Submitted:
01 March 2024
Posted:
04 March 2024
You are already at the latest version
Abstract
Proteins need to be located in appropriate spatiotemporal contexts to carry out their diverse biological functions. Mislocalized proteins may lead to a broad range of diseases, such as cancer and Alzheimer’s disease. Knowing where a target protein resides within a cell will give insights into tailored drug design for a disease. As the gold validation standard, the conventional wet lab uses fluorescent microscopy imaging, immunoelectron microscopy, and fluorescent biomarker tags for protein subcellular location identification. However, the booming era of proteomics and high-throughput sequencing generates tons of newly discovered proteins, making protein subcel-lular localization by wet-lab experiments a mission impossible. To tackle this concern, in the past decades, artificial intelligence (AI) and machine learning (ML), especially deep learning methods, have made significant progress in this research area. In this article, we review the latest advances in AI-based method development in three typical types of approaches, including sequence-based, knowledge-based, and image-based methods. We also elaborately discuss existing challenges and future directions in AI-based method development in this research field.
Keywords:
Protein Subcellular Localization
; Machine Learning
; Deep Learning
; Artificial Intelligence
; Gene Ontology
; Sequence Analysis
1. Introduction
Within a cell, mature proteins must reside in specific subcellular structures to properly perform their biological roles, as different cellular compartments provide distinct chemical environments (e.g., pH and redox conditions), potential interacting partners, or substrates for diverse functions [1,2]. Most cellular biological processes, such as the nucleocytosolic shuttling of transcription factors [3], the re-localization of mitochondrial proteins during apoptosis [4], and the endocytic uptake of cell-surface cargo receptors, all rely on precise protein localization. Conversely, mislocalization is often associated with cellular dysfunction and diseases, such as cancer [5,6], neurodegenerative diseases [7,8], and metabolic disorders [9,10].
Conventionally, identifying subcellular localization of proteins primarily relies on wet lab experimental methods. Fluorescence microscopy imaging, which apply fluorescent dyes or fluorescent protein tags to label target proteins, has commonly been used for observing their distribution within cells [11,12]. This method has become one of the preferred tools for studying protein subcellular localization due to its high resolution and real-time observation advantages [13]. By using labeled antibodies against target proteins, immunoelectron microscopy technique is regarded as a gold standard to provide the high resolution of electron microscopy [14]. Another method involves the use of fluorescent biomarker tags [15] like the protein A-GFP tag, which fuses a fluorescent protein with the target protein, allowing it to emit a fluorescent signal among different cell compartments [16]. These experimental methods yield high-resolution location of targeted proteins for researchers, enabling direct observation to uncover biological processes and metabolic mechanisms.
However, these wet lab experimental methods also have some significant drawbacks: they often require expensive equipment and time-consuming steps, making them costly for large-scale studies. These problems are exacerbated given that the number of newly discovered proteins has increased exponentially in the post-genomic era. Take the UniProt Database [17] as an example. The gap between the reviewed and unreviewed proteins has significantly expanded during the past decade (Figure 1A). Specifically, as shown in Figure 1B, In the latest 2024_01.version of UniProt, a notable majority of data entries are unreviewed proteins in TrEMBL. In this case, implementing wet lab experiments alone for subcellular localization determination for remarkably large amounts of data from different species (Figure 1C) becomes an impossible mission. Moreover, the rich collection of accurately annotated protein data in databases (Figure 1D) can facilitate the development of robust prediction methods. Computational models, especially AI-assisted approaches known for their proficiency in handling large-scale datasets, can be effectively applied in this context.
Recent decades have witnessed the booming of in-silico methods for protein subcellular location prediction. Based on features used for computational modeling, most of existing methods can be generally divided into three main categories: (1) sequence-based methods, which only use the amino acid sequence of the query protein as inputs; (2) knowledge-based methods, using protein annotations from multiple databases to correlate the information with their subcellular locations; (3) image-based methods, extracting subcellular location features from bioimages and then identify the likelihood of proteins being located in various subcellular compartments. The primary sequence for a protein is much easier to obtain with existing sequencing technologies. With remarkable advances in machine learning and deep learning, coupled with an increasing number of proteins with experimentally determined localization information as well as functional annotations and imaging records in publicly available databases, accurate and efficient computational frameworks provide a promising way for protein subcellular localization.
In this review, we will first present some remarkable progress in in-silico models, including three major types of models mentioned above. In Section 2, we will introduce common features and algorithms used in sequence-based methods, and so for knowledge-based and image-based frameworks in Section 3 and Section 4, respectively. The simplified flowchart for the prediction frameworks mentioned is illustrated in Figure 2. Then, we will give an overview of location prediction models that are specially designed for different species for more accurate prediction with specific data inputs. Lastly, we will explore the existing challenges and future trajectories of this research domain and propose our expectations.
2. Sequence-Based Methods
2.1. Sequence-Based Features
In protein primary sequences, the 20 standard amino acids (AA) exert different biochemical properties such as hydrophobicity, hydrophilicity, side chain characters, etc. Sequence-based methods intend to make predictions out of the correlations between protein subcellular locations and the information embedded in amino acid sequences. There are three major types of features used for model construction: AA composition information, sorting signal information and evolutionary information.
The composition-based features, which include AA occurrences and order in the query sequence, were commonly used in the earliest subcellular prediction methods. Moreover, previous studies have confirmed a better performance of the model by combining AA original sequence, gapped amino acid composition (GapAA) [18], and amino-acid pair composition (PairAA) [19]. Based on AA-composition features, Chou [20] proposed pseudo-amino-acid composition (PseAA) using the sequence-order correlation factor for more biomedical properties discovery when avoiding the high-dimensional vector formation. The simplicity of composition features helps the generalization and interpretation of the computational models since they capture the most basic trends in protein sequences associated with their locations. However, they may not provide sufficient resolution for a high accuracy rate since there’s a loss of information about important sequences or structural motifs highly related to proteins’ subcellular location.
The sorting signal sequences or signal peptides, including transit peptides like mitochondrial transit peptides (mTPs) and chloroplast transit peptides (cTPs) [21], are short and cleavable segment of amino acid sequences added to a newly synthesized proteins, determining their destination of the transportation process. These short peptides possess the directions mature proteins should be transported, reflecting the possible location event for one protein [22]. Available approaches with signal peptides for protein localization mainly refer to finding their cleavage sites [23]. As described in previous studies, sorting-signal sequences vary in length and composition but have similar structures: the N-terminal flanking region, also known as the n-region, the central hydrophobic region (h-region), and the C-terminal flanking region (c-region) [24]. The hydrophobicity in the h-region and a large proportion of nonpolar residues in the c-region are used to label the cleavage sites by computational methods [25,26]. According to the location signal embedded in those short peptides, one can mimic the de facto information processing in cells and find the target spot of the test protein.
In addition, based on the fact that homologous sequences are likely to share the same subcellular location, the unknown protein can be assigned the same subcellular location as its homologs generated from PSI-BLAST [27]. Moreover, the evolutionary similarity profiles extracted from the position-specific scoring matrix (PSSM) and position-specific frequency matrix (PSFM) derived from multiple sequences alignment results can contribute as classification features providing valuable information such as conserved motifs or targeting signals among different protein families. This representation can also be extended by integrating pseudo-analysis [28]. Once aligned with known homologs in the database, this method can achieve high accuracy. However, as one amino acid change can directly influence the characters of one protein sequence, this method is more likely to be one of the sources of feature basis of prediction models.
2.2. Sequences-Based AI Approaches
Most computational frameworks include three major steps: feature extraction, feature selection, and final classification. Considering common features discussed above, the complexity of the models developed also increases with the amount of data processed and the dimension of input features, from traditional machine learning classification to complex deep learning analytical models. Besides the development of computational frameworks, we will also introduce techniques that are used to improve the algorithms dealing with multi-location proteins in the following.
For conventional classification, Support Vector Machine (SVM) [29], K-Nearest Neighbor (KNN) [30], and Random Forest (RF) [31,32] are widely chosen classifiers for training. Their simplicity makes them easy to use for prediction protocols with fast speed and low computational cost, suitable for limited data and low-dimensional inputs. Combined with highly efficient feature extraction methods, these frameworks will work well in most cases [33]. For instance, Du et al. [34] proposed two novel feature extraction methods that utilize evolutionary information via the transition matrix of the consensus sequence (CTM) and PSSM before adopting SVM, which in the end reach an overall accuracy of 99.7% in CL317 dataset. A feature extraction-based hierarchical extreme learning machine (H-ELM) introduced by Zhang et al. [35] can handle high-dimension feature inputs directly without demanding dimension reduction for acceptable results. Alaa et al. [36] exploits an extended Markov chain to provide the latent feature vector, which records micro-similarities between the given sequence and their counterparts in reference models. These methods help extract more abundant features of query sequences and provide better performance.
However, these conventional models may not perform well in complex scenarios [1], especially multi-locational protein prediction [28]. Though many proteins only stay in one subcellular space, studies have discovered many multi-location proteins that have special functions or are involved in crucial biological steps [37]. Moreover, rather than staying in one place, proteins move from one subcellular compartment to another or simultaneously reside at two locations and participate in different cellular processes [38]. Recent studies have also shown the remarkable significance of multilocation proteins in cell growth and development [39]. For instance, phosphorylation-related multilocation proteins can function as a “needle and thread” via protein-protein interactions (PPI), thus playing an important role in organelle communication and regulating plant growth [40]. Under these circumstances, there are mainly two ways for predicting multi-location proteins based on conventional classifiers: algorithm-adaption and problem transformation. The former method extends existing algorithms to deal with multi-label problems. Jiang et al. [41] considers weighted prior probabilities with a multi-label KNN algorithm to increase the model accuracy. Library of SVM (LIBSVM) toolbox [34,42], instead, uses a one-versus-one (OVO) strategy to solve multi-class classification problems. Customization of well-known algorithms enhances their ability for specific requirements, but there is a risk of overfitting and may require significant computational resources. The problem transformation approach focuses on transforming the original problem into a different representation or formulation that is solvable with existing algorithms [43] [44], such as converting a multi-location classification problem into multiple single-label classification problems [45]. Shen et al. [28] introduces multi-kernel SVM by training multiple independent SVM classifiers to solve single-label problems before combining their results, one classifier for each class. Following this idea, an algorithm can be easily extended to solve multi-label classification.
In summary, traditional machine learning algorithms can achieve fast training times and high accuracy in scenarios with well-organized feature spaces and clear decision boundaries, their performance may degrade quickly when faced with large-scale data inputs, even with tailored classifiers featuring more selected features. Dimension reduction [46] and parallel processing [47] can be applied to mitigate the challenges, allowing an improved computational method scalability.
As multi-layered structure provides better performance compared to traditional approaches [31], more methods based on deep networks especially neural networks have become increasingly popular in protein subcellular localization research [48,49]. Starting as effective feature extractors which automatically obtain deep features embedded in sequences [50], convolutional neural network (CNN) is widely implanted in multi-locus protein localization framework. Mining deeper, Kaleel et al. [51] ensemble Deep N-to-1 Convolutional Neural Networks that predict the location of endomembrane system and secretory pathway versus all others and outperform many state-of-the-art web servers. Cong et al. [52] proposed a self-evoluting deep convolutional neural network (DCNN) protocol to solve the difficulties in feature correlation between sites and avoid the impact of unknown data distribution while using the self-attention mechanism [53] and a customized loss function to ensure the model performance. In addition, long short-term memory network (LSTM) which combines the previous states and current inputs is also commonly used [54,55], with Generative Adversarial Network (GAN) [56] and Synthetic Minority Over-sampling Technique (SMOTE) [57] used for synthesizing minority samples to deal with data imbalance. Developing data augmentation methods by deep learning algorithms has also made protein language model construction possible [58,59]. Through transfer learning [60], pre-trained models can be fine-tuned on different downstream tasks, reduces the need for large amounts of labeled data for training. For example, Heinzinger et al. [61] proposed Sequence-to-Vector (SeqVec) that embeds biophysical properties of protein sequences as continuous vectors by using the natural language processing model ELMo on unlabeled big data. This represents a way to speed up the prediction process independent of the size of inputs. Details of computational models mentioned above can be found in Table 1.
Deep learning will demonstrate exceptional outcomes dealing with high-dimensional inputs with deep feature extraction, eliminating the need for manual feature engineering and capturing intricate patterns in sequences. However, large, labeled, and high-quality datasets are still needed for original model training, which results in too many hyper-parameters and makes it hard to interpret the model itself [31].
