
Submitted:
21 February 2024
Posted:
23 February 2024
You are already at the latest version
Abstract
A drug repositioning effort supported the possible use of the anti-HIV drug etravirine as a treatment for Friedreich Ataxia (FRDA). Etravirine increases frataxin protein and corrects the biochemical defects in cells derived from FRDA patients. Because of these findings, and since etravirine displays a favorable safety profile, we conducted this pilot open-label phase 2 clinical trial assessing the safety and potential efficacy of etravirine in FRDA patients. Thirty-five patients were stratified into 3 severity groups and randomized to etravirine 200 mg/day or 400 mg/day. They were treated for 4 months. Efficacy endpoints were represented by changes in peak oxygen uptake and workload as measured by incremental exercise test, SARA score, cardiac measures, measures of QoL and disability. Data were collected 4 months before the start of the treatment (T-4), at the start (T0), at the end (T4) and 4 months after the termination of the treatment (T+4). Etravirine was well tolerated. Etravirine completely stopped the progression of the SARA score during the 4-months treatment period, compared to the 4 months pre and post treatment. It increased peak workload, while the improvement of peak oxygen uptake was not statistically significant. No changes in the cardiac measures were observed. Health and QoL measures showed a worsening at the suspension of the drug. In this open trial etravirine significantly improved neurological function and was generally safe and reasonably tolerated. This suggests that etravirine represents a potential therapeutic agent in FRDA deserving testing in a randomized placebo controlled clinical trial.
Keywords:
Friedreich ataxia
; etravirine
; treatment
; safety
; efficacy
1. Introduction
1.1. Background and Rationale
Friedreich’s ataxia (FRDA) is the most common inherited ataxia with a prevalence of approximately 1 in 50,000 people affected. It is an autosomal recessive neurodegenerative disorder. The disease is caused by the defective synthesis of frataxin, a mitochondrial protein synthesized by the frataxin gene (FXN) [1]. The role of this protein has not yet been fully clarified. We know from the literature that it is a protein responsible for regulating iron-sulfur cluster enzymes within the mitochondria. The defect in frataxin causes a dysfunction of adenosine triphosphate synthesis, accumulation of mitochondrial iron and increased sensitivity to oxidative stress with a consequent further reduced activity of the mitochondrial respiratory chain. Free radicals production and reduced cell respiration lead to cell death especially in highly energy dependent tissues such as neurons, cardiomyocytes, Langerhans pancreatic cells [2,3]. Approximately 96% of patients carry a homozygous intronic GAA expansion that impairs FXN gene transcription and translation. The persistence of an intact reading frame within the genome of most FRDA patients opens the way to treatment attempts that target FXN gene transcription and messenger RNA (mRNA) translation to increase endogenous frataxin levels and balance the complex metabolic consequences of frataxin deficiency [4]. The age of onset of the disease is typically within the first two decades of life and wheelchair use is necessary ten to fifteen years after disease onset [5]. The disease is characterized by a relentless progression involving motor coordination (ataxia), heart function and structure (hypertrophic cardiomyopathy), exercise capacity (impaired bioenergetics with reduced aerobic capacity), endocrine function (diabetes) visual and auditory function (optic atrophy and hearing loss) [5,6,7].
Recently, it has been shown that the drug etravirine (ETR), a low-molecular-weight agent currently used in human immunodeficiency virus (HIV) therapy with an excellent safety profile and tolerability, stimulates the translation of the FXN gene and promotes cellular accumulation of frataxin.
By screening a library of 853 Food and Drug Association (FDA)—approved drugs, few drugs were identified capable of upregulating frataxin in vitro. Among them, ETR was selected for its generally favorable safety profile and its significant effect on frataxin levels.
Preclinical studies in cells derived from FRDA patients have confirmed a role of ETR in promoting the accumulation of frataxin, correcting the biochemical damage caused by frataxin deficiency and protecting cells from oxidative damage. ETR induces an overexpression of the frataxin protein by a mechanism of action that presumably involves the redistribution of the frataxin mRNA onto polyribosomes and consequently improved efficiency of protein translation [8]. In addition, ETR is able to up-regulate the frataxin protein in the heart and skeletal muscle of frataxin-defective YG8 mice after oral administration for 5 days (unpublished). These results assume even greater significance considering that reconstitution of frataxin levels in vitro was obtained with ETR concentrations corresponding to those achieved in plasma during anti-HIV ETR therapy at standard doses (200-400 mg/day).
ETR is a diarylpyrimidine that acts as a non-nucleoside reverse transcriptase inhibitor. It was approved in 2008 by the FDA (trade name Intelence) for the treatment of patients affected by HIV that are resistant to other drugs. In 2018 the label was extended to include patients 2 years of age and older. The FDA label reports no contraindications. ETR is administered orally in tablets.
Treatment options are currently very limited for FRDA patients, with omaveloxolone being the first and only drug approved in 2023 by FDA for adult and adolescent FRDA patients 16 years of age and older, as capable of improving mFARS score after chronic treatment. Omav is a Nuclear erythroid-derived 2-related factor 2 (Nrf2) activator. In FRDA there is reduced activation of Nrf2 in cells. Nrf2 is a transcription factor essential for counteracting oxidative stress and the consequent cellular damage, reducing the expression of the inflammatory cytokines TNF-alpha and IL-6 [9,10].
1.2. Objectives
Because of its ability to upregulate frataxin protein and rescue phenotypic defects in FRDA patient-derived cells and because of its safety and approved use in children, ETR represents an attractive potential therapy for FRDA. Our goal is to test this drug in patients with FRDA through a pilot open phase 2 clinical study. The study aims to explore the transferability from laboratory to clinical practice of the encouraging results obtained on human cells and animal models. The questions that the study intends to address are the following:
1. Will ETR in FRDA patients continue to maintain the same safety and tolerability profile shown so far in licensed clinical use?
2. Will in vivo ETR in FRDA patients with the doses proposed in this study, capable of ensuring serum concentrations similar to those tested in vitro, be associated with significant variation of the indicators used as efficacy endpoints?
2. Materials and Methods
2.1. Study Design
We designed a monocenter randomized pilot open label Phase 2 clinical trial to evaluate the safety and potential efficacy of ETR in FRDA at two doses. The patients were stratified by severity and assigned randomly to two different groups treated with different dosages (200 mg/die or 400 mg/die). The 400 mg/day dose was chosen as this is the full dose recommended in the current label. The dose of 200 mg/day was chosen based on feedback from many patients with FRDA who are already using ETR off-label.
The study includes 5 visits comprising a period of treatment of 4 months, preceded by 4 months and followed by an additional 4 months of study in absence of treatment for measure and considers intra-individual disease progression.
This design and this treatment time were chosen on the basis of the experience gained in the study “Safety and efficacy of treatment with γIFN in subjects with FRDA” recently concluded by the promoter, in which the comparison between disease progression in absence of treatment and in the presence of treatment provided significant data and in which this variation was already found in the third month of treatment [11].
2.2. Setting
The study was performed at the “Eugenio Medea’’ Scientific Institute in Pieve di Soligo and Conegliano (Treviso, Italy) from September 2020 to January 2023, with recruitment concluded by January 2022.
2.3. Participants
A total of 35 individuals with FRDA were recruited. All participants and their parents/legal tutors were informed regarding the experimental nature of the study and signed the informed consent in accordance with the Declaration of Helsinki (World Medical Association, 1964). The study has been reviewed and approved by the competent Ethics Committee (Prot. No 0103439, 01/07/2020). This study was registered at www.clinicaltrial.gov (NCT04273165).
Enrollment was done by advertising the study through the patients’ associations and among FRDA patients currently followed at the site.
Patients were required to have molecularly defined FRDA diagnosis (at least one expanded FXN allele) and be ≥ 10 years of age and ≤ 40 years of age. They also had to be able to complete an incremental exercise testing defined by the ability to ride a cycle ergometer with upper limbs at least 45 rpm with no added resistance for 3 minutes. Moreover, they had to weigh >30 kg.
Patients were excluded if they had HIV, HBV or HCV infection, severe organ failure, history of clinically significant cardiac disease and uncontrolled diabetes. Another exclusion criteria was known hypersensibility to ETR or to any component of the study drug. Patients were required to discontinue other experimental therapeutics for FRDA at least 4 weeks before study entry. No participation in other experimental studies was allowed. Patients were then asked to maintain their typical exercise routine.
2.4. Interventions
Patients received a dose of 200 mg/day (1 tablet/day in the morning) or 400 mg/day (2 tablets/day morning and evening) for 4 months.
