 874–888. [Google Scholar] [CrossRef] [PubMed]
- Zaccard, C.R.; Rinaldo, C.R.; Mailliard, R.B. Linked in: immunologic membrane nanotube networks. J Leukoc Biol 2016, 100, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Shi, Y.; You, J. Immune Cell Connection by Tunneling Nanotubes: The Impact of Intercellular Cross-Talk on the Immune Response and Its Therapeutic Applications. Mol Pharm 2021, 18, 772–786. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, Y.; Qi, Z.; Cao, L.; Ding, S. Mitochondrial transfer/transplantation: an emerging therapeutic approach for multiple diseases. Cell Biosci 2022, 12, 66. [Google Scholar] [CrossRef]
- Driscoll, J.; Gondaliya, P.; Patel, T. Tunneling Nanotube-Mediated Communication: A Mechanism of Intercellular Nucleic Acid Transfer. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- McCoy-Simandle, K.; Hanna, S.J.; Cox, D. Exosomes and nanotubes: Control of immune cell communication. Int J Biochem Cell Biol 2016, 71, 44–54. [Google Scholar] [CrossRef]
- Zurzolo, C. Tunneling nanotubes: Reshaping connectivity. Curr Opin Cell Biol 2021, 71, 139–147. [Google Scholar] [CrossRef]
- Dubois, F.; Bénard, M.; Jean-Jacques, B.; Schapman, D.; Roberge, H.; Lebon, A.; Goux, D.; Monterroso, B.; Elie, N.; Komuro, H.; et al. Investigating Tunneling Nanotubes in Cancer Cells: Guidelines for Structural and Functional Studies through Cell Imaging. Biomed Res Int 2020, 2020, 2701345. [Google Scholar] [CrossRef]
- Sontheimer, H. Tumour cells on neighbourhood watch. Nature 2015, 528, 49–50. [Google Scholar] [CrossRef]
- Zhang, H.; Yu, X.; Ye, J.; Li, H.; Hu, J.; Tan, Y.; Fang, Y.; Akbay, E.; Yu, F.; Weng, C.; et al. Systematic investigation of mitochondrial transfer between cancer cells and T cells at single-cell resolution. Cancer Cell 2023, 41, 1788–1802 e1710. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. Journal of Experimental Medicine 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Sperb, N.; Tsesmelis, M.; Wirth, T. Crosstalk between tumor and stromal cells in pancreatic ductal adenocarcinoma. International journal of molecular sciences 2020, 21, 5486. [Google Scholar] [CrossRef] [PubMed]
- Sahu, P.; Jena, S.R.; Samanta, L. Tunneling Nanotubes: A Versatile Target for Cancer Therapy. Curr Cancer Drug Targets 2018, 18, 514–521. [Google Scholar] [CrossRef]
- Chauveau, A.; Aucher, A.; Eissmann, P.; Vivier, E.; Davis, D.M. Membrane nanotubes facilitate long-distance interactions between natural killer cells and target cells. Proc Natl Acad Sci U S A 2010, 107, 5545–5550. [Google Scholar] [CrossRef]
- Jash, E.; Prasad, P.; Kumar, N.; Sharma, T.; Goldman, A.; Sehrawat, S. Perspective on nanochannels as cellular mediators in different disease conditions. Cell Communication and Signaling 2018, 16, 76. [Google Scholar] [CrossRef]
- Linares, J.; Marin-Jimenez, J.A.; Badia-Ramentol, J.; Calon, A. Determinants and Functions of CAFs Secretome During Cancer Progression and Therapy. Front Cell Dev Biol 2020, 8, 621070. [Google Scholar] [CrossRef]
- Luo, M.; Luo, Y.; Mao, N.; Huang, G.; Teng, C.; Wang, H.; Wu, J.; Liao, X.; Yang, J. Cancer-Associated Fibroblasts Accelerate Malignant Progression of Non-Small Cell Lung Cancer via Connexin 43-Formed Unidirectional Gap Junctional Intercellular Communication. Cell Physiol Biochem 2018, 51, 315–336. [Google Scholar] [CrossRef]
