
Submitted:
07 November 2023
Posted:
08 November 2023
You are already at the latest version
Abstract
Background: Hereditary Hemorrhagic Telangiectasia (HHT) is a vascular autosomically inherited rare disease. Epistaxis (nose bleeds) is the most common symptom in HHT, leading to anemia and affecting patient’s quality of life. In addition to epistaxis, gastrointestinal bleeding (GI), more often at older ages, may lead to severe anemia and need for blood transfussions. Thus, finding drugs to control both types of bleeding is a primary necessity in HHT. Methods: A cross-sectional observational study was conducted in a series of 11 HHT patients treated with low tacrolimus doses (0.5-1mg) on an off-label prescription basis. Patients showed refractory bleeding to previous treatments. Epistaxis severity score (ESS) and hemoglobin levels were parameters used to evaluate tacrolimus impact. The occurrence of side effects was also recorded. Results: Tacrolimus was well tolerated in most of the patients, except in 2 which abandoned the treatment. The remaining patients tolerated the treatment, with a general improvement in their health condition. Epistaxis was significantly reduced comparing the ESS before and after treatment. Hemoglobin levels were significantly increased overcoming the anemia in the course of the treatment. Conclusion: Tacrolimus at low doses should be considered as a promising treatment for bleeding derived of epistaxis and GI in HHT.
Keywords:
HHT
; bleeding
; epistaxis
; GI bleeding
; tacrolimus
; ESS
; Hemoglobin
1. Introduction
Hereditary haemorrhagic telangiectasia (HHT) or Rendu-Osler-Weber syndrome is an autosomal dominant inherited vascular disease.
In 2000, the Curaçao criteria for clinical diagnosis of HHT were agreed upon. These criteria include multisystem symptoms such as spontaneous and recurrent epistaxis (nosebleeds), mucocutaneous telangiectasias, involvement of the visceral vasculature as gastrointestinal telangiectasis, arteriovenous malformations (AVMs), mainly in the lung, brain or liver, and a first-degree relative with a definite diagnosis of HHT [1,2,3]. The prevalence of HHT is considered to be 1:5,000 on average, although due to founder and isolation effects, the prevalence is higher in some regions such as the Jura region of France, the island of Funen in Denmark and the Dutch Caribbean Netherlands Antilles [3,4,5]. Heterozygous mutations in the Endoglin (ENG) or ACVRL1/ALK1 genes are responsible for pathogenesis in about 85% of patients with HHT [6,7].
Less common mutations responsible for HHT are detected in the MADH4/SMAD4 gene, leading to a combined syndrome, Juvenile Polyposis and HHT (JPHT) [8] where in addition to HHT symptoms, colon polyps and thoracic aneurysms appear [9]. In addition, chromosomes 5 and 7 have been described to possess two loci with unknown genes, which cause HHT3 [10] and HHT4, respectively [11]. An HHT-like syndrome called HHT5 is associated with mutations in BMP9/GDF2 [12]. Importantly, all proteins encoded by these genes belong to the BMP9/TGF-β signalling pathway.
In addition, another HHT-related pathology is capillary malformation (CM)/AVM syndrome, phenotypically similar to HHT, characterised by the appearance of multiple randomly distributed CMs. These are small and red, round/oval in shape with a peripheral white halo, in addition to internal arterio-venous malformations. This disease is associated with heterozygous pathogenic variants in EPHB2, 4 or RASA1 identified by molecular genetic testing [13].
Nosebleeds are the most frequent clinical symptom in HHT. Up to 93% of patients suffer from mild to moderate bleeding [14,15], which interferes with their quality of life [16]. Epistaxis is due to telangiectasis of the nasal mucosa [17]. In addition to epistaxis, gastrointestinal (GI) bleeding as consequence of telangiectasias in the digestive tract is observed in up to 80% of patients with HHT especially in older age [18]. GI bleeding represents a major clinical problem, which may lead to dependence on blood transfusions [19].
Typically, pharmaceutical therapies or minor surgical procedures treat clinical symptoms. However, it should be noted that telangiectasias and AVMs may be mosaics in which some or many of the endothelial cells have undergone a second hit and have become homozygous mutants for the pathogenic gene, as has recently been shown [20].
There is currently no optimal treatment for gastrointestinal bleeding, however some of the systemic treatments for epistaxis may also be useful for gastrointestinal bleeding. This paper will present a small case series using tacrolimus as a pharmacological treatment for very severe bleeding in patients with HHT, including epistaxis and gastrointestinal bleeding.
The background for the interest in Tacrolimus or FK506, historically comes from a case report of a patient with HHT undergoing liver transplantation. To prevent rejection, the immunosuppressant FK506 was administered in combination with Aspirin and sirolimus to prevent rejection. One month after starting this treatment, the telangiectasias (both internal mucosa and external skin), epistaxis and anaemia were cured [21].
This clinical case was the starting point for an in vitro study by Albiñana et al [22,23] in an attempt to elucidate the molecular mechanism underlying this beneficial clinical effect on HHT. Endothelial cells were treated with tacrolimus alone, resulting in increased protein and mRNA expression of endoglin and ALK1. At the same time, stimulation of the BMP9/TGF-β1/ALK1 signalling pathway as measured by nuclear translocation of Smad4 and increased expression of downstream genes such as Id1, were obtained. These molecular findings were accompanied by enhanced endothelial cell functions such as tubulogenesis and wound healing [22,23] (Figure 1).