3. Knowledge-Based Methods
3.1. Legitimacy of Using Gene Ontology (GO) Features
Knowledge-based methods tend to dig into the correlation between the annotation of one protein and its subcellular location to establish predictors. Compared to Swiss-Prot keywords [69,70] or PubMed abstracts [71,72], Gene Ontology (GO) terms-based methods are more attractive for the following reasons.
GO terms describe reviewed knowledge of the biological domain in from three aspects: (1) Molecular Function, representing activities that can be performed by individual or by assembled complexes of gene products at the molecular level; (2) Cellular Component, labeling locations relative to cellular compartments; (3) Biological Process, describing the events achieved by one or more ordered assemblies of molecular functions. These well-organized information can be used for protein subcellular localization because (1) Instead of table-lookup, which is dependent on cellular component GO terms, they perform deeper mining into items to accumulate every related GO category to improve prediction results, (2) The methods outperform previous sequence-based methods without compromising either inputs or outputs [73]. Mining deeper, the GO term itself is structurally organized but loosely hierarchical consisting of cellular components, biological processes, and molecular functions of gene products. The relationship between GO terms can be “part-of” (part and whole), which may embed some similarity information, and “is-a” (parent and child), which may result in more than one parent term. Starting from semantic similarity measurement, SS-Loc [74] incorporates a richer source of homologs and generates more features for prediction. Make use of the loosely hierarchical structure, relevance similarity (RS) consider the “distance” between the parent and child nodes. Take HybridGO-Loc [2] for example, it combines the frequency of occurrences of GO terms and semantic similarity between extracted GO terms to form a hybridized vector as input features, giving outstanding performance.
Mapping AA entries of query protein or accession number (AC) of its homologous to the GO database [75] will result in a list of GO items representing the possible functions and biological metabolism process this protein is involved in. For further computational methods implementation, reorganizing and transferring the list of data into numerical vectors is of high significance. Gneg-mPLoc [76], Euk-PLoc [77], and Hum-PLoc [78] consider GO terms as the basis of forming an Euclidean space, which only consists of 0 or 1 for coordinates. ProLoc-GO [43] on the other hand, represented the hit of annotated GO terms mined from Gene Ontology Annotation (GOA) with a n-dimensional binary feature vector. The constructed GO vectors are used for the following training.
3.2. Knowledge-Based AI Approaches
Originally, most machine learning methods used GO terms as the only input sources in simple classification model [79,80]. Given the growing richness of comprehensive protein annotation like related metabolism pathways and structural information, the integration of various input sources, including annotations, interaction networks, and pathway enrichment knowledge, contributes to a multi-view foundation for model improvement [81,82,83]. Applying deep learning algorithms enables a more comprehensive understanding of these high-dimensional and complex features, and further the combination of sequence and knowledge as input sources. According to the number of input sources, the methods can be roughly divided into GO terms only and fusion methods.
For single input source, mGOASVM [84] introduces a new decision scheme in SVM multi-class classifiers to collect all the positive decisions, enabling both single and multi-label localization. AD-SVM [85] enhances the binary relevance methods by integrating an adaptive decision scheme, thereby transforming the linear SVMs into piecewise linear SVMs, reducing the over-prediction instances. By using the frequency of the appearance of one protein in different places, Euk-mPLoc 2.0 [86] creates a virtual sample counting the appearance of protein to separate the total sequences input and the number of locations. However, a large number of proteins especially new discovered proteins have not been functionally annotated yet, and directly using homologs cannot guarantee the availability of enough GO terms to be found in the GOA Database. Moreover, the GO is not related to the representation of dynamics or pathway dependencies for protein, which will result in the risk of noise and overestimation of the novel proteins [87]. More details of the methods mentioned can be found in Table 2.
To improve the interpretability of the proposed model, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways is also considered as functional annotation that can be incorporate in the computational approaches [88]. Since in vivo protein interaction is likely to reside within the same subcellular locations, it is possible to reveal protein subcellular localization with protein-protein interaction (PPI) network [89,90,91], which is sensitive to mis-localization events [92].
The fusion methods can basically be divided into two categories: feature-level fusion [93,94,95] and decision-level fusion [96]. Feature level fusion is mostly based on average pooling, weighted combination [97], serial combination or concatenation of selected values. Liu et al. [95] utilized the latent semantic index method to represent multi-label information, while Yu et al. [47] constructed a novel parallel framework of attribute fusion to avoid the impact of duplicated information. This fusion level enhances the information from multiple sources and allows flexibility in fusion techniques, such as early integration, intermediate integration, and late integration [98]. But low data quality and difficulty in feature selection will affect building one efficient computational model. At the decision level, basic classifiers are used for different data sources first for selecting the suitable ones, then the results of each chosen method are ensembled as part of the determination protocol [99], as for the decision voting process [96]. Though the integration strategy is simple, this method can help create various decision-making systems that lead to more robust and accurate predictors. For instance, multi-view model like ML-FGAT [100], incorporates most of the feature types (e.g., sequence, evolutionary information, physicochemical property, etc.), which minimizes the perturbation of extraneous data in predictive tasks while concurrently enhancing the descriptive capability.

Table 2.
A summary of state-of-the-art knowledge-based and fusion models for protein subcellular localization prediction. S: Single-Location, M: Multi-Location. PSSM: Position-Specific Scoring Matrix, PsePSSM: Pseudo Position-Specific Scoring Matrix, PC: Physicochemical Properties, KNN: K-Nearest Neighbor, NN: Nearest Neighbor, RF: Random Forest, CDD: Conserved Functional Domain, PseAAC: Pseudo Amino Acid Composition, PPI: Protein-Protein Interaction Network, KEGG: KEGG (Kyoto Encyclopedia of Genes and Genomes) Pathway, SVM: Support Vector Machine, EBGW: OET-KNN: Optimized Evidence-Theoretic K nearest neighbor.
Table 2.
A summary of state-of-the-art knowledge-based and fusion models for protein subcellular localization prediction. S: Single-Location, M: Multi-Location. PSSM: Position-Specific Scoring Matrix, PsePSSM: Pseudo Position-Specific Scoring Matrix, PC: Physicochemical Properties, KNN: K-Nearest Neighbor, NN: Nearest Neighbor, RF: Random Forest, CDD: Conserved Functional Domain, PseAAC: Pseudo Amino Acid Composition, PPI: Protein-Protein Interaction Network, KEGG: KEGG (Kyoto Encyclopedia of Genes and Genomes) Pathway, SVM: Support Vector Machine, EBGW: OET-KNN: Optimized Evidence-Theoretic K nearest neighbor.
| Method | Features | Algorithm | Single-/Multi-Location | Species | Availability | Year |
|---|---|---|---|---|---|---|
| ML-FGAT | GO terms, Sequence Information, PsePSSM, PC | KNN | M | Human, Virus, Gram-negative bacteria, plant, SARS-CoV-2 | [101] | 2024 |
| PMPSL-GRAKEL | GO terms | RF | M | Human, Bacteria, animal | [102] | 2024 |
| Wang et al. | GO Terms, CDD, PseAAC, PSSM | NN | M | Human | [83] | 2023 |
| Zhang et al. | PPI, KEGG features, Functional GO | RF, SVM | M | Human | [94] | 2022 |
| ML-locMLFE | GO terms, PseAAC, PSSM | MLFE | M | Bacteria, Plant, Virus | [103] | 2021 |
| Chen et al. | GO, KEGG, PPI | RF, SVM, KNN, DT | S | Human | [88] | 2021 |
| Gpos-ECC-mPLoc | GO terms | SVM | M | Gram-positive Bacteria | [104] | 2015 |
| mGOASVM | GO terms | SVM | M | Virus, Plant | [105] | 2012 |
| iLoc-Euk | GO terms | KNN | M | Eukaryote | [106] | 2011 |
| Gneg-mPLoc | GO terms, Functional Domain, Evolutional Information | OET-KNN | M | Gram-negative bacteria | [107] | 2010 |
| PSORTb 3.0 | Swissprot Annotation | SVM | S | Prokaryotes | [108] | 2010 |
4. Bioimage-Based Methods
4.1. Bioimage-Based Features
Compared to amino acid sequences, representing proteins with 2D images is more interpretable and concise when determining the subcellular localization. With the rapid improvement in microscopic imaging technology, scientists have paid more attention to bioimage-based methods. Computer hardware improvement, especially in graphics processing units (GPUs) makes it possible to deal with more complex calculation problems. The development of neural network structure also accelerates deep learning algorithm architecture improvement for image analysis significantly. For high equality data, with the mission of mapping all human proteins in cells, tissues and organs, the human protein atlas (HPA) program [109] was initialized in 2003 as an open-access database that consists of imaging, mass spectrometry-based proteomics, transcriptomics, etc. The subcellular section of HPA shows detailed expression and spatial distribution conditions of proteins encoded by 13147 genes. As recently updated to version 23, it is the most powerful training data source for computational method development [110,111].
The subcellular location features (SLF) collected can be divided into two categories, namely, global features and local features [112]. Composed of DNA distribution information and global textures, the global features such as morphological features, local binary patterns (LBP) [113] and Zernike features [114] mainly describe the spatial structure of the whole image. The Haralick [115] texture feature, which obtains statistical features including contrast, correlation, and entropy from the gray-level co-occurrence matrix of input images, is one well-known global image descriptor in pattern recognition. Local features, instead, can describe the micro-patterns ignored in global features. Take scale-invariant feature transform (SIFT) [116] as an example. SIFT was originally used for salient points detection and is suitable for fluorescence object description, which guarantees good performance in fluorescence image studies, especially when combined with global features.
4.2. Bioimage-Based AI Methods
Image-related methods can be roughly organized into three phases based on the algorithms and the number of data types used, namely conventional or traditional machine learning methods, deep learning methods, and complex fusion methods, respectively. Figure 3 shows the development of these models from simple to complicated.
Traditional machine learning methods construct the prediction models with the aforementioned hand-crafted features for classification [117,118,119]. For instance, Li et al. [120] extended a logistic regression algorithm with structured latent variables for underlying components in different image regions for further classification. With two-layer deep learned feature selection, Ulah et al. [121] established a SVM model based on both radial basis function and linear kernel for location prediction. However, these convolutional methods can be sensitive to noise and variability of imaging data collected, resulting in decreased model robustness. Spatial relationships that embedded in images are rarely detected as well, due to manual feature engineering. As deep learning predictors employed and have achieved high performance on various image-based tasks, recent advances in protein subcellular location rely more on deep learning methods [117].
Deep neural network implementation is the starting point, which increases the inner feature extraction power and the model’s learning ability for large and complicated datasets. In addition to selecting and integrating key features during the image preprocessing steps, most of the deep neural networks consider processed image segmentation as inputs for multi-layer convolutional neural networks (ML-CNN) [122]. Moreover, some predictors can integrate both low and high-level features embedded in bioimages for a more in-depth view. For multi-label prediction, traditional CNN is extended with a criterion learning strategy to leverage label-attribute relevancy and label-label relevancy to determine the final location [123,124]. To be more specific, the diversity in input data types across various dimensions contributes to shaping the complexity of the entire model. From image datasets, DeepPSL automatically learn meaningful features and their correlations for prediction improvement [125]. Xue et al. [126] unmixed the IHC images into protein and DNA channels for representation construction while segmenting the images into patches for fine-tuning network training. Ding et al. [127] ensemble different classification models using different depth of feature vectors constructed from images as inputs to achieve high accuracy outputs. By collecting different imaging types, Wei et al. [128] built another parallel integrative deep network for label-free cell optical images. More details about the models can be found in Table 3. Though further techniques can be applied during the pretraining step [129,130,131,132,133], image-only methods still lack generalization capability and external validation. When incorporating more modality of data which are not directly observable from imaging alone but related to protein subcellular localization during model establishment will take more contextual information into consideration and overcome the limitations in model performance.
5. Protein Subcellular Localization in Different Species
Analyzing species separately allows a more accurate model generalization, since specific proteins and their subcellular localization patterns may differ in various cell organizations and organelle structures. Multi-species database Compartments [138], fungal database FunSecKB2 [139], plant database PlantSecKB [140], human and animal database MetazSecKB [141] mostly obtained and arranged from UniProt provide efficient search for each organism and high-quality protein subcellular location annotation datasets across species.
