
Submitted:
22 November 2023
Posted:
23 November 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Objective: To identify and characterise genomic and phylogenetically isolated SARS-CoV-2 viral isolates in patients from Lambayeque, Peru. Methods: nasopharyngeal swabs were taken from patients from the Almanzor Aguinaga Asenjo Hospital, Chiclayo, Lambayeque, Peru, which have been considered mild, moderate and severe cases of COVID-19. Patients had to have tested positive for COVID-19, using a positive RT-PCR for SARS-CoV-2. Subsequently, the SARS-CoV-2 complete viral genome sequencing was carried out using Illumina MiSeq®. The sequences obtained from the sequence were analysed in Nextclade V1.10.0 to assign the corresponding clades, identify mutations in the SARS-CoV-2 genes and perform quality control of the sequences obtained. All sequences were aligned using MAFFT v7.471. The SARS-CoV-2 isolate Wuhan NC 045512.2 was used as a reference sequence to analyse mutations at the amino acid level. The construction of the phylogenetic tree model was achieved with IQ-TREE v1.6.12. Results: it was determined that during the period December 2020 and January 2021, the lineages s C.14, C.33, B.1.1.485, B.1.1, B.1.1.1, B.1.111 circulated, lineage C.14 the most predominant with 76.7% (n=23/30), these lineages were classified in clade 20D mainly and also within clade 20B and 20A. On the contrary, the variants found in the second batch of samples of the period September – October 2021 were Delta Variant (72.7%), Gamma (13.6%), Mu (4.6%), Lambda (9.1%), distributed between clades 20J, 21G, 21H, 21J and 21I. Conclusions: This study reveals updated information on the viral genomics of SARS-CoV-2 in the Lambayeque region, Peru, which is crucial to understanding the origins and dispersion of the virus and provides information on viral pathogenicity, transmission and epidemiology.
Keywords:
SARS-CoV-2
; genome
; sequencing
; phylogenetic analysis
; Peru
Introduction
COVID-19 is a respiratory disease caused by the SARS-CoV-2 virus, declared a pandemic by the World Health Organization (WHO) in early 2020. This disease has caused a health and economic emergency worldwide. Currently, research on SARS-CoV-2 is booming and great efforts are being made to characterise SARS-CoV-2 molecularly. The genomic and molecular variability of SARS-CoV-2 can be the basis for glimpses of etiological and pathological aspects of this virus, understanding that the virus can accumulate mutations of importance while expanding worldwide, as well as also be able to establish antiviral strategies designed and based on the molecular specificities of SARS-CoV-2.
One of the most striking aspects of COVID-19 is the marked difference in the evolution of the disease in patients. The spread and manifestations of COVID-19, an infectious disease, are influenced by multiple interrelated factors. These include the virus itself (SARS-CoV-2), the human host (comorbidities and genetics), and the environment (physical conditions, social interactions, containment measures). All of these play a role in determining the course of the disease and the pandemic (1). By elucidating and obtaining these genomic data, it would be possible to reveal the evolutionary events of SARS-CoV-2, establish the types of circulating genomes, and determine in which parts of the genome these viral isolates differ (2,3).
In Peru, lineages of regional and global relevance variants have emerged; some researchers detected the circulation of SARS-CoV-2 strains with the D614G mutation in the Lambayeque region at the beginning of 2020. This mutation had already spread widely in Europe at that time. However, other uncommon mutations demonstrate the virus’s rapid evolutionary processes and adaptive capabilities (4). Subsequent investigations corroborated the presence of a variant endemic to the region, which was designated the Lambda variant (5).
The genetic variability of SARS-CoV-2 requires continuous study to elucidate various aspects of its molecular biology. Due to this, various modifications or changes in the nucleotide sequence of the viral genome have been reported worldwide, causing the appearance of variants, which have been grouped into differentiated clades. Among the variants of interest of SARS-CoV-2 are Lambda and Mu, first identified in Peru and Colombia, respectively. Meanwhile, the variants of concern of SARS-CoV-2 identified and reported globally, in chronological order, are Alpha (British), Beta (South African), Gamma (Brazilian), Delta (Indian) and recently Omicron (6).
For this reason, sequencing of the SARS-CoV-2 viral genome in Peru is urgently required; this will provide information on the prevalence of viral clades belonging to SARS-CoV-2, which could lead to a better understanding of transmission patterns, outbreak monitoring and formulation of effective containment measures. Mutation data may also provide important clues for developing vaccines, antiviral drugs, and effective diagnostic assays.
The present research aimed to investigate the genomic variation of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in Peru through the whole genome sequencing of SARS-CoV-2 strains and compare their evolutionary trajectories with global strains through phylogenetic analysis.
Methods
Sample collection and complete sequencing of the SARS-CoV-2 genome
Nasopharyngeal swabs were obtained from positive cases of COVID-19, and the first 30 sequenced samples were obtained at the Almanzor Aguinaga Asenjo Hospital belonging to Essalud, Chiclayo, Lambayeque, Peru, during the period December 2020 and January 2021. Those samples with a CT ≤28 were selected for genome sequencing; these samples (n = 30) were sent to the Microbial Genomics laboratory of the Universidad Peruana Cayetano Heredia for subsequent analysis and sequencing. Another 44 samples of nasopharyngeal swabs from positive cases of COVID-19 confirmed by the reference laboratory of the Regional Health Management Lambayeque were included; these biological samples belong to the period September–October 2021, thus completing 74 genomes sequenced by the authors of the present research.
Whole genome sequencing of SARS-CoV-2 isolates was performed with a MiSeq (Illumina) using the COVIDseq Illumina kit. In detail, the samples were processed using the Sansure brand RNA kit and a Sansure Natch 48 automated equipment. Subsequently, genomic libraries were prepared with the Illumina COVIDSeq kit. The library sequencing was on an instrument of the Illumina MiSeq model and the 300-cycle v2 kit to achieve an average coverage of 1500X. Once the sequences were obtained, they were processed and assembled with Illumina’s DRAGEN pipeline through its Base Space application. The Illumina MiSeq sequencing procedure was performed at the Microbial Genomics Laboratory, Department of Cellular and Molecular Sciences Faculty of Sciences and Philosophy (Universidad Peruana Cayetano Heredia, Peru).
Bioinformatics analysis
All bioinformatic analyses used in the development of this research were based on the protocols proposed and designed for the genomic surveillance of SARS-COV-2 in Costa Rica (7).
