Submitted:
04 November 2023
Posted:
07 November 2023
You are already at the latest version
Abstract
Inflammatory bowel diseases (IBD), including Crohn's disease and ulcerative colitis, is a disease of chronic inflammatory conditions of the intestinal tract due to disturbance of the inflammation and immune system. Symptoms of IBD include abdominal pain, diarrhea, bleeding, reduced weight, and fatigue. In IBD, the immune system attacks the intestinal tract's inner wall, causing chronic inflammation and tissue damage. In particular, IL-6 and IL-17 act on immune cells, including T cells and macrophages, to amplify the immune responses so that tissue damage and morphological changes occur. Of note, excessive calorie intake and obesity also affect the immune system due to inflammation caused by lipotoxicity and changes in lipids supply. Similarly, individuals with IBD have alterations in liver function after sustained high-fat diet feeding. In addition, excess dietary fat intake, along with alterations in primary and secondary bile acids in the colon, can affect the onset and progression of IBD because inflammatory cytokines contribute to insulin resistance; the factors include the release of inflammatory cytokines, oxidative stress, and changes in intestinal microflora, which may also contribute to the disease progression. However, interfering with de novo fatty acid synthase by deleting the enzyme acetyl-CoA-carboxylase 1 in intestinal epithelial cells leads to the deficiency of epithelial crypt structures and tissue regeneration, which seems to be due to Lgr5+ intestinal stem cell function. Thus, conflicting reports exist regarding high-fat diet effects on IBD animal models. This review will focus on the pathological basis of the link between dietary lipids intake and IBD and cover currently available pharmacological approaches.
Keywords:
IBD
; IECs
; Lipid intake
; Stem cells
; Inflammation
1. Background Information
Inflammatory bowel disease (IBD) is the immune-associated inflammation of the gastrointestinal tract. IBD includes ulcerative colitis (UC) and Crohn’s disease (CD) with persistently increased co-morbidities [1]. The exact pathological mechanism for the progression of IBD is still a debate and includes both environmental and genetic factors [1,2]. Studies have shown that polymorphic loci regulate cytokines, chemokines signaling, and antibacterial peptides, further modulating autophagic and immune cell activity by elevating the risk of ileal or colonic CD and UC [2,3]. Clinically, IBD characteristics vary in age groups and on a gender basis; i.e., >60% of female patients exhibit CD with more rectal bleeding, whereas 62% of males have UC with comparatively less rectal bleeding and abdominal pain [4]. More than 80% of the reported CD cases affect the distal part of the small intestine. The prevalence data estimates that ~1 million people in the USA suffer from CD [5]. However, inflammatory lesions were observed in the distal colon of patients suffering from UC [6].
1.1. Liver to the gut pathway
Gut-associated lymphoid tissue (GALT) plays a role in strengthening the mucosal immune system by acting as both protective and tolerant tissues against any pathogenic response. The liver also abundantly contains innate immune cells and acts as a primary immunological organ because of its continuous exposure to the circulating antigens and endotoxins from the gut microbiota. The portal vein takes gut-derived materials to the liver and feedback of bile from the liver to the intestine, suggestive of a reciprocal association between the microbiomes and the liver. In addition, bile acids affect the gut microbiota and interact with nuclear receptors in hepatocytes and intestinal epithelial cells to modulate metabolic activities [7]. The liver abundantly contains innate immune cells and acts as a primary immunological organ because of its continuous exposure to the circulating antigens and endotoxins from the gut microbiota. The liver-to-gut disorders are correlated with gut and liver immune system abnormalities [8] because the liver is linked to the GALT and synergizes its immune reconnaissance [8,9]. Thus, hepatic-gut disorders comprise abnormalities of the gut and liver immune systems [8].