The first dose was given under direct medical supervision in the facility. The subsequent treatment was carried on at home following the prescription given to the patients.
2.5. Study Endpoints
Primary endpoint was the safety and tolerability of ETR at 200 or 400 mg daily dosage in FRDA subjects aged 10-40 years. To monitor it, the patients were subjected to physical examination including vital parameters assessment, skin inspection, blood tests (clinical chemistry, hematology) and urinalysis. These assessments were done 4 months before the start of the treatment (T-4), at the start of the treatment (T0), after 2 months (T2), at the end of the treatment (T4) and 4 months after the termination of the treatment (T+4). In addition, a checklist listing all known adverse events (AE) associated with ETR was distributed to all patients and reviewed at each follow up visit. Any additional AE was registered irrespective of its likely connection with the treatment.
Secondary endpoint was the assessment of the efficacy of the treatment. The main outcome was peak oxygen uptake (peak VO2) attained during incremental exercise test. Other outcomes were:
- peak workload (peak W);
- neurological progression measured with Scale for the assessment and Rating of Ataxia (SARA) [12];
- echocardiography (septal wall thickness) and electrocardiogram (Sokolow-Lyon index) parameters;
- frataxin protein levels in peripheral blood mononuclear cells and molecular analysis of frataxin mRNA translation efficiency;
- quality of life evaluated with Short Form Health Survey 36 (SF-36) [13];
- disability, assessed by World Health Organization Disability Assessment Schedule 2.0 (WHODAS 2.0) 36-item self-administered version [14].
These measures were assessed at T-4, T0, T4 and T+4, while SARA scale was also at T2.
The study scheme is represented in Figure 1.
2.5.1. Incremental Exercise Test
It has been shown that peak VO2 normalized by weight is a very stable objective measure of aerobic capacity [15,16]. FRDA is associated with significant decrease (about 50% less) aerobic fitness as measured by standardized exercise testing and its measure has been employed as endpoint in FRDA intervention trials several times [9,17,18,19]. Incremental exercise test was performed on a cycle ergometer for upper limb (Cosmed arm ergometer 400 P) and the assessments included peak VO2 (in mL/min/kg) and peak W (in Watt).
The test included 1 minute warm-up period at 0 W workload, maintaining a cadence of at least 45 rpm, followed by a 2 W increase in work rate every one minute. Subjects were encouraged to work to exhaustion. This mild exercise protocol was chosen to include patients with a wide range of severity (SARA score).
2.5.2. Neurological Measure
SARA scale was used as the neurological measure. It is based on a semi-quantitative clinical assessment of cerebellar ataxia (spinocerebellar, Friedreich’s and sporadic ataxia) on an impairment level. SARA has 8 items (gait, stance, sitting, speech, finger-chase, nose-finger, fast alternating movements, heel-shin). It has a maximum score of 40 (severe ataxia) and a minimum of 0 (no ataxia).
2.5.3. Cardiac Parameters
Heart is primarily affected in FRDA showing progressive cardiac hypertrophy. Cardiomyopathy is the main determinant of survival in this population. The Electrocardiogram (ECG) changes summarized by the Sokolow-Lyon index (∆%QRS-voltage) are reliable indicators of ventricular hypertrophy. Echocardiography (EchoCG) can detect and measure very precisely the extent and severity of heart hypertrophic changes. We measured interventricular septal wall thickness (in mm) and Sokolow-Lyon index (in mm).
2.5.4. Health and Disability Measures
SF-36 is a self-administered questionnaire, completed by the patient, which aims to quantify the state of health and measure the health-related quality of life. The questionnaire explores 8 domains (Physical functioning; Physical role limitations; Bodily pain; General health; Vitality; Social functioning; Emotional role limitations; Mental health) that are scored separately. The scores are transformed to range from 0 (worst possible health) to 100 (best possible health).
WHODAS 2.0 is an internationally validated widely applied disease non-specific measure of disability developed by WHO within the framework of the biopsychosocial model of functioning and disability. It asks the subject to score in terms of severity and duration any difficulty experienced in the 30 previous days in 6 main life areas (Cognition; Mobility; Self-care; Getting along; Life; Participation). Self-administered version was used. Domain-specific scores and a summary score are calculated and the results were computed as normalized scores that range from 0 (no disability) to 100 (full disability).
These measures have been administered only to subjects ≥16 years of age.
2.6. Sample Size Calculation
Sample size was calculated considering the primary safety endpoint (no Serious Adverse Events - SAE - related to ETR therapy) and the principal efficacy outcome. The VO2max test on the cycle ergometer in normal subjects shows a value > 16.2 ml/min/kg with a test-retest variability of ± 4 ml/min/Kg. This parameter shows values reduced by about 50% in patients with FRDA. Assuming an expected variation of the range of 0-2% between time points T-4 and T0 and of +20% between T0 and T4 (α error of 0.05, statistical power of 80%), the required number of participants was 30. Assuming approximately 15% of the trial dropped out due to non-safety issues (eg, failure to complete assessments, non-compliance with drug regimen, or withdrawal of consent), the enrollment goal was set at 35 patients.
2.7. Randomization
After being stratified into 3 severity groups defined by the SARA score obtained at T-4 (<20, 20-30, >30), the recruited subjects were assigned to the dose of 200 mg/day or 400 mg/day randomly by computer-generated sequencing, with a 1:1 allocation ratio using blocks of 4 within each group to ensure that the intervention was balanced within each stratum.
2.8. Statistical Analysis
Descriptive statistics were obtained for the population characteristics. Continuous variables were described as mean (SD) or median (IQr), while categorical variables were expressed as number (percentage). The comparison between the two dose groups was performed using t-test or Mann–Whitney U test for continuous variables and Fisher’s Exact test for categorical variables. The effect of the treatment on the considered outcomes was studied using linear mixed-effect models with subject as random effect. Post hoc tests were performed for the significant models’ effects. All models were adjusted for age, severity, and dosage. Finally, multiple linear models were used to study the effect of dosage and severity (independent variables) on differences in outcomes (dependent variables) during the treatment period (between T0 and T4) and in the post-treatment period (between T4 and T+4), adjusting for age.
Values of p<0.05 were considered statistically significant. Statistical analyses were performed using R [20].
3. Results
3.1. Participant Flow
With the goal of enrolling 35 participants, 38 patients were screened. 2 of them were excluded because they did not meet the criteria of inclusion and 1 withdrew the consent. The enrolled patients were divided into 3 severity classes: 19 with SARA <20, 8 with SARA 20-30 and 8 with SARA >30. In the first group 9 patients were randomized to ETR at doses of 200 mg/day, and 10 patients were randomized at doses of 400 mg/day. In the other two groups 4 patients received 200 mg and 4 assumed 400 mg. One participant withdrew after completing the T4 visit. Another patient dropped out during the treatment for reasons unrelated to the drug (see Figure 2).
3.2. Patient Features
The patients are 30 adults and 5 children, 19 females and 16 males. The average age was 25.0 (7.1) (mean (SD)) years and disease duration of 13.2 (6.0) years, with an age at onset of 11.8 (5.7) years. The mean overall SARA score at T-4 was 20.7 (8.2), while for the first group of severity was 14.4 (3.6), for the second one 24.8 (2.6) and the third one 32.2 (1.1). Thirty-three patients were homozygous for GAA repeat expansion and 2 had a repeat expansion on one allele and a point mutation on the other. For 5 patients the precise number of triplets was not available, while for the others the mean GAA repeat expansion in the short allele (GAAsr) was 665.5 (189.0) while in the long allele (GAAlr) 862.9 (203.9).
Patient demographics and clinical data are presented in Table 1.
3.3. Safety
One event of symptomatic supraventricular tachycardia (SAE), judged unrelated to study drug, occurred. There were no treatment interruptions or discontinuations due to tolerability issues.
Overall, AE were generally mild in severity and were consistent with the Summary of Product Characteristics.
Table 2 shows the AE reported by the patients or detected in the physical examination, dividing them into the first two months and the last two months of treatment. The most commonly reported AE was headache, followed by diarrhea, nausea, skin rash, fatigue and hypotension. In particular, it should be noted that the most frequent AE appeared and resolved in the first two weeks of treatment, with the exception of 4 cases that presented sporadic episodes of headaches, diarrhea and nausea even in the last two months, but also after the suspension in the follow-up period. Most events resolved spontaneously, except for 2 cases of diarrhea and 7 cases of headache, in which targeted drug therapy was taken, respectively lactic acid ferments and paracetamol or other non-steroid anti-inflammatory drugs.