- Aasen, T.; Leithe, E.; Graham, S.V.; Kameritsch, P.; Mayán, M.D.; Mesnil, M.; Pogoda, K.; Tabernero, A. Connexins in cancer: bridging the gap to the clinic. Oncogene 2019, 38, 4429–4451. [Google Scholar] [CrossRef]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: communicating for 50 years. Nat Rev Cancer 2016, 16, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Finetti, F.; Cassioli, C.; Baldari, C.T. Transcellular communication at the immunological synapse: a vesicular traffic-mediated mutual exchange. F1000Res 2017, 6, 1880. [Google Scholar] [CrossRef]
- Belardi, B.; Son, S.; Felce, J.H.; Dustin, M.L.; Fletcher, D.A. Cell-cell interfaces as specialized compartments directing cell function. Nat Rev Mol Cell Biol 2020, 21, 750–764. [Google Scholar] [CrossRef]
- Bertolotto, C.; Rosillo, J.C.; Botti, B.; Fernández, A.S. “Synapse-like” Connections between Adipocytes and Stem Cells: Morphological and Molecular Features of Human Adipose Tissue. J Stem Cell Dev Biol 2018, 1, 100003. [Google Scholar]
- Garcia-Parajo, M.F.; Cambi, A.; Torreno-Pina, J.A.; Thompson, N.; Jacobson, K. Nanoclustering as a dominant feature of plasma membrane organization. J Cell Sci 2014, 127, 4995–5005. [Google Scholar] [CrossRef]
- Wright, G.J. Signal initiation in biological systems: the properties and detection of transient extracellular protein interactions. Molecular bioSystems 2009, 5, 1405–1412. [Google Scholar] [CrossRef]
- Santos, A.K.; Tonelli, F.M.; Silva, D.A.; Gomes, K.N.; Ladeira, L.O.; Resende, R.R. The role of cell adhesion, cell junctions, and extracellular matrix in development and carcinogenesis. Trends in Stem Cell Proliferation and Cancer Research 2013, 13–49. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. 2020, 367. [CrossRef]
- Naito, Y.; Yoshioka, Y.; Ochiya, T. Intercellular crosstalk between cancer cells and cancer-associated fibroblasts via extracellular vesicles. Cancer Cell Int 2022, 22, 367. [Google Scholar] [CrossRef]
- Liu, Y.J.; Wang, C. A review of the regulatory mechanisms of extracellular vesicles-mediated intercellular communication. Cell Commun Signal 2023, 21, 77. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nature Cell Biology 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Margolis, L.; Sadovsky, Y. The biology of extracellular vesicles: The known unknowns. PLoS Biol 2019, 17, e3000363. [Google Scholar] [CrossRef] [PubMed]
- Somiya, M. Where does the cargo go?: Solutions to provide experimental support for the “extracellular vesicle cargo transfer hypothesis”. Journal of Cell Communication and Signaling 2020, 14, 135–146. [Google Scholar] [CrossRef]
- Del Monte, U. Does the cell number 109 still really fit one gram of tumor tissue? Cell Cycle 2009, 8, 505–506. [Google Scholar] [CrossRef]
- Wen, Y.; Chen, Y.; Wang, G.; Abhange, K.; Xue, F.; Quinn, Z.; Mao, W.; Wan, Y. Factors influencing the measurement of the secretion rate of extracellular vesicles. Analyst 2020, 145, 5870–5877. [Google Scholar] [CrossRef]
- Auber, M.; Svenningsen, P. An estimate of extracellular vesicle secretion rates of human blood cells. Journal of Extracellular Biology 2022, 1, e46. [Google Scholar] [CrossRef]
- Castillo-Sanchez, R.; Churruca-Schuind, A.; Martinez-Ival, M.; Salazar, E.P. Cancer-associated Fibroblasts Communicate with Breast Tumor Cells Through Extracellular Vesicles in Tumor Development. Technol Cancer Res Treat 2022, 21, 15330338221131647. [Google Scholar] [CrossRef] [PubMed]
- Sverdlov, E.D. Amedeo Avogadro’s cry: what is 1 microg of exosomes? Bioessays 2012, 34, 873–875. [Google Scholar] [CrossRef] [PubMed]
