Submitted:
22 November 2023
Posted:
23 November 2023
You are already at the latest version
Abstract
8-oxoguanine glycosylase 1 (OGG1), which was initially identified as the enzyme that catalyzes the first step in the DNA base excision repair pathway, is now known also as a modulator of gene expression. What is important for cancer, is that OGG1 acts as a modulator of NFκB-driven gene expression. Specifically, oxidant stress in the cell transiently halts the enzymatic activity of substrate-bound OGG1. The stalled OGG1 facilitates DNA binding of transactivators, including NFκB, to their cognate sites to enable expression of cytokines and chemokines, with ensuing recruitment of inflammatory cells. Recently, we highlighted chief aspects of OGG1 involvement in regulation of gene expression, which have a significance in lung cancer development. However, OGG1 has also been implicated in the molecular underpinning of acute myeloid leukemia. In general, the capacity of cancer cells to adapt to oxidative stress depends on molecular systems such as the interface of OGG1 with NFκB, which bestows a cancer cell with the molecular mechanism of transformation of its microenvironment to enable adaptation and survival of malignant clones.
Keywords:
OGG1
; NFκB
; gene regulation
; microenvironment
; EMT
; innate immunity
; cancer stem cells
; lung cancer
; acute myeloid leukemia
; oxidant stress
1. Introduction
Initial event in cancer, is a set of drastic molecular changes that drives disruption in tissue homeostasis of a specific niche [1,2]. This disruption redirects the normal succession of inflammation-regeneration into a process that perturbs immunity and enables growth of malignant cell clones with a tumor-initiating potential [3,4]. This disruption is rooted in changes in cellular chromatin that alter patterns of gene expression, increasing thereby production of molecules that facilitate cancer progression and interfere with the function of the immune system [5].
Oxidative stress is a key factor that influences gene expression [6]. All types of antineoplastic treatment lead to increased cellular stress that is aimed to kill tumor cells. Cellular stress, however triggers also adaptation mechanisms that may enhance adaptability of malignant tumors. At least one of the adaptation mechanisms triggered by oxidative stress is the induction of inflammatory gene expression. Inflammatory genes include cytokines that mediate interactions between tumor and stroma, leading to cellular phenotypes that enable metastasis, protection of malignant cells from the immune system, and drug resistance.
Oxidant stress has been recognized as a key factor of immunosuppression as both myeloid-derived suppressor cells and immunosuppressive extramedullary erythroid progenitor cells employ reactive oxygen species (ROS) to impair CD8+ T cell responses against pathogens and tumors [7,8]. ROS promote cancer in diverse ways that affect multiple cell types that include both malignant cells, as well as stromal and immune cells [9].
We here explore and discuss the intricate connections between redox imbalance, oxidative DNA base modifications, and the interactions of OGG1 with NFκB and Myc in the tumor microenvironment. Further research and understanding of the specific molecular mechanisms and their roles in tumor biology will be essential for the development of targeted and effective therapeutic interventions.
2. Oxidative Stress Produces DNA Lesions, Turning OGG1 into an Epigenetic Eeader
Nucleobases in DNA and RNA react differently with reactive entities, guanines (Gua) being the most sensitive due to their lowest oxidation potential among the five nucleobases. The interaction of Gua in RNA or DNA with reactive entities most often generates 7,8-dihydro-8-oxoguanine (8-oxoGua) and 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyGua), which are the most abundant lesions in nucleic acids [10]. For 8-oxoGua the anti-conformation is preferred in DNA, under replication or transcription (single stranded) it can take the syn conformation and pair with adenine (Hoogsteen base pairing). The 8-oxoGua: adenine base pair can produce a mutation in the following round of DNA replication, resulting in a G:C to T:A mutation [11].
8-oxoGua differs from guanine by only two atoms, but OGG1 can efficiently recognize it when paired with cytosine or 5-methylcytosine. 8-oxoGua is also recognized by human homologs of Nei-like glycosylases, primarily in single-stranded DNA [12,13,14]. After excision, the resulting apurinic/apyrimidinic (AP) sites are processed by AP endonuclease1/Ref1 (APE1/Ref1) or the polynucleotide kinase-phosphatase (PNKP) to form polymerase-ready 3'OH residues [15,16]).
A question for these glycosylases is how they recognize modified bases in chromatin. Recent studies show, for example that OGG1, a relatively low molecular weight polypeptide, searches for DNA base damage with the aid of its asymmetric surface charge distribution, requiring nearly no energy for migration (sliding) over the DNA duplex [17]. Substrate recognition and binding are accomplished via a complex set of molecular alterations, associating residues within the active site center with the oxidatively altered nucleotide to cleave the glycosyl bond. The removal of modified DNA base lesions ensures genome integrity and serves as a signal for cellular responses to oxidative stress [18]. It has been documented that oxidative stress induces the translocation of OGG1 (and other BER proteins) to open chromatin, which contains cis elements for transacting factors. These data imply that the searching activity of OGG1 for substrates is synchronized with chromatin remodelers that are transcriptionally active [19]. Cofactors such as histone marks, protein cofactors, or 8-oxoGua and FapyGua can increase the effective recruitment of OGG1 in chromatinized DNA [20,21,18].
Early studies documented that the OGG1 substrate, 8-oxoGua, is located within or in close proximity to cis elements for hypoxia-inducible factor-1 (HIF-1) within the promoter of vascular endothelial cell growth factor (VEGF), and it plays a crucial role in gene expression in hypoxic signaling [22]. Subsequent research identified 8-oxoGua as an epigenetic-like mark in the context of promoters within potential G-quadruplex, Z-DNA forming and i-motif-forming sequences, as well as in promoters of proinflammatory genes and genes of high importance in epithelial-to-mesenchymal transitions [23,24,25,26. These results imply site-specific generation of 8-oxoGua, which is difficult to envision. However, studies have revealed a role for a nuclear flavoenzyme, lysine-specific demethylase 1 (LSD1) in mediating site-specific generation of ROS and consequently 8-oxoGua during demethylation of H3 lysine [27,28]. Thus studies on LSD1 provided an integrated mechanism by which 8-oxoGua is specifically generated at both enhancer and promoter sites. This, in turn, recruits OGG1 and DNA repair components, thereby activating estrogen-induced gene expression [28,29]. This is significant, as histone H3 Lys4 trimethylation sites are suggested to be oxidant-sensitive epigenetic marks, which modulate gene expression under stress conditions [30,31]. Consequently, the distinction between modification(s) to DNA base(s), DNA repair, and epigenetics is now blurred, as studies have clearly identified changes in gene expression [32].
The identification of 8-oxoGua as an epigenetic mark and the understanding of OGG1's trans-regulatory function were advanced by technologies that mapped 8-oxoGua in the genome. These studies linked 8-oxoGua with the regulation of transcription, especially from enhancers, promoters and untranslated regions (UTR) on a genome-wide scale [33,34,35,36]. High-throughput sequencing analysis found OGG1 in chromatinized DNA at 8-oxoGua over thousands of promoters, UTRs including genes implicated in innate immune responses (IIR; e.g., cytokines, chemokines, interleukins) and those responding to oxidative stress [37]. Further supporting the regulatory (epigenetic) role of 8-oxoGua in promoters, in-depth studies demonstrated its key role during the transcriptional program of epithelial-to-mesenchymal transition (EMT), triggered by transforming growth factor β1 (TGFβ-1) [38,39].
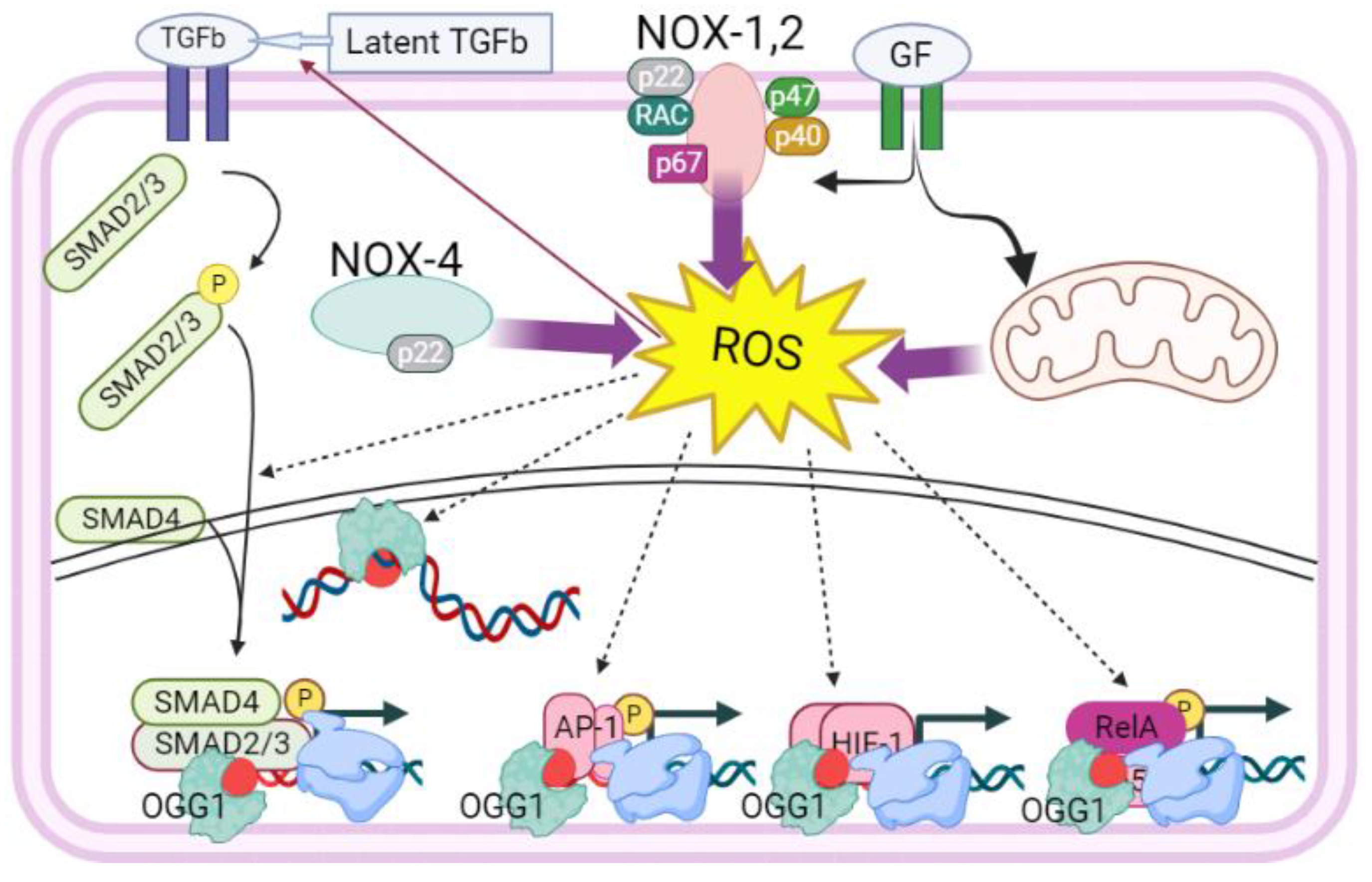
Figure 1.