Table 3 shows the main alterations detected in blood and urine tests that occurred while taking the drug. Five patients had a lipid alteration, in particular 2 patients had an increase of total cholesterol, 1 of triglycerides and 2 of both. There was 1 case of liver enzymes transient elevation (AST/ALT), 1 case of amylase rise and 1 case of mild leukopenia. There were also 2 cases (6%) of slight increase in glycated hemoglobin, but within the limits of 10%. These values normalized when the treatment was stopped.
These effects were not dose-dependent. They were observed in both high and low dose treatment.
3.4. Efficacy
3.4.1. Variations in Clinical and Functional Outcomes
Regarding clinical and functional outcomes, Figure 3 represents the trend of the mean scores in all patients in timepoints (scores in Table S1). Except for the septal wall thickness, the variations were in the direction of a functional improvement taking the drug (T4 vs. T0) that reversed at drug discontinuation (T+4 vs. T4).
In particular, from mixed effect models results, described in Table 4, peak VO2 showed an average increase, although not statistically significant, with the drug of 0.959 ml/min/kg while the decrease with the suspension (-1.145 ml/min/kg) approached statistical significance (p.value=0.088). We observed a slight improvement from T-4 to T0 which can be considered physiological or a consequence of familiarization with the test.
Among the secondary endpoints, it should be noted that SARA scale showed the evolution of the disease from T-4 to T0 with a mean worsening of 0.456 points, while a significant improvement with the treatment (mean reduction score -1.132, p.value<0.001). There was also a significant worsening when the drug was discontinued (average increase score 1.229, p.value<0.001).
For peak W there was a significant improvement while on drug treatment (average increase 2.941 Watt, p.value=0.021) and a subsequent worsening but not significant decrease with the suspension (-1.037 Watt).
For cardiac measurements there were no significant differences between timepoints.
Considering the mean differences by severity and dose (Table 5), we found the same trend in all three severity groups and for both doses, that is an improvement in the scores during treatment and a worsening at suspension, except for the septal wall thickness.
Specifically, on the SARA scale, at lower severity there appears to be a greater average improving effect of the drug, but also a greater worsening effect upon discontinuation. Regarding peak VO2 and Sokolow-Lyon index, no particular trends are noted for different severities, while for peak W the effect appears greater in more severe patients.
These results are supported by the regression model results (Table 6). We observed a significant difference for the SARA score between the high severity and low severity groups both during treatment (mean difference of 1.398 points, p.value=0.010) and after treatment (mean difference of -1.552 points, p.value=0.002). In contrast, for peak VO2 and peak W, no significant differences were observed between the severity groups. Regarding dosage, the observed mean effects appeared more pronounced for high dosages, both in terms of improvement during the treatment and in terms of worsening at suspension, but not significant. Also for cardiac parameters no significant differences were observed between severity and dosage groups.
Models are adjusted for age. Reference groups: SARA < 20 for severity, 200 mg/day for dose.
3.4.2. Variations in SF-36 and WHODAS 2.0 Questionnaires
In all the 8 domains of the SF-36 the average score was fairly stable before the start of treatment; although not statistically significant, in Emotional Role limitations e in Mental Health there was an improvement, which also continued while taking the drug. The domain that improved significantly during the treatment was Vitality, which then worsened significantly by suspending the drug, together with the domain of Social Functioning. The trend of all domains in the four months following the end of treatment was worsening (Figure 4).
We then considered the trend of the WHODAS 2.0 summary score and its 6 domains. With the exception of the Cognition domain that remained stable, also these scores showed a trend of worsening in the 4 months before taking the drug, of improvement during treatment and of subsequent worsening after withdrawal. In particular, the latter was statistically significant in the Mobility and Life Activities domains. (Figure 5).
3.4.3. Variations in Frataxin
The sample for the analysis of the frataxin protein and mRNA was shipped to the laboratory identified for the extraction and preparation of the material and then to the laboratory identified for the analysis. The shipments were to be completed within strict time frames but the logistics complications linked to the concurring pandemic condition caused fatal deterioration of the samples preventing any reliable analytical result.
4. Discussion
We report here the results of a pilot open-label clinical trial testing ETR in a group of 35 FRDA patients. This is the first study to evaluate the effect of ETR in patients with FRDA and it demonstrates that ETR represents a relatively safe potential therapeutic option in the treatment of FRDA.
The choice of the drug was driven by the pre-clinical research results, reinforcing the strength of the translational potential of the drug repurposing research, particularly in the context of rare diseases such as FRDA [21,22]. The design was inspired by the goal of appreciating the capacity of the tested drug to modify the disease’s natural history, while the duration was chosen to meet the preliminary results obtained on the animal model and to keep the trial duration at the minimum maximizing efficiency.
This phase 2 study was aimed primarily at addressing the safety and tolerability of ETR in FRDA patients. We found only minor adverse effects, both qualitatively and quantitatively similar to what already observed in other studies on ETR for other diseases [23], which indicates that ETR is generally safe and reasonably tolerated by FRDA patients. The single SAE, albeit deemed not related to treatment, calls for special attention to cardiac parameters in FRDA subjects taking ETR.
In addition to the overall safety of ETR in FRDA, we evaluated efficacy.
This protocol, which provides for a total duration of one year (4 months pre-treatment, 4 months with treatment and 4 months post-treatment), made it possible to evaluate the patient in a year of disease and how ETR affects the disease trend. We document the expected progression of the disease in the pre- and post-treatment period, from T-4 to T0 and from T4 to T+4. The extension of the study to 4 months before treatment start and 4 months after treatment termination gives the possibility to study and compare, for each patient, the disease progression in the absence of therapy and the disease course after termination of therapy. In this design, each subject is an effective control for him/herself. During treatment, from T0 to T4, disease progression was stopped and even reversed. This study was able to record a remarkable average decrease of about 1.1 points in the SARA scale, after 4 months of treatment. This improvement is significant, given that 1 SARA point would be the expected disease progression in one year [24].
In addition, at the incremental exercise test, there was a significant increase in peak W, but not in peak VO2 although the trend is improving.
Given the open nature of the trial we cannot exclude a placebo effect, but the significance of the observed SARA and workload variations support the need for a longer placebo-controlled trial.
No significant change was observed in the cardiac parameters. From a cardiological point of view, there is a stability of the parameters while taking the drug. The short duration of treatment does not allow us to draw conclusions.
In the SF-36 there is an improvement in some items related to emotional aspects. At WHODAS 2.0 there is an improvement especially for aspects of daily life and mobility. Of notable relevance was the worsening after discontinuation of the drug in all domains of both questionnaires. This trend could be linked to the expectation of healing related to drug intake. However, the trend of these PROMs is the same as the other efficacy indicators.
In support of these findings, patients and caregivers reported additional notations not captured by any formal tool: reduced fatigue and increased energy and endurance; less help in moving and transferring, greater stability with reduction of falls; improved manual skills and writing; reduced dysphagia episodes, improved swallowing; improved mood; improved voice quality and speech; reduced urinary urgency. These informal reports may guide the development of more tailored PROMs. The perceived efficacy was such that 22 patients requested to start an off-label treatment with ETR.
We originally planned to also quantitate the amount of frataxin mRNA and protein in peripheral blood cells, as an additional efficacy measure. However, the limited amount of blood that could be drawn from most patients, combined with sample shipment issues due to the pandemic, did not allow a consistent extraction of frataxin mRNA and protein from all samples, preventing a complete and reliable analysis. In future trials the attempt to study frataxin protein or mRNA may consider targeting tissues with higher frataxin content such as cells in urinary sediments or skeletal muscle obtained by needle biopsy.
Patients of all stages of the disease were enrolled in our study. The study cohort consisted of a heterogeneous patient population, including ambulatory patients at disease onset and patients in more advanced clinical stages who had already been in a wheelchair for years. Our study shows that there is a greater response to ETR in subjects with mild severity, while in severely affected subjects with SARA >30 the effect is smaller. This result could be explained by the ceiling effect of the scale. We observed a ceiling effect in particular for the items concerning gait, sitting position and heel-shin slip. The ceiling effect of SARA has already been observed for patients with FRDA by several authors [24,25]. The ceiling effect of SARA may have underestimated the clinical variations in severely affected patients.