- Piffoux, M.; Volatron, J.; Silva, A.K.A.; Gazeau, F. Thinking Quantitatively of RNA-Based Information Transfer via Extracellular Vesicles: Lessons to Learn for the Design of RNA-Loaded EVs. Pharmaceutics 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- de Jong, O.G.; Murphy, D.E.; Mäger, I.; Willms, E.; Garcia-Guerra, A.; Gitz-Francois, J.J.; Lefferts, J.; Gupta, D.; Steenbeek, S.C.; van Rheenen, J.; et al. A CRISPR-Cas9-based reporter system for single-cell detection of extracellular vesicle-mediated functional transfer of RNA. Nature Communications 2020, 11, 1113. [Google Scholar] [CrossRef] [PubMed]
- Inaba, M.; Ridwan, S.M.; Antel, M. Removal of cellular protrusions. Semin Cell Dev Biol 2022, 129, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Wang, A.; Yuan, Y.; Zhu, B.; Long, H. Heterogeneity of the tumor immune microenvironment and its clinical relevance. Exp Hematol Oncol 2022, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Mezheyeuski, A.; Micke, P. The Immune Landscape of Colorectal Cancer. 2021, 13. [CrossRef]
- Yin, Y.; Sakakibara, R.; Honda, T.; Kirimura, S.; Daroonpan, P.; Kobayashi, M.; Ando, K.; Ujiie, H.; Kato, T.; Kaga, K.; et al. High density and proximity of CD8(+) T cells to tumor cells are correlated with better response to nivolumab treatment in metastatic pleural mesothelioma. Thorac Cancer 2023, 14, 1991–2000. [Google Scholar] [CrossRef] [PubMed]
- Gordon-Weeks, A.; Yuzhalin, A.E. Cancer Extracellular Matrix Proteins Regulate Tumour Immunity. Cancers 2020, 12, 3331. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Nogawa, D.; Kobayashi, M.; Asakawa, A.; Ohata, Y.; Kitagawa, S.; Kubota, K.; Takahashi, H.; Yamada, M.; Oda, G.; et al. Quantitative high-throughput analysis of tumor infiltrating lymphocytes in breast cancer. Front Oncol 2022, 12, 901591. [Google Scholar] [CrossRef]
- Finotello, F.; Eduati, F. Multi-Omics Profiling of the Tumor Microenvironment: Paving the Way to Precision Immuno-Oncology. Front Oncol 2018, 8, 430. [Google Scholar] [CrossRef]
- Minini, M.; Fouassier, L. Cancer-Associated Fibroblasts and Extracellular Matrix: Therapeutical Strategies for Modulating the Cholangiocarcinoma Microenvironment. Current Oncology 2023, 30, 4185–4196. [Google Scholar] [CrossRef]
- Aden, D.; Zaheer, S.; Ahluwalia, H.; Ranga, S. Cancer-associated fibroblasts: Is it a key to an intricate lock of tumorigenesis? Cell Biology International 2023, 47, 859–893. [Google Scholar] [CrossRef]
- Sarkar, M.; Nguyen, T.; Gundre, E.; Ogunlusi, O.; El-Sobky, M.; Giri, B.; Sarkar, T.R. Cancer-associated fibroblasts: The chief architect in the tumor microenvironment. Front Cell Dev Biol 2023, 11, 1089068. [Google Scholar] [CrossRef]
- Goenka, A.; Khan, F.; Verma, B.; Sinha, P.; Dmello, C.C.; Jogalekar, M.P.; Gangadaran, P.; Ahn, B.-C. Tumor microenvironment signaling and therapeutics in cancer progression. Cancer Communications 2023, 43, 525–561. [Google Scholar] [CrossRef]
- Wright, K.; Ly, T.; Kriet, M.; Czirok, A.; Thomas, S.M. Cancer-Associated Fibroblasts: Master Tumor Microenvironment Modifiers. Cancers (Basel) 2023, 15. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Sakai, R. Direct Interaction between Carcinoma Cells and Cancer Associated Fibroblasts for the Regulation of Cancer Invasion. Cancers (Basel) 2015, 7, 2054–2062. [Google Scholar] [CrossRef] [PubMed]
- Asif, P.J.; Longobardi, C.; Hahne, M.; Medema, J.P. The Role of Cancer-Associated Fibroblasts in Cancer Invasion and Metastasis. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef]
