Submitted:
18 December 2023
Posted:
19 December 2023
You are already at the latest version
Abstract
This review centers on the pivotal role of the oncoprotein FOSL1, within the dimeric AP-1 transcription factor consisting of FOS-related elements. FOSL1 emerges as a key regulator governing invasion and metastatic dissemination, therefore making it a promising target for therapeutic intervention in cancer patients. The review provides a comprehensive survey of the regulatory mechanisms that exert influence over FOSL1. These encompass a spectrum of transcriptional and post-translational modifications. Notably, we unveiled the intricate involvement of diverse non-coding RNAs, specifically microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), in the modulation of FOSL1 expression through targeted interactions with its mRNA transcripts. Furthermore, an exploration into the functional dimensions of FOSL1 unfolds, encompassing apoptosis, proliferation, and migration. This investigation is accompanied by a thorough examination of the elaborate signaling pathways that underpin these multifaced roles. Additionally, emphasis is placed on the role of FOSL1 in orchestrating the stimulation of various cytokines, including TGF-beta, and the initiation of IL-6 and VEGF production in tumor-associated macrophages (TAMs) that transverse into the tumor microenvironment. A distinct focus is dedicated to appraising the prognostic implications associated with FOSL1. Finally, insights emerge into the evolving roles of FOSL1 in the context of mechanisms governing resistance and dependency on targeted therapeutic modalities.
Keywords:
glioblastoma
; FOSL1
; glioma stem cells
; drug resistance
; prognosis
; biomarker
1. Introduction
Glioma is a type of tumor that develops from glial cells in the brain or spine. Glioblastoma multiforme (GBMs) are aggressive and malignant brain tumors that often recur despite intensive therapy. This recurrence is often associated with resistance to treatment and can be attributed to various factors, with glioblastoma stem cells (GSCs), playing a significant role. GSCs are a small subpopulation of cells within the tumor that exhibit stem cell-like properties, such as self-renewal and the capacity to differentiate into various cell types found within the tumor. GSCs contribute to the recurrence and resilience of glioblastoma in many ways such as therapy resistance, heterogeneity, microenvironment interaction, invasion, migration, and tumor regrowth [1]. CD133, also known as prominin-1, is a glycoprotein, that has been associated with stem cell-like properties in various cancers, including brain tumors; in many studies involving brain cancer stem cells, researchers utilize the CD133 as a cell surface marker to isolate and analyze these cells based on their functional traits. A recent study uncovered that four specific transcription factors (SOX2, SALL2, OLIG2, and POU3F2) can induce a conversion of differentiated glioblastoma cells into cells with stem-like qualities [2]. FOSL1 manages the activity of these four factors by binding to their promoter regions, thereby influencing various markers associated with stemness. Additionally, it diminishes the cells’ ability to aggregate by increasing the presence of fibronectin1 FN1 in the extracellular matrix. FOSL1 orchestrates changes in stemness by regulating the behavior of these four transcription factors [2].
FOSL1 is a gene that encodes Fos-related-antigen-1(FRA-1) protein consisting of 271 amino acids. FOSL1 is a member of the FOSL1 family protein belonging to the activator protein-1(AP-1) transcription factor superfamily. AP-1 complex results from the dimerization between members of the JUN (c-Jun, JunB, and JunD), FOS ( FOS, FOSB, FOSL1, and FOSL2) family member, and other activating transcription factor protein families (ATF and Maf) [3,4]. JUN can form both JUN-JUN homodimers and JUN-FOS heterodimers, while FOS can only form JUN-FOS heterodimers with JUN. The formation of AP-1 dimer is dependent on the basic leucine zipper (bZIP) domain on JUN and FOS. The bZIP domain of FOSL1 consists of two regions: a basic region and a leucine zipper region. Activator Protein 1 (AP-1) constitutes a dimeric transcription factor composed of members of the Fos, Jun, Maf, and ATF multigene families. These AP-1 dimers bind to different types of palindromic sequences depending on their compositions [5,6]. The two main types of AP-1 sites are TPA-responsive elements (TREs) and cyclic AMP response elements (CREs). TREs are palindromic sequences that are responsive to the phorbol ester 12-O-tetradecanoylphorbol-13-acetate (TPA) and are commonly found in the promoters of genes involved in cell proliferation, differentiation, and survival. On the other hand, CREs are palindromic sequences that respond to cyclic AMP (cAMP) and play a crucial role in regulating genes associated with metabolism, inflammation, and stress response [7,8].
FOSL1 exhibits varying levels of expression in different cell types and tissues. FOSL1 has been found to exhibit high expression levels in numerous cancer types including thyroid tumors, gastric cancer (GC), breast cancer, nephroblastoma, liver cancer, and laryngeal squamous cell carcinoma [8,9]. However, its expression is decreased in NSCLC and cervical cancer [10]. The precise role of AP-1 in tumorigenesis is complex and likely depends on the specific tumor type, stage, and genetic background of the tumor. Thus, the distinct structural features of FOSL1 can be attributed to both its unique structural characteristics and the various specific dimers it forms.
In this paper, we provide a comprehensive summary of recent studies focusing on the transcriptional, epigenetic regulation, and signal transduction mechanism of FOSL1. We explore its significant role in various aspects of cancer biology, including cell proliferation, survival, epithelial-mesenchymal transition (EMT), metastasis, prognosis, and drug resistance in glioma and other tumor types. Moreover, we discuss the critical implications of FOSL1 in the diagnosis and treatment of tumors with a special view on glioblastoma, emphasizing its relevance in targeted therapy approaches.
2. Regulation of FOSL1
2.1. Transcriptional regulation
The expression and activity of FOSL1 can be regulated by various factors, including extracellular signals, transcriptional and post-transcriptional mechanisms, and epigenetic modifications [11]. The transcription of the FOSL1 gene is regulated by multiple transcription factors that interact with the FOSL1 promoter region (AP-1, beta-catenin, TWIST1, SNAIL1, Elk, SRF, ATF, and CREB) or the intronic enhancer (AP-1, MYC, TWIST1, and SNAIL1). The positive autoregulation of FOSL1 is mediated by multiple binding sites of the AP-1 transcription factor. This positive feedback loop is further amplified by the posttranslational accumulation of FOSL1. The BRD4 epigenetic reader, which is associated with the enhancer, plays a key role in the recruitment of p-TEFb, which in turn phosphorylates the Carboxy-Terminal Domain (CTD) of RNA polymerase II (RNAPII). This triggers transcriptional elongation by releasing the RNAPII paused on the FOSL1 promoter [11,12]. Interestingly, the FOSL1 intronic enhancer is part of a much larger Super Enhancer (SE) region, which has been identified by genome-wide analyses in GBM [13,14], pancreatic, and colorectal cancer cells. SEs are clusters of enhancers that drive high levels of transcription of genes that are important for cell identity and disease phenotype [12]. We previously reported that TRPM7 regulates glioma cells’ stemness through STAT3 [15]. To further gain insight into the mechanism by which TRPM7 activates transcription of the FOSL1 gene to contribute to glioma stemness, we constructed a FOSL1 promoter and its GAS mutants followed by luciferase reporter assays and ChIP-qPCR. These combined results demonstrated that TRPM7 induced FOSL1 transcriptional activation, which is mediated by the action of STAT3, a mechanism shown to be important in glioma stemness [16].
2.2. Non-coding RNAs regulate FOSL1
The two main types of regulatory non-coding RNAs (ncRNAs) are long non-coding RNAs (lncRNA, >200 nts) and microRNAs (miRNAs, <30 nts). The function of miRNAs is to regulate gene expression by binding to the untranslated region (3’-UTR) of the target mRNAs. On the other hand, lncRNA can act as competing endogenous RNAs (ceRNAs) and sponge miRNAs subsequently reduce the function of miRNAs. The cross-talk between lncRNAs, miRNAs, and mRNAs has a pivotal role in the pathogenesis of malignancy [17,18]. Recently, there has been a growing interest in investigating the posttranscriptional regulation of FOSL1 by ncRNAs. Studies have revealed that several non-coding RNAs, including miRNAs and lncRNAs, are involved in regulating FOSL1 expression by targeting its mRNA transcripts [19,20,21].
2.2.1. miRNAs and FOSL1
The ectopic expression of FOSL1 was reported in several types of malignancy. Recent studies have shown that several miRNAs target FOSL1 and regulate its expression, leading to the modulation of various cellular functions. It has been reported that the downregulation of miRNAs can result in increased expression of FOSL1 in various cancers [22,23,24]. 1) Breast cancer (BC): The expression levels of miR34a/c in patients significantly decreased in patients with metastatic breast tumors as well as in metastatic cancerous cells MDA-MB-231 and Hs578T. In vitro and in vivo studies reveal that ectopic expression of two miR34a/c suppressed invasion and migration of cancerous breast cells. Yang et al. reported that overexpression of miR34a/c can directly bind to 3ؘ UTR- FOSL1, consequently inhibiting its expression at both mRNA and protein levels [25]. It has been reported that elevated levels of FOSL1 in triple-negative breast cancer (TNBC) patients are associated with poor survival. miR-4516 target genes such as FOSL1 are involved in the regulation of proliferation [26]. They demonstrated a significant downregulation of miR-4516 expression in patients with invasive ductal carcinoma [26]. Furthermore, their findings revealed that the overexpression of miR-4516 significantly inhibited the proliferation of TNBC cells by targeting the FOSL1 gene [26]. In addition, ectopic expression of miR-130a targets FOSL1 and suppresses invasion and migration in invasive breast cancer Hs578T and MDA-MB-231 cell lines. The overexpression of miR-130a led to an increase in ZO-1 expression, a tight-junction protein, through the downregulation of FOSL1 [24]. miR-19a-3p has also been implicated in the regulation of FOSL1 in tumor-associated macrophages (TAMs). The downregulation of miR-19a-3p results in the accumulation of FOSL1 in TAMs, which in turn promotes the invasion and metastasis of breast cancer cells [27]. 2) Colorectal cancer (CRC): The downregulation of miR34a/c and miR-15/16-family member miR-497 has been observed in CRC. This downregulation plays a significant role in promoting neoplastic cell invasion and EMT driven by FOSL1. These miRNAs are known to target FOSL1, leading to its upregulation and subsequently, the activation of genes involved in EMT and metastasis [28,29]. 3) Gallbladder carcinoma (GC): In gallbladder cancer, miR-195-5p has been identified as downregulated, and this downregulation promotes cell proliferation, migration, and invasion. It achieves this effect by targeting FOSL1, which, in turn, activates the Wnt/β-catenin signaling pathway [23]. In vitro investigations revealed that overexpression of miR-195-5p in NOZ and GBC-SD gallbladder carcinoma cell lines not only suppressed proliferation, invasion and migration but also induced cell cycle arrest in the G0/G1 phase. miR-195-5p exerts this function via targeting FOSL1 and inhibiting the expression of proteins including β-catenin, CCND1, and c-Myc that are involved in the Wnt signaling pathway [23]. 4) Squamous cell carcinoma (SCC): miR-138 is a tumor suppressor that regulates the expression of various oncogenes, including FOSL1. In squamous cell carcinoma, the down-regulation of FOSL1 by microRNA-138 may inhibit cell proliferation and invasion [30]. In this paper, Yi et al. reported that miR-138 binds directly to the 3’-UTR of FOSL1, leading to a decrease in its expression. Moreover, miR-138 was found to indirectly regulate FOSL1 target genes, including Snail2. Subsequently, Snail2 was observed to have effects on E-cadherin expression. They also discovered that miR-138 can bind not only to the 3’-UTR of FOSL1 but also to the 5’-UTR and coding region of FOSL1. In summary, the miR-138/ FOSL1/ Snail2 axis plays a pivotal role in the progression of squamous cell carcinoma [30]. 5) Glioma: FOSL1, functioning as a transcriptional factor, has exhibited upregulation in several tumors, including glioma. Rong et al. demonstrated that radiotherapy treatment leads to the induction of apoptosis and inhibition of cell viability in glioma cancer stem cell (CSC) lines CSC-U-251MG and CSC-A172. After radiotherapy, the expression of FOSL1, miR-27a-5p, and Bcl-2 remarkably decreased, while Bax and cleaved caspase-3 increased. Using bioinformatics tools, they have demonstrated that FOSL1 exhibits a significant signal in promoter region of miR-27a-5p. Remarkably, with the silencing of FOSL1 sensitizes glioma cancer stem cells through the downregulation of miR-27a-5p, whereas the ectopic expression of FOSL1 reduces the effectiveness of radiotherapy on apoptosis and cell viability by modulating miR-27a-5p [22]. (Figure 1).