Take bacteria as an example. As prokaryotes, they exhibit significant structural differences from eukaryotic organisms, like lacking common cellular organelles such as mitochondria, endoplasmic reticulum, and Golgi apparatus. However, within bacteria, a notable class of self-assembling microstructures, known as bacterial microcompartments (BMCs), consist of a protein shell encapsulating an enzymatic core [142,143], creating an internally enclosed space for protein resides. Furthermore, bacteria possess special cell walls that can be classified as Gram-positive and Gram-negative bacteria [144], which closely associated with different protein localization modes. For real-world application [145], the subcellular localization changes in host cells like plants that need precise localization after viral infection can give insights into the interactions of host cells and viruses, which helps in genetic resistance target identification [146]. Many models have specially designed for distinct species (e.g., iLoc-Euk [147], iLoc-Virus [148] and iLoc-Plant [149], mPLR-Loc [150]. As for knowledge-based, Gram-LocEN [151] is a predictor for large-scale dataset of both single and multi-location proteins in bacteria. It created two databases called ProSeq and ProSeq-GO for query protein from Swiss-Prot and GOA databases [152], respectively, to guarantee the effectiveness and decrease storage complexity. After defining GO space and constructing GO vectors, the model demonstrated elastic net (EN) to enable automatic feature selection and further classification.
6. Current Challenges and Future Directions
6.1. Challenges
Despite the significant advances, challenges still exist for AI-based method development in protein subcellular localization field. The interpretability of the model will be one of the big concerns. As deep learning algorithms have complicated training process that generates high dimensional deep features for prediction, it is of great importance to interpret the decision-making procedures of the model for a better understanding of the essential factors that influence protein localization. SHAP [153], DeepExplainer [154] based on DeepLift [155] and other methodologies major in capturing the importance of features for overall prediction tasks are implemented in recent studies for increasing model interpretability. Luo et al. [156] have also reduced the dimensionality of feature vectors by constructing autoencoders to obtain a better feature representation for downstream analysis. In ML-FGAT [100], the interpretability is strengthened by analyzing the attention weight parameters. Explainable and understandable frameworks will give more reliable predictions that benefits further studies from biological perspective.
Moreover, protein subcellular location is influenced by multiple factors. AI-based methods mostly rely on original sequences or images as inputs, which lack the information after protein biosynthesis. There is also a chance that the prediction model provides the same subcellular location when the mutant protein resides in a different place [146]. Post-translational modifications (PTMs), which refer to amino acid side chain modification after the synthesis of some proteins, can contribute significant changes to their subcellular location [157]. There are many kinds of PTMs, such as phosphorylation, glycosylation, and acetylation, which dynamically regulate the protein within the cell simultaneously [158], resulting in sparse and incomplete experimental data for model training. As more post-translational positions are discovered [158], AI-based prediction proteins that consider PTMs as key features can also be further investigated [158,159].
Establishing models to leverage both annotated and unannotated proteins for localization can also be a challenge, with a large proportion of unreviewed data reported each year (Figure 1A-B). Though data augmentation methods like SMOTE and GAN are widely used to handle data imbalance, semi-supervised learning can also be established to solve the problem [133,160]. To be more specific, EnTrans-Chlo [161] incorporates multi-modal features and converts them into sample-to-sample similarity features with assigned weights, for feeding a high-efficient learning model. LNP-Chlo [162] extended the previous approach by adopting a quadratic programming algorithm to optimize the weights of nearest neighbors. These semi-supervised models remarkably outperformed state-of-the-art supervised methods, while integrating different data modalities and dimensionalities with less requirement of sufficient labeled data.
6.2. Future Directions
Currently, cutting-edge research directions in subcellular localization mainly lie in spatial proteomics [9], and RNA subcellular localization.
With the blooming of single-cell research, it is possible to gain a full understanding of disease from cell and tissue heterogeneity. Since the exact location of proteins at the subcellular, cellular, or tissue levels directly links to their functions, it is essential for protein localization with a single-cell and spatial resolution [163]. Zhu et al. [164] have created cell-based methods with a pseudo-label assignment to discover protein subcellular localization results across distinct cells with heterogeneity among single cells. Husain et al. [135] presents the Hybrid subCellular Protein Localiser (HCPL) that robustly localizes single-cell subcellular protein patterns. Wang’s work with mass spectrometry (MS)-based spatial proteomics [165] shows the possibility of larger dimensional feature maps and higher learning ability of computational models.
System-wide studies of RNA subcellular localization (e.g., mRNA [166]) have also paved the way for a more comprehensive analysis of the cellular dynamics [167,168], as proteins are usually transcribed by RNA molecules. Moreover, except for RNA transcripts for protein, other RNAs, like long non-coding RNAs (lncRNAs), may also be involved in many biological functions [169]. Predicting their subcellular locations with AI-based methods [169] can significantly reduce costs and time expenditure, enabling the investigation of their functionalities with limited data [167]. Moreover, common [170] and rare cellular-compartment-specific prediction models can be further explored [171].
7. Conclusion
In this review, we have reviewed three types of computational methods using machine learning or deep learning models to construct predictors for protein subcellular localization. For different kinds of inputs such as protein sequence, GO terms or IHC images, the predictors will first convey the biological data to numerical or mathematical representations of essential features embedded in the source and apply widely used classifiers for single or multi-class tasks. When low-dimension data like sequence and texture the performance become more reliable. Traditional machine learning methods can combine various features and manage the high dimensional data by dimensionality reduction techniques like random projection [172] to avoid the curse of dimensionality and achieve interpretable outcomes under large data scales. Alternatively, they can combine the results of different classifiers, which run the calculation parallelly, to improve the overall performance. Deep learning methods that are mostly based on neural networks will learn and extract high-level features and their correlations from the inputs before the classification. When dealing with large-scale datasets, prediction with a language model is also available with deep learning. For future direction, in addition to faster and more effective algorithm development, we also assume that the localization prediction will incorporate more biochemical interactions like protein-protein interaction networks (PPI), metabolic networks, gene co-expression interaction, etc., into consideration, since proteins intricately engage in complex physiological reactions within the cellular space. Above all, we are confident that the computational methods will raise more and more attention for (1) for systematic research like proteomics and metabolomics, (2) to provide dynamic insights into cells, and to see what the influence will be when the target protein is muted; (3) to assist the experimental side with data analysis, experimental design and so on. In the long run, this research area will benefit clinical drug development and contribute to disease detection, diagnosis, prognosis, and treatment.
Author Contributions
HX, YZ, JW, and SW wrote the manuscript. SW supervised the manuscript. The manuscript was approved by all authors.
Funding
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number P30CA036727. This work was supported by the American Cancer Society under award number IRG-22-146-07-IRG, and by the Buffett Cancer Center, which is supported by the National Cancer Institute under award number CA036727. This work was supported by the Buffet Cancer Center, which is supported by the National Cancer Institute under award number CA036727, in collaboration with the UNMC/Children’s Hospital & Medical Center Child Health Research Institute Pediatric Cancer Research Group. This study was supported, in part, by the National Institute on Alcohol Abuse and Alcoholism (P50AA030407-5126, Pilot Core grant). This study was also supported by the Nebraska EPSCoR FIRST Award (OIA-2044049). The content is solely the responsibility of the authors and does not necessarily represent the official views from the funding organizations.
Acknowledgments
Some figures in the manuscript are generated by BioRender.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Xu, Q.; Hu, D.H.; Xue, H.; Yu, W.; Yang, Q. Semi-Supervised Protein Subcellular Localization. BMC Bioinformatics 2009, 10, S47. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Mak, M.-W.; Kung, S.-Y. HybridGO-Loc: Mining Hybrid Features on Gene Ontology for Predicting Subcellular Localization of Multi-Location Proteins. PLoS ONE 2014, 9, e89545. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M. Molecular Mechanism of the Nuclear Protein Import Cycle. Nat Rev Mol Cell Biol 2007, 8, 195–208. [Google Scholar] [CrossRef]
- Mayor, S.; Pagano, R.E. Pathways of Clathrin-Independent Endocytosis. Nat Rev Mol Cell Biol 2007, 8, 603–612. [Google Scholar] [CrossRef]
- Lee, K.; Byun, K.; Hong, W.; Chuang, H.-Y.; Pack, C.-G.; Bayarsaikhan, E.; Paek, S.H.; Kim, H.; Shin, H.Y.; Ideker, T.; et al. Proteome-Wide Discovery of Mislocated Proteins in Cancer. Genome Res. 2013, 23, 1283–1294. [Google Scholar] [CrossRef]
- Wang, X.; Li, S. Protein Mislocalization: Mechanisms, Functions and Clinical Applications in Cancer. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 2014, 1846, 13–25. [Google Scholar] [CrossRef]
- Barmada, S.J.; Skibinski, G.; Korb, E.; Rao, E.J.; Wu, J.Y.; Finkbeiner, S. Cytoplasmic Mislocalization of TDP-43 Is Toxic to Neurons and Enhanced by a Mutation Associated with Familial Amyotrophic Lateral Sclerosis. J. Neurosci. 2010, 30, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Ziff, O.J.; Harley, J.; Wang, Y.; Neeves, J.; Tyzack, G.; Ibrahim, F.; Skehel, M.; Chakrabarti, A.M.; Kelly, G.; Patani, R. Nucleocytoplasmic mRNA Redistribution Accompanies RNA Binding Protein Mislocalization in ALS Motor Neurons and Is Restored by VCP ATPase Inhibition. Neuron 2023, 111, 3011–3027.e7. [Google Scholar] [CrossRef]
- Lundberg, E.; Borner, G.H.H. Spatial Proteomics: A Powerful Discovery Tool for Cell Biology. Nat Rev Mol Cell Biol 2019, 20, 285–302. [Google Scholar] [CrossRef]
- Xiang, L.; Yang, Q.-L.; Xie, B.-T.; Zeng, H.-Y.; Ding, L.-J.; Rao, F.-Q.; Yan, T.; Lu, F.; Chen, Q.; Huang, X.-F. Dysregulated Arginine Metabolism Is Linked to Retinal Degeneration in Cep250 Knockout Mice. Invest. Ophthalmol. Vis. Sci. 2023, 64, 2. [Google Scholar] [CrossRef]
