
Submitted:
01 December 2023
Posted:
04 December 2023
You are already at the latest version
Abstract
Inflammation is a conserved process that involve the activation of immune and non-immune cells aiming at protecting the host from bacteria, viruses, toxins and injury. However, unresolved inflammation and permanent release of pro-inflammatory mediators are responsible for the promotion of a condition called “low-grade systemic chronic inflammation”, characterized by tissue and organ damages, metabolic changes and increased susceptibility to non-communicable diseases. Several studies have demonstrated that different dietary components may influence modifiable risk factors for diverse chronic human pathologies. Marine n-3 polyunsaturated fatty acids (n-3 PUFAs), mainly eicosapentaenoic (EPA) and docosahexaenoic acid (DHA), are well recognized anti-inflammatory and immunomodulatory agents able to influence many aspects of the inflammatory process. The aim of this article is to review the recent literature that relates to the modulation of human disease, such as rheumatoid arthritis, by n-3 PUFAs.
Keywords:
Fish oil
; Inflammation
; n-3 PUFAs
; Rheumatoid arthritis
; Specialized pro-resolving mediators
1. Introduction
Chronic non-communicable diseases (NCDs) are still the most common global cause of morbidity and mortality. These illnesses are characterized by the presence of elevated levels of inflammatory markers, mainly cytokines and chemokines, both at the inflammatory site and in the systemic circulation, and a huge infiltration of inflammatory cells at the site of disease activity [1,2]. NCDs include metabolic syndrome, type 2 diabetes, hypertension, cardiovascular disease (CVD), non-infectious respiratory disease, chronic kidney disease, neurodegenerative and autoimmune diseases (i.e., rheumatoid arthritis), depression, osteoporosis, age-related macular degeneration and various type of cancers [2,3].
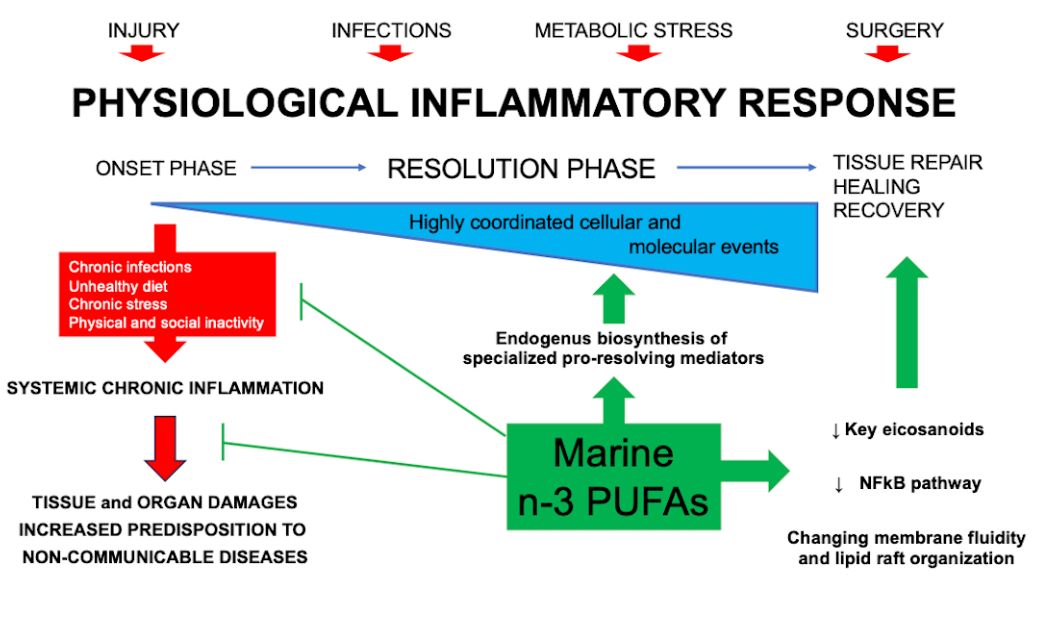
Inflammation is a conserved process that involve the activation of immune and non-immune cells aiming at protecting the host from bacteria, viruses, toxins and injury, and, as a consequence, at removing the pathogens and supporting tissue repair and recovery [4,5]. The acute inflammatory response starts within minutes of recognition of a harmful signal with an “onset phase” that involves the production of chemokines, cytokines, eicosanoids, proteases, vasoactive amines, neuropeptides and neurotransmitters by resident immune and structural cells. Moreover, this process is characterized by the recruitment of different cell types, such as granulocytes, from blood to the tissue inflammatory site [6]. In physiological condition, the inflammatory response is programmed to evolve in an active “resolution phase” characterized by highly coordinated cellular and molecular events, including the release of anti-inflammatory cytokines and pro-resolving mediators, the loss of receptors for pro-inflammatory stimuli and the activation of regulatory cells that weaken the activity of pro-inflammatory cells [7]. The effectiveness of the “resolution phase” is conditional on specific cellular mechanisms that are orchestrated by pro-resolving mediators engaged of halting the inflammatory response, and initiating tissue repair and healing. Specifically, the recruitment of non-phlogistic monocytes and their differentiation into macrophages able to clear the local leukocytes by apoptosis and subsequent phagocytosis is one of the key events in determining the initiation of the “resolution phase” [8]. In addition, the apoptotic cells “inform” the phagocytosing macrophages that the inflammatory response is ending, and trigger the macrophage mediator production from a pro-inflammatory (M1) to an anti-inflammatory and pro-resolving phenotype (M2) [9]. It has been observed that the balance between the M1 and M2 macrophages is fundamental for proper resolution of inflammation [10]. Others cellular actors of the “resolution phase” are: 1) regulatory T cells (Treg), which release anti-inflammatory cytokines, such as Interleukin 10 (IL-10) and Transforming Growth Factor-beta (TGF-beta), and remove IL-2, essential for the activation of T cells [11]; 2) innate lymphoid cells (ILCs), such as ILC2, ILC3, and NK cells [12]; 3) myeloid-derived suppressor cells (MDSCs), which possess immunomodulatory activities through the induction of Treg cell expansion, and the production of IL-10 and TGF-beta. Moreover, the MDSCs mediate the efferocytosis of apoptotic neutrophils [13,14]. Meanwhile, the lipid mediator class switching promotes the shift from the synthesis of prostaglandins (PGs) and leukotrienes (LTs) via 5-lipoxygenase (LOX) in inflammatory exudates to the production of Lipoxin A4 via 15-LOX, and the following reprogramming of granulocytes to initiate the “resolution phase” [15]. As stated above, during the “resolution phase” several mediators are produced to prevent the exacerbation of acute inflammatory mechanisms, and eventually restore tissue homeostasis. These anti-inflammatory and pro-resolving mediators display peculiar activities: i) halting or inhibiting neutrophil recruitment; ii) promoting the influx of non-phlogistic monocytes; iii) inducing neutrophil apoptosis and efferocytosis by macrophages; iv) promoting the shifting from M1 to M2 macrophage phenotypes; v) organizing the coming back of non-apoptotic cells to the blood or the egress via the lymphatic vasculature; vi) stimulating the cellular repopulation of the tissue, aiming to adapted homeostasis [16].
Nevertheless, unresolved inflammation and permanent release of pro-inflammatory mediators are responsible for the promotion of a condition called “low-grade systemic chronic inflammation”, characterized by tissue and organ damages, metabolic changes and increased predisposition to NCDs [2,17]. In the last twenty years, the therapeutic approach has been focused on the possibility to positively impact on the “resolution phase” of the inflammatory process [18].
Rheumatoid arthritis (RA) is an autoimmune disease that leads progressive joint damage, threatening the quality of life, and increasing functional disability. The pathogenesis of RA is unknown, but starting from the beginning of XXI century, dramatic improvements in understanding the basic mechanisms have been made, leading to significant changes in RA therapies [19,20]. Additionally, diverse studies have demonstrated that different dietary components may influence modifiable risk factors for diverse chronic human pathologies [21]. Particular attention has been drawn to fish consumption since it lowers plasma lipid concentrations and attenuates inflammation. Indeed, fish is the main source of omega-3 long-chain polyunsaturated fatty acids (n-3 PUFAs) [22].
The aim of this article is to review the recent literature that relates to the modulation of inflammatory-based disease, such as RA, by n-3 PUFAs.
2. Omega-3 polyunsaturated fatty acids (n-3 PUFAs) and specialized pro-resolving mediators (SPMs)
Omega-3 fatty acids are a family of n-3 PUFAs characterized by the presence of the last double bond between carbon 3 and 4. The main components of this family are eicosapentaenoic (EPA), docosapentaenoic (DPA), and docosahexaenoic acid (DHA). EPA and DHA are abundant in the flesh of both lean and oily fish (with greater amount being for DHA) and in supplements, such as fish oils, cod liver oil and krill oil, and in some algal oils [23]. It is widely recognized that the anti-inflammatory activity of n-3 PUFAs involves their incorporation into cell membrane’s phospholipids at the expense of arachidonic acid (ARA). This effect causes the decrease of the amount of AA in the membranes. Therefore, inhibiting ARA metabolism and expression of cyclooxygenase (COX) gene, and eventually decreasing the ARA-derived eicosanoids produced [24]. These processes are triggered by inflammatory stimuli, which firstly active the enzyme phospholipase A2 responsible for the hydrolysis of the sn-2 acyl chain of glycerol phospholipids, originating free AA or free EPA or DHA. Three main enzymatic pathways are responsible for eicosanoids production from ARA: 1) COX (COX1 and COX2); 2) 5-LOX, 12-LOX and 15-LOX; and 3) cytochrome 450 mixed function oxidase enzymes (CYP450). The classical eicosanoids are PGs, thromboxanes (TXs), and LTs, which are the best-known mediators and regulators of inflammation [25].