- Mezheyeuski, A.; Bergsland, C.H.; Backman, M.; Djureinovic, D.; Sjöblom, T.; Bruun, J.; Micke, P. Multispectral imaging for quantitative and compartment-specific immune infiltrates reveals distinct immune profiles that classify lung cancer patients. The Journal of pathology 2018, 244, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Gide, T.N.; Silva, I.P.; Quek, C.; Ahmed, T.; Menzies, A.M.; Carlino, M.S.; Saw, R.P.; Thompson, J.F.; Batten, M.; Long, G.V. Close proximity of immune and tumor cells underlies response to anti-PD-1 based therapies in metastatic melanoma patients. Oncoimmunology 2020, 9, 1659093. [Google Scholar] [CrossRef] [PubMed]
- Feichtenbeiner, A.; Haas, M.; Büttner, M.; Grabenbauer, G.G.; Fietkau, R.; Distel, L.V. Critical role of spatial interaction between CD8+ and Foxp3+ cells in human gastric cancer: the distance matters. Cancer Immunology, Immunotherapy 2014, 63, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Tabassum, D.P.; Janiszewska, M.; Place, A.E.; Trinh, A.; Rozhok, A.I.; Pyne, S.; Guerriero, J.L.; Shu, S.; Ekram, M.; et al. Spatial Proximity to Fibroblasts Impacts Molecular Features and Therapeutic Sensitivity of Breast Cancer Cells Influencing Clinical Outcomes. Cancer Res 2016, 76, 6495–6506. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, A.L.; Turley, S.J. Who am I? (re-)Defining fibroblast identity and immunological function in the age of bioinformatics. Immunol Rev 2021, 302, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Qiu, Y.; Zhang, Y. Research Progress on Therapeutic Targeting of Cancer-Associated Fibroblasts to Tackle Treatment-Resistant NSCLC. Pharmaceuticals 2022, 15, 1411. [Google Scholar] [CrossRef]
- Yao, H.; Wang, H.; Li, C.; Fang, J.Y.; Xu, J. Cancer Cell-Intrinsic PD-1 and Implications in Combinatorial Immunotherapy. Front Immunol 2018, 9, 1774. [Google Scholar] [CrossRef]
- Pauken, K.E.; Torchia, J.A.; Chaudhri, A.; Sharpe, A.H.; Freeman, G.J. Emerging concepts in PD-1 checkpoint biology. Seminars in Immunology 2021, 52, 101480. [Google Scholar] [CrossRef]
- Lu, J.; Wu, J.; Mao, L.; Xu, H.; Wang, S. Revisiting PD-1/PD-L pathway in T and B cell response: Beyond immunosuppression. Cytokine & Growth Factor Reviews 2022, 67, 58–65. [Google Scholar] [CrossRef]
- Xu-Monette, Z.Y.; Zhang, M.; Li, J.; Young, K.H. PD-1/PD-L1 Blockade: Have We Found the Key to Unleash the Antitumor Immune Response? Front Immunol 2017, 8, 1597. [Google Scholar] [CrossRef]
- Nishimura, C.D.; Pulanco, M.C.; Cui, W.; Lu, L.; Zang, X. PD-L1 and B7-1 Cis-Interaction: New Mechanisms in Immune Checkpoints and Immunotherapies. Trends in molecular medicine 2021, 27, 207–219. [Google Scholar] [CrossRef]
- Barrett, R.L.; Pure, E. Cancer-associated fibroblasts and their influence on tumor immunity and immunotherapy. Elife 2020, 9. [Google Scholar] [CrossRef]
- Giuliani, A.; Fais, S. Proposal to Consider Chemical/Physical Microenvironment as a New Therapeutic Off-Target Approach. Pharmaceutics 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Sverdlov, E.D. Multidimensional Complexity of Cancer. Simple Solutions Are Needed. Biochemistry (Mosc) 2016, 81, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.P.; Sharma, A.; Patil, V.M. Molecular Processes Exploited as Drug Targets for Cancer Chemotherapy. Anticancer Agents Med Chem 2021, 21, 1638–1649. [Google Scholar] [CrossRef]
- Ma, Y.; Conforti, R.; Aymeric, L.; Locher, C.; Kepp, O.; Kroemer, G.; Zitvogel, L. How to improve the immunogenicity of chemotherapy and radiotherapy. Cancer Metastasis Rev 2011, 30, 71–82. [Google Scholar] [CrossRef]
- Pomeroy, A.E.; Schmidt, E.V.; Sorger, P.K.; Palmer, A.C. Drug independence and the curability of cancer by combination chemotherapy. Trends Cancer 2022, 8, 915–929. [Google Scholar] [CrossRef] [PubMed]