- Kohnhorst, C.L.; Schmitt, D.L.; Sundaram, A.; An, S. Subcellular Functions of Proteins under Fluorescence Single-Cell Microscopy. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2016, 1864, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Sekine, S.; Pessino, V.; Li, H.; Leonetti, M.D.; Huang, B. Improved Split Fluorescent Proteins for Endogenous Protein Labeling. Nat Commun 2017, 8, 370. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, R.S.; White, M.L.; Eswara, P.J. Live-Cell Fluorescence Microscopy to Investigate Subcellular Protein Localization and Cell Morphology Changes in Bacteria. 2020. [CrossRef]
- Liang, F.-X.; Sall, J.; Petzold, C.; Van Opbergen, C.J.M.; Liang, X.; Delmar, M. Nanogold Based Protein Localization Enables Subcellular Visualization of Cell Junction Protein by SBF-SEM. In Methods in Cell Biology; Elsevier, 2023; Volume 177, pp. 55–81. ISBN 978-0-323-91607-3. [Google Scholar] [CrossRef]
- Schornack, S.; Fuchs, R.; Huitema, E.; Rothbauer, U.; Lipka, V.; Kamoun, S. Protein Mislocalization in Plant Cells Using a GFP-binding Chromobody. The Plant Journal 2009, 60, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Orbán, T.I.; Seres, L.; Özvegy-Laczka, C.; Elkind, N.B.; Sarkadi, B.; Homolya, L. Combined Localization and Real-Time Functional Studies Using a GFP-Tagged ABCG2 Multidrug Transporter. Biochemical and Biophysical Research Communications 2008, 367, 667–673. [Google Scholar] [CrossRef]
- The UniProt Consortium. Bateman, A.; Martin, M.-J.; Orchard, S.; Magrane, M.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bye-A-Jee, H.; et al. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Research 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Park, K.-J.; Kanehisa, M. Prediction of Protein Subcellular Locations by Support Vector Machines Using Compositions of Amino Acids and Amino Acid Pairs. Bioinformatics 2003, 19, 1656–1663. [Google Scholar] [CrossRef]
- Chou, K.-C. Using Pair-Coupled Amino Acid Composition to Predict Protein Secondary Structure Content. J Protein Chem 1999, 18, 473–480. [Google Scholar] [CrossRef]
- Chou, K. Prediction of Protein Cellular Attributes Using Pseudo-amino Acid Composition. Proteins 2001, 43, 246–255. [Google Scholar] [CrossRef]
- Von Heijne, G.; Steppuhn, J.; Herrmann, R.G. Domain Structure of Mitochondrial and Chloroplast Targeting Peptides. European Journal of Biochemistry 1989, 180, 535–545. [Google Scholar] [CrossRef]
- Wan, S.; Mak, M.-W. Machine Learning for Protein Subcellular Localization Prediction:; DE GRUYTER, 2015; ISBN 978-1-5015-1048-9. [CrossRef]
- Xue, S.; Liu, X.; Pan, Y.; Xiao, C.; Feng, Y.; Zheng, L.; Zhao, M.; Huang, M. Comprehensive Analysis of Signal Peptides in Saccharomyces Cerevisiae Reveals Features for Efficient Secretion. Advanced Science 2023, 10, 2203433. [Google Scholar] [CrossRef]
- Martoglio, B.; Dobberstein, B. Signal Sequences: More than Just Greasy Peptides. Trends in Cell Biology 1998, 8, 410–415. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Salvatore, M.; Emanuelsson, O.; Winther, O.; von Heijne, G.; Elofsson, A.; Nielsen, H. Detecting Sequence Signals in Targeting Peptides Using Deep Learning. Life Sci Alliance 2019, 2, e201900429. [Google Scholar] [CrossRef]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; Von Heijne, G.; Nielsen, H. SignalP 6.0 Predicts All Five Types of Signal Peptides Using Protein Language Models. Nat Biotechnol 2022, 40, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Hirano, Y.; Ohno, Y.; Kubota, Y.; Fukagawa, T.; Kihara, A.; Haraguchi, T.; Hiraoka, Y. Ceramide Synthase Homolog Tlc4 Maintains Nuclear Envelope Integrity via Its Golgi Translocation. Journal of Cell Science 2023, 136, jcs260923. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Tang, J.; Guo, F. Identification of Protein Subcellular Localization via Integrating Evolutionary and Physicochemical Information into Chou’s General PseAAC. Journal of Theoretical Biology 2019, 462, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Man-Wai Mak; Jian Guo; Sun-Yuan Kung PairProSVM: Protein Subcellular Localization Based on Local Pairwise Profile Alignment and SVM. IEEE/ACM Trans. Comput. Biol. and Bioinf. 2008, 5, 416–422. [CrossRef] [PubMed]
- Tahir, M.; Khan, F.; Hayat, M.; Alshehri, M.D. An Effective Machine Learning-Based Model for the Prediction of Protein–Protein Interaction Sites in Health Systems. Neural Comput & Applic 2024, 36, 65–75. [Google Scholar] [CrossRef]
- Wang, J.; Nabil, M.; Zhang, J. Deep Forest-Based Prediction of Protein Subcellular Localization. Current Gene Therapy 2018, 18. [Google Scholar] [CrossRef]
- Sui, J.; Chen, Y.; Cao, Y.; Zhao, Y. Accurate Identification of Submitochondrial Protein Location Based on Deep Representation Learning Feature Fusion. In Proceedings of the Advanced Intelligent Computing Technology and Applications; Huang, D.-S., Premaratne, P., Jin, B., Qu, B., Jo, K.-H., Hussain, A., Eds.; Springer Nature Singapore: Singapore, 2023; pp. 587–596. [Google Scholar] [CrossRef]
- Javed, F.; Hayat, M. Predicting Subcellular Localization of Multi-Label Proteins by Incorporating the Sequence Features into Chou’s PseAAC. Genomics 2019, 111, 1325–1332. [Google Scholar] [CrossRef]
- Du, L.; Meng, Q.; Chen, Y.; Wu, P. Subcellular Location Prediction of Apoptosis Proteins Using Two Novel Feature Extraction Methods Based on Evolutionary Information and LDA. BMC Bioinformatics 2020, 21, 212. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, T.; Liu, C. Prediction of Apoptosis Protein Subcellular Localization via Heterogeneous Features and Hierarchical Extreme Learning Machine. SAR and QSAR in Environmental Research 2019, 30, 209–228. [Google Scholar] [CrossRef]
- Alaa, A.; Eldeib, A.M.; Metwally, A.A. Protein Subcellular Localization Prediction Based on Internal Micro-Similarities of Markov Chains. In Proceedings of the 2019 41st Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC); IEEE: Berlin, Germany, July 2019; pp. 1355–1358. [Google Scholar] [CrossRef]
- Murphy, R.F. Communicating Subcellular Distributions. Cytometry Pt A 2010, 77A, 686–692. [Google Scholar] [CrossRef]
- Cohen, S.E.; Erb, M.L.; Selimkhanov, J.; Dong, G.; Hasty, J.; Pogliano, J.; Golden, S.S. Dynamic Localization of the Cyanobacterial Circadian Clock Proteins. Current Biology 2014, 24, 1836–1844. [Google Scholar] [CrossRef]
- Kim, W.; Jeon, T.J. Dynamic Subcellular Localization of DydA in Dictyostelium Cells. Biochemical and Biophysical Research Communications 2023, 663, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Xiong, E.; Cao, D.; Qu, C.; Zhao, P.; Wu, Z.; Yin, D.; Zhao, Q.; Gong, F. Multilocation Proteins in Organelle Communication: Based on Protein–Protein Interactions. Plant Direct 2022, 6, e386. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Wang, D.; Wu, P.; Chen, Y.; Shang, H.; Wang, L.; Xie, H. Predicting Subcellular Localization of Multisite Proteins Using Differently Weighted Multi-Label k-Nearest Neighbors Sets. THC 2019, 27, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-C.; Lin, C.-J. LIBSVM: A Library for Support Vector Machines. ACM Trans. Intell. Syst. Technol. 2011, 2. [Google Scholar] [CrossRef]
- Ding, Y.; Tang, J.; Guo, F. Human Protein Subcellular Localization Identification via Fuzzy Model on Kernelized Neighborhood Representation. Applied Soft Computing 2020, 96, 106596. [Google Scholar] [CrossRef]
- Hasan, Md.A.M.; Ahmad, S.; Molla, Md.K.I. Protein Subcellular Localization Prediction Using Multiple Kernel Learning Based Support Vector Machine. Mol. BioSyst. 2017, 13, 785–795. [CrossRef]
- Boutell, M.R.; Luo, J.; Shen, X.; Brown, C.M. Learning Multi-Label Scene Classification. Pattern Recognition 2004, 37, 1757–1771. [Google Scholar] [CrossRef]
- Wang, S.; Liu, S. Protein Sub-Nuclear Localization Based on Effective Fusion Representations and Dimension Reduction Algorithm LDA. IJMS 2015, 16, 30343–30361. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Wu, X.; Shen, H.; Yang, J.; Tang, Z.; Qi, Y.; Yang, J. Enhancing Membrane Protein Subcellular Localization Prediction by Parallel Fusion of Multi-View Features. IEEE Trans.on Nanobioscience 2012, 11, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Jing, R.; Li, Y.; Xue, L.; Liu, F.; Li, M.; Luo, J. autoBioSeqpy: A Deep Learning Tool for the Classification of Biological Sequences. J. Chem. Inf. Model. 2020, 60, 3755–3764. [Google Scholar] [CrossRef] [PubMed]
- Semwal, R.; Varadwaj, P.K. HumDLoc: Human Protein Subcellular Localization Prediction Using Deep Neural Network. CG 2020, 21, 546–557. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Wang, J.; Zhao, L.; Wang, C.; Zhan, H. A Novel Protein Subcellular Localization Method With CNN-XGBoost Model for Alzheimer’s Disease. Front. Genet. 2019, 9, 751. [Google Scholar] [CrossRef]
- Kaleel, M.; Zheng, Y.; Chen, J.; Feng, X.; Simpson, J.C.; Pollastri, G.; Mooney, C. SCLpred-EMS: Subcellular Localization Prediction of Endomembrane System and Secretory Pathway Proteins by Deep N-to-1 Convolutional Neural Networks. Bioinformatics 2020, 36, 3343–3349. [Google Scholar] [CrossRef]
- Cong, H.; Liu, H.; Chen, Y.; Cao, Y. Self-Evoluting Framework of Deep Convolutional Neural Network for Multilocus Protein Subcellular Localization. Med Biol Eng Comput 2020, 58, 3017–3038. [Google Scholar] [CrossRef]
- Cong, H.; Liu, H.; Cao, Y.; Chen, Y.; Liang, C. Multiple Protein Subcellular Locations Prediction Based on Deep Convolutional Neural Networks with Self-Attention Mechanism. Interdiscip Sci Comput Life Sci 2022, 14, 421–438. [Google Scholar] [CrossRef]
- Liao, Z.; Pan, G.; Sun, C.; Tang, J. Predicting Subcellular Location of Protein with Evolution Information and Sequence-Based Deep Learning. BMC Bioinformatics 2021, 22, 515. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, D.; Yao, Y.; Eubel, H.; Künzler, P.; Møller, I.M.; Xu, D. MULocDeep: A Deep-Learning Framework for Protein Subcellular and Suborganellar Localization Prediction with Residue-Level Interpretation. Computational and Structural Biotechnology Journal 2021, 19, 4825–4839. [Google Scholar] [CrossRef]
- Wu, L.; Gao, S.; Yao, S.; Wu, F.; Li, J.; Dong, Y.; Zhang, Y. Gm-PLoc: A Subcellular Localization Model of Multi-Label Protein Based on GAN and DeepFM. Front. Genet. 2022, 13, 912614. [Google Scholar] [CrossRef]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. SMOTE: Synthetic Minority over-Sampling Technique. J. Artif. Int. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Elnaggar, A.; Heinzinger, M.; Dallago, C.; Rehawi, G.; Wang, Y.; Jones, L.; Gibbs, T.; Feher, T.; Angerer, C.; Steinegger, M.; et al. ProtTrans: Toward Understanding the Language of Life Through Self-Supervised Learning. IEEE Trans. Pattern Anal. Mach. Intell. 2022, 44, 7112–7127. [Google Scholar] [CrossRef]
- Wang, X.; Han, L.; Wang, R.; Chen, H. DaDL-SChlo: Protein Subchloroplast Localization Prediction Based on Generative Adversarial Networks and Pre-Trained Protein Language Model. Briefings in Bioinformatics 2023, 24, bbad083. [Google Scholar] [CrossRef] [PubMed]
- Hosna, A.; Merry, E.; Gyalmo, J.; Alom, Z.; Aung, Z.; Azim, M.A. Transfer Learning: A Friendly Introduction. J Big Data 2022, 9, 102. [Google Scholar] [CrossRef]
- Heinzinger, M.; Elnaggar, A.; Wang, Y.; Dallago, C.; Nechaev, D.; Matthes, F.; Rost, B. Modeling Aspects of the Language of Life through Transfer-Learning Protein Sequences. BMC Bioinformatics 2019, 20, 723. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Han, L.; Wang, R.; Chen, H. DaDL-SChlo: Protein Subchloroplast Localization Prediction Based on Generative Adversarial Networks and Pre-Trained Protein Language Model. Briefings in Bioinformatics 2023, 24, bbad083. [Google Scholar] [CrossRef]
- Thumuluri, V.; Almagro Armenteros, J.J.; Johansen, A.R.; Nielsen, H.; Winther, O. DeepLoc 2.0: Multi-Label Subcellular Localization Prediction Using Protein Language Models. Nucleic Acids Research 2022, 50, W228–W234. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Jiang, L.; Akhil, C.S.; Wang, D.; Zhang, Z.; Zhang, W.; Xu, D. MULocDeep Web Service for Protein Localization Prediction and Visualization at Subcellular and Suborganellar Levels. Nucleic Acids Res 2023, 51, W343–W349. [Google Scholar] [CrossRef]