Several in vitro and in vivo studies demonstrated that n-3 PUFAs decrease cell surface expression of adhesion molecules, production of inflammatory cytokines (such as tumor necrosis factor (TNF)-alpha, IL-1 beta and IL-6) and COX-2 metabolites [26]. These effects are mediated by the ability of n-3 PUFAs to impact on the nuclear factor kappa B (NFkB) pathway [23]. Specifically, NFkB is one of the transcription factors engaged in the up-regulation of genes encoding inflammatory proteins. The cytosolic and inactive form of the NFkB is a trimer and its activation is promoted by inflammatory stimuli through a signaling cascade, including endotoxin (for example, lipopolysaccharides, LPS) binding to toll-like receptor (TLR) 4. This cascade starts with the phosphorylation of the inhibitory subunit of NFkB, which then dissociates from the trimer and the remaining dimer is able to translocate into the nucleus, where it binds to response elements and up-regulates inflammatory genes [27,28]. Nowadays, three alternative mechanisms have been highlighted to explain the ability of n-3 PUFAs to dampen inflammatory signaling via NFkB: 1) activation of peroxisome proliferator activated receptor (PPAR)-gamma which physically interacts with the dimeric form of NFkB preventing its nuclear translocation; 2) interfering with the raft formation in the membrane of inflammatory cells via TLR4 and myeloid differentiation primary response gene 88 (MyD88); 3) binding to G-protein coupled cell membrane receptor (GPR120) which trigger an anti-inflammatory cascade stimuli able to inhibit NFkB activation [23].
Besides exerting these anti-inflammatory activities, n-3 PUFAs have been indicated as substrates for the synthesis of specialized pro-resolving lipid mediators (SPMs). These SPMs are molecules recognized having a central role in inflammation resolution and in the protection of the organism against the harmful consequences of uncontrolled inflammatory process [29]. SPMs, synthesized by the action of COX, LOX and CYP450 enzymes, include resolvins (Rvs), protectins (aka as neuroprotectins, PD1/NPD1), maresins (MaRs), and the novel cysteinyl-SPMs (cys-SPMs). In addition, in the presence of aspirin, different epimers of SPMs are produced and are called aspirin-triggered (AT) SPMs [7]. These biosynthetic pathways may occur within a single cell type (if all the enzymes are expressed) or in a transcellular way, with the early steps occurring in one cell type and the following once in a different cell that synthetizes the final biologically active lipid mediator [30]. Specifically, SPMs play a key role in several pro-resolving mechanisms: 1) limiting granulocytes chemotaxis and infiltration in vivo; 2) stimulating macrophage phagocytosis and efferocytosis; 3) enhancing macrophage M2 polarization; 4) accelerating wound healing; 5) reducing pro-inflammatory cytokine production (TNF-alpha and IL-1beta) and lipid mediators (PGs and LTs); 6) promoting Treg response and release of IL-10; 7) reducing platelet aggregation and inflammasome formation [7]. Moreover, studies performed in animal models of both acute and chronic inflammation have reported the therapeutic efficacy of SPMs [30]. These data could open a new strategy for the treatment of diverse inflammatory-based pathologies, being, at least in part, free of the adverse effects commonly observed in clinical trials using standard of care therapies [31].
DHA is the precursor of D-series Rvs, PD1/NPD1, MaR1 and cys-SPMs (Figure 1).
Studies have demonstrated that RvDs are strong immunoresolvents agents, acting through mainly two G-couple receptors, ALX/DRV1/GPR32[32] and DRV2/GPR18 that bind specifically RvD1 and RvD2, respectively [33]. PD1/NPD1, through its receptor GPR37, exerts its anti-inflammatory functions in neural (NPD1) and immune systems (PD1). It has been demonstrated its ability to protect the host from ischemic stroke, retina degenerative disease, and traumatic brain injury [34]. Structural studies have identified a positional isomer of PD1, called PDX, able to inhibit platelet activation, enhance insulin sensitivity and reduce atherosclerosis, even though a specific receptor still needs to be identified [35]. MaR1, a DHA-macrophage derived maresins, synthetized by 12-LOX pathways promotes fundamental pro-resolving functions of macrophages, by interacting with a cell surface leucin-rich repeat-containing G protein-coupled receptor, LGR6 [36]. Cys-SPMs is a group of pro-resolving and pro-repair mediators containing three series of peptide-lipid conjugated SPMs, such as resolving conjugates in tissue regeneration (RCTRs) [37], protectin conjugates in tissue regeneration (PCTRs) [38], and maresin conjugates in tissue regeneration (MCTRs) [39].
E-series Rvs (RvE1, RvE2, RvE3, and RvE4) are instead biosynthesized from EPA, and RvE1 and RvE2 act interacting with their receptors, ERV1/ChemR23 and BLT1, respectively (Figure 2A) [40]. RvE1 binding to ERV1/ChemR23 promotes the activation of intracellular signals, such as phosphorylation of kinase. Experimental studies using agonist to RVE1 receptor highlighted that the stimulation of the endogenous resolution mechanisms may control both inflammation and cancer progression [34].
Alongside the above cited and well characterized DHA and EPA-derived SPMs, almost ten years ago Dalli et al identified analogous compounds synthetized from the third marine n-3 PUFAs, i.e. DPA (Figure 2B), that have been assigned to the Rv, PD and MaR families [41]. Specifically, they were designated as RvDn-3 DPA (RvD1,2,5n-3 DPA), PDn-3 DPA (PD1,2,3n-3 DPA) and MaRn-3 DPA (MaR1,2,3n-3 DPA) (Figure 2) [41]. In addition, few years later, the same authors characterized the structure and function of novel 13-series resolvins, named RvT1, RvT2, RvT3 and RvT4, specifically produced in the circulation during the interaction between vascular endothelial cells and neutrophils (Figure 2B) [42]. Interestingly, the synthesis of these new immunomodulatory compounds is stimulated by statins, particularly atorvastatin and pravastatin, via nitrosylation of COX-2, which enhances its enzymatic activity. A mouse model of arthritis it has been demonstrated that both atorvastatin and pravastatin increase the tissue and plasma concentrations of RvTs and are able to diminish the joint disease as well as the leukocytes trafficking and activation [43].
3. Rheumatoid arthritis (RA)
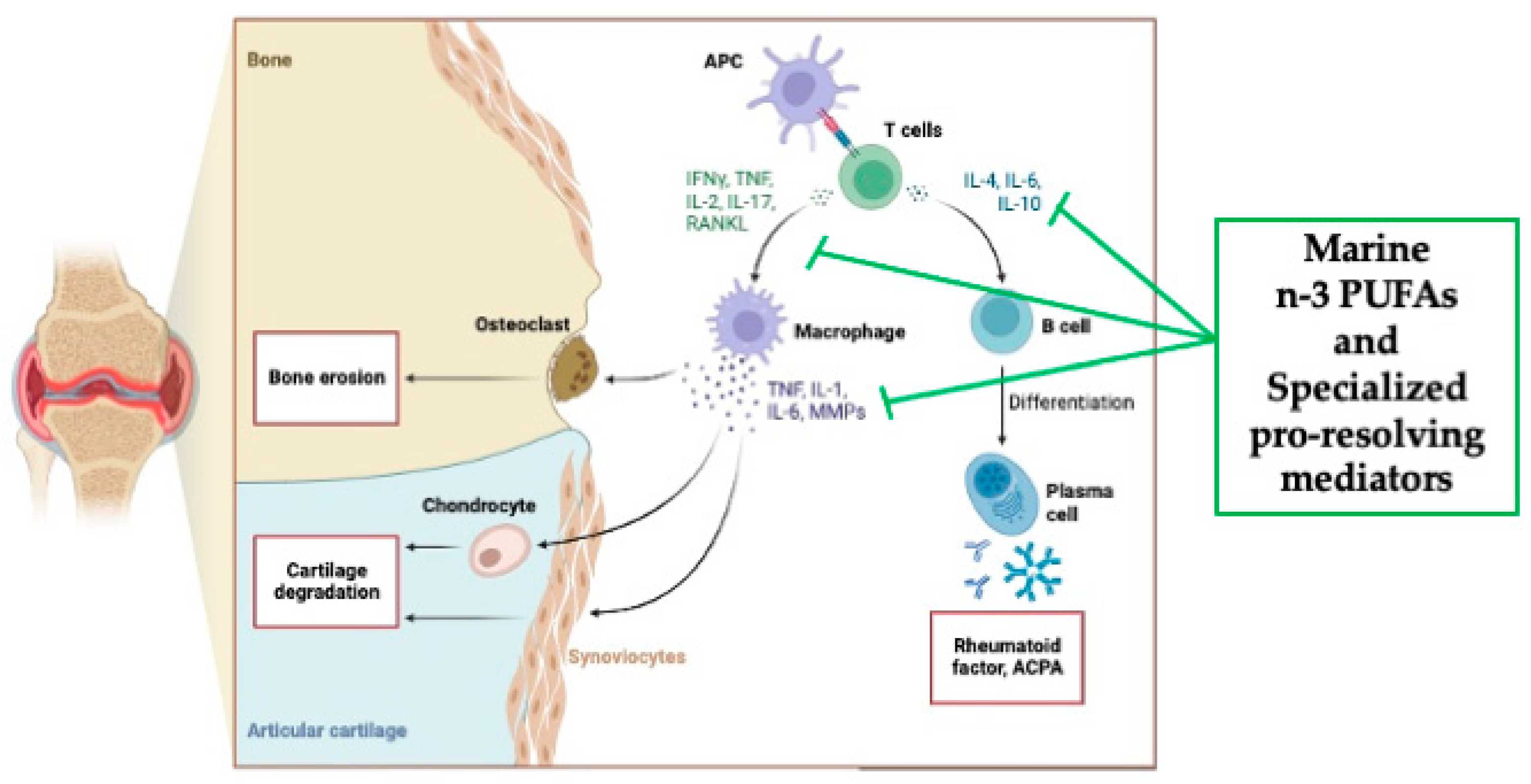
RA is a systemic autoimmune disorder, characterized by chronic inflammation of the joints, which manifests as swelling, pain, functional impairment and morning stiffness, and autoantibody production, i.e. rheumatoid factor (a high affinity autoantibody against the Fc portion of immunoglobulin) and anti-citrullinated protein antibody (ACPA) [44]. Additionally, RA includes systemic features, such as cardiovascular disease (CVD), and pulmonary, psychological, and skeletal disorders [45,46,47]. Any peripheral joint could be affected, but most commonly affected joints are within the feet, knees and hands. The prevalence of RA is 0.5% to 1% of the population worldwide, with the predominant percentage being women [48]. The etiological agent responsible for triggering the onset of RA is unknown. However, it is well recognized that RA comprises a complex interplay among genotype, environmental triggers, and change [45]. Conventional and genome-wide approaches have been applied to identify more than 100 loci associated with RA risk and progression [49,50]. Specifically, the majority of these loci are genes encoding: i) MHC class II molecules, mainly HLADR locus which is implicated in T-cell recognition of autoantigens [51]; ii) co-stimulatory pathways (CD28, CD40, chemokines, and cytokine receptors) [52], post-translational modification enzymes [i.e., peptidyl arginine deiminase, type IV (PADI4), responsible for the conversion of peptidylarginine to citrulline] [53], and intracellular regulatory pathways (i.e. PTPN22, TNFAIP3, STAT3) [54,55], all potentially able to modify the threshold for immune activation or failed regulation. These observations have been confirmed by studies of gene-environment interactions. Indeed, smoking and other forms of bronchial stress (e.g., exposure to silica dust) are directly associated to the RA risk’s increase among persons with susceptibility HLA-DR4 alleles [56,57]. While, smoking and HLA-DRB1 alleles work together in raising one’s risk of having ACPA [58]. Additionally, environmental stressors of pulmonary and other barrier tissue could trigger the post-translational activity of PADI4, eventually leading to quantitative or qualitative alteration in citrullination of mucosal proteins [59]. Among the citrullinate self-proteins recognized through the diagnostic assay are alpha-enolase, keratin, fibrinogen, fibronectin, collagen and vimentin [60]. Infection diseases, caused by diverse virus and bacteria species as well as by their products, have been correlated to the onset of RA [61]. The formation of immune complexes during infection may indeed stimulate the synthesis of rheumatoid factor [62]. Since it has been observed that Porphyromonas gingivalis expresses PADI4, RA seems to be associated with periodontal disease [63]. Lastly, the gastrointestinal microbiota has been identified as a factor influencing the development of autoimmunity, because it is involved in maintaining immune homeostasis and acts as a marker of the host’s health status [64,65]. Perturbation of this interaction can affect mucosal and systemic immunity, and promote diverse inflammatory and autoimmune disease, such as RA [66]. Many critical questions still need an answer, mainly why the systemic loss of tolerance transforms a pauci-cellular synovium into a chronically inflamed tissue? However, progresses in understanding the pathogenesis of the disease have promoted the discovery of new pharmacological treatments, leading to improved outcomes [45].