Currently Omav is the only drug approved in the USA for FRDA. Lynch et al. [9] conducted a phase II, double-blind, randomized, placebo-controlled, dose-ranging study for Omav. Lynch’s work demonstrated that Omav treatment significantly improved neurological function, measured with the modified Friedreich’s Ataxia Rating Scale (mFARS), from baseline in a dose-dependent manner. In our study, the clinical outcomes of ETR treatment response were assessed with SARA. Similar to our study, the magnitude of the improvements Lynch and co-workers observed is equivalent to approximately 1 year of FRDA disease progression. The population selected for our study includes children and severe subjects, while in the Lynch’s study the patients are milder (90% ambulatory) and there are no children.
Possible use of ETR in combination as already suggested [26] could be considered, since its mechanism differs from Omav and also from other proposed or tested treatments (γIFN, Vatiquinone, Elamipretide, Leriglitazone).
4.1. Limitations of the Study
A major limitation of this study, as already stated, is the absence of a placebo group.
Although the number of participants in the study is relevant considering that FRDA is a rare disease, the cohort was too small to be stratified according to expansion size and disease duration. Moreover, the chosen duration of the trial, while convenient in terms of sustainability, it reduces the opportunity to detect medium term effects of ETR treatment. The unavailability of the frataxin and frataxin mRNA measures weakens the study relevance, preventing the demonstration of the biological changes associated with treatment.
5. Conclusions
This pilot open study shows promises for ETR use as disease modifying drug in FRDA and could offer the basis for the design of a larger and longer placebo-controlled trial. The drug does not appear to have significantly modified the main measure of efficacy, but there has been a significant improvement in the functional scale of disease.
The future study should consider a larger cohort to favor stratification.
Clinical biomarkers of disease progression such as FARS and SARA score are unable to detect small changes in neurological function in short lasting studies. Power calculations on solely clinical measures indicate that it takes 2 years and > 150 subjects to detect a 50% reduction in disease progression [27,28,29]. Therefore, the markers to be used in the study must be carefully considered.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: Overall and by dose average outcome measures scores.
Author Contributions
Conceptualization, Andrea Martinuzzi, Roberto Testi and Alessandra Rufini; Methodology, Andrea Martinuzzi, Roberto Testi and Alessandra Rufini; Validation, Andrea Martinuzzi, Gabriella Paparella and Cristina Stragà; Formal analysis, Nicola Pesenti; Investigation, Gabriella Paparella, Cristina Stragà, Valentina Dal Molin, Gian Antonio Martorel, Vasco Merotto, Cristina Genova, Arianna Piazza, Giuseppe Piccoli, Elena Panzeri, Alessandra Rufini and Roberto Testi; Data curation, Gabriella Paparella and Cristina Stragà; Writing—original draft preparation, Gabriella Paparella and Cristina Stragà; Writing—review and editing, Andrea Martinuzzi, Gabriella Paparella, Cristina Stragà, Roberto Testi, Alessandra Rufini and Nicola Pesenti; Visualization, Andrea Martinuzzi, Gabriella Paparella and Cristina Stragà; Supervision, Andrea Martinuzzi; Project administration, Cristina Stragà; Funding acquisition, Andrea Martinuzzi. All authors have read and agreed to the published version of the manuscript. Conceptualization, Alessandra Rufini, Roberto Testi and Andrea Martinuzzi; Data curation, Gabriella Paparella and Cristina Stragà; Formal analysis, Nicola Pesenti; Funding acquisition, Andrea Martinuzzi; Investigation, Gabriella Paparella, Cristina Stragà, Valentina Dal Molin, Gian Martorel, Vasco Merotto, Cristina Genova, Arianna Piazza, Giuseppe Piccoli, Elena Panzeri, Alessandra Rufini and Roberto Testi; Methodology, Alessandra Rufini, Roberto Testi and Andrea Martinuzzi; Project administration, Cristina Stragà; Supervision, Andrea Martinuzzi; Validation, Gabriella Paparella and Cristina Stragà; Visualization, Gabriella Paparella, Cristina Stragà and Andrea Martinuzzi; Writing – original draft, Gabriella Paparella and Cristina Stragà; Writing – review & editing, Gabriella Paparella, Cristina Stragà, Nicola Pesenti, Alessandra Rufini, Roberto Testi and Andrea Martinuzzi.
Funding
This study was funded by the following Associations: “OGNI GIORNO” - Per Emma Onlus, “aCHaf Association”, “Babel Family”, “A.I.S.A. Onlus”, “Un petalo per Margherita Onlus”, “Per il sorriso di Ilaria di Montebruno - Onlus” and “CUREFA Foundation”. This research received funds from the Italian Ministry of Health, grant number RC 2022-23.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee for Clinical Trials for Provinces of Treviso and Belluno (Prot. No 0103439, 01/07/2020).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data supporting reported results are stored in the institutional servers and might be shared in anonymized form upon motivated request.
Acknowledgments
We thank all the patients for participating in the clinical trial and their families for the support provided. We thank the Associations for providing financial support of the study.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. Specifically, they do not own any intellectual property asset or rights, or any commercial interest of any kind, concerning any possible use of ETR in FRDA. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Campuzano, V.; Montermini, L.; Moltò, M.D.; Pianese, L.; Cossée, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; Zara, F.; Cañizares, J.; Koutnikova, H.; Bidichandani, S.I.; Gellera, C.; Brice, A.; Trouillas, P.; De Michele, G.; Filla, A.; De Frutos, R.; Palau, F.; Patel, P.I.; Di Donato, S.; Mandel, J.L.; Cocozza, S.; Koenig, M.; Pandolfo, M. Friedreich’s Ataxia: Autosomal Recessive Disease Caused by an Intronic GAA Triplet Repeat Expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.B.; Dehmer, T.; Schöls, L.; Mende, H.; Hardt, C.; Vorgerd, M.; Bürk, K.; Matson, W.; Dichgans, J.; Beal, M.F.; Bogdanov, M.B. Oxidative Stress in Patients with Friedreich Ataxia. Neurology 2000, 55, 1719–1721. [Google Scholar] [CrossRef]
- Clark, E.; Johnson, J.; Dong, Y.N.; Mercado-Ayon, E.; Warren, N.; Zhai, M.; McMillan, E.; Salovin, A.; Lin, H.; Lynch, D.R. Role of Frataxin Protein Deficiency and Metabolic Dysfunction in Friedreich Ataxia, an Autosomal Recessive Mitochondrial Disease. Neuronal Signal. 2018, 2, NS20180060. [Google Scholar] [CrossRef] [PubMed]
- Galea, C.A.; Huq, A.; Lockhart, P.J.; Tai, G.; Corben, L.A.; Yiu, E.M.; Gurrin, L.C.; Lynch, D.R.; Gelbard, S.; Durr, A.; Pousset, F.; Parkinson, M.; Labrum, R.; Giunti, P.; Perlman, S.L.; Delatycki, M.B.; Evans-Galea, M.V. Compound Heterozygous FXN Mutations and Clinical Outcome in Friedreich Ataxia. Ann. Neurol. 2016, 79, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, M.H.; Boesch, S.; Nachbauer, W.; Mariotti, C.; Giunti, P. Clinical Features of Friedreich’s Ataxia: Classical and Atypical Phenotypes. J. Neurochem. 2013, 126 Suppl 1, 103–117. [Google Scholar] [CrossRef]
- Schulz, J.B.; Boesch, S.; Bürk, K.; Dürr, A.; Giunti, P.; Mariotti, C.; Pousset, F.; Schöls, L.; Vankan, P.; Pandolfo, M. Diagnosis and Treatment of Friedreich Ataxia: A European Perspective. Nat. Rev. Neurol. 2009, 5, 222–234. [Google Scholar] [CrossRef]