2.2.2. LncRNA and FOSL1
Several studies reported the involvement of lncRNAs such as HOTAIR [21], AGAP2-AS1, LINC01503, and HOXA11-AS1 in the regulation of FOSL1 in cancer [19,20,31]. In our study, we found that lnc RNA HOTAIR acts as a competing endogenous RNA (ceRNA) by sponging miR-301a-3p and indirectly regulates FOSL1 expression [21] 1) GBM: TRPM7 is one of the members of channel TRP superfamily that have a pivotal role in malignancy. Our group reported that TRPM7 serves as a positive regulator of lncRNA HOTAIR and contributes to gliomagenesis. In glioblastoma, HOTAIR accelerates tumor progression by acting as a sponge for miR-301a-3p, leading to an increase in the expression of FOSL1, a key player in glioma progression. Silencing of HOTAIR results in increased miR-301a-3p expression, leading to the suppression of FOSL1 and subsequently reducing cell proliferation, invasion, and migration [21]. 2) In esophageal cancer (EC), AGAP2-AS1 sponges miR-195-5p subsequently upregulates FOSL1, which in turn increases the proliferation of cancerous cells, metastasis, and invasion. The Knockdown of AGAP2-AS1 leads to the promotion of miR-195-5p expression, which, in turn, inhibits FOSL1 expression, subsequently impeding tumor progression. Moreover, this knockdown also increases apoptosis and causes cell cycle arrest in the G0/G1 phase [32]. 3) In nasopharyngeal carcinoma (NPC), LINC01503 recruited splicing factor proline-and glutamine-rich (SFPQ) and colocalizes in the nucleus. They jointly bind to the promoter region of FOSL1 genes, thereby promoting its expression. It can be concluded that the LINC01503/SFPQ/FOSL1 axis is involved in different aspects of NPC development including cell proliferation, migration, invasion, tumor growth, and metastasis [33]. HOXA11-AS1 lncRNA plays a critical role in metastasis in hypopharyngeal squamous cell carcinoma (HSCC). It achieves this function by regulating the mRNA stability of FOSL1 through its binding to PTBP1, which acts as a key regulator of tumorigenesis. On the other hand, FOSL1 binds to the PD-L1 promoter, leading to increased PD-L1 expression and promoting immune escape. Taken together, the interaction between HOXA11-AS1, FOSL1, PTBP1, and PD-L1 forms a crucial axis that collectively enhances proliferation, metastasis, immune escape, and tumor growth [34].
2.3. Post-translational regulation
In addition to transcriptional regulation, post-translational modifications also play a crucial role in the regulation of FOSL1 accumulation. These modifications affect the half-life of the protein and involve processes such as phosphorylation, acetylation, and ubiquitination. The phosphorylation of FOSL1 by various kinases, such as ERK and JNK, can stabilize or destabilize the protein depending on the context of the signaling pathways involved. Acetylation of FOSL1 can increase its stability and transcriptional activity, while ubiquitination can target FOSL1 for degradation by the proteasome [14,35,36]. 1) FOSL1 Phosphorylation: FOSL1 has several phosphorylation sites at serine and threonine residues. ERK2 and Rsk1 kinases phosphorylate serine residues at positions S252 and S265, respectively, while PKC-theta phosphorylates threonine residues at positions T223 and T230 [4,37]. Phosphorylation at these specific sites protects FRA-1 from degradation in proteasomes and consequently stabilizes its expression levels. Furthermore, phosphorylation of threonine residues also enhances FOSL1’s biological activity, leading to increased transcriptional activation of downstream target genes [38,39]. For example, when Thr231 (T223) is substituted with Asp, it generates a dominant negative variant of FOSL1. This mutated form of FOSL1 loses its ability to activate FOSL1 target genes, including Mmp1 [39,40]. 2) FOSL1 acetylation/deacetylation: acetylation can also regulate the function of FOSL1. The IL6/STAT3 axis is involved in the deacetylation of FOSL1 lysine-116 residue by the deacetylase HDAC6 in CRCs. This modification results in the cancer cells acquiring stemness properties, which represents a critical step in the development and progression of cancer [41]. Our most recent studies demonstrated that deacetylation of FOSL1 at the Lys-16 residue located within its DNA binding domain leads to an increase in FOSL1 transcriptional activity[16].
3. The general roles of FOSL1 in glioma and other tumor types
The general roles of FOSL1 in cancer are diverse and significant. FOSL1 is known to promote tumor cell proliferation, survival, and invasion. It also plays a crucial role in EMT, which is essential for cancer cell migration and metastasis. Additionally, FOSL1 has been implicated in the regulation of genes involved in angiogenesis, immune evasion, and resistance to chemotherapy and targeted therapies. Hence, FOSL1 emerges as a key transcription factor contributing to various aspects of tumor development and progression in glioma and other tumor types (Table 1). FOSL1 has been extensively studied by various groups including our own, specifically for its significant role in promoting proneural (PN) to mesenchymal (MSN) states in gliomagenesis. It achieves this through pathways involving NF1 [42] and the UBC9/CYD/NF-κB axis [43] contributing to its widespread oncogenic roles. Even when EGFR and MET were inhibited, the suppression of FOSL1 phosphorylation was temporary, rebounding by 48 hours. Consequently, the rebound led to increased transcription levels of five FOSL1 target genes, SPRY2, MMP2, JUN, PLAUR, MMP1, and IL-6, contributing to resistance against kinase inhibitors [44,45,46,47,48,49,50]. FOSL1 serves as a clinical prognostic marker for glioma [16,21,51].

Table 1.
The role of FOS1 in different cancers.
| Cancer type | Clinical samples | Assessed cell line | FOSL1 expression | Effects: in vitro/ in vivo | Regulatory mechanism | Ref |
|---|---|---|---|---|---|---|
| BC |
51 paired tissues | MDA-MB-231, Hs578T | Up | ∆ miR34a/c: ↓Invasion, ↓Migration | miR-34a/c-Fra1 |
19 |
| 30 paired tissues | MDA-MB-231, MCF7, BT-474, SK-BR-3, BT-20, NF, CAF | UP | ∆ miR4516: ↓Proliferation | miR4516/FOSL1 | 20 | |
| 33 TNBC and 34 non-TNBC patients | MDA-MB-231, Hs578T | UP | ∆ miR-130a: ↑Tight-junction protein, ↓Invasion, ↓Migration | miR-130a/ FOSL1/ ZO1 | 18 | |
| - | 4T1, 4T07, RAW264.7 | UP | ∆ miR-19a-3p: ↓Metastasis, ↓Invasion, ↓Migration, ↑Macrophage polarization, ↓Tumor growth | miR-19a-3p /Fra-1/STAT3 | 21 | |
| MCF7, 4T1, ZR-75-1 | UP | ∆ FOSL1: Invasion ↓, Migration ↓ | MLK3–FRA-1–MMP-1 | 50 | ||
| MDA-MB-231, BT549 | UP | ∆ FOSL1: Invasion ↓, Migration ↓ | ERK1/2, SPAK | 51 | ||
| MDA-MB-231, MDA-MB-436, MCF-7 | UP | ∆ FOSL1: Invasion ↓, Migration ↓ | HMGA1 | 53 | ||
| fourth node mice | BRC-17, BRC-31, BRC-32, BRC-36, BRC- | UP | ∆ FOSL1: Invasion ↓, Migration ↓ | Integrin-uPAR | 53 | |
| 4T1 | UP | ∆ FOSL1: Invasion ↓, Migration ↓ | IL-6/JAK/Stat3 | 60 | ||
| CRC |
- | CRL-1831, LoVo, RKO, HCT15, HCT28, HCT116, and SW480 | UP | ∆ miR-497: ↓EMT, ↓Migration, ↓Invasion | miR-497/ Fra-1 | 27 |
| 40 paired tissues | HEK293T, RKO, HCT116, HCT116 | UP | ▼Fra-1 or ∆ miR-34a: ↓Migration, ↓Invasion | miR-34a/p53/ Fra-1 | 26 | |
| HCT116 |
UP | ∆ FOSL1: Invasion ↓, Migration | miR-34a/Fra1 |
|||
| HT-29, HCT116 |
UP | ∆ FOSL1: Invasion ↓, Migration ↓ |
USP21 |
47 | ||
| SW620, HCT116, DLD1 HT29 |
UP | ∆ FOSL1: Invasion ↓, Migration ↓ | RAS | 48 | ||
| paired tissues, PDX mouse model |
UP |
∆ FOSL1: Proliferation↓, Invasion ↓, Migration ↓, |
EGFRMAPK-FRA-1 |
49 | ||
| GC | 20 paired tissues |
AGS | UP | ∆ FOSL1: Proliferation↓, apoptosis | PI3K/Akt ,p53 |
35 |
| NPC |
20 NPC and 16 non-NPC patients | NP69, N2-Tert, C666-1, CNE1, CNE2, HK1, HNE1, HONE1, SUNE1, 5–8F and 6–10B | UP | ▼LINC01503: ↓Proliferation, ↓Invasion , ↓Migration , ↓Metastasis, ↓Tumor growth | LINC01503/ SFPQ/ FOSL1 | 24 |
| 53 paired tissues | CNE1 | UP | ∆ FOSL1: Proliferation↓, Growth tumors ↓ | MSK1, LMP1 | 38 | |
| LC | 55 paired tissues | H460 | UP | ∆ FOSL1: apoptosis↓ | p53 |
40 |
| OS | Saos2 , MG63 | UP | ∆ FOSL1: Proliferation↓, Invasion ↓, Migration ↓, |
ERK/AP-1 |
37 | |
| GBC | - | NOZ , GBC-SD | UP | ∆ miR-195-5p: ↓Proliferation, ↑G0/G1 phase , ↓Invasion , ↓Migration , ↓Tumor growth | miR-195-5p /FOSL1/Wnt pathway | 17 |
| TC |
FRTL-5K-Ra | UP | ∆ FOSL1: Proliferation↓ |
cyclin A, Jun B |
38 | |
| paired tissues | BHP10-3, BCPAP, SNU790 | UP | ∆ FOSL1: Invasion ↓, Migration ↓ | ERK/Fra-1/ZEB1 | 42 | |
| HSCC | 40 paired tissues | FaDu, Detroit 562, NP69 | UP | ▼HOXA11-AS1: ↓Immune escape, ↓ Metastasis, ↓Proliferation, ↓Tumor growth | HOXA11-AS1/FOSL1/PTBP1/PD-L1 | 25 |
| EC | 53 paired tissues | E70, KYSE-510, EC9706, HEECs | UP | ▼AGAP2-AS1: ↓Proliferation, ↓Invasion , ↓Migration , ↑Apoptosis, ↑G0/G1 phase, ↓Tumor growth | AGAP2-AS1/ miR-195-5p/FOSL1 | 23 |
| Glioma | - | A172, T98G, U87MG | UP | ▼FOSL1: ↑Apoptosis, ↓Tumor growth | FOSL1/ miR-28-5p | 16 |
| 22 | U251 | UP | miR-33a/FOSL1 | 51 |
(∆: Over-expression, ▼: Knockdown). BC: Breast cancer, CRC: Colorectal cancer, GBC: Gallbladder carcinoma, HSCC: Hypopharyngeal squamous cell carcinoma, NPC: Nasopharyngeal carcinoma, PC: Prostate cancer, EC: Esophageal cancer, GBM: Glioblastoma, HNSCC: Head and neck squamous cell carcinoma, TNBC: Triple-negative breast cancer, TC Thyroid Cancer, OS Osteosarcoma, LC Lung Cancer, GC Gastric Cancer.
3.1. The role of FOSL1 in apoptosis and proliferation and its associated signaling pathways.
FOSL1 is a transcription factor that can either promote or inhibit apoptosis and proliferation depending on the cellular context and signaling pathway interactions. Many tumor types have overexpression of FOSL1. CRISPR-induced FOSL1 transcription led to FOSL1 overexpression, causing a decrease in cell adhesion to fibronectin and collagen, consequently impacting cell cycle progression [52]. Its overexpression contributes to tumor progression by regulating several cell signaling pathways. In GC, FOSL1 has been shown to inhibit apoptosis and promote cell proliferation. The upregulation of FOSL1 in GC cells leads to the activation of the PI3K/AKT pathway and the upregulation of MDM2. MDM2, in turn, inhibits the activity of the p53 tumor suppressor gene, which plays a key role in regulating apoptosis and cell cycle arrest. Taken together, the overexpression of FOSL1 in GC cells leads to the inhibition of apoptosis and the promotion of cell proliferation by activating the PI3K/AKT pathway and inhibiting the activity of the p53 tumor suppressor gene [53]. Silencing FOSL1 leads to the suppression of the ERK/AP-1 signaling pathway, effectively inhibiting osteosarcoma (OS) cell proliferation, invasion, and migration [54]. In nasopharyngeal carcinoma cells (CNE1), there is an upregulation of mitogen and stress-activated kinase 1 (MSK1) expression. Furthermore, the induction of EBV latent membrane protein 1 (LMP1) leads to an increase in phosphorylation of histone H3 at Ser10. These mechanisms are believed to mediate the effects of FOSL1 on cell proliferation and apoptosis inhibition in these cancer cells [55]. The FOSL1 gene is commonly found to be overexpressed in thyroid cell transformations. When FOSL1 is knocked out, cell proliferation is arrested, leading to apoptosis, the majority of cells accumulating in the G2 phase, while a few undergo abnormal cell division. FOSL1 binds to previously unidentified AP-1 site and CRE on the cyclin A promoter during the cell cycle, promoting cell proliferation [56]. Additionally, FOSL1 induces the expression of Jun B, which interacts with the cyclin A promoter, subsequently enhancing cell growth and survival in RAS-transformed thyroid cells[57]. Therefore, FOSL1 is a critical regulator of cell cycle progression [58]. Psoralen has been discovered to display antiproliferative effects on breast cancer cells due to its ability to increase the expression of Axin2 and decrease the expression of FOSL1, suggesting that psoralen’s mechanism of action in breast cancer involves the regulation of the cell cycle by modulating the expression of Axin2 and FOSL1 [59]. Breast cancer cells, specifically MCF-7 and MDA-MB-231, have high FOSL1 levels, enhancing their proliferative and metastatic ability [60,61].