These results would explain the improvement in the aforementioned patient, through a partial compensation of endoglin and ALK1 insufficiency, since the amount of both proteins and the signalling of the BMP9/TGF-β1/ALK1 pathway was stimulated.
Supporting this view, five years later, Ruiz et al. reported that tacrolimus rescued gene expression dysregulations associated with ALK1 inhibition by increasing the ALK1 signalling pathway in endothelial cells derived from HHT patients. Furthermore, in an animal model of HHT immunotreated with antibodies against BMP9/10, tacrolimus improved vascular pathology by inhibiting VEGF signalling and thereby decreasing retinal hypervascularisation [24].
The same group more recently reported that in the same neonatal mouse model of HHT (BMP9/BMP10 immunosupressed mouse) the mTOR inhibitor sirolimus and the receptor tyrosine kinase inhibitor nintedanib could synergistically completely block, and also reverse, retinal AVMs. Sirolimus plus nintedanib prevented vascular pathology in the oral mucosa, lungs and liver of BMP9/10ib mice, and significantly reduced gastrointestinal bleeding and anaemia in adult mice with inducible ALK1 deficiency [25].
In a more clinical context, Sommer et al. published in 2019 that low-dose FK506/Advagraf decreased bleeding in a patient with HHT who also had pulmonary arterial hypertension [26]. This case report points to low-dose tacrolimus (0.5-1.5 mg/day) as the optimal range for patients with refractory nasal or GI bleeding, rather than high doses (5-10 mg/day) normally used for immunosuppression in transplantation [26]. An additional report by Hosman et al. involving two HHT transfusion-dependent patients due to severe bleeding demonstrated improvement after treatment with low-dose tacrolimus [27].
In addition, results regarding the efficacy and safety of tacrolimus 0.1% topically applied as nasal ointment from the TACRO clinical trial were published in 2020 [28]. Tacrolimus nasal ointment did not improve 6 weeks after the end of treatment, but the good tolerability and significant improvement in the duration of epistaxis during treatment prompted the investigators to conduct a phase 3 trial in a larger patient population and with a longer treatment time, with the main outcome being a reduction in the duration of epistaxis during treatment.
The present study shows the results of 11 patients treated off-label with low doses of systemic tacrolimus, prescribed by HHT referral physicians in Spain. In all cases, patients suffered from severe epistaxis and/or gastrointestinal bleeding refractory to other treatments. The results are not part of a clinical trial, but a collection of cases that may be useful to illustrate the efficacy and safety of systemic tacrolimus treatment.
2. Materials and Methods
2.1. Patients and Study Design
This work represents a cross-sectional study performed from 2018. It includes HHT patients treated at the Hospital Universitario Fundación Alcorcón (HUFA) and patients treated at the HHT reference unit of Hospital Universitario Ramón y Cajal. All patients were older than 18 years, had a confirmed diagnosis of HHT (clinical and/or genetic), and were followed by the HUFA Otorhinolaryngology Unit and Ramón y Cajal Hospital, Internal Medicine Unit. Treatment with low doses of tacrolimus was recommended in all cases to patients with epistaxis or GI bleeding, refractory to other previous treatment. The patients were treated on an off-label a compassionate basis, they signed informed consent and the treatment for each patient received approval from the Pharmaceutical Committee and the Clinical Research Ethics Committee from HUFA and Ramón y Cajal Hospitals, respectively.
2.2. Outcomes and Assessments
The primary outcome was the impact on frequency and severity of epistaxis, as measured by the HHT epistaxis severity score (HHT-ESS). HHT-ESS is an objective, standardized and internationally validated reference tool to estimate the severity of epistaxis in HHT patients [29]. HHT-ESS is calculated from six parameters related to epistaxis: frequency, duration, and intensity of nosebleed, presence/absence of anaemia, need for blood transfusion, and need to seek medical care to treat the haemorrhage. In addition, hemoglobin levels were measured at the beginning and in the course of the follow up. Other effects as the measure of hemoglobin levels, collateral side effects and drop-out rate were evaluated.
3. Results
Table 1 shows the characteristics of 11 patients treated with oral tacrolimus, as an off label treatment. It is a sample with a median age of 55.8 [38.0] years, 5 women. All patients presented epistaxis, as other manifestations and complications were described gastro-intestinal (GI) bleeding, pulmonary arteriovenous malformations (AVMs) and anaemia. Patients were suffering from epistaxis, GI bleeding, or both, and the hemorrhages had been refractory to previous treatments, listed also in the same table. It is important to mention that the treatment with tacrolimus was combined with on demand sclerotherapy in several cases. However, sclerotherapy became less frequent in those cases where tacrolimus was used. At least, 4 out of 5 patients, treated on demand by sclerotherapy, reduced the need for this procedure, more than 1 year, since the beginning of tacrolimus treatment.
Patients were prescribed with a low dose of tacrolimus, ranging from 0.5-2 mg/day, depending on the tolerance and the severity of the hemorrhages. In no case patients were treated with doses leading to immunosuppression. Tacrolimus shows reduced epistaxis when used for immunosuppression in HHT patients subjected to organ transplants [21].