- Delatycki, M.B.; Corben, L.A. Clinical Features of Friedreich Ataxia. J. Child Neurol. 2012, 27, 1133–1137. [Google Scholar] [CrossRef]
- Alfedi, G.; Luffarelli, R.; Condò, I.; Pedini, G.; Mannucci, L.; Massaro, D.S.; Benini, M.; Toschi, N.; Alaimo, G.; Panarello, L.; Pacini, L.; Fortuni, S.; Serio, D.; Malisan, F.; Testi, R.; Rufini, A. Drug Repositioning Screening Identifies Etravirine as a Potential Therapeutic for Friedreich’s Ataxia. Mov. Disord. Off. J. Mov. Disord. Soc. 2019, 34, 323–334. [Google Scholar] [CrossRef]
- Lynch, D.R.; Farmer, J.; Hauser, L.; Blair, I.A.; Wang, Q.Q.; Mesaros, C.; Snyder, N.; Boesch, S.; Chin, M.; Delatycki, M.B.; Giunti, P.; Goldsberry, A.; Hoyle, C.; McBride, M.G.; Nachbauer, W.; O’Grady, M.; Perlman, S.; Subramony, S.H.; Wilmot, G.R.; Zesiewicz, T.; Meyer, C. Safety, Pharmacodynamics, and Potential Benefit of Omaveloxolone in Friedreich Ataxia. Ann. Clin. Transl. Neurol. 2018, 6, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.R.; Chin, M.P.; Delatycki, M.B.; Subramony, S.H.; Corti, M.; Hoyle, J.C.; Boesch, S.; Nachbauer, W.; Mariotti, C.; Mathews, K.D.; Giunti, P.; Wilmot, G.; Zesiewicz, T.; Perlman, S.; Goldsberry, A.; O’Grady, M.; Meyer, C.J. Safety and Efficacy of Omaveloxolone in Friedreich Ataxia (MOXIe Study). Ann. Neurol. 2021, 89, 212–225. [Google Scholar] [CrossRef]
- Vavla, M.; D’Angelo, M.G.; Arrigoni, F.; Toschi, N.; Peruzzo, D.; Gandossini, S.; Russo, A.; Diella, E.; Tirelli, S.; Salati, R.; Scarpazza, P.; Luffarelli, R.; Fortuni, S.; Rufini, A.; Condò, I.; Testi, R.; Martinuzzi, A. Safety and Efficacy of Interferon γ in Friedreich’s Ataxia. Mov. Disord. Off. J. Mov. Disord. Soc. 2020, 35, 370–371. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Hübsch, T.; du Montcel, S.T.; Baliko, L.; Berciano, J.; Boesch, S.; Depondt, C.; Giunti, P.; Globas, C.; Infante, J.; Kang, J.-S.; Kremer, B.; Mariotti, C.; Melegh, B.; Pandolfo, M.; Rakowicz, M.; Ribai, P.; Rola, R.; Schöls, L.; Szymanski, S.; van de Warrenburg, B.P.; Dürr, A.; Klockgether, T.; Fancellu, R. Scale for the Assessment and Rating of Ataxia: Development of a New Clinical Scale. Neurology 2006, 66, 1717–1720. [Google Scholar] [CrossRef]
- Apolone, G.; Mosconi, P. The Italian SF-36 Health Survey: Translation, Validation and Norming. J. Clin. Epidemiol. 1998, 51, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Ustün, T.B.; Chatterji, S.; Kostanjsek, N.; Rehm, J.; Kennedy, C.; Epping-Jordan, J.; Saxena, S.; von Korff, M.; Pull, C.; WHO/NIH Joint Project. Developing the World Health Organization Disability Assessment Schedule 2.0. Bull. World Health Organ. 2010, 88, 815–823. [Google Scholar] [CrossRef]
- Herdy, A.H.; Uhlendorf, D. Reference Values for Cardiopulmonary Exercise Testing for Sedentary and Active Men and Women. Arq. Bras. Cardiol. 2011, 96, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Lintu, N.; Viitasalo, A.; Tompuri, T.; Veijalainen, A.; Hakulinen, M.; Laitinen, T.; Savonen, K.; Lakka, T.A. Cardiorespiratory Fitness, Respiratory Function and Hemodynamic Responses to Maximal Cycle Ergometer Exercise Test in Girls and Boys Aged 9-11 Years: The PANIC Study. Eur. J. Appl. Physiol. 2015, 115, 235–243. [Google Scholar] [CrossRef]
- Drinkard, B.E.; Keyser, R.E.; Paul, S.M.; Arena, R.; Plehn, J.F.; Yanovski, J.A.; Di Prospero, N.A. Exercise Capacity and Idebenone Intervention in Children and Adolescents with Friedreich Ataxia. Arch. Phys. Med. Rehabil. 2010, 91, 1044–1050. [Google Scholar] [CrossRef]
- Saccà, F.; Puorro, G.; Marsili, A.; Antenora, A.; Pane, C.; Casali, C.; Marcotulli, C.; Defazio, G.; Liuzzi, D.; Tatillo, C.; Cambriglia, D.M.; Schiano di Cola, G.; Giuliani, L.; Guardasole, V.; Salzano, A.; Ruvolo, A.; De Rosa, A.; Cittadini, A.; De Michele, G.; Filla, A. Long-Term Effect of Epoetin Alfa on Clinical and Biochemical Markers in Friedreich Ataxia. Mov. Disord. Off. J. Mov. Disord. Soc. 2016, 31, 734–741. [Google Scholar] [CrossRef]
- Pane, C.; Salzano, A.; Trinchillo, A.; Del Prete, C.; Casali, C.; Marcotulli, C.; Defazio, G.; Guardasole, V.; Vastarella, R.; Giallauria, F.; Puorro, G.; Marsili, A.; De Michele, G.; Filla, A.; Cittadini, A.; Saccà, F. Safety and Feasibility of Upper Limb Cardiopulmonary Exercise Test in Friedreich Ataxia. Eur. J. Prev. Cardiol. 2022, 29, 445–451. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing., 2022. https://www.R-project.org/.
- Roessler, H.I.; Knoers, N.V.A.M.; van Haelst, M.M.; van Haaften, G. Drug Repurposing for Rare Diseases. Trends Pharmacol. Sci. 2021, 42, 255–267. [Google Scholar] [CrossRef]
- Rufini, A.; Malisan, F.; Condò, I.; Testi, R. Drug Repositioning in Friedreich Ataxia. Front. Neurosci. 2022, 16, 814445. [Google Scholar] [CrossRef]
- Allavena, C.; Katlama, C.; Cotte, L.; Roger, P.M.; Delobel, P.; Cheret, A.; Duvivier, C.; Poizot-Martin, I.; Hoen, B.; Cabie, A.; Cheret, A.; Lahoulou, R.; Raffi, F.; Pugliese, P.; Dat’AIDS Study group. Long-Term Efficacy and Safety of Etravirine-Containing Regimens in a Real-Life Cohort of Treatment-Experienced HIV-1-Infected Patients. Infect. Dis. Lond. Engl. 2016, 48, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Marelli, C.; Figoni, J.; Charles, P.; Anheim, M.; Tchikviladze, M.; Vincitorio, C.-M.; du Montcel, S.T.; Brice, A.; Golmard, J.L.; Dürr, A. Annual Change in Friedreich’s Ataxia Evaluated by the Scale for the Assessment and Rating of Ataxia (SARA) Is Independent of Disease Severity. Mov. Disord. Off. J. Mov. Disord. Soc. 2012, 27, 135–138. [Google Scholar] [CrossRef]
- Tanguy Melac, A.; Mariotti, C.; Filipovic Pierucci, A.; Giunti, P.; Arpa, J.; Boesch, S.; Klopstock, T.; Müller Vom Hagen, J.; Klockgether, T.; Bürk, K.; Schulz, J.B.; Reetz, K.; Pandolfo, M.; Durr, A.; Tezenas du Montcel, S.; EFACTS group. Friedreich and Dominant Ataxias: Quantitative Differences in Cerebellar Dysfunction Measurements. J. Neurol. Neurosurg. Psychiatry 2018, 89, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.R.; Schadt, K.; Kichula, E. Etravirine in Friedreich’s Ataxia: Lessons from HIV? Mov. Disord. Off. J. Mov. Disord. Soc. 2019, 34, 305–306. [Google Scholar] [CrossRef] [PubMed]
- Reetz, K.; Dogan, I.; Hilgers, R.-D.; Giunti, P.; Mariotti, C.; Durr, A.; Boesch, S.; Klopstock, T.; de Rivera, F.J.R.; Schöls, L.; Klockgether, T.; Bürk, K.; Rai, M.; Pandolfo, M.; Schulz, J.B.; EFACTS Study Group. Progression Characteristics of the European Friedreich’s Ataxia Consortium for Translational Studies (EFACTS): A 2 Year Cohort Study. Lancet Neurol. 2016, 15, 1346–1354. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Isaacs, C.J.; Seyer, L.; Brigatti, K.; Gelbard, S.; Strawser, C.; Foerster, D.; Shinnick, J.; Schadt, K.; Yiu, E.M.; Delatycki, M.B.; Perlman, S.; Wilmot, G.R.; Zesiewicz, T.; Mathews, K.; Gomez, C.M.; Yoon, G.; Subramony, S.H.; Brocht, A.; Farmer, J.; Lynch, D.R. Progression of Friedreich Ataxia: Quantitative Characterization over 5 Years. Ann. Clin. Transl. Neurol. 2016, 3, 684–694. [Google Scholar] [CrossRef]
- Reetz, K.; Dogan, I.; Hilgers, R.-D.; Giunti, P.; Parkinson, M.H.; Mariotti, C.; Nanetti, L.; Durr, A.; Ewenczyk, C.; Boesch, S.; Nachbauer, W.; Klopstock, T.; Stendel, C.; Rodríguez de Rivera Garrido, F.J.; Rummey, C.; Schöls, L.; Hayer, S.N.; Klockgether, T.; Giordano, I.; Didszun, C.; Rai, M.; Pandolfo, M.; Schulz, J.B.; EFACTS study group. Progression Characteristics of the European Friedreich’s Ataxia Consortium for Translational Studies (EFACTS): A 4-Year Cohort Study. Lancet Neurol. 2021, 20, 362–372. [Google Scholar] [CrossRef]
Figure 1.