It is conceivable that infections or traumatic insults could induce the initial inflammatory response into the synovium. Synovium is the tissue lining the joints, and it is the place where the infiltration of neutrophils, B and T lymphocytes, monocyte-derived macrophages and the proliferation of fibroblast-like synovial cells, called synoviocytes, occurs [67]. As the disease develops, the cells of the synovial lining proliferate, forming together with new blood vessels an invasive pannus, which leads to progressive destruction of cartilage and bone [68]. This condition can lead to muscle wasting and increased risk of osteoporosis [69]. Synovial B cells are mainly localized in T-cell-B-cell-aggregates, and their activation is endorsed by the expression of factors including a proliferation-inducing ligand (APRIL), B-lymphocyte stimulator (BLyS), and CC and CXC chemokines [70]. Of note, a multicenter, randomized, double-blind, controlled study demonstrated that a selective depletion of B cells throughout the administration of rituximab, a genetically engineered chimeric anti-CD20 monoclonal antibody, to patients with active RA, proved efficacy in ameliorating disease symptoms [71]. Importantly, macrophages play a key role in synovitis. Clinical effective biological agents, indeed, aim at hampering macrophage infiltration in the synovium [72]. Specifically, by looking at the pattern of pro-inflammatory cytokine’s expression the hypothesis is that the M1 macrophage phenotype is the predominant one [45]. Diverse are the macrophage activating pathways, mainly i) toll-like receptors (TLRs) 2/6, 3, 4 and 8; ii) nucleotide-associated molecular patterns; iii) damage-associated molecular patterns, such as bacterial, viral, and putative endogenous ligand; iv) cytokines; v) cognate interactions with T cells; vi) immune complexes; vii) lipoprotein particles; viii) liver X-receptors agonists, such as oxysterols, oxidized low-density lipoproteins (LDL), ix) serum amyloid A-rich high-density lipoproteins (HDL); x) protease-rich microenvironment through protease-activated receptor 2 [73]. High concentrations of different cytokines and chemokines, including IL-1beta, IL-6, IL-8, IL-21, IL-23, IL-17A, IL-17F and TNF-alpha, have been detected in the synovial fluid of patients with RA and have been associated with the development of the disease [74]. Of note, TNF-alpha and IL-6 play a fundamental role in the RA, confirmed by successful therapeutic blockade of membrane and soluble TNF-alpha and the IL-6 receptor in patients affected by RA [45]. TNF-alpha, indeed, triggers the expression of cytokines, chemokines, and endothelial-cell adhesion molecules; protects the synovial fibroblasts; promotes the angiogenesis; suppresses the regulatory T cells; induces the pain sensation [75,76]. While, IL-6 orchestrates local leukocyte activation and autoantibody production, together with systemic effects that promote acute-phase responses, anemia, cognitive disfunction, and lipid-metabolism dysregulation [77]. An additional pathogenic pathway includes antigen-nonspecific, T-cell contact-mediated activation of macrophages and fibroblasts, acting through their ligand’s interactions, i.e., CD40-CD40L, Cd200-CD200L, intracellular adhesion molecule 1 (ICAM-1) and leukocyte-function-associated antigen 1 [78]. Moreover, the AA-derived lipid mediators of inflammation, PGE2, LTB4, 5-hydroxyeicosatretraenic acid and platelet-activating factor (PAF) are also found in the synovial fluid and are believed to be involved in the recruitment of leukocytes into the diseased tissue [79]. Finally, as stated above RA is characterized by bone erosion, that occurs rapidly (affecting 80% of patients within 1 year after diagnosis [80]) and is correlated with prolonged, increased inflammation [81]. Synovial cytokines, specifically macrophage colony-stimulating factor and receptor activator of NF-kB ligands (RANKL), promote osteoclast differentiation and migration of the periosteal surface adjacent to articular cartilage [82]. It is well demonstrated that osteoclasts possess the acidic enzymatic machinery necessary to demolish mineralized tissues, including mineralized cartilage and subchondral bone. This osteoclast’s acidic activity leads to deep resorption pits, which are replaced by inflammatory tissue [82]. Clinical studies have confirmed the pivotal role of these cytokines because their inhibition alts erosion in RA [83].
The clinical diagnosis of RA is based on criteria developed by the European and American rheumatology association [84] and treatment are focused on disease-modifying antirheumatic drugs (DMARDs), that target remission of the disease [84]. Several studies have showed that starting the treatment with DMARDs at the early stages of RA is associated with a positive change in the progression of the disease, leading to a slow-down in the joint destruction [85]. The number of the therapeutic options available for the treatment of RA is increased dramatically in the past 30 years [86]. Specifically, non-steroidal anti-inflammatory drugs, glucocorticoids [87], and DMARDs of synthetic origin [i.e. methotrexate or janus kinase (JAK)-inhibitors] or of biological origin (i.e., TNF-inhibitors, costimulation modifiers, IL-6-inhibitors, and B cell depleting drugs) are included in the first-line protocols for RA patients [88,89]. Nevertheless, due to the heterogeneity of RA pathophysiology many patients do not attain the clinical remission, causing reduced mobility, disability, and low quality of life [90]. Additionally, life style changes (i.e., smoking cessation, dental care, weight control), assessment of vaccination status, and management of comorbidities should be included in the standard of care therapies [91].
Among the modifiable factors influencing the development and the progression of RA, smoking, alcohol and unsaturated fatty acids (FAs) consumption have been indicated as the most important predictors [92]. For this reason, even if the inflammation would be controlled by the biologic agents, appropriate nutritional intervention should still be considered for these patients. In this context, nutrition plays both direct roles in disease development, carrying pro or anti-inflammatory food components, and indirect roles through effects on the body mass index, visceral fat accumulation, and by contributing to the onset or the prevention of diabetes and CVD [93]. In addition, changes in lifestyle, of which healthy eating is one pillar, physical activity, management of stress, avoiding risky substances and social connections, may be effective in preventing RA. The most popular nutritional approaches include the Mediterranean diet and the dietary supplements, especially fish oil or n-3 PUFAs [94].
Importantly, n-3 PUFAs and their SPMs lipid mediators (Figure 3), being recognized as anti-inflammatory molecules through their ability to suppress inflammatory signaling via NFkB [20], have been widely investigated in the context of RA. Two experimental studies showed that n-3 PUFAs, i.e. EPA and DHA, reduced the incidence and severity of arthritis in two different animal models of arthritis [20,95,96], with EPA being the more effective compared with DHA. Furthermore, Ierna et al [97] demonstrated that in mice, subjected to collagen-induced arthritis, the administration of krill oil, which provides marine n-3 PUFAs partly in the form of phospholipids, was more efficacious than fish oil, mainly composed of n-3 PUFAs in triacylglycerol form, in slowing the onset of arthritis, reducing its severity, decreasing paw swelling, and reducing knee joint pathology. These results are in line with studies showing a more effective delivery of marine n-3 PUFAs from phospholipids than from triacylglycerols [98].
The dietary consumption of n-3 PUFAs could play a key role in the development of RA. An observational study by Di Giuseppe et al found that dietary n-3 PUFAs intake greater than 0.21 g/day was associated with a 35% lower risk of developing RA, as compared with women consuming a reduced amount [99]. Moreover, long-term consumption of fish >1 serving/week was correlated with a 29% reduction of RA risk compared with <1 serving/week [99]. Similar results were reported by Rosell [100] and Pedersen [101], confirming the association between n-3 PUFAs consumption and reduction in the development of RA. In addition, a recent systematic review and meta-analysis, including seven cohort and six-case control studies, demonstrated that for every 100 g/day increment in fish intake there was a 15% lower risk of developing RA [102].