- Finetti, F.; Baldari, C.T. The immunological synapse as a pharmacological target. Pharmacol Res 2018, 134, 118–133. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.J.; Nelson, W.J. Secret handshakes: cell-cell interactions and cellular mimics. Curr Opin Cell Biol 2018, 50, 14–19. [Google Scholar] [CrossRef]
- Ebnet, K.; Kummer, D.; Steinbacher, T.; Singh, A.; Nakayama, M.; Matis, M. Regulation of cell polarity by cell adhesion receptors. Semin Cell Dev Biol 2018, 81, 2–12. [Google Scholar] [CrossRef]
- Bausch-Fluck, D.; Milani, E.S.; Wollscheid, B. Surfaceome nanoscale organization and extracellular interaction networks. Curr Opin Chem Biol 2019, 48, 26–33. [Google Scholar] [CrossRef]
- Han, X.; Wang, X. Opportunities and Challenges in Tunneling Nanotubes Research: How Far from Clinical Application? Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Han, Q.; Huang, B.; Huang, Z.; Cai, J.; Gong, L.; Zhang, Y.; Jiang, J.; Dong, W.; Wang, Z. Tumor cell-fibroblast heterotypic aggregates in malignant ascites of patients with ovarian cancer. Int J Mol Med 2019, 44, 2245–2255. [Google Scholar] [CrossRef]
- Dustin, M.L.; Choudhuri, K. Signaling and Polarized Communication Across the T Cell Immunological Synapse. Annu Rev Cell Dev Biol 2016, 32, 303–325. [Google Scholar] [CrossRef] [PubMed]
- Hatherley, D.; Lea, S.M.; Johnson, S.; Barclay, A.N. Structures of CD200/CD200 receptor family and implications for topology, regulation, and evolution. Structure 2013, 21, 820–832. [Google Scholar] [CrossRef]
- Padhan, K.; Varma, R. Immunological synapse: a multi-protein signalling cellular apparatus for controlling gene expression. Immunology 2010, 129, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, P.A.; Carreno, L.J.; Cespedes, P.F.; Bueno, S.M.; Riedel, C.A.; Kalergis, A.M. Modulation of tumor immunity by soluble and membrane-bound molecules at the immunological synapse. Clin Dev Immunol 2013, 2013, 450291. [Google Scholar] [CrossRef]
- Yamada, S.; Nelson, W.J. Synapses: sites of cell recognition, adhesion, and functional specification. Annu Rev Biochem 2007, 76, 267–294. [Google Scholar] [CrossRef]
- Bayliss, R.J.; Piguet, V. Masters of manipulation: Viral modulation of the immunological synapse. Cell Microbiol 2018, 20, e12944. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L. Modular design of immunological synapses and kinapses. Cold Spring Harb Perspect Biol 2009, 1, a002873. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L.; Baldari, C.T. The Immune Synapse: Past, Present, and Future. Methods Mol Biol 2017, 1584, 1–5. [Google Scholar] [CrossRef]
- Garber, K. Beyond ipilimumab: new approaches target the immunological synapse. J Natl Cancer Inst 2011, 103, 1079–1082. [Google Scholar] [CrossRef]
- Klein, I.A.; Boija, A.; Afeyan, L.K.; Hawken, S.W.; Fan, M.; Dall’Agnese, A.; Oksuz, O.; Henninger, J.E.; Shrinivas, K.; Sabari, B.R.; et al. Partitioning of cancer therapeutics in nuclear condensates. Science 2020, 368, 1386–1392. [Google Scholar] [CrossRef]
- Conner, M.; Hance, K.W.; Yadavilli, S.; Smothers, J.; Waight, J.D. Emergence of the CD226 Axis in Cancer Immunotherapy. Front Immunol 2022, 13, 914406. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, A.; Nakanishi, H.; Kimura, K.; Matsubara, K.; Ozaki-Kuroda, K.; Katata, T.; Honda, T.; Kiyohara, Y.; Heo, K.; Higashi, M.; et al. Nectin: an adhesion molecule involved in formation of synapses. J Cell Biol 2002, 156, 555–565. [Google Scholar] [CrossRef]
