
Submitted:
03 January 2024
Posted:
04 January 2024
You are already at the latest version
Abstract
The dual leucine zipper kinase (DLK) alias mitogen-activated protein 3 kinase 12 (MAP3K12) has gained much attention in recent years. DLK belongs to the mixed lineage kinases, characterized by homology to serine/threonine and tyrosine kinase but exerts serine/threonine kinase activity. DLK has been implicated in many diseases including several neurodegenerative diseases, glaucoma, and diabetes mellitus. As a MAP3K, it is generally assumed that DLK becomes phosphorylated and activated by upstream signals and phosphorylates and activates itself, the downstream serine/threonine MAP2K, and ultimately MAPK. In addition, other mechanisms such as protein-protein interactions, proteasomal degradation, dephosphorylation by various phosphatases, palmitoylation, and subcellular localization have been shown to be involved in the regulation of DLK activity or its fine-tuning. In the present review, the diverse mechanisms regulating DLK activity will be summarized to provide a better insight into DLK action and possibly new targets to modulate DLK function.
Keywords:
dual leucine zipper kinase
; phosphorylation
; protein-protein interaction
; proteasomal degradation
; palmitoylation
1. Introduction
The dual leucine zipper kinase (DLK) alias mitogen-activated protein 3 kinase 12 (MAP3K12) has gained much attention in recent years due to its involvement in several neurodegenerative diseases such as amyotrophic lateral sclerosis and Alzheimer’s disease [1,2,3,4,5,6], glaucoma [7] and diabetes mellitus type 2 [8,9,10]. DLK belongs to the class of the mixed lineage kinases (MLK), characterized by sequence homology to both serine/threonine and tyrosine kinases in their primary structure, but functioning as serine/threonine kinase [11,12,13]. The class of the MLK can be subdivided into three subclasses: The largest class consists of MLK1-4 (MAP3K9-11 and 21, respectively), sharing 75 % sequence identity in their kinases domains and displaying an amino-terminal SH3 domain, followed by the kinase domain, a leucine zipper, Cdc42/Rac Interacting Binding (CRIB) motif and a large C-terminal region. Another subgroup consists of the leucine zipper and sterile-alpha motif (SAM) ZAK or MLK7 (MAP3K20), containing the dimerizing SAM in addition to the leucine zipper domain. DLK forms another MLK subgroup with leucine zipper kinase (LZK; MAP3K13). The kinases share 90% amino acid sequence identity within their enzymatic and dual leucine zipper domain (Figure 1).
DLK and LZK are highly conserved orthologues of Wallenda/DLK in Drosophila melanogaster and DLK-1 in Caenorhabditis elegans, suggesting an important role for DLK in evolution and development. Indeed, while mice lacking Mlk1, Mlk2, Mlk3 or Lzk are viable [12,14,15], mice lacking Dlk die perinatally and show signs of impaired neuronal development [16]. In line with this, the genome aggregation database (gnomAD v 2.1), a repository of data on human genes and their single nucleotide variants (SNV), calculated for MAP3K12 (gene) 41.3 Loss of function (LoF) SNV, but observed none of them in their different cohorts. Thus, in species as diverse as mice and humans, intact DLK appears to be essential for prenatal development. Yet, the conditional deletion of Dlk in mice aged 10 to 12 weeks did not result in gross phenotypic changes, suggesting that in adults, the absence of DLK or its function does not interfere with vital functions under normal conditions [17]. Inhibition of DLK has been proposed as a promising drug target to treat neurodegenerative diseases like amyotrophic lateral sclerosis, Alzheimer’s disease, Parkinson’s disease [1,2,4], glaucoma [7] and diabetes mellitus type 2 [8,9,10]. This suggests that abnormal DLK activity in adults contributes to pathological signaling in various tissues. However, DLK signaling is required for the induction of the pro-regenerative transcriptional program in peripheral nerves after injury [18,19], and has been shown to be constitutively active in the adult mouse brain, exerting both homeostatic and stress-induced functions [3]. These findings suggest that indeed DLK acts as a “double-edged sword” [20]. Acting as a MAP3K, DLK activates mainly the MAP2Ks MKK4 and 7 leading to the phosphorylation and activation of the MAPK c-Jun N-terminal kinase (JNK) [21]. In a refined model, DLK under basal conditions is tethered to the scaffold protein JNK-Interacting protein/Islet Brain 1 (JIP/IB1), rendering the kinase inactive. Upon phosphorylation of JIP/IB1 by JNK on Thr103, DLK dissociates from the scaffold, homodimerizes, autophosphorylates in trans, and becomes active [22,23,24,25]. This model implies that activation of JNK leads to the activation of DLK, which in turn stimulates JNK activity, thereby amplifying possibly cell-toxic signals. In addition to activating JNK and being (indirectly) activated by this kinase, DLK also activates other MAPKs [2]. Furthermore, different posttranslational modifications which might result in DLK’s proteasomal degradation or enhanced protein stability, protein-protein interactions, subcellular localization, and microRNAs contribute to DLK activity. To better understand DLK function and the regulation of its activity, this review summarizes the regulation of DLK activity by various mechanisms at transcriptional, translational and post-translational levels.
2. Results
2.1. Regulation of DLK at the transcriptional level
Not much is known about the regulation of DLK gene expression: The TATA-box - less core promoter regions of human and mouse DLK gene upstream of exon 1 share 88% identity with completely conserved xenobiotic responsive element-like site, GC-boxes, and exon 1 in between both species. Using electrophoretic mobility shift assays (EMSA) and reporter gene assays with 5’-deleted promoter fragments, the transcription Sp3 factor was shown to bind to and activate the core promoter in the human neuroblastoma cell line SH-SY5Y [26]. In the 3T3-L1 cell line, the ligand of the peroxisome-proliferator-activated receptor γ (PPARγ) rosiglitazone increased DLK expression, whereas inhibition of PPARγ either by small hairpin RNA or by the receptor antagonist GW9662 suppressed DLK protein and mRNA expression. Two binding-sites for PPARγ and its heterodimer retinoic X receptor were identified by EMSA and chromatin immunoprecipitation assays [27]. In human adipose stromal/stem cells, precursors of mature adipocytes, bisphenol A increased DLK expression, presumably after binding to estrogen receptors [28], but it was not investigated whether the DLK promoter contains an estrogen receptor responsive element. Thus, DLK gene expression is regulated by Sp3, the nuclear receptors PPARγ, and estrogen receptor. Of note, DLK itself has been shown to increase PPARγ gene expression in 3T3-L1 and adipose stromal/stem cells [28,29]. Finally, reduced Dlk expression was observed in the lenses of mice deficient in the transcription factors Mafg and Mafk [30], suggesting that these transcription factors may be involved in Dlk expression.
2.2. Regulation of DLK at the posttranscriptional level
The human MAP3K12 gene spans 21.871 nucleotides (nt) and is located on the complementary strand of chromosome 12, GRCh38.p14 Primary Assembly. It is flanked by the gene encoding for the TARBP2 subunit of RISC loading complex (TARBP2) and the poly(rC) binding protein 2 (PCBP2) gene, both located on the positive strand in opposite direction relative to the DLK gene. The transcripts of the human DLK Isoforms 1 (NM_001193511.2) and 2 (NM_006301.4) differ in a 99 nt stretch that is absent in Isoform 2, resulting in a slightly shorter protein of 859 amino acids (aa) and a calculated molecular weight of 93.2 kDa instead of 892 aa and 96.3 kDa (ncbi, https://www.ncbi.nlm.nih.gov/gene/7786, 30.06.2023, 14:33. conserved) (Figure 2).
In various additional model organism different isoforms of DLK are expressed, probably due to differential splicing (Table 1).
It is not known how the differential splicing of DLK RNA is regulated, and - with exception of the C. elegans DLK-1 gene [31] - whether the isoforms exert distinct functions or are differentially expressed in tissues.
MicroRNAs (miRNAs) provide another mechanism of regulating gene expression at the posttranscriptional level. MiRNAs are small non-coding RNAs of about 22 nucleotides. By directing the RNA-induced silencing complex (RISC) to specific target mRNAs, miRNA can repress target genes and affect various biological responses [32]. In turn, expression of miRNAs is regulated by several physiological and pathophysiological conditions. DLK mRNA is predicted to be a target for miRNAs (TargetScan v8.0; targetscan.org) [33] and several interactions have been validated experimentally (miRTarBase) [34]. In neuroblasts, downregulation of the entire miR-17 family during neuronal differentiation and upregulation of DLK mRNA was observed, and the overexpression of miR-17 and miR-20a reduced DLK mRNA [35]. In endothelial progenitor cells (EPC) from type 2 diabetic patients, the expression of miRNA-130a was reduced, and increased DLK expression was observed compared to EPC from healthy controls. In line, the overexpression of miRNA-130a decreased DLK protein expression in EPC, suggesting that DLK expression is regulated by miRNA-130a [36]. Regulation of DLK via miRNA was also demonstrated in a mouse model of Alzheimer’s disease (AD). MicroRNA-191-5p was shown to target the 3ʹ-untranslated region of MAP3K12 downregulating DLK expression and alleviating microglial cell injury in the AD mouse model [37]. Yu et al. found in various prostate cancer cell lines that tumor suppressor miR-150-5p downregulates MAP3K12 [38]. These studies show that DLK expression is subject to miRNA regulation. Of note, miRNAs do not target mRNAs of selective proteins.