The proposed role of OGG1 in gene expression regulation related to tumor cell proliferation. In response to growth factors, hormones, metabolites, or environmental factors ROS are generated by NADPH oxidases and mitochondrial respiratory complexes. ROS signaling is crucial for activating growth factors (e.g., TGFβ1), transacting factors (e.g., NF-κB, AP-1), and also for modifying DNA bases (e.g., guanine → 8-oxoGua, an epigenetic mark). ROS also induce the oxidation of OGG1 at cysteine residues, inhibiting its glycosylase activity without affecting its ability to recognize/interact with intrahelical 8-oxoGua and the complementary cytosine. OGG1's interaction with 8-oxoGua induces topographical rearrangements in DNA, resulting in an approximately 70-degree bending. This transient topographical change facilitates the DNA occupancy of transcription factors (e.g., SMADs, AP-1, HIF-1, NF-κB) and components of the transcriptional machinery. Abbreviations: TGFβ, transforming growth factor beta; GF, growth factor; NOX, NADPH oxidoreductase; ROS, reactive oxygen species, SMAD, Mothers against decapentaplegic homolog; AP-1, Jun activation domain binding protein; HIF, hypoxia inducible factor; NF-κB, nuclear factor kappa B. Created by Bio-Render: https://app.biorender.com.
Figure 1.
The proposed role of OGG1 in gene expression regulation related to tumor cell proliferation. In response to growth factors, hormones, metabolites, or environmental factors ROS are generated by NADPH oxidases and mitochondrial respiratory complexes. ROS signaling is crucial for activating growth factors (e.g., TGFβ1), transacting factors (e.g., NF-κB, AP-1), and also for modifying DNA bases (e.g., guanine → 8-oxoGua, an epigenetic mark). ROS also induce the oxidation of OGG1 at cysteine residues, inhibiting its glycosylase activity without affecting its ability to recognize/interact with intrahelical 8-oxoGua and the complementary cytosine. OGG1's interaction with 8-oxoGua induces topographical rearrangements in DNA, resulting in an approximately 70-degree bending. This transient topographical change facilitates the DNA occupancy of transcription factors (e.g., SMADs, AP-1, HIF-1, NF-κB) and components of the transcriptional machinery. Abbreviations: TGFβ, transforming growth factor beta; GF, growth factor; NOX, NADPH oxidoreductase; ROS, reactive oxygen species, SMAD, Mothers against decapentaplegic homolog; AP-1, Jun activation domain binding protein; HIF, hypoxia inducible factor; NF-κB, nuclear factor kappa B. Created by Bio-Render: https://app.biorender.com.
3. OGG1-Induced DNA Remodeling in Proximity of Transacting Factor’s cis Elements
As detailed above, OGG1 scans DNA through diffusion, allowing it to search for modified Gua(s) (reviewed in [20]). Structural studies have shown that OGG1's association with the epigenetic mark 8-oxoGua involves two major steps: 1) an initial enzyme-substrate interaction, and 2) a fundamental remodeling of DNA [40], reviewed in [41]. During step 1, 8-oxoGua is extruded from the helix and placed into the OGG1 active-site pocket. In step 2, OGG1 interacting with the opposite cytosine caused it to become unstacked from adjacent bases, resulting in a sharp bending of the duplex DNA [40]. These slight but substantial changes in DNA structure around 8-oxoGua, in the context of chromatin, reduce the energy needed for transcription factors to occupy DNA and for the association of OGG1 with transacting factors and other proteins [13,42].
To better understand the role of OGG1 at 8-oxoGua in TFs DNA occupancy, it was shown that binding of NF-κB homodimers (p50-p50) and heterodimers (p50-RelA/p65) to 8-oxoGua containing DNA are 15 to 25-fold higher in the presence of OGG1 [32]. In the context of chromatin, OGG1 binding was mapped in close proximity to NF-κB [37]. The binding of OGG1 to 8-oxoGua in promoters enhanced NF-κB/RelA binding to cis-elements and facilitated the recruitment to the transcriptional complex of specificity protein 1 (SP1), transcription initiation factor II-D (TFIID), and phosphorylated-RNA polymerase II (p-RNA Pol-II), leading to increased expression of pro-inflammatory chemokines and cytokines in cultured cells and lungs of experimental animals [43].
Additionally, it was shown that DNA-bound OGG1 at 8-oxoGua interacted with mitogen-and stress-activated kinase 1, which was essential for phosphorylation of NF-κB/RelA at serine 276 and expression of innate cytokines and chemokines [44]. A recent study documented that OGG1 at 8-oxoGua in the context of chromatin recruited phosphorylated SMAD2/3 complex and NF-κB to their cognate binding sequence located in promoters of tissue remodeling genes including alpha-smooth muscle actin (α-SMA), collagen (COL), and fibronectin (FN) for excessive gene expression [39,45]. A recent study revealed that Myc oncogene binding to the E-box promoter recognition sequence required OGG1 in oxidative stress conditions [46]. Specifically, fluorescence polarization experiments discovered that interaction with the Myc oncogene conformationally modified hOGG1 at 8-oxoGua lesions, and suspended OGG1’ glycosylase activity, while in turn OGG1 facilitated loading of Myc to E-box sequences [46]. The pathophysiological implication of OGG1 in promoting NF-κB, SP1, TFIID, SMADs, Myc, as well as p-RNA Pol-II and transcription, for immune responses and Myc-induced tumor progression, has unforeseen significance.
4. Direct Impact of the OGG1 Interaction with NFκB
OGG1 stimulates DNA binding of NFκB to several inflammatory gene promoters, increasing thereby expression of several cytokines, chemokines, and a number of other factors; this has been implicated in inflammatory syndromes in the lung and other organs [47,48]. Activated NFκB alters the landscape of gene regulatory sequences in chromatin, enabling drastic changes in the phenotype of the cell [5]. In part, these changes occur by switching off numerous enhancer sequences and switching on other enhancers; this phenomenon is especially evident in extended enhancer sequences that are rich in transcription factor binding sites, and are termed stretch enhancers, or superenhancers [49,50]. Superenhancer changes are associated with cancer progression; in leukemia, specific feed-forward loops are proposed to drive disease progression by switching on expression of NFκB downstream genes that include but are not limited to Myc [51,52,53]. After stimulation of embryonic kidney cells with TNFa, OGG1 and NFκB were found to bind to DNA recognition sites of organic cation transporter (OCT2), sex determining region Y box transcription factor (SOX) and other families of regulators of cellular development by chromatin immunoprecipitation [54]. OGG1 and NFκB also activate RAS signaling, which is a known oncogenic driver [55,56].
Drastic changes in chromatin facilitate profound alterations in the cellular phenotype. While such events are crucial for the effective function of most cell types that are involved in inflammation, during cancer the induction of chromatin alterations enables cancer cells to acquire novel properties.
During inflammation, a sequential activation of different types of leukocytes takes place, induced by cytokines, chemokines, and adhesion factors. Malignant cells express subsets of genes that are normally induced in cell subtypes that are essential for survival of the organism [3]. As result, the sequence of events between inflammation and tissue regeneration is disrupted. Although cellular components of innate and adaptive immunity may be active, cancer cell niches are protected due to expression of immunosuppressive molecules in tumor nest [54].
5. A Convoluted Mode of Regulation for NFκB Leads to a Complex Function in Cancer
NFκB is held inactive in the cytoplasm of most normal cells, bound by the polypeptide IkBa. Upon a cell receiving an inflammatory stimulus IkBa is degraded and NFκB enters the nucleus to regulate gene expression (reviewed in [57]); the result of NFκB activation depends on the assortment of posttranslational modifications and interactions with other proteins, including also OGG1 [32]. Although in most cell types the degradation of IkBa occurs at the proteasome, it may be accomplished by other proteolytic systems, making pharmacological inhibition of NFκB in this step in some cases unsuccessful.
In particular, the lysosome and calpain may also degrade IkBa [58,59,60]. Especially in advanced cancer cells after exposure to chemotherapy, cell stress may activate biomolecule degradation at the lysosome to prevent cell death and provide essential metabolic intermediates. Most drugs cause cellular stress, and through the lysosome cell stress itself becomes a driver for cancer progression [61].
NFκB activity in normal cells induces a number of negative feedback mechanisms that protect tissues from aberrant initiation of inflammatory cascades, which would cause damage to functional tissue components though the immune system. However in cancer cells the network of feedback signal pathways to NFκB is not intact, leading on the one hand to protection of cancer cells from the immune system, and on the other hand to destruction of non-cancerous tissue [3,5,54]. Recent data indicate that NFκB is often aberrantly active in malignant cells, and this activity can be also initiated or maintained by cell stress, enabling at least some tumor cell clones to survive under conditions that would be lethal to the original tumor.
In an impressive demonstration, restoring negative feedback to NFκB by a myeloid cell-targeted miR-146a mimic that prevented excessive NFκB activation in myeloid cells, did not only alleviate lethal inflammation in a chimeric antigen receptor (CAR) T-cell-induced cytokine release syndrome model of xenotransplanted B-cell lymphoma. It also was cytotoxic to leukemia cells in vitro and in vivo, it inhibited NFκB target genes expression and thereby thwarted progression of disseminated HL-60 promyeocytic leukemia [62]. Conversely, chronic activation of NFκB and expression of inflammatory genes in mouse hematopoietic stem cells leads to a “myeloid bias” during aging [63] and predisposes to leukemias [64].
In diverse types of cancer, constitutive NF-κB activity enables malignant cells to survive oncogene activation, tumor suppressors, radiation, drug treatments, extensive genetic alterations and the surveillance of both innate and adaptive immune cells [4,5]. However, more important is the fact that NFκB confers cancer cells flexibility in the capacity to acquire or relinquish stem cell attributes. Specifically, on the one hand NFκB facilitates efficient induction of expression for antiapoptotic genes, and for the protooncogene encoding transactivator c-Myc, which prepares a cell for rapid growth by drastic changes in expression of metabolic enzymes [3]. On the other hand NFκB confers to cancer cells the capacity to adopt stem cell features [65,66] by multiple indirect pathways that culminate in the acquisition of immortality and resistance to oxidative stress [5,67]. Malignant cells thereby can switch between different metabolic and phenotypic states.
6. Oxidative Stress as a Driver for Cancer Plasticity
Cancer progression succeeds after adaptation of malignant cells to the physiological, pathological, and pharmacological conditions of host tissue, which result from a combination of endocrine factors, drug treatment, and the resulting cellular stress: cancer cells that survive cytotoxic conditions show adaptation to cell stress [68]. Exposure of cells to increased oxidative stress provides a powerful stimulus to NFκB-driven gene regulation [69]. This is exemplified by NFκB interaction with OGG1 [32,45,70], which redirects NFκB activity and impairs immunity [70]. In contrast, enzymatically active OGG1 guards genome integrity through either lesion repair or elimination of cells with malignant potential, to maintain the homeostasis of the host, which might depend on the magnitude of guanine oxidation [71].
The impact of OGG1 on cancer development can thus be dissociated from its function in DNA repair [54]. This is a fact that can be best illustrated by the OGG1 genetic allele variant that carries a C-to-G substitution at codon 326 giving rise to a protein product with a cysteine in place of serine in amino acid residue position 326. This allelic variant of OGG1 (Ser326Cys) is identified in over 50% and 40 % in Asian and Caucasian populations, respectively [72]. This naturally-occurring polymorphic OGG1 variant has decreased activity, increased capacity to dimerize via Cys-Cys disulfide bond formation [73,74], it is more sensitive to inactivation by oxidative stress, and does not recover from oxidant stress after stimulation of cells with physiological concentration of TNFα [75] and therefore may provide a more effective enhancement of NFκB-driven gene expression. Specifically, the enrichment of OGG1 on chromatinized DNA likely at its substrate for a prolonged period of time is due to the loss of its enzymatic activity by ROS in cellulo, due to the lower threshold of OGG1 repair activity inhibition by oxidant stress.