- Hermans, D.; Rodriguez-Mogeda, C.; Kemps, H.; Bronckaers, A.; de Vries, H.E.; Broux, B. Nectins and Nectin-like molecules drive vascular development and barrier function. Angiogenesis 2023, 26, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. Cancer trial results show power of weaponized antibodies. Nature 2023, 623, 231–232. [Google Scholar] [CrossRef]
- Portsmouth, D.; Hlavaty, J.; Renner, M. Suicide genes for cancer therapy. Mol Aspects Med 2007, 28, 4–41. [Google Scholar] [CrossRef]
- Alekseenko, I.V.; Snezhkov, E.V.; Chernov, I.P.; Pleshkan, V.V.; Potapov, V.K.; Sass, A.V.; Monastyrskaya, G.S.; Kopantzev, E.P.; Vinogradova, T.V.; Khramtsov, Y.V.; et al. Therapeutic properties of a vector carrying the HSV thymidine kinase and GM-CSF genes and delivered as a complex with a cationic copolymer. J Transl Med 2015, 13, 78. [Google Scholar] [CrossRef]
- Alekseenko, I.; Kuzmich, A.; Kondratyeva, L.; Kondratieva, S.; Pleshkan, V.; Sverdlov, E. Step-by-Step Immune Activation for Suicide Gene Therapy Reinforcement. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Aznar, M.A.; Tinari, N.; Rullan, A.J.; Sanchez-Paulete, A.R.; Rodriguez-Ruiz, M.E.; Melero, I. Intratumoral Delivery of Immunotherapy-Act Locally, Think Globally. J Immunol 2017, 198, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Castanon, E.; Alvarez, M.; Champiat, S.; Marabelle, A. Intratumoural administration and tumour tissue targeting of cancer immunotherapies. Nat Rev Clin Oncol 2021, 18, 558–576. [Google Scholar] [CrossRef] [PubMed]
- First two CAR-T cell medicines recommended for approval in the European Unio. Available online: https://www.ema.europa.eu/en/news/first-two-car-t-cell-medicines-recommended-approval-european-union (accessed on 24.11.2023).
- Andrews, M. Insurers And Government Are Slow To Cover Expensive CAR-T Cancer Therapy. Available online: https://www.npr.org/sections/health-shots/2018/07/17/629543151/insurers-and-government-are-slow-to-cover-expensive-car-t-cancer-therapy (accessed on 24.11.2023).
- LifeSciencesIntelligence. The Top 5 Most Expensive FDA-Approved Gene Therapies. Available online: https://lifesciencesintelligence.com/features/the-top-5-most-expensive-fda-approved-gene-therapies (accessed on 12.11.2023).
- Ahmed, L.; Constantinidou, A.; Chatzittofis, A. Patients’ perspectives related to ethical issues and risks in precision medicine: a systematic review. Front Med (Lausanne) 2023, 10, 1215663. [Google Scholar] [CrossRef]
Figure 1.
Tumor microtube network. In tumors, structures called microtubes and tunneling nanotubes (see text) connect tumor cells and cells from microenvironments such as CAFs, allowing them to act as a single, organism-like unit. Healthy cells possibly do not participate in this network. The network also involves immune cells forming synapses with cancer cells and, possibly, with CAFs [76]. Recent studies have confirmed the existence of TNTs in various types of cells, especially in different kinds of immune cells [77]. Direct contact of CAFs with cancer cells has been proposed [36,78,79].
Figure 1.
Tumor microtube network. In tumors, structures called microtubes and tunneling nanotubes (see text) connect tumor cells and cells from microenvironments such as CAFs, allowing them to act as a single, organism-like unit. Healthy cells possibly do not participate in this network. The network also involves immune cells forming synapses with cancer cells and, possibly, with CAFs [76]. Recent studies have confirmed the existence of TNTs in various types of cells, especially in different kinds of immune cells [77]. Direct contact of CAFs with cancer cells has been proposed [36,78,79].