1.2. Association of metabolic disorder and IBD
Any pathological disruption due to chronic caloric intake results in metabolic syndrome (i.e., diabetes, obesity, and fatty liver disease) [10] (Table 1). Since the liver regulates lipid metabolism by fatty acid oxidation and lipogenesis and maintains the human body’s energy under normal physiological conditions, the metabolic disorder is often accompanied by non-alcoholic fatty liver disease (NAFLD), accounting for ~25% of all cases worldwide [11]. NAFLD comprises clinico-pathological abnormalities and leads to steatosis with or without mild inflammation (nonalcoholic fatty liver) and a neuroinflammatory variant (nonalcoholic steatohepatitis), distinguished by the presence of hepatocellular damage such as hepatocyte ballooning [12]. The pathophysiology of liver symptoms includes lipotoxicity, autophagy dysregulation, endoplasmic reticulum stress, and IR [13]. Thus, NAFLD, known as MAFLD, may cause systemic metabolic dysfunction [14].
The complications of NAFLD are confined to not only T2D but also IBD [13,22]. For the first time, Thomas documented the link between colon ulceration and fatty liver early in 1873 [23]. However, the frequency of NAFLD in IBD patients varies greatly, ranging from 1.5% to 40%, depending on the diagnostic criteria [24,25]. The recent era of NAFLD progression belongs to the intake of chronic caloric intake, while based on hospitalization diagnosis, IBD patients with high body mass index because of high-fat diet (HFD) consumption exhibit deleterious symptoms [26]. The effect of liver disease on IBD progression is still unclear because a few dilemmas still support the theory that HFD intake damages the liver and exacerbates IBD, while others believe that HFD deleteriously affects the liver only and protects the intestine from its progression to IBD [27]. This review aims to summarize the molecular basis as to how dietary lipids intake acts as a dual sword in the case of IBD.
2. IBD Pathophysiology
2.1. Intestinal permeability and barrier
Human health depends on the structural veracity of epithelial and endothelial barriers in the body. The intestine accompanies the largest internal barrier and takes part in body protection against the harmful chemicals and bacteria found in the gut. The barrier comprises the mucus layer, commensal bacteria, epithelial cells, and immune cells in the lamina propria [28]. Intestinal epithelial goblet cells conceal mucus glycoproteins and inhibit the microorganism and colonocytes’ direct contact with the gut, whereas mucus released in the small intestine allows bacteria to move freely [29]. Paneth cells are responsible for releasing anti-microbial proteins, which neutralize the effects of bacterial cells in the small intestine, whereas B cells secrete IgA in the lamina propria, which binds to bacteria, and its related toxins hinder their entry into the body [30].
Recent scientific advancements have significantly improved our understanding of IEC functions and their subtypes, such as enterocytes and goblet cells [31]. However, intestinal enteroendocrine cells comprise various subgroups, including enterochromaffin cells, D cells, and G cells [31,32]. Gunnar C. Hansson and his colleagues discovered a new subtype of goblet cells and named it sentinel goblet cells found at the apex of colonic crypts [33]. Unlike traditional goblet cells, the cells have a distinct ability to sense bacteria and respond by secreting mucin, which results in the adjacent environment becoming red in response to noxious stimuli [33]. Tuft cells exhibit cellular differentiation and exist into two subtypes: one type of epithelial cell expresses cytokine Tslp, whereas the other one articulates the immunological marker CD45 [34].
Germ-free animal studies reveal that microbes play a role in fostering appropriate intestinal development and function. Germ-free mice have thin mucosa, resulting in diminished IEC proliferation and compromised fabrication of mucins and other IEC-producing mediators [35]. Because of the loss of the mucin layer, germ-free mice result in the candid disclosure of colitogenic toxins, emphasizing the gut microbiome’s function in intestinal tissue protection and healing [36]. The reduction of microbiota and their substitution by pathogens, known as dysbiosis, may have the capability to alter the gut barrier. Intestinal nutrients and water absorption occur through the transcellular and paracellular pathways and junctions. Intestinal pore channels are charged and size-selective, so pore size varies; the lowest limit is 8 Å diameter, whereas the largest diameter is ~100 Å, a non-selective leaky pathway [37,38].