Although cancer is a highly heterogeneous disease, at least two powerful examples demonstrate that OGG1 S326C can be implicated in malignant progression.
I) Lung cancer. In advanced inoperable non-small cell lung cancer (NSCLC) patients who received treatment with platinum-based chemotherapy, the OGG1 genetic allele variant carrying a C-to-G substitution at codon 326 was associated with poor progression-free survival [76,77]. The imbalance between the Ser326Cys OGG1-enhanced inflammation and the delayed Ser326Cys OGG1 repair activity would be expected to impact lung pathology, especially in neoplastic tissue. Indeed, Ser326Cys Ogg1 was linked to shorter progression -free survival in inoperable NSCLC [76]. It should also be noted that the monoallelic Ser/Cys and biallelic Cys/Cys variants of OGG1 are potentially implicated in carcinogenesis, as they, as they have previously been linked to increased susceptibility to various human malignancies including endometrial [78], colorectal [79], breast [80], oropharyngeal squamous cell carcinoma [81] and prostate cancer [82,83].
II) Acute myeloid leukemia (AML). OGG1 S326C was observed more frequently in patients with AML relapse when compared to other patients [28.9 vs. 8.9%, odds ratio = 4.10, 95% confidence interval = 1.35–12.70, p = 0.01], and the patients with S326C exhibited a shorter relapse-free survival [84]. Remarkably, oxidative stress appears to have a key role in the outcomes of acute erythroid leukemia, which is a subtype of AML with particularly poor survival. This is suggested by the following facts:
a) in erythroleukemia cells (in vitro) the lipid peroxidation byproduct 4-hydroxynonenal, a reactive aldehyde [85], modulates Myc expression [86], This provides a downstream target for stalled OGG1, with relevance for AML: increased oxidant stress stalls OGG1, which becomes an activator for NFκB-regulated gene expression; Myc is a known target gene of OGG1 [87]. Besides of being a downstream NFκB target gene Myc is a key regulator in malignant cells, and AML cells in particular [88,89,90]. In AML, NFκB transcriptional activity has an established role in fostering tumor progression by paracrine secretion of cytokines in the microenvironment; however constitutive activation of NFκB due to aberrant feedback is associated with diverse advantages for AML cells, and leukemia stem cells in particular [91,92,93]. This makes NFκB-activating modules, such as stalled, substrate-bound OGG1, a very strong candidate intervention target in AML.
b) the RNA expression of ALDH1A1, which encodes a key 4-hydroxynonenal detoxifying enzyme, is increased in bone marrow samples from erythroleukemia patients [94]. Therefore upstream of stalled OGG1 there exist candidate targets for AML treatment too: the cause of OGG1 stalling, being oxidative stress, affects the intracellular milieu through lipid peroxidation products, namely reactive aldehydes. An enzyme that protects cells from reactive aldehydes, is ALDH1A1. It is therefore not surprising that ALDH1A1 RNA is overexpressed in erythroleukemia and in AML poor prognosis patients, in general.
What is important in terms of disease outcome, is that AML patients of the poor prognosis risk group show frequent elevation of RNA expression for the enzymes ALDH1A1 and ALDH2, and this elevation is associated with an increased risk of death [94]. In the same study, ALDH1A1 encoding RNA was linked to an AML stemness signature. These findings are consistent with research that shows ALDH enzymatic activity protecting AML cells from chemotherapy and ALDH1A1 null AML cells susceptible to chemotherapy, while ALDH1A1 null patients belong to the favorable risk [95,96]. Therefore for patients, the capacity of AML cells to survive increases in reactive aldehydes, is associated with a poor prognosis and disease progression, and can be traced at least in part, to increased expression of ALDH enzymes [97,98,99,100].
In AML there is a solid connection of NFκB with malignant progression, but nevertheless, interventions based on inhibiting NFκB or its associated mediators such as bromodomain and extra-terminal domain (BET) protein BRD4 [101] have not proven clinically effective [102,103]. This could be associated with the convoluted mode of NFκB regulation in cancer cells. This mode, however, exposes a number of potential intervention targets, both upstream and downstream from NFκB activation.
Leukemia stem cells exhibited resistance to BET-inhibitors both ex vivo and in vivo, independently of Myc expression, and in connection with an upregulation of WNT/b-catenin signaling, which appeared essential to the manifestation of resistance [104]. Also non-stem cells of leukemia could develop resistance to BET-inhibition, yet this time the resistance was linked to restoring Myc expression and required transcriptional plasticity. Chromatin immunoprecipitation sequencing and self-transcribing active regulatory region sequencing of enhancer profiles revealed that BET-resistant states are characterized by remodeled regulatory landscapes, involving the activation of a focal Myc enhancer that recruits WNT machinery in response to BET inhibition [105].
Thus depending on the leukemia cell type, both BRD4 and Myc can become dispensable under certain conditions.
7. A Model for Malignant Progression for Cancers Driven by Changes in Oxidative Stress Response of OGG1
A key aim in research for cancer treatment is to target mechanisms that permit malignant cells to withstand cytotoxic chemotherapy. One general aspect of cancer cell resistance mechanisms to chemotherapy is the development of clones with increased capacity to respond to cellular stress. Chemotherapy is administered to kill cancer cells, and proves especially effective in killing cancer cells that operate error-prone systems of biomolecule synthesis and processing. However, exposure of cancer cells to chemotherapy or any other cytotoxic conditions tends to select for clones that operate efficient cell stress adaptation mechanisms, which often act by inducing the removal of mediators of cell death, or by preventing accumulation of cytotoxic metabolites. The latter is particularly important for leukemia cells exposed to chemotherapy [67,106].
Therefore we may work with a model for malignant progression for cancers driven by changes in oxidative stress response. Specifically, increased oxidative stress initially inactivates several enzymes by the formation of adducts with lipid peroxidation products such as 4-HNE [107] ; one such enzyme is OGG1, which during increased oxidative stress becomes a transcriptional coactivator for transactivators such as NFκB [32], enhancing expression of several inflammatory genes, including cytokines that enable transformation of the tumor microenvironment, and other proteins that protect cancer cells from the immune system, despite of activation of several types of immune cells [54]. While a number of tumor cell clones may die, activation of biomolecule recycling by lysosomes can rescue malignant cells that adapt to increased oxidative stress. Enhanced activity of enzymes such as ALDH1A1 that detoxifies reactive aldehydes, and of enzymes that participate in glutathione reduction allows adapted cancer cells to escape apoptosis, necrosis, ferroptosis, and cuproptosis [67].
Cellular stress itself becomes thereby a driver of tumor cell escape from destruction.
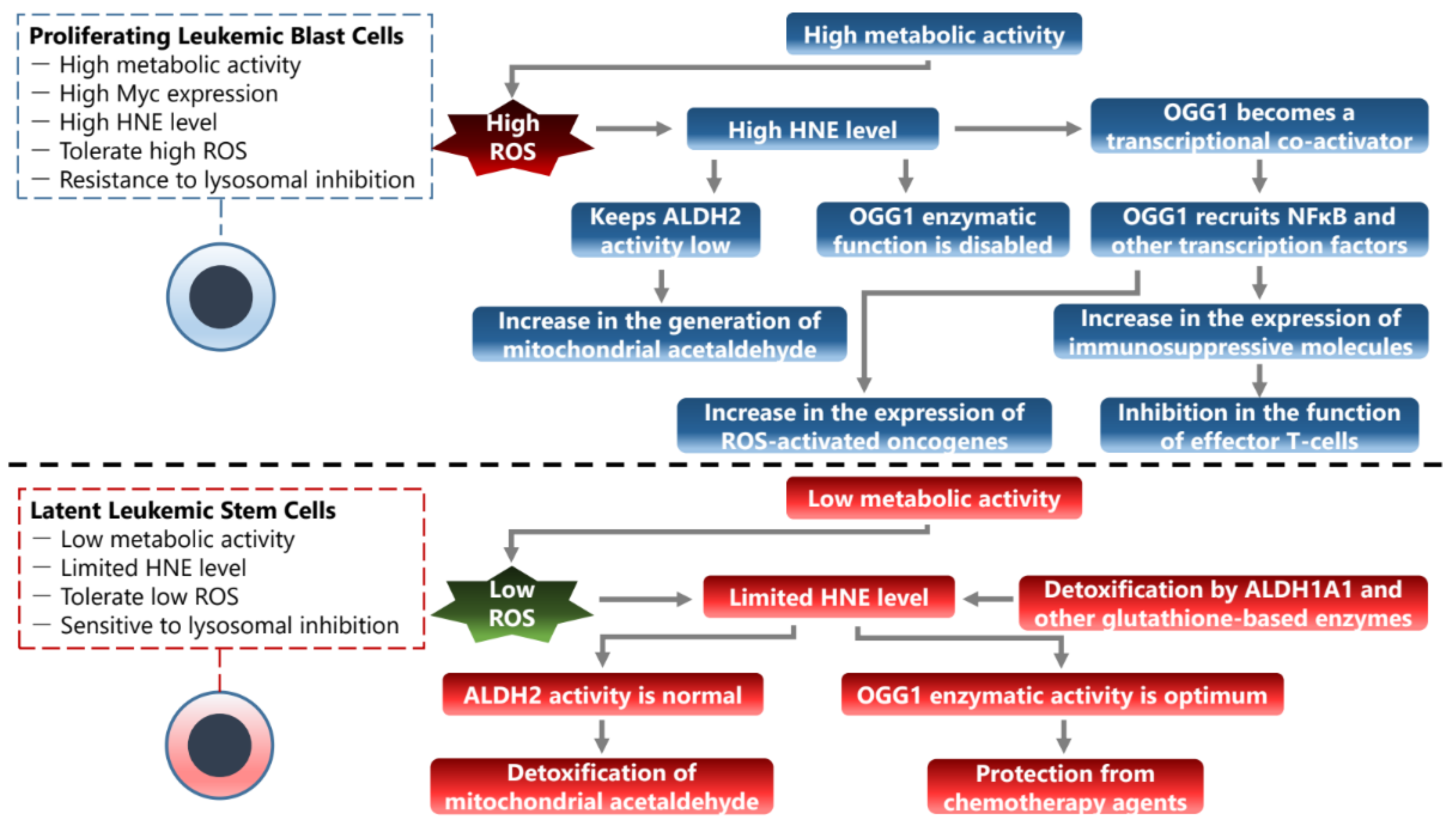
In the example of AML progression, when viewed from the angle of ALDH enzymes, a refinement of the model is possible: ALDH1A1 is crucial in specific AML stages. It coincides with specific challenges to a cells’ malignant function and survival. ALDH2 is not stage-specific. I general, aldh2 overexpression coincides with bad outcome in AML. ALDH2 functions more efficiently to detoxify mitochondrial acetaldehyde, which is rather constitutively produced. This means that specific stages of AML, which can be viewed as distinct phases when a given leukemic cell phenotype dominates, need increased ALDH1A1 activity. HNE can offer a plausible explanation for this exclusivity of specific AML stages for the hazard of ALDH1A1 overexpression:
a) proliferating leukemic blast cells have high metabolic activity and Myc expression, and may tolerate higher ROS [108], with accompanied high levels of HNE. These cells are resistant to lysosomal inhibition because they are not dependent on the recycling of biomolecules. OGG1 enzymatic function is disabled by HNE [107], and OGG1 transcriptional activity via recruited transcription factors such as NFκB delivers high expression of inflammatory mediators, feed-forward ROS-activated oncogenes [54,55,109,110], as well as immunosuppressive molecules that in cooperation with increased oxidant stress in the microenvironment, inhibit the function of effector T-cells. HNE also keeps ALDH2 activity low by forming adducts with ALDH2 [111], which allows increased generation of mitochondrial acetaldehyde. In general however, as increased ROS damage chromatin [112], these cells may generate increased genetic diversity for AML, even if a large portion of AML cell clones undergoes cell death. To note: studies have revealed that the DNA occupancy of Myc to its E-box in the chromatin is facilitated by OGG1. In oxidative stress conditions present in AML cells, OGG1 undergoes dimerization via cys28 residues. However, it can still recognize 8-oxoGua, and through its interaction with Myc it promotes binding at 8-oxoGua in the promoter. Additionally, data indicate that Myc significantly decreases the glycosylase activity of OGG1, resulting in a prolonged binding to the epigenetic mark. Myc recruitment to E-box DNA motifs by OGG1 in oxidizing cellular milieu can lead to inflammatory conditions, and to enhanced gene expression of Myc target genes [46].