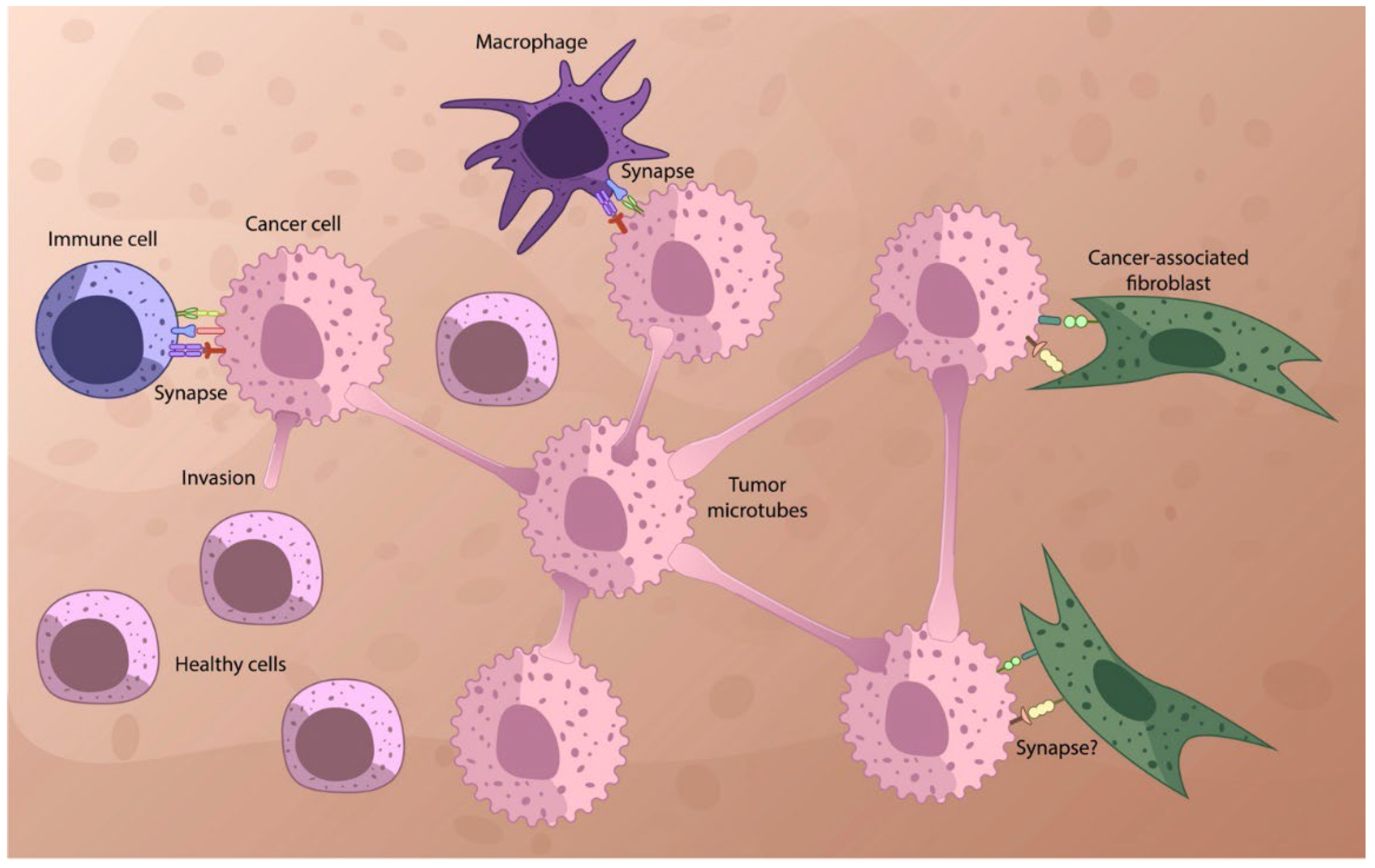
Figure 3.
A) A simple illustration of a two-stage reaction of a bifunctional cross-linking reagent with a protein cluster: the first stage, searching for a cluster, is slow; the second reaction inside a cluster with neighbors is fast. B) Examples of crosslinking agents. C) A simple illustration of a bifunctional cross-linking reagent containing the (photo) activated group, shown as a star, and a cleavable link. The cleavable link can be used to deliver an actable agent into the cell (see text).
Figure 3.