2.2. Inflammatory mediators
Although several variables are engaged in the pathophysiology of IBD, a disruption in the epithelial barrier is found primarily. The initial injury causes inflammation, causing additional damage and a vicious spiral. Tumor necrosis factor α (TNF-α) is a prototype proinflammatory cytokine released by activated macrophages, monocytes, and T lymphocytes. Study results on CD patients found enhanced TNF-α proteins and mRNA levels in mucosal biopsies [39].
TNF is majorly found via actuated macrophages and T lymphocytes having 26 kDa. TNF binds to TNF receptor 1 and leads to the generation of TNF receptor signaling complex (complex-I), which encompasses the core proteins, TRADD [40], TRAF2, RIPK1, cIAP1/2 and the linear ubiquitin chain assembly complex [41]. The results of the experimental studies have proven that TNF-α is involved in mucosal inflammation in CD [42]. TNF-α modulates gut inflammation in CD patients via a variety of mechanisms. In vitro experiments using patient specimens in clinical trials of anti-TNF-α therapy, the levels of TNF-α were found to be reduced with subsequential downregulation of IFN-γ in the mucosa [43]. Hence, TNF-neutralizing monoclonal antibodies (e.g., vedolizumab) are used to deal with CD and UC [44], whereas antibodies to IL-12/IL-23 p40 are for the management of CD [45,46].
The interleukin family of cytokines (i.e., IL-1α, IL-β IL-18, IL-33, and IL-36) play roles in the modulation of the proinflammatory pathway, resulting in intestinal inflammation through NF-κB [47,48,49]. Studies have reported a significant drop in the IL-1 receptor antagonists to IL-1 ratio in IBD patients, suggestive of the significance of the IL-1 pathway in the exacerbation of IBD [50]. Furthermore, in severe infant-onset IBD, as well as animal experiments, decreased IL-10 signaling, which may account for enhanced production of IL-1 in macrophages, also results in CD4+ T cell activation [51,52,53]. IL-10-related cytokines, such as IL-19, IL-20, IL-22, IL-24, IL-26, IL-28 and IL-29, are all involved in the modulation of inflammatory and immune responses (Figure 1) [54]. In mice studies, IL-1β may be a potential inducer of Helicobacter hepaticus–mediated colitis by stimulating innate lymphoid cells and conscription of neutrophils via the IL-1 receptor signaling pathway [55], which modulates mucosal aggregation of T cells and produces TH17, resulting in colitis [56,57] and further carcinogenesis [58]. The results from other studies reveal that IL-1β activation leads to chronic intestinal inflammation by endorsing the accretion of IL-17A-secreting innate lymphoid cells and CD4+ Th17 cells, resulting in intestinal pathology [59]. Consistently, IL-1 receptor antagonist treatment in mice ameliorates acute colitis [60].
IL-6 is generated by several cells present inside the tumor, such as tumor-infiltrating cells, and stromal cells. IL-6 in normal blood concentration (1.6 pg/ml) facilitates a mild immune response against the defense of incessant pathogens [61]. Studies have proven that IL-6 association with the vagus nerve may have effects on the smooth muscle cells or secretory cells, which results in intestinal motility and secretion [61,62]. The classic pathway of IL-6 signaling includes the binding of IL-6 with the membrane-bound receptor IL-6 receptor-α, also known as IL-6R. This binding results in the development of a heterohexameric complex comprising two IL-6, IL-6R, and the β subunit of IL-6 receptor (gp130) [63,64]. This complex then leads to the stimulation of the JAK/STAT3 pathway, consequently integrating STAT3 target genes (Figure 2). Interestingly, the complex also triggers the PI3K/AKT/mTOR and RAS/RAF/MEK/ERK pathways [65]. The major role of the classical pathway is to provoke anti-inflammatory impacts during the acute-phase response [66].