In 2 cases patients left the treatment for gastric intolerance, but for this side effect, the absence of side effects and general tolerance was quite good.
All patients were following the Curaçao clinical criteria, 3 patients did not have a genetic diagnosis and in 1, mutation was not found in ENG nor in ACVRL1/ALK1. Four patients were diagnosed as HHT1 and three as HHT2. In the case of the 27 years old patient, the problem was unsual bleeding from oral cavity and throat. This case is an exception, due to the bleeding origin. Therefore, in this case, no ESS is shown, and the patient did not suffer from anemia.
The Epistaxis Severity Score (ESS) was moderate in 2 cases (around 4), however the reason to be prescribed with tacrolimus was anemia due mainly to GI bleeding. In the remaining 8 patients ESSs were severe. In all cases, ESSs decreased. Altogether, the ESS decrease was highly significative from a mean of 7.3 before tacrolimus treatment, to 3 after the treatment. This means a transition from severe to moderate/light epistaxis.
In all cases, where tacrolimus was well tolerated, the hemoglobin increased significantly. Before tacrolimus, the mean of hemoglobin was 7.9 g/dL. After tacrolimus, the mean hemoglobin value increased to 12 g/dL, very close to normality. Of note the case of P11, where the improvement was astonishing. It was a special case since the patient refused blood transfusions for religious reasons. Before treatments, the ESS was the highest possible, and the hemoglobin was extremely low. The sclerotherapy first, and tacrolimus combined with sclerotherapy, reversed the extremely severe situation of this patient.
The evolution of ESS and Hemoglobin after initiation of tacrolimus are illustrated in Figure 2.
4. Discussion
This study presents clinical results of 11 HHT patients treated with low dose of oral tacrolimus with no immunosuppressive activity [22]
The main objective of this work is to propose tacrolimus as a drug with potential benefit in HHT patients with severe and refractory bleeding (epistaxis or GI), who in some cases are transfusion dependent.
A significant decrease in the severity of epistaxis was found as evidenced by the ESS in the majority of the patients treated with tacrolimus. On the second hand, these patients also showed an increase in hemoglobin values after initiation of tacrolimus treatment. As this treatment acts on a systemic basis, the improvement of anaemia could be related to decrease in both nasal and GI tract bleeding.
Epistaxis is the most commonly experienced symptom of HHT and significantly impacts patients' quality of life. Thus, to find promising drugs to control nose bleeds alone, or in combination with other current treatments is extremely important [30].
To date, there are few pharmaceutical strategies to prevent this complication. Most clinicians recommend the use of local treatments as moisturizing ointments, oral tranexamic acid, pazopanib, bevacizumab, even more recently, as off label treatment propranolol, although for very severe bleeding, and severe anemia the management is difficult and depends on the clinician expertise and surgical procedures [31].
The benefits of tacrolimus have been noted in isolated cases of liver-transplanted HHT patients. However, most of these patients receive immunosuppressive doses of tacrolimus indicated to avoid transplant rejection [22].
Currently, one active clinical trial for this drug (NCT04646356) is in progress, with the Unit Health of Toronto as sponsor. This study investigates the effectiveness of oral low-dose tacrolimus for the treatment of recurrent nasal hemorrhage in HHT subjects. The primary outcome for the trials will be the reduction of epistaxis severity (minutes of bleeding per week). The trial started in 2020 and the estimated study completion date is September 2024.
Recently Dupuis Girod et al, [28] published a phase II study with tacrolimus as a nasal ointment showing encouraging results that could lead to a phase III study with a large population.
More recently, Hessels et al. [32] conducted a clinical trial based on 20 patients with HHT who received tacrolimus between 1-2 mg daily for 20 weeks. The primary end point was improvement of hemoglobin values. At the end of the study, they found encouraging results; however, 64% of the patients presented minor adverse effects such as headache, diarrhea, abdominal pain and insomnia. The patients in this study were closely followed and treated with the standard care during the study. The study participants exhibited quite severe anemia, and advanced age, potentially hindering efficacy of tacrolimus. Probably patients starting with tacrolimus at earlier ages, and without such a low hemoglobin baseline values (on average 6.1 g/dL, range 5.2-6.9 g/dL) could lead to improved outcomes as it is shown in the present study. It would be worthwhile to explore potential beneficial effects of administering tacrolimus at varying doses and durations based on patient based characteristics (personalized treatments) in a future.
On the present study, we have noticed that lower doses of tacrolimus (0.5 to 2 mg per day) can provide clinical benefits without causing immunosuppression. Of note, the adverse effects of this drug are also related to higher doses. As reported in the current study, most of the patients tolerated appropriately the medication with few significant side effects. It would is therefore important to deepen our knowledge of this drug for HHT in relation to its pharmacodynamics, pharmacokinetics, and potential benefits versus adverse effects.
We can highlight that tacrolimus is a drug with potential use in HHT and that the clinical and molecular results are encouraging. Therefore, it would be interesting to perform a clinical trial with more participants to analyze the clinical benefit, quality of life and cost-benefit of tacrolimus in this type of patients.