- Kaleel, M.; Zheng, Y.; Chen, J.; Feng, X.; Simpson, J.C.; Pollastri, G.; Mooney, C. SCLpred-EMS: Subcellular Localization Prediction of Endomembrane System and Secretory Pathway Proteins by Deep N-to-1 Convolutional Neural Networks. Bioinformatics 2020, 36, 3343–3349. [Google Scholar] [CrossRef]
- Zhang, N.; Rao, R.S.P.; Salvato, F.; Havelund, J.F.; Møller, I.M.; Thelen, J.J.; Xu, D. MU-LOC: A Machine-Learning Method for Predicting Mitochondrially Localized Proteins in Plants. Front. Plant Sci. 2018, 9, 634. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, W.; Zhang, Q.; Li, G.-Z. MultiP-SChlo: Multi-Label Protein Subchloroplast Localization Prediction with Chou’s Pseudo Amino Acid Composition and a Novel Multi-Label Classifier. Bioinformatics 2015, 31, 2639–2645. [Google Scholar] [CrossRef]
- Ryngajllo, M.; Childs, L.; Lohse, M.; Giorgi, F.M.; Lude, A.; Selbig, J.; Usadel, B. SLocX: Predicting Subcellular Localization of Arabidopsis Proteins Leveraging Gene Expression Data. Front Plant Sci 2011, 2, 43. [Google Scholar] [CrossRef]
- Lu, Z.; Szafron, D.; Greiner, R.; Lu, P.; Wishart, D.S.; Poulin, B.; Anvik, J.; Macdonell, C.; Eisner, R. Predicting Subcellular Localization of Proteins Using Machine-Learned Classifiers. Bioinformatics 2004, 20, 547–556. [Google Scholar] [CrossRef]
- Nair, R.; Rost, B. Sequence Conserved for Subcellular Localization. Protein Science 2002, 11, 2836–2847. [Google Scholar] [CrossRef]
- Fyshe, A.; Liu, Y.; Szafron, D.; Greiner, R.; Lu, P. Improving Subcellular Localization Prediction Using Text Classification and the Gene Ontology. Bioinformatics 2008, 24, 2512–2517. [Google Scholar] [CrossRef]
- Brady, S.; Shatkay, H. EPILOC: A (WORKING) TEXT-BASED SYSTEM FOR PREDICTING PROTEIN SUBCELLULAR LOCATION. In Proceedings of the Biocomputing 2008; WORLD SCIENTIFIC: Kohala Coast, Hawaii, USA, December 2007; pp. 604–615. [Google Scholar] [CrossRef]
- Huang, W.-L.; Tung, C.-W.; Ho, S.-W.; Hwang, S.-F.; Ho, S.-Y. ProLoc-GO: Utilizing Informative Gene Ontology Terms for Sequence-Based Prediction of Protein Subcellular Localization. BMC Bioinformatics 2008, 9, 80. [Google Scholar] [CrossRef]
- He, L.; Liu, X. The Development and Progress in Machine Learning for Protein Subcellular Localization Prediction. TOBIOIJ 2022, 15, e187503622208110. [Google Scholar] [CrossRef]
- Wan, S.; Mak, M.-W.; Kung, S.-Y. GOASVM: A Subcellular Location Predictor by Incorporating Term-Frequency Gene Ontology into the General Form of Chou’s Pseudo-Amino Acid Composition. Journal of Theoretical Biology 2013, 323, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.-B.; Chou, K.-C. Gneg-mPLoc: A Top-down Strategy to Enhance the Quality of Predicting Subcellular Localization of Gram-Negative Bacterial Proteins. Journal of Theoretical Biology 2010, 264, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.-B.; Yang, J.; Chou, K.-C. Euk-PLoc: An Ensemble Classifier for Large-Scale Eukaryotic Protein Subcellular Location Prediction. Amino Acids 2007, 33, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.-C.; Shen, H.-B. Hum-PLoc: A Novel Ensemble Classifier for Predicting Human Protein Subcellular Localization. Biochemical and Biophysical Research Communications 2006, 347, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Mei, S.; Fei, W.; Zhou, S. Gene Ontology Based Transfer Learning for Protein Subcellular Localization. BMC Bioinformatics 2011, 12, 44. [Google Scholar] [CrossRef]
- Chen, L.; Qu, R.; Liu, X. Improved Multi-Label Classifiers for Predicting Protein Subcellular Localization. MBE 2023, 21, 214–236. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, S.; Yu, B.; Zhang, Q.; Han, Y.; Zhang, Y.; Ma, Q. DMLDA-LocLIFT: Identification of Multi-Label Protein Subcellular Localization Using DMLDA Dimensionality Reduction and LIFT Classifier. Chemometrics and Intelligent Laboratory Systems 2020, 206, 104148. [Google Scholar] [CrossRef]
- Chen, C.; Chen, L.-X.; Zou, X.-Y.; Cai, P.-X. Predicting Protein Structural Class Based on Multi-Features Fusion. Journal of Theoretical Biology 2008, 253, 388–392. [Google Scholar] [CrossRef]
- Wang, S.; Zou, K.; Wang, Z.; Zhu, S.; Yang, F. A Novel Multi-Label Human Protein Subcellular Localization Model Based on Gene Ontology and Functional Domain. In Proceedings of the Proceedings of the 2023 3rd International Conference on Bioinformatics and Intelligent Computing; Association for Computing Machinery: New York, NY, USA, 2023; pp. 376–380. [Google Scholar] [CrossRef]
- Wan, S.; Mak, M.-W.; Kung, S.-Y. mGOASVM: Multi-Label Protein Subcellular Localization Based on Gene Ontology and Support Vector Machines. BMC Bioinformatics 2012, 13, 290. [Google Scholar] [CrossRef]
- Wan, S.; Mak, M.-W. Predicting Subcellular Localization of Multi-Location Proteins by Improving Support Vector Machines with an Adaptive-Decision Scheme. Int. J. Mach. Learn. & Cyber. 2018, 9, 399–411. [Google Scholar] [CrossRef]
- Chou, K.-C.; Shen, H.-B. A New Method for Predicting the Subcellular Localization of Eukaryotic Proteins with Both Single and Multiple Sites: Euk-mPLoc 2.0. PLoS ONE 2010, 5, e9931. [Google Scholar] [CrossRef]
- Mei, S. Multi-Label Multi-Kernel Transfer Learning for Human Protein Subcellular Localization. PLoS ONE 2012, 7, e37716. [Google Scholar] [CrossRef]
- Chen, L.; Li, Z.; Zeng, T.; Zhang, Y.-H.; Zhang, S.; Huang, T.; Cai, Y.-D. Predicting Human Protein Subcellular Locations by Using a Combination of Network and Function Features. Front. Genet. 2021, 12, 783128. [Google Scholar] [CrossRef]
- Garapati, H.S.; Male, G.; Mishra, K. Predicting Subcellular Localization of Proteins Using Protein-Protein Interaction Data. Genomics 2020, 112, 2361–2368. [Google Scholar] [CrossRef]
- Jiang, J.Q.; Wu, M. Predicting Multiplex Subcellular Localization of Proteins Using Protein-Protein Interaction Network: A Comparative Study. BMC Bioinformatics 2012, 13, S20. [Google Scholar] [CrossRef]
- Li, M.; Li, W.; Wu, F.-X.; Pan, Y.; Wang, J. Identifying Essential Proteins Based on Sub-Network Partition and Prioritization by Integrating Subcellular Localization Information. Journal of Theoretical Biology 2018, 447, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-H.; Luo, T.; Zhang, H.-L.; Du, P.-F. PLA-GNN: Computational Inference of Protein Subcellular Location Alterations under Drug Treatments with Deep Graph Neural Networks. Computers in Biology and Medicine 2023, 157, 106775. [Google Scholar] [CrossRef]
- Li, B.; Cai, L.; Liao, B.; Fu, X.; Bing, P.; Yang, J. Prediction of Protein Subcellular Localization Based on Fusion of Multi-View Features. Molecules 2019, 24, 919. [Google Scholar] [CrossRef]
- Zhang, Y.-H.; Ding, S.; Chen, L.; Huang, T.; Cai, Y.-D. Subcellular Localization Prediction of Human Proteins Using Multifeature Selection Methods. BioMed Research International 2022, 2022, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jin, S.; Gao, H.; Wang, X.; Wang, C.; Zhou, W.; Yu, B. Predicting the Multi-Label Protein Subcellular Localization through Multi-Information Fusion and MLSI Dimensionality Reduction Based on MLFE Classifier. Bioinformatics 2022, 38, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhai, Y.-J.; Xue, Z.-Z.; Xu, Y.-Y. Improving Protein Subcellular Location Classification by Incorporating Three-Dimensional Structure Information. Biomolecules 2021, 11, 1607. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, Y.; Li, S.; Han, Y.; Jin, S.; Gu, H.; Yu, B. Accurate Prediction of Multi-Label Protein Subcellular Localization through Multi-View Feature Learning with RBRL Classifier. Briefings in Bioinformatics 2021, bbab012. [Google Scholar] [CrossRef]
- Picard, M.; Scott-Boyer, M.-P.; Bodein, A.; Périn, O.; Droit, A. Integration Strategies of Multi-Omics Data for Machine Learning Analysis. Computational and Structural Biotechnology Journal 2021, 19, 3735–3746. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Liu, F.; Ju, Y.; Wang, Z.; Wang, C. Human Protein Subcellular Localization with Integrated Source and Multi-Label Ensemble Classifier. Sci Rep 2016, 6, 28087. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, Y.; Ding, P.; Li, S.; Yu, X.; Yu, B. ML-FGAT: Identification of Multi-Label Protein Subcellular Localization by Interpretable Graph Attention Networks and Feature-Generative Adversarial Networks. Computers in Biology and Medicine 2024, 170, 107944. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, Y.; Ding, P.; Li, S.; Yu, X.; Yu, B. ML-FGAT: Identification of Multi-Label Protein Subcellular Localization by Interpretable Graph Attention Networks and Feature-Generative Adversarial Networks. Computers in Biology and Medicine 2024, 170, 107944. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Qu, R.; Liu, X. Improved Multi-Label Classifiers for Predicting Protein Subcellular Localization. MBE 2023, 21, 214–236. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jin, S.; Gao, H.; Wang, X.; Wang, C.; Zhou, W.; Yu, B. Predicting the Multi-Label Protein Subcellular Localization through Multi-Information Fusion and MLSI Dimensionality Reduction Based on MLFE Classifier. Bioinformatics 2022, 38, 1223–1230. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, J.; Li, G.-Z. Multi-Location Gram-Positive and Gram-Negative Bacterial Protein Subcellular Localization Using Gene Ontology and Multi-Label Classifier Ensemble. BMC Bioinformatics 2015, 16 Suppl 12, S1. [Google Scholar] [CrossRef]
- Wan, S.; Mak, M.-W.; Kung, S.-Y. mGOASVM: Multi-Label Protein Subcellular Localization Based on Gene Ontology and Support Vector Machines. BMC Bioinformatics 2012, 13, 290. [Google Scholar] [CrossRef]
- Chou, K.-C.; Wu, Z.-C.; Xiao, X. iLoc-Euk: A Multi-Label Classifier for Predicting the Subcellular Localization of Singleplex and Multiplex Eukaryotic Proteins. PLoS ONE 2011, 6, e18258. [Google Scholar] [CrossRef]
- Shen, H.-B.; Chou, K.-C. Gneg-mPLoc: A Top-down Strategy to Enhance the Quality of Predicting Subcellular Localization of Gram-Negative Bacterial Proteins. Journal of Theoretical Biology 2010, 264, 326–333. [Google Scholar] [CrossRef]
- Yu, N.Y.; Wagner, J.R.; Laird, M.R.; Melli, G.; Rey, S.; Lo, R.; Dao, P.; Sahinalp, S.C.; Ester, M.; Foster, L.J.; et al. PSORTb 3.0: Improved Protein Subcellular Localization Prediction with Refined Localization Subcategories and Predictive Capabilities for All Prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Ouyang, W.; Winsnes, C.F.; Hjelmare, M.; Cesnik, A.J.; Åkesson, L.; Xu, H.; Sullivan, D.P.; Dai, S.; Lan, J.; Jinmo, P.; et al. Analysis of the Human Protein Atlas Image Classification Competition. Nat Methods 2019, 16, 1254–1261. [Google Scholar] [CrossRef]
- Thul, P.J.; Lindskog, C. The Human Protein Atlas: A Spatial Map of the Human Proteome. Protein Science 2018, 27, 233–244. [Google Scholar] [CrossRef]
- Xu, Y.-Y.; Yao, L.-X.; Shen, H.-B. Bioimage-Based Protein Subcellular Location Prediction: A Comprehensive Review. Front. Comput. Sci. 2018, 12, 26–39. [Google Scholar] [CrossRef]
- Nanni, L.; Lumini, A.; Brahnam, S. Survey on LBP Based Texture Descriptors for Image Classification. Expert Systems with Applications 2012, 39, 3634–3641. [Google Scholar] [CrossRef]
- Tahir, M.; Khan, A.; Majid, A. Protein Subcellular Localization of Fluorescence Imagery Using Spatial and Transform Domain Features. Bioinformatics 2012, 28, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Haralick, R.M.; Shanmugam, K.; Dinstein, I. Textural Features for Image Classification. IEEE Trans. Syst., Man, Cybern. 1973, SMC-3, 610–621. [Google Scholar] [CrossRef]
- Godil, A.; Lian, Z.; Wagan, A. Exploring Local Features and the Bag-of-Visual-Words Approach for Bioimage Classification. In Proceedings of the Proceedings of the International Conference on Bioinformatics, Computational Biology and Biomedical Informatics; ACM: Wshington DC USA September 22. , 2013; pp. 694–695. [Google Scholar] [CrossRef]
- Liu, G.-H.; Zhang, B.-W.; Qian, G.; Wang, B.; Mao, B.; Bichindaritz, I. Bioimage-Based Prediction of Protein Subcellular Location in Human Tissue with Ensemble Features and Deep Networks. IEEE/ACM Trans. Comput. Biol. and Bioinf. 2020, 17, 1966–1980. [Google Scholar] [CrossRef]