SARS-CoV-2 sequences
Until April 28, 2022, all SARS-CoV-2 sequences available from the Lambayeque – Peru region were recovered and retrieved from the GISAID (Global Initiative on Sharing All Influenza Data, www.gisaid.org). A total of 714 sequences were recovered from the Lambayeque region, of which 74 were sequenced and uploaded to GISAID by the authors of this research.
Multi-sequence alignment
All sequences were aligned using MAFFT v7.471(8). The SARS-CoV-2 isolate Wuhan NC 045512.2 was used as a reference sequence to analyse mutations at the amino acid level.
Phylogenetic analyses
The construction of the phylogenetic tree model was achieved with IQ-TREE v1.6.12 (9), including ModelFinder (10) to select the best nucleotide substitution model (using the Bayesian Information Criterion BIC, the best model was TN+F+I). The visualisation was performed using the iTOL v4 tool (11)
Selection and identification of mutations of epidemiological relevance of SARS-COV-2 and their geographical association
According to the top 5% table of mutations observed in the 714 genomes analysed in the Lambayeque region, Peru (Figure 2), a manual search was performed for the missense or non-synonymous mutations of each SARS-COV-2 gene on the Nextstrain® (https://nextstrain.org/) online server, identifying the lineage to which this genomic sequence belonged. Subsequently, this lineage was verified in Outbreak.info® (https://outbreak.info/) to know the geographical distribution of said mutation according to the GISAID database. Only those mutations predominately in Peru and mainly in the Lambayeque region were selected for detailed characterisation.
Ethical considerations
Ethical approval for sample collection and analysis protocols was granted by the ethics committee of the Almanzor Aguinaga Asenjo Hospital, Chiclayo, Peru, through the ICIS-RPL code 066-DEC-2021. Participation in the study was voluntary, with the signing of an informed consent approved by the same ethics committee; in the case of patients, the consent was signed by their family member or proxy. All information obtained from participants will be used only for this research. Therefore, such information will not be stored or used for further studies.
Results
Seventy-four nasopharyngeal swab samples were collected from SARS-CoV-2 positive patients (cycle threshold values [CT] obtained by qPCR, ≤28). The Illumina MiSeq sequencing procedure has been performed in the Microbial Genomics Laboratory, Department of Cellular and Molecular Sciences Faculty of Sciences and Philosophy (Universidad Peruana Cayetano Heredia, Peru). The viral genomes were assembled by mapping using as a reference the genome of the Wuhan Hu-1 strain deposited in GenBank (acc number NC_045512).
Seventy-four genomic sequences were obtained from SARS-CoV-2 viral isolates from patients at the Almanzor Aguinaga Asenjo Hospital, Chiclayo, Peru. These sequences were divided into two batches of samples: the first 30 sequences were obtained from COVID-19-positive nasopharyngeal swabs from December 2020 and January 2021 after performing the analysis of these sequences in Nextclade V1.10.0 (https://clades.nextstrain.org/) and PANGOLIN V3.1.16 (https://pangolin.cog-uk.io)
The second batch of samples consisted of 44 nasopharyngeal swabs positive for COVID-19 from September and October 2021, after analysing these sequences in Nextclade V1.10.0 (https://clades.nextstrain.org/) and PANGOLIN V3.1.16 (https://pangolin.cog-uk.io/) it was determined that the predominant variants in this sampling period were Delta Variant (72.7%), Gamma (13.6%), Mu (4.6%), Lambda (9.1%), distributed between clades 20J, 21G, 21H, 21J and 21I.
Another variant of interest is C.37 (Lambda Variant); the present investigation determined that 04 sequences (n = 4/44) of the second batch of samples analysed belonged to C.37, classified within clade 21G. C.37 is considered a variant native to Peru, also called the Andean variant; the first reports of C.37 began in Lima, Peru, approximately in August 2020. Subsequently, this variant has been predominant in the sequencing results detected in Peru since its first report, and it has spread to most countries in South America (12,13)
Subsequently, the genomes of the first batch of sequenced samples (n=30) were analysed using different bioinformatics tools such as PANGOLIN V3.1.16 and Nextclade of Nextstrain V1.10.0.
According to PANGOLIN (Phylogenetic assignment of epidemic lines with global name), the analysed sequences were classified in the lineage C.14, C.33, B.1.1.485, B.1.1, B.1.1.1, B.1.111. The C.14 lineage is the most predominant, with 76.7% (n=23) and the other lineages ranging from 3.3% to 6.7%, respectively. The sequences fasta of the first and second batch of sequenced samples were analysed in the online program NEXTCLADE V1.10.0 (https://clades.nextstrain.org/) to assign the corresponding clades, identify the mutations in each SARS-CoV-2 gene and also to perform quality control of these sequences.
Characteristic mutations that have been found in the C.14 lineage include the T1246I and G3278S mutation in the ORFIa gene; P314L in the ORFIb gene; D614G in spike protein; R203K and G204R in gene N (Table 1).
In addition, in the C.14 lineage, it was possible to identify that the most frequent and relevant amino acid change in the SARS-CoV-2 Nucleocapsid gene was R203K and G204R (N gene, n = 34/35). Likewise, in the case of the changes of nearby nucleotides GGG>AAC at positions 28881-28883, the triplet was also present in n=34/35 of the sequences analysed.
Likewise, once the FASTA sequences were obtained from the second batch of processed samples (n = 44), the genomes were analysed using the bioinformatics tool of PANGOLIN (Phylogenetic assignment of epidemic lines with global name), the variants found in this second batch were: Delta variant (72.7%), Gamma (13.6%), Mu (4.6%), Lambda (9.1%). According to Nextstrain’s NextClade tool, the last batch of 44 sequences analysed was classified into clade 20J, 21G, 21H, 21J and 21I, as seen in the following phylogenetic tree.
Using the NextClade tool of Nextstrain, the Delta variant of SARS-CoV-2 (n=32) was classified into clade 21J (n=27) and 21I (n=05). It was observed that within the Delta variant of SARS-CoV-2, there were several sublineages, among which are AY.26, AY.39.2, AY.100, AY.122, AY.43, AY.102, B.1.617.2. The characteristic mutations found in the Delta variant and its sublineages can be seen in Table 2.