Of note, nutrition has been shown to have an influence on disease flares, overall management, and clinical outcomes. One of the first study was conducted by Kremer et al [103] in RA patients treated with 1.8 g of EPA for 12 weeks. The results showed that patients reported both a reduction in joint stiffness in the morning and in the number of tender joints. In a study by Espersen et al, the plasma concentrations of IL-1 beta were reduced (p<0.001) in patients treated with 3.6 g/day of n-3 PUFAs for 12 weeks [104]. Sperling et al demonstrated that fish oil supplementation was able to decrease the AA/EPA ratio by 30% and the mean LTB4 production by 33%, together with a 37% reduction of PAF production [105]. These data were later confirmed by Dawczynski [106]. In this study has been also observed a significant decrease in the AA/EPA ratio in the erythrocyte membranes. The same authors showed that n-3 PUFAs were able to inhibit the immune response through the reduction of the number of lymphocytes and monocytes recruited [107]. Diverse studies revealed that patients taking n-3 PUFAs achieved significant improvements in global assessment and pain, and reduction in antirheumatic medication (about 30%) [108,109,110]. In addition, in the study by Geusens et al [109] it was observed that high-dose (2.6 g /day) proved more efficacy in reducing RA pain than low-dose (1.3 g /day). Meta-analyses have been published to provide information concerning the influence of n-3 PUFAs on clinical manifestation on RA patients. Goldberg et al [111] evaluated the pain-relieving effects of EPA/DHA in RA patients. Seventeen randomized controlled trials (RCTs) were included in the final meta-analysis. Patients receiving different doses of n-3 PUFAs, ranging from 1.8 to 9.6 g/day, were compared with individuals receiving different type of placebo (such as: soy oil, linoleic acid capsules, air-filled capsules, olive oil, coconut oil, corn oil, water). Six different pain outcomes were identified: 1) patient assessed pain; 2) physician assessed pain; 3) duration of morning stiffness; 4) number of painful and/or tender joints; 5) Ritchie articular index [112]; 6) nonselective nonsteroidal anti-inflammatory drugs (NSAIDs) consumption. The results of this meta-analysis suggest that n-3 PUFAs supplementation reduces patient assessed pain, morning stiffness, number of painful and/or tender joints, and NSAIDs consumption, but has no effect on physician assessed pain and Ritchie articular index [111]. In order to maximize the therapeutic efficacy, the authors suggested for the future trials that: i) all studies clearly stated the doses and type of NSAIDs used and ii) a non-olive oil placebo will be included as control. The reason is based on the anti-inflammatory proprieties of olive oil previously demonstrated. Indeed, oleic acid, its main constituent, may compete with AA for incorporation into phospholipids, and minor components, such as tyrosol and beta-sitosterol, possess anti-oxidant and anti-inflammatory effects [111,113]. In 2012, Lee et al published a meta-analysis considering only the ten RCTs, already included in the previous study, in which RA patients were treated with a dose of n-3 PUFAs above 2.7 g/day for a minimum duration of three months [114]. No clinical outcome measures were improved by n-3 PUFAs supplementation, with only a trend towards tender joint count, swollen joint count, morning stiffness, and physical function recovery observed in n-3 PUFAs treated group. The authors highlighted that NSAIDs requirements were significantly different between the n-3 PUFAs and the placebo groups. However, this association was gathered from only two case-control studies. Among the outcomes evaluated in this meta-analysis, no results were reported concerning the impact of n-3 PUFAs on erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) [112]. A more recent meta-analysis was published by Gioxari [48]. This meta-analysis included 20 RCTs with the following inclusion criteria: written in English; conducted in humans; oral intake of n-3 PUFAs (either as a supplement or from food source); duration of the study greater or equal three months; the maintenance of conventional drug treatment for all the study course. The outcomes evaluated were: i) changes in RA disease activity (through the monitoring of 16 RA markers), ii) inflammation (measuring CRP, IL-1, IL-6, TNF-alpha, and LTB4), and iii) risk for CVD (assessing CVD-related risk factors, such as TG, TC, LDL and HDL). Oral treatment with n-3 PUFAs proved efficacy in ameliorating several RA disease markers, namely early morning stiffness, tender joint count, pain scale, Ritchie articular index, health assessment questionnaire, grip strength, ESR, and in reducing plasma levels of LTB4 and TG. No effects were detected on TC, LDL and HDL plasma concentrations [48]. These data were also confirmed by a randomized, single-blind intervention study. RA patients were treated with 1800 mg/d of EPA and 1200 mg/d of DHA for 90 days. Compared with the baseline values, RA patients showed reductions in diverse RA disease markers and CRP levels [115]. Additionally, an in vitro study found that Rvd5 suppressed Th17 cell differentiation and CD4+ T cell proliferation, facilitated Treg differentiation, and inhibited osteoclastogenesis [116]. Of note, a cross-sectional study demonstrated that taking oral over-the-counter fish oil supplementation increases DHA and EPA plasma concentrations as well as SPMs precursors levels. Specifically, they have detected a rise in the concentration of both 18-hydroxy-EPA and 17-hydroxy-DHA that have been proved to be potent inhibitors of macrophage-mediated pro-inflammation, in addition of being substrate for SPMs [117]. Finally, two systematic reviews highlighted from one hand an inverse correlation between the highest compared the lowest category of fish consumption and RA risk, and from the other the efficacy of a dose >2g/d of PUFAs at improving tender and swollen joints, morning stiffness, and a reduction on NSAIDs use [118]. While, a meta-analysis of 23 randomized placebo-controlled trials revealed only a trend toward benefit following n-3 PUFAs supplementation for improving RA disease parameters [119].
4. Conclusions and Perspectives
Inflammation is a physiological process, within the immune response, involved in the protection of the organism against pathogens and in the reaction to injury and in wound healing. The inflammatory response is normally a self-limiting and resolving process, thanks to the activation of diverse negative feedback mechanisms. Indeed, in parallel to an appropriate functioning of the inflammatory response are numerous pro-resolving lipid mediators, such as n-3 PUFA-derived SPMs, implicated in the active “resolution phase” of the inflammation. However, excessive or uncontrolled inflammation can be harmful and responsible of tissue damage and of the pathogenesis of several human disease. Marine-derived n-3 PUFAs have shown potential efficacy in reducing inflammation and promoting resolution, thus preventing or modulating several NCDs, such as rheumatoid arthritis. Altogether the data reported in this review show that anti-inflammatory interventions, i.e. high fish consumption or supplements containing n-3 PUFAs, should be the standard of care, along with pharmacotherapy, in treating patients with RA. However, all the investigators strengthened the need of longer, larger and well-designed clinical studies to achieve a final statement. Finally, it is important to underline that researchers may need to focus on the diversity of lipid-derived mediator classes to better understand the therapeutic potential of n-3 PUFAs supplements.
Author Contributions
C.P. conceived the idea and reviewed the published literature. C.P. wrote, revised and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by MIUR Progetto Eccellenza.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- World Health Organization, Available online: https://www.who.int/news-room/fact-sheets/detail/noncommunicable-diseases.