- Newberg, J.; Murphy, R.F. A Framework for the Automated Analysis of Subcellular Patterns in Human Protein Atlas Images. J. Proteome Res. 2008, 7, 2300–2308. [Google Scholar] [CrossRef]
- Zou, K.; Wang, S.; Wang, Z.; Zou, H.; Yang, F. Dual-Signal Feature Spaces Map Protein Subcellular Locations Based on Immunohistochemistry Image and Protein Sequence. Sensors 2023, 23, 9014. [Google Scholar] [CrossRef]
- Li, J.; Xiong, L.; Schneider, J.; Murphy, R.F. Protein Subcellular Location Pattern Classification in Cellular Images Using Latent Discriminative Models. Bioinformatics 2012, 28, i32–i39. [Google Scholar] [CrossRef]
- Ullah, M.; Han, K.; Hadi, F.; Xu, J.; Song, J.; Yu, D.-J. PScL-HDeep: Image-Based Prediction of Protein Subcellular Location in Human Tissue Using Ensemble Learning of Handcrafted and Deep Learned Features with Two-Layer Feature Selection. Briefings in Bioinformatics 2021, 22, bbab278. [Google Scholar] [CrossRef]
- Pärnamaa, T.; Parts, L. Accurate Classification of Protein Subcellular Localization from High-Throughput Microscopy Images Using Deep Learning. [CrossRef]
- Wang, F.; Wei, L. Multi-Scale Deep Learning for the Imbalanced Multi-Label Protein Subcellular Localization Prediction Based on Immunohistochemistry Images. Bioinformatics 2022, 38, 2602–2611. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; He, L.; Liu, T.; Liu, X.; Wei, L. Protein Subcellular Localization Based on Deep Image Features and Criterion Learning Strategy. Briefings in Bioinformatics 2021, 22, bbaa313. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Ding, Y.; Su, R.; Tang, J.; Zou, Q. Prediction of Human Protein Subcellular Localization Using Deep Learning. Journal of Parallel and Distributed Computing 2018, 117, 212–217. [Google Scholar] [CrossRef]
- Xue, Z.-Z.; Wu, Y.; Gao, Q.-Z.; Zhao, L.; Xu, Y.-Y. Automated Classification of Protein Subcellular Localization in Immunohistochemistry Images to Reveal Biomarkers in Colon Cancer. BMC Bioinformatics 2020, 21, 398. [Google Scholar] [CrossRef]
- Ding, J.; Xu, J.; Wei, J.; Tang, J.; Guo, F. A Multi-Scale Multi-Model Deep Neural Network via Ensemble Strategy on High-Throughput Microscopy Image for Protein Subcellular Localization. Expert Systems with Applications 2023, 212, 118744. [Google Scholar] [CrossRef]
- Wei, Z.; Liu, W.; Yu, W.; Liu, X.; Yan, R.; Liu, Q.; Guo, Q. Multiple Parallel Fusion Network for Predicting Protein Subcellular Localization from Stimulated Raman Scattering (SRS) Microscopy Images in Living Cells. IJMS 2022, 23, 10827. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, M.; Liu, H.; Yang, Y. Prediction of Protein Subcellular Localization Based on Microscopic Images via Multi-Task Multi-Instance Learning. Chinese J of Electronics 2022, 31, 888–896. [Google Scholar] [CrossRef]
- Hu, J.; Yang, Y.; Xu, Y.; Shen, H. Incorporating Label Correlations into Deep Neural Networks to Classify Protein Subcellular Location Patterns in Immunohistochemistry Images. Proteins 2022, 90, 493–503. [Google Scholar] [CrossRef]
- Tu, Y.; Lei, H.; Shen, H.-B.; Yang, Y. SIFLoc: A Self-Supervised Pre-Training Method for Enhancing the Recognition of Protein Subcellular Localization in Immunofluorescence Microscopic Images. Briefings in Bioinformatics 2022, 23, bbab605. [Google Scholar] [CrossRef]
- Long, W.; Yang, Y.; Shen, H.-B. ImPLoc: A Multi-Instance Deep Learning Model for the Prediction of Protein Subcellular Localization Based on Immunohistochemistry Images. Bioinformatics 2020, 36, 2244–2250. [Google Scholar] [CrossRef]
- Xu, Y.-Y.; Yang, F.; Zhang, Y.; Shen, H.-B. Bioimaging-Based Detection of Mislocalized Proteins in Human Cancers by Semi-Supervised Learning. Bioinformatics 2015, 31, 1111–1119. [Google Scholar] [CrossRef]
- Liu, S.; Huang, M.; Liu, X.; Han, K.; Wang, Z.; Sun, G.; Guo, Q. Swin Transformer Based Neural Network for Organelles Prediction from Quantitative Label-Free Imaging with Phase and Polarization (Qlipp) in Unlabeled Live Cells and Tissue Slices; SSRN, 2023.
- Husain, S.S.; Ong, E.-J.; Minskiy, D.; Bober-Irizar, M.; Irizar, A.; Bober, M. Single-Cell Subcellular Protein Localisation Using Novel Ensembles of Diverse Deep Architectures. Commun Biol 2023, 6, 489. [Google Scholar] [CrossRef]
- Ullah, M.; Hadi, F.; Song, J.; Yu, D.-J. PScL-DDCFPred: An Ensemble Deep Learning-Based Approach for Characterizing Multiclass Subcellular Localization of Human Proteins from Bioimage Data. Bioinformatics 2022, 38, 4019–4026. [Google Scholar] [CrossRef]
- Tahir, M.; Anwar, S.; Mian, A.; Muzaffar, A.W. Deep Localization of Subcellular Protein Structures from Fluorescence Microscopy Images. Neural Comput & Applic 2022, 34, 5701–5714. [Google Scholar] [CrossRef]
- Binder, J.X.; Pletscher-Frankild, S.; Tsafou, K.; Stolte, C.; O’Donoghue, S.I.; Schneider, R.; Jensen, L.J. COMPARTMENTS: Unification and Visualization of Protein Subcellular Localization Evidence. Database 2014, 2014, bau012–bau012. [Google Scholar] [CrossRef] [PubMed]
- Meinken, J.; Asch, D.K.; Neizer-Ashun, K.A.; Chang, G.-H.; Cooper, C.R., Jr.; Min, X.J. FunSecKB2: A Fungal Protein Subcellular Location Knowledgebase. cmb 2014. [Google Scholar] [CrossRef]
- Lum, G.; Meinken, J.; Orr, J.; Frazier, S.; Min, X. PlantSecKB: The Plant Secretome and Subcellular Proteome KnowledgeBase. cmb 2014. [Google Scholar] [CrossRef]
- Meinken, J.; Walker, G.; Cooper, C.R.; Min, X.J. MetazSecKB: The Human and Animal Secretome and Subcellular Proteome Knowledgebase. Database 2015, 2015, bav077. [Google Scholar] [CrossRef] [PubMed]
- Kerfeld, C.A.; Aussignargues, C.; Zarzycki, J.; Cai, F.; Sutter, M. Bacterial Microcompartments. Nat Rev Microbiol 2018, 16, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Yeates, T.O.; Crowley, C.S.; Tanaka, S. Bacterial Microcompartment Organelles: Protein Shell Structure and Evolution. Annu. Rev. Biophys. 2010, 39, 185–205. [Google Scholar] [CrossRef] [PubMed]
- Forster, B.M.; Marquis, H. Protein Transport across the Cell Wall of Monoderm Gram-positive Bacteria. Molecular Microbiology 2012, 84, 405–413. [Google Scholar] [CrossRef]
- Yadav, A.K.; Singla, D. VacPred: Sequence-Based Prediction of Plant Vacuole Proteins Using Machine-Learning Techniques. J Biosci 2020, 45, 106. [Google Scholar] [CrossRef]
- Rodriguez-Peña, R.; Mounadi, K.E.; Garcia-Ruiz, H. Changes in Subcellular Localization of Host Proteins Induced by Plant Viruses. Viruses 2021, 13, 677. [Google Scholar] [CrossRef]
- Chou, K.-C.; Wu, Z.-C.; Xiao, X. iLoc-Euk: A Multi-Label Classifier for Predicting the Subcellular Localization of Singleplex and Multiplex Eukaryotic Proteins. PLoS ONE 2011, 6, e18258. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Wu, Z.-C.; Chou, K.-C. iLoc-Virus: A Multi-Label Learning Classifier for Identifying the Subcellular Localization of Virus Proteins with Both Single and Multiple Sites. Journal of Theoretical Biology 2011, 284, 42–51. [Google Scholar] [CrossRef]
- iLoc-Plant: A Multi-Label Classifier for Predicting the Subcellular Localization of Plant Proteins with Both Single and Multiple Sites. Molecular BioSystems 2011, 7, 3287–3297. [CrossRef]
- Wan, S.; Mak, M.-W.; Kung, S.-Y. mPLR-Loc: An Adaptive Decision Multi-Label Classifier Based on Penalized Logistic Regression for Protein Subcellular Localization Prediction. Analytical Biochemistry 2015, 473, 14–27. [Google Scholar] [CrossRef]
- Wan, S.; Mak, M.-W.; Kung, S.-Y. Gram-LocEN: Interpretable Prediction of Subcellular Multi-Localization of Gram-Positive and Gram-Negative Bacterial Proteins. Chemometrics and Intelligent Laboratory Systems 2017, 162, 1–9. [Google Scholar] [CrossRef]
- Camon, E. The Gene Ontology Annotation (GOA) Database: Sharing Knowledge in Uniprot with Gene Ontology. Nucleic Acids Research 2004, 32, 262D–266. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, S.M.; Lee, S.-I. A Unified Approach to Interpreting Model Predictions. [CrossRef]
- Ancona, M.; Ceolini, E.; Öztireli, C.; Gross, M. Towards Better Understanding of Gradient-Based Attribution Methods for Deep Neural Networks 2018. [CrossRef]
- Shrikumar, A.; Greenside, P.; Kundaje, A. Learning Important Features Through Propagating Activation Differences 2019. [CrossRef]
- Luo, Z.; Wang, R.; Sun, Y.; Liu, J.; Chen, Z.; Zhang, Y.-J. Interpretable Feature Extraction and Dimensionality Reduction in ESM2 for Protein Localization Prediction. Briefings in Bioinformatics 2024, 25, bbad534. [Google Scholar] [CrossRef]
- Zecha, J.; Gabriel, W.; Spallek, R.; Chang, Y.-C.; Mergner, J.; Wilhelm, M.; Bassermann, F.; Kuster, B. Linking Post-Translational Modifications and Protein Turnover by Site-Resolved Protein Turnover Profiling. Nat Commun 2022, 13, 165. [Google Scholar] [CrossRef] [PubMed]
- Ramazi, S.; Zahiri, J. Post-Translational Modifications in Proteins: Resources, Tools and Prediction Methods. Database 2021, 2021, baab012. [Google Scholar] [CrossRef]
- Nickchi, P.; Jafari, M.; Kalantari, S. PEIMAN 1.0: Post-Translational Modification Enrichment, Integration and Matching ANalysis. Database 2015, 2015. [Google Scholar] [CrossRef]
- Pacharawongsakda, E.; Theeramunkong, T. Predict Subcellular Locations of Singleplex and Multiplex Proteins by Semi-Supervised Learning and Dimension-Reducing General Mode of Chou’s PseAAC. IEEE Trans.on Nanobioscience 2013, 12, 311–320. [Google Scholar] [CrossRef]
- Wan, S.; Mak, M.-W.; Kung, S.-Y. Transductive Learning for Multi-Label Protein Subchloroplast Localization Prediction. IEEE/ACM Trans. Comput. Biol. and Bioinf. 2017, 14, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Mak, M.-W.; Kung, S.-Y. Ensemble Linear Neighborhood Propagation for Predicting Subchloroplast Localization of Multi-Location Proteins. J. Proteome Res. 2016, 15, 4755–4762. [Google Scholar] [CrossRef]
- Digre, A.; Lindskog, C. The Human Protein Atlas—Spatial Localization of the Human Proteome in Health and Disease. Protein Science 2021, 30, 218–233. [Google Scholar] [CrossRef]
- Zhu, X.-L.; Bao, L.-X.; Xue, M.-Q.; Xu, Y.-Y. Automatic Recognition of Protein Subcellular Location Patterns in Single Cells from Immunofluorescence Images Based on Deep Learning. Briefings in Bioinformatics 2023, 24, bbac609. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, X.; Xu, C.; Han, X.; Wang, Y.; Situ, C.; Li, Y.; Guo, X. DeepSP: A Deep Learning Framework for Spatial Proteomics. J. Proteome Res. 2023, 22, 2186–2198. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Li, F.; Guo, X.; Wang, Z.; Pan, T.; Guo, Y.; Webb, G.I.; Yao, J.; Jia, C.; Song, J. Clarion Is a Multi-Label Problem Transformation Method for Identifying mRNA Subcellular Localizations. Briefings in Bioinformatics 2022, 23, bbac467. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zou, Q.; Yuan, L. A Review from Biological Mapping to Computation-Based Subcellular Localization. Molecular Therapy - Nucleic Acids 2023, 32, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, E.; Smith, T.; Pizzinga, M.; Elzek, M.; Queiroz, R.M.L.; Harvey, R.F.; Breckels, L.M.; Crook, O.M.; Monti, M.; Dezi, V.; et al. System-Wide Analysis of RNA and Protein Subcellular Localization Dynamics. Nat Methods 2024, 21, 60–71. [Google Scholar] [CrossRef]
- Cai, J.; Wang, T.; Deng, X.; Tang, L.; Liu, L. GM-lncLoc: LncRNAs Subcellular Localization Prediction Based on Graph Neural Network with Meta-Learning. BMC Genomics 2023, 24, 52. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; He, L.; Wang, Q.; Wang, Z.; Bao, W.; Cheng, H. Mit Protein Transformer: Identification Mitochondrial Proteins with Transformer Model. In Proceedings of the Advanced Intelligent Computing Technology and Applications; Huang, D.-S., Premaratne, P., Jin, B., Qu, B., Jo, K.-H., Hussain, A., Eds.; Springer Nature Singapore: Singapore, 2023; pp. 607–616. [Google Scholar] [CrossRef]
- Liu, M.-L.; Su, W.; Guan, Z.-X.; Zhang, D.; Chen, W.; Liu, L.; Ding, H. An Overview on Predicting Protein Subchloroplast Localization by Using Machine Learning Methods. Current Protein & Peptide Science 2020, 21, 1229–1241. [Google Scholar] [CrossRef]
- Wan, S.; Kim, J.; Won, K.J. SHARP: Hyperfast and Accurate Processing of Single-Cell RNA-Seq Data via Ensemble Random Projection. Genome Res. 2020, 30, 205–213. [Google Scholar] [CrossRef]
Figure 1.