As for the Gamma variant of SARS-CoV-2, according to the NextClade tool of Nextstrain, the Gamma variant (n=06) was classified in clade 20J. It was observed that within the Gamma variant, there were two sublineages, among which stand out: P.1 (n = 02) and P.1.12 (N = 04). The characteristic mutations found in the Gamma variant and its sublineages can be seen in Table 3.
With respect to the Lambda variant of SARS-CoV-2, according to the NextClade tool of Nextstrain, the Gamma variant (n=04) was classified in clade 21G. The characteristic mutations found in the Lambda variant (C.37) can be seen in Table 4.
The phylogenetic tree of the 714 genomes obtained on April 28, 2022, from the GISAID International Base (www.gisaid.org) of the Lambayeque region – Peru, was carried out. It can be seen in the phylogenetic tree that the sequences belong to the variants Delta, Omicron, Mu, Lambda, and Gamma of SARS-CoV-2 mostly, as shown in Figure 1.
A potency law pattern was recognised in the analysed genomes, and the presence/absence of variants in the 714 is evident. Few variants are widely distributed across genomes, and many are uniquely present in a single genome (Figure 2). This means that only a few variants are present in several genomes, and further analysis can focus on those variants.
Figure 2.
Law of potency of mutations observed in the analysis of 714 genomes from the Lambayeque region – Peru. The presence/absence of variants in the 714 genomes analysed is evident. Few variants are widely distributed among genomes, and many are uniquely present in a single genome.
Figure 2.
Law of potency of mutations observed in the analysis of 714 genomes from the Lambayeque region – Peru. The presence/absence of variants in the 714 genomes analysed is evident. Few variants are widely distributed among genomes, and many are uniquely present in a single genome.

The identification of variants of the SARS-COV-2 genome observed in more than 5% of the genomes analysed in the Lambayeque region – Peru was also carried out; specific analysis of these variants can be seen in Table N°05. The identification of relevant mutations of SARS – CoV – 2 and their geographical association was also carried out; for this, a manual review was made, mutation by mutation in the nextstrain online tool of those mutations with a frequency greater than 5% to determine where they had been reported according to the international database of GISAID.
Discussion
Genomic surveillance of SARS-CoV-2 plays a critical role in understanding and responding to the pandemic. Tracking the emergence of mutations and variants through whole genome sequencing enables early detection of novel variants of concern and monitoring their spread, allowing public health officials to implement timely tailored containment measures. Study of the biological and pathogenic properties of new variants, including their transmissibility, virulence, and immune evasion ability. Improve existing diagnostics and treatments to ensure they remain effective against new variants. Identifying key mutations that correlate with concerning properties provides valuable insights into the virus’s adaptation. Thus, genomic surveillance is essential to keep pace with the evolution of SARS-CoV-2 and adapt strategies to combat current and future variants, thus improving our response to the pandemic (6).
The present study allowed us to identify and characterise the genome and lineage of 74 viral strains of the SARS-CoV-2 virus obtained from patients at the Almanzor Aguinaga Asenjo Hospital in Chiclayo, Peru through next-generation sequencing (NGS) with the Illumina MiSeq system. NGS sequencing allows us to know the molecular epidemiology of SARS-CoV-2 and obtain knowledge about the virus’s evolution, transmission, virulence and pathology. Before the arrival of the COVID-19 pandemic, several researchers worldwide began to sequence the complete genome of SARS-CoV-2 to genetically understand this virus, try to elucidate its origin and find a molecular target that serves as a basis for the development of a biological product or vaccine against SARS-CoV-2. The implementation of bioinformatics and genomic tools allowed the active surveillance of SARS-CoV-2, as well as the identification of new lineages and the registration of new mutations in the viral genome, which will allow a better understanding of the evolution and replacement rates of the virus. Several countries in Latin America, Asia, Europe and Africa have published their sequencing results of the whole genome of SARS-CoV-2 in the GISAID international database. (14,15).
The predominant lineage in this sampling period was lineage C.14 in 76.7% (n = 23) belonging to clade 20D, which also detected the circulation of lineages C.33, B.1.1.485, B.1.1, B.1.1.1, B.1.111 in a percentage ranging between 3.3% - 6.7% respectively, distributed between clades 20B and 20A. The results agreed with those reported by (4), who sequenced 5 genomes obtained from patients from the Lambayeque region at the end of April 2020, reporting the circulation of lineage B.1.1.1, classified according to Nextclade of Nextstrain in clade 20B. Also, our results agree with what was described by (3); these authors indicate that SARS-CoV-2 isolates during the initial period of the pandemic in Peru belong or are grouped mainly in clade 20B; this clade is very characteristic of isolates obtained from patients with COVID-19 in the European continent. Likewise, these authors identified nine predominant lineages: A.1, A.2, A.5, B.1, B.1.1, B.1.1.1, B.1.5, B.1.8, and B.2, the most predominant being B.1, B.1.1.1.
According to the analyses carried out in GISAID and Pangolin (16) (B1.1.1, B1.5), highlighting that most Peruvian SARS-CoV-2 sequences are classified within clade B.1 and within subclade B.1.1.1. The results described above differ from our results because the predominant lineage in our first sequences was C.14, this difference in results can be attributed to the sampling period in which the samples of nasopharyngeal swabs positive for COVID-19 were collected. Although there are no reports of the C.14 lineage, the GISAID-enabled outbreak.info mutation tracker (https://outbreak.info/situation-reports?pango=C.14) indicates that this lineage has been reported in the following countries: Peru (93.0%), United States (2.0%), Japan (2.0%), Democratic Republic of Congo (1.0%), Brazil (1.0%) and was first reported on 2020-03-20. (CITA)
The study reports that the principal variant detected in the second batch was B.1.617.2 (Delta), which has greater transmissibility virulence and can cause cases of reinfection and outbreaks due to the presence of a high number of mutations in the spike protein that allow more significant resistance to the action of antibodies or immune escape. The Delta variant has been reported in several countries worldwide and can replace other regional variants in circulation. The high infectivity of Delta is linked to its high viral load and the short incubation period until the appearance of symptoms. The study also reports that the Delta variant has been found to have immune evasion in patients who received doses of Pfizer®, Moderna®, and Covax®, suggesting that the variant may require updated vaccines to provide better protection (17,18).