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. 2019, 25, 1822-1832. [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. 2018, 15, 505-522. [CrossRef]
- Calder, P.C.; Albers, R.; Antoine, J-M.; Blum, S.; Bourdet-Sicard, R.; Ferns, G.A.; Folkerts, G.; Friedmann, P.S.; Frost, G.S.; Guarner, F.; et al. Inflammatory disease processes and interactions with nutrition. Br J Nutr. 2009, 101, S1-45. [CrossRef]
- Netea, M.G.; Balkwill, F.; Chonchol, M.F.; Cominelli, F.; Donath, M.Y.; Giamarellos-Bourboulis, E.J.; Golenbock, D.; Gresnigt, M.S.; Heneka, M.T.; Hoffman, H.M.; et al. A guiding map for inflammation. Nat Immunol. 2017, 18, 826-831. [CrossRef]
- Larsen, G.L.; Henson, P.M. Mediators of inflammation. Annu Rev Immunol. 1993, 1, 335-359. [CrossRef]
- Serhan, C.N. Treating inflammation and infection in the 21st century: new hints from decoding resolution mediators and mechanisms. Faseb J. 2017, 31, 1273-1288. [CrossRef]
- Barnig, C.; Bezema, T.; Calder, P.C.; Charloux, A.; Frossard, N.; Garssen, J.; Haworth, O.; Dilevskaya, K.; Levi-Schaffer, F.; Lonsdorfer, E.; et al. Activation of Resolution Pathways to Prevent and Fight Chronic Inflammation: Lessons from Asthma and Inflammatory Bowel Disease. Front Immunol. 2019, 10, 1699. [CrossRef]
- Dalli, J.; Serhan, C.N. Pro-Resolving Mediators in Regulating and Conferring Macrophage Function. Front Immunol. 2017, 8, 1400. [CrossRef]
- Smith, T.D.; Nagalla, R.R.; Chen, E.Y.; Liu, W.F. Harnessing macrophage plasticity for tissue regeneration. Adv Drug Deliv Rev. 2017, 114, 193-205. [CrossRef]
- Li, J.; Tan, J.; Martino, M.M.; Lui, K.O. Regulatory T-Cells: Potential Regulator of Tissue Repair and Regeneration. Front Immunol. 2018, 9 585. [CrossRef]
- Rauber, S.; Luber. M.; Weber, S.; Maul, L.; Soare, A.; Wohlfahrt, T.; Lin, N-Y.; Dietel, K.; Bozec, A.; Herrmann, M.; et al. Resolution of inflammation by interleukin-9-producing type 2 innate lymphoid cells. Nat Med. 2017, 23, 938-944. [CrossRef]
- Poe, S.L.; Arora, M.; Oriss, T.B.; Yarlagadda, M.; Isse, K.; Khare, A.; Levy, D.E.; Lee, J.S.; Mallampalli, R.K.; Chanet, Y.R. et al. STAT1-regulated lung MDSC-like cells produce IL-10 and efferocytose apoptotic neutrophils with relevance in resolution of bacterial pneumonia. Mucosal Immunol. 2013, 6, 189-199. [CrossRef]
- Ray, A.; Chakraborty, K.; Ray, P. Immunosuppressive MDSCs induced by TLR signaling during infection and role in resolution of inflammation. Front Cell Infect Microbiol. 2013, 3, 52. [CrossRef]
- Levy, B.D.; Clish, C.B.; Schmidt, B.; Gronert, K.; Serhan, C.N. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol. 2001, 2, 612-619. [CrossRef]
- Sugimoto, M.A.; Vago, J.P.; Perretti, M.; Teixeira, M.M. Mediators of the Resolution of the Inflammatory Response. Trends Immunol. 2019, 40, 212-227. [CrossRef]
- Fullerton, J.N.; Gilroy, D.W. Resolution of inflammation: a new therapeutic frontier. Nat Rev Drug Discov. 2016, 15, 551-567. [CrossRef]
- Gilroy, D.W.; Lawrence, T.; Perretti, M.; Rossi, A.G. Inflammatory resolution: new opportunities for drug discovery. Nat Rev Drug Discov. 2004, 3, 401-416. [CrossRef]
- Aletaha, D.; Smolen, J.S. Diagnosis and Management of Rheumatoid Arthritis: A Review. JAMA. 2018, 320, 1360-1372. [CrossRef]
- Figus, F.A.; Piga, M.; Azzolin, I.; McConnell, R.; Iagnocco, A. Rheumatoid arthritis: Extra-articular manifestations and comorbidities. Autoimmun Rev. 2021, 20, 102776. [CrossRef]
- Illiano, P.; Brambilla, R.; Parolini, C. The mutual interplay of gut microbiota, diet and human disease. Febs J. 2020, 287, 833-855. [CrossRef]
- Parolini, C. Effects of Fish n-3 PUFAs on Intestinal Microbiota and Immune System. Mar Drugs. 2019, 17, 374. [CrossRef]
- Calder, P.C. Marine omega-3 fatty acids and inflammatory processes: Effects, mechanisms and clinical relevance. Biochim Biophys Acta. 2015, 4, 469-484. [CrossRef]
- Parolini, C. Marine n-3 polyunsaturated fatty acids: Efficacy on inflammatory-based disorders. Life Sci. 2020, 263, 118591. [CrossRef]
- Lee, J.Y.; Sohn, K.H.; Rhee, S.H.; Hwang, D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem. 2001, 276, 16683-16689. [CrossRef]
- Kumar, A.; Takada, Y.; Boriek, A.M.; Aggarwal, B.B. Nuclear factor-kappaB: its role in health and disease. Mol Med (Berl). 2004, 82, 434-448. [CrossRef]
- Perkins, N.D. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007, 8, 49-62. [CrossRef]
- Gabbs, M.; Leng, S.; Devassy, J.G.; Monirujjaman, M.; Aukema, H.M. Advances in Our Understanding of Oxylipins Derived from Dietary PUFAs. Adv Nutr. 2015, 6, 513-540. [CrossRef]
- Spite, M.; Norling, L.V.; Summers, L.; Yang, R.; Cooper, D.; Petasis, N.A.; Flower, R.J.; Perretti, M.; Serhan, C.N. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature. 2009, 461, 1287-1291. [CrossRef]
- Panigrahy, D.; Gilligan, M.M.; Serhan, C.N.; Kashfi, K. Resolution of inflammation: An organizing principle in biology and medicine. Pharmacol Ther. 2021, 227, 107879. [CrossRef]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014, 510, 92-101. [CrossRef]
- Serhan, C.N.; Brain, S.D.; Buckley, C.D.; Gilroy, D.W.; Haslett, C.; O’Neill, L.A.J.; Perretti, M.; Rossi, A.G.; Wallace, J.L.; Resolution of inflammation: state of the art, definitions and terms. Faseb J. 2007, 21, 325-332. [CrossRef]
- Zhang, L.; Qiu, C.; Yang, L.; Zhang, Z.; Zhang, Q.; Wang, B.; Wang, X. GPR18 expression on PMNs as biomarker for outcome in patient with sepsis. Life Sci. 2019, 217, 49-56. [CrossRef]
- Dyall, S.C.; Balas, L.; Bazan, N.G.; Brenna, J.T.; Chiang, N.; da Costa Souza, F.; Dalli, J.; Durand, T.; Galano, J-M.; Lein, P.J. Polyunsaturated fatty acids and fatty acid-derived lipid mediators: Recent advances in the understanding of their biosynthesis, structures, and functions. Prog Lipid Res. 2022, 86, 101165. [CrossRef]
- Serhan CN, Gotlinger K, Hong S, et al. (2006) Anti-inflammatory actions of neuroprotection D1/protectin D1 and its natural stereoisomers: assignments of dihydroxy-containing docosatrienes. J Immunol 176, 1848. [CrossRef]
- Schwarz, B.; Sharma, L.; Robert, L.; Peng, X.; Bermejo, S.; Leighton, J.; Casanovas-Massana, A.; Minasyan, M.; Farhadian, S.; Ko, A.I. Cutting Edge: Severe SARS-CoV-2 Infection in Humans Is Defined by a Shift in the Serum Lipidome, Resulting in Dysregulation of Eicosanoid Immune Mediators. J Immunol. 2021, 206, 329-334. [CrossRef]
- de la Rosa, X.; Norris, P.C.; Chiang, N.; Rodriguez, A.R.; Spur, B.W.; Serhan, C.N.; Identification and Complete Stereochemical Assignments of the New Resolvin Conjugates in Tissue Regeneration in Human Tissues that Stimulate Proresolving Phagocyte Functions and Tissue Regeneration. Am J Pathol. 2018, 188, 950-966. [CrossRef]
- Ramon, S.; Dalli, J.; Sanger, J.M.; Winkler, J.W.; Aursnes, M.; Tungen, J.E.; Hansen, T.V.; Serhan, C.N. The Protectin PCTR1 Is Produced by Human M2 Macrophages and Enhances Resolution of Infectio us Inflammation. Am J Pathol. 2016, 186, 962-973. [CrossRef]
- Dalli, J.; Sanger, J.M.; Rodriguez, A.R.; Chiang, N.; Spur, B.W.; Serhan, C.N. Identification and Actions of a Novel Third Maresin Conjugate in Tissue Regeneration: MCTR3. PloS One. 2016, 11, e0149319. [CrossRef]
- Chiang, N.; Serhan, C.N. Structural elucidation and physiologic functions of specialized pro-resolving mediators and their receptors. Mol Aspects Med. 2017, 58, 114-129. [CrossRef]
- Dalli, J.; Colas, R.A.; Serhan, C.N. Novel n-3 immunoresolvents: structures and actions. Sci Rep. 2013, 3, 1940. [CrossRef]
- Dalli, J.; Chiang, N.; Serhan, C.N. Elucidation of novel 13-series resolvins that increase with atorvastatin and clear infections. Nat Med. 2015, 21, 1071. [CrossRef]
- Walker, M.E.; Souza, P.R.; Colas, R.A.; Dalli, J. 13-Series resolvins mediate the leukocyte-platelet actions of atorvastatin and pravastatin in inflammatory arthritis. Faseb J. 2017, 31, 3636-3648. [CrossRef]
- Firestein, G.S. Evolving concept of rheumatoid arthritis. Nature. 2003, 423, 356-361. [CrossRef]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011, 365, 2205-2219. [CrossRef]
- Myasoedova, E.; Davis, J.MIII.; Crowson, C.S.; Gabriel, S.E. Epidemiology of rheumatoid arthritis: rheumatoid arthritis and mortality. Curr Rheumatol Rep. 2010, 5, 379-385. [CrossRef]