Author Contributions
S.M., L-M.B., V.A., L.L.-H., L.R.-P. and J.L.P., A.V.-N., V.G.O. Conceptualisation, S.M., J.L.P; methodology, S.M., J.L.P., A.V.-N., V.G.O; software, V.A.; A.V.-N.; validation, S.M., J.L.P., A.V.-N., V.G.O; formal analysis, S.M., L-M.B., V.A., L.L.-H., L.R.-P. and J.L.P,. A.V.-N., V.G.O investigation, L-M.B., V.A., L.L.-H., L.R.-P.; resources, L-M.B.; data curation, L-M.B., V.A.; A.V.-N.; writing—original draft preparation, L.M.B.; writing—review and editing, S.M., J.L.P., A.V.-N., V.G.O., L.M.B. and V.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding
Institutional Review Board Statement
Data collected from patients and informed consent form were evaluated and approved by the Research Ethics Committee of the Hospital Universitario Ramón y Cajal. The study complies with the recommendations of good clinical practice (GCP) and the Declaration of Helsinki, as well as the current Spanish Legislation as well as the current Spanish legislation on biomedical research. All data necessary for the study are treated in an encrypted form and based on the security measures established in compliance with the General Data Protection Regulation Regulation (EU) 2016/679 of the European Parliament, of the Council of 27 April 2016 on Data Protection (RGPD) and the Organic Law 3/2018, of December 5, on the Protection of Personal Data and guarantee of digital rights. The drug was administered after acceptance by the Pharmacy and Therapeutics Committee of the Ramón y Cajal University Hospital as a drug for use in special situations.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study, as well as from the patients to publish this paper.
Data Availability Statement
Reported results can be found in the files Hospital Universitario Fundación Alcorcón (HUFA) and Hospital Universitario Ramón y Cajal, Madrid, Spain.
Acknowledgements
We thank all patients for their help and Spanish HHT Association.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Shovlin, C.L.; Guttmacher, A.E.; Buscarini, E.; Faughnan, M.E.; Hyland, R.H.; Westermann, C.J.J.; Kjeldsen, A.D.; Plauchu, H. Diagnostic criteria for Hereditary Hemorrhagic Telangiectasia (RenduOsler-Weber Syndrome). Am J Med Genet 2000, 91, 66–67. [Google Scholar] [CrossRef]
- Shovlin, C.L. Hereditary haemorrhagic telangiectasia: Pathophysiology, diagnosis and treatment. Blood Rev 2010, 24, 203–219. [Google Scholar] [CrossRef]
- Shovlin, C.L. Pulmonary arteriovenous malformations. Am J Respir Crit Care Med 2014, 190, 1217–1228. [Google Scholar] [CrossRef]
- Kjeldsen, A.D.; Vase, P.; Green, A. Hereditary haemorrhagic telangiectasia: A population-based study of prevalence and mortality in Danish patients. J Intern Med 1999, 245, 31–39. [Google Scholar] [CrossRef]
- Jessurun, G.A.J.; Kamphuis, D.J.; Van der Zande, F.H.R.; Nossent, J.C. Cerebral arteriovenous malformations in the Netherlands Antilles. High prevalence of hereditary hemorrhagic telangiectasia-related single and multiple cerebral arteriovenous malformations. Clin Neurol Neurosurg 1993, 95, 193–198. [Google Scholar] [CrossRef]