In addition, our sequences assigned or classified as Delta variant (21I, 21J) presented various mutations in the Spike gene (S gene) such as L452R, T478K, D614G, P618R, and these mutations have been reported worldwide by various researchers, and indicate that they provide biological advantages, among which are: an increase in binding to the ACE-2 receptor, increased transmissibility, risk of hospitalisation, immune escape or resistance to specific antibodies (19,20). Some reports indicate that the delta variant has undergone another mutation, K417N, T95I, and W258L, calling it the Delta Plus variant; however, in our results, we have not found this mutation in any of the analysed sequences belonging to the Delta variant. Some research indicates that the K417N, T95I, and W258L mutation of the spike protein increases the viral ability to achieve immune evasion; however, little is still known about the pathogenicity and virulence of this new variant of SARS-CoV-2(21)
The study identified that the most frequent and relevant mutation in the SARS-CoV-2 Spike gene of the C.14 lineage was D614G, which is associated with more significant pathogenesis and virulence, and evidence suggests that it can improve the transmission of the virus by increasing the amount of viral load in the upper respiratory tract. The sequences analysed and classified as C.37 contain a characteristic deletion in the gene S and mutations not synonymous in the gene Spike. These could provide biological advantages such as increased transmissibility, virulence, viral invasion into host cells, and immune escape properties. The Gamma variant of SARS-CoV-2 was also detected in the study, which has lineage-defining mutations, including K417T, E484K, and N501Y, and mutations that allow this variant to increase ACE-2 receptor binding affinity, cause reinfection, increased transmissibility, higher viral load, and immune evasion. (22) The study also cites reports that patients infected with SARS-CoV-2 who carried the D614G mutation developed a moderate/severe COVID-19 condition, while patients infected with SARS-CoV-2 who did not carry this mutation developed mild symptoms and that the Gamma variant was the predominant lineage in the second wave of COVID-19 cases in Brazil.
During the third wave of the COVID-19 pandemic in Peru, the Delta and Gamma variants predominated until the emergence of the Omicron variant. Studies from other countries, such as one from Pakistan, report that the Delta, Beta and Gamma variants had specific mutations that could provide various biological advantages to these variants. The simultaneous coexistence of highly transmissible SARS-CoV-2 variants could lead to evolutionary competition, where specific variants with mutations that improve their infectious capacity compete with others characterised by their immunological evasion capacity. In Peru, the Lambda variant (C.37) became the predominant variant in the coastal and Andean region, surpassing other circulating Variants of Concern (VOC) such as Gamma and Delta, despite Gamma having a higher frequency during the second wave in the Northwest region due to its proximity to Brazil. (23).
In our results, we can also observe that the N gene of SARS-CoV-2 presents several amino acid mutations that confer various changes or biological advantages. Worldwide, several reports indicate that mutations in the N gene of SARS-CoV-2 reduce the sensitivity of molecular tests (RT-PCR) for the detection of SARS-CoV-2, thus causing the appearance of false negative results for this gene. The N gene of SARS-CoV-2 is of vital importance in the structure and viral cycle, as it is involved in viral assembly, replication and the immune response of the host; also, this SARS-CoV-2 gene is a gene not conserved due to its mutation rate. All these characteristics described above make this gene a target or target to update tests that allow viral diagnosis through and for the development of vaccines(24,25)
The natural evolution of SARS-CoV-2 has led to the emergence of multiple genetic variants with various biological properties, including increased transmission, immune escape, infectivity, and lethality. The initiation of mass vaccination could also be associated with an increase in selective pressure, leading to the appearance of escape mutants. Large-scale whole genome sequencing of SARS-CoV-2 is vital to track the spread of the virus, study local outbreaks, and identify critical mutations in SARS-CoV-2 genes. However, sharing sequencing results in the GISAID database is crucial for almost real-time genomic surveillance worldwide, providing a better understanding of the transmission and viral evolution dynamics of SARS-CoV-2. (26,27)
Although this study provides valuable information, it has some limitations related mainly to the sample size and the lack of clinical data. Thus, only 74 viral sequences were analysed in two groups, which may limit the ability to detect some low-frequency circulating variants. The lack of clinical and epidemiological data associated with the cases analysed could be considered a limitation since the study focused on the genomic analysis of the samples but did not report data on the severity of the cases, hospitalisations, contacts, etc. Although it was not the study’s objective, this information is essential to determine the clinical and epidemiological impact of the detected variants. Lastly, it is necessary to consider a possible geographic bias since the study focused on a single city in the region, so it may not capture all the diversity of variants circulating in other areas. Future genomic surveillance studies should ideally include representative samples from the entire region. Genomic surveillance in Peru, as in other countries of Latin America, has been vital in the understanding of the COVID-19 pandemic evolution during these almost four years (28-30).
Genomic surveillance is a powerful tool for monitoring and understanding the dynamics of infectious diseases, enabling a more proactive and effective response to emerging threats, such as SARS-CoV-2 and future pandemic pathogens.
In conclusion, the study of SARS-CoV-2 genomes in Chiclayo, Peru, highlights the presence of multiple lineages and variants of concern circulating in the region. The emergence of Delta as the most common variant in later samples from 2021, along with other variants like Gamma, Mu, and Lambda, is particularly alarming due to their potential increased transmission and virulence and their possible ability to escape the immune response. The use of whole genome sequencing and data sharing in databases like GISAID is crucial for understanding the evolution and epidemiology of SARS-CoV-2, which can inform response measures and aid in detecting emerging strains. The findings underscore the importance of genomic surveillance in tracking the spread of SARS-CoV-2 variants and developing tailored public health strategies to limit their transmission. Continued monitoring and sequencing efforts are necessary to stay ahead of the virus’s evolution and ensure effective pandemic control.

Table 5.
SARS-COV-2 genome variants were observed in more than 5% of genomes analysed from the Lambayeque region, Peru.
Table 5.