The effects of FOSL1 on apoptosis are contradictory in different types of cancer. 1) Studies have shown that in cervical cancer, the expression of FOSL1 is reduced in tumor cells compared to normal tissue. Consequently, downregulation of MDM2 and consequent p53 accumulation favors apoptosis, indicating that FOSL1 functions as a pro-apoptotic factor [10,62]. The reasons for these inconsistencies remain unclear. 2) FOSL1 is significantly upregulated in lung cancer. Studies have shown that lung cancer cells with elevated FOSL1 expression demonstrated reduced apoptosis rates compared to control cells. The decline in apoptosis is closely related to a decrease in mitochondrial membrane potential (Δψm) and an increase in intracellular reactive oxygen species (ROS) and calcium ion (Ca2+) concentration. Conversely, overexpression of FOSL1 leads to an increase in mitochondrial membrane potential, a decrease in intracellular ROS and Ca2+ concentration, and ultimately leads to the inhibition of apoptosis in lung cancer cells [63]. Yet, it’s crucial to approach the data explanation cautiously as the study solely relied on the Hela cell line [64] or H460 lung cancer cell line [65] and included data from only 20 [64] or 10 [65] glioma patients. Therefore, deriving a broad conclusion about all cervical cancers from this paper might be challenging. Studying the pathways implicated in cancer development is of great value as it helps identify potential targets for therapy, which is crucial in effectively managing medical conditions.
3.2. The role of FOSL1 in migration and its associated signaling pathways.
FOSL1 plays a pivotal role in migration, and its mechanisms within the signaling pathways are crucial to understanding its function. FOSL1 is responsible for inducing changes in the structure and arrangement of the cytoskeleton, loss of polarization in epithelial cells, increased ability to move, and invasion of tumor cells. These changes indicate different degrees of mesenchymal transformation that are dependent on the context, ranging from partial to complete EMT [66]. Overexpression of FOSL1 in immortalized epithelial cells leads to a suppression of epithelial markers, such as cadherin E (Cdh1), and induction of endothelial markers, such as vimentin (Vim), Ascdh2, fibronectin (Fn1), Cdh3, S100a4, and Spp1. Additionally, FOSL1 overexpression leads to an upregulation of the genes encoding integrins 5 and 1 (Itgba5 and Itgb1, respectively) [67]. Cells with silenced FOSL1 exhibit the opposite effects compared to cells with FOSL1 overexpression. Silenced FOSL1 leads to the downregulation of genes associated with the mesenchymal phenotype, including Snail, Slug/Snai2, Zeb1, and Zeb2. These genes are transcription factors that promote the process of EMT [67,68].
3.2.1. CRC
FOSL1 expression levels are associated with the extent of local invasion, lymph node involvement, and liver metastases in CRC patients. The pro-inflammatory cytokine, interleukin-6 (IL-6), is a crucial factor in the development and progression of CRC. In CRC cell lines, activation of STAT3 during IL-6-mediated EMT promotes the upregulation of FOSL1 gene expression, which in turn induces the expression of EMT-promoting factors (ZEB1, Snail, Slug, MMP-2, and MMP-9), leading to increased invasiveness of CRC cells. Hence, the IL-6/STAT3/FOSL1 signaling axis triggers EMT, thereby promoting the invasiveness of CRC [69]. In another study, it was discovered that FOSL1 downregulates the expression of FBXL2, an E3 ligase complex known for its role in inhibiting tumorigenesis by ubiquitinating Aurora B. The downregulation of FBXL2 is believed to be facilitated by FOSL1 through the regulation of the transcriptional activities of Smurf1. These events are thought to be mediated by the Wnt/β-catenin signaling pathway, implying that targeting FOSL1 using shRNA could be a promising therapeutic approach for treating CRC [70]. MicroRNA-34a (miR-34a) is a tumor suppressor gene and a transcriptional target of p53, which plays a crucial role in inhibiting the migration and invasion of colon cancer cells. This is achieved through the upregulation of miR-34a by p53 DNA activity, leading to the downregulation of FOSL1 expression, which in turn reduces the expression of matrix metalloproteinases MMP-1 [71] . The stability of FOSL1 in CRC is increased by the action of ubiquitin-specific protease 21 (USP21), which removes ubiquitin molecules from FOSL1, leading to enhanced expression of genes targeted by FOSL1. Additionally, USP21 plays a role in controlling the migration and invasion activities of CRCs through its effect on FOSL1 [72]. The overexpression of FOSL1 may regulate vimentin during the EMT induced by Ha-RAS and contribute to colon cell migration. These findings suggest that in colon cells, the upregulation of proteins such as FOSL1 and vimentin may play a role in inducing epithelial-mesenchymal transition through Ha-RAS oncogenic signaling [73]. PDAP1 plays a role in both mucosal restitution and carcinogenesis in colitis-associated cancer. The upregulation of PDAP1, driven by c-Myc, promotes the proliferation, migration, invasion, and metastasis of CRC cells through the EGFR-MAPK-FOSL1 signaling axis. Therefore, inhibiting PDAP1 may be beneficial for patients with CRC who have high levels of PDAP1 expression [74].
3.2.2. Breast cancer
FOSL1 has the potential to undergo phosphorylation by various kinases, including mitogen-activated protein kinase (MAPK), protein kinase C (PKC), cAMP-dependent kinase (PKA), and cyclin-dependent kinase 1-cdc2 (CDC2). The continuous stimulation of the MAPK/extracellular signal-regulated kinase (ERK) signaling pathway triggers the migration of breast cancer cells [38]. MLK3 expression induced a significant increase in the oncogenic transcription factor FOSL1, along with elevated levels of MMP-1 and MMP-9, in non-metastatic ER+ BC cells. This increase in invasion was found to be dependent on FOSL1, as silencing FOSL1 reversed the effect of MLK3. In contrast, high levels of FOSL1 in metastatic TNBC were reduced upon MLK3 depletion through gene silencing or CRISPR/Cas9n editing, and both MMP-1 and MMP-9 expression were reduced by inhibiting MLK3 or using an MLK inhibitor [75]. SPAK STE20-related proline/alanine-rich kinase SPAK and ERK1/2 participate in the stabilization of FOSL1 induced by PKCθ, but their involvement varies among different cell lines. The ERK1/2 pathway primarily contributes to the accumulation of FRA-1/FOSL1 in ER-MDA-MB-231 cells, whereas in ER-BT549 cells, SPAK signaling is the main mechanism responsible for the accumulation of FRA-1/FOSL1. PKCθ signaling is a crucial regulator of Fra-1/FOSL1 accumulation in ER-negative breast cancer cells. Furthermore, Fra-1/FOSL1 is essential for PKCθ-induced cell migration and invasion. Taken together, these results indicate that PKCθ may play a role in the progression of certain breast cancers and could potentially serve as a novel therapeutic target [75,76]. FOSL1 plays a role in regulating the expression of High Mobility Group A1(HMGA1) mRNA at the transcriptional level by binding to enhancer elements in breast cancer cells. This promotes the invasion and migration of triple-negative breast cancer cells through RNA polymerase II recruitment [77]. FOSL1 also has other signaling pathways that induce invasion and migration of breast cancer cells, such as the increased expression of integrins αVβ3 and uPAR activating the FAK-SRC-ERK2 signal, leading to increased phosphorylation and activation of FOSL1[78].
3.2.3. Other cancers
ABCA1 plays a role in regulating EMT through the ERK/FOSL1/ZEB1 pathway and has been identified as a new predictor of response to thyroid cancer with lung metastasis. These discoveries highlight the potential of ABCA1 as an important oncogene and a promising therapeutic target for the treatment of thyroid cancer (TC) with lung metastasis [79]. High expression of FOSL1 was found to be significantly linked to positive lymphatic invasion in GC. This finding suggests that FOSL1 might play a crucial part in the migration and invasion of GC cells, but more research is required to confirm this hypothesis [80]. The loss of the neurofibromatosis type 1 gene (NF1) leads to an increase in RAS/MAPK activity, causing changes in the expression of FOSL1. Deletion of FOSL1 prompts a change from a mesenchymal to a proneural transcriptional signature, and results in a reduction of both stemness and tumor growth. The up-regulation of FOSL1 expression and induction of the EMT program in glioma cells can be attributed to the activation of Wnt/β-catenin signaling. This signaling pathway activates the transcription of FOSL1 by direct binding of β-catenin/Tcf/Lef transcriptional complex to the Wnt response element (WRE) situated in the proximal region of the FOSL1 promoter[81] (Figure 2).
3.3. The involvement of FOSL1 in the tumor microenvironment.
The interaction between TAMs and neoplastic cells within the tumor microenvironment (TME) including stromal cells, tumor cells, fibroblasts, endothelial cells, cytokines, and others, plays a critical role in promoting tumor cell invasion and progression [82,83]. The role of macrophages in cancer is not completely clear, as the M1 phenotype is associated with the ability to kill tumors, while the M2 phenotype of TAMs has a tumor-promoting effect. Interactions between mouse breast tumor cells and TAMs can change the tumor TME, causing an increase in the expression of FOSL1. The function of the FOSL1 proto-oncogene extends to regulate the expression of IL-6 in macrophages, leading to the development of M2d macrophages [84]. FOSL1 triggers the activation of the IL-6/JAK/Stat3 signaling pathway, resulting in a malignant switch in breast tumor cells that leads to increased release of pro-angiogenic factors such as MMP-9, VEGF, and TGF-β from the tumor cells. Consequently, the activated transcription factor FOSL1 and the IL-6/JAK/Stat3 signaling pathway play a significant role in altering the TME [85].
FOSL1 is highly concentrated in cancer-associated fibroblasts (CAFs) obtained from colorectal cancer (CRC) tissues. Moreover, FOSL1 can be transferred from CAFs to CRC cells via exosomes, which leads to increased CRC cell proliferation and resistance to oxaliplatin. In addition, FOSL1 promotes the expression of integrin β4 (ITGB4) by binding directly to its promoter region [86]. Tumor-initiating cells (T-ICs) in liver cancer are influenced by factors from the TME, just like normal stem cells. Since liver cancer often develops in the context of cirrhosis, which involves activated fibroblasts, if CAFs regulate T-ICs researchers were widely investigated in the liver. The presence of a-SMA (+) CAFs is linked to poor clinical outcomes. CAFs produce HGF, which activates FOSL1 in T-ICs through the Erk1,2 pathway. Further analysis revealed that HEY1 is a direct downstream mediator of FOSL1. In a mouse model of liver cancer, HGF-induced FOSL1 activation is associated with fibrosis-related HCC development. Therefore, targeting the CAF-derived, HGF-mediated c-Met/FOSL1/HEY1 pathway could be a promising therapeutic strategy for treating HCC [87]. miR-19a-3p, responsible for regulating TAMs within the breast TME, could influence the phenotype of TAMs by targeting the FOSL1 gene and other genes within its downstream signaling pathways. The decreased expression of miR-19a-3p in TAMs is likely caused by TME induction, which encourages the shift from M1 to M2 and subsequently enhances the migration and invasion of breast cancer cells [27].
3.4. Cancer stem cells
Cancer stem-like cells possess the ability to self-renew and play significant roles in cancer growth, treatment resistance, and metastasis. Among these cells, glioblastoma stem-like cells have been the subject of the most extensive research.
FOSL1 can transform mature cancer cells into stem-like cells that can propagate the tumor by controlling transcription factors related to stemness [88]. FOSL1 regulates SOX2, SALL2, OLIG2, and POU3F2 by binding to their promoter regions and influences a number of stemness indicators [2]. It also reduces the ability of cells to form aggregates by increasing FN1 in the extracellular matrix. FOSL1 changes stemness by regulating these four transcription factors [89].