- McAllister, K.A.; Grogg, K.M.; Johnson, D.W.; Gallione, C.J.; Baldwin, M.A.; Jackson, C.E.; Helmbold, E.A.; Markel, D.S.; McKinnon, W.C.; Murrel, J.; et al. Endoglin, a TGF- β binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet 1994, 8, 345–351. [Google Scholar] [CrossRef]
- Johnson, D.W.; Berg, J.N.; Baldwin, M.A.; Gallione, C.J.; Marondel, I.; Yoon, S.J.; Stenzel, T.T.; Speer, M.; Pericak-Vance,M.A.; Diamond, A.; et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type. Nat Genet 1996, 13, 189–195. [CrossRef]
- Gallione, C.J.; Repetto, G.M.; Legius, E.; Rustgi, A.K.; Schelley, S.L.; Tejpar, S.; Mitchell, G.; Drouin, É.; Westermann, C.J.J.; Marchuk, D.A. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet 2004, 363, 852–859. [Google Scholar] [CrossRef]
- Duan, X.Y.; Guo, D.C.; Regalado, E.S.; Shen, H.; Coselli, J.S.; Estrera, A.L.; Safi, H.J.; Bamshad, M.J.; Nickerson, D.A.; LeMaire, S.A.; et al. SMAD4 rare variants in individuals and families with thoracic aortic aneurysms and dissections. Eur J Hum Genet 2019, 27, 1054–1060. [Google Scholar] [CrossRef]
- Cole, S.G.; Begbie, M.E.; Wallace, G.M.F.; Shovlin, C.L.L. A new locus for hereditary haemorrhagic telangiectasia (HHT3) maps to chromosome 5. J Med Genet 2005, 42, 577–582. [Google Scholar] [CrossRef]
- Bayrak-Toydemir, P.; McDonald, J.; Akarsu, N.; Toydemir, R.M.; Calderon, F.; Tuncali, T.; Tang, W.; Miller, F.; Mao, R. A fourth locus for hereditary hemorrhagic telangiectasia maps to chromosome 7. Am. J Med Genet 2006, 140, 2155–2162. [Google Scholar] [CrossRef]
- Wooderchak-Donahue, W.L.; McDonald, J.; O’Fallon, B.; Upton, P.D.; Li, W.; Roman, B.L.; Young, S.; Plant, P.; Fülöp, G.T.; Langa, C.; et al. BMP9 mutations cause a vascular-anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia. Am J Hum Genet 2013, 93, 530–537. [Google Scholar] [CrossRef]
- Bayrak-Toydemir, P.; Stevenson, D. Capillary Malformation-Arteriovenous Malformation Syndrome. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Morales-Angulo, C.; Del Valle-Zapico, A. Hereditary hemorrhagic telangiectasia. Otolaryngol. Head Neck Surg 1998, 119, 293. [Google Scholar] [CrossRef]
- Assar, O.S.; Friedman, C.M.; White, R.I. The Natural History of Epistaxis in Hereditary Hemorrhagic Telangiectasia. Laryngoscope 1991, 101, 977–980. [Google Scholar] [CrossRef]
- Geisthoff, U.W.; Schneider, G.; Fischinger, J.; Plinkert, P.K. Hereditäre hämorrhagische teleangiektasie (Morbus Osler). Eine interdisziplinäre herausforderung. HNO 2002, 50, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Guttmacher, A.E.; Marchuk, D.A.; White, R.I. Hereditary hemorrhagic telangiectasia. N Engl J Med 1995, 333, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Ingrosso, M.; Sabb à, C.; Pisani, A.; Principi, M.; Gallitelli, M.; Cirulli, A.; Francavilla, A. Evidence of small-bowel involvement in hereditary hemorrhagic telangiectasia: A capsule-endoscopic study. Endoscopy 2004, 36, 1074–1079. [Google Scholar] [CrossRef] [PubMed]