SARS-COV-2 genome variants were observed in more than 5% of genomes analysed from the Lambayeque region, Peru.
| Mutation Number | POS | REF | ALT | Total of genomes with mutation | Clase of mutation | Effect | Gene | Transcript | AA | Sequence in transcript | Sequence protein | Patterns in the world | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 53 | 3037 | C | T | 712 | synonymous | Low | ORF1ab | c.2772C>T | p.Phe924Phe | 2772/21291 | 924/7096 | N/A | |
| 490 | 23403 | A | G | 712 | missense | Moderate | S | c.1841A>G | p.Asp614Gly | 1841/3822 | 614/1273 | Diseminado en el mundo | |
| 236 | 10029 | C | T | 518 | missense | Moderate | ORF1ab | c.9764C>T | p.Thr3255Ile | 9764/21291 | 3255/7096 | Diseminado en el mundo | |
| 373 | 15451 | G | A | 330 | missense | Moderate | ORF1ab | c.15187G>A | p.Gly5063Ser | 15187/21291 | 5063/7096 | Diseminado en el mundo | |
| 541 | 25469 | C | T | 328 | missense | Moderate | ORF3a | c.77C>T | p.Ser26Leu | 77/828 | 26/275 | Diseminado en el mundo | |
| 644 | 28461 | A | G | 328 | missense | Moderate | N | c.188A>G | p.Asp63Gly | 188/1260 | 63/419 | Diseminado en el mundo | |
| 213 | 8986 | C | T | 309 | synonymous | Low | ORF1ab | c.8721C>T | p.Asp2907Asp | 8721/21291 | 2907/7096 | N/A | |
| 216 | 9053 | G | T | 309 | missense | Moderate | ORF1ab | c.8788G>T | p.Val2930Leu | 8788/21291 | 2930/7096 | Diseminado en el mundo | |
| 244 | 11332 | A | G | 309 | synonymous | Low | ORF1ab | c.11067A>G | p.Val3689Val | 11067/21291 | 3689/7096 | N/A | |
| 97 | 4181 | G | T | 308 | missense | Moderate | ORF1ab | c.3916G>T | p.Ala1306Ser | 3916/21291 | 1306/7096 | Diseminado en el mundo | |
| 624 | 28311 | C | T | 200 | missense | Moderate | N | c.38C>T | p.Pro13Leu | 38/1260 | 13/419 | Diseminado en el mundo | |
| 91 | 4002 | C | T | 194 | missense | Moderate | ORF1ab | c.3737C>T | p.Thr1246Ile | 3737/21291 | 1246/7096 | Muy poco diseminado en el mundo | |
| 157 | 5716 | G | T | 121 | missense | Moderate | ORF1ab | c.5451G>T | p.Lys1817Asn | 5451/21291 | 1817/7096 | Muy poco diseminado en el mundo | |
| 226 | 9867 | T | C | 115 | missense | Moderate | ORF1ab | c.9602T>C | p.Leu3201Pro | 9602/21291 | 3201/7096 | Muy poco diseminado en el mundo | |
| 225 | 9857 | C | T | 111 | synonymous | Low | ORF1ab | c.9592C>T | p.Leu3198Leu | 9592/21291 | 3198/7096 | N/A | |
| 508 | 25000 | C | T | 87 | synonymous | Low | S | c.3438C>T | p.Asp1146Asp | 3438/3822 | 1146/1273 | N/A | |
| 564 | 25584 | C | T | 87 | synonymous | Low | ORF3a | c.192C>T | p.Thr64Thr | 192/828 | 64/275 | N/A | |
| 137 | 5386 | T | G | 86 | synonymous | Low | ORF1ab | c.5121T>G | p.Ala1707Ala | 5121/21291 | 1707/7096 | N/A | |
| 259 | 11537 | A | G | 86 | missense | Moderate | ORF1ab | c.11272A>G | p.Ile3758Val | 11272/21291 | 3758/7096 | Diseminado en el mundo | |
| 338 | 13195 | T | C | 86 | synonymous | Low | ORF1ab | c.12930T>C | p.Val4310Val | 12930/21291 | 4310/7096 | N/A | |
| 604 | 26270 | C | T | 86 | missense | Moderate | E | c.26C>T | p.Thr9Ile | 26/228 | Set-75 | Diseminado en el mundo | |
| 406 | 17259 | G | T | 72 | missense | Moderate | ORF1ab | c.16995G>T | p.Glu5665Asp | 16995/21291 | 5665/7096 | Muy poco diseminado en el mundo | |
| 153 | 5648 | A | C | 71 | missense | Moderate | ORF1ab | c.5383A>C | p.Lys1795Gln | 5383/21291 | 1795/7096 | Muy poco diseminado en el mundo | |
| 514 | 25088 | G | T | 71 | missense | Moderate | S | c.3526G>T | p.Val1176Phe | 3526/3822 | 1176/1273 | Muy poco diseminado en el mundo | |
| 14 | 733 | T | C | 70 | synonymous | Low | ORF1ab | c.468T>C | p.Asp156Asp | 468/21291 | 156/7096 | N/A | |
| 312 | 12778 | C | T | 70 | synonymous | Low | ORF1ab | c.12513C>T | p.Tyr4171Tyr | 12513/21291 | 4171/7096 | N/A | |
| 347 | 13860 | C | T | 70 | synonymous | Low | ORF1ab | c.13596C>T | p.Asp4532Asp | 13596/21291 | 4532/7096 | N/A | |
| 646 | 28512 | C | G | 70 | missense | Moderate | N | c.239C>G | p.Pro80Arg | 239/1260 | 80/419 | Muy poco diseminado en el mundo | |
| 31 | 1048 | G | T | 66 | missense | Moderate | ORF1ab | c.783G>T | p.Lys261Asn | 783/21291 | 261/7096 | Diseminado en el mundo | |
| 477 | 20937 | G | T | 58 | synonymous | Low | ORF1ab | c.20673G>T | p.Thr6891Thr | 20673/21291 | 6891/7096 | N/A | |
| 598 | 25844 | C | T | 44 | missense | Moderate | ORF3a | c.452C>T | p.Thr151Ile | 452/828 | 151/275 | Muy poco diseminado en el mundo | |
| 145 | 5515 | G | T | 41 | synonymous | Low | ORF1ab | c.5250G>T | p.Val1750Val | 5250/21291 | 1750/7096 | N/A | |
| 566 | 25613 | C | T | 38 | missense | Moderate | ORF3a | c.221C>T | p.Ser74Phe | 221/828 | 74/275 | Muy poco diseminado en el mundo |
Funding
Universidad Continental, Huancayo, Peru, covered the APC of this article.
Conflicts of Interest
AJRM has been consultant/speaker of AstraZeneca, Valneva and Moderna in relationship with COVID-19 vaccines and long COVID-19. Rest of authors, no conflicts.