A) A simple illustration of a two-stage reaction of a bifunctional cross-linking reagent with a protein cluster: the first stage, searching for a cluster, is slow; the second reaction inside a cluster with neighbors is fast. B) Examples of crosslinking agents. C) A simple illustration of a bifunctional cross-linking reagent containing the (photo) activated group, shown as a star, and a cleavable link. The cleavable link can be used to deliver an actable agent into the cell (see text).
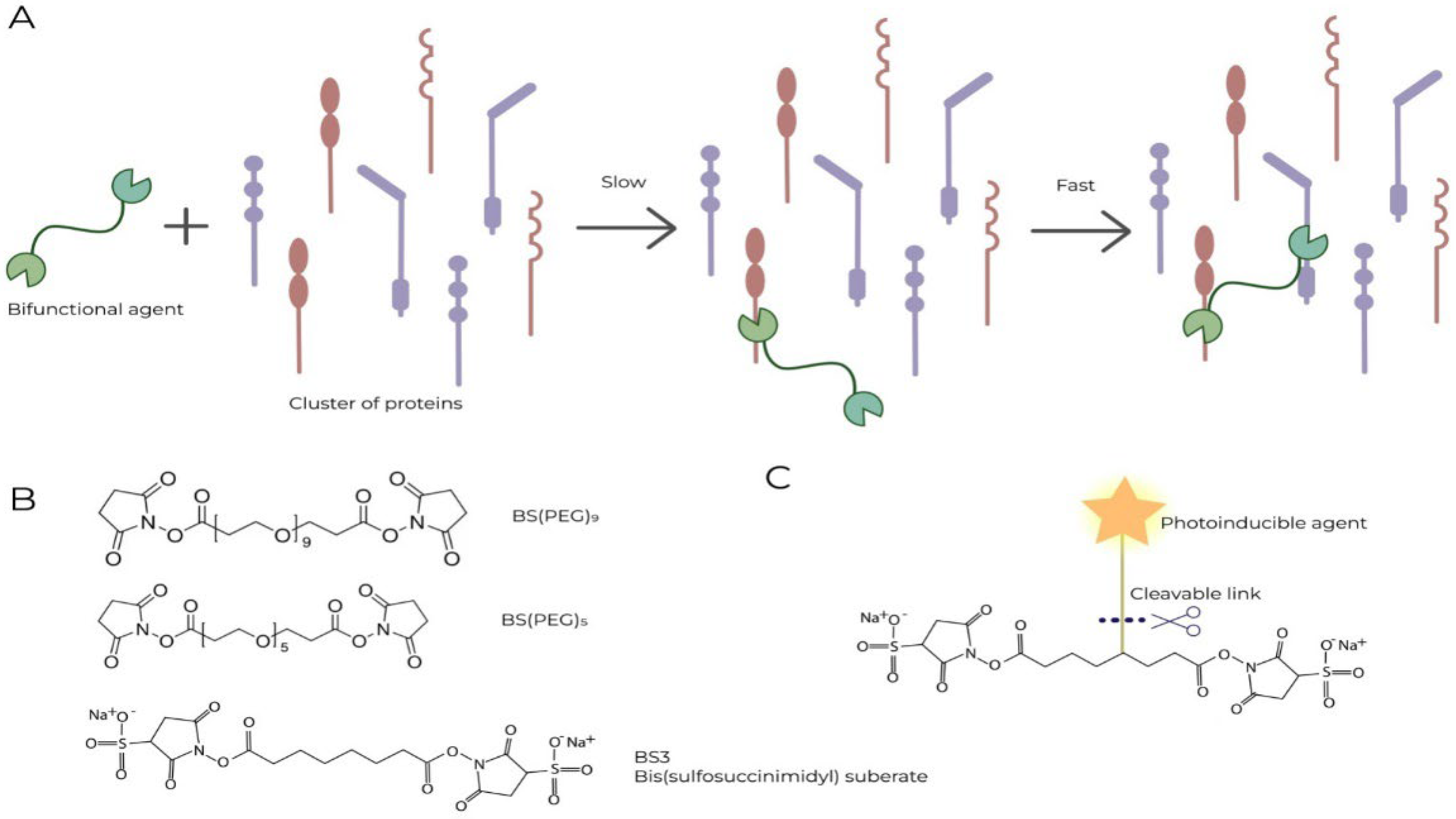
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