- Mason, J.C.; Libby, P. Cardiovascular disease in patients with chronic inflammation: mechanisms underlying premature cardiovascular events in rheumatologic conditions. Eur Heart J. 2015, 36, 482-489. [CrossRef]
- Gioxari, A.; Kaliora, A.C.; Marantidou, F.; Panagiotakos, D.P. Intake of ω-3 polyunsaturated fatty acids in patients with rheumatoid arthritis: A systematic review and meta-analysis. Nutrition. 2018, 45, 114. [CrossRef]
- MacGregor, A.J.; Snieder, H.; Rigby, A.S.; Koskenvuo, M.; Kaprio, J.; Aho, K.; Silman, A.J. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum. 2000, 43, 30-37. [CrossRef]
- Okada, Y.; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S.; et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014, 506, 376-381. [CrossRef]
- Gregersen, P.K.; Silver, J.; Winchester, R.J. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987, 30, 1205-1213. [CrossRef]
- Felix, N.J.; Suri, A.; Salter-Cid, L.; Nadler, S.G.; Gujrathi, S.; Corbo, M.; Aranda, R. Targeting lymphocyte co-stimulation: from bench to bedside. Autoimmunity. 2010, 43, 514-525. [CrossRef]
- De Rycke, L.; Peene, I.; Hoffman, I.E.A.; Kruithof, E.; Union, A.; Meheus, L.; Lebeer K.; Wyns, B.; Vincent, C.; Mielants, H.; et al. Rheumatoid factor and anticitrullinated protein antibodies in rheumatoid arthritis: diagnostic value, associations with radiological progression rate, and extra-articular manifestations. Ann Rheum Dis. 2004, 63, 1587-1593. [CrossRef]
- Kallberg, H.; Padyukov, L.; Plenge, R.M.; Ronnelid, J.; Gregersen, P.K.; van der Helm-van Mil, A.H.M.; Toes, R.E.M.; Huizinga, T.W.; Klareskog, L.; Alfredsson, L.; et al. Gene-gene and gene-environment interactions involving HLA-DRB1, PTPN22, and smoking in two subsets of rheumatoid arthritis. Am J Hum Genet. 2007, 80, 867-875. [CrossRef]
- Remmers, E.F.; Plenge, R.M.; Lee, A.T.; Graham, R.R.; Hom, G.; Behrens, T.W.; de Bakker, P.I.W.; Le, J.M.; Lee, H-S.; Batliwalla, F.; et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med. 2007, 357, 977-986. [CrossRef]
- Klareskog, L.; Stolt, P.; Lundberg, K.; Källberg, H.; Bengtsson, C.; Grunewald, J.; Rönnelid, J.; Harris, H.E.; Ulfgren, A-K.; Rantapää-Dahlqvist, S.; et al. A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. 2006, 54, 38-46. [CrossRef]
- Stolt, P.; Yahya, A.; Bengtsson, C.; Källberg, H.; Rönnelid, J.; Lundberg, I.; Klareskog, L.; Alfredsson, L.; EIRA Study Group. Silica exposure among male current smokers is associated with a high risk of developing ACPA-positive rheumatoid arthritis. Ann Rheum Dis. 2010, 69, 1072-1076. [CrossRef]
- Källberg, H.; Ding, B.; Padyukov, L.; Bengtsson, C.; Rönnelid, J.; Klareskog, L.; Alfredsson; L.; EIRA Study Group. Smoking is a major preventable risk factor for rheumatoid arthritis: estimations of risks after various exposures to cigarette smoke. Ann Rheum Dis. 2011, 70, 508-511. [CrossRef]
- van der Woude, D.; Rantapää-Dahlqvist, S.; Ioan-Facsinay, A.; Onnekink, C.; Schwarte, C.M.; Verpoort, K.N.; Drijfhout, J.W.; Huizinga, T.W.J.; Toes, R.E.M.; Pruijn, G.J.M. Epitope spreading of the anti-citrullinated protein antibody response occurs before disease onset and is associated with the disease course of early arthritis. Ann Rheum Dis. 2010, 69, 1554-1561. [CrossRef]
- Mahdi, H.; Fisher, B.A.; Källberg, H.; Plant, D.; Malmström, V.; Rönnelid, J.; Charles, P.; Ding, B.; Alfredsson, L.; Padyukov, L.; et al. Specific interaction between genotype, smoking and autoimmunity to citrullinated alpha-enolase in the etiology of rheumatoid arthritis. Nat Genet. 2009, 41, 1319-1324. [CrossRef]
- Takei, M.; Kitamura, N.; Nagasawa, Y.; Tsuzuki, H.; Iwata, M.; Nagatsuka, Y.; Nakamura, H.; Imai, K.; Fujiwara, S. Are Viral Infections Key Inducers of Autoimmune Diseases? Focus on Epstein-Barr Virus. Viruses. 2022, 14, 1900. [CrossRef]
- Kamphuis, S.; Kuis, W.; de Jager, W.; Teklenburg, G.; Massa, M.; Gordon, G.; Boerhof, M.; Rijkers, G.T.; Uiterwaal, C.S.; Otten, H.G.; et al. Tolerogenic immune responses to novel T-cell epitopes from heat-shock protein 60 in juvenile idiopathic arthriti. Lancet. 2005, 366, 50-56. [CrossRef]
- Wegner, N.; Wait, R.; Sroka, A.; Eick, S.; Nguyen, K-A.; Lundberg, K.; Kinloch, A.; Culshaw, S.; Potempa, J.; Venables, P.J. Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and α-enolase: implications for autoimmunity in rheumatoid arthritis. Arthritis Rheum. 2010, 62, 2662-2672. [CrossRef]
- Manasson, J.; Blank, R.B.; Scher, J.U. The microbiome in rheumatology: Where are we and where should we go? Ann Rheum Dis. 2020, 79, 727-733. [CrossRef]
- Scher, J.U.; Sczesnak, A.; Longman, R.S.; Segata, N.; Ubeda, C.; Bielski, C.; Rostron, T.; Cerundolo, V.; Pamer, E.G.; Abramson, S.B.; et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife. 2013, 2, e01202. [CrossRef]
- Attur, M.; Scher, J.U.; Abramson, S.B.; Attur, M. Role of Intestinal Dysbiosis and Nutrition in Rheumatoid Arthritis. Cells. 2022, 11, 2436. [CrossRef]
- Sweeney, S.E.; Firestein, G.S. Rheumatoid arthritis: regulation of synovial inflammation. Int J Biochem Biol. 2004, 36, 372-378. [CrossRef]
- Komatsu, N.; Takayanagi, H. Mechanisms of joint destruction in rheumatoid arthritis – immune cell-fibroblast-bone interactions. Nat Rev Rheumatol. 2022, 18, 415-429. [CrossRef]
- Feldmann, M.; Maini, R.N. The role of cytokines in the pathogenesis of rheumatoid arthritis. Rheumatology. 1999, 38 (Suppl. 2) 3-7.
- Ohata, J.; Zvaifler, N.J.; Nishio, M.; Boyle, D.L.; Kalled, S.L.; Carson, D.A.; Kipps, T.J. Fibroblast-like synoviocytes of mesenchymal origin express functional B cell-activating factor of the TNF family in response to proinflammatory cytokines. J Immunol. 2005, 174, 864-870. [CrossRef]
- Edwards, J.C.W.; Szczepanski, L.; Szechinski, J.; Filipowicz-Sosnowska, A.; Emery, P.; Close, D.R.; Stevens, R.M., Shaw, T. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004, 350, 2572-2581. [CrossRef]
- Haringman, J.J.; Gerlag, D.H.; Zwinderman, A.H.; Smeets, T.J.M.; Kraan, M.C.; Baeten, D.; McInnes, I.B.; Bresnihan, B.; Tak, P.P. Synovial tissue macrophages: a sensitive biomarker for response to treatment in patients with rheumatoid arthritis. Ann Rheum Dis. 2005, 64, 834-838. [CrossRef]
- Liew, F.Y.; McInnes, I.B. The role of innate mediators in inflammatory response. Mol Immunol. 2002, 38, 887-890. [CrossRef]
- Krishnamurthy, A.; Joshua, V.; Hensvold, A.H.; Jin, T.; Sun, M.; Vivar, N.; Ytterberg, A.J.; Engström, M.; Fernandes-Cerqueira, C.; Amara, K.; et al. Identification of a novel chemokine-dependent molecular mechanism underlying rheumatoid arthritis-associated autoantibody-mediated bone loss. Ann Rheum Dis. 2016, 75, 721-729. [CrossRef]
- McInnes, I.B.; Schett, G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet. 2017, 389, 2328-2337. [CrossRef]
- Hess, A.; Axmann, R.; Rech, J.; Finzel, S.; Heindl, C.; Kreitz, S.; Sergeeva, M.; Saake, M.; Garcia, M.; Kollias, G.; et al. Blockade of TNF-α rapidly inhibits pain responses in the central nervous system. Proc Natl Acad Sci U S A. 2011, 108, 3731-3736. [CrossRef]
- Li, F.; Ai, W.; Ye, J.; Wang, C.; Yuan, S.; Xie, Y.; Mo, X.; Li, W.; He, Z.; Chen, Y.; et al. Inflammatory markers and risk factors of RA patients with depression and application of different scales in judging depression. Clin Rheumatol. 2022, 41, 2309-2317. [CrossRef]
- McInnes, I.B.; Leung, B.P.; Liew, F.Y. Cell-cell interactions in synovitis. Interactions between T lymphocytes and synovial cells. Arthritis Res. 2000, 2, 374-378. [CrossRef]
- Marcouiller, P.; Pelletier, J-P.; Guévremont, M.; Martel-Pelletier, J.; Ranger, P.; Laufer, S.; Reboul, P. Leukotriene and prostaglandin synthesis pathways in osteoarthritic synovial membranes: regulating factors for interleukin 1beta synthesis. J Rheumatol. 2005, 32, 704-712.
- van der Heijde, D.M. Joint erosions and patients with early rheumatoid arthritis. Br J Rheumatol. 1995, 34, 74-78.
- Visser, H.; le Cessie, S.; Vos, K.; Breedveld, F.C.; Hazes, J.M.W. How to diagnose rheumatoid arthritis early: a prediction model for persistent (erosive) arthritis. Arthritis Rheuml. 2002, 46, 357-365. [CrossRef]
- Schett, G.; Teitelbaum, S.L. Osteoclasts and Arthritis. J Bone Miner Res. 2009, 24, 1142-1146.