IL-6 also induces a trans-signaling cascade, including soluble IL-R6 (sIL-R6) binding to IL-6. sIL-6 is produced due to alternative splicing of IL-6R mRNA or via the breakdown of membrane-bound IL-R6 through ADAM 10 or ADAM 17 [67,68]. The interaction of IL-6 to sIL-6R results in complex formation, provoking the dimerization of gp130 and stimulating the downstream signaling cascade (Figure 2). The complex of IL-6/IL-6R is bound by disulfide bonds and activates Box-1 and Box-2 in the cytoplasmic domain of gp130; this results in JAK activation leading to the phosphorylation at tyrosine residue of gp130 cytoplasmic domain [69,70]. The phosphorylated pTyr-X-X-Gln motif (X = amino acid) on gp130 conscripts Src homology domain in STAT3. Phosphorylation of STAT3 in response to IL-6 impedes the binding of Suppressors of Cytokine Signaling 3 (SOCS3) to STAT3 (Figure 1) [71]. Phosphorylation of STAT3 by JAK at tyrosine residue leads to STAT3 dimerization and nuclear translocation and target genes transcription (i.e., intestinal inflammation and cancer) [72,73,74]. The result of another study shows that STAT3 and STAT4 act reciprocally on intestinal inflammation. The stimulation of STAT4 via either IL-12 or leukemia inhibitory factor via STAT3 inhibits the action of Th17, and promotes the repair of intestinal epithelial damage in IBD (Figure 1)
Interestingly, gp130 chain dimerization by the IL-6-IL6R complex stimulates the non-overlapping intracellular signaling pathway via phosphorylation of the cytoplasmic region of gp130 linked with the Janus kinase family (Figure 1). The resultant stimulation leads to activator protein 1 (AP-1) phosphorylation and induces the inflammatory genes [75,76].
IL-9 is initially found as a synergistic growth factor for T and mast cells and plays a role in asthma [77]. Studies have shown that IL-9 mRNA levels were raised in UC patients. Mechanistically, IL-9 is secreted by peripheral blood lymphocytes and binds to its receptor in the gut and polymorphonuclear leukocytes. Astonishingly, IL-9 stimulation potentiates IL-8. IL-9 is overexpressed in epithelial cells and activates STAT5 [78].
In addition, IL-18 is normally in a pro-active form and is stimulated by the action of cleavage enzyme caspase-1 into the stimulation of NLRP3, which enhances the risk of metabolic and autoimmune disorders [79,80]. NLRP3 inflammasome activation has been demonstrated in patients with IBD. Systematically, IL-18 is produced via overstimulated intestinal epithelial cells or macrophages, leading to goblet-cell depletion and churning out proinflammatory cytokines, including IFN-γ and TNF-α [81,82].
Recently, IL-36α and IL-36γ levels have been shown to be significantly increased in patients with IBD. In experimental studies, IL-36γ was identified in intestinal epithelium nuclei, while IL-36α was detected in CD+14 inflammatory macrophages in the cytoplasm [83]. Reduced IL-36 levels potentiated the dextran sodium sulfate (DSS)-induced acute colitis by impairing the cell proliferation and enhancing the effect of IL-22 associated with fibroblast stimulation [83,84]. In another study of humans with IBD, fibrotic intestinal tissues showed enhanced levels of IL36A, which is responsible for the regulation of the genes involved in the fibrogenesis in fibroblast. However, a reciprocal effect was observed in IL-36 knockout mice treated with either 2,4,6-trinitrobenzene sulfonic acid (TNBS) or DSS, showing diminished chronic colitis and intestinal fibrosis [85].
Interleukin-37 has anti-inflammatory and innate immunity suppressor activity, while IL-37b epithelial expression level was raised in IBD patients. Experimentally, IL-37b blocks the TNF-α-induced-interferon-γ-inducible protein-10 expression in human colonic subepithelial myofibroblasts [86]. Overexpression of IL-37 reduces DSS-induced colitis in transgenic mice [87]. Another study’s results demonstrate that the IL-37b gene transfer by an adenovirus vector causes the potentiation of mesenchymal stem cells against DSS-induced colitis via stimulating the Treg cell activity and inhibiting cytokines release [88].