References
- Molina-Mora JA, Reales-González J, Camacho E, Duarte-Martínez F, Tsukayama P, Soto-Garita C, et al. Overview of the SARS-CoV-2 genotypes circulating in Latin America during 2021. Front Public Health [Internet]. 2023 Mar 2 [cited 2023 Apr 14];11. Available from: /PMC/articles/PMC10018007/. [CrossRef]
- Chong YM, Sam IC, Ponnampalavanar S, Omar SFS, Kamarulzaman A, Munusamy V, et al. Complete Genome Sequences of SARS-CoV-2 Strains Detected in Malaysia. Microbiol Resour Announc [Internet]. 2020 May 14 [cited 2021 Nov 17];9(20). Available from: /PMC/articles/PMC7225546/. [CrossRef]
- Juscamayta-López E, Tarazona D, Valdivia F, Rojas N, Carhuaricra D, Maturrano L, et al. Phylogenomics reveals multiple introductions and early spread of SARS-CoV-2 into Peru. bioRxiv [Internet]. 2020 Sep 21 [cited 2021 Oct 3];2020.09.14.296814. Available online: https://www.biorxiv.org/content/10.1101/2020.09.14.296814v2. [CrossRef]
- Aguilar-Gamboa FR, Salcedo-Mejía LA, Serquén-López LM, Mechan-Llontop ME, Tullume-Vergara PO, Bonifacio-Briceño JJ, et al. Genomic Sequences and Analysis of Five SARS-CoV-2 Variants Obtained from Patients in Lambayeque, Peru. Microbiol Resour Announc [Internet]. 2021 Jan 7 [cited 2021 Oct 3];10(1). Available from: /pmc/articles/PMC8407726/. [CrossRef]
- Romero PE, Dávila-Barclay A, Salvatierra G, González L, Cuicapuza D, Solis L, et al. The Emergence of SARS-CoV-2 Variant Lambda (C.37) in South America. medRxiv [Internet]. 2021 Jul 3 [cited 2021 Oct 3];2021.06.26.21259487. Available online: https://www.medrxiv.org/content/10.1101/2021.06.26.21259487v1. [CrossRef]
- Aguilar-Gamboa FR, Suclupe-Campos DO, Vega-Fernández JA, Silva-Diaz H. Diversidad genómica en SARS-CoV-2: Mutaciones y variantes. Rev Cuerpo Méd Hosp Nac Almanzor Aguinaga Asenjo [Internet]. 2021 Oct 1 [cited 2023 May 14];14(4):572–82. Available online: http://cmhnaaa.org.pe/ojs/index.php/rcmhnaaa/article/view/1465/556. [CrossRef]
- Molina-Mora JA, Cordero-Laurent E, Godínez A, Calderón-Osorno M, Brenes H, Soto-Garita C, et al. SARS-CoV-2 genomic surveillance in Costa Rica: Evidence of a divergent population and an increased detection of a spike T1117I mutation. Infection, Genetics and Evolution. 2021 Aug 1;92:104872. [CrossRef]
- Katoh K, Standley DM. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol Biol Evol. 2013 Apr;30(4):772. [CrossRef]
- Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015 Jan 1;32(1):268–74. [CrossRef]
- Kalyaanamoorthy S, Minh BQ, Wong TKF, Von Haeseler A, Jermiin LS. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat Methods [Internet]. 2017 May 30 [cited 2023 Feb 8];14(6):587. Available from: /pmc/articles/PMC5453245/. [CrossRef]
- Letunic I, Bork P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res [Internet]. 2019 Jul 7 [cited 2023 Feb 8];47(W1):W256. Available from: /pmc/articles/PMC6602468/. [CrossRef]
- Darvishi, M.; Rahimi, F.; Abadi, A.T.B. SARS-CoV-2 Lambda (C.37): An emerging variant of concern? Gene Rep. 2021, 25, 101378. [Google Scholar] [CrossRef]
- Padilla-Rojas, C.; Jimenez-Vasquez, V.; Hurtado, V.; Mestanza, O.; Molina, I.S.; Barcena, L.; et al. Genomic analysis reveals a rapid spread and predominance of lambda (C.37) SARS-COV-2 lineage in Peru despite circulation of variants of concern. J Med Virol. 2021, 93, 6845–9. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.E. Escasa información genómica en bases de datos públicas para investigar el SARS-CoV-2 en Latinoamérica. Rev Peru Med Exp Salud Publica. 2020, 37, 374–374. [Google Scholar] [CrossRef] [PubMed]
- Mahmood TBin Saha, A.; Hossan, M.I.; Mizan, S.; Arman, S.M.A.S.; Chowdhury, A.S. A next generation sequencing (NGS) analysis to reveal genomic and proteomic mutation landscapes of SARS-CoV-2 in South Asia. Curr Res Microb Sci. 2021, 100065. [Google Scholar]
- Padilla-Rojas, C.; Vega-Chozo, K.; Galarza-Perez, M.; Calderon, H.B.; Lope-Pari, P.; Balbuena-Torres, J.; et al. Genomic analysis reveals local transmission of SARS-CoV-2 in early pandemic phase in Peru. bioRxiv. 2020 Sep 6;2020.09.05.284604. [CrossRef]