2.3. Regulation of DLK at the posttranslational level
2.3.1. Phosphorylation of DLK
Already the first studies on DLK showed that this kinase is heavily phosphorylated, and overexpression of DLK alone is sufficient to activate this kinase [11,39]. At least some phosphorylation-sites and some DLK phosphorylating kinases have been identified in the meantime. Upon homodimerization via its leucine zipper domains, DLK becomes auto-phosphorylated in trans at Ser-302 (Figure 3B) [22,40,41].
Indeed, the phosphorylation at this residue is crucial for DLK activation, since mutation of Ser-302 renders the kinase inactive [40,41]. Hence, phosphorylation at this residue is a pre-requisite for DLK activation and can serve as a marker for DLK activity [10,41,42]. In addition to DLK, protein kinase A (PKA) phosphorylates DLK at Ser-302 and activates the kinase, linking DLK to the evolutionary conserved mechanism of cyclic AMP induced axonal regeneration in mammals, D. melanogaster and C. elegans [42,43] (Figure 3B). Furthermore, DLK is a substrate for its non-downstream kinase JNK: using stable isotope labeling with amino acids in the HEK 293T cell line (SILAC) followed by mass spectrometry analysis, Huntwork-Rodriguez et al. (2013) identified Thr-43 and Ser-533 as residues becoming phosphorylated by JNK (Figure 3B). These findings were confirmed in a murine model of neuronal stress. Phosphorylations at these residues prevent the interaction of DLK with the E3 ubiquitin ligase Pam/Highwire/RPM-1 (PHR), thus stabilizing DLK (Figure 3A) [40]. In an attempt to identify kinases that activate neurodegenerative DLK/JNK signaling in neurons, inhibition of the MAP4K subfamily of germinal center kinase-IV (GCK-IV), MAP4K4, misshapen-like kinase 1 (MINK, MAP4K6) and Traf2- and Nck-interacting kinase (TNIK, MAP4K7) reduced DLK activation, phosphorylation on Thr-43 and protein stability upon nerve growth factor (NGF) withdrawal in murine dorsal root ganglia (Figure 3B). However, the combined knock-down of all three MAP4K was needed to protect against NGF withdrawal induced DLK/JNK signaling [44]. Using the optic nerve crush model, inhibitors of the GCK-IV kinase family enhanced the survival of retinal ganglia cells but, in contrast to DLK inhibition, did not interfere with axon regeneration [45]. Notably, the overexpression of MAP4K3 (hematopoietic progenitor kinase 1, HPK1), shown to activate JNK signaling [46], phosphorylated MLK3 on Ser-281, corresponding to Ser-302 in DLK [47]. Thus, at least some MAP4K might activate DLK, but not all functions of DLK overlap with those of MAP4K, suggesting additional upstream signals for DLK or an additional regulation of DLK action by other mechanisms, such as (tissue-dependent) dephosphorylation, (tissue-dependent) interaction with various scaffolds or other proteins, palmitoylation or changing DLK subcellular localization [3,5,9,10,39,40,48,49,50,51]. Investigations of Daviau et al. (2009) indicate that DLK might also be activated by tyrosine phosphorylation, and thus be involved in a separate pathway. Various experiments showed that platelet-derived growth factor (PDGF) induced tyrosine phosphorylation and subsequent activation of DLK, which was dependent on the cytosolic tyrosine kinase Src. PDGF dependent phosphorylation and activation of ERK and Akt was abolished by RNA silencing of DLK and rescued by re-introduction of recombinant wild-type DLK, suggesting that PDGF signal propagation depends on DLK. However, the tyrosine residue within DLK phosphorylated by PDGF induced signaling was not identified [52]. So far, the phosphorylation of DLK that have been described are activating phosphorylations. In mouse embryonic stem cells, two Akt phosphorylation sites within DLK, Ser584 and Thr659 in murine DLK, were identified. Akt-induced phosphorylation of these residues reduced DLK kinase activity, whereas the overexpression of these DLK mutants rendered the kinase more active, suppressing the self-renewal of mouse embryonic stem cells [53].
2.3.2. Dephosphorylation of DLK
Kinase-phosphatase interactions are a well-known component of cellular responses and signaling pathways, affecting kinase phosphorylation, expression levels, and interactions with other proteins. In invertebrates, DLK’s activity has been shown to be regulated by the protein phosphatases Mg2+/Mn2+ dependent (PPM)-1 and PPM-2, and the protein phosphatase 2A (PP2A) [5,54,55]. In mammals, inhibition of protein phosphatases 2A [5,39,52] and 2B [10,39,56,57] affected DLK activity.
Several studies described the role of the PHR proteins as key regulators of presynaptic differentiation and function, thereby fundamentally affecting neuronal development [58,59,60]. Among the PHR proteins, the Regulator of Presynaptic Morphology (RPM)-1 has been shown to negatively regulate DLK-1 as part of an ubiquitin ligase complex in C. elegans [61,62]. In 2011, Tulgren et al. provided additional evidence that PPM-1, a serine/threonine phosphatase homologous to human PPM1A, acts as a second negative regulatory mechanism downstream of RPM-1 to control the DLK-1 pathway in C. elegans [54]. However, the involvement of PPM-1 was shown to act at the level of PMK-3 (p38 MAPK) and not directly at the level of DLK-1 (MAPKKK) as previously described by Takekawa et al. in mammalian cells [63]. In contrast, the serine/threonine Protein Phosphatase Magnesium/Manganese dependent 2 (PPM-2) has been described in transgenic animals, genetic, and biochemical approaches to directly regulate DLK-1 [55]. Baker et al. (2014) demonstrated that PPM-2 acts on DLK-1 at the Ser-874 regulating its phosphorylation and activation. However, the authors note that the activation of RPM-1 is a prerequisite for the activity of PPM-2 on DLK-1 and this PHR protein employs ubiquitination and phosphatase-based mechanisms to inhibit DLK-1. This observation is based on immunoprecipitation approaches followed by mass spectrometry, immunoblot, and immunofluorescence analysis in which Baker et al. showed that RPM-1 binds to and positively regulates PPM-2. As RPM-1 has previously been shown to be a part of a neuronal complex involving multiple proteins [61,62], more precise approaches were lacking to confirm that RPM-1 directly binds to PPM-2. In addition, the small segment of the C. elegans DLK-1 containing Ser-874 is not conserved with mammalian DLK, and no functional orthologue of PPM-2 is known in vertebrates (Yan et al., 2012; WormBase WS290).
In D.melanogaster, Valakh et al. (2013; 2015) showed that cytoskeletal dysregulation activates Wallenda/DLK [64,65]. Hayne and DiAntionio (2022) hypothesized that disruption of the cytoskeletal structure is mediated by inhibition of the serine/threonine protein phosphatase 2A (PP2A), which ultimately triggers DLK activation [5]. In addition to cytoskeletal perturbations, dysregulation of PP2A function leads to a variety of cell type-specific cellular dysfunctions, including defects in mitotic processes, and cell death [66]. In mammalian cells, pharmaceutical intervention with okadaic acid, an inhibitor of serine/threonine phosphatases including PP2A, showed an increase in the abundance of phosphorylated DLK [39] as well as in DLK activity [52]. However, these experiments have not definitively shown whether DLK is a direct target of PP2A. Daviau et al. (2009) also showed that the protein phosphotyrosyl-phosphatase inhibitor vanadate induced an approximately 3.5-fold increase in DLK activation compared to premeasurement activity and approximately 1.5-fold greater than the effect of okadaic acid. In contrast to PP2A, the authors state that the effect of vanadate on DLK is not a direct effect on the kinase, but is mediated through a Src kinase-dependent pathway, and showed similar results by induction of the PDGF receptor signaling pathway [11,52].