- Vase, P.; Grove, O. Gastrointestinal Lesions in Hereditary Hemorrhagic Telangiectasia. Gastroenterology 1986, 91, 1079–1083. [Google Scholar] [CrossRef]
- Snellings, D.A.; Gallione, C.J.; Clark, D.S.; Vozoris, N.T.; Faughnan, M.E.; Marchuk, D.A. Somatic Mutations in Vascular Malformations of Hereditary Hemorrhagic Telangiectasia Result in Bi-allelic Loss of ENG or ACVRL1. Am J Hum Genet 2019, 105, 894–906. [Google Scholar] [CrossRef]
- Skaro, A.I.; Marotta, P.J. Regression of cutaneous and gastrointestinal telangiectasia with sirolimus and aspirin in a patient with hereditary hemorrhagic telangiectasia. Ann Intern Med 2006, 144, 226–227. [Google Scholar] [CrossRef]
- Albiñana, V.; Sanz-Rodríguez, F.; Recio-Poveda, L.; Bernabéu, C.; Botella, L.M. Immunosuppressor FK506 increases endoglin and activin receptor-like kinase 1 expression and modulates transforming growth factor- β1 signaling in endothelial cells. Mol Pharmacol 2011, 79, 833–843. [Google Scholar] [CrossRef]
- Albiñana, V.; Velasco, L.; Zarrabeitia, R.; Botella, L.M. Tacrolimus as a therapeutic drug in hereditary hemorrhagic telangiectasia (HHT). In Tacrolimus: Effectiveness, Safety and Drug Interactions; Nova Science Publishers: Hauppauge, NY, USA, 2013; pp. 163–172. ISBN 9781628083668. [Google Scholar]
- Ruiz, S.; Chandakkar, P.; Zhao, H.; Papoin, J.; Chatterjee, P.K.; Christen, E.; Metz, C.N.; Blanc, L.; Campagne, F.; Marambaud, P. Tacrolimus rescues the signaling and gene expression signature of endothelial ALK1 loss-of-function and improves HHT vascular pathology. Hum Mol Genet 2017, 26, 4786–4798. [Google Scholar] [CrossRef] [PubMed]
- Ruiz S, Zhao H, Chandakkar P, Papoin J, Choi H, Nomura-Kitabayashi A, Patel R, Gillen M, Diao L, Chatterjee PK, He M, Al-Abed Y, Wang P, Metz CN, Oh SP, Blanc L, Campagne F, Marambaud P. Correcting Smad1/5/8, mTOR, and VEGFR2 treats pathology in hereditary hemorrhagic telangiectasia models. J Clin Invest 2020; 130, 942-957. [CrossRef]
- Sommer, N.; Droege, F.; Gamen, K.E.; Geisthoff, U.; Gall, H.; Tello, K.; Richter, M.J.; Deubner, L.M.; Schmiedel, R.; Hecker, M.; et al. Treatment with low-dose tacrolimus inhibits bleeding complications in a patient with hereditary hemorrhagic telangiectasia and pulmonary arterial hypertension. Pulm Circ 2019, 9, 2045894018805406. [Google Scholar] [CrossRef] [PubMed]
- Hosman, A.; Kroon, S.; Vorselaars, V.; Doef, H.; Post, M.; Snijder, R.; Mager, J. Tacrolimus treatment for two rare complications caused by hereditary haemorrhagic telangiectasia: A description of two cases. Angiogenesis 2019, 22, 629. [Google Scholar]