The activation of the EMT program during malignant progression can lead to the enrichment of cancer stem cells (CSCs). Inhibiting protein kinase C a (PKCa) can specifically target CSCs with little effect on non-CSCs. The conversion of non-stem cells into CSCs involves a change from EGFR to PDGFR signaling and triggers PKCa-dependent activation of FOSL1. An AP-1 molecular switch has been identified where c-FOS and FOSL1 are used differently in non-CSCs and CSCs. PKCa and FOSL1 expression is linked to aggressive triple-negative breast cancers, and reducing FOSL1 levels leads to a mesenchymal-epithelial transition. Identifying molecular features that shift between cell states can be used to target critical signaling components in CSCs [90]. The transition from proneural (PN) to mesenchymal (MES) in glioblastoma stem cells (GSCs) is an important shift. FOSL1 plays a key role in regulating this transition. FOSL1 is mainly expressed in the MES subtype of GSCs and not in the PN subtype. Knocking down FOSL1 expression in MES GSCs results in the loss of MES characteristics and tumorigenicity, while overexpression of FOSL1 in PN GSCs promotes the PN to MES transition and maintains MES characteristics.
FOSL1 also facilitates the induction of the transition to the mesenchymal state by ionizing radiation (IR), leading to an increase in radio resistance in PN GSCs. Blocking FOSL1 can improve the effectiveness of radiotherapy against tumors by preventing the PMT triggered by irradiation. Mechanistically, through the modulation of the IKKb-IkBa-NF-kB signaling pathway, FOSL1 can induce the process of PMT. The introduction of FOSL1 in PN GSCs leads to multiple processes such as the phosphorylation of IKKb on Ser181, degradation of IkBa, and relocation of NF-kB p65 to the nucleus. [91]. In the presence of inflammation and IL-6 secretion in the microenvironment, the activation of the STAT3 pathway occurs. This activation, through transcriptional (promoter binding) and post-translational (K116 deacetylation) upregulation of FOSL1/FOSL1, promotes the stemness and malignancy of CRC. HDAC6 is responsible for deacetylating the Lys-116 residue in FOSL1. The initiation of the EMT program in CRC cells can be triggered by the pro-inflammatory cytokine IL-6 stimulation. Further investigation into the mechanism demonstrated that IL-6 induced the transactivation of FOSL1 by activating the phosphorylation and acetylation of STAT3 simultaneously. Clinical sample analyses provided evidence that FOSL1, which is aberrantly induced by IL-6/STAT3, plays a crucial role in mediating the EMT and aggressiveness of CRC. In addition to promoting EMT features, IL-6 induces stem-like properties in colorectal cancer cells, such as the expression of CSC markers CD44+/CD133+, the formation of spheroids, and increased drug-resistance, through FOSL1-dependent pathways. These pathways involve the transactivation of the stemness master gene NANOG, which is mediated by the binding of the K116-deacetylated FOSL1 to the NANOG promoter [92].
In summary, FOSL1 is emerging as a key regulator of CSCs in several cancer types. Their dysregulation contributes to CSC self-renewal, survival, and resistance to therapy. Therefore, targeting FOSL1 and their downstream effectors represents a promising approach to preventing tumor recurrence and improving patient outcomes.
4. FOSL1 serves as an independent and prognostic factor
The lack of reliable prognostic biomarkers for certain tumors has led to a significant demand for novel and effective therapeutic targets for cancer patients. Additionally, there is a growing interest in developing new prognostic indicators for patients. FOSL1 holds promise as a prognostic tool, as tumors with increased FOSL1 levels are often associated with aggressive cancer forms. In addition, overexpression of FOSL1 can result in chemotherapy resistance [93]. Mutated KRAS activation is a primary cause of lung cancer deaths with inadequate knowledge of its downstream targets and growth mechanisms of tumor cells. FOSL1 was identified as a crucial factor in regulating the survival and proliferation of human lung adenocarcinoma cells, particularly in the presence of KRAS mutations. Therefore, FOSL1 activation may serve as a potential target for human lung cancer therapy and a prognostic marker, particularly in cases with KRAS mutations [94]. FOSL1/AP-1 transcription factor plays a role in causing lung damage and death in mice when exposed to LPS. FOSL1 achieves this by controlling the expression of proinflammatory cytokine genes through the regulation of c-Jun and NF-kB signaling. This study highlights the potential of targeting FOSL1 to reduce lung injury and inflammation in patients with acute lung injury and adult respiratory distress syndrome, as well as to minimize the impact of endotoxin exposure in clinical settings [95].
FOSL1 expression was found to be increased in both CRC tissues and cells and was strongly associated with a higher risk of lymph node metastasis, advanced TNM staging, and poorer tumor differentiation. Additionally, there was a demonstrated correlation between FOSL1 expression and lymphatic invasion, TNM stage, and lower 5-year survival rates in patients with gastric adenocarcinoma [70]. Depletion of FOSL1 results in a significant reduction of tumor burden, up to 200-fold. Additionally, a FOSL1 classifier was created by comparing RNA profiles of CRC cells that were either parental or FOSL1-depleted. This classifier can predict the prognosis of colon cancer patients. Through functional pathway analysis, it was discovered that the Wnt pathway is central in the classifier, indicating a possible mechanism of how FOSL1 functions in colon cancer metastasis. These findings demonstrate that FOSL1 plays a crucial role in determining the metastatic potential of human colon cancer cells. Moreover, the FOSL1 classifier has the potential to serve as a prognostic predictor for colon cancer patients [96].
The significance of FOSL1 as a prognostic factor for patients with ER(+) breast cancer is highlighted, and it is suggested that FOSL1 has distinct roles in breast tumor cells depending on the ER status. In addition to its previously known effects on promoting cell proliferation and invasion, FOSL1 may also impact breast cancer progression by modulating the adhesion of tumor cells to endothelial surfaces [97]. Recent research has shown FOSL1 to be a valuable clinical outcome indicator for breast cancer patients who are ER-negative. The study revealed that patients with higher levels of FOSL1 expression had shorter overall survival rates, although this correlation was not observed in those with ER-positive breast cancer. Additionally, the study discovered that TNBC cells had higher levels of FOSL1 expression than ER-positive cells due to more enhancers present in the FOSL1 sequence in TNBC cells. The reliance on copy number alterations (CNAs) in the FOSL1 gene was noticeably higher in the TNBC subtype compared to luminal tumors, indicating a link with more aggressive phenotypes of breast tumors.
The risk of developing oral squamous cell carcinoma (OSCC) is influenced by the transcription factors c-Jun and FOSL1, which also play a significant role in the cancer’s progression. A poor prognosis is linked to the high expression of c-Jun or FOSL1, with FOSL1 expression resulting in a worse prognosis than c-Jun expression. The interaction between c-Jun and FOSL1 is antagonistic, implying that their activities may impact each other. Both c-Jun and FOSL1 are high-risk predictors of death from OSCC. These findings offer a new perspective on the prognosis and treatment of OSCC [98]. To promote tumorigenicity and metastasis, FOSL1 selectively associates with mediators to establish super-enhancers (SEs) at various cancer stemness and pro-metastatic genes, such as SNAI2 and FOSL1 itself. FOSL1 depletion disrupts SEs and inhibits the expression of these critical oncogenes, leading to tumor initiation and metastasis suppression. Additionally, a positive correlation between FOSL1 and SNAI2 abundance in HNSCC, and high expression levels of FOSL1 and SNAI2 corresponded with shorter overall and disease-free survival. These results demonstrate that FOSL1 functions as a master regulator in promoting HNSCC metastasis through SE-driven transcription and is a promising therapeutic target [99]. Knocking down FOSL1 significantly decreased cell adhesion and migration, while overexpressing an active FOSL1 mutant (FOSL1DD) increased these processes in a JNK/c-Jun-dependent manner. KIND1 was identified as a cytoskeletal regulator of the cell adhesion molecule β1-integrin, as a new transcriptional target of FOSL1. The study also found that recovery of migratory defects that resulted from the loss of FOSL1 was achieved by restoring the expression of KIND1. In line with the in vitro data, HNSCC cells with FOSL1 loss showed decreased rates of subcutaneous tumor growth and pulmonary metastasis. The findings suggest that FOSL1 has a role in promoting cancer growth via AKT and enhancing cancer cell migration through JNK/c-Jun, identifying FOSL1 as a significant integrator of JNK and AKT signaling pathways, and a promising therapeutic target for treating cSCC and HNSCC [100].
The outlook for individuals with hepatocellular carcinoma (HCC) is very poor, and there are only a few drugs available that specifically target this type of cancer. Despite the application of new techniques such as high-throughput sequencing, there has been little progress in the identification of prognostic biomarkers for HCC. FOSL1 expression was analyzed in 20 pairs of fresh HCC tissues and 114 paraffin-embedded HCC tissues. The findings indicated that there is a strong correlation between high FOSL1 expression and larger tumor size, advanced T stage, and lower survival rates. Elevated levels of FOSL1 are a significant predictor of poor prognosis in HCC patients and serve as an independent risk factor, suggesting that detecting FOSL1 expression can help identify high-risk patients, and targeting FOSL1 could be a viable therapeutic strategy for HCC treatment [101].
FOSL1 plays a crucial role in maintaining stem cell properties of the MES subtype of GBM, making it an attractive target for therapy. Although inhibiting FOSL1/FOSL1 pharmacologically may be challenging due to its oncogenic function, targeting therapy using techniques such as CRISPR/Cas9 or PROTAC that target FOSL1/FOSL1 expression could be a promising strategy to treat patients with mesenchymal GBM [102]. In our previous publication, the TCGA database, which contained information on 454 glioblastoma patients, classified based on WHO classification as GBM, was analyzed for the clinical significance of FOSL1 mRNA expression. FOSL1 mRNA expression levels in GBM patients were significantly higher than those in normal individuals as detected by Affymetrix HT HG U133A; furthermore, classical, mesenchymal, and neural molecular subtypes in glioma tissue were higher than those in the normal brain (p<0.05). Although a similar tendency was displayed in the proneural subtype, it did not reach a significant difference. To determine whether FOSL1 gene expression is related to patient survival, all GBM patients from The Human Protein Atlas were analyzed in a pooled setting; the 7-year overall survival (OS) rate, as revealed by the Kaplan-Meier survival curves, was significantly higher (p<0.001; log-rank test) in those with low FOSL1 transcript levels compared with those with high expression [16]. Recently, several new binding partners of FOSL1were identified in human Th17 cells, which include HDAC2, XRN1, AP2A1, PCBP1, ILF3, TRIM21, and HNRNPH1 [103]. Using Affymetrix HT HG U133A data from TCGA, we observed that TRIM21 is associated with an unfavorable outcome, whereas HDAC2, PCBP1, and HNRNPH1 are favorable factors for glioma patients’ survival.
Taken together, FOSL1 is a prognostic factor in various types of cancer, including breast, lung, liver, and colorectal cancer. Its expression has been associated with tumor progression and poor prognosis, and it has been identified as an independent prognostic factor in several studies. Cancer drug resistance is a key factor in the failure of efficacy treatment with chemotherapy agents. Increased resistance to chemotherapy agents in cancer cells has several reasons, including CSC, multidrug resistance transporter 1(MDR1), defect in DNA repair, ABC transporters, and the elevated expression of anti-apoptotic proteins [104,105,106]. Past studies report that transcription factors such as FOSL1 have a pivotal role in resistance to chemotherapy agents [105]. There are several reasons why FOSL1 can serve as a useful prognostic tool. Firstly, solid tumors with a higher grade usually show intense staining for FOSL1. Secondly, aggressive forms of cancer are often associated with tumors that have higher levels of FOSL1. Lastly, increased expression of FOSL1 has been linked to resistance to chemotherapy. By monitoring the levels of FOSL1, it could be possible to determine the grade of the tumor [107].
5. The role of FOSL1 in drug resistance
It has been reported that the expression levels of FOSL1 in drug-resistant breast cancer cells, specifically MCF-7/ADR and MDA-MB-231/ADR, remarkably increase, leading to the induction of resistance to doxorubicin in vitro and in vivo. They revealed that FOSL1 promotes the expression of dual specificity phosphatase 7 (DUSP7) via binding to its promoter. Subsequently, FOSL1 through DUSP7 induces dephosphorylation of PEA15 (proliferation and apoptosis adaptor protein 15). Dephosphorylated PEA15 promotes cell proliferation, invasion, and migration while inhibiting apoptosis. Taken together, the FOSL1/DUSP7/DEA15 axis promotes drug resistance in breast cancer [108]. In another study, it was reported that breast cancer patients who were treated with epirubicin-based neoadjuvant chemotherapy (NCT) showed a significant suppression in the expression levels of FOSL1. Duan et al. reported that the levels of FOSL1 in NCT-sensitive patients were significantly lower than those in NCT-resistant patients. Knockdown of FOSL1 in the MCF-7/ADR cancerous cell line sensitized the cells to doxorubicin, resulting in decreased cell proliferation and cell growth [109]. Dan et al. paradoxically reported that there is a negative correlation between the expression of FOSL1 and clinical chemoresistance in breast cancer patients. They indicated that the recurrence of the tumor is delayed when the expression of FOSL1 is increased. They demonstrated that silencing FOSL1 expression led to a significant increase in the population of CSCs, while ectopic expression of FOSL1 resulted in a decrease in the CSC population. Bothe in vitro and in vivo data revealed that FOSL1 enhances chemosensitivity to doxorubicin and cyclophosphamide by driving CSCs from dormancy [110].