Statistical analysis of UniProtKB [17] (2024_01.version). (A) The trend of protein number growth in TrEMBL (unreviewed proteins) and Swiss-Prot (reviewed proteins). The number of newly discovered unannotated proteins far exceeds that of newly added experimentally validated proteins. (B) The proportion of new-added protein counts between the two databases in the 2024_01.version. (C) Taxonomic distribution of protein sequences. (D) Number of proteins in the Top 10 subcellular locations.
Figure 1.
Statistical analysis of UniProtKB [17] (2024_01.version). (A) The trend of protein number growth in TrEMBL (unreviewed proteins) and Swiss-Prot (reviewed proteins). The number of newly discovered unannotated proteins far exceeds that of newly added experimentally validated proteins. (B) The proportion of new-added protein counts between the two databases in the 2024_01.version. (C) Taxonomic distribution of protein sequences. (D) Number of proteins in the Top 10 subcellular locations.
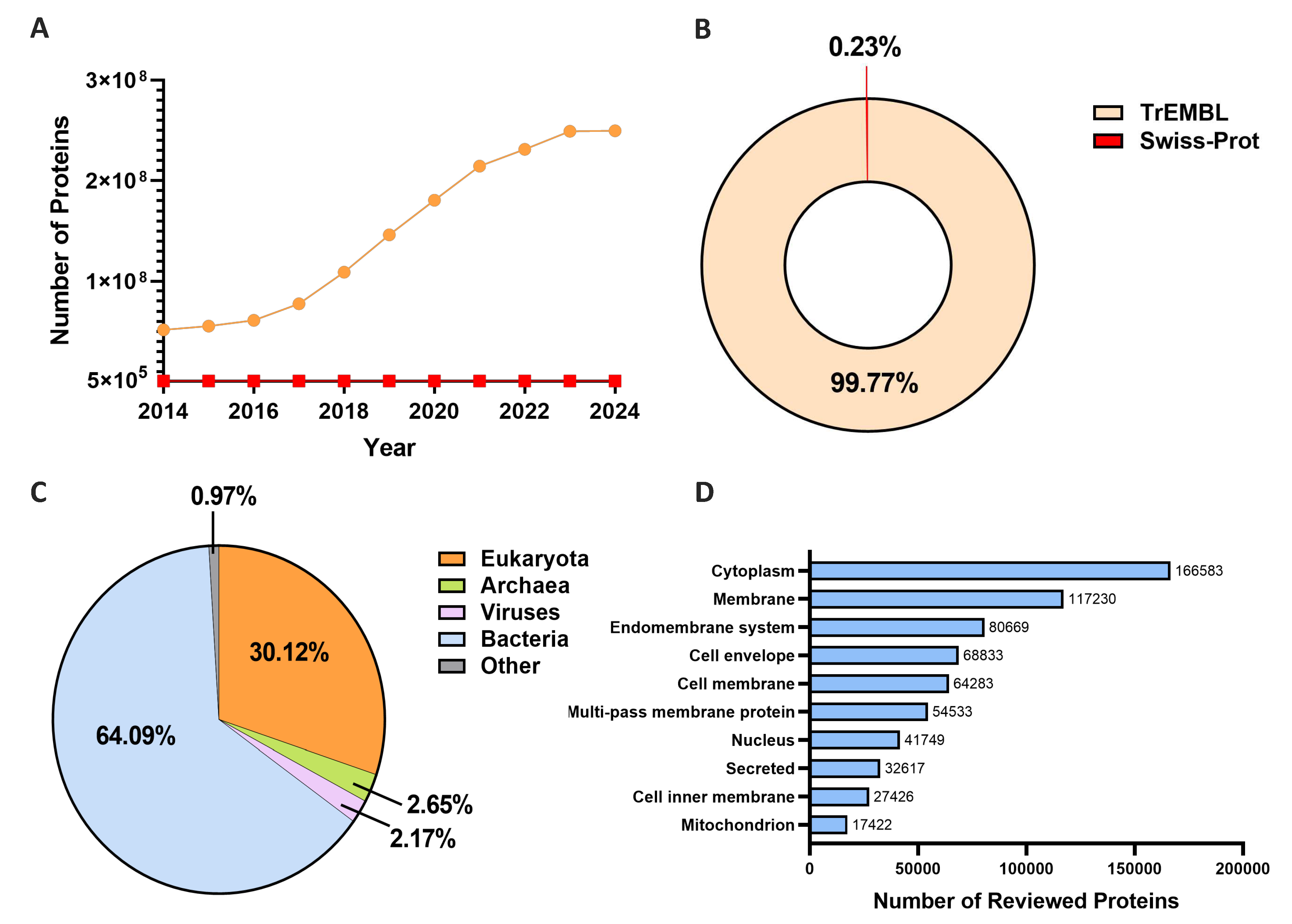
Figure 2.
The flowchart of three major types of AI-based prediction methods. The procedures include sequences or images as input, feature extraction, model prediction, and subcellular location output. (A) Key features extracted from sequences, annotations, and image inputs. Different classifiers extract composition information, encompassing AA order and frequency, physicochemical properties, and identifying signal peptide cleavage sites from sequence inputs. In addition to straightforward data, evolutionary profiles are also considered through homology alignment with the Position-Specific Scoring Matrix (PSSM) and the Position-Specific Frequency Matrix (PSFM). Knowledge-based methods involve the establishment of Gene Ontology (GO) vectors, derived from GO terms collected from specific databases with protein sequences or accession numbers as keywords. Other functional annotations, such as protein-protein interaction (PPI) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway information, can also be fused as input features. Imaging features mainly consist of morphological, haralick data and information from different channels, namely hand-crafted features, and deep features captured by deep learning algorithms. (B) Three types of algorithms used for prediction modules in computational models. (C) Major subcellular locations in a plant cell as an example of potential outputs, for proteins with single or multiple locations.
Figure 2.
The flowchart of three major types of AI-based prediction methods. The procedures include sequences or images as input, feature extraction, model prediction, and subcellular location output. (A) Key features extracted from sequences, annotations, and image inputs. Different classifiers extract composition information, encompassing AA order and frequency, physicochemical properties, and identifying signal peptide cleavage sites from sequence inputs. In addition to straightforward data, evolutionary profiles are also considered through homology alignment with the Position-Specific Scoring Matrix (PSSM) and the Position-Specific Frequency Matrix (PSFM). Knowledge-based methods involve the establishment of Gene Ontology (GO) vectors, derived from GO terms collected from specific databases with protein sequences or accession numbers as keywords. Other functional annotations, such as protein-protein interaction (PPI) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway information, can also be fused as input features. Imaging features mainly consist of morphological, haralick data and information from different channels, namely hand-crafted features, and deep features captured by deep learning algorithms. (B) Three types of algorithms used for prediction modules in computational models. (C) Major subcellular locations in a plant cell as an example of potential outputs, for proteins with single or multiple locations.
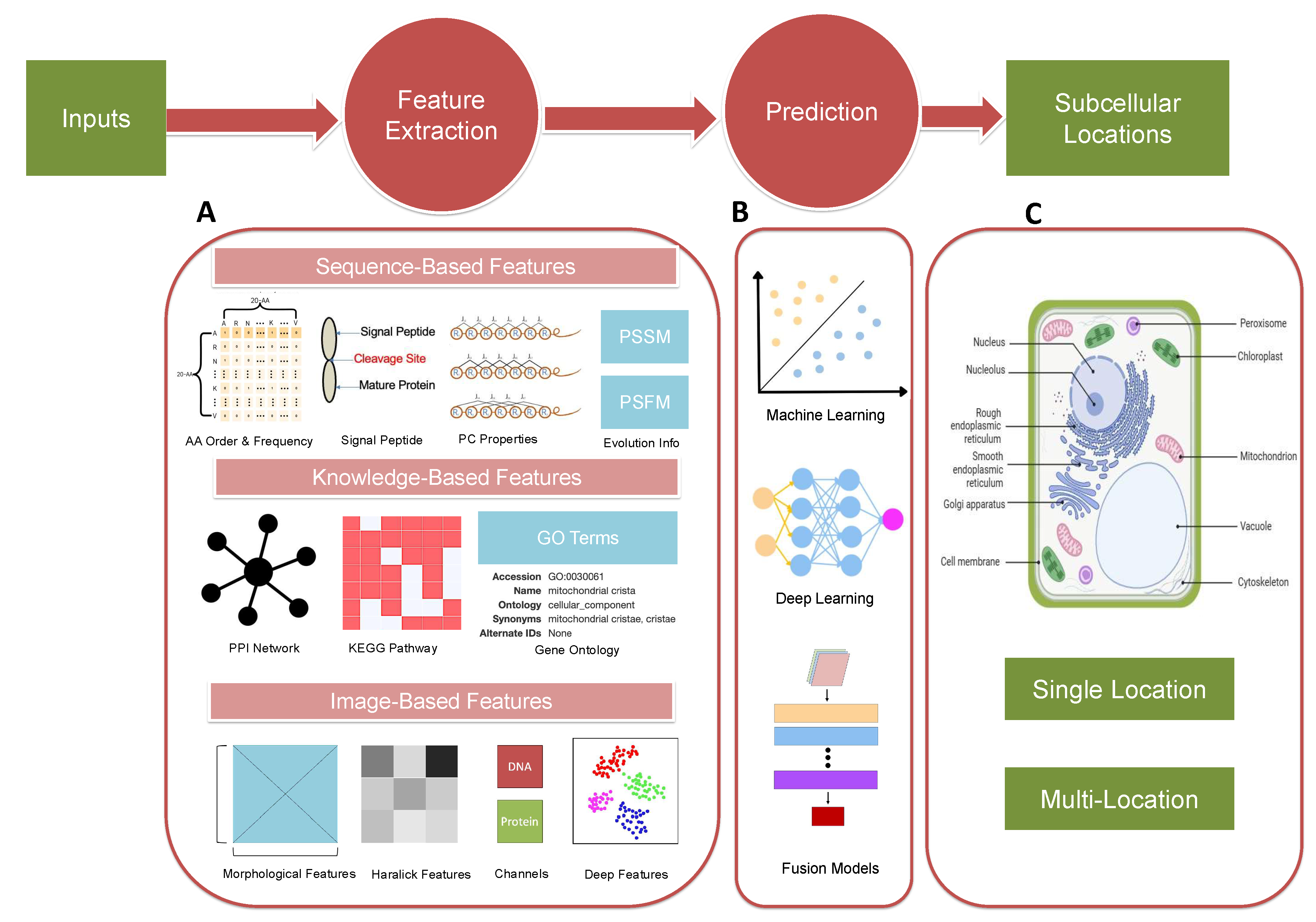
Figure 3.