b) latent leukemic stem cells have low metabolic activity, tolerate low ROS levels, and keep limited levels of HNE, which is readily detoxified by enzymes such as ALDH1A1 and other systems, such as glutathione-based enzymes [113,114]. These cells are dependent on lysosomal activity and therefore are sensitive to lysosomal inhibition [115]. OGG1 enzymatic function in these cells is within optimum range [84,116] and protects from some chemotherapy agents such as cytarabine [117], and ALDH2 activity detoxifies mitochondrial acetaldehyde, further protecting cellular macromolecules. Latent cells offer a low profile to the immune system, probably by expression of “don’t eat me” signals and suppression of key antigen exposure [118,119]. Excessive increase in HNE abrogates the colony-forming capacity of AML cells [120]. Increased expression of enzymes such as ALDH1A1, which form a first-line defense from HNE protects cells from chemotherapy-triggered lipid peroxidation and enables the emergence of chemoresistant AML [67]. ALDH-high leukemia cells are also associated with a poor patient prognosis [121].
It is important to note that the latent leukemic cells described in (b) are not propagated as a fixed cellular phenotype. These cells rather represent a key phenotypic intermediate, which easily converts into proliferating blast cells when exposed to excessive cell stress: the presence of a substantial antioxidant capacity simply offers to leukemia cell clones the critical opportunity to grow long enough to generate new resistant subclones, which under hostile conditions may become the dominant cellular phenotype. It is therefore important to bear this in mind when adjusting treatment design, and it should become evident experimentally when research models are monitored for a time window that is long enough to allow for the development of phenomena that require lengthy periods of cellular evolution, which correspond to the development of cell subclones that occurs in a living mammalian organism.
Such phenomena should be anticipated or expected to occur also in the organism of a patient that experiences leukemia relapse, or leukemia that is refractory to treatment. This conceptual model may provide an explanation on why the ROS/Myc/BRD4 axis is not indispensable for leukemia: while Myc overexpression and activation “burns the brakes” of tissue homeostasis AND creates the conditions that enable Myc-driven carcinogenesis, excessive cellular stress and ROS enable alternative transcriptional programs that allow development of leukemic stem cell clones. It would not be surprising if such observations emerge also for solid tumors in respect to transcriptional flexibility and the growth of resistance to anti-cancer treatment.
Figure 2.
A proposed model for two basic phenotypes of AML stem-like cells that may also coexist in a form of dynamic equilibrium that is regulated by the metabolic state of their microenvironment.
Figure 2.
A proposed model for two basic phenotypes of AML stem-like cells that may also coexist in a form of dynamic equilibrium that is regulated by the metabolic state of their microenvironment.
8. Phenotypic Transitions in the Microenvironment
In addition to its effects on cancer and stromal cells, oxidative stress can modulate endothelial cell function, which is suggested by research on the OGG1-NFκB interaction in chromatin [122]. Dysfunction of the vascular endothelial barrier in cancer has been implicated in disease pathology, progression and outcome [6,123] as vascular endothelial cells have a key role during tumor metastasis [124] and the interaction between cancer cell and endothelium has been documented in research models for migration through the blood-brain barrier [125]. In particular, endothelial-mesenchymal transition has been proposed to occur after anthracycline treatment and to initiate vascular remodelling [126] ; anthracyclines are known triggers of NFκB activation [127] and specifically initiate endothelial-to-mesenchymal transition [128] on the one hand, and on the other hand NFκB activation protects cancer cells from apoptosis [129]. However, endothelial dysfunction in cancer has also severe symptoms that may prove that endothelial injury is lethal on its own [130]. It is critical to restore endothelial function in cancer by controlling oxidative stress, as an optimum level of endothelial redox normalization is required to allow extravasation of effector T-cells into the tumor microenvironment [131].
Not only too high oxidative stress, but also too low may prove critically detrimental in cancer: cyclic adenosine monophosphate (cAMP) activators by inducing oxidative stress impaired glioma-derived endothelial cell differentiation in vivo, normalized the tumor vessels, and altered the tumor immune profile, especially increasing the influx and function of CD8+ effector T cells [132].
Lastly, OGG1 is a mediator of pulmonary fibrosis sparked by bleomycin; the underlying mechanism is alveolar epithelial-mesenchymal transition (EMT) and can be inhibited by small-molecule OGG1 inhibitor TH5487 or enhanced by OGG1 overexpression [133,134]. EMT is a process involved in lung fibrosis and cancer metastasis associated with NFκB and SMAD activation [135,136]. It was shown that NFκB activation and EMT were essential for the expression of immunosuppressive molecule PD-L1 by TNFα plus TGFβ1 - stimulated A549 lung adenocarcinoma cells [137]. Specifically, TGFβ1 decreased the expression of DNA methyltransferase DNMT1 and that resulted in PD-L1 promoter demethylation whereas TNFα induced the NF-κB pathway that promoted expression of the demethylated PD-L1 promoter. In a previous study, platelet-derived TGFβ activated the Smad and NF-κB pathways in cancer cells, resulting in EMT and enhanced metastasis in vivo; inhibition of NF-κB signaling in cancer cells or ablation of TGFβ1 expression in platelets protected against lung metastasis in vivo [138,139]. A model for cancer metastasis proposes that it is initiated by EMT that is possibly spurred by TGFβ1 -expressing macrophages, and in the destination niche it is accomplished by a mesenchymal-to-epithelial transition that is facilitated by resident cells such as Ly6C+ myeloid progenitor that have been shown in the metastatic lungs to express extracellular matrix proteoglycan versican [140]. What is important, is that inhibiting OGG1 suppressed the numbers of M2-macrophages [141,134], which is a phenotype shown to promote angiogenesis [142] and immunosuppression [143].
9. Conclusions
These findings suggest that OGG1-enabled NFκB activity can promote cancer progression via a number of key mechanisms that include phenotypic transitions of cancer and stromal cells, expression and secretion of molecules that modulate innate immunity, and alteration of the vascular network. Mounting evidence supports the idea that increased levels of oxidative stress play a crucial role in the onset and progression of solid and lymphoid malignancies. This is achieved by inducing genomic instability, promoting cell survival, signaling for cell proliferation, and increasing resistance to treatment modalities (reviewed in [144]). Multiple mechanisms are described for aberrant ROS generation in malignant cells, particularly of myeloid origin. This could involve dysfunction of the mitochondrial respiratory chain, activation/dysregulation of various oxidoreductases/antioxidant machineries, and/or changes in metabolism, allowing cell survival and proliferation under constant oxidative stress. Under oxidative stress, the supraphysiological levels of non-genotoxic 8-oxoGua, along with redox inactivation of OGG1 via cysteine oxidation and/or dimerization, provide multiple interfaces with redox-activated/modulated NFκB and other transacting factors, including Myc. This promotes not only cell proliferation, the expression of innate immune genes, and angiogenesis but also the metastatic capability of cells involving the expression of epithelial-mesenchymal transition (EMT) genes. Taken together, facilitation by OGG1 of NFκB and Myc binding to their cognate motifs on DNA can alter the course of malignancy and render cancer cells resistant to chemo/radio therapy. Therefore, we speculate that small molecules that inhibit generation of the epigenetic-like mark 8-oxoGua, and/or recognition of its genomic substrate by OGG1 may have clinical utility as this was suggested by pioneering work [117]. A deeper understanding of the interactions between ROS, OGG1, NFκB and Myc could help understand and ultimately treat malignant disease.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gregory, C.D. Hijacking Homeostasis: Regulation of the Tumor Microenvironment by Apoptosis. Immunol. Rev. 2023. [Google Scholar] [CrossRef] [PubMed]
- Tabu, K.; Taga, T. Cancer Ego-System in Glioma: An Iron-Replenishing Niche Network Systemically Self-Organized by Cancer Stem Cells. Inflamm. Regen. 2022, 42, 54. [Google Scholar] [CrossRef] [PubMed]