The calcium/calmodulin-dependent serine/threonine protein phosphatase 2B (calcineurin) has also been implicated in the regulation of DLK activity in several studies [10,39,56,57]. Mata et al. (1996) showed in rat aggregating-glial cell cultures that inhibition of calcineurin by its selective inhibitor cyclosporin A (CsA) affected DLK electrophoretic mobility only after membrane depolarisation by veratridine and not in unstimulated conditions, highlighting membrane depolarisation as a prerequisite for the effect of CsA action on DLK [39]. Consistent with this, Daviau et al. also observed no effect of CsA on DLK activity under unstimulated conditions in COS-7 cells transfected with a vector encoding a wild-type T7-DLK sequence [52]. Controversially, other studies in insulin-producing beta-cells have observed an effect of CsA on DLK activity: i) enhancement of DLK-dependent c-Jun phosphorylation [56], ii) increase of DLK kinase activity by an in vitro assay using casein as a substrate and induction of beta-cell apoptosis [57], iii) augmented DLK-dependent phosphorylation of the c-Jun N-terminal kinase (JNK), increased phosphorylation and activation of DLK at Ser-302, increased nuclear translocation of DLK with concomitant increase in beta-cell apoptosis [10]. Most of these DLK/calcineurin-dependent effects were also observed after calcineurin inhibition by tacrolimus (FK506), another structurally distinct, selective calcineurin inhibitor. It should be noted that Mata et al. (1996) and Daviau et al. (2009) used higher concentration of CsA (30 µM) and a longer treatment time (4 h) than the other studies (5 or 10 µM for 10 or 30 min). Although there was considerable evidence that DLK activity is partly regulated by the serine/protein phosphatase 2B, it was not known whether DLK and calcineurin interact directly. Duque Escobar et al. (2021) revealed a distinct φLxVP motif (aa 362-365) within DLK for interacting with the calcineurin A and B subunit interface, as already described for the NFAT transcription factor [10,67]. The authors used several mutations to show that Val-364 prevented the protein-protein interaction and exhibited increased DLK activity, measured as phosphorylation of the downstream JNK, inhibition of CRE-dependent gene transcription and induction of beta-cell apoptosis [10]. Furthermore, the activation of DLK by the calcineurin inhibitors might contribute to the pathogenesis of post-transplant diabetes mellitus observed after treatment with these immunosuppressant drugs [68].
2.3.3. Palmitoylation of DLK
The Dual Leucine Zipper Kinase is crucial for retrograde signaling after injury in neurons and important in brain development, but the activation of DLK also can lead to apoptosis and neuronal degeneration in different disease models such as ALS or Alzheimer’s. Due to the importance of the DLK for cell fate the activity of DLK at least in neurons needs to be highly restricted in temporal and spatial manners and confined to local events. In a search for an explanation on how a bioinformatically predicted soluble protein could travel from distant axonal regions back to the nucleus in a controlled way, Holland et al. found and validated an evolutionary conserved cysteine residue (C127, in human DLK) in the DLK as a site for post-translational modification via palmitoylation (Figure 4) [51].
Palmitoylation is a fast, dynamic posttranslational modification which is catalyzed by palmitoyl acyl transferases such as zinc aspartate-histidine-histidine-cysteine (zDHHC) family members and reversed by acyl protein thioesterases. Palmitoylated proteins are tethered to membranous structures like vesicle, the golgi, plasma membrane or the outer mitochondrial membrane [69]. Holland et al. (2016) showed that palmitoylated DLK is targeted to motile trafficking vesicles allowing it to traffic retrogradely in the event of axonal injury [51]. In addition, palmitoylation was essential for interaction of DLK with the scaffold protein JNK-interacting protein-3 (JIP3) resulting in the phosphorylation of DLK’s direct and indirect substrates like MKK4/7, JNK3 and c-Jun, rendering palmitoylation as novel mechanism for regulating DLK activity [70]. DLK palmitoylation does not require the phosphorylation or homodimerization of DLK, rather it brings DLK and its substrates in close proximity [51,71]. A model was proposed that palmitoylation tethers DLK and JNK3 to the same axon vesicle membranes and leads to a forward loop, whereby DLK through activation of MKK4/7 phosphorylates JNK3 which in turn activates DLK, thus perpetuating DLK signaling [70]. This model implies that JNK contributes to the activation of DLK, which has been shown before in non-neuronal and neuronal cells [23,40,41]. DLK subcellular localization is also palmitoylation dependent in non-neuronal cells (HEK293T) suggesting that inhibitors of palmitoylation might inhibit DLK activation [72]. Although not all MLKs are palmitoylated [70], many more proteins besides DLK undergo this posttranslational modification, thus, interfering with palmitoylation is expected to be very unselective. In an optic nerve crush (ONC) model Niu et al. (2020) identified the palmitoyltransferase ZDHHC17 as DLK palmitoylating enzyme [73]. Taken together, palmitoylation of DLK represents a mechanism to bring DLK and its substrates in proximity and to direct the subcellular localization of this kinase. Stress signals are reported to increase the palmitoylation of DLK, but it remains unknown how palmitoyl acyl transferases become activated.
2.3.4. Regulation by protein-protein interactions
DLK protein content and therefore activity is regulated by the interaction with diverse proteins. The Regulator of Presynaptic Morphology 1 (RPM-1) in C. elegans and Highwire (Hiw) in D. melanogaster were the first proteins demonstrated to interact with DLK-1 and Wallenda, respectively. Together with the human Protein Associated with Myc (PAM), these proteins are termed PHR (PAM, Highwire, RPM-1) proteins, which are huge proteins with more than 3700 aa containing diverse enzymatic activities and mainly regulate synapse formation and axon termination [74]. In C. elegans, in D. melanogaster and in the dorsal root ganglia of mammals, RPM-1, Highwire, and PHR, respectively, ubiquitinate DLK-1/Wallenda/DLK leading to the kinase’s proteasomal degradation thus terminating kinase activity [74]. However, in mammals, PHR does function independent of DLK in some neuronal contexts [74]. Using primary dorsal root ganglia cells as model, Huntwork-Rodriguez et al. (2013) showed that the interaction between DLK and the E3 ubiquitin ligase PHR is regulated by JNK-induced phosphorylation of DLK: Phosphorylation of DLK on Thr-43 and Ser-533 stabilize DLK, presumably be preventing the interaction with PHR1. Under basal conditions, when DLK is not phosphorylated, its abundance is regulated by a balance between PHR1 and the deubiquitinase ubiquitin-specific peptidase 9, X-linked (USP9X) [40]. In addition to PHR, the FK506-binding protein-like (FKBPL) and the FK506-binding protein 8 (FKBP8) were identified as DLK interacting proteins [50]. By interacting with the N-terminus of DLK, containing its kinase domain, the N-terminus of FKBPL, containing its peptidyl-prolyl isomerase domain, inhibited DLK activity and reduced its protein stability. Both, FKBPL and FKBP8, induced DLK degradation by the lysosomal pathway. Additionally, FKBP8 mediated DLK degradation was prevented when Lys-271 was mutated to Arg, which was shown to function as an ubiquitination and SUMOylation site. Thus, FKBP8 induced DLK lysosomal and proteasomal degradation [50].
Whereas the interactions of DLK with PHR, FKBP8, and FKBPL result in the degradation of the kinase and thus in its inactivation, the interaction with the scaffold protein JIP brings DLK and its substrates in proximity, thereby increasing and perpetuating DLK activity [24,25]. In a thorough mutational analysis, Mooney and Whitmarsh (2004) identified two JIP1-interaction sites within DLK, whereby the mutations of Phe-117, Leu-397 and Asp-398 to alanine severely impaired the DLK – JIP1 interaction and JNK phosphorylation [75]. The DLK – JIP – JNK interaction is regulated by phosphorylation of JIP: Phosphorylation of JIP1 on Thr-103 by JNK induces the dissociation of DLK from the scaffold protein, DLK homodimerization, activation and ultimately the phosphorylation of JNK. In murine brain cell lysates, phosphorylation of JIP1 on tyrosine residues by Src kinases increased the affinity of DLK to JIP1, thereby strengthening this interaction and preventing DLK activation [23,24]. Of note, palmitoylation contributes to bringing DLK, JIP and JNK in proximity [70]. The interaction of DLK with specific JIPs seems to be tissue dependent and contributes to selective DLK action. In the cerebellum of adult mice, where JIP1 was preferentially expressed, DLK was constitutively active in the absence of injury signals. In the murine forebrain, where JIP3 and another scaffold protein of DLK, Plenty of SH (POSH), are expressed, DLK becomes activated only by neuronal injury [3,76]. Further analysis revealed that in the cerebellum but not in the forebrain after neuronal injury, DLK regulated insulin growth factor 1 signaling. Hence, the regulation of DLK action seems to depend on the interaction with particular scaffold proteins and on the tissue [3].
The heat shock proteins (HSP) are another group of proteins regulating DLK protein abundance and thereby enzymatic activity. The HSP90 acts as a chaperone which in contrast to other HSPs facilitates maturation, complex assembly, localization and ligand binding of signal transduction proteins like kinases and nuclear receptors [48,77]. Using HSP90 and DLK inhibition and co-immunoprecipitation assays, an interaction between DLK and HSP90 was shown to occur in D. melanogaster, embryonic DRG neurons and in the HEK cell line, whereby inhibition or loss of HSP90 (or its fly ortholog Hsp83) reduced DLK protein content [48]. Hence, the interaction between HSP90 and DLK preserves DLK, possibly by preventing the interaction between DLK and PHR1 leading to DLK proteasomal degradation. In contrast, in the cell line COS7, the interaction of activated DLK with HSP70 results in DLK proteasomal degradation, which was dependent on the HSP70 co-chaperone CHIP (C-terminus of HSP70 interacting protein), an E3 ubiquitin ligase [49]. Notably, both chaperones, HSP90 and HSP70, can interact with the co-chaperone CHIP and induce the proteasomal degradation of their respective clients [78]. This suggests, that the outcome of the interaction of DLK with HSP chaperones does depend on the expression of the co-chaperone CHIP.