- Dupuis-Girod S, Fargeton AE, Grobost V, Rivière S, Beaudoin M, Decullier E, Bernard L, Bréant V, Colombet B, Philouze P, Bailly S, Faure F, Hermann R. Efficacy and Safety of a 0.1% Tacrolimus Nasal Ointment as a Treatment for Epistaxis in Hereditary Hemorrhagic Telangiectasia: A Double-Blind, Randomized, Placebo-Controlled, Multicenter Trial. J Clin Med 2020, 9, 1262. [Google Scholar] [CrossRef]
- Hoag, J.B. , Terry P., Mitchell S., Reh D., Merlo C.A. An epistaxis severity score for hereditary hemorrhagic telangiectasia. Laryngoscope 2010, 120, 838–843. [Google Scholar] [CrossRef]
- Peterson AM, Kallogjeri D, Spitznagel E, Chakinala MM, Schneider JS, Piccirillo JF. Development and Validation of the Nasal Outcome Score for Epistaxis in Hereditary Hemorrhagic Telangiectasia (NOSE HHT). JAMA Otolaryngol Head Neck Surg 2020, 146, 999–1005. [Google Scholar] [CrossRef]
- Albiñana V, Cuesta AM, Rojas-P I, Gallardo-Vara E, Recio-Poveda L, Bernabéu C, Botella LM. Review of Pharmacological Strategies with Repurposed Drugs for Hereditary Hemorrhagic Telangiectasia Related Bleeding. J Clin Med 2020, 9, 1766. [Google Scholar] [CrossRef]
- Hessels, J.; Kroon, S.; Boerman, S.; Nelissen, R.C.; Grutters, J.C.; Snijder, R.J.; Lebrin, F.; Post, M.C.; Mummery, C.L.; Mager, J.-J. Efficacy and Safety of Tacrolimus as Treatment for Bleeding Caused by Hereditary Hemorrhagic Telangiectasia: An Open-Label, Pilot Study. J Clin Med 2022, 11, 5280. [Google Scholar] [CrossRef]
Figure 1.
Hypotetic signaling pathway triggered by FK506. If cell does not receive TGF-β/BMP9 angiogenic signals, the TRβ-I is inhibited by FKBP12, and the pathway is not initiated. In the presence of TGF-β/BMP9, the pathway will be activated with consequent phosphorylation of Smad and expression of the target genes. After treatment with the immunosuppressant FK506, it is observed the same activation of the pathway, as it sequesters the inhibitor of the TRβ-I, FKBP12, which can phosphorylate Smad.
Figure 1.
Hypotetic signaling pathway triggered by FK506. If cell does not receive TGF-β/BMP9 angiogenic signals, the TRβ-I is inhibited by FKBP12, and the pathway is not initiated. In the presence of TGF-β/BMP9, the pathway will be activated with consequent phosphorylation of Smad and expression of the target genes. After treatment with the immunosuppressant FK506, it is observed the same activation of the pathway, as it sequesters the inhibitor of the TRβ-I, FKBP12, which can phosphorylate Smad.
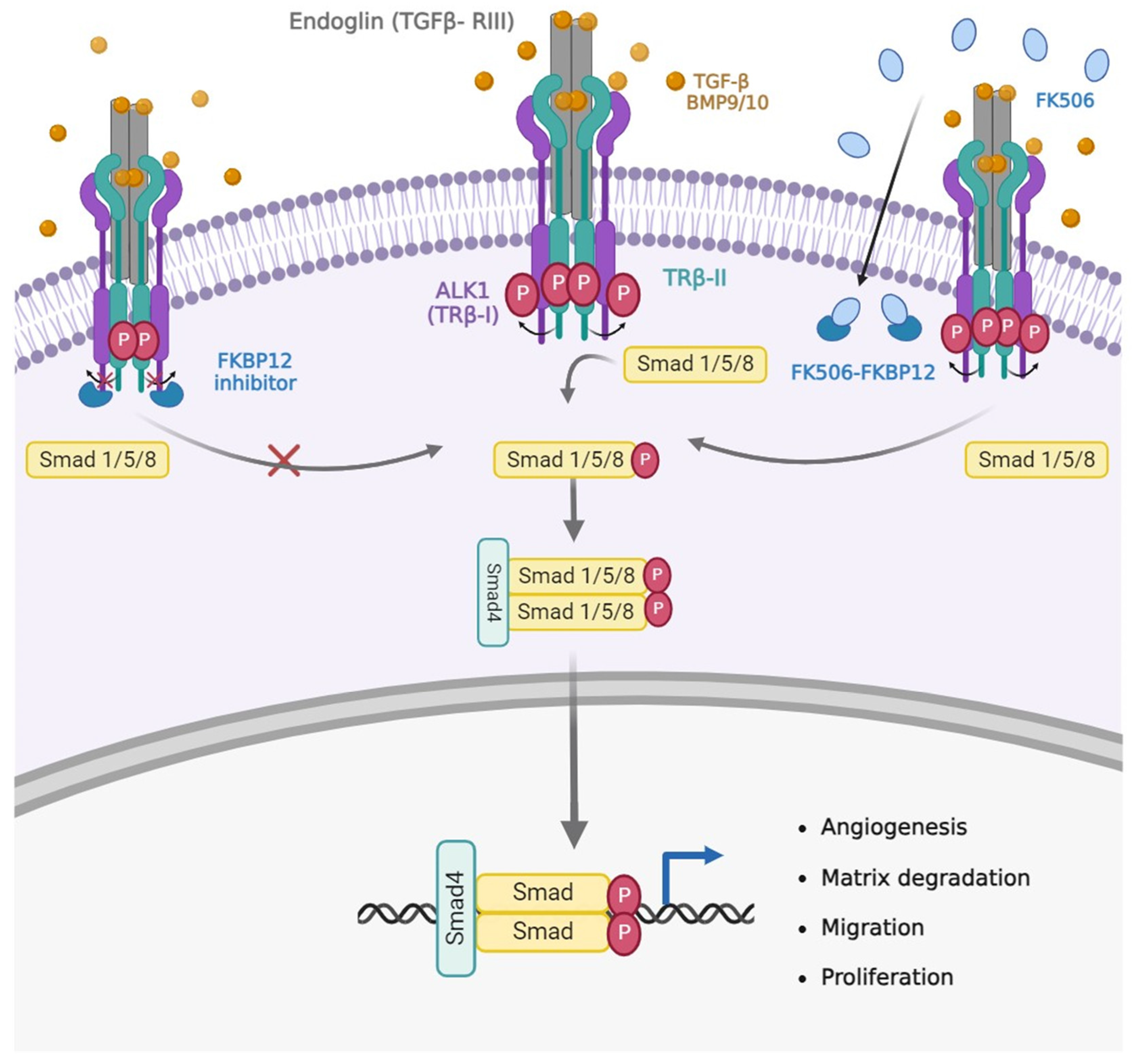
Figure 2.
(A) Evolution of the Epistaxis severity score in all 11 patients before and after tracrolimus treatment. (B) Hemoglobin levels of each patient before and after tacrolimus treatment. (C) Mean values of ESS and Hemoglobin levels. * p < 0.05, *** p < 0.001.
Figure 2.
(A) Evolution of the Epistaxis severity score in all 11 patients before and after tracrolimus treatment. (B) Hemoglobin levels of each patient before and after tacrolimus treatment. (C) Mean values of ESS and Hemoglobin levels. * p < 0.05, *** p < 0.001.
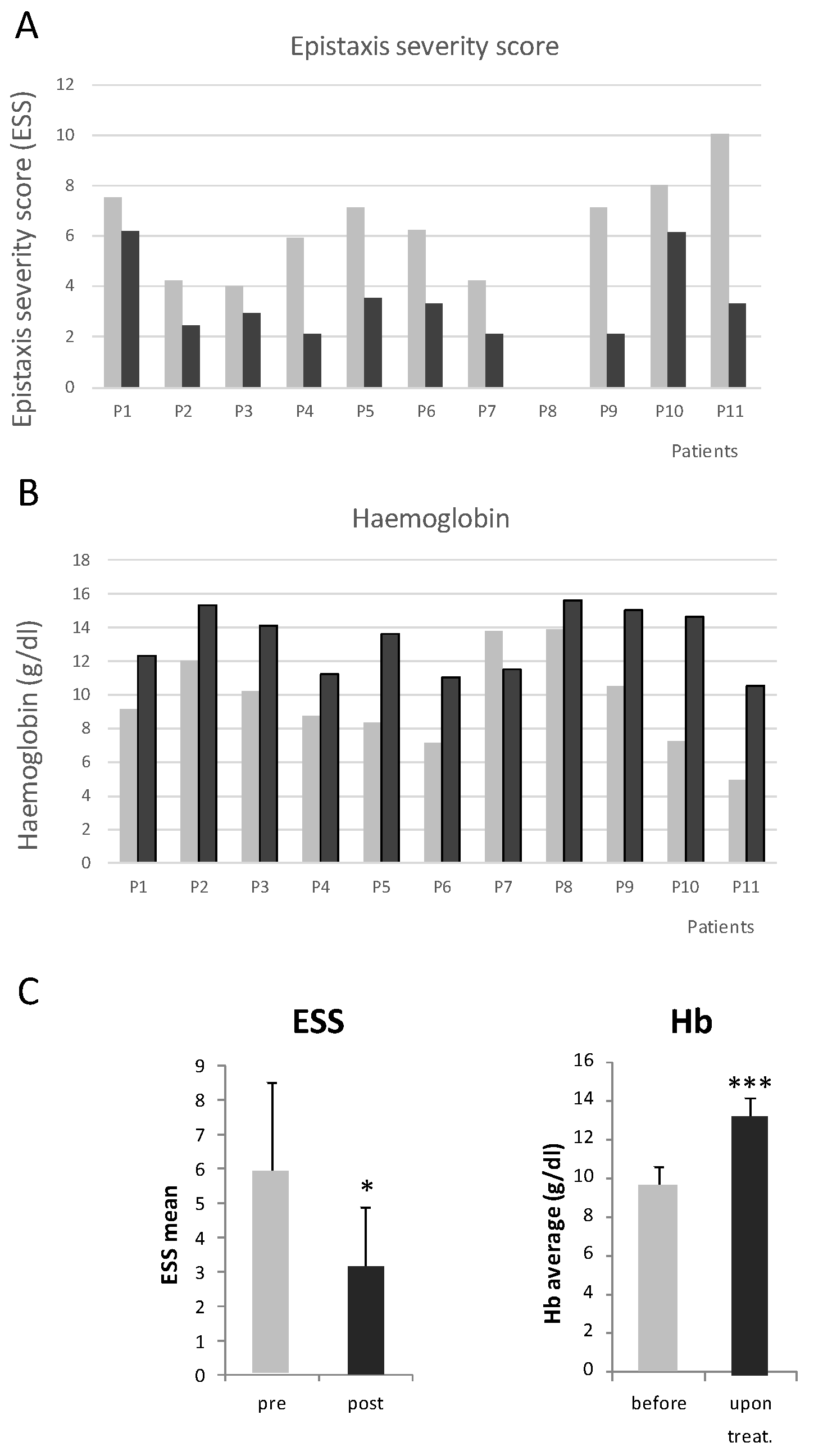

Table 1.