- Tareq AM, Emran T Bin, Dhama K, Dhawan M, Tallei TE. Impact of SARS-CoV-2 delta variant (B.1.617.2) in surging second wave of COVID-19 and efficacy of vaccines in tackling the ongoing pandemic. Hum Vaccin Immunother. 2021;1. [CrossRef]
- Luo, C.H.; Morris, C.P.; Sachithanandham, J.; Amadi, A.; Gaston, D.; Li, M.; et al. Infection with the SARS-CoV-2 Delta Variant is Associated with Higher Infectious Virus Loads Compared to the Alpha Variant in both Unvaccinated and Vaccinated Individuals. medRxiv. 2021 Aug 20. [CrossRef]
- Liu Y, Liu J, Johnson BA, Xia H, Ku Z, Schindewolf C, et al. Delta spike P681R mutation enhances SARS-CoV-2 fitness over Alpha variant. bioRxiv. 2021. [CrossRef]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; et al. Sars-cov-2 spike mutations, l452r, t478k, e484q and p681r, in the second wave of covid-19 in Maharashtra, India. Microorganisms 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, F.; Talebi Bezmin Abadi, A. Emergence of the Delta Plus variant of SARS-CoV-2 in Iran. Gene Rep. 2021, 25, 101341. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, C.K.V.; Gräf, T.; Barcia CA de, L.; Costa, V.F.; de Oliveira, J.L.; Passos R da, H.; et al. SARS-CoV-2 variant of concern P.1 (Gamma) infection in young and middle-aged patients admitted to the intensive care units of a single hospital in Salvador, Northeast Brazil, February 2021. International Journal of Infectious Diseases 2021, 111, 47. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Herrera, N.; Araujo-Castillo, R.V.; Mestanza, O.; Galarza, M.; Rojas-Serrano, N.; Solari-Zerpa, L. SARS-CoV-2 Lambda and Gamma variants competition in Peru, a country with high seroprevalence. Lancet Regional Health Americas 2022, 6, 100112. [Google Scholar] [CrossRef] [PubMed]
- Leelawong, M.; Mitchell, S.L.; Fowler, R.C.; Gonzalez, E.; Hughes, S.; Griffith, M.P.; et al. SARS-CoV-2 N gene mutations impact detection by clinical molecular diagnostics: reports in two cities in the United States. Diagn Microbiol Infect Dis. 2021, 101, 115468. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Won, D.; Kim, C.K.; Ahn, J.; Lee, Y.; Na, H.; et al. Novel indel mutation in the N gene of SARS-CoV-2 clinical samples that were diagnosed positive in a commercial RT-PCR assay. Virus Res. 2021, 297, 198398. [Google Scholar] [CrossRef] [PubMed]
- Chiara, M.; D’Erchia, A.M.; Gissi, C.; Manzari, C.; Parisi, A.; Resta, N.; et al. Next generation sequencing of SARS-CoV-2 genomes: challenges, applications and opportunities. Brief Bioinform. 2021, 22, 616–30. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Díaz, D.A.; Laiton-Donato, K.; Franco-Muñoz, C.; Mercado-Reyes, M. Secuenciación del SARS-CoV-2: la iniciativa tecnológica para fortalecer los sistemas de alerta temprana ante emergencias de salud pública en Latinoamérica y el Caribe. Biomédica 2020, 40 (Suppl 2), 188. [Google Scholar] [CrossRef]
- Rodriguez-Morales, A.J.; Balbin-Ramon, G.J.; Rabaan, A.A.; Sah, R.; Dhama, K.; Paniz-Mondolfi, A.; Pagliano, P.; Esposito, S. Genomic Epidemiology and its importance in the study of the COVID-19 pandemic. Infez Med. Ahead of print Jun 1. 2020, 28, 139–142. [Google Scholar] [PubMed]
- Rabaan AA, Al-Ahmed SH, Sah R, Al-Tawfiq JA, Haque S, Harapan H, Arteaga-Livias K, Aldana DKB, Kumar P, Dhama K, Rodriguez-Morales AJ. Genomic Epidemiology and Recent Update on Nucleic Acid-Based Diagnostics for COVID-19. Curr Trop Med Rep. 2020;7(4):113-119. Epub 2020 Sep 24. PMID: 32989413; PMCID: PMC7513458. [CrossRef]
- Rodriguez-Morales AJ, Rodriguez-Morales AG, Méndez CA, Hernández-Botero S. Tracing New Clinical Manifestations in Patients with COVID-19 in Chile and Its Potential Relationship with the SARS-CoV-2 Divergence. Curr Trop Med Rep. 2020;7(3):75-78. Epub 2020 Apr 18. PMID: 32313804; PMCID: PMC7165999. [CrossRef]
Figure 1.
Phylogenetic tree created in IQ-TREE v1.6.12 of the 714 genomes of the Lambayeque region – Peru (Until April 28, 2022). The genomes are classified within the variants Mu, Delta, Gamma, Omicron, and Lambda.
Figure 1.
Phylogenetic tree created in IQ-TREE v1.6.12 of the 714 genomes of the Lambayeque region – Peru (Until April 28, 2022). The genomes are classified within the variants Mu, Delta, Gamma, Omicron, and Lambda.
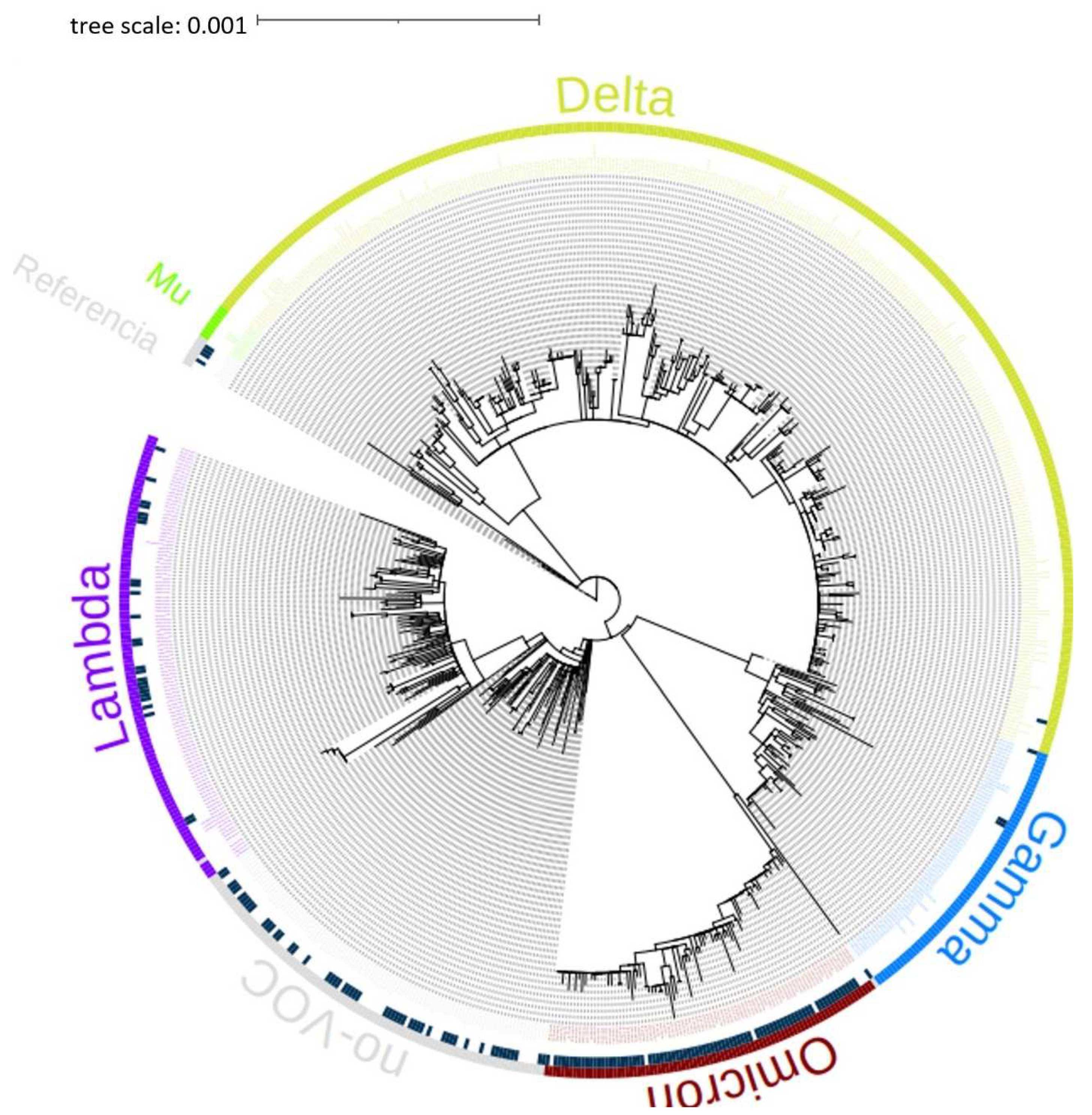
Table 1.