In addition, a direct interaction DLK and calcineurin has been demonstrated [10].
2.3.5. Regulation of DLK by its oligomerization
The homodimerization of DLK via its leucine zipper has been shown to be essential for DLK activity [22]. Experiments conducted in different cell lines (NIH3T3, COS-1) indicated that the formation of high molecular DLK polymers occurred in response to the apoptosis-inducing agent Calphostin C independent of its ability to inhibit protein kinase C (PKC) [79,80,81]. DLK oligomerization was abolished by the tissue-transglutaminase (tTG) inhibitor monodansylcadaverine, indicating that a tTG-catalyzed protein crosslinking reaction was the underlying cause. A model emerged whereby Calphostin-C induced intracellular rise of Ca2+ stimulates the Ca2+ dependent tTG2 crosslinking-activity, leading to an increase in DLK polymers. Oligomerization then increases DLK activity and hence activation of the JNK-Pathway. After JNK-activation the apoptosis regulator Bax translocates to mitochondria and induces caspase activation ultimately leading to apoptosis [79,80,81]. It remains unknown which domains of DLK mediate the oligomerization. Nevertheless, bringing single DLK molecules together either as homo- or oligomers, leads to increased DLK activity.
3. Conclusions
DLK protein levels, enzymatic activity and localization are tightly regulated at different levels and via distinct mechanisms, possibly tissue dependent. In addition, DLK activity and proteins levels are subject to further fine tuning via interactions with several other proteins and/or via posttranslational modifications. Furthermore, transcriptional and posttranscriptional mechanisms contribute to DLK regulation. Further elucidation of these diverse mechanisms will contribute to the identification of drug targets to specifically regulate DLK action.
Author Contributions
Conceptualization, KAK, MD, JDE, EO. writing—original draft preparation, KAK, EO.; writing—review and editing, KAK, MD, JDE, EO.; visualization, KAK.; KAK, EO. All authors have read and agreed to the published version of the manuscript.
Funding
Research about DLK in our lab was funded by DFG, grant number OE 181/5-1 and DDG to KAK.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Le Pichon, C.E.; Meilandt, W.J.; Dominguez, S.; Solanoy, H.; Lin, H.; Ngu, H.; Gogineni, A.; Sengupta Ghosh, A.; Jiang, Z.; Lee, S.-H.; et al. Loss of dual leucine zipper kinase signaling is protective in animal models of neurodegenerative disease. Science Translational Medicine 2017, 9, eaag0394. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-W.A.; Zhou, B.; Wernig, M.; Südhof, T.C. ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Aβ Secretion. Cell 2017, 168, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Goodwani, S.; Fernandez, C.; Acton, P.J.; Buggia-Prevot, V.; McReynolds, M.L.; Ma, J.; Hu, C.H.; Hamby, M.E.; Jiang, Y.; Le, K.; et al. Dual Leucine Zipper Kinase Is Constitutively Active in the Adult Mouse Brain and Has Both Stress-Induced and Homeostatic Functions. International Journal of Molecular Sciences 2020, 21, 4849. [Google Scholar] [CrossRef] [PubMed]
- Katz, J.S.; Rothstein, J.D.; Cudkowicz, M.E.; Genge, A.; Oskarsson, B.; Hains, A.B.; Chen, C.; Galanter, J.; Burgess, B.L.; Cho, W. A Phase 1 study of GDC-0134, a dual leucine zipper kinase inhibitor, in ALS. Annals of Clinical and Translational Neurology. [CrossRef] [PubMed]
- Hayne, M.; DiAntonio, A. Protein phosphatase 2A restrains DLK signaling to promote proper Drosophila synaptic development and mammalian cortical neuron survival. Neurobiology of Disease 2022, 163, 105586. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Roy, E.R.; Wang, Y.; Watkins, T.; Cao, W. DLK-MAPK Signaling Coupled with DNA Damage Promotes Intrinsic Neurotoxicity Associated with Non-Mutated Tau. Molecular Neurobiology, 2023. [Google Scholar] [CrossRef]
- Welsbie, D.S.; Yang, Z.; Ge, Y.; Mitchell, K.L.; Zhou, X.; Martin, S.E.; Berlinicke, C.A.; Hackler, L., Jr.; Fuller, J.; Fu, J.; et al. Functional genomic screening identifies dual leucine zipper kinase as a key mediator of retinal ganglion cell death. Proc Natl Acad Sci U S A 2013, 110, 4045–4050. [Google Scholar] [CrossRef] [PubMed]
- Oetjen, E. Regulation of Beta-Cell Function and Mass by the Dual Leucine Zipper Kinase. Archiv der Pharmazie 2016, 349, 410–413. [Google Scholar] [CrossRef] [PubMed]
- Wallbach, M.; Duque Escobar, J.; Babaeikelishomi, R.; Stahnke, M.-J.; Blume, R.; Schröder, S.; Kruegel, J.; Maedler, K.; Kluth, O.; Kehlenbach, R.H.; et al. Distinct functions of the dual leucine zipper kinase depending on its subcellular localization. Cellular Signalling 2016, 28, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Duque Escobar, J.; Kutschenko, A.; Schröder, S.; Blume, R.; Köster, K.-A.; Painer, C.; Lemcke, T.; Maison, W.; Oetjen, E. Regulation of dual leucine zipper kinase activity through its interaction with calcineurin. Cellular Signalling 2021, 82, 109953. [Google Scholar] [CrossRef]
- Holzman, L.B.; Merritt, S.E.; Fan, G. Identification, molecular cloning, and characterization of dual leucine zipper bearing kinase. A novel serine/threonine protein kinase that defines a second subfamily of mixed lineage kinases. Journal of Biological Chemistry 1994, 269, 30808–30817. [Google Scholar] [CrossRef]
- Gallo, K.A.; Johnson, G.L. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat Rev Mol Cell Biol 2002, 3, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Gallo, K.A.; Ellsworth, E.; Stoub, H.; Conrad, S.E. Therapeutic potential of targeting mixed lineage kinases in cancer and inflammation. Pharmacology & Therapeutics 2020, 207, 107457. [Google Scholar] [CrossRef]
- Bisson, N.; Tremblay, M.; Robinson, F.; Kaplan, D.R.; Trusko, S.P.; Moss, T. Mice lacking both mixed-lineage kinase genes Mlk1 and Mlk2 retain a wild type phenotype. Cell Cycle 2008, 7, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Geoffroy, C.G.; Wong, H.N.; Tress, O.; Nguyen, M.T.; Holzman, L.B.; Jin, Y.; Zheng, B. Leucine Zipper-bearing Kinase promotes axon growth in mammalian central nervous system neurons. Scientific Reports 2016, 6, 31482. [Google Scholar] [CrossRef] [PubMed]
- Hirai, S.-i.; Feng Cui, D.; Miyata, T.; Ogawa, M.; Kiyonari, H.; Suda, Y.; Aizawa, S.; Banba, Y.; Ohno, S. The c-Jun N-Terminal Kinase Activator Dual Leucine Zipper Kinase Regulates Axon Growth and Neuronal Migration in the Developing Cerebral Cortex. The Journal of Neuroscience 2006, 26, 11992–12002. [Google Scholar] [CrossRef]
- Pozniak, C.D.; Sengupta Ghosh, A.; Gogineni, A.; Hanson, J.E.; Lee, S.H.; Larson, J.L.; Solanoy, H.; Bustos, D.; Li, H.; Ngu, H.; et al. Dual leucine zipper kinase is required for excitotoxicity-induced neuronal degeneration. J Exp Med 2013, 210, 2553–2567. [Google Scholar] [CrossRef] [PubMed]
- Asghari Adib, E.; Smithson, L.J.; Collins, C.A. An axonal stress response pathway: degenerative and regenerative signaling by DLK. Current Opinion in Neurobiology 2018, 53, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.E.; Ha, H.; Kim, Y.K.; Cho, Y.; DiAntonio, A. DLK regulates a distinctive transcriptional regeneration program after peripheral nerve injury. Neurobiology of Disease 2019, 127, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, A.; Bradke, F. The DLK signalling pathway--a double-edged sword in neural development and regeneration. EMBO Rep 2013, 14, 605–614. [Google Scholar] [CrossRef]
- Jin, Y.; Zheng, B. Multitasking: Dual Leucine Zipper–Bearing Kinases in Neuronal Development and Stress Management. Annual Review of Cell and Developmental Biology 2019, 35, 501–521. [Google Scholar] [CrossRef]