- Vlahopoulos, S.A.; Cen, O.; Hengen, N.; Agan, J.; Moschovi, M.; Critselis, E.; Adamaki, M.; Bacopoulou, F.; Copland, J.A.; Boldogh, I.; et al. Dynamic Aberrant NF-κB Spurs Tumorigenesis: A New Model Encompassing the Microenvironment. Cytokine Growth Factor Rev. 2015, 26, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Lambrou, G.I.; Hatziagapiou, K.; Vlahopoulos, S. Inflammation and Tissue Homeostasis: The NF-κB System in Physiology and Malignant Progression. Mol. Biol. Rep. 2020. [Google Scholar] [CrossRef] [PubMed]
- Vlahopoulos, S.A. Aberrant Control of NF-κB in Cancer Permits Transcriptional and Phenotypic Plasticity, to Curtail Dependence on Host Tissue: Molecular Mode. Cancer Biol. Med. 2017, 14, 254–270. [Google Scholar] [CrossRef] [PubMed]
- Neganova, M.; Liu, J.; Aleksandrova, Y.; Klochkov, S.; Fan, R. Therapeutic Influence on Important Targets Associated with Chronic Inflammation and Oxidative Stress in Cancer Treatment. Cancers 2021, 13, 6062. [Google Scholar] [CrossRef] [PubMed]
- Beury, D.W.; Carter, K.A.; Nelson, C.; Sinha, P.; Hanson, E.; Nyandjo, M.; Fitzgerald, P.J.; Majeed, A.; Wali, N.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cell Survival and Function Are Regulated by the Transcription Factor Nrf2. J. Immunol. Baltim. Md 1950 2016, 196, 3470–3478. [Google Scholar] [CrossRef]
- Zhao, L.; He, R.; Long, H.; Guo, B.; Jia, Q.; Qin, D.; Liu, S.-Q.; Wang, Z.; Xiang, T.; Zhang, J.; et al. Late-Stage Tumors Induce Anemia and Immunosuppressive Extramedullary Erythroid Progenitor Cells. Nat. Med. 2018, 24, 1536–1544. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Chen, H.; Liang, J.; Li, Y.; Yang, J.; Luo, C.; Tang, Y.; Ding, Y.; Liu, X.; Yuan, Q.; et al. Dual Role of Reactive Oxygen Species and Their Application in Cancer Therapy. J. Cancer 2021, 12, 5543–5561. [Google Scholar] [CrossRef]
- Burrows, C.J.; Muller, J.G. Oxidative Nucleobase Modifications Leading to Strand Scission. Chem. Rev. 1998, 98, 1109–1152. [Google Scholar] [CrossRef]
- Cheng, K.C.; Cahill, D.S.; Kasai, H.; Nishimura, S.; Loeb, L.A. 8-Hydroxyguanine, an Abundant Form of Oxidative DNA Damage, Causes G----T and A----C Substitutions. J. Biol. Chem. 1992, 267, 166–172. [Google Scholar] [CrossRef]
- Hazra, T.K.; Das, A.; Das, S.; Choudhury, S.; Kow, Y.W.; Roy, R. Oxidative DNA Damage Repair in Mammalian Cells: A New Perspective. DNA Repair 2007, 6, 470–480. [Google Scholar] [CrossRef]
- McCullough, A.K.; Dodson, M.L.; Lloyd, R.S. Initiation of Base Excision Repair: Glycosylase Mechanisms and Structures. Annu. Rev. Biochem. 1999, 68, 255–285. [Google Scholar] [CrossRef]
- Mitra, S.; Izumi, T.; Boldogh, I.; Bhakat, K.K.; Hill, J.W.; Hazra, T.K. Choreography of Oxidative Damage Repair in Mammalian Genomes. Free Radic. Biol. Med. 2002, 33, 15–28. [Google Scholar] [CrossRef]
- Bessho, T.; Roy, R.; Yamamoto, K.; Kasai, H.; Nishimura, S.; Tano, K.; Mitra, S. Repair of 8-Hydroxyguanine in DNA by Mammalian N-Methylpurine-DNA Glycosylase. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 8901–8904. [Google Scholar] [CrossRef]
- Izumi, T.; Hazra, T.K.; Boldogh, I.; Tomkinson, A.E.; Park, M.S.; Ikeda, S.; Mitra, S. Requirement for Human AP Endonuclease 1 for Repair of 3’-Blocking Damage at DNA Single-Strand Breaks Induced by Reactive Oxygen Species. Carcinogenesis 2000, 21, 1329–1334. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.J.; Wilson, S.H. DNA Scanning by Base Excision Repair Enzymes and Implications for Pathway Coordination. DNA Repair 2018, 71, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Sampath, H.; Lloyd, R.S. Roles of OGG1 in Transcriptional Regulation and Maintenance of Metabolic Homeostasis. DNA Repair 2019, 81, 102667. [Google Scholar] [CrossRef] [PubMed]
- Lebraud, E.; Pinna, G.; Siberchicot, C.; Depagne, J.; Busso, D.; Fantini, D.; Irbah, L.; Robeska, E.; Kratassiouk, G.; Ravanat, J.-L.; et al. Chromatin Recruitment of OGG1 Requires Cohesin and Mediator and Is Essential for Efficient 8-oxoG Removal. Nucleic Acids Res. 2020, 48, 9082–9097. [Google Scholar] [CrossRef]
- D’Augustin, O.; Huet, S.; Campalans, A.; Radicella, J.P. Lost in the Crowd: How Does Human 8-Oxoguanine DNA Glycosylase 1 (OGG1) Find 8-Oxoguanine in the Genome? Int. J. Mol. Sci. 2020, 21, 8360. [Google Scholar] [CrossRef]
- Dizdaroglu, M.; Coskun, E.; Jaruga, P. Repair of Oxidatively Induced DNA Damage by DNA Glycosylases: Mechanisms of Action, Substrate Specificities and Excision Kinetics. Mutat. Res. Rev. Mutat. Res. 2017, 771, 99–127. [Google Scholar] [CrossRef] [PubMed]
- Grishko, V.; Solomon, M.; Breit, J.F.; Killilea, D.W.; Ledoux, S.P.; Wilson, G.L.; Gillespie, M.N. Hypoxia Promotes Oxidative Base Modifications in the Pulmonary Artery Endothelial Cell VEGF Gene. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2001, 15, 1267–1269. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.M.; Ding, Y.; Burrows, C.J. Oxidative DNA Damage Is Epigenetic by Regulating Gene Transcription via Base Excision Repair. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 2604–2609. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.M.; Burrows, C.J. 8-Oxo-7,8-Dihydroguanine, Friend and Foe: Epigenetic-like Regulator versus Initiator of Mutagenesis. DNA Repair 2017, 56, 75–83. [Google Scholar] [CrossRef]
- Fleming, A.M.; Zhu, J.; Ding, Y.; Burrows, C.J. 8-Oxo-7,8-Dihydroguanine in the Context of a Gene Promoter G-Quadruplex Is an On-Off Switch for Transcription. ACS Chem. Biol. 2017, 12, 2417–2426. [Google Scholar] [CrossRef]
- Ba, X.; Boldogh, I. 8-Oxoguanine DNA Glycosylase 1: Beyond Repair of the Oxidatively Modified Base Lesions. Redox Biol. 2018, 14, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Forneris, F.; Binda, C.; Vanoni, M.A.; Mattevi, A.; Battaglioli, E. Histone Demethylation Catalysed by LSD1 Is a Flavin-Dependent Oxidative Process. FEBS Lett. 2005, 579, 2203–2207. [Google Scholar] [CrossRef]
- Perillo, B.; Ombra, M.N.; Bertoni, A.; Cuozzo, C.; Sacchetti, S.; Sasso, A.; Chiariotti, L.; Malorni, A.; Abbondanza, C.; Avvedimento, E.V. DNA Oxidation as Triggered by H3K9me2 Demethylation Drives Estrogen-Induced Gene Expression. Science 2008, 319, 202–206. [Google Scholar] [CrossRef]
- Amente, S.; Bertoni, A.; Morano, A.; Lania, L.; Avvedimento, E.V.; Majello, B. LSD1-Mediated Demethylation of Histone H3 Lysine 4 Triggers Myc-Induced Transcription. Oncogene 2010, 29, 3691–3702. [Google Scholar] [CrossRef]
- Bazopoulou, D.; Knoefler, D.; Zheng, Y.; Ulrich, K.; Oleson, B.J.; Xie, L.; Kim, M.; Kaufmann, A.; Lee, Y.-T.; Dou, Y.; et al. Developmental ROS Individualizes Organismal Stress Resistance and Lifespan. Nature 2019, 576, 301–305. [Google Scholar] [CrossRef]
- Fleming, A.M.; Burrows, C.J. DNA Modifications Walk a Fine Line between Epigenetics and Mutagenesis. Nat. Rev. Mol. Cell Biol. 2023, 24, 449–450. [Google Scholar] [CrossRef]
- Pan, L.; Zhu, B.; Hao, W.; Zeng, X.; Vlahopoulos, S.A.; Hazra, T.K.; Hegde, M.L.; Radak, Z.; Bacsi, A.; Brasier, A.R.; et al. Oxidized Guanine Base Lesions Function in 8-Oxoguanine DNA Glycosylase-1-Mediated Epigenetic Regulation of Nuclear Factor κB-Driven Gene Expression. J. Biol. Chem. 2016, 291, 25553–25566. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Fleming, A.M.; Burrows, C.J. Sequencing the Mouse Genome for the Oxidatively Modified Base 8-Oxo-7,8-Dihydroguanine by OG-Seq. J. Am. Chem. Soc. 2017, 139, 2569–2572. [Google Scholar] [CrossRef] [PubMed]
- Amente, S.; Di Palo, G.; Scala, G.; Castrignanò, T.; Gorini, F.; Cocozza, S.; Moresano, A.; Pucci, P.; Ma, B.; Stepanov, I.; et al. Genome-Wide Mapping of 8-Oxo-7,8-Dihydro-2’-Deoxyguanosine Reveals Accumulation of Oxidatively-Generated Damage at DNA Replication Origins within Transcribed Long Genes of Mammalian Cells. Nucleic Acids Res. 2019, 47, 221–236. [Google Scholar] [CrossRef]
- Gorini, F.; Scala, G.; Ambrosio, S.; Majello, B.; Amente, S. OxiDIP-Seq for Genome-Wide Mapping of Damaged DNA Containing 8-Oxo-2’-Deoxyguanosine. Bio-Protoc. 2022, 12, e4540. [Google Scholar] [CrossRef] [PubMed]
- Gorini, F.; Scala, G.; Cooke, M.S.; Majello, B.; Amente, S. Towards a Comprehensive View of 8-Oxo-7,8-Dihydro-2’-Deoxyguanosine: Highlighting the Intertwined Roles of DNA Damage and Epigenetics in Genomic Instability. DNA Repair 2021, 97, 103027. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Qi, T.; Pan, L.; Wang, R.; Zhu, B.; Aguilera-Aguirre, L.; Radak, Z.; Hazra, T.K.; Vlahopoulos, S.A.; Bacsi, A.; et al. Effects of the Stimuli-Dependent Enrichment of 8-Oxoguanine DNA Glycosylase1 on Chromatinized DNA. Redox Biol. 2018, 18, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Pezone, A.; Taddei, M.L.; Tramontano, A.; Dolcini, J.; Boffo, F.L.; De Rosa, M.; Parri, M.; Stinziani, S.; Comito, G.; Porcellini, A.; et al. Targeted DNA Oxidation by LSD1-SMAD2/3 Primes TGF-Β1/ EMT Genes for Activation or Repression. Nucleic Acids Res. 2020, 48, 8943–8958. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Hao, W.; Xue, Y.; Wang, K.; Zheng, X.; Luo, J.; Ba, X.; Xiang, Y.; Qin, X.; Bergwik, J.; et al. 8-Oxoguanine Targeted by 8-Oxoguanine DNA Glycosylase 1 (OGG1) Is Central to Fibrogenic Gene Activation upon Lung Injury. Nucleic Acids Res. 2023, 51, 1087–1102. [Google Scholar] [CrossRef]