Clinical and laboratory characteristics of hereditary hemorrhagic telangiectasia patients under tacrolimus treatment.
Table 1.
Clinical and laboratory characteristics of hereditary hemorrhagic telangiectasia patients under tacrolimus treatment.
| Patient | Dose | Start | End | Tolerance | Sex | Age | Genetics | ESS pre | ESS post | Hb Before (g/dL) | Hb upon treatment (g/dL) | Epistaxis, telangiectasis | Internal Arteriovenous Malformations | Previous Procedures | Previous Drugs | ||
| P1 | 1mg/24h | 2019 | - | Good | F | 61 | Unknown | 7.5 | 6.15 | 9.1 | 12.3 | Yes | - | HAVMs | CAVM | Embolization right and left external carotid artery branches | Propranolol, raloxifen. IV iron |
| P2 | 1mg/24h | 2019 | 2022 | Good | M | 61 | ACVRL1/ALK-1 c.1208T>C p.L403P Missense | 4.2 | 2.42 | 12 | 15.3 | Yes | GI AVMs | HAVMs | - | - | Propranolol, bevacizumab IV iron |
| P3 | 1mg/24h | 2019 | - | Good | F | 64 | ENG c360+1G>A | 4 | 2.9 | 10.2 | 14.1 | Yes | GI AVMs | HAVM (arterio-portal), (Portal Hypertension) | PAVMs. CAVMs. Anemia | Nasal sclerosisPulmonar embolization | Amchafibrin. Octeotride and oral tamoxifene |
| P4 | 0.5mg/24h | 2019 | - | Good | F | 54 | unknown | 5.9 | 2.1 | 8.7 | 11.2 | Yes | - | HAVMs | Anemia | Nasal sclerosis | Propranolol, amchafibrin. Oral iron |
| P5 | 0.5mg/24h | 2019 | 2021 | Digestive Intolerant | M | 55 | unknown | 7.1 | 3.5 | 8.3 | 13.6 | Yes | - | HAVMs | PAVMs | - | IV iron |
| P6 | 1mg/24h | 2018 | - | Good | M | 64 | Not found in ENG. ALK1/ACVRL1 | 6.2 | 3.3 | 7.1 | 11 | Yes | - | - | GI AVMsAnemia | Laser for GI AVMs. | IV iron |
| P7 | 1mg/12h | 2018 | 2020 | Digestive Intolerant | M | 65 | ENG c.663C>G p.W221X nonsense | 4.2 | 2.1 | 13.8 | 11.5 | Yes | - | - | - | Inner Maxilar Artery embolization. Young procedure. Sphenopalatine artery embolization.Nasal sclerosis | Anchafibrin Oral iron, blood transfusions, 5 Bevacizumab cycles in 2017 |
| P8 | 1.5mg/24h | 2018 | - | Good | M | 27 | ACVRL1/ALK1. Intron 9 c.1377+45T>C & c.1377+65A>G | 0 | 0 | 13.9 | 15.6 | Yes | Buccal AVMs | - | - | Nasal Sclerosis | Propranolol |
| P9 | 0.5mg/12h | 2018 | Good | F | 51 | ENG/HHT1 | 7.1 | 2.1 | 10.5 | 15 | Yes | PAVMs | - | Anemia | Nasal Sclerosis | Amchafibrin blood Transfusions | |
| P10 | 0.5mg/12h | 2021 | - | Good | F | 47 | ACVRL1/ALK1 ex 8 c.1129 G>A p.A377T | 8 | 6.1 | 7.2 | 14.6 | Yes | - | - | Anemia | Nasal Sclerosis | Oral iron |
| P11 | 1mg /12h | 2018 | - | Good | M | 65 | ENG ex 5 c.646 A>G p.K216Q | 10 | 3.3 | 4.9 | 10.5 | Yes | GI AVM | - | Anemia | Nasal Sclerosis | - |
* ESS: Epistaxis Severity Score; Hb: Hemoglobin; AVM: Arteriovenous malformations; HAVM: Hepatic arteriovenous malformation; PAVM: Pulmonary arteriovenous malformation; F: female; M: male; GI: Gastrointestinal; IV: Intravenous.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