- Nihalani, D.; Meyer, D.; Pajni, S.; Holzman, L.B. Mixed lineage kinase-dependent JNK activation is governed by interactions of scaffold protein JIP with MAPK module components. The EMBO Journal 2001, 20, 3447–3458. [Google Scholar] [CrossRef] [PubMed]
- Nihalani, D.; Wong, H.N.; Holzman, L.B. Recruitment of JNK to JIP1 and JNK-dependent JIP1 Phosphorylation Regulates JNK Module Dynamics and Activation. Journal of Biological Chemistry 2003, 278, 28694–28702. [Google Scholar] [CrossRef] [PubMed]
- Nihalani, D.; Wong, H.; Verma, R.; Holzman, L.B. Src Family Kinases Directly Regulate JIP1 Module Dynamics and Activation. Molecular and Cellular Biology 2007, 27, 2431–2441. [Google Scholar] [CrossRef] [PubMed]
- Blouin, R. DLK (Dual Leucine Zipper-Bearing Kinase). In Encyclopedia of Signaling Molecules; Choi, S., Ed.; Springer International Publishing: Cham, 2018. [Google Scholar]
- Itoh, A.; Wang, Z.; Ito, Y.; Reddy, U.R.; Itoh, T. SP3 acts as a positive regulator on the core promoter of human ZPK gene. Biochemical and Biophysical Research Communications 2004, 313, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Couture, J.P.; Blouin, R. The DLK gene is a transcriptional target of PPARgamma. Biochem J 2011, 438, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Ohlstein, J.F.; Strong, A.L.; McLachlan, J.A.; Gimble, J.M.; Burow, M.E.; Bunnell, B.A. Bisphenol A enhances adipogenic differentiation of human adipose stromal/stem cells. Journal of Molecular Endocrinology 2014, 53, 345–353. [Google Scholar] [CrossRef]
- Couture, J.P.; Daviau, A.; Fradette, J.; Blouin, R. The mixed-lineage kinase DLK is a key regulator of 3T3-L1 adipocyte differentiation. PLoS One 2009, 4, e4743. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.D.; Anand, D.; Motohashi, H.; Katsuoka, F.; Yamamoto, M.; Lachke, S.A. Deficiency of the bZIP transcription factors Mafg and Mafk causes misexpression of genes in distinct pathways and results in lens embryonic developmental defects. Frontiers in Cell and Developmental Biology 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Jin, Y. Regulation of DLK-1 kinase activity by calcium-mediated dissociation from an inhibitory isoform. Neuron 2012, 76, 534–548. [Google Scholar] [CrossRef]
- Heeyoung, S.; Juyoung, H.; Eun-Sook, J.; Sung Wook, C. MicroRNA Target Recognition: Insights from Transcriptome-Wide Non-Canonical Interactions. Mol. Cells 2016, 39, 375–381. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Huang, H.-Y.; Lin, Y.-C.-D.; Cui, S.; Huang, Y.; Tang, Y.; Xu, J.; Bao, J.; Li, Y.; Wen, J.; Zuo, H.; et al. miRTarBase update 2022: an informative resource for experimentally validated miRNA–target interactions. Nucleic Acids Research 2021, 50, D222–D230. [Google Scholar] [CrossRef]
- Beveridge, N.J.; Tooney, P.A.; Carroll, A.P.; Tran, N.; Cairns, M.J. Down-regulation of miR-17 family expression in response to retinoic acid induced neuronal differentiation. Cellular Signalling 2009, 21, 1837–1845. [Google Scholar] [CrossRef]
- Ye, M.; Li, D.; Yang, J.; Xie, J.; Yu, F.; Ma, Y.; Zhu, X.; Zhao, J.; Lv, Z. MicroRNA-130a Targets MAP3K12 to Modulate Diabetic Endothelial Progenitor Cell Function. Cell Physiol Biochem 2015, 36, 712–726. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Liu, G.; Li, X.; Liu, Y.; Wang, Y.; Pan, H.; Hu, J. MiR-191-5p alleviates microglial cell injury by targeting Map3k12 (mitogen-activated protein kinase kinase kinase 12) to inhibit the MAPK (mitogen-activated protein kinase) signaling pathway in Alzheimer’s disease. Bioengineered 2021, 12, 12678–12690. [Google Scholar] [CrossRef]
- Yu, J.; Feng, Y.; Wang, Y.; An, R. Aryl hydrocarbon receptor enhances the expression of miR-150-5p to suppress in prostate cancer progression by regulating MAP3K12. Archives of Biochemistry and Biophysics 2018, 654, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Mata, M.; Merritt, S.E.; Fan, G.; Yu, G.G.; Holzman, L.B. Characterization of Dual Leucine Zipper-bearing Kinase, a Mixed Lineage Kinase Present in Synaptic Terminals Whose Phosphorylation State Is Regulated by Membrane Depolarization via Calcineurin. Journal of Biological Chemistry 1996, 271, 16888–16896. [Google Scholar] [CrossRef] [PubMed]
- Huntwork-Rodriguez, S.; Wang, B.; Watkins, T.; Ghosh, A.S.; Pozniak, C.D.; Bustos, D.; Newton, K.; Kirkpatrick, D.S.; Lewcock, J.W. JNK-mediated phosphorylation of DLK suppresses its ubiquitination to promote neuronal apoptosis. J Cell Biol 2013, 202, 747–763. [Google Scholar] [CrossRef]
- Börchers, S.; Babaei, R.; Klimpel, C.; Duque Escobar, J.; Schröder, S.; Blume, R.; Malik, M.N.H.; Oetjen, E. TNFα-induced DLK activation contributes to apoptosis in the beta-cell line HIT. Naunyn-Schmiedeberg’s Archives of Pharmacology 2017, 390, 813–825. [Google Scholar] [CrossRef]
- Hao, Y.; Frey, E.; Yoon, C.; Wong, H.; Nestorovski, D.; Holzman, L.B.; Giger, R.J.; DiAntonio, A.; Collins, C. An evolutionarily conserved mechanism for cAMP elicited axonal regeneration involves direct activation of the dual leucine zipper kinase DLK. eLife 2016, 5, e14048. [Google Scholar] [CrossRef]
- Ghosh-Roy, A.; Wu, Z.; Goncharov, A.; Jin, Y.; Chisholm, A.D. Calcium and Cyclic AMP Promote Axonal Regeneration in <span class="named-content project" id="named-content-1">Caenorhabditis elegans</span> and Require DLK-1 Kinase. The Journal of Neuroscience 2010, 30, 3175–3183. [Google Scholar] [CrossRef] [PubMed]
- Larhammar, M.; Huntwork-Rodriguez, S.; Rudhard, Y.; Sengupta-Ghosh, A.; Lewcock, J.W. The Ste20 Family Kinases MAP4K4, MINK1, and TNIK Converge to Regulate Stress-Induced JNK Signaling in Neurons. The Journal of Neuroscience 2017, 37, 11074–11084. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.K.; Broyer, R.M.; Lee, C.D.; Lu, T.; Louie, M.J.; La Torre, A.; Al-Ali, H.; Vu, M.T.; Mitchell, K.L.; Wahlin, K.J.; et al. Inhibition of GCK-IV kinases dissociates cell death and axon regeneration in CNS neurons. Proceedings of the National Academy of Sciences 2020, 117, 33597–33607. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Qiu, W.R.; Wang, X.; Meyer, C.F.; Tan, T.H. Human HPK1, a novel human hematopoietic progenitor kinase that activates the JNK/SAPK kinase cascade. Genes & Development 1996, 10, 2251–2264. [Google Scholar] [CrossRef]
- Leung, I.W.-L.; Lassam, N. The Kinase Activation Loop Is the Key to Mixed Lineage Kinase-3 Activation via Both Autophosphorylation and Hematopoetic Progenitor Kinase 1 Phosphorylation*. Journal of Biological Chemistry 2001, 276, 1961–1967. [Google Scholar] [CrossRef] [PubMed]
- Karney-Grobe, S.; Russo, A.; Frey, E.; Milbrandt, J.; DiAntonio, A. HSP90 is a chaperone for DLK and is required for axon injury signaling. Proceedings of the National Academy of Sciences 2018, 115, E9899–E9908. [Google Scholar] [CrossRef] [PubMed]
- Daviau, A.; Proulx, R.; Robitaille, K.; Di Fruscio, M.; Tanguay, R.M.; Landry, J.; Patterson, C.; Durocher, Y.; Blouin, R. Down-regulation of the mixed-lineage dual leucine zipper-bearing kinase by heat shock protein 70 and its co-chaperone CHIP. J Biol Chem 2006, 281, 31467–31477. [Google Scholar] [CrossRef]
- Lee, B.; Oh, Y.; Cho, E.; DiAntonio, A.; Cavalli, V.; Shin, J.E.; Choi, H.W.; Cho, Y. FK506-binding protein-like and FK506-binding protein 8 regulate dual leucine zipper kinase degradation and neuronal responses to axon injury. Journal of Biological Chemistry 2022, 298, 101647. [Google Scholar] [CrossRef] [PubMed]