- Bruner, S.D.; Norman, D.P.; Verdine, G.L. Structural Basis for Recognition and Repair of the Endogenous Mutagen 8-Oxoguanine in DNA. Nature 2000, 403, 859–866. [Google Scholar] [CrossRef]
- Boiteux, S.; Radicella, J.P. The Human OGG1 Gene: Structure, Functions, and Its Implication in the Process of Carcinogenesis. Arch. Biochem. Biophys. 2000, 377, 1–8. [Google Scholar] [CrossRef]
- Hitomi, K.; Iwai, S.; Tainer, J.A. The Intricate Structural Chemistry of Base Excision Repair Machinery: Implications for DNA Damage Recognition, Removal, and Repair. DNA Repair 2007, 6, 410–428. [Google Scholar] [CrossRef]
- Ba, X.; Bacsi, A.; Luo, J.; Aguilera-Aguirre, L.; Zeng, X.; Radak, Z.; Brasier, A.R.; Boldogh, I. 8-Oxoguanine DNA Glycosylase-1 Augments Proinflammatory Gene Expression by Facilitating the Recruitment of Site-Specific Transcription Factors. J. Immunol. Baltim. Md 1950 2014, 192, 2384–2394. [Google Scholar] [CrossRef]
- Xue, Y.; Li, C.; Deng, S.; Chen, X.; Han, J.; Zheng, X.; Tian, M.; Hao, W.; Pan, L.; Boldogh, I.; et al. 8-Oxoguanine DNA Glycosylase 1 Selectively Modulates ROS-Responsive NF-κB Targets through Recruitment of MSK1 and Phosphorylation of RelA/P65 at Ser276. J. Biol. Chem. 2023, 299, 105308. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Vlahopoulos, S.; Tanner, L.; Bergwik, J.; Bacsi, A.; Radak, Z.; Egesten, A.; Ba, X.; Brasier, A.R.; Boldogh, I. Substrate-Specific Binding of 8-Oxoguanine DNA Glycosylase 1 (OGG1) Reprograms Mucosal Adaptations to Chronic Airway Injury. Front. Immunol. 2023, 14, 1186369. [Google Scholar] [CrossRef]
- Bangalore, D.M.; Tessmer, I. Direct hOGG1-Myc Interactions Inhibit hOGG1 Catalytic Activity and Recruit Myc to Its Promoters under Oxidative Stress. Nucleic Acids Res. 2022, 50, 10385–10398. [Google Scholar] [CrossRef] [PubMed]
- Visnes, T.; Cázares-Körner, A.; Hao, W.; Wallner, O.; Masuyer, G.; Loseva, O.; Mortusewicz, O.; Wiita, E.; Sarno, A.; Manoilov, A.; et al. Small-Molecule Inhibitor of OGG1 Suppresses Proinflammatory Gene Expression and Inflammation. Science 2018, 362, 834–839. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wu, S.; Wu, G.; Yang, J. Inhibition of 8-Oxoguanine DNA Glycosylase (OGG1) Expression Suppresses Polycystic Ovarian Syndrome via the NF-κB Signaling Pathway. Reprod. Biol. 2022, 22, 100679. [Google Scholar] [CrossRef]
- Wibisana, J.N.; Inaba, T.; Shinohara, H.; Yumoto, N.; Hayashi, T.; Umeda, M.; Ebisawa, M.; Nikaido, I.; Sako, Y.; Okada, M. Enhanced Transcriptional Heterogeneity Mediated by NF-κB Super-Enhancers. PLoS Genet. 2022, 18, e1010235. [Google Scholar] [CrossRef]
- Xu, J.-W.; Ling, S.; Liu, J. Higher-Order Chromatin Regulation of Inflammatory Gene Expression. Mediators Inflamm. 2017, 2017, 7848591. [Google Scholar] [CrossRef]
- Wong, R.W.J.; Tan, T.K.; Amanda, S.; Ngoc, P.C.T.; Leong, W.Z.; Tan, S.H.; Asamitsu, K.; Hibi, Y.; Ueda, R.; Okamoto, T.; et al. Feed-Forward Regulatory Loop Driven by IRF4 and NF-κB in Adult T-Cell Leukemia/Lymphoma. Blood 2020, 135, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Zhao, M.; Long, B.; Li, H. Super-Enhancer-Associated Gene CAPG Promotes AML Progression. Commun. Biol. 2023, 6, 622. [Google Scholar] [CrossRef] [PubMed]
- Daenthanasanmak, A.; Bamford, R.N.; Yoshioka, M.; Yang, S.-M.; Homan, P.; Karim, B.; Bryant, B.R.; Petrus, M.N.; Thomas, C.J.; Green, P.L.; et al. Triple Combination of BET plus PI3K and NF-κB Inhibitors Exhibit Synergistic Activity in Adult T-Cell Leukemia/Lymphoma. Blood Adv. 2022, 6, 2346–2360. [Google Scholar] [CrossRef] [PubMed]
- Vlahopoulos, S.; Adamaki, M.; Khoury, N.; Zoumpourlis, V.; Boldogh, I. Roles of DNA Repair Enzyme OGG1 in Innate Immunity and Its Significance for Lung Cancer. Pharmacol. Ther. 2018. [Google Scholar] [CrossRef] [PubMed]
- Aguilera-Aguirre, L.; Bacsi, A.; Radak, Z.; Hazra, T.K.; Mitra, S.; Sur, S.; Brasier, A.R.; Ba, X.; Boldogh, I. Innate Inflammation Induced by the 8-Oxoguanine DNA Glycosylase-1-KRAS-NF-κB Pathway. J. Immunol. Baltim. Md 1950 2014, 193, 4643–4653. [Google Scholar] [CrossRef] [PubMed]
- Gerke, M.B.; Christodoulou, I.; Karantanos, T. Definitions, Biology, and Current Therapeutic Landscape of Myelodysplastic/Myeloproliferative Neoplasms. Cancers 2023, 15, 3815. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-H.; Lee, J.; Woo, J.; Lee, C.-H.; Yoo, C.-G. Proteasome Inhibitor-Induced IκB/NF-κB Activation Is Mediated by Nrf2-Dependent Light Chain 3B Induction in Lung Cancer Cells. Mol. Cells 2018, 41, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Y.; Li, L.; Zhang, M.; Song, N.; Zhao, Q.; Liu, Z.; Diao, A. TMEPAI Promotes Degradation of the NF-κB Signaling Pathway Inhibitory Protein IκBα and Contributes to Tumorigenesis. Int. J. Biol. Macromol. 2023, 235, 123859. [Google Scholar] [CrossRef]
- Han, Y.; Weinman, S.; Boldogh, I.; Walker, R.K.; Brasier, A.R. Tumor Necrosis Factor-Alpha-Inducible IkappaBalpha Proteolysis Mediated by Cytosolic m-Calpain. A Mechanism Parallel to the Ubiquitin-Proteasome Pathway for Nuclear Factor-Kappab Activation. J. Biol. Chem. 1999, 274, 787–794. [Google Scholar] [CrossRef]
- Varisli, L.; Cen, O.; Vlahopoulos, S. Dissecting Pharmacological Effects of Chloroquine in Cancer Treatment: Interference with Inflammatory Signaling Pathways. Immunology 2019. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.-L.; Wang, X.; Mann, M.; Adamus, T.P.; Wang, D.; Moreira, D.F.; Zhang, Z.; Ouyang, C.; He, X.; Zhang, B.; et al. Myeloid Cell-Targeted miR-146a Mimic Inhibits NF-κB-Driven Inflammation and Leukemia Progression in Vivo. Blood 2020, 135, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Mehta, A.; de Boer, C.G.; Kowalczyk, M.S.; Lee, K.; Haldeman, P.; Rogel, N.; Knecht, A.R.; Farouq, D.; Regev, A.; et al. Heterogeneous Responses of Hematopoietic Stem Cells to Inflammatory Stimuli Are Altered with Age. Cell Rep. 2018, 25, 2992–3005.e5. [Google Scholar] [CrossRef] [PubMed]
- Jost, P.J.; Höckendorf, U. Necroinflammation Emerges as a Key Regulator of Hematopoiesis in Health and Disease. Cell Death Differ. 2019, 26, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Ghatak, D.; Datta, A.; Roychowdhury, T.; Chattopadhyay, S.; Roychoudhury, S. MicroRNA-324-5p-CUEDC2 Axis Mediates Gain-of-Function Mutant P53-Driven Cancer Stemness. Mol. Cancer Res. MCR 2021, 19, 1635–1650. [Google Scholar] [CrossRef]
- Wang, J.; Liu, X.; Li, P.; Wang, J.; Shu, Y.; Zhong, X.; Gao, Z.; Yang, J.; Jiang, Y.; Zhou, X.; et al. Long Noncoding RNA HOTAIR Regulates the Stemness of Breast Cancer Cells via Activation of the NF-κB Signaling Pathway. J. Biol. Chem. 2022, 298, 102630. [Google Scholar] [CrossRef]
- Dancik, G.M.; Varisli, L.; Vlahopoulos, S.A. The Molecular Context of Oxidant Stress Response in Cancer Establishes ALDH1A1 as a Critical Target: What This Means for Acute Myeloid Leukemia. Int. J. Mol. Sci. 2023, 24, 9372. [Google Scholar] [CrossRef]
- Moschovi, M.; Critselis, E.; Cen, O.; Adamaki, M.; Lambrou, G.I.; Chrousos, G.P.; Vlahopoulos, S. Drugs Acting on Homeostasis: Challenging Cancer Cell Adaptation. Expert Rev. Anticancer Ther. 2015, 15, 1405–1417. [Google Scholar] [CrossRef]
- Vlahopoulos, S.; Boldogh, I.; Casola, A.; Brasier, A.R. Nuclear Factor-kappaB-Dependent Induction of Interleukin-8 Gene Expression by Tumor Necrosis Factor Alpha: Evidence for an Antioxidant Sensitive Activating Pathway Distinct from Nuclear Translocation. Blood 1999, 94, 1878–1889. [Google Scholar] [CrossRef]
- Xue, Y.; Pan, L.; Vlahopoulos, S.; Wang, K.; Zheng, X.; Radak, Z.; Bacsi, A.; Tanner, L.; Brasier, A.R.; Ba, X.; et al. Epigenetic Control of Type III Interferon Expression by 8-Oxoguanine and Its Reader 8-Oxoguanine DNA Glycosylase1. Front. Immunol. 2023, 14, 1161160. [Google Scholar] [CrossRef]
- Wang, R.; Li, C.; Qiao, P.; Xue, Y.; Zheng, X.; Chen, H.; Zeng, X.; Liu, W.; Boldogh, I.; Ba, X. OGG1-Initiated Base Excision Repair Exacerbates Oxidative Stress-Induced Parthanatos. Cell Death Dis. 2018, 9, 628. [Google Scholar] [CrossRef] [PubMed]
- Hung, R.J.; Hall, J.; Brennan, P.; Boffetta, P. Genetic Polymorphisms in the Base Excision Repair Pathway and Cancer Risk: A HuGE Review. Am. J. Epidemiol. 2005, 162, 925–942. [Google Scholar] [CrossRef]
- Bravard, A.; Vacher, M.; Moritz, E.; Vaslin, L.; Hall, J.; Epe, B.; Radicella, J.P. Oxidation Status of Human OGG1-S326C Polymorphic Variant Determines Cellular DNA Repair Capacity. Cancer Res. 2009, 69, 3642–3649. [Google Scholar] [CrossRef]
- Hill, J.W.; Evans, M.K. Dimerization and Opposite Base-Dependent Catalytic Impairment of Polymorphic S326C OGG1 Glycosylase. Nucleic Acids Res. 2006, 34, 1620–1632. [Google Scholar] [CrossRef]
- Morreall, J.; Limpose, K.; Sheppard, C.; Kow, Y.W.; Werner, E.; Doetsch, P.W. Inactivation of a Common OGG1 Variant by TNF-Alpha in Mammalian Cells. DNA Repair 2015, 26, 15–22. [Google Scholar] [CrossRef]