- Schett, G.; Stach, C.; Zwerina, J.; Voll, R.; Manger, B. How antirheumatic drugs protect joints from damage in rheumatoid arthritis. Arthritis Rheuml. 2008, 58, 2936-2948. [CrossRef]
- Smolen, J.S.; Landewé, R.B.M.; Bergstra, S.A.; Kerschbaumer, A.; Sepriano, A.; Aletaha, D.; Caporali, R.; Edwards, C.J.; Hyrich, K.L.; Pope, J.E.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2022 update. Ann Rheum Dis. 2023, 82, 3-18. [CrossRef]
- van Nies, J.A.; Krabben, A.; Huizinga, J.W.; Huizinga, T.W.J.; Kloppenburg, M.; van der Helm-van Mil, A.H.M. What is the evidence for the presence of a therapeutic window of opportunity in rheumatoid arthritis? A systematic literature review. Ann Rheum Dis. 2014, 73, 861-870. [CrossRef]
- Burmester, G.R.; Bijlsma, J.W.J.; Cutolo, M.; McInnes, I.B. Managing rheumatic and musculoskeletal diseases - past, present and future. Nat Rev Rheumatol. 2017, 13, 443-448. [CrossRef]
- Bergstra, S.A.; Sepriano, A.; Kerschbaumer, A.; van der Heijde, D.; Caporali, R.; Edwards, C.J.; Verschueren, P.; de Souza, S.; Pope, J.E.; Takeuchi, T.; et al. Efficacy, duration of use and safety of glucocorticoids: a systematic literature review informing the 2022 update of the EULAR recommendations for the management of rheumatoid arthritis. Ann Rheum Dis. 2023, 82, 81-94. [CrossRef]
- Sepriano, A.; Kerschbaumer, A.; Bergstra, S.A.; Smolen, J.S.; van der Heijde, D.; Caporali, R.; Edwards, C.J.; Verschueren, P.; de Souza, S.; Pope, J.E.; et al. Safety of synthetic and biological DMARDs: a systematic literature review informing the 2022 update of the EULAR recommendations for the management of rheumatoid arthritis. Ann Rheum Dis. 2023, 82, 107-118. [CrossRef]
- Kerschbaumer, A.; Sepriano, A.; Bergstra, S.A.; Smolen, J.S.; van der Heijde, D.; Caporali, R.; Edwards, C.J.; Verschueren, P.; de Souza, S.; Pope, J.E.; et al. Efficacy of synthetic and biological DMARDs: a systematic literature review informing the 2022 update of the EULAR recommendations for the management of rheumatoid arthritis. Ann Rheum Dis. 2023, 82, 95-106. [CrossRef]
- Weng, H.H.; Ranganath, V.K.; Khanna, D.; Oh, M.; Furst, D.E.; Park, G.S.; Elashoff, D.A.; Sharp, J.T.; Gold, R.H.; Peter, J.B.; et al. Equivalent responses to disease-modifying antirheumatic drugs initiated at any time during the first 15 months after symptom onset in patients with seropositive rheumatoid arthritis. J Rheumatol. 2010, 37, 550-557. [CrossRef]
- Burmester, G.R.; Pope, J.E. Novel treatment strategies in rheumatoid arthritis. Lancet. 2017, 389, 2338-2348. [CrossRef]
- Tanski, W.; Swiatoniawska-Lonc, N.; Tabin, M.; Jankowska-Polanska, B. The Relationship between Fatty Acids and the Development, Course and Treatment of Rheumatoid Arthritis. Nutrients. 2022, 14, 1030. [CrossRef]
- Scott, D.L.; Wolfe, F.; Huizinga, T.W. Rheumatoid arthritis. Lancet. 2010, 9746, 1094-10108. [CrossRef]
- Nikiphorou. E.; Philippou, E. Nutrition and its role in prevention and management of rheumatoid arthritis. Autoimmun Rev. 2023, 7, 103333. [CrossRef]
- Leslie, C.A.; Gonnerman, W.A.; Ullman, M.D.; Hayes, K.C.; Franzblau, C.; Cathcart, E.S. Dietary fish oil modulates macrophage fatty acids and decreases arthritis susceptibility in mice. J Exp Med. 1985, 4, 1336-1349. [CrossRef]
- Volker, D.H.; FitzGerald, P.E.; Garg, M.L. The eicosapentaenoic to docosahexaenoic acid ratio of diets affects the pathogenesis of arthritis in Lew/SSN rats. J Nutr. 2000, 3, 559-565. [CrossRef]
- Ierna, M.; Kerr, A.; Scales, H.; Berge, K.; Griinari, M. Supplementation of diet with krill oil protects against experimental rheumatoid arthritis. BMC Musculoskelet Disord. 2010, 11, 136. [CrossRef]
- Ramprasath, V.R.; Eyal, I.; Zchut, S.; Jones, P.J.H. Enhanced increase of omega-3 index in healthy individuals with response to 4-week n-3 fatty acid supplementation from krill oil versus fish oil. Lipid Health Dis. 2013, 12, 178. [CrossRef]
- Di Giuseppe, D.; Vallin, A.; Bottai, M.; Askling, J.; Wolk, A. Long-term intake of dietary long-chain n-3 polyunsaturated fatty acids and risk of rheumatoid arthritis: a prospective cohort study of women. Ann Rheum Dis. 2014, 11, 1949-1953. [CrossRef]
- Rosell, M.; Wesley, A.M.; Rydin, K.; Klareskog, L.; Alfredsson, L.; EIRA study group. Dietary fish and fish oil and the risk of rheumatoid arthritis. Epidemiology. 2009, 6, 896. Doi.10.1097/EDE.0b013e3181b5f0ce.
- Pedersen, M.; Stripp, C.; Klarlund, M.; Olsen, S.F.; Tjønneland, A.M.; Frisch, M. Diet and risk of rheumatoid arthritis in a prospective cohort. J Rheumatol. 2005, 7, 1249-1252.
- Asoudeh, F.; Jayedi, A.; Kavian, Z.; Ebrahimi-Mousavi, S.; Nielsen, S.M.; Mohammadi, H. A systematic review and meta-analysis of observational studies on the association between animal protein sources and risk of rheumatoid arthritis. Clin Nut. 2021, 7, 4644. [CrossRef]
- Kremer, J.M.; Bigauoette, J.; Michalek, A.V.; Timchalk, M.A.; Lininger, L.; Rynes, R.I.; Huyck, C.; Zieminski, J.; Bartholomew L.E. Effects of manipulation of dietary fatty acids on clinical manifestations of rheumatoid arthritis. Lancet. 1985, 8422, 184-187. [CrossRef]
- Espersen, G.T.; Grunnet, N.; Lervang, H.H.; Nielsen, G.L.; Thomsen, B.S.; Faarvang, K.L.; Dyerberg, J.; Ernst, E. Decreased interleukin-1 beta levels in plasma from rheumatoid arthritis patients after dietary supplementation with n-3 polyunsaturated fatty acids. Clin Rheumatol. 1992, 11, 393-395. [CrossRef]
- Sperling, R.I.; Weinblatt, M.; Robin, J.L.; Ravalese, J3rd.; Hoover, R.L.; House, F.; Coblyn, J.S.; Fraser, P.A.; Spur, B.W.; Robinson, D.R.; et al. Effects of dietary supplementation with marine fish oil on leukocyte lipid mediator generation and function in rheumatoid arthritis. Arthritis Rheum. 1987, 9, 988-997. [CrossRef]
- Dawczynski, C.; Hackermeier, U.; Viehweger, M.; Stange, R.; Springer, M.; Jahreis, G. Incorporation of n-3 PUFA and γ-linolenic acid in blood lipids and red blood cell lipids together with their influence on disease activity in patients with chronic inflammatory arthritis--a randomized controlled human intervention trial. Lipids Health Dis. 2011, 10, 130. [CrossRef]
- Dawczynski, C.; Schubert, R.; Hein, G.; Müller, A.; Eidner, T.; Vogelsang, H.; Basu, S.; Jahreis, G. Long-term moderate intervention with n-3 long-chain PUFA-supplemented dairy products: effects on pathophysiological biomarkers in patients with rheumatoid arthritis. Br J Nutr. 2009, 10, 1517-1526. [CrossRef]
- Belch, J.J.; Ansell, D.; Madhok, R.; O'Dowd, A.; Sturrock, R.D. Effects of altering dietary essential fatty acids on requirements for non-steroidal anti-inflammatory drugs in patients with rheumatoid arthritis: a double blind placebo controlled study. Ann Rheum Dis. 1988, 2, 96-104. [CrossRef]
- Geusens, P.; Wouters, C.; Nijs, J.; Jiang, Y.; Dequeker, J. Long-term effect of omega-3 fatty acid supplementation in active rheumatoid arthritis. A 12-month, double-blind, controlled study. Arthritis Rheum. 1994, 6, 824. [CrossRef]
- Galarraga, B.; Ho, M.; Youssef, H.M.; Hill, A.; McMahon, H.; Hall, C.; Ogston, S.; Nuki, G.; Belch, J.J.F. Cod liver oil (n-3 fatty acids) as an non-steroidal anti-inflammatory drug sparing agent in rheumatoid arthritis. Rheumatology (Oxford). 2008, 5, 665-669. [CrossRef]
- Goldberg, R.J.; Katz, J. A meta-analysis of the analgesic effects of omega-3 polyunsaturated fatty acid supplementation for inflammatory joint pain. Pain. 2007, 129, 210. [CrossRef]
- Ritchie, D.M.; Boyle, J.A.; Mclnnes, J.M.; Jasani, M.K.; Dalakos, T.G.; Grieveson, P.; Buchanan, W.W. Clinical studies with an articular index for the assessment of joint tenderness in patients with rheumatoid arthritis. Q J Med. 1968, 37, 393-406.
- Moreno, J.J.; Carbonell, T.; Sanchez, T.; Miret, S.; Mitjavila, M.T. Olive oil decreases both oxidative stress and the production of arachidonic acid metabolites by the prostaglandin G/H synthase pathway in rat macrophages. J Nutr. 2001, 131, 2145-2149. [CrossRef]
- Lee, Y-H.; Bae, S-C.; Song, G.G. Omega-3 polyunsaturated fatty acids and the treatment of rheumatoid arthritis: a meta-analysis. Arch Med Res. 2012, 43, 356-362. [CrossRef]
- Fatel, E.C.S.; Rosa, F.T.; Alfieri, D.F.; Flauzino, T.; Scavuzzi, B.M.; Lozovoy, M.A.B.; Iriyoda, T.M.V.; Simão, A.N.C.; Dichi, I. Beneficial effects of fish oil and cranberry juice on disease activity and inflammatory biomarkers in people with rheumatoid arthritis. Nutrition. 2021, 86, 111183. [CrossRef]
- Yamada, H.; Saegusa, J.; Sendo, S.; Ueda, Y.; Okano, T.; Shinohara, M.; Morinobu, A. Effect of resolvin D5 on T cell differentiation and osteoclastogenesis analyzed by lipid mediator profiling in the experimental arthritis. Sci Rep. 2021, 11, 17312. [CrossRef]
- Marchand, N.E.; Choi, M.Y.; Oakes, E.G.; Cook, N.R.; Stevens, E.; Gomelskaya, N.; Kotler, G.; Manson, J.E.; Lasky-Su, J.; Mora, S.; et al. Over-the-counter fish oil supplementation and pro-resolving and pro-inflammatory lipid mediators in rheumatoid arthritis. Prostaglandins Leukot Essent Fatty Acid. 2023, 190, 102542. [CrossRef]
- Sigaux, J.; Mathieu, S.; Nguyen, Y.; Sanchez, P.; Letarouilly, J-G.; Soubrier, M.; Czernichow, S.; Flipo, R-M.; Sellam, J.; Daïen C. Impact of type and dose of oral polyunsaturated fatty acid supplementation on disease activity in inflammatory rheumatic diseases: a systematic literature review and meta-analysis. Arthritis Res Ther. 2022, 24, 100. [CrossRef]
- Gkiouras, K.; Grammatikopoulou, M.G.; Myrogiannis, I.; Papamitsou, T.; Rigopoulou, E.I.; Sakkas, L.I.; Bogdanos, D.P. Efficacy of n-3 fatty acid supplementation on rheumatoid arthritis' disease activity indicators: a systematic review and meta-analysis of randomized placebo-controlled trials. Crit Rev Food Sci Nutr. 2022, 28, 1-15. [CrossRef]
Figure 1.