Wu et al. reveal that the axis involving FOSL1– miR-134–SDS22- JNK/ERK enhances drug resistance in ovarian cancerous cells. They demonstrated that FOSL1 induces miR-134 expression while suppressing SDS22 expression, as SDS22 is one of the targets of miR-134. Consequently, this leads to an increase in the activity of JNK/ERK signaling pathway, which further promotes FOSL1 expression. Taken together it can be concluded that FOSL1– miR-134 axis drives a positive feedback loop, promoting ERK/JNK signaling and enhancing chemoresistance in ovarian cancer cells [111].
Cancer-associated fibroblasts (CAFs) constitute a significant portion of cells present Fin the TEM. Lin and Zhu reported that exosomes derived from CAFs isolated from CRC stromal cells are enriched with FOSL1. Upon transferring these to CRC cells, they observed a significant increase in stemness, cell proliferation, and resistance to oxaliplatin. FOSL1 facilitates these effects by promoting integrin β4 (ITGB4) transcription [112].
After subjecting colorectal cancer cell lines (SW480 and SW620) to X-ray or c-ion radiation, the expression level of FOSL1 remarkably decreases. Endo et al. reported that the sensitivity of cancerous cells to radiation increases after knockdown of FOSL1 [113]. In pancreatic ductal adenocarcinoma (PDAC) cell lines, Bosutinib, a small molecule inhibitor, when used alone and in combination with chemotherapy agent (5′-fluorouracil and gemcitabine), decreases proliferation and tumorigenesis of cancerous cells. Bosutinib sensitizes cancer cell lines to 5′-fluorouracil and gemcitabine by disrupting the Src-FOSL1 axis, resulting in decreased expression of mucins (MUC4 and MUC5AC) [114].
In HCC cancer cells, ectopic expression of αB-Crystallin promotes resistance to sorafenib by inducing EMT and subsequently activation of the ERK1/2/FOSL1/slug signaling pathway [115]. Inhibition of the PI3K signaling pathway decreases the expression of FOSL1 as an AP-1 subunit and enhances sensitivity to γ-radiation in prostate cancer cells [116]. In cervical cancer stem-like cells (CaCxSLCs), there was an increased expression of AP-1 family members (c-Fos, c-Jun, Jun B, and Jun D) at both proteins and mRNA levels, but the FOSL1 transcript was lacking. Upon exposure of CaCxSLCs to UV-irradiation, the expression level of c-Fos and c-Jun remarkably increased, resulting in radioresistance. However, pre-treatment of CaCxSLCs with curcumin followed by UV-irradiation led to the suppression of c-Fos and c-Jun while upregulating FOSL1. Targeting the AP-1 family sensitized CaCxSLCs to UV-irradiation by increasing FOSL1 [106]. In summary, targeting FOSL1 has emerged as a potential strategy for overcoming drug resistance in cancer. Its overexpression is associated with poor prognosis and resistance to chemotherapy and targeted therapies.
FOSL1 has emerged as a potential contributor to GBM resistance against erlotinib, an EGFR tyrosine kinase inhibitor. This identification suggests that targeting FOSL1 could enhance the effectiveness of erlotinib in combating GBMs [117]. when both EGFR and MET were inhibited, the transient suppression of FOSL1 phosphorylation was observed, followed by a rebound effect, leading to elevated transcription levels of five FOSL1 target genes [44,45,46,47,48,49,50]: SPRY2, MMP2, JUN, PLAUR, MMP1, and IL-6. This cascade contributed to resistance against kinase inhibitors. Our unpublished data showed that silencing FOSL1 potentiates GBM cell response to TMZ. Having established that FOSL1 signaling is active in GBM cell models and contributes to cell proliferation, we then asked whether FOSL1 activity plays a role in cellular resistance to TMZ. To test this, U87MG (sensitive to TMZ) and T98G cells (resistant to TMZ) were transiently transfected with siFOSL1 or siCtrl with a range of TMZ doses (0-500 µM). Treatment efficacy was assessed using an MTT assay to measure cell viability 72 h after transfection. A dose-response experiment showed TMZ produced an additive decrease in cell viability with siFOSL1 for all TMZ doses tested in TMZ-sensitive U87MG cells. In TMZ-resistant T98G cells, siFOSL1 alone had a strong inhibitory effect, while the addition of TMZ had a more inhibitory effect. A further 4-day time-course experiment of T98G cells treated with the highest dose of TMZ (500 µM) confirmed additive effects on cell proliferation in a time-dependent pattern.
6. Conclusions
FOSL1, a transcription factor from the activator protein-1 (AP-1) superfamily, plays a significant role in various cancers by promoting tumor growth, survival, differentiation, and invasion. Its expression level is high in many cancer cells and correlates with the severity of the disease and patient survival rates. FOSL1 has been implicated in the development and progression of several types of cancer, including breast, lung, prostate, gastric, colorectal cancer, and glioma. It exerts its oncogenic effects by regulating various signaling pathways, including MAPK, PI3K/Akt, Wnt/β-catenin, and TGF-β, which are involved in cell proliferation, survival, apoptosis, and metastasis. Recently, it has been revealed that noncoding RNAs (ncRNAs) play important roles in the regulation of FOSL1 expression and function. Additionally, FOSL1 has been found to modulate the expression of various cytokines, chemokines, and extracellular matrix (ECM) proteins that hold crucial functions in the TME. For example, FOSL1 has been reported to upregulate the expression of IL-6 in breast cancer cells, which can promote tumor growth and invasion by activating the STAT3 signaling pathway. FOSL1 has also been shown to play a role in cancer stemness. CSCs are a subpopulation of tumor cells that are capable of self-renewal, differentiation, and tumor initiation. FOSL1 is overexpressed in CSCs of various cancers, including breast, colorectal, and lung cancers. Inhibition of FOSL1 in colorectal cancer cells has been found to reduce the population of cancer stem cells and inhibit tumor growth. There is growing evidence suggesting that FOSL1 may contribute to drug resistance in cancer cells. One study found that overexpression of FOSL1 in breast cancer cells led to increased resistance to the chemotherapy drug doxorubicin, while downregulation of FOSL1 increased sensitivity to the drug. FOSL1 has been identified as a potential therapeutic target and prognostic factor in various types of cancer. Despite its potential as a therapeutic target, there are currently no targeted inhibitors of FOSL1 available. Therefore, further research is needed to identify and develop effective inhibitors of FOSL1. Additionally, the specific mechanism by which FOSL1 contributes to tumor development and progression in different types of cancer is not yet fully understood and requires further investigation.
Author Contributions
AK: Conceptualization, writing the original draft. SG: Conceptualization, writing the original draft, and critical review. VR: Analyzing bioinformatic data. BH: Analyzing bioinformatic data. ML: Conceptualization, writing original draft and critical review, supervision, project administration.
Funding
This research was funded by grants NIH/NIGMS SC1 GM144021 (ML) and NIH NIGMS U54GM104940.
Data Availability Statement
Data will be made available on reasonable request.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. The funder has no role in the design of the study, in the collection, analysis, or interpretation of data, in the writing of the manuscript, or in the decision to publish the results.
References
- Rong, L.; Li, N.; Zhang, Z. Emerging therapies for glioblastoma: Current state and future directions. Journal of Experimental & Clinical Cancer Research 2022, 41, 1–18. [Google Scholar]
- Pecce, V.; Verrienti, A.; Fiscon, G.; Sponziello, M.; Conte, F.; Abballe, L.; Durante, C.; Farina, L.; Filetti, S.; Paci, P. The role of FOSL1 in stem-like cell reprogramming processes. Scientific reports 2021, 11, 14677. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.R.; Curran, T. fra-1: a serum-inducible, cellular immediate-early gene that encodes a fos-related antigen. Molecular and Cellular Biology 1988, 8, 2063–2069. [Google Scholar] [PubMed]
- Talotta, F.; Casalino, L.; Verde, P. The nuclear oncoprotein Fra-1: a transcription factor knocking on therapeutic applications’ door. Oncogene 2020, 39, 4491–4506. [Google Scholar] [CrossRef] [PubMed]
- Curran, T.; Franza Jr, B.R. Fos and Jun: the AP-1 connection. Cell 1988, 55, 395–397. [Google Scholar] [CrossRef] [PubMed]
- Angel, P.; Imagawa, M.; Chiu, R.; Stein, B.; Imbra, R.J.; Rahmsdorf, H.J.; Jonat, C.; Herrlich, P.; Karin, M. Phorbol ester-inducible genes contain a common cis element recognized by a TPA-modulated trans-acting factor. Cell 1987, 49, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Eferl, R.; Wagner, E.F. AP-1: a double-edged sword in tumorigenesis. Nature Reviews Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; He, J.; Jin, X.; Liao, Q.; Chen, Z.; Peng, H.; Zhou, Y. FRA-1: A key factor regulating signal transduction of tumor cells and a potential target molecule for tumor therapy. Biomedicine & Pharmacotherapy 2022, 150, 113037. [Google Scholar]
- Jiang, X.; Xie, H.; Dou, Y.; Yuan, J.; Zeng, D.; Xiao, S. Expression and function of FRA1 protein in tumors. Molecular Biology Reports 2020, 47, 737–752. [Google Scholar] [CrossRef]
- Zhang, M.; Liang, L.; He, J.; He, Z.; Yue, C.; Jin, X.; Gao, M.; Xiao, S.; Zhou, Y. Fra-1 inhibits cell growth and the Warburg effect in cervical cancer cells via STAT1 regulation of the p53 signaling pathway. Frontiers in cell and developmental biology 2020, 8, 579629. [Google Scholar] [CrossRef]
- Basbous, J.; Chalbos, D.; Hipskind, R.; Jariel-Encontre, I.; Piechaczyk, M. Ubiquitin-independent proteasomal degradation of Fra-1 is antagonized by Erk1/2 pathway-mediated phosphorylation of a unique C-terminal destabilizer. Molecular and cellular biology 2007, 27, 3936–3950. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef]
- Casalino, L.; Talotta, F.; Cimmino, A.; Verde, P. The Fra-1/AP-1 Oncoprotein: From the “Undruggable” Transcription Factor to Therapeutic Targeting. Cancers 2022, 14, 1480. [Google Scholar] [CrossRef]
- Liu, M.; Inoue, K.; Leng, T.; Guo, S.; Xiong, Z.G. TRPM7 channels regulate glioma stem cell through STAT3 and Notch signaling pathways. Cell Signal 2014, 26, 2773–2781. [Google Scholar] [CrossRef]