- Peng, Y.; Li, Z.; Zhang, S.; Xiong, Y.; Cun, Y.; Qian, C.; Li, M.; Ren, T.; Xia, L.; Cheng, Y.; et al. Association of DNA Base Excision Repair Genes (OGG1, APE1 and XRCC1) Polymorphisms with Outcome to Platinum-Based Chemotherapy in Advanced Nonsmall-Cell Lung Cancer Patients. Int. J. Cancer 2014, 135, 2687–2696. [Google Scholar] [CrossRef] [PubMed]
- Le Marchand, L.; Donlon, T.; Lum-Jones, A.; Seifried, A.; Wilkens, L.R. Association of the hOGG1 Ser326Cys Polymorphism with Lung Cancer Risk. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 2002, 11, 409–412. [Google Scholar]
- Smolarz, B.; Michalska, M.M.; Samulak, D.; Wójcik, L.; Romanowicz, H. Studies of Correlations Between Single Nucleotide Polymorphisms of DNA Repair Genes and Endometrial Cancer in Polish Women. Anticancer Res. 2018, 38, 5223–5229. [Google Scholar] [CrossRef]
- Kabzinski, J.; Walczak, A.; Dziki, A.; Mik, M.; Majsterek, I. Impact of the Ser326Cys Polymorphism of the OGG1 Gene on the Level of Oxidative DNA Damage in Patients with Colorectal Cancer. Pol. Przegl. Chir. 2018, 90, 13–15. [Google Scholar] [CrossRef]
- Alanazi, M.; Pathan, A.A.K.; Shaik, J.P.; Alhadheq, A.; Khan, Z.; Khan, W.; Al Naeem, A.; Parine, N.R. The hOGG1 Ser326Cys Gene Polymorphism and Breast Cancer Risk in Saudi Population. Pathol. Oncol. Res. POR 2017, 23, 525–535. [Google Scholar] [CrossRef]
- Costa, E.F.D.; Santos, E.S.; Liutti, V.T.; Leal, F.; Santos, V.C.A.; Rinck-Junior, J.A.; Mariano, F.V.; Coutinho-Camillo, C.M.; Altemani, A.; Lima, C.S.P.; et al. Association between Polymorphisms in Genes Related to DNA Base-Excision Repair with Risk and Prognosis of Oropharyngeal Squamous Cell Carcinoma. J. Cancer Res. Clin. Oncol. 2016, 142, 1917–1926. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Elahi, A.; Pow-Sang, J.; Lazarus, P.; Park, J. Association between Polymorphism of Human Oxoguanine Glycosylase 1 and Risk of Prostate Cancer. J. Urol. 2003, 170, 2471–2474. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zheng, S.L.; Turner, A.; Isaacs, S.D.; Wiley, K.E.; Hawkins, G.A.; Chang, B.; Bleecker, E.R.; Walsh, P.C.; Meyers, D.A.; et al. Associations between hOGG1 Sequence Variants and Prostate Cancer Susceptibility. Cancer Res. 2002, 62, 2253–2257. [Google Scholar] [PubMed]
- Gotoh, N.; Saitoh, T.; Takahashi, N.; Kasamatsu, T.; Minato, Y.; Lobna, A.; Oda, T.; Hoshino, T.; Sakura, T.; Shimizu, H.; et al. Association between OGG1 S326C CC Genotype and Elevated Relapse Risk in Acute Myeloid Leukemia. Int. J. Hematol. 2018, 108, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Fazio, V.M.; Barrera, G.; Martinotti, S.; Farace, M.G.; Giglioni, B.; Frati, L.; Manzari, V.; Dianzani, M.U. 4-Hydroxynonenal, a Product of Cellular Lipid Peroxidation, Which Modulates c-Myc and Globin Gene Expression in K562 Erythroleukemic Cells. Cancer Res. 1992, 52, 4866–4871. [Google Scholar] [PubMed]
- Wang, W.; Ma, Y.; Huang, M.; Liang, W.; Zhao, X.; Li, Q.; Wang, S.; Hu, Z.; He, L.; Gao, T.; et al. Asymmetrical Arginine Dimethylation of Histone H4 by 8-Oxog/OGG1/PRMT1 Is Essential for Oxidative Stress-Induced Transcription Activation. Free Radic. Biol. Med. 2021, 164, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.-Y.; Wang, Y.; Cui, S.-Y.; Wu, X.-L.; Guo, Y.; Xu, R.-R. MicroRNA-125a Regulates Proliferation and Apoptosis of Acute Myeloid Leukemia through Targeting NF-κB Pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 3594–3601. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.-H.; Zhang, W.-J.; Zhu, J.-F.; Cui, D.; Song, K.-D.; Qiang, P.; Mei, C.-Z.; Nie, Z.-C.; Ding, B.-S.; Han, Z.; et al. CaMKIIγ Regulates the Viability and Self-Renewal of Acute Myeloid Leukaemia Stem-like Cells by the Alox5/NF-κB Pathway. Int. J. Lab. Hematol. 2021, 43, 699–706. [Google Scholar] [CrossRef]
- Chen, X.-J.; Zhang, W.-N.; Chen, B.; Xi, W.-D.; Lu, Y.; Huang, J.-Y.; Wang, Y.-Y.; Long, J.; Wu, S.-F.; Zhang, Y.-X.; et al. Homoharringtonine Deregulates MYC Transcriptional Expression by Directly Binding NF-κB Repressing Factor. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 2220–2225. [Google Scholar] [CrossRef]
- Reikvam, H. Inhibition of NF-κB Signaling Alters Acute Myelogenous Leukemia Cell Transcriptomics. Cells 2020, 9, 1677. [Google Scholar] [CrossRef] [PubMed]
- Di Francesco, B.; Verzella, D.; Capece, D.; Vecchiotti, D.; Di Vito Nolfi, M.; Flati, I.; Cornice, J.; Di Padova, M.; Angelucci, A.; Alesse, E.; et al. NF-κB: A Druggable Target in Acute Myeloid Leukemia. Cancers 2022, 14, 3557. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, Q.; Zou, Z.; Huang, Z.; Chen, Y. TRIM10 Is Downregulated in Acute Myeloid Leukemia and Plays a Tumor Suppressive Role via Regulating NF-κB Pathway. Cancers 2023, 15, 417. [Google Scholar] [CrossRef] [PubMed]
- Dancik, G.M.; Varisli, L.; Tolan, V.; Vlahopoulos, S. Aldehyde Dehydrogenase Genes as Prospective Actionable Targets in Acute Myeloid Leukemia. Genes 2023, 14, 1807. [Google Scholar] [CrossRef] [PubMed]
- Gasparetto, M.; Pei, S.; Minhajuddin, M.; Khan, N.; Pollyea, D.A.; Myers, J.R.; Ashton, J.M.; Becker, M.W.; Vasiliou, V.; Humphries, K.R.; et al. Targeted Therapy for a Subset of Acute Myeloid Leukemias That Lack Expression of Aldehyde Dehydrogenase 1A1. Haematologica 2017, 102, 1054–1065. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yao, R.; Wang, H. Update of ALDH as a Potential Biomarker and Therapeutic Target for AML. BioMed Res. Int. 2018, 2018, 9192104. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Gasparetto, M.; Humphries, K.; Pollyea, D.A.; Vasiliou, V.; Jordan, C.T. Aldehyde Dehydrogenases in Acute Myeloid Leukemia. Ann. N. Y. Acad. Sci. 2014, 1310, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Gasparetto, M.; Smith, C.A. ALDHs in Normal and Malignant Hematopoietic Cells: Potential New Avenues for Treatment of AML and Other Blood Cancers. Chem. Biol. Interact. 2017, 276, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Dancik, G.M.; Voutsas, I.F.; Vlahopoulos, S. Aldehyde Dehydrogenase Enzyme Functions in Acute Leukemia Stem Cells. Front. Biosci. Sch. Ed. 2022, 14, 8. [Google Scholar] [CrossRef]
- Dancik, G.M.; Voutsas, I.F.; Vlahopoulos, S. Lower RNA Expression of ALDH1A1 Distinguishes the Favorable Risk Group in Acute Myeloid Leukemia. Mol. Biol. Rep. 2022, 49, 3321–3331. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, Y.; Toubai, T.; Oravecz-Wilson, K.; Liu, C.; Mathewson, N.; Wu, J.; Rossi, C.; Cummings, E.; Wu, D.; et al. BET Bromodomain Inhibition Suppresses Graft-versus-Host Disease after Allogeneic Bone Marrow Transplantation in Mice. Blood 2015, 125, 2724–2728. [Google Scholar] [CrossRef] [PubMed]
- Latif, A.-L.; Newcombe, A.; Li, S.; Gilroy, K.; Robertson, N.A.; Lei, X.; Stewart, H.J.S.; Cole, J.; Terradas, M.T.; Rishi, L.; et al. BRD4-Mediated Repression of P53 Is a Target for Combination Therapy in AML. Nat. Commun. 2021, 12, 241. [Google Scholar] [CrossRef] [PubMed]
- Snyder, K.J.; Choe, H.K.; Gao, Y.; Sell, N.E.; Braunreiter, K.M.; Zitzer, N.C.; Neidemire-Colley, L.; Kalyan, S.; Dorrance, A.M.; Keller, A.; et al. Inhibition of Bromodomain and Extra Terminal (BET) Domain Activity Modulates the IL-23R/IL-17 Axis and Suppresses Acute Graft-Versus-Host Disease. Front. Oncol. 2021, 11, 760789. [Google Scholar] [CrossRef] [PubMed]
- Fong, C.Y.; Gilan, O.; Lam, E.Y.N.; Rubin, A.F.; Ftouni, S.; Tyler, D.; Stanley, K.; Sinha, D.; Yeh, P.; Morison, J.; et al. BET Inhibitor Resistance Emerges from Leukaemia Stem Cells. Nature 2015, 525, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Rathert, P.; Roth, M.; Neumann, T.; Muerdter, F.; Roe, J.-S.; Muhar, M.; Deswal, S.; Cerny-Reiterer, S.; Peter, B.; Jude, J.; et al. Transcriptional Plasticity Promotes Primary and Acquired Resistance to BET Inhibition. Nature 2015, 525, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Moreb, J.; Schweder, M.; Suresh, A.; Zucali, J.R. Overexpression of the Human Aldehyde Dehydrogenase Class I Results in Increased Resistance to 4-Hydroperoxycyclophosphamide. Cancer Gene Ther. 1996, 3, 24–30. [Google Scholar] [PubMed]
- Pan, G.; Deshpande, M.; Pang, H.; Stemmer, P.M.; Carruthers, N.J.; Shearn, C.T.; Backos, D.S.; Palaniyandi, S.S. 4-Hydroxy-2-Nonenal Attenuates 8-Oxoguanine DNA Glycosylase 1 Activity. J. Cell. Biochem. 2020. [Google Scholar] [CrossRef]
- Costa, R.G.A.; Silva, S.L.R.; Dias, I.R.S.B.; Oliveira, M. de S.; Rodrigues, A.C.B. da C.; Dias, R.B.; Bezerra, D.P. Emerging Drugs Targeting Cellular Redox Homeostasis to Eliminate Acute Myeloid Leukemia Stem Cells. Redox Biol. 2023, 62, 102692. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; He, N.; Gu, D.; Wickliffe, J.; Salazar, J.; Boldogh, I.; Xie, J. Genetic Evidence for XPC-KRAS Interactions During Lung Cancer Development. J. Genet. Genomics Yi Chuan Xue Bao 2015, 42, 589–596. [Google Scholar] [CrossRef]
- Cogoi, S.; Ferino, A.; Miglietta, G.; Pedersen, E.B.; Xodo, L.E. The Regulatory G4 Motif of the Kirsten Ras (KRAS) Gene Is Sensitive to Guanine Oxidation: Implications on Transcription. Nucleic Acids Res. 2018, 46, 661–676. [Google Scholar] [CrossRef]