2.3. Immune mechanisms
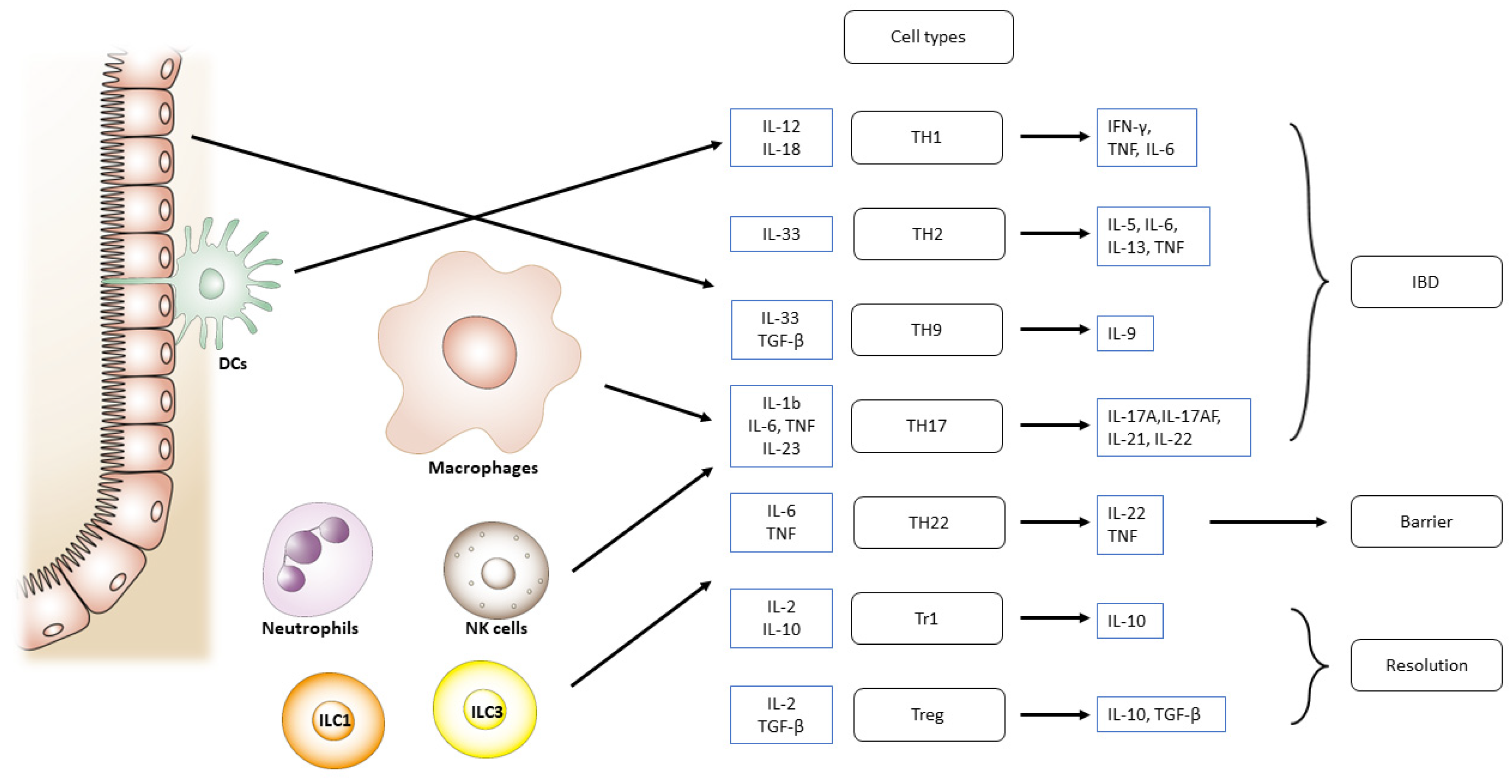
Patients with IBD lack resistance to enteric commensal bacteria and show macrophage, neutrophil, and T/B cell responses [89,90]. Resistance is facilitated in normal hosts by governing T/B lymphocytes, NK cells, and dendritic cells [42,91]. TNF and IL-12 p40 have been related to the etiology of CD in antibody-neutralization studies, whereas T cells have been associated with UC by T-cell-ablative medications [92] such as cyclosporin and tacrolimus [93,94]. Recent study findings show that reduced epithelial expression of microbiota-sensitive histone deacetylase 3 (HDAC3) leads to the elevated accretion of commensal-specific CD4+ T cells in the intestine (Figure 1) [95]. In both CD and UC, the cells engaged in innate responses are triggered, resulting in the enhanced production of cytokines and chemokines. In all types of IBD, macrophages and dendritic cells in the lamina propria are augmented in an absolute quantity. However, in CD, TH1- and TH17-related cytokines implicated in innate immunity are preferentially activated and have been rarely reported in UC [96] (Figure 3). Moreover, TH17-associated transcription factor RORγt levels were elevated in the lamina propria of IBD patients [97].