- Guo, S.; Ramar, V.; Guo, A.A.; Saafir, T.; Akpobiyeri, H.; Hudson, B.; Li, J.; Liu, M. TRPM7 transactivates the FOSL1 gene through STAT3 and enhances glioma stemness. Cell Mol Life Sci 2023, 80, 270. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; He, X.; Jia, L.; Zhang, X. Circular RNAs in glioma: Molecular functions and pathological implications. Noncoding RNA Res 2024, 9, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Mallela, V.R.; Rajtmajerová, M.; Trailin, A.; Liška, V.; Hemminki, K.; Ambrozkiewicz, F. miRNA and lncRNA as potential tissue biomarkers in hepatocellular carcinoma. Noncoding RNA Res 2024, 9, 24–32. [Google Scholar] [CrossRef]
- He, S.W.; Xu, C.; Li, Y.Q.; Li, Y.Q.; Zhao, Y.; Zhang, P.P.; Lei, Y.; Liang, Y.L.; Li, J.Y.; Li, Q.; et al. AR-induced long non-coding RNA LINC01503 facilitates proliferation and metastasis via the SFPQ-FOSL1 axis in nasopharyngeal carcinoma. Oncogene 2020, 39, 5616–5632. [Google Scholar] [CrossRef]
- Shen, S.; Li, K.; Liu, Y.; Liu, X.; Liu, B.; Ba, Y.; Xing, W. Silencing lncRNA AGAP2-AS1 Upregulates miR-195-5p to Repress Migration and Invasion of EC Cells via the Decrease of FOSL1 Expression. Molecular therapy. Nucleic acids 2020, 20, 331–344. [Google Scholar] [CrossRef]
- Guo, S.; King, P.; Liang, E.; Guo, A.A.; Liu, M. LncRNA HOTAIR sponges miR-301a-3p to promote glioblastoma proliferation and invasion through upregulating FOSL1. Cell Signal 2022, 94, 110306. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Che, W.; Liang, N.; Deng, S.; Song, Z.; Yang, L. Silent FOSL1 enhances the Radiosensitivity of glioma stem cells by Down-regulating miR-27a-5p. Neurochemical Research 2021, 46, 3222–3246. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Chen, Z.; Yu, J.; Wu, J.; Zhuo, X.; Chen, Q.; Liang, Y.; Li, G.; Wan, Y. MiR-195-5p suppresses the proliferation, migration, and invasion of gallbladder cancer cells by targeting FOSL1 and regulating the Wnt/β-catenin pathway. Annals of Translational Medicine 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhao, M.; Huang, J.; Li, Y.; Wang, S.; Harrington, C.A.; Qian, D.Z.; Sun, X.X.; Dai, M.S. MicroRNA-130a suppresses breast cancer cell migration and invasion by targeting FOSL1 and upregulating ZO-1. Journal of cellular biochemistry 2018, 119, 4945–4956. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Li, Y.; Gao, J.; Zhang, T.; Li, S.; Luo, A.; Chen, H.; Ding, F.; Wang, X.; Liu, Z. MicroRNA-34 suppresses breast cancer invasion and metastasis by directly targeting Fra-1. Oncogene 2013, 32, 4294–4303. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Kim, B.G.; Jang, Y.; Kang, S.; Lee, J.H.; Cho, N.H. The stromal loss of miR-4516 promotes the FOSL1-dependent proliferation and malignancy of triple negative breast cancer. Cancer letters 2020, 469, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Z.; Chen, C.; Liu, Y.; Si, Q.; Chuang, T.; Li, N.; Gomez-Cabrero, A.; Reisfeld, R.; Xiang, R. MicroRNA-19a-3p inhibits breast cancer progression and metastasis by inducing macrophage polarization through downregulated expression of Fra-1 proto-oncogene. Oncogene 2014, 33, 3014–3023. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wu, G.; Lv, L.; Ren, Y.F.; Zhang, X.J.; Xue, Y.F.; Li, G.; Lu, X.; Sun, Z.; Tang, K.F. MicroRNA-34a inhibits migration and invasion of colon cancer cells via targeting to Fra-1. Carcinogenesis 2012, 33, 519–528. [Google Scholar] [CrossRef]
- Zhang, N.; Shen, Q.; Zhang, P. miR-497 suppresses epithelial-mesenchymal transition and metastasis in colorectal cancer cells by targeting fos-related antigen-1. OncoTargets and therapy 2016, 9, 6597–6604. [Google Scholar] [CrossRef]
- Jin, Y.; Wang, C.; Liu, X.; Mu, W.; Chen, Z.; Yu, D.; Wang, A.; Dai, Y.; Zhou, X. Molecular characterization of the microRNA-138-Fos-like antigen 1 (FOSL1) regulatory module in squamous cell carcinoma. Journal of Biological Chemistry 2011, 286, 40104–40109. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, Q.; Zhang, G.; Mohammed, D.; Amadou, S.; Tan, G.; Zhang, X. HOXA11-AS1 Promotes PD-L1-Mediated Immune Escape and Metastasis of Hypopharyngeal Carcinoma by Facilitating PTBP1 and FOSL1 Association. Cancers 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Li, K.; Liu, Y.; Liu, X.; Liu, B.; Ba, Y.; Xing, W. Silencing lncRNA AGAP2-AS1 upregulates miR-195-5p to repress migration and invasion of EC cells via the decrease of FOSL1 expression. Molecular Therapy-Nucleic Acids 2020, 20, 331–344. [Google Scholar] [CrossRef] [PubMed]
- He, S.-W.; Xu, C.; Li, Y.-Q.; Li, Y.-Q.; Zhao, Y.; Zhang, P.-P.; Lei, Y.; Liang, Y.-L.; Li, J.-Y.; Li, Q. AR-induced long non-coding RNA LINC01503 facilitates proliferation and metastasis via the SFPQ-FOSL1 axis in nasopharyngeal carcinoma. Oncogene 2020, 39, 5616–5632. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Liu, Q.; Zhang, G.; Mohammed, D.; Amadou, S.; Tan, G.; Zhang, X. HOXA11-AS1 promotes PD-L1-mediated immune escape and metastasis of hypopharyngeal carcinoma by facilitating PTBP1 and FOSL1 association. Cancers 2022, 14, 3694. [Google Scholar] [CrossRef] [PubMed]
- Casalino, L.; De Cesare, D.; Verde, P. Accumulation of Fra-1 in ras-transformed cells depends on both transcriptional autoregulation and MEK-dependent posttranslational stabilization. Molecular and cellular biology 2003, 23, 4401–4415. [Google Scholar] [CrossRef] [PubMed]
- Doehn, U.; Hauge, C.; Frank, S.R.; Jensen, C.J.; Duda, K.; Nielsen, J.V.; Cohen, M.S.; Johansen, J.V.; Winther, B.R.; Lund, L.R. RSK is a principal effector of the RAS-ERK pathway for eliciting a coordinate promotile/invasive gene program and phenotype in epithelial cells. Molecular cell 2009, 35, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Belguise, K.; Cherradi, S.; Sarr, A.; Boissière, F.; Boulle, N.; Simony-Lafontaine, J.; Choesmel-Cadamuro, V.; Wang, X.; Chalbos, D. PKCθ-induced phosphorylations control the ability of Fra-1 to stimulate gene expression and cancer cell migration. Cancer Letters 2017, 385, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Gruda, M.C.; Kovary, K.; Metz, R.; Bravo, R. Regulation of Fra-1 and Fra-2 phosphorylation differs during the cell cycle of fibroblasts and phosphorylation in vitro by MAP kinase affects DNA binding activity. Oncogene 1994, 9, 2537–2547. [Google Scholar] [PubMed]
- Terasawa, K.; Okazaki, K.; Nishida, E. Regulation of c-Fos and Fra-1 by the MEK5-ERK5 pathway. Genes to Cells 2003, 8, 263–273. [Google Scholar] [CrossRef]
- Tower, G.B.; Coon, C.I.; Belguise, K.; Chalbos, D.; Brinckerhoff, C.E. Fra-1 targets the AP-1 site/2G single nucleotide polymorphism (ETS site) in the MMP-1 promoter. European journal of biochemistry 2003, 270, 4216–4225. [Google Scholar] [CrossRef]
- Basbous, J.; Jariel-Encontre, I.; Gomard, T.; Bossis, G.; Piechaczyk, M. Ubiquitin-independent-versus ubiquitin-dependent proteasomal degradation of the c-Fos and Fra-1 transcription factors: is there a unique answer? Biochimie 2008, 90, 296–305. [Google Scholar] [CrossRef]
- Marques, C.; Unterkircher, T.; Kroon, P.; Oldrini, B.; Izzo, A.; Dramaretska, Y.; Ferrarese, R.; Kling, E.; Schnell, O.; Nelander, S.; et al. NF1 regulates mesenchymal glioblastoma plasticity and aggressiveness through the AP-1 transcription factor FOSL1. Elife 2021, 10. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, S.; Li, H.L.; Luo, H.; Wu, X.; Lu, J.; Wang, H.W.; Chen, Y.; Chen, D.; Wu, W.T.; et al. FOSL1 promotes proneural-to-mesenchymal transition of glioblastoma stem cells via UBC9/CYLD/NF-κB axis. Mol Ther 2022, 30, 2568–2583. [Google Scholar] [CrossRef] [PubMed]
- Geer, L.Y.; Marchler-Bauer, A.; Geer, R.C.; Han, L.; He, J.; He, S.; Liu, C.; Shi, W.; Bryant, S.H. The NCBI BioSystems database. Nucleic Acids Res 2010, 38, D492–496. [Google Scholar] [CrossRef] [PubMed]
- Wykosky, J.; Hu, J.; Gomez, G.G.; Taylor, T.; Villa, G.R.; Pizzo, D.; VandenBerg, S.R.; Thorne, A.H.; Chen, C.C.; Mischel, P.S.; et al. A urokinase receptor-Bim signaling axis emerges during EGFR inhibitor resistance in mutant EGFR glioblastoma. Cancer research 2015, 75, 394–404. [Google Scholar] [CrossRef]
- Ramsdale, R.; Jorissen, R.N.; Li, F.Z.; Al-Obaidi, S.; Ward, T.; Sheppard, K.E.; Bukczynska, P.E.; Young, R.J.; Boyle, S.E.; Shackleton, M.; et al. The transcription cofactor c-JUN mediates phenotype switching and BRAF inhibitor resistance in melanoma. Sci Signal 2015, 8, ra82. [Google Scholar] [CrossRef]
- Kim, S.M.; Kwon, O.J.; Hong, Y.K.; Kim, J.H.; Solca, F.; Ha, S.J.; Soo, R.A.; Christensen, J.G.; Lee, J.H.; Cho, B.C. Activation of IL-6R/JAK1/STAT3 signaling induces de novo resistance to irreversible EGFR inhibitors in non-small cell lung cancer with T790M resistance mutation. Mol Cancer Ther 2012, 11, 2254–2264. [Google Scholar] [CrossRef]
- Kesanakurti, D.; Chetty, C.; Rajasekhar Maddirela, D.; Gujrati, M.; Rao, J.S. Functional cooperativity by direct interaction between PAK4 and MMP-2 in the regulation of anoikis resistance, migration and invasion in glioma. Cell death & disease 2012, 3, e445. [Google Scholar] [CrossRef]
- Saito, R.; Miki, Y.; Ishida, N.; Inoue, C.; Kobayashi, M.; Hata, S.; Yamada-Okabe, H.; Okada, Y.; Sasano, H. The Significance of MMP-1 in EGFR-TKI-Resistant Lung Adenocarcinoma: Potential for Therapeutic Targeting. International journal of molecular sciences 2018, 19. [Google Scholar] [CrossRef]
- Day, E.K.; Sosale, N.G.; Xiao, A.; Zhong, Q.; Purow, B.; Lazzara, M.J. Glioblastoma Cell Resistance to EGFR and MET Inhibition Can Be Overcome via Blockade of FGFR-SPRY2 Bypass Signaling. Cell Rep 2020, 30, 3383–3396. [Google Scholar] [CrossRef]
- Wang, W.; Liu, W.; Xu, J.; Jin, H. MiR-33a targets FOSL1 and EN2 as a clinical prognostic marker for sarcopenia by glioma. Front Genet 2022, 13, 953580. [Google Scholar] [CrossRef] [PubMed]
- Al-Khayyat, W.; Pirkkanen, J.; Dougherty, J.; Laframboise, T.; Dickinson, N.; Khaper, N.; Lees, S.J.; Mendonca, M.S.; Boreham, D.R.; Tai, T.C.; et al. Overexpression of FRA1 (FOSL1) Leads to Global Transcriptional Perturbations, Reduced Cellular Adhesion and Altered Cell Cycle Progression. Cells 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhu, G.; Gao, L.; Chen, P.; Long, Y.; Liao, S.; Yi, H.; Yi, W.; Pei, Z.; Wu, M. Fra-1 is upregulated in gastric cancer tissues and affects the PI3K/Akt and p53 signaling pathway in gastric cancer. International journal of oncology 2015, 47, 1725–1734. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Zhao, X.; Sun, Y.; Sui, Y.; Liu, J. Retracted: Effects of FOSL1 silencing on osteosarcoma cell proliferation, invasion and migration through the ERK/AP-1 signaling pathway. 2019.