- Mali, V.R.; Ning, R.; Chen, J.; Yang, X.-P.; Xu, J.; Palaniyandi, S.S. Impairment of Aldehyde Dehydrogenase-2 by 4-Hydroxy-2-Nonenal Adduct Formation and Cardiomyocyte Hypertrophy in Mice Fed a High-Fat Diet and Injected with Low-Dose Streptozotocin. Exp. Biol. Med. Maywood NJ 2014, 239, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Kreuz, S.; Fischle, W. Oxidative Stress Signaling to Chromatin in Health and Disease. Epigenomics 2016, 8, 843–862. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.S.; Singh, S.P.; Singhal, P.; Horne, D.; Singhal, J.; Awasthi, S. Antioxidant Role of Glutathione S-Transferases: 4-Hydroxynonenal, a Key Molecule in Stress-Mediated Signaling. Toxicol. Appl. Pharmacol. 2015, 289, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Milkovic, L.; Zarkovic, N.; Marusic, Z.; Zarkovic, K.; Jaganjac, M. The 4-Hydroxynonenal-Protein Adducts and Their Biological Relevance: Are Some Proteins Preferred Targets? Antioxid. Basel Switz. 2023, 12, 856. [Google Scholar] [CrossRef] [PubMed]
- Folkerts, H.; Hilgendorf, S.; Wierenga, A.T.J.; Jaques, J.; Mulder, A.B.; Coffer, P.J.; Schuringa, J.J.; Vellenga, E. Inhibition of Autophagy as a Treatment Strategy for P53 Wild-Type Acute Myeloid Leukemia. Cell Death Dis. 2017, 8, e2927. [Google Scholar] [CrossRef] [PubMed]
- Liddiard, K.; Hills, R.; Burnett, A.K.; Darley, R.L.; Tonks, A. OGG1 Is a Novel Prognostic Indicator in Acute Myeloid Leukaemia. Oncogene 2010, 29, 2005–2012. [Google Scholar] [CrossRef] [PubMed]
- Owen, N.; Minko, I.G.; Moellmer, S.A.; Cammann, S.K.; Lloyd, R.S.; McCullough, A.K. Enhanced Cytarabine-Induced Killing in OGG1-Deficient Acute Myeloid Leukemia Cells. Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2016833118. [Google Scholar] [CrossRef] [PubMed]
- Radpour, R.; Stucki, M.; Riether, C.; Ochsenbein, A.F. Epigenetic Silencing of Immune-Checkpoint Receptors in Bone Marrow- Infiltrating T Cells in Acute Myeloid Leukemia. Front. Oncol. 2021, 11, 663406. [Google Scholar] [CrossRef] [PubMed]
- Paczulla, A.M.; Rothfelder, K.; Raffel, S.; Konantz, M.; Steinbacher, J.; Wang, H.; Tandler, C.; Mbarga, M.; Schaefer, T.; Falcone, M.; et al. Absence of NKG2D Ligands Defines Leukaemia Stem Cells and Mediates Their Immune Evasion. Nature 2019, 572, 254–259. [Google Scholar] [CrossRef]
- Hassane, D.C.; Guzman, M.L.; Corbett, C.; Li, X.; Abboud, R.; Young, F.; Liesveld, J.L.; Carroll, M.; Jordan, C.T. Discovery of Agents That Eradicate Leukemia Stem Cells Using an in Silico Screen of Public Gene Expression Data. Blood 2008, 111, 5654–5662. [Google Scholar] [CrossRef]
- Karantanos, T.; Jones, R.J. Acute Myeloid Leukemia Stem Cell Heterogeneity and Its Clinical Relevance. Adv. Exp. Med. Biol. 2019, 1139, 153–169. [Google Scholar] [CrossRef]
- Vlahopoulos, S.; Wang, K.; Xue, Y.; Zheng, X.; Boldogh, I.; Pan, L. Endothelial Dysfunction through Oxidatively Generated Epigenetic Mark in Respiratory Viral Infections. Cells 2021, 10, 3067. [Google Scholar] [CrossRef] [PubMed]
- Terwoord, J.D.; Beyer, A.M.; Gutterman, D.D. Endothelial Dysfunction as a Complication of Anti-Cancer Therapy. Pharmacol. Ther. 2022, 237, 108116. [Google Scholar] [CrossRef]
- Feng, Y.; Luo, S.; Fan, D.; Guo, X.; Ma, S. The Role of Vascular Endothelial Cells in Tumor Metastasis. Acta Histochem. 2023, 125, 152070. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, X.; Tang, K.; Xin, Y.; Hu, G.; Zheng, Y.; Li, K.; Zhang, C.; Tan, Y. Adhesion to the Brain Endothelium Selects Breast Cancer Cells with Brain Metastasis Potential. Int. J. Mol. Sci. 2023, 24, 7087. [Google Scholar] [CrossRef] [PubMed]
- Podyacheva, E.; Danilchuk, M.; Toropova, Y. Molecular Mechanisms of Endothelial Remodeling under Doxorubicin Treatment. Biomed. Pharmacother. Biomedecine Pharmacother. 2023, 162, 114576. [Google Scholar] [CrossRef]
- Wang, S.; Kotamraju, S.; Konorev, E.; Kalivendi, S.; Joseph, J.; Kalyanaraman, B. Activation of Nuclear Factor-kappaB during Doxorubicin-Induced Apoptosis in Endothelial Cells and Myocytes Is pro-Apoptotic: The Role of Hydrogen Peroxide. Biochem. J. 2002, 367, 729–740. [Google Scholar] [CrossRef]
- Xu, A.; Deng, F.; Chen, Y.; Kong, Y.; Pan, L.; Liao, Q.; Rao, Z.; Xie, L.; Yao, C.; Li, S.; et al. NF-κB Pathway Activation during Endothelial-to-Mesenchymal Transition in a Rat Model of Doxorubicin-Induced Cardiotoxicity. Biomed. Pharmacother. Biomedecine Pharmacother. 2020, 130, 110525. [Google Scholar] [CrossRef] [PubMed]
- Mi, J.; Zhang, X.; Rabbani, Z.N.; Liu, Y.; Reddy, S.K.; Su, Z.; Salahuddin, F.K.; Viles, K.; Giangrande, P.H.; Dewhirst, M.W.; et al. RNA Aptamer-Targeted Inhibition of NF-Kappa B Suppresses Non-Small Cell Lung Cancer Resistance to Doxorubicin. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 66–73. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, C.; Zuo, N.; Yang, H.; Fang, S.; Shi, J. Extracellular Traps Increase Burden of Bleeding by Damaging Endothelial Cell in Acute Promyelocytic Leukaemia. Front. Immunol. 2022, 13, 841445. [Google Scholar] [CrossRef]
- Martínez-Rey, D.; Carmona-Rodríguez, L.; Fernández-Aceñero, M.J.; Mira, E.; Mañes, S. Extracellular Superoxide Dismutase, the Endothelial Basement Membrane, and the WNT Pathway: New Players in Vascular Normalization and Tumor Infiltration by T-Cells. Front. Immunol. 2020, 11, 579552. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Huang, Y.; Li, Z.; Pan, G.; Zheng, L.; Xiao, X.; Wang, F.; Chen, J.; Chen, X.; Lin, X.; et al. Glioblastoma Vascular Plasticity Limits Effector T-Cell Infiltration and Is Blocked by cAMP Activation. Cancer Immunol. Res. 2023, 11, 1351–1366. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Song, C.; Fang, Y.; Yin, Y.; Wu, Z.; Wang, Y.; Xu, Z.; Gao, S.; Li, A.; Liu, G. TH5487, a Small Molecule Inhibitor of OGG1, Attenuates Pulmonary Fibrosis by NEDD4L-Mediated OGG1 Degradation. Chem. Biol. Interact. 2022, 362, 109999. [Google Scholar] [CrossRef] [PubMed]
- Tanner, L.; Single, A.B.; Bhongir, R.K.V.; Heusel, M.; Mohanty, T.; Karlsson, C. a. Q.; Pan, L.; Clausson, C.-M.; Bergwik, J.; Wang, K.; et al. Small-Molecule-Mediated OGG1 Inhibition Attenuates Pulmonary Inflammation and Lung Fibrosis in a Murine Lung Fibrosis Model. Nat. Commun. 2023, 14, 643. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-Y.; Jeon, S.; Yoo, Y.J.; Jin, H.; Won, H.Y.; Yoon, K.; Hwang, E.S.; Lee, Y.-J.; Na, Y.; Cho, J.; et al. The Hsp27-Mediated IkBα-NFκB Signaling Axis Promotes Radiation-Induced Lung Fibrosis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 5364–5375. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, P.; Dey, P.; Ghosh, S.; Sarma, A.; Ghosh, U. Reduction of Metastatic Potential by Inhibiting EGFR/Akt/P38/ERK Signaling Pathway and Epithelial-Mesenchymal Transition after Carbon Ion Exposure Is Potentiated by PARP-1 Inhibition in Non-Small-Cell Lung Cancer. BMC Cancer 2019, 19, 829. [Google Scholar] [CrossRef] [PubMed]
- Asgarova, A.; Asgarov, K.; Godet, Y.; Peixoto, P.; Nadaradjane, A.; Boyer-Guittaut, M.; Galaine, J.; Guenat, D.; Mougey, V.; Perrard, J.; et al. PD-L1 Expression Is Regulated by Both DNA Methylation and NF-kB during EMT Signaling in Non-Small Cell Lung Carcinoma. Oncoimmunology 2018, 7, e1423170. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct Signaling between Platelets and Cancer Cells Induces an Epithelial-Mesenchymal-like Transition and Promotes Metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef]
- Gao, D.; Vahdat, L.T.; Wong, S.; Chang, J.C.; Mittal, V. Microenvironmental Regulation of Epithelial-Mesenchymal Transitions in Cancer. Cancer Res. 2012, 72, 4883–4889. [Google Scholar] [CrossRef]
- Gao, D.; Joshi, N.; Choi, H.; Ryu, S.; Hahn, M.; Catena, R.; Sadik, H.; Argani, P.; Wagner, P.; Vahdat, L.T.; et al. Myeloid Progenitor Cells in the Premetastatic Lung Promote Metastases by Inducing Mesenchymal to Epithelial Transition. Cancer Res. 2012, 72, 1384–1394. [Google Scholar] [CrossRef]
- Tanner, L.; Bergwik, J.; Bhongir, R.K.V.; Pan, L.; Dong, C.; Wallner, O.; Kalderén, C.; Helleday, T.; Boldogh, I.; Adner, M.; et al. Pharmacological OGG1 Inhibition Decreases Murine Allergic Airway Inflammation. Front. Pharmacol. 2022, 13, 999180. [Google Scholar] [CrossRef] [PubMed]
- Jetten, N.; Verbruggen, S.; Gijbels, M.J.; Post, M.J.; De Winther, M.P.J.; Donners, M.M.P.C. Anti-Inflammatory M2, but Not pro-Inflammatory M1 Macrophages Promote Angiogenesis in Vivo. Angiogenesis 2014, 17, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Mia, S.; Warnecke, A.; Zhang, X.-M.; Malmström, V.; Harris, R.A. An Optimized Protocol for Human M2 Macrophages Using M-CSF and IL-4/IL-10/TGF-β Yields a Dominant Immunosuppressive Phenotype. Scand. J. Immunol. 2014, 79, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Trombetti, S.; Cesaro, E.; Catapano, R.; Sessa, R.; Lo Bianco, A.; Izzo, P.; Grosso, M. Oxidative Stress and ROS-Mediated Signaling in Leukemia: Novel Promising Perspectives to Eradicate Chemoresistant Cells in Myeloid Leukemia. Int. J. Mol. Sci. 2021, 22, 2470. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