- Holland, S.M.; Collura, K.M.; Ketschek, A.; Noma, K.; Ferguson, T.A.; Jin, Y.; Gallo, G.; Thomas, G.M. Palmitoylation controls DLK localization, interactions and activity to ensure effective axonal injury signaling. Proc Natl Acad Sci U S A 2016, 113, 763–768. [Google Scholar] [CrossRef]
- Daviau, A.; Di Fruscio, M.; Blouin, R. The mixed-lineage kinase DLK undergoes Src-dependent tyrosine phosphorylation and activation in cells exposed to vanadate or platelet-derived growth factor (PDGF). Cellular Signalling 2009, 21, 577–587. [Google Scholar] [CrossRef]
- Wu, C.-C.; Wu, H.-J.; Wang, C.-H.; Lin, C.-H.; Hsu, S.-C.; Chen, Y.-R.; Hsiao, M.; Schuyler, S.C.; Lu, F.L.; Ma, N.; et al. Akt suppresses DLK for maintaining self-renewal of mouse embryonic stem cells. Cell Cycle 2015, 14, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Tulgren, E.D.; Baker, S.T.; Rapp, L.; Gurney, A.M.; Grill, B. PPM-1, a PP2Cα/β phosphatase, Regulates Axon Termination and Synapse Formation in Caenorhabditis elegans. Genetics 2011, 189, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.T.; Opperman, K.J.; Tulgren, E.D.; Turgeon, S.M.; Bienvenut, W.; Grill, B. RPM-1 Uses Both Ubiquitin Ligase and Phosphatase-Based Mechanisms to Regulate DLK-1 during Neuronal Development. PLOS Genetics 2014, 10, e1004297. [Google Scholar] [CrossRef] [PubMed]
- Oetjen, E.; Lechleiter, A.; Blume, R.; Nihalani, D.; Holzman, L.; Knepel, W. Inhibition of membrane depolarisation-induced transcriptional activity of cyclic AMP response element binding protein (CREB) by the dual-leucine-zipper-bearing kinase in a pancreatic islet beta cell line. Diabetologia 2006, 49, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Plaumann, S.; Blume, R.; Börchers, S.; Steinfelder, H.J.; Knepel, W.; Oetjen, E. Activation of the Dual-Leucine-Zipper-Bearing Kinase and Induction of β-Cell Apoptosis by the Immunosuppressive Drug Cyclosporin A. Molecular Pharmacology 2008, 73, 652–659. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, J.; Hendricks, M.; Le Guyader, S.; Subburaju, S.; Grunewald, B.; Scholich, K.; Jesuthasan, S. Formation of the retinotectal projection requires Esrom, an ortholog of PAM (protein associated with Myc). Development 2005, 132, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Lewcock, J.W.; Genoud, N.; Lettieri, K.; Pfaff, S.L. The Ubiquitin Ligase Phr1 Regulates Axon Outgrowth through Modulation of Microtubule Dynamics. Neuron 2007, 56, 604–620. [Google Scholar] [CrossRef] [PubMed]
- Hammarlund, M.; Nix, P.; Hauth, L.; Jorgensen, E.M.; Bastiani, M. Axon Regeneration Requires a Conserved MAP Kinase Pathway. Science 2009, 323, 802–806. [Google Scholar] [CrossRef] [PubMed]
- Liao, E.H.; Hung, W.; Abrams, B.; Zhen, M. An SCF-like ubiquitin ligase complex that controls presynaptic differentiation. Nature 2004, 430, 345–350. [Google Scholar] [CrossRef]
- Nakata, K.; Abrams, B.; Grill, B.; Goncharov, A.; Huang, X.; Chisholm, A.D.; Jin, Y. Regulation of a DLK-1 and p38 MAP Kinase Pathway by the Ubiquitin Ligase RPM-1 Is Required for Presynaptic Development. Cell 2005, 120, 407–420. [Google Scholar] [CrossRef]
- Takekawa, M.; Saito, H. A Family of Stress-Inducible GADD45-like Proteins Mediate Activation of the Stress-Responsive MTK1/MEKK4 MAPKKK. Cell 1998, 95, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Valakh, V.; Walker, L.J.; Skeath, J.B.; DiAntonio, A. Loss of the Spectraplakin Short Stop Activates the DLK Injury Response Pathway in <em>Drosophila</em>. The Journal of Neuroscience 2013, 33, 17863–17873. [Google Scholar] [CrossRef]
- Valakh, V.; Frey, E.; Babetto, E.; Walker, L.J.; DiAntonio, A. Cytoskeletal disruption activates the DLK/JNK pathway, which promotes axonal regeneration and mimics a preconditioning injury. Neurobiol Dis 2015, 77, 13–25. [Google Scholar] [CrossRef]
- JANSSENS, V.; GORIS, J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochemical Journal 2001, 353, 417–439. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Roy, J.; Martinez-Martinez, S.; Lopez-Maderuelo, M.D.; Nino-Moreno, P.; Orti, L.; Pantoja-Uceda, D.; Pineda-Lucena, A.; Cyert, M.S.; Redondo, J.M. A conserved docking surface on calcineurin mediates interaction with substrates and immunosuppressants. Mol Cell 2009, 33, 616–626. [Google Scholar] [CrossRef]
- Ahmed, S.H.; Biddle, K.; Augustine, T.; Azmi, S. Post-Transplantation Diabetes Mellitus. Diabetes Therapy 2020, 11, 779–801. [Google Scholar] [CrossRef] [PubMed]
- Dennis, K.M.J.H.; Heather, L.C. Post-translational palmitoylation of metabolic proteins. Frontiers in Physiology 2023, 14. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Holland, S.M.; Ketschek, A.; Collura, K.M.; Hesketh, N.L.; Hayashi, T.; Gallo, G.; Thomas, G.M. Palmitoylation couples the kinases DLK and JNK3 to facilitate prodegenerative axon-to-soma signaling. Science Signaling 2022, 15, eabh2674. [Google Scholar] [CrossRef]
- Tortosa, E.; Sengupta Ghosh, A.; Li, Q.; Wong, W.R.; Hinkle, T.; Sandoval, W.; Rose, C.M.; Hoogenraad, C.C. Stress-induced vesicular assemblies of dual leucine zipper kinase are signaling hubs involved in kinase activation and neurodegeneration. The EMBO Journal 2022, 41, e110155. [Google Scholar] [CrossRef]
- Martin, D.D.O.; Kanuparthi, P.S.; Holland, S.M.; Sanders, S.S.; Jeong, H.-K.; Einarson, M.B.; Jacobson, M.A.; Thomas, G.M. Identification of Novel Inhibitors of DLK Palmitoylation and Signaling by High Content Screening. Scientific Reports 2019, 9, 3632. [Google Scholar] [CrossRef]
- Niu, J.; Sanders, S.S.; Jeong, H.-K.; Holland, S.M.; Sun, Y.; Collura, K.M.; Hernandez, L.M.; Huang, H.; Hayden, M.R.; Smith, G.M.; et al. Coupled Control of Distal Axon Integrity and Somal Responses to Axonal Damage by the Palmitoyl Acyltransferase ZDHHC17. Cell Reports 2020, 33, 108365. [Google Scholar] [CrossRef] [PubMed]
- Grill, B.; Murphey, R.K.; Borgen, M.A. The PHR proteins: intracellular signaling hubs in neuronal development and axon degeneration. Neural Development 2016, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Mooney, L.M.; Whitmarsh, A.J. Docking Interactions in the c-Jun N-terminal Kinase Pathway*. Journal of Biological Chemistry 2004, 279, 11843–11852. [Google Scholar] [CrossRef]
- Xu, Z.; Kukekov, N.V.; Greene, L.A. POSH acts as a scaffold for a multiprotein complex that mediates JNK activation in apoptosis. The EMBO Journal 2003, 22, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nature Reviews Molecular Cell Biology 2010, 11, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Quintana-Gallardo, L.; Martín-Benito, J.; Marcilla, M.; Espadas, G.; Sabidó, E.; Valpuesta, J.M. The cochaperone CHIP marks Hsp70- and Hsp90-bound substrates for degradation through a very flexible mechanism. Scientific Reports 2019, 9, 5102. [Google Scholar] [CrossRef] [PubMed]
- Hébert, S.S.; Daviau, A.; Grondin, G.; Latreille, M.; Aubin, R.A.; Blouin, R. The Mixed Lineage Kinase DLK Is Oligomerized by Tissue Transglutaminase during Apoptosis. Journal of Biological Chemistry 2000, 275, 32482–32490. [Google Scholar] [CrossRef]
- Robitaille, K.; Daviau, A.; Tucholski, J.; Johnson, G.V.W.; Rancourt, C.; Blouin, R. Tissue transglutaminase triggers oligomerization and activation of dual leucine zipper-bearing kinase in calphostin C-treated cells to facilitate apoptosis. Cell Death & Differentiation 2004, 11, 542–549. [Google Scholar] [CrossRef]
- Robitaille, K.; Daviau, A.; Lachance, G.; Couture, J.P.; Blouin, R. Calphostin C-induced apoptosis is mediated by a tissue transglutaminase-dependent mechanism involving the DLK/JNK signaling pathway. Cell Death & Differentiation 2008, 15, 1522–1531. [Google Scholar] [CrossRef]
Figure 1.