Three primary categories of computational methodologies for processing imaging data. The red arrow depicts the progressive complexity of prediction models, reflecting advancements toward more sophisticated computational frameworks. Blue rectangle: Features used for model training; Green rectangle: Algorithms for location prediction. (A) Conventional Machine Learning Methods. Hand-crafted figures representing global and local information of images are extracted and trained for simple models. (B) Deep Learning Methods. Coupled with hand-crafted features, deep image features are obtained by deep neuro networks. (C) Complex Fusion Models. This method integrates multi-modality data like sequence, annotation texts and imaging data as model inputs to gain a more comprehensive and interpretable model for protein subcellular localization. SVM: Support Vector Machine. KNN: K-Nearest Neighbor. RF: Random Forest. LASSO: Least Absolute Shrinkage and Selection Operator. CNN: Convolutional Neural Network. DNN: Deep Neural Network. GAN: Generative Adversarial Network. LSTM: Long Short-Term Memory.
Figure 3.
Three primary categories of computational methodologies for processing imaging data. The red arrow depicts the progressive complexity of prediction models, reflecting advancements toward more sophisticated computational frameworks. Blue rectangle: Features used for model training; Green rectangle: Algorithms for location prediction. (A) Conventional Machine Learning Methods. Hand-crafted figures representing global and local information of images are extracted and trained for simple models. (B) Deep Learning Methods. Coupled with hand-crafted features, deep image features are obtained by deep neuro networks. (C) Complex Fusion Models. This method integrates multi-modality data like sequence, annotation texts and imaging data as model inputs to gain a more comprehensive and interpretable model for protein subcellular localization. SVM: Support Vector Machine. KNN: K-Nearest Neighbor. RF: Random Forest. LASSO: Least Absolute Shrinkage and Selection Operator. CNN: Convolutional Neural Network. DNN: Deep Neural Network. GAN: Generative Adversarial Network. LSTM: Long Short-Term Memory.
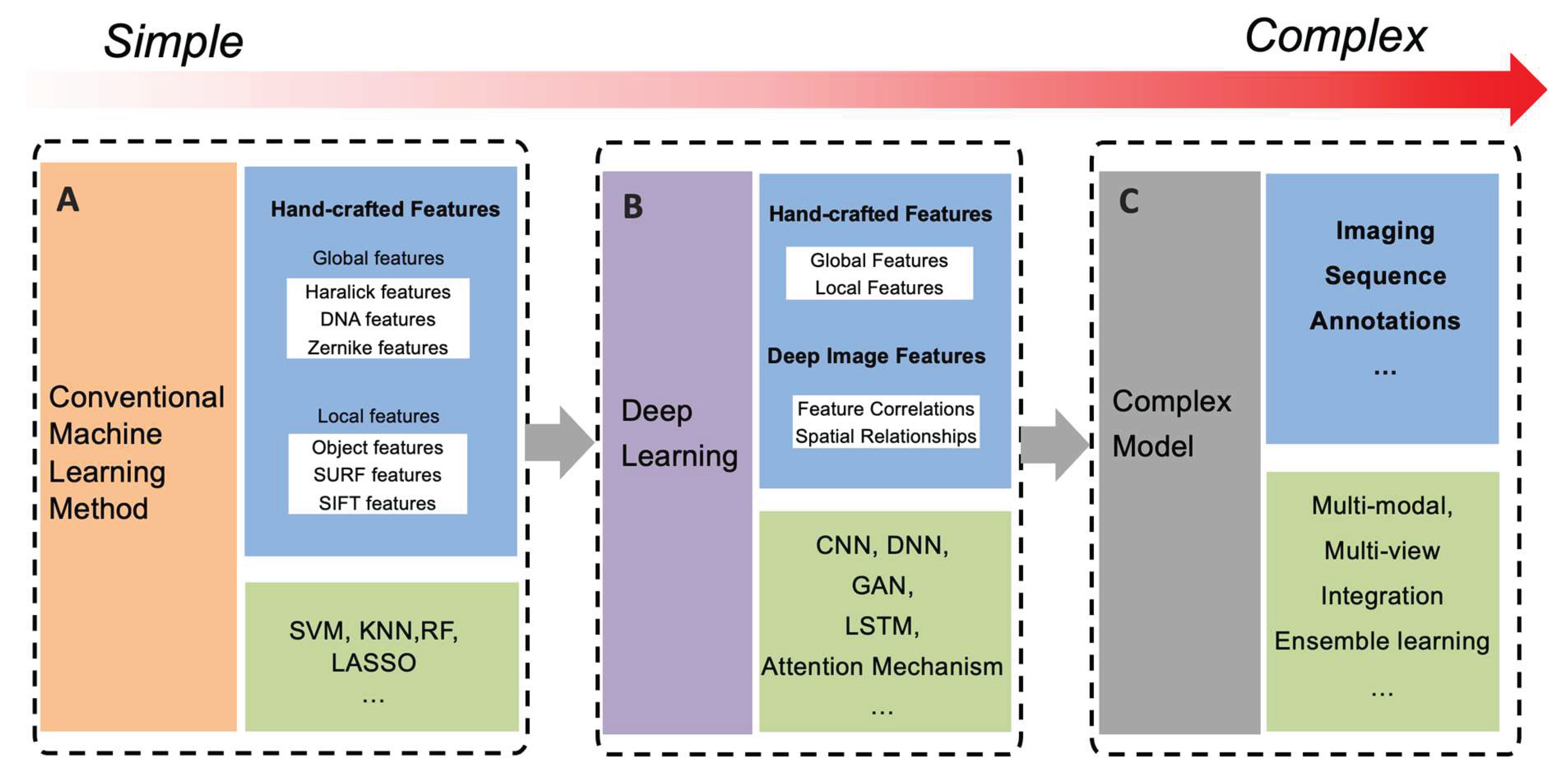
Table 1.
A summary of state-of-the-art sequence-based protein subcellular localization frameworks. S: Single-Location, M: Multi-location, GAN: Generative Adversarial Network, CNN: Convolutional Neural Network, LM: Language Model, SP: Signal Peptide, PSSM: Protein-Specific Scoring Matrix, LSTM: Long Short-Term Memory, AAC: Amino Acid Composition, LDA: Linear Discriminant Analysis, PseAA: Pseudo Amino Acid, PseAAC: Pseudo Amino Acid Composition, PPWM: Protein Position Weight Matrix, DNN: Deep Neural Network. KNN: K-Nearest Neighbor. SVM: Support Vector Machine.
Table 1.
A summary of state-of-the-art sequence-based protein subcellular localization frameworks. S: Single-Location, M: Multi-location, GAN: Generative Adversarial Network, CNN: Convolutional Neural Network, LM: Language Model, SP: Signal Peptide, PSSM: Protein-Specific Scoring Matrix, LSTM: Long Short-Term Memory, AAC: Amino Acid Composition, LDA: Linear Discriminant Analysis, PseAA: Pseudo Amino Acid, PseAAC: Pseudo Amino Acid Composition, PPWM: Protein Position Weight Matrix, DNN: Deep Neural Network. KNN: K-Nearest Neighbor. SVM: Support Vector Machine.
| Method | Features | Algorithm | Single-/Multi-Location | Species | Availability | Year |
|---|---|---|---|---|---|---|
| DaDL-SChlo | Handcrafted Features, Deep Features | ProtBERT, GAN, CNN | M | Plants | [62] | 2023 |
| DeepLoc – 2.0 | Masked-LM Objective | Multilayer Perceptron, Protein LM | M | Eukaryote | [63] | 2022 |
| SignalP – 6.0 | SP | Transformer Protein LM | M | Archaea, Gram-positive Bacteria, Gram-negative Bacteria and Eukarya | [26] | 2022 |
| MULocDeep | Physico-chemical Properties, PSSM | LSTM | M | Viridiplantae, Metazoa, Fungi | [64] | 2021 |
| SCLpred-EMS | AA Frequency | Deep N-to-1 CNN | S | Eukaryote | [65] | 2020 |
| CTM-AECA-PSSM-LDA | PSSM, LDA | SVM | S | Apoptosis Proteins on CL317 & ZW225 datasets | [34] | 2020 |
| TargetP – 2.0 | SP | LSTM | S | Plants and Non-plants | [25] | 2019 |
| Javed and Hayat | PseAA | KNN | M | Bacteria, Virus | [33] | 2019 |
| MU-LOC | AAC, PPWM, Functional Features | DNN, SVM | S | Plants (Mitochondrian) | [66] | 2018 |
| MultiP-SChlo | PseAAC | SVM | M | Plants (Subchloroplast) | [67] | 2015 |
| SLocX | AA Order, Gene Expression Profile | SVM | S | Plants | [68] | 2011 |
Table 3.
A summary of state-of-the-art image-based methods for protein subcellular localization prediction. S: Single-Location, M: Multi-Location. PSSM: Position-Specific Scoring Matrix, PseACC: Pseudo Amino Acid Composition, PC: Physicochemical Properties, LASSO: Least Absolute Shrinkage and Selection Operator, CNN: Convolutional Neural Network, DNN: Deep Neural Network, ResNet: Residual Network, DenseNet: Dense Convolutional Network, SRS: Stimulated Raman Scattering, MPFNet: Multiple parallel Fusion Network, DCF: Deep-cascade Forest, IF: Immunofluorescence Microscopic.
Table 3.
A summary of state-of-the-art image-based methods for protein subcellular localization prediction. S: Single-Location, M: Multi-Location. PSSM: Position-Specific Scoring Matrix, PseACC: Pseudo Amino Acid Composition, PC: Physicochemical Properties, LASSO: Least Absolute Shrinkage and Selection Operator, CNN: Convolutional Neural Network, DNN: Deep Neural Network, ResNet: Residual Network, DenseNet: Dense Convolutional Network, SRS: Stimulated Raman Scattering, MPFNet: Multiple parallel Fusion Network, DCF: Deep-cascade Forest, IF: Immunofluorescence Microscopic.
| Method | Features | Algorithm | Single-/Multi-Location | Species | Availability | Year |
|---|---|---|---|---|---|---|
| Zou et al. | Haralick, LBP, PSSM, PseAAC, PC | LASSO | S | Human | [119] | 2023 |
| ST-Net | Image Features | CNN, Transformer-networks | S | Human | [134] | 2023 |
| HCPL | Deep, Handcrafted Features of images | DNN | M | Human | [135] | 2023 |
| Ding et al. | Abstract Features with Different Depth | DNN | M | Yeast | [127] | 2023 |
| Muti-task Learning Strategy | Features Generated from ResNet or DenseNet | ResNet, DenseNet, CNN | M | Human | [129] | 2022 |
| MPFnetwork | SRS images | MPFNet | S | Human | [128] | 2022 |
| PScL-DDCFPred | Global & Local Features, Integrative Features | DNN, DCF | M | Human | [136] | 2022 |
| PLCNN | Raw Fluorescence Microscopy | CNN | M | Human, Yeast | [137] | 2022 |
| SIFLoc | IF images | ResNet18 | M | Human | [131] | 2022 |
| Deep-Yeast | Haralick, Gabor, Zernike Features | DNN | M | Yeast | [122] | 2017 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