Study scheme. Timeline of the study.
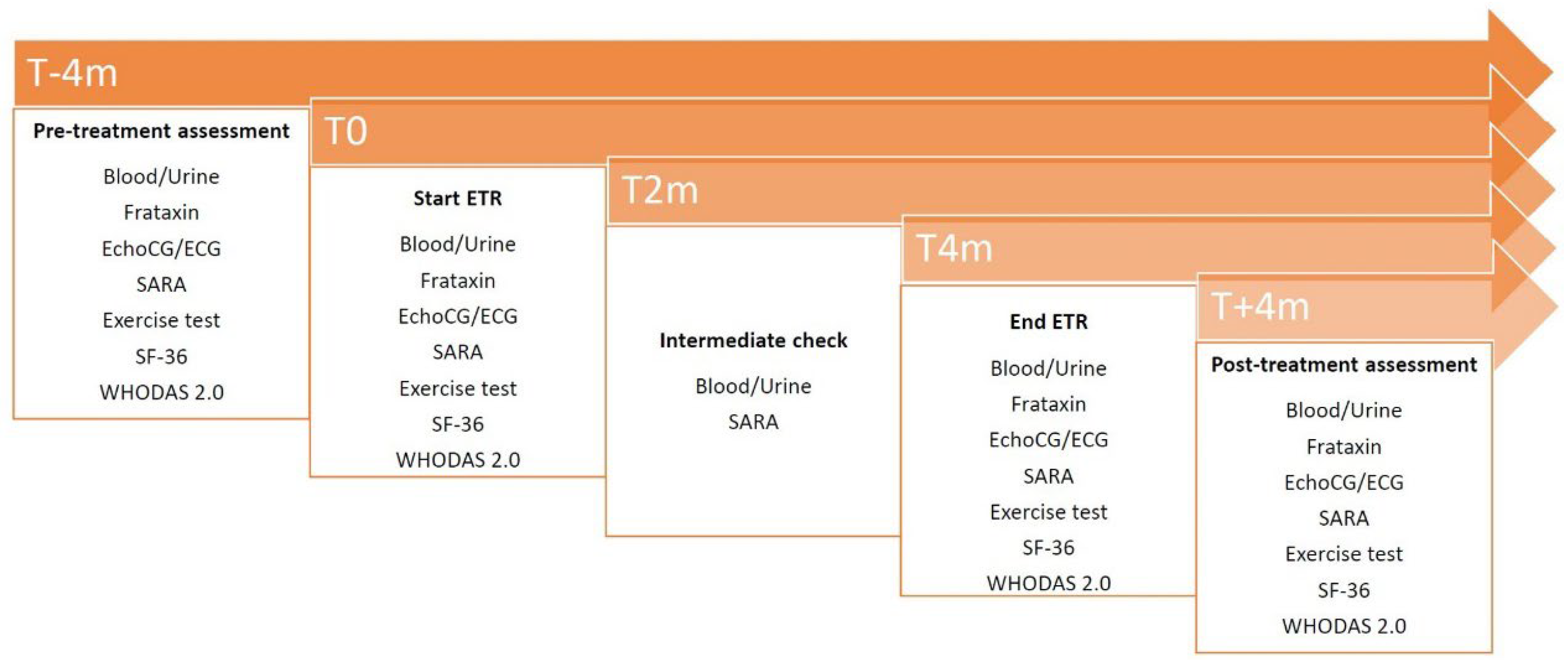
Figure 2.
CONSORT flowchart of the study.
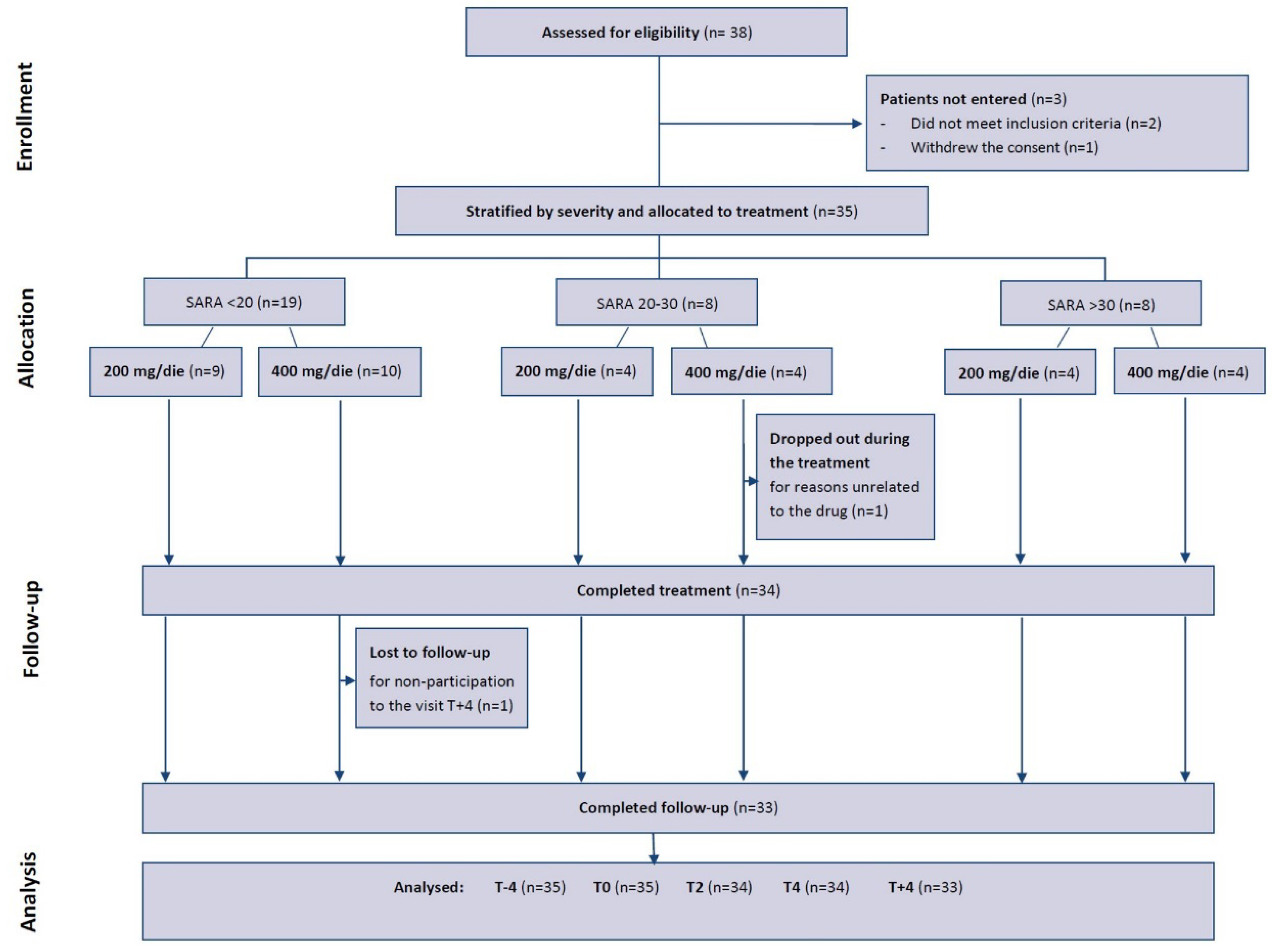
Figure 3.
Trend of clinical and functional outcomes over time in the entire cohort of patients. Sig: *** <0.001, ** <0.01, * <0.05, . <0.1.
Figure 3.