Mixed-lineage kinase (MLK) Subfamilies. Schematic depiction of functional domains of different MLK-family-members (not to scale). MLK (Mixed-lineage kinase), DLK (Dual Leucine Zipper Kinase), LZK (Leucine Zipper Kinase), ZAK (Sterile alpha motif and leucine zipper containing kinase AZK), SH3 (Src homology 3 domain), LZ (Leucine Zipper), CRIB (Cdc42/Rac interactive-binding), SAM (sterile-α motif). NCBI RefSeq accession numbers (if applicable): MLK1/MAP3K9 (NP_001271159.1, Isoform 2), MLK2/MAP3K10 (NP_002437.2), MLK3/MAP3K11 (NP_002410.1), MLK 4/MAP3K21 (CAC84639.1, Isoform 1/α; NP_115811.2, Isoform 2/β), DLK/MAP3K12 (NP_001180440.1, Isoform 1), LZK/MAP3K13 (NP_004712.1, Isoform 1), ZAK/MAP3K20 (NP_057737.2, Isoform 1/α; NP_598407.1, Isoform 2/β).
Figure 1.
Mixed-lineage kinase (MLK) Subfamilies. Schematic depiction of functional domains of different MLK-family-members (not to scale). MLK (Mixed-lineage kinase), DLK (Dual Leucine Zipper Kinase), LZK (Leucine Zipper Kinase), ZAK (Sterile alpha motif and leucine zipper containing kinase AZK), SH3 (Src homology 3 domain), LZ (Leucine Zipper), CRIB (Cdc42/Rac interactive-binding), SAM (sterile-α motif). NCBI RefSeq accession numbers (if applicable): MLK1/MAP3K9 (NP_001271159.1, Isoform 2), MLK2/MAP3K10 (NP_002437.2), MLK3/MAP3K11 (NP_002410.1), MLK 4/MAP3K21 (CAC84639.1, Isoform 1/α; NP_115811.2, Isoform 2/β), DLK/MAP3K12 (NP_001180440.1, Isoform 1), LZK/MAP3K13 (NP_004712.1, Isoform 1), ZAK/MAP3K20 (NP_057737.2, Isoform 1/α; NP_598407.1, Isoform 2/β).

Figure 2.
The human MAP3K12 gene spans 21.871 nucleotides (nt) and is located on the complementary strand of chromosome 12, GRCh38.p14 Primary Assembly. It is flanked by the gene encoding for the TARBP2 subunit of RISC loading complex (TARBP2) and the poly(rC) binding protein 2 (PCBP2) gene, both located on the positive strand in opposite direction relative to the DLK gene. The transcripts of the human DLK Isoforms 1 (NM_001193511.2) and 2 (NM_006301.4) differ in a 99 nt stretch absent in Isoform 2, resulting in a slightly shorter protein of 859 amino acids (aa) and a calculated molecular weight of 93,2 kDa instead of 892 aa and 96,3 kDa. Data retrieved from ncbi, https://www.ncbi.nlm.nih.gov/gene/7786, 30.06.2023, 14:33. Figure created with BioRender.
Figure 2.
The human MAP3K12 gene spans 21.871 nucleotides (nt) and is located on the complementary strand of chromosome 12, GRCh38.p14 Primary Assembly. It is flanked by the gene encoding for the TARBP2 subunit of RISC loading complex (TARBP2) and the poly(rC) binding protein 2 (PCBP2) gene, both located on the positive strand in opposite direction relative to the DLK gene. The transcripts of the human DLK Isoforms 1 (NM_001193511.2) and 2 (NM_006301.4) differ in a 99 nt stretch absent in Isoform 2, resulting in a slightly shorter protein of 859 amino acids (aa) and a calculated molecular weight of 93,2 kDa instead of 892 aa and 96,3 kDa. Data retrieved from ncbi, https://www.ncbi.nlm.nih.gov/gene/7786, 30.06.2023, 14:33. Figure created with BioRender.
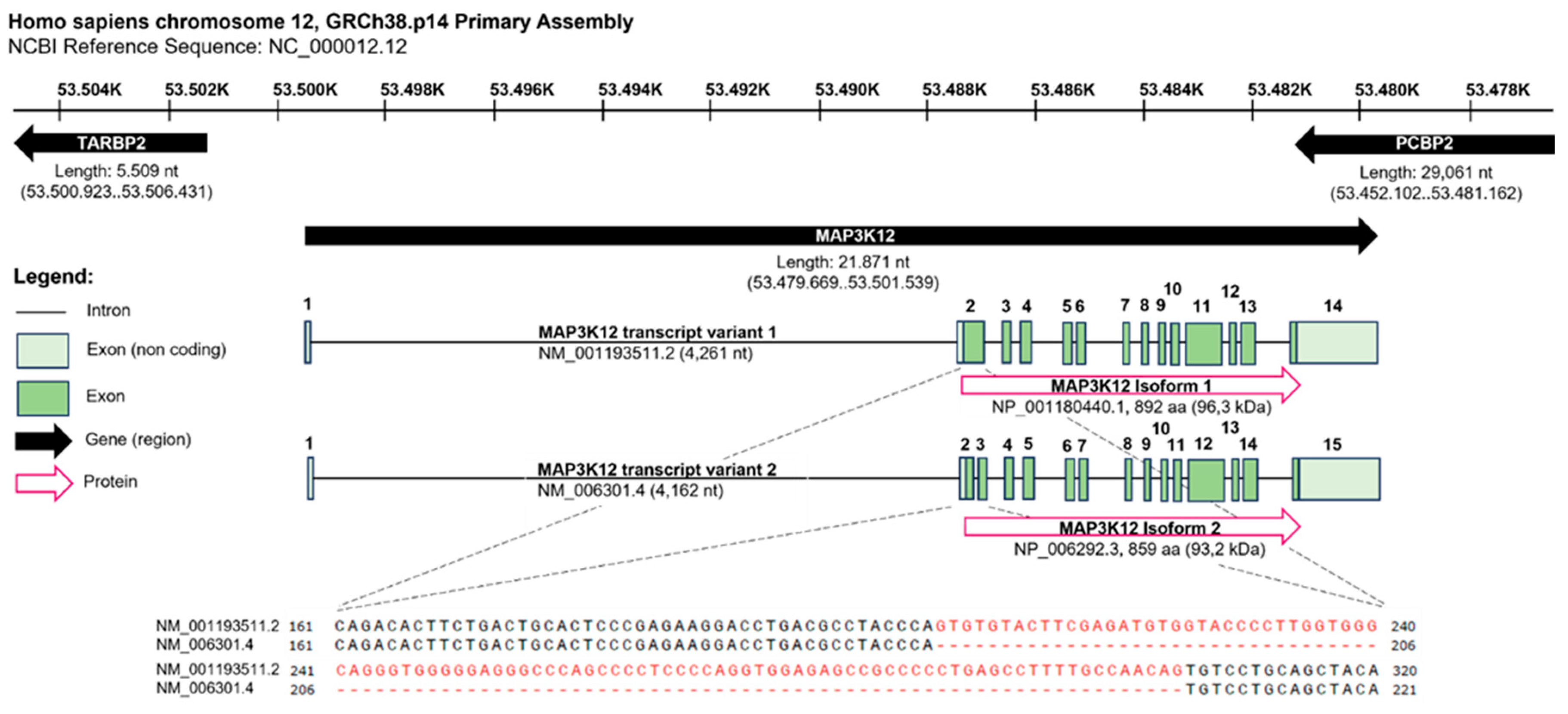
Figure 3.
Examples for regulation of DLK activity. A, under basal unstimulated conditions unphosphorylated DLK protein abundance is regulated by the E3 ubiquitin ligase PHR1 and the deubiquitinase USP9X. B, Signals activating cAMP and PKA phosphorylate dimerized DLK on Ser-302, leading to the activation of MKK4/7 and JNK. JNK in turn phosphorylates DLK on Thr-43 and Ser-535, preventing the interaction with PHR1 thereby stabilizing DLK. Other signals activating MAP4K phosphorylate DLK on Thr-43 and stabilize DLK. C, upon an increase in the intracellular calcium concentration, calcineurin interacts with monomeric or dimeric DLK and dephosphorylates the kinase. Inhibition of calcineurin by ROS prevents the dephosphorylation of DLK, whereas the interaction of immunophilin bound CsA or FK506 displace DLK from the calcineurin interaction site, DLK dimerizes and autophosphorylates in trans. For further information, please, see the text.