- Li, B.; Wan, Z.; Huang, G.; Huang, Z.; Zhang, X.; Liao, D.; Luo, S.; He, Z. Mitogen-and stress-activated Kinase 1 mediates Epstein-Barr virus latent membrane protein 1-promoted cell transformation in nasopharyngeal carcinoma through its induction of Fra-1 and c-Jun genes. BMC cancer 2015, 15, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Casalino, L.; Bakiri, L.; Talotta, F.; Weitzman, J.B.; Fusco, A.; Yaniv, M.; Verde, P. Fra-1 promotes growth and survival in RAS-transformed thyroid cells by controlling cyclin A transcription. The EMBO journal 2007, 26, 1878–1890. [Google Scholar] [CrossRef] [PubMed]
- Vallone, D.; Battista, S.; Pierantoni, G.M.; Fedele, M.; Casalino, L.; Santoro, M.; Viglietto, G.; Fusco, A.; Verde, P. Neoplastic transformation of rat thyroid cells requires the junB and fra-1 gene induction which is dependent on the HMGI-C gene product. The EMBO journal 1997, 16, 5310–5321. [Google Scholar] [CrossRef] [PubMed]
- Chiappetta, G.; Tallini, G.; De Biasio, M.C.; Pentimalli, F.; de Nigris, F.; Losito, S.; Fedele, M.; Battista, S.; Verde, P.; Santoro, M. FRA-1 expression in hyperplastic and neoplastic thyroid diseases. Clinical Cancer Research 2000, 6, 4300–4306. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, C.; Hua, Y.; Cheng, K.; Zhang, Y.; Liu, J.; Han, Y.; Liu, S.; Zhang, G.; Xu, S. Psoralen induced cell cycle arrest by modulating Wnt/β-catenin pathway in breast cancer cells. Scientific reports 2018, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Belguise, K.; Kersual, N.; Galtier, F.; Chalbos, D. FRA-1 expression level regulates proliferation and invasiveness of breast cancer cells. Oncogene 2005, 24, 1434–1444. [Google Scholar] [CrossRef]
- Ibrahim, S.A.E.; Abudu, A.; Johnson, E.; Aftab, N.; Conrad, S.; Fluck, M. The role of AP-1 in self-sufficient proliferation and migration of cancer cells and its potential impact on an autocrine/paracrine loop. Oncotarget 2018, 9, 34259–34278. [Google Scholar] [CrossRef]
- Xiao, S.; Zhou, Y.; Yi, W.; Luo, G.; Jiang, B.; Tian, Q.; Li, Y.; Xue, M. Fra-1 is downregulated in cervical cancer tissues and promotes cervical cancer cell apoptosis by p53 signaling pathway in vitro. International journal of oncology 2015, 46, 1677–1684. [Google Scholar] [CrossRef] [PubMed]
- Zhong, G.; Chen, X.; Fang, X.; Wang, D.; Xie, M.; Chen, Q. Fra-1 is upregulated in lung cancer tissues and inhibits the apoptosis of lung cancer cells by the P53 signaling pathway. Oncology reports 2016, 35, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liang, L.; He, J.; He, Z.; Yue, C.; Jin, X.; Gao, M.; Xiao, S.; Zhou, Y. Fra-1 Inhibits Cell Growth and the Warburg Effect in Cervical Cancer Cells via STAT1 Regulation of the p53 Signaling Pathway. Front Cell Dev Biol 2020, 8, 579629. [Google Scholar] [CrossRef] [PubMed]
- Zhong, G.; Chen, X.; Fang, X.; Wang, D.; Xie, M.; Chen, Q. Fra-1 is upregulated in lung cancer tissues and inhibits the apoptosis of lung cancer cells by the P53 signaling pathway. Oncol Rep 2016, 35, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Tulchinsky, E. FRA-1 as a driver of tumour heterogeneity: a nexus between oncogenes and embryonic signalling pathways in cancer. Oncogene 2015, 34, 4421–4428. [Google Scholar] [CrossRef] [PubMed]
- Bakiri, L.; Macho-Maschler, S.; Custic, I.; Niemiec, J.; Guío-Carrión, A.; Hasenfuss, S.; Eger, A.; Müller, M.; Beug, H.; Wagner, E. Fra-1/AP-1 induces EMT in mammary epithelial cells by modulating Zeb1/2 and TGFβ expression. Cell Death & Differentiation 2015, 22, 336–350. [Google Scholar]
- Zhao, C.; Qiao, Y.; Jonsson, P.; Wang, J.; Xu, L.; Rouhi, P.; Sinha, I.; Cao, Y.; Williams, C.; Dahlman-Wright, K. Genome-wide profiling of AP-1–regulated transcription provides insights into the invasiveness of triple-negative breast cancer. Cancer research 2014, 74, 3983–3994. [Google Scholar] [CrossRef]
- Liu, H.; Ren, G.; Wang, T.; Chen, Y.; Gong, C.; Bai, Y.; Wang, B.; Qi, H.; Shen, J.; Zhu, L. Aberrantly expressed Fra-1 by IL-6/STAT3 transactivation promotes colorectal cancer aggressiveness through epithelial–mesenchymal transition. Carcinogenesis 2015, 36, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yue, M.; Li, Z. FOSL1 promotes tumorigenesis in colorectal carcinoma by mediating the FBXL2/Wnt/β-catenin axis via Smurf1. Pharmacological Research 2021, 165, 105405. [Google Scholar] [CrossRef]
- Wu, J.; Wu, G.; Lv, L.; Ren, Y.-F.; Zhang, X.-J.; Xue, Y.-F.; Li, G.; Lu, X.; Sun, Z.; Tang, K.-F. MicroRNA-34a inhibits migration and invasion of colon cancer cells via targeting to Fra-1. Carcinogenesis 2012, 33, 519–528. [Google Scholar] [CrossRef]
- Yun, S.-I.; Hong, H.K.; Yeo, S.-Y.; Kim, S.-H.; Cho, Y.B.; Kim, K.K. Ubiquitin-specific protease 21 promotes colorectal cancer metastasis by acting as a Fra-1 deubiquitinase. Cancers 2020, 12, 207. [Google Scholar] [CrossRef]
- Andreolas, C.; Kalogeropoulou, M.; Voulgari, A.; Pintzas, A. Fra-1 regulates vimentin during Ha-RAS-induced epithelial mesenchymal transition in human colon carcinoma cells. International journal of cancer 2008, 122, 1745–1756. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.Y.; Wei, W.; Qian, M.R.; Tian, R.F.; Fu, X.; Li, H.W.; Nan, G.; Yang, T.; Lin, P.; Chen, X. PDGFA-associated protein 1 is a novel target of c-Myc and contributes to colorectal cancer initiation and progression. Cancer Communications 2022, 42, 750–767. [Google Scholar] [CrossRef]
- Rattanasinchai, C.; Llewellyn, B.; Conrad, S.; Gallo, K. MLK3 regulates FRA-1 and MMPs to drive invasion and transendothelial migration in triple-negative breast cancer cells. Oncogenesis 2017, 6, e345–e345. [Google Scholar] [CrossRef]
- Belguise, K.; Milord, S.; Galtier, F.; Moquet-Torcy, G.; Piechaczyk, M.; Chalbos, D. The PKCθ pathway participates in the aberrant accumulation of Fra-1 protein in invasive ER-negative breast cancer cells. Oncogene 2012, 31, 4889–4897. [Google Scholar] [CrossRef]
- Tolza, C.; Bejjani, F.; Evanno, E.; Mahfoud, S.; Moquet-Torcy, G.; Gostan, T.; Maqbool, M.A.; Kirsh, O.; Piechaczyk, M.; Jariel-Encontre, I. AP-1 signaling by Fra-1 directly regulates HMGA1 oncogene transcription in triple-negative breast cancers. Molecular cancer research 2019, 17, 1999–2014. [Google Scholar] [CrossRef] [PubMed]
- Annis, M.G.; Ouellet, V.; Rennhack, J.P.; L’Esperance, S.; Rancourt, C.; Mes-Masson, A.-M.; Andrechek, E.R.; Siegel, P.M. Integrin-uPAR signaling leads to FRA-1 phosphorylation and enhanced breast cancer invasion. Breast Cancer Research 2018, 20, 1–15. [Google Scholar] [CrossRef]
- Park, J.-H.; Myung, J.-K.; Lee, S.-J.; Kim, H.; Kim, S.; Lee, S.-B.; Jang, H.; Jang, W.-I.; Park, S.; Yang, H. ABCA1-Mediated EMT Promotes Papillary Thyroid Cancer Malignancy through the ERK/Fra-1/ZEB1 Pathway. Cells 2023, 12, 274. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Liu, H.; Xu, Z.; Zhang, Y. Expression and clinical significance of FOS-like antigen 1 in gastric adenocarcinoma. Pathology-Research and Practice 2019, 215, 152394. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, H.; Mu, X.; Cui, J.; Peng, Z. Dysregulation of Fra1 expression by Wnt/β-catenin signalling promotes glioma aggressiveness through epithelial–mesenchymal transition. Bioscience reports 2017, 37. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K. Role of tumor microenvironment in tumorigenesis. Journal of Cancer 2017, 8, 761. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Charles, K.A.; Mantovani, A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer cell 2005, 7, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ni, H.; Lan, L.; Wei, X.; Xiang, R.; Wang, Y. Fra-1 protooncogene regulates IL-6 expression in macrophages and promotes the generation of M2d macrophages. Cell research 2010, 20, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zhou, H.; Krueger, J.; Kaplan, C.; Liao, D.; Markowitz, D.; Liu, C.; Chen, T.; Chuang, T.-H.; Xiang, R. The role of proto-oncogene Fra-1 in remodeling the tumor microenvironment in support of breast tumor cell invasion and progression. Oncogene 2010, 29, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Jiang, X.; Wang, G.; Zhai, C.; Liu, Y.; Li, H.; Zhang, Y.; Yu, W.; Zhao, Z. ITGB4 is a novel prognostic factor in colon cancer. Journal of Cancer 2019, 10, 5223. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.Y.T.; Lo, J.; Cheng, B.Y.L.; Ma, M.K.F.; Lee, J.M.F.; Ng, J.K.Y.; Chai, S.; Lin, C.H.; Tsang, S.Y.; Ma, S. Cancer-associated fibroblasts regulate tumor-initiating cell plasticity in hepatocellular carcinoma through c-Met/FRA1/HEY1 signaling. Cell reports 2016, 15, 1175–1189. [Google Scholar] [CrossRef] [PubMed]
- Jing, N.; Gao, W.-Q.; Fang, Y.-X. Regulation of formation, stemness and therapeutic resistance of cancer stem cells. Frontiers in Cell and Developmental Biology 2021, 9, 641498. [Google Scholar] [CrossRef]
- Pecce, V.; Verrienti, A.; Fiscon, G.; Sponziello, M.; Conte, F.; Abballe, L.; Durante, C.; Farina, L.; Filetti, S.; Paci, P. The role of FOSL1 in stem-like cell reprogramming processes. Scientific reports 2021, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.L.; Lu, H.; Buikhuisen, J.; Soh, B.S.; Lim, E.; Reinhardt, F.; Wu, Z.J.; Krall, J.A.; Bierie, B.; Guo, W. Protein kinase C α is a central signaling node and therapeutic target for breast cancer stem cells. Cancer cell 2013, 24, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, S.; Li, H.-L.; Luo, H.; Wu, X.; Lu, J.; Wang, H.-W.; Chen, Y.; Chen, D.; Wu, W.-T. FOSL1 promotes proneural-to-mesenchymal transition of glioblastoma stem cells via UBC9/CYLD/NF-κB axis. Molecular Therapy 2022, 30, 2568–2583. [Google Scholar] [CrossRef]
- Wang, T.; Song, P.; Zhong, T.; Wang, X.; Xiang, X.; Liu, Q.; Chen, H.; Xia, T.; Liu, H.; Niu, Y. The inflammatory cytokine IL-6 induces FRA1 deacetylation promoting colorectal cancer stem-like properties. Oncogene 2019, 38, 4932–4947. [Google Scholar] [CrossRef] [PubMed]
- Sobolev, V.V.; Khashukoeva, A.Z.; Evina, O.E.; Geppe, N.A.; Chebysheva, S.N.; Korsunskaya, I.M.; Tchepourina, E.; Mezentsev, A. Role of the transcription factor FOSL1 in organ development and tumorigenesis. International journal of molecular sciences 2022, 23, 1521. [Google Scholar] [CrossRef] [PubMed]
- Elangovan, I.M.; Vaz, M.; Tamatam, C.R.; Potteti, H.R.; Reddy, N.M.; Reddy, S.P. FOSL1 promotes Kras-induced lung cancer through amphiregulin and cell survival gene regulation. American journal of respiratory cell and molecular biology 2018, 58, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Vaz, M.; Reddy, N.M.; Rajasekaran, S.; Reddy, S.P. Genetic disruption of Fra-1 decreases susceptibility to endotoxin-induced acute lung injury and mortality in mice. American journal of respiratory cell and molecular biology 2012, 46, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Iskit, S.; Schlicker, A.; Wessels, L.; Peeper, D.S. Fra-1 is a key driver of colon cancer metastasis and a Fra-1 classifier predicts disease-free survival. Oncotarget 2015, 6, 43146. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Ferrer, L.; Kürschner, M.; Labitzky, V.; Wicklein, D.; Müller, V.; Lüers, G.; Schumacher, U.; Milde-Langosch, K.; Schröder, C. Prognostic impact of transcription factor Fra-1 in ER-positive breast cancer: contribution to a metastatic phenotype through modulation of tumor cell adhesive properties. Journal of cancer research and clinical oncology 2015, 141, 1715–1726. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Jin, X.; Yuan, Y.; Deng, P.; Jiang, L.; Zeng, X.; Li, X.-S.; Wang, Z.-Y.; Chen, Q.-M. Prognostic value from integrative analysis of transcription factors c-Jun and Fra-1 in oral squamous cell carcinoma: a multicenter cohort study. Scientific reports 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Hoyle, R.G.; Ma, Z.; Sun, B.; Cai, W.; Cai, H.; Xie, N.; Zhang, Y.; Hou, J.; Liu, X. FOSL1 promotes metastasis of head and neck squamous cell carcinoma through super-enhancer-driven transcription program. Molecular Therapy 2021, 29, 2583–2600. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, J.; Luo, S.; Lechler, T.; Zhang, J.Y. FRA1 promotes squamous cell carcinoma growth and metastasis through distinct AKT and c-Jun dependent mechanisms. Oncotarget 2016, 7, 34371. [Google Scholar] [CrossRef]
- Li, L.; Zhang, W.; Zhao, S.; Sun, M. FOS-like antigen 1 is a prognostic biomarker in hepatocellular carcinoma. Saudi Journal of Gastroenterology: Official Journal of the Saudi Gastroenterology Association 2019, 25, 369. [Google Scholar]
- Marques, C.; Unterkircher, T.; Kroon, P.; Oldrini, B.; Izzo, A.; Dramaretska, Y.; Ferrarese, R.; Kling, E.; Schnell, O.; Nelander, S. NF1 regulates mesenchymal glioblastoma plasticity and aggressiveness through the AP-1 transcription factor FOSL1. Elife 2021, 10, e64846. [Google Scholar] [CrossRef] [PubMed]
- Shetty, A.; Bhosale, S.D.; Tripathi, S.K.; Buchacher, T.; Biradar, R.; Rasool, O.; Moulder, R.; Galande, S.; Lahesmaa, R. Interactome Networks of FOSL1 and FOSL2 in Human Th17 Cells. ACS omega 2021, 6, 24834–24847. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resistance 2019, 2, 141. [Google Scholar] [CrossRef] [PubMed]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef]
- Tyagi, A.; Vishnoi, K.; Kaur, H.; Srivastava, Y.; Roy, B.G.; Das, B.C.; Bharti, A.C. Cervical cancer stem cells manifest radioresistance: Association with upregulated AP-1 activity. Scientific reports 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chiappetta, G.; Ferraro, A.; Botti, G.; Monaco, M.; Pasquinelli, R.; Vuttariello, E.; Arnaldi, L.; Di Bonito, M.; D’Aiuto, G.; Pierantoni, G.M. FRA-1 protein overexpression is a feature of hyperplastic and neoplastic breast disorders. BMC cancer 2007, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, N.; Xiong, Y.; Guo, G.; Zhu, M.; Gu, Y. Transcription factor FOSL1 enhances drug resistance of breast cancer through DUSP7-mediated dephosphorylation of PEA15. Molecular Cancer Research 2022, 20, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Zhao, M.; Ang, L.; Hu, H.; Wu, Z.; Wang, J.; Huang, J.; Zheng, L.; Dong, W. Down-regulation of FOS-like antigen 1 enhances drug sensitivity in breast cancer. International journal of clinical and experimental pathology 2020, 13, 2092. [Google Scholar] [PubMed]
- Lu, D.; Chen, S.; Tan, X.; Li, N.; Liu, C.; Li, Z.; Liu, Z.; Stupack, D.G.; Reisfeld, R.A.; Xiang, R. Fra-1 Promotes Breast Cancer Chemosensitivity by Driving Cancer Stem Cells from DormancyFra-1 Drives Breast Cancer Cells from Dormancy. Cancer research 2012, 72, 3451–3456. [Google Scholar] [CrossRef]
- Wu, J.; Sun, Y.; Zhang, P.-Y.; Qian, M.; Zhang, H.; Chen, X.; Ma, D.; Xu, Y.; Chen, X.; Tang, K.-F. The Fra-1–miR-134–SDS22 feedback loop amplifies ERK/JNK signaling and reduces chemosensitivity in ovarian cancer cells. Cell death & disease 2016, 7, e2384–e2384. [Google Scholar]
- Lin, S.; Zhu, B. Exosome-transmitted FOSL1 from cancer-associated fibroblasts drives colorectal cancer stemness and chemoresistance through transcriptionally activating ITGB4. 2023.