Trend of clinical and functional outcomes over time in the entire cohort of patients. Sig: *** <0.001, ** <0.01, * <0.05, . <0.1.
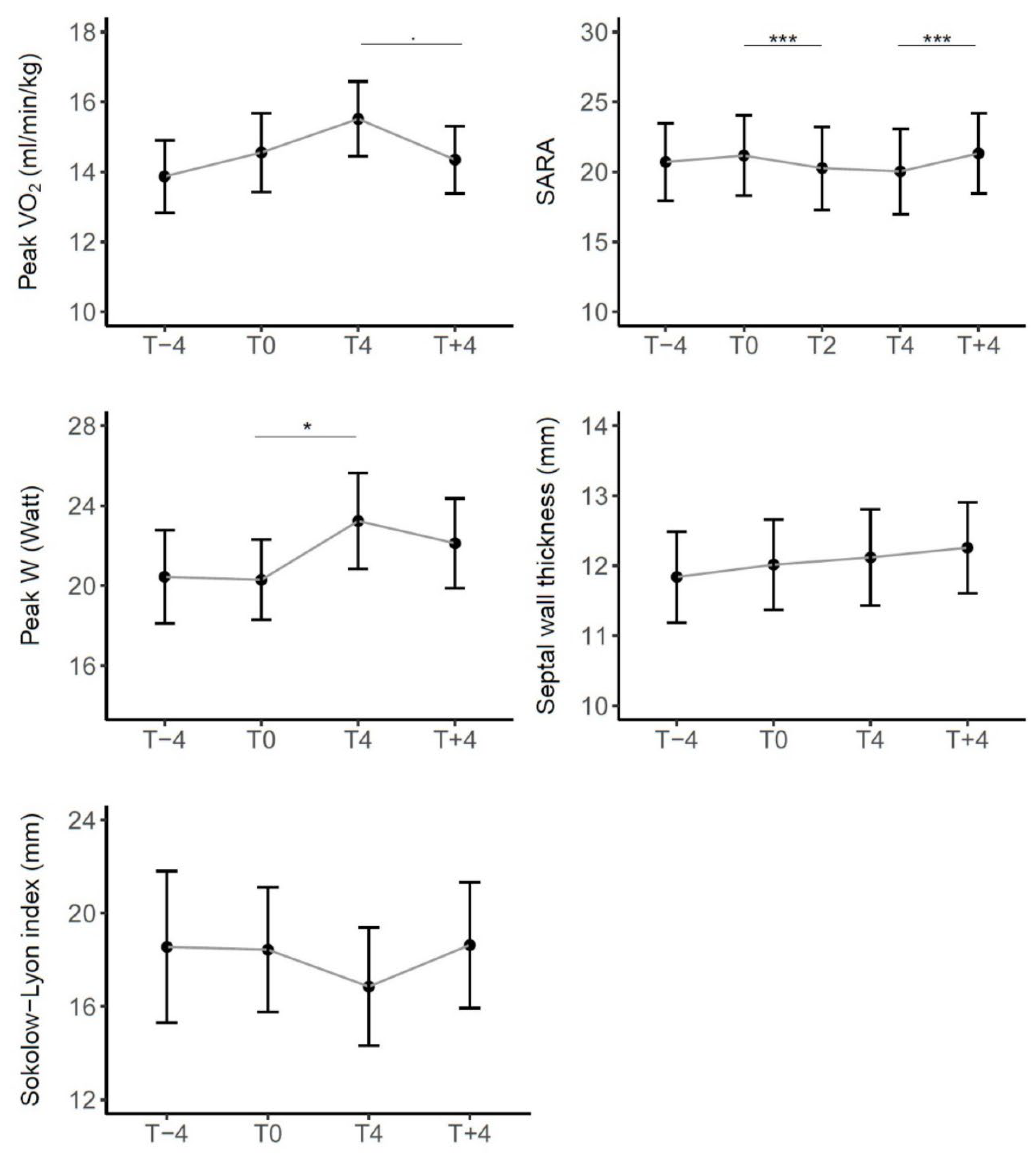
Figure 4.
Trend of scores in SF-36 over time in the entire cohort of patients. Scores range from 0 (worst possible health) to 100 (best possible health). Sig: *** <0.001, ** <0.01, * <0.05, . <0.1.
Figure 4.
Trend of scores in SF-36 over time in the entire cohort of patients. Scores range from 0 (worst possible health) to 100 (best possible health). Sig: *** <0.001, ** <0.01, * <0.05, . <0.1.
Figure 5.
Trend of scores in WHODAS 2.0 over time in the entire cohort of patients. Scores range from 0 (no disability) to 100 (full disability). Sig: *** <0.001, ** <0.01, * <0.05, . <0.1.
Figure 5.
Trend of scores in WHODAS 2.0 over time in the entire cohort of patients. Scores range from 0 (no disability) to 100 (full disability). Sig: *** <0.001, ** <0.01, * <0.05, . <0.1.
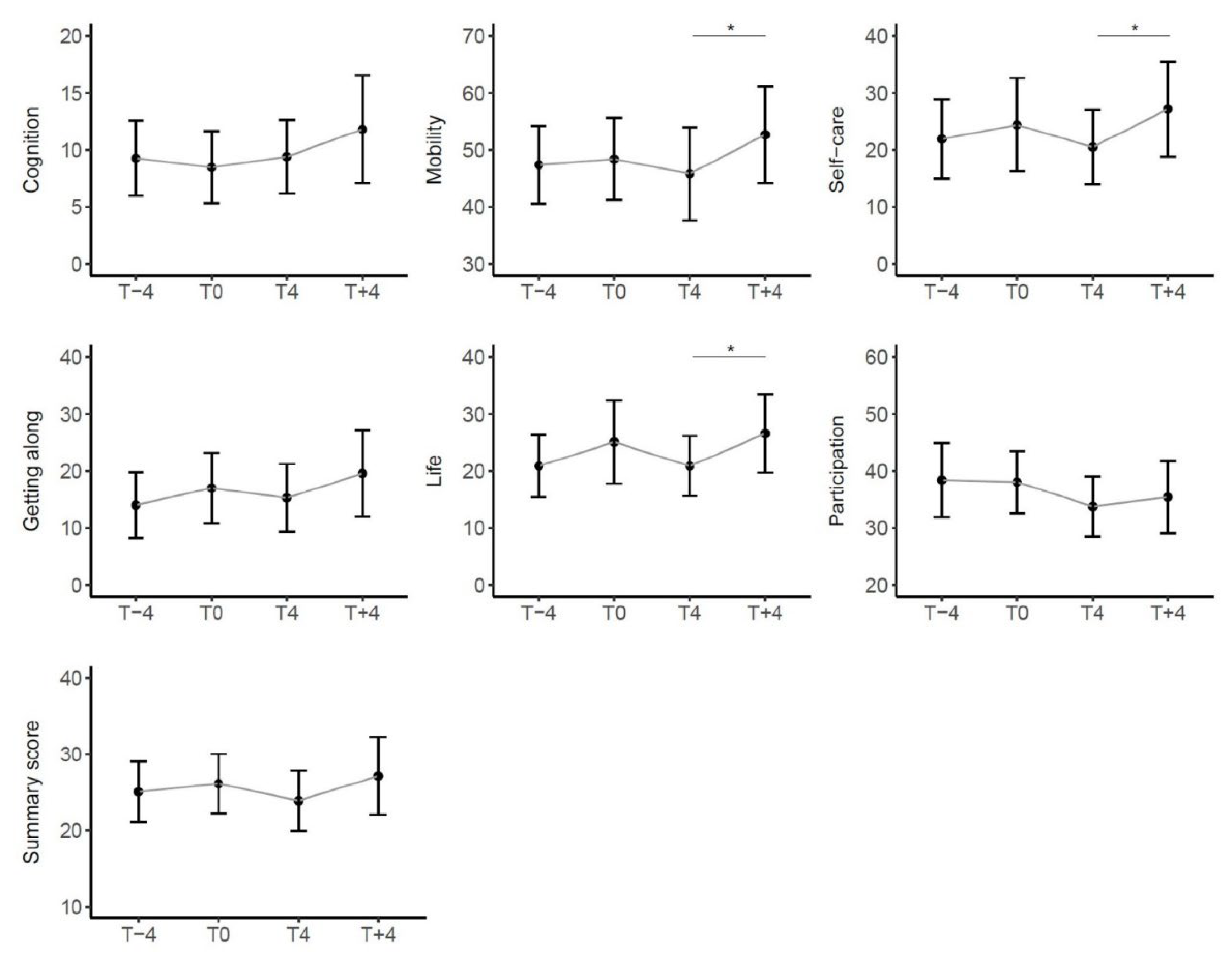

Table 1.