Figure 3.
Examples for regulation of DLK activity. A, under basal unstimulated conditions unphosphorylated DLK protein abundance is regulated by the E3 ubiquitin ligase PHR1 and the deubiquitinase USP9X. B, Signals activating cAMP and PKA phosphorylate dimerized DLK on Ser-302, leading to the activation of MKK4/7 and JNK. JNK in turn phosphorylates DLK on Thr-43 and Ser-535, preventing the interaction with PHR1 thereby stabilizing DLK. Other signals activating MAP4K phosphorylate DLK on Thr-43 and stabilize DLK. C, upon an increase in the intracellular calcium concentration, calcineurin interacts with monomeric or dimeric DLK and dephosphorylates the kinase. Inhibition of calcineurin by ROS prevents the dephosphorylation of DLK, whereas the interaction of immunophilin bound CsA or FK506 displace DLK from the calcineurin interaction site, DLK dimerizes and autophosphorylates in trans. For further information, please, see the text.
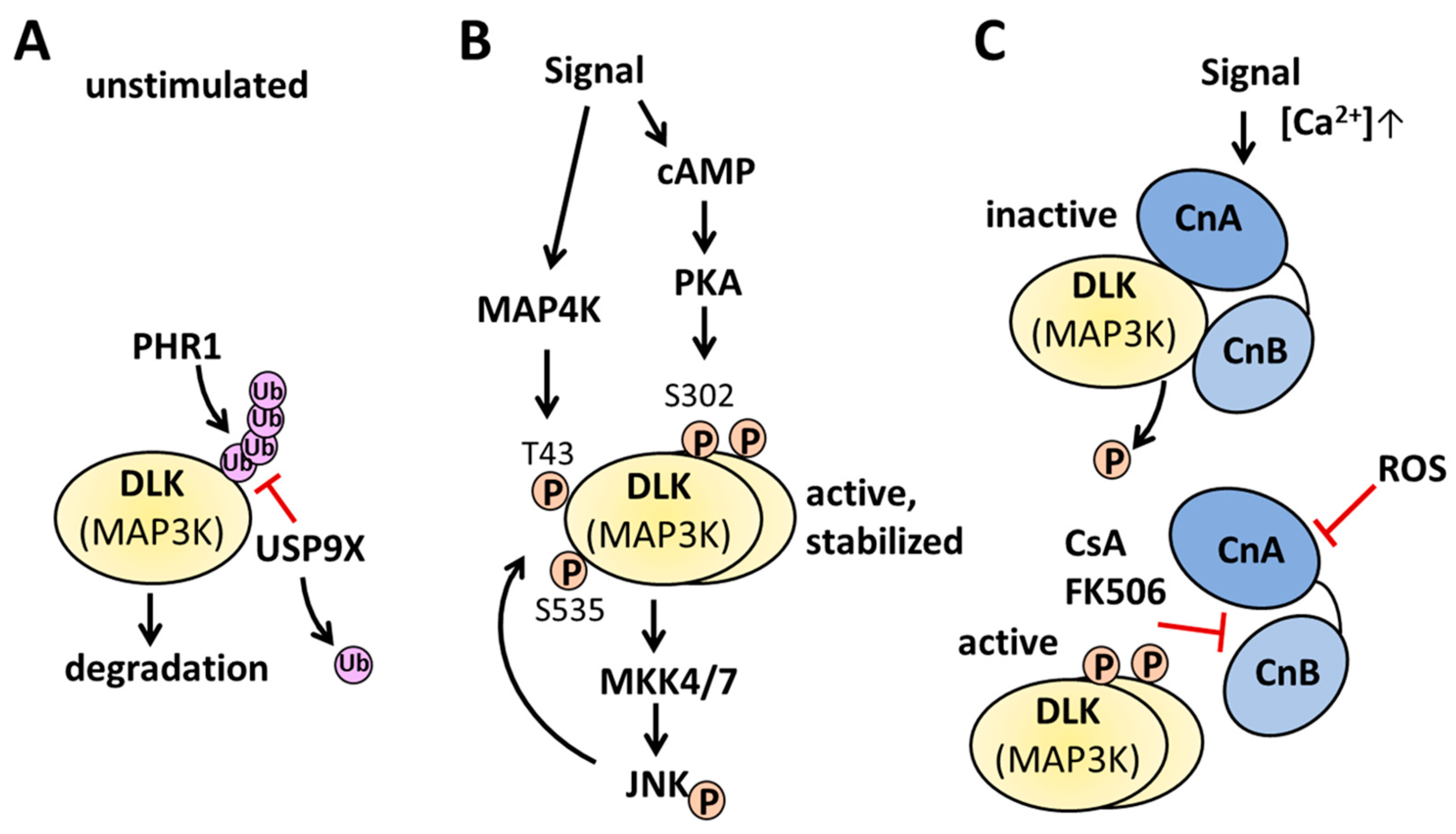
Figure 4.
DLK amino acid residues which are modulated on the post translational level. Isoform 1 of human DLK is depicted; T – threonin; C – cystein; K – lysin; S – serin; orange dots – phosphorylation and possibly dephosphorylation sites; violet dot – palmitoylation site; blue dot – site for ubiquitylation and SUMOylation. KD – kinase domain; AL - activation loop within the kinase domain; LZ – leucine zipper for homodimerization. For further information, please, see the text.
Figure 4.
DLK amino acid residues which are modulated on the post translational level. Isoform 1 of human DLK is depicted; T – threonin; C – cystein; K – lysin; S – serin; orange dots – phosphorylation and possibly dephosphorylation sites; violet dot – palmitoylation site; blue dot – site for ubiquitylation and SUMOylation. KD – kinase domain; AL - activation loop within the kinase domain; LZ – leucine zipper for homodimerization. For further information, please, see the text.
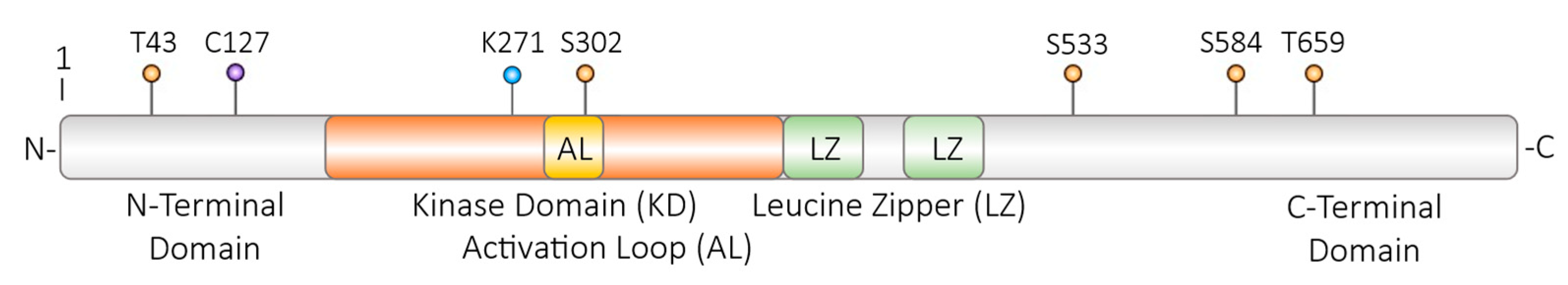

Table 1.
MAP3K12 Isoforms in different model organisms.
| Model Organism | Protein | Accession Number | Isoforms | Length |
|---|---|---|---|---|
| Homo sapiens | MAP3K12, ZPK | NP_001180440.1 | Isoform 1 | 892 aa |
| Homo sapiens | MAP3K12, ZPK | NP_006292.3 | Isoform 2 | 859 aa |
| Mus musculus | MAP3K12, DLK | NP_001157115.1 | (not described) | 888 aa |
| Rattus norvegicus | MAP3K12, MUK | NP_037187.1 | (not described) | 888 aa |
| Mesocricetus auratus | MAP3K12, DLK | XP_040609646.1 | Isoform X1 | 920 aa |
| Mesocricetus auratus | MAP3K12, DLK | XP_012966775.1 | Isoform X2 | 862 aa |
| Mesocricetus auratus | MAP3K12, DLK | XP_005067383.1 | Isoform X3 | 892 aa |
| Caenorhabditis elegans | DLK-1 | NP_001021443.1 | long | 928 aa |
| Caenorhabditis elegans | DLK-1 | NP_001021445.1 | short | 577 aa |
| Drosophila melanogaster | Wallenda, wnd | NP_649137.3 | Isoform (A) | 977 aa |
| Drosophila melanogaster | Wallenda, wnd | NP_788540.1 | Isoform (B) | 977 aa |
| Drosophila melanogaster | Wallenda, wnd | NP_788541.1 | Isoform (C) | 977 aa |
| Drosophila melanogaster | Wallenda, wnd | NP_001189132.1 | Isoform (D) | 950 aa |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