Mutations found in the C.14 SARS-CoV-2 lineage of patients from the Lambayeque Region, Peru.
Table 1.
Mutations found in the C.14 SARS-CoV-2 lineage of patients from the Lambayeque Region, Peru.
| Lineage C.14 | Genes affected by mutations | |||||
|---|---|---|---|---|---|---|
| ORF1a | ORF1b | S | ORF3a | ORF9b | N | |
|
C.14 |
P2144L T1246I G3278S P2685T |
P314L S638I H1087Y V2073L |
A222V D253E D614G |
L101F L140F S171L V225F |
T83I | H145Y R203K G204K |
Table 2.
Mutations found in the sublineages of the Delta SARS-CoV-2 variant of patients from the Lambayeque Region, Peru.
Table 2.
Mutations found in the sublineages of the Delta SARS-CoV-2 variant of patients from the Lambayeque Region, Peru.
|
Gene |
Sublineages variant Delta SARS-CoV-2 | ||||||
|---|---|---|---|---|---|---|---|
| AY.26 | AY.39.2 | AY.122 | AY.100 | AY.43 | AY.102 | B.1.617.2 |
|
| ORF1a | P1640L A3209V V3718A T3750I |
E743D A1306S K1817N P2046L P2287S V2930L T3255I T3646A |
K261N A1306S P2046L P2287S V2930L T3255I T3646A |
T403I A1306S P2046L P2287S V2930L T3255I T3646A |
A1306S P2046L P2287S V2930L T3255I T3646A |
A1306S P2046L P2287S V2930L T3255I T3646A |
A1306S T3255I T3646A |
| ORF1b | P314L G662S P1000L |
P314L G662S P1000L A1918V Q2635H |
P314L G662S P1000L A1918V |
P314L G662S P1000L A1219S A1918V |
P314L G662S L829I P1000L A1918V |
P314L G662S P1000L A1918V |
P314L G662S P1000L A1918V |
| S | T19R R158G Δ156/157 A222V L452R T478K D614G P681R D950N V1264L |
T19R R158G Δ156/157 L452R T478K D614G P681R D950N K1073N |
T19R R158G Δ156/157 L452R T478K D614G P681R D950N |
T19R R158G Δ156/157 L452R T478K D614G P681R G769V D950N |
T19R R158G Δ156/157 L452R T478K D614G P681R D950N |
T19R R158G Δ156/157 L452R T478K D614G P681R D950N |
T19R R158G Δ156/157 L452R T478K D614G P681R D950N |
| ORF3a | S26L | S26L | S26L | S26L |
S26L T34A |
S26L | S26L |
| M | I82T | I82T | I82T | I82T | I82T | I82T | I82T |
| ORF6 | K48N | ---- | ---- | ---- | ---- | ---- | ---- |
| ORF7a | V82A T120I |
V71I V82A T120I |
V82A T120I |
V82A T120I |
V82A T120I |
V82A T120I |
V82A T120I |
| ORF7b | ---- | T40I | T40I | T40I | T40I | T40I | T40I |
| ORF 8 | S84L Δ119/120 |
S84L Δ119/120 |
Δ119/120 | Δ119/120 | Δ119/120 | Δ119/120 | Δ119/120 |
| N | D63G R203M D377Y |
D63G R203M G215C D377Y |
D63G R203M G215C D377Y |
D63G R195K R203M G215C D377Y |
Q9L D63G R203M G215C D377Y |
D63G R203M G215C D377Y |
D63G R203M G215C D377Y |
Table 3.
Mutations found in the sublineages of the SARS-CoV-2 Gamma variant of patients from the Lambayeque Region, Peru.
Table 3.
Mutations found in the sublineages of the SARS-CoV-2 Gamma variant of patients from the Lambayeque Region, Peru.
| Sublinajes variante Gamma | Genes affected by mutations | |||||
| ORF1a | ORF1b | S | ORF3a | ORF8 | N | |
|
P.1.12 |
S1118L K1795Q Δ3675/3677 |
P314L E1264D |
L18F T20N P26S D138Y R190S K417T N501Y D614G H655Y T1027I V1176F |
S253P | E92K | P80R |
|
P.1 |
S1118L K1795Q Δ3675/3677 |
P314L E1264D |
L18F T20N P26S D138Y R190S K417T E484K N501Y D614G H655Y T1027I V1176F |
S253P |
E92K |
P80R R203K G204R |
Table 4.
Mutations found in the Lambda (C.37) SARS-CoV-2 variant from patients in the Lambayeque Region, Peru.
Table 4.
Mutations found in the Lambda (C.37) SARS-CoV-2 variant from patients in the Lambayeque Region, Peru.
| Lambda Variant | Genes affected by mutations | ||||||
|---|---|---|---|---|---|---|---|
| ORF1a | ORF1b | S | ORF3a | ORF9b | M | N | |
|
C.37 |
T1246I P1659T P2287S F2387V P2483S L3201P T3255I G3278S A3620V Δ3675/3677 |
S59F P314L T1137I A1643V Y1784C K2385E K2674R |
L5F G75V Δ246-252 L452Q A475V E484K P499R N501T D614G H655Y P681R T859N |
P240H | P10S | I82T | P13L R203K G204R G214C |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