Demographic and clinical data of patients included in this study and comparisons between dose groups.
Table 1.
Demographic and clinical data of patients included in this study and comparisons between dose groups.
| Overall (N=35) | Dose 200 (N=17) | Dose 400 (N=18) | p-value | |
|---|---|---|---|---|
| Patient type, n (%) | >0.999 | |||
| Adult | 30 (85.7) | 15 (88.2) | 15 (83.3) | |
| Children | 5 (14.3) | 2 (11.8) | 3 (16.7) | |
| Male, n (%) | 16 (45.7) | 5 (29.4) | 11 (61.1) | 0.123 |
| Severity, n (%) | 0.988 | |||
| SARA <20 | 19 (54.3) | 9 (52.9) | 10 (55.6) | |
| SARA 20-30 | 8 (22.9) | 4 (23.5) | 4 (22.2) | |
| SARA >30 | 8 (22.9) | 4 (23.5) | 4 (22.2) | |
| GAA sr | 665.50 (189.02) | 670.87 (197.10) | 660.13 (187.35) | 0.880 |
| GAA lr | 862.89 (203.86) | 917.64 (226.81) | 808.14 (168.56) | 0.159 |
| Age at onset (years), mean (SD) | 11.83 (5.70) | 10.65 (4.20) | 12.94 (6.76) | 0.239 |
| Age (years), mean (SD) | 25.00 (7.14) | 23.53 (5.01) | 26.39 (8.61) | 0.242 |
| Disease duration (years), mean (SD) | 13.17 (6.00) | 12.88 (5.29) | 13.44 (6.75) | 0.786 |
| Years of education, median [IQR] | 13.00 [13.00, 16.50] | 14.00 [13.00, 17.00] | 13.00 [13.00, 15.50] | 0.458 |
P-values were obtained using t test, Mann-Whitney U Test, or Fisher’s exact test. GAAsr, GAA short repeat; GAAlr, GAA long repeat.
Table 2.
Number (%) of subjects with AE detected during the study.
| Months 1 e 2 | Months 3 e 4 | |
|---|---|---|
| Headache | 16 (47%) | 3 (9%) |
| Diarrhea | 7 (21%) | 1 (3%) |
| Nausea | 4 (12%) | 1 (3%) |
| Skin Rash | 4 (12%) | 0 |
| Other: | ||
| Fatigue | 3 (9%) | 0 |
| Hypotension | 2 (6%) | 0 |
| Tachycardia | 1 (3%) | 1 (3%) |
| Vomit | 1 (3%) | 1 (3%) |
| Constipation | 1 (3%) | 0 |
| Blurred vision | 1 (3%) | 0 |
| Sweating | 1 (3%) | 0 |
| Urinary urgency | 1 (3%) | 0 |
| Calf cramps | 1 (3%) | 0 |
Table 3.
Number (%) of alterations in blood tests during treatment.
| N (%) | |
|---|---|
| Total cholesterol + | 4 (12%) |
| Triglyceride + | 3 (9%) |
| AST/ALT + | 1 (3%) |
| Amylase + | 1 (3%) |
| White blood cells - | 1 (3%) |
Table 4.
Mixed models’ post hoc differences for clinical and functional outcomes between timepoints.
Table 4.
Mixed models’ post hoc differences for clinical and functional outcomes between timepoints.
| T0 vs T-4 | T2 vs T0 | T4 vs T2 | T4 vs T0 | T+4 vs T4 | |
|---|---|---|---|---|---|
| Start ETR vs Pre-treatment | Intermediate check vs Start ETR | End ETR vs Intermediate check |
End ETR vs Start ETR |
Post-treatment vs End ETR | |
| Peak VO2 | 0.685 (0.619) | 0.959 (0.224) | -1.145 (0.088) | ||
| SARA | 0.456 (0.243) | -0.897 (<0.001) | -0.235 (0.914) | -1.132 (<0.001) | 1.229 (<0.001) |
| Peak W | -0.147 (>0.999) | 2.941 (0.021) | -1.037 (0.905) | ||
| Septal wall thickness | 0.176 | 0.103 | 0.111 | ||
| Sokolow-Lyon index | -0.118 | -1.582 | 1.808 |
Estimated difference between times (p.values). P.values for post-hoc are shown only for outcomes with significant effect at mixed model (peak VO2, SARA and peak W).
Table 5.
Mean (SD) differences in scores during treatment and after suspension by severity and dosage.
Table 5.
Mean (SD) differences in scores during treatment and after suspension by severity and dosage.
| SARA < 20 (N=19) | SARA 20-30 (N=7) | SARA > 30 (N=8) | Dose 200 (N=17) | Dose 400 (N=17) | |
|---|---|---|---|---|---|
| Peak VO2 | |||||
| delta T4 - T0 | 0.51 (2.26) | 1.81 (1.88) | 1.29 (2.45) | 0.80 (2.80) | 1.12 (1.55) |
| delta T+4 - T4 | -1.42 (2.84) | -1.21 (2.15) | -0.36 (1.18) | -0.99 (2.60) | -1.25 (2.19) |
| SARA | |||||
| delta T4 - T0 | -1.53 (1.03) | -1.21 (1.82) | -0.12 (0.35) | -0.97 (1.21) | -1.29 (1.30) |
| delta T+4 - T4 | 1.78 (1.23) | 1.14 (1.14) | 0.06 (0.50) | 0.94 (1.14) | 1.53 (1.36) |
| Peak W | |||||
| delta T4 - T0 | 2.95 (4.34) | 1.43 (2.76) | 4.25 (8.45) | 2.71 (3.24) | 3.18 (6.82) |
| delta T+4 - T4 | -1.00 (2.77) | 1.43 (1.90) | -3.00 (11.36) | -0.71 (2.91) | -1.25 (8.13) |
| Septal wall thickness | |||||
| delta T4 - T0 | 0.11 (0.86) | 0.43 (0.84) | -0.19 (1.07) | 0.06 (0.58) | 0.15 (1.16) |
| delta T+4 - T4 | 0.19 (0.86) | 0.07 (0.61) | 0.06 (0.68) | -0.09 (0.73) | 0.38 (0.72) |
| Sokolow-Lyon index | |||||
| delta T4 - T0 | -1.31 (3.64) | -3.29 (6.21) | -0.75 (2.82) | -2.00 (4.09) | -1.16 (4.17) |
| delta T+4 - T4 | 2.06 (3.40) | 2.00 (4.36) | 1.25 (2.49) | 1.65 (2.94) | 2.06 (3.82) |
Delta T+4 - T4: N=18 in SARA <20, N=16 in Dose 400.
Table 6.
Multiple linear models’ estimated differences (p.values) in changes overtime between severity or dose and the reference group.
Table 6.
Multiple linear models’ estimated differences (p.values) in changes overtime between severity or dose and the reference group.
| T4 vs T0 | T+4 vs T4 | |
|---|---|---|
| Peak VO2 | ||
| Severity (SARA 20-30) | 1.347 (0.196) | 0.212 (0.850) |
| Severity (SARA >30) | 0.715 (0.478) | 1.242 (0.268) |
| Dose (400) | 0.354 (0.661) | -0.205 (0.815) |
| SARA | ||
| Severity (SARA 20-30) | 0.282 (0.589) | -0.569 (0.234) |
| Severity (SARA >30) | 1.398 (0.010) | -1.552 (0.002) |
| Dose (400) | -0.304 (0.459) | 0.637 (0.094) |
| Peak W | ||
| Severity (SARA 20-30) | -1.485 (0.546) | 2.290 (0.403) |
| Severity (SARA >30) | 0.962 (0.689) | -2.742 (0.315) |
| Dose (400) | 0.189 (0.922) | -0.627 (0.770) |
| Septal wall thickness | ||
| Severity (SARA 20-30) | 0.335 (0.422) | -0.100 (0.769) |
| Severity (SARA >30) | -0.220 (0.589) | -0.198 (0.559) |
| Dose (400) | 0.147 (0.653) | 0.441 (0.108) |
| Sokolow-Lyon index | ||
| Severity (SARA 20-30) | -1.911 (0.317) | -0.034 (0.983) |
| Severity (SARA >30) | 0.444 (0.810) | -0.864 (0.588) |
| Dose (400) | 0.651 (0.662) | 0.408 (0.746) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