Overview of the pathways of the endogenous synthesis of specialized pro-resolving mediators (SPMs) from docosahexaenoic acid (DHA). LOX, lipoxygenase. To be highlighted, not all intermediates and enzymes are indicated.
Figure 1.
Overview of the pathways of the endogenous synthesis of specialized pro-resolving mediators (SPMs) from docosahexaenoic acid (DHA). LOX, lipoxygenase. To be highlighted, not all intermediates and enzymes are indicated.
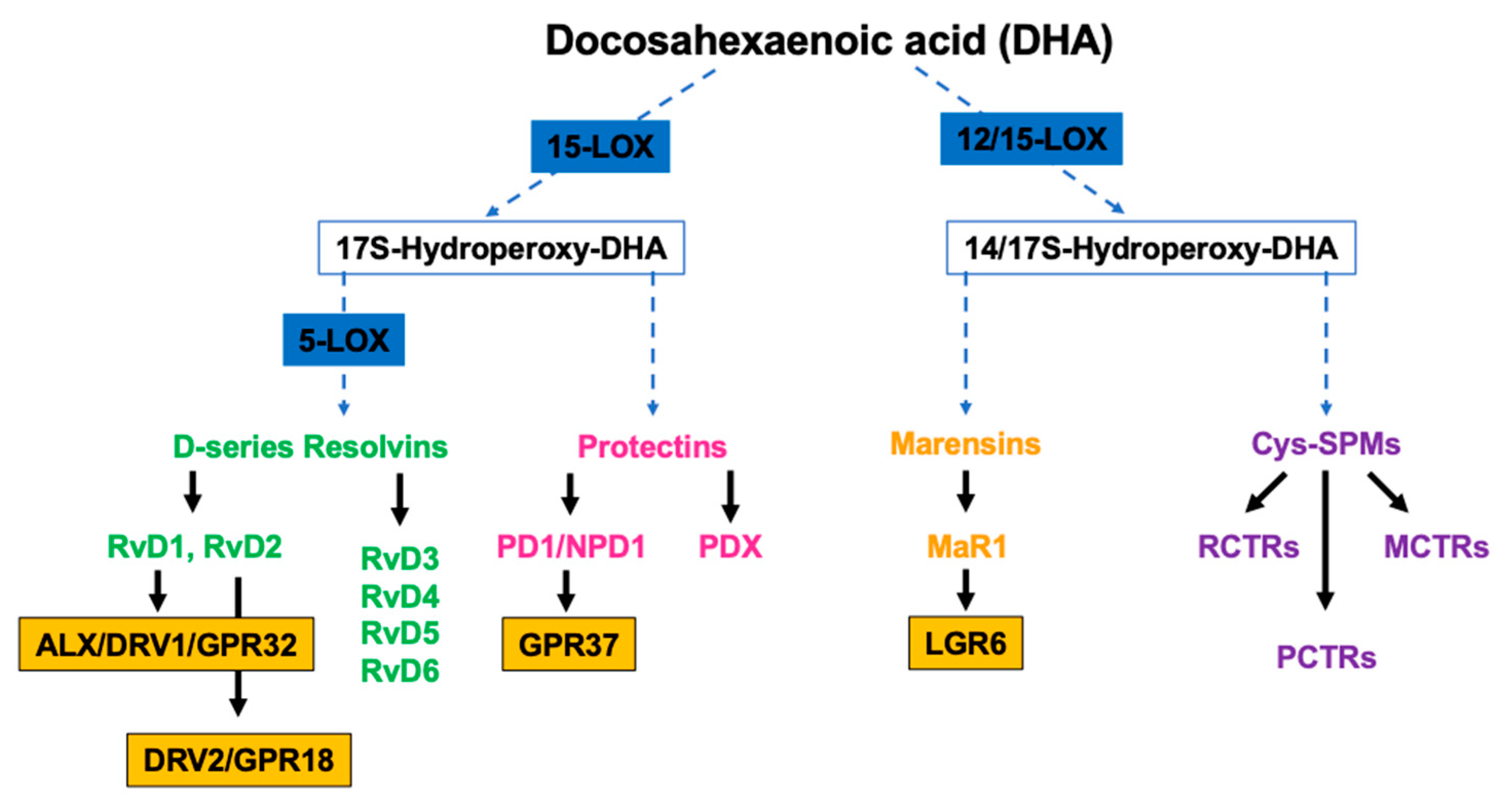
Figure 2.
Overview of the pathways of the endogenous synthesis of specialized pro-resolving mediators (SPMs) from (A) eicosapentaenoic (EPA) and (B) docosapentaenoic acid (DHA). CYP450, cytochrome 450 mixed function oxidase enzymes; COX, cyclooxygenase; LOX, lipoxygenase. To be highlighted, not all intermediates and enzymes are indicated.
Figure 2.
Overview of the pathways of the endogenous synthesis of specialized pro-resolving mediators (SPMs) from (A) eicosapentaenoic (EPA) and (B) docosapentaenoic acid (DHA). CYP450, cytochrome 450 mixed function oxidase enzymes; COX, cyclooxygenase; LOX, lipoxygenase. To be highlighted, not all intermediates and enzymes are indicated.
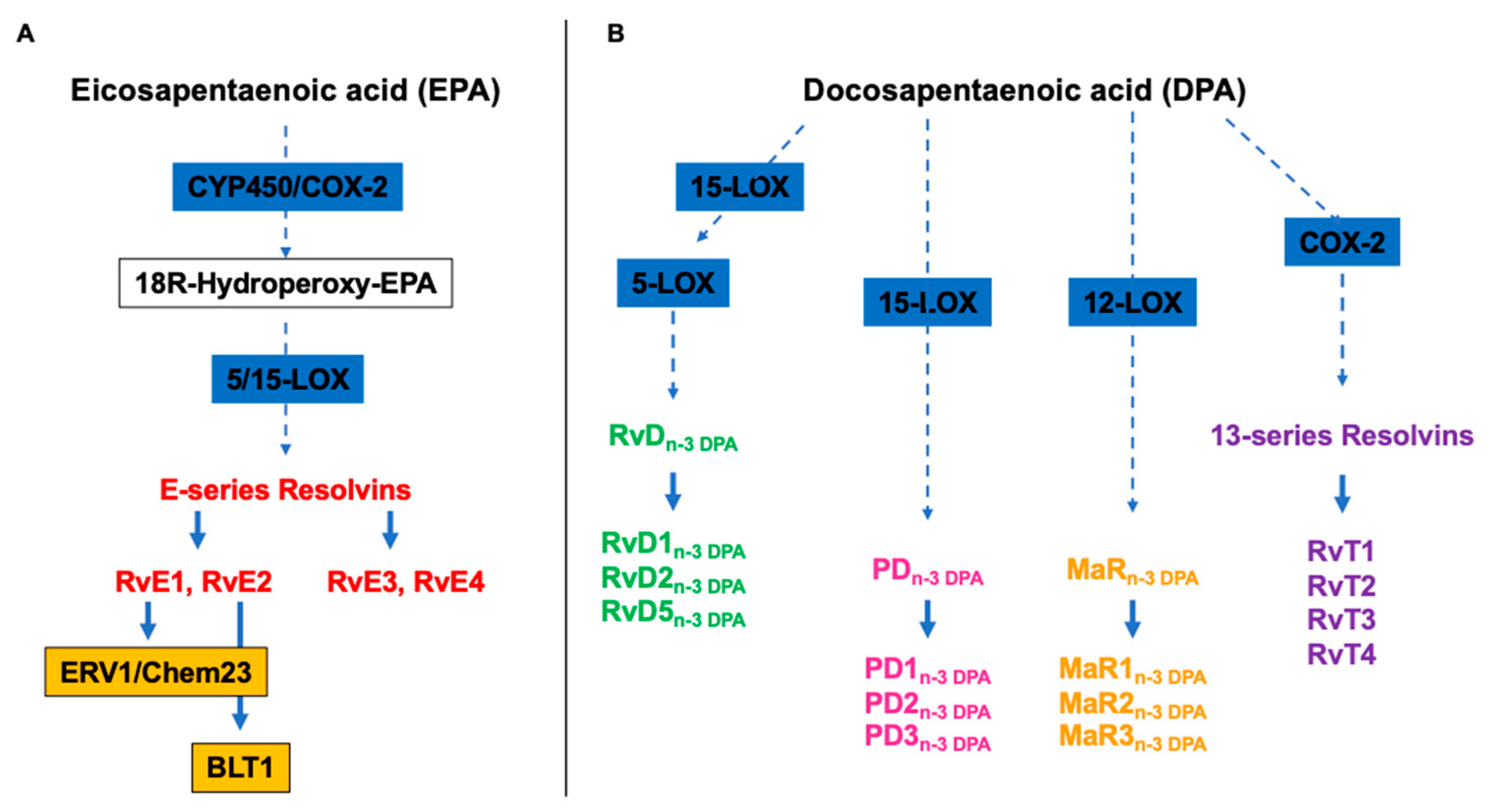
Figure 3.
Overview of the Rheumatoid arthritis’s pathological pathways and the potential anti-inflammatory mechanism of actions of marine n-3 PUFAs as well specialized pro-resolving mediators (SPMs) (created with Biorender.com, accessed on 22 November, 2023).
Figure 3.
Overview of the Rheumatoid arthritis’s pathological pathways and the potential anti-inflammatory mechanism of actions of marine n-3 PUFAs as well specialized pro-resolving mediators (SPMs) (created with Biorender.com, accessed on 22 November, 2023).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