- Endo, S.; Fujita, M.; Yamada, S.; Imadome, K.; Nakayama, F.; Isozaki, T.; Yasuda, T.; Imai, T.; Matsubara, H. Fra-1 enhances the radioresistance of colon cancer cells to X-ray or C-ion radiation. Oncology Reports 2018, 39, 1112–1118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Atri, P.; Nallasamy, P.; Parte, S.; Rauth, S.; Nimmakayala, R.K.; Marimuthu, S.; Chirravuri-Venkata, R.; Bhatia, R.; Halder, S. Small molecule inhibitor against onco-mucins disrupts Src/FosL1 axis to enhance gemcitabine efficacy in pancreatic ductal adenocarcinoma. Cancer Letters 2022, 551, 215922. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.Y.; Ke, A.W.; Shi, G.M.; Zhang, X.; Zhang, C.; Shi, Y.H.; Wang, X.Y.; Ding, Z.B.; Xiao, Y.S.; Yan, J. αB-crystallin complexes with 14-3-3ζ to induce epithelial-mesenchymal transition and resistance to sorafenib in hepatocellular carcinoma. Hepatology 2013, 57, 2235–2247. [Google Scholar] [CrossRef] [PubMed]
- Kajanne, R.; Miettinen, P.; Tenhunen, M.; Leppä, S. Transcription factor AP-1 promotes growth and radioresistance in prostate cancer cells. International journal of oncology 2009, 35, 1175–1182. [Google Scholar]
- Halatsch, M.E.; Löw, S.; Mursch, K.; Hielscher, T.; Schmidt, U.; Unterberg, A.; Vougioukas, V.I.; Feuerhake, F. Candidate genes for sensitivity and resistance of human glioblastoma multiforme cell lines to erlotinib. Laboratory investigation. Journal of neurosurgery 2009, 111, 211–218. [Google Scholar] [CrossRef]
Figure 1.
A schematic representation illustrates the role of miRNAs in the regulation of FOSL1. Different miRNAs act to modulate (inhibit) the expression levels of FOSL1, thus exerting an influence on various aspects of cancer behavior. Through the downregulation of FOSL1, these miRNAs influence the Wnt signaling pathway, apoptosis, cell proliferation, cell cycle, invasion, and migration, ultimately resulting in the reduction of tumor growth across a range of types [23,24,25,26,30].
Figure 1.
A schematic representation illustrates the role of miRNAs in the regulation of FOSL1. Different miRNAs act to modulate (inhibit) the expression levels of FOSL1, thus exerting an influence on various aspects of cancer behavior. Through the downregulation of FOSL1, these miRNAs influence the Wnt signaling pathway, apoptosis, cell proliferation, cell cycle, invasion, and migration, ultimately resulting in the reduction of tumor growth across a range of types [23,24,25,26,30].
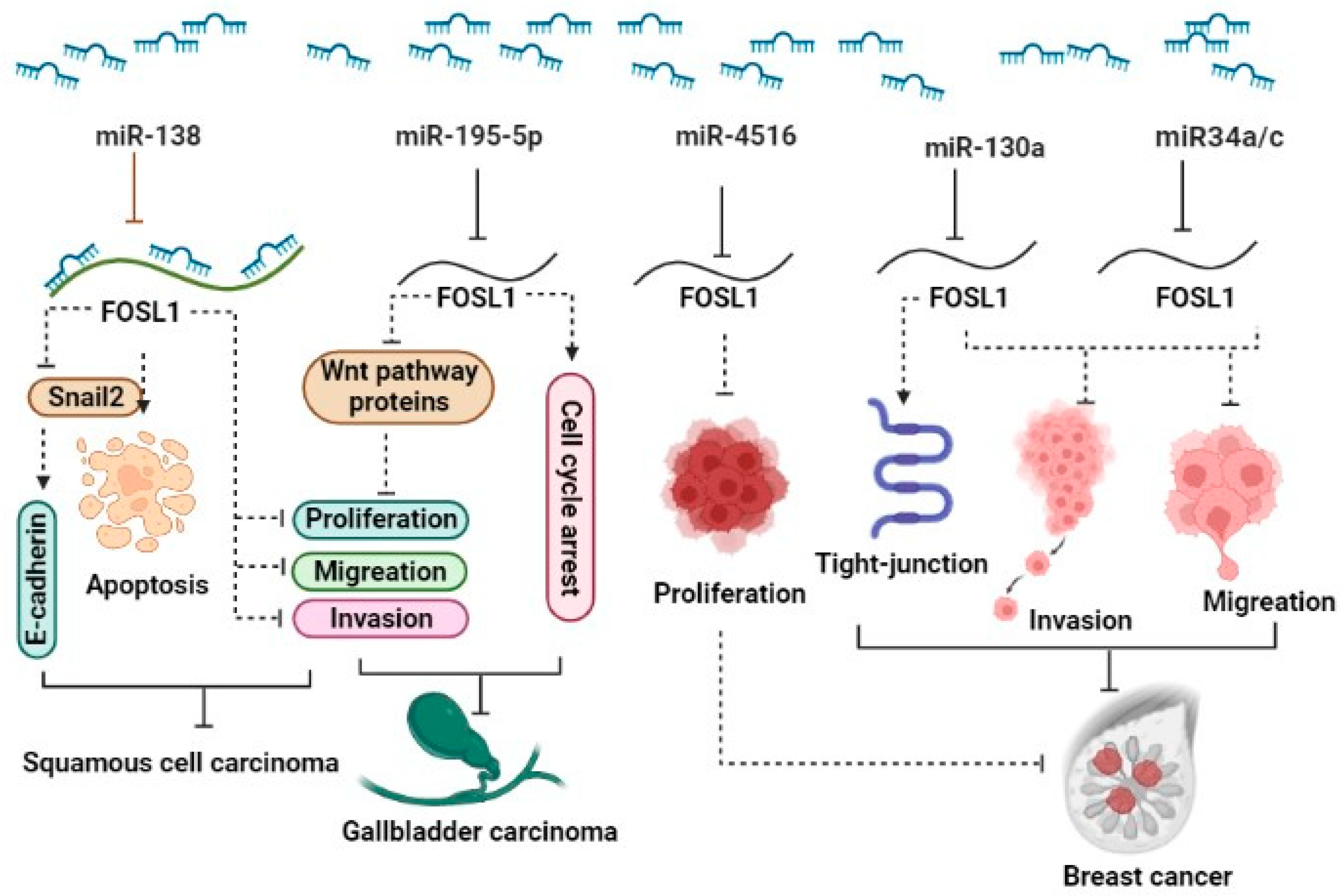
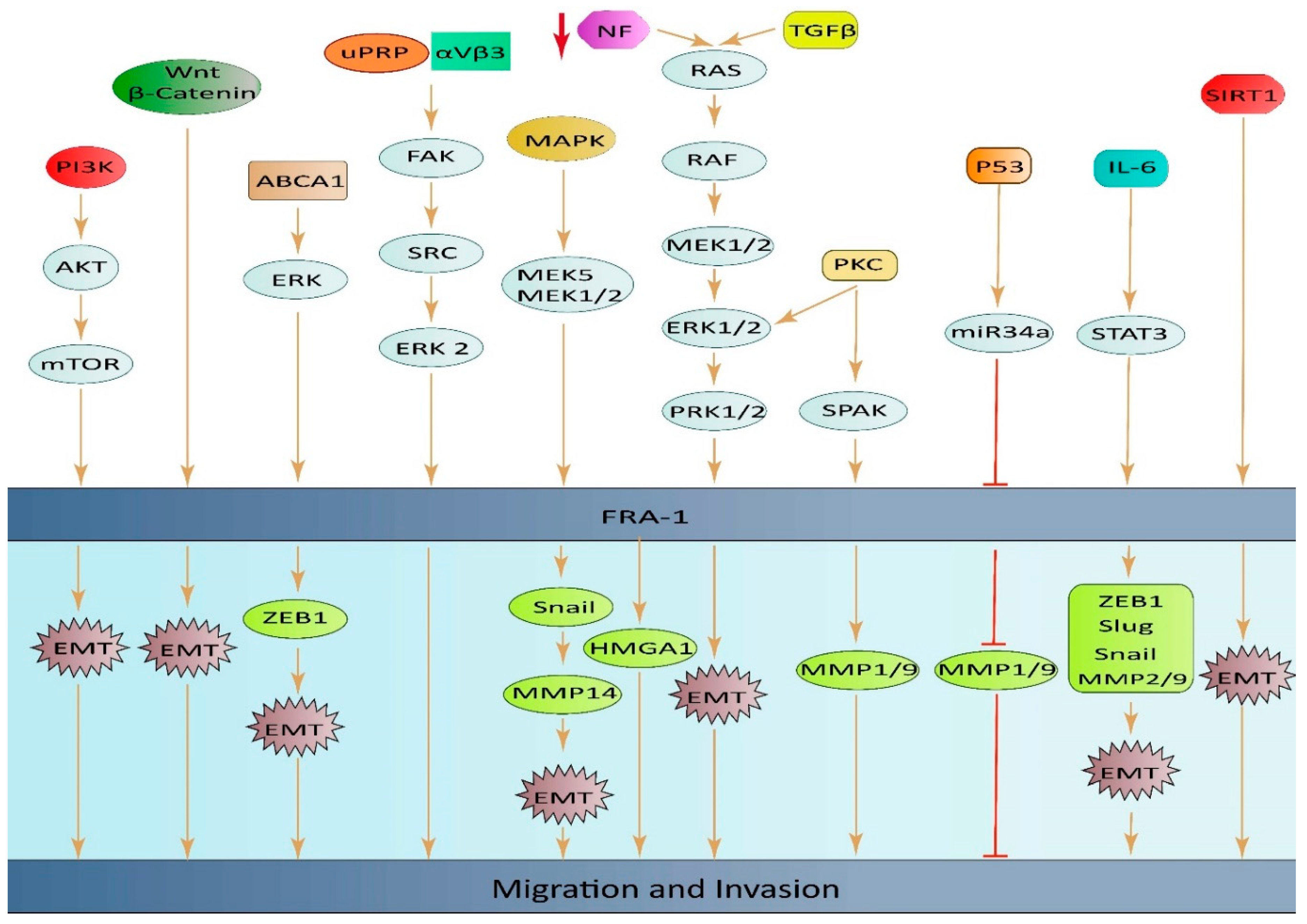
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