TLRs on the cell membrane bind to bacterial and viral targets. TLRs are least expressed in the normal physiological intestinal environment, whereas in the case of pathogenesis, TLRs are expressed in intestinal, respiratory, and urogenital epithelial cells. Overexpression of TLRs in the epithelial layer leads to the enhanced release of cytokines, chemokines, and anti-microbial peptides. Ligand activation of TLR then activates NF-κB and MAPK signaling pathways [91,98]; the transcription factors promote pro- and anti-inflammatory gene expression. CARD4 (also known as NOD1) and CARD15 (formerly NOD2) homologous intracellular receptors bind to diaminopimelic acid and muramyl dipeptide to activate NF-κB (Figure 2). TLR2 activation affects CARD15 and NF-κB activation [99,100]. Most of the cytokines can be selectively inhibited to delay colitis.
The epithelial layer is the primary line of defense against infections. Epithelial chemokines can be found on the luminal surface of vascular endothelium in both local tissue and draining lymph nodes, and there, they contribute to cell recruitment [101]. Monocytes and PMNs bind to injury sites, generating more proinflammatory mediators than resident macrophages. IBD patients show increases in proinflammatory cytokines and overexpression of adhesion molecules and co-stimulatory molecules [94]. To facilitate the migration of the cells, ICAM1 is required for cells in the blood to adhere and activate endothelium. It is worth noting that adhesion molecules, such as ICAM-1, have been shown to bind CD11b/CD18. However, ICAM-1 is produced only on the apical epithelial surface during inflammation [102]. Intestinal macrophages that reside in the gut have a reduced capacity to respond to bacterial components. This leads to the dysregulation of bacterial eminent receptors, including TLRs and CD14, which act as co-ligands of LPS [103].
Figure 3.
Roles of infiltrating inflammatory cells for the activation of different T cell subsets engaged in intestinal injury, barrier, and inflammation resolution in IBD. IBD increases the production of inflammatory mediators. In IBD, cells engaged in innate immune responses are triggered. In all types of IBD, macrophages and dendritic cells are augmented in an absolute quantity.Abbreviations: IBD, inflammatory bowel disease.
Figure 3.
Roles of infiltrating inflammatory cells for the activation of different T cell subsets engaged in intestinal injury, barrier, and inflammation resolution in IBD. IBD increases the production of inflammatory mediators. In IBD, cells engaged in innate immune responses are triggered. In all types of IBD, macrophages and dendritic cells are augmented in an absolute quantity.Abbreviations: IBD, inflammatory bowel disease.
2.4. Lipids and inflammation
The dysregulation of lymphocyte trafficking and immune cell migration can lead to the dissemination of chronic inflammation, prompting researchers to investigate potential agents responsible for lymphocyte migration and infiltration via the bloodstream to inflamed targets in the intestinal mucosa. One area of interest is sphingolipids (S1Ps) which are active metabolic products involved in the inflammatory cascade and immune response. S1Ps contribute to the maintenance of structural components of eukaryotic membranes, and their cascades participate in de novo synthesis and catabolic recycling with various physiological functions, leading to the recruitment of lymphocytes in injurious areas of the intestine, which intensifies inflammation by enhancing proinflammatory cytokines [23].
2.5. Intestinal stem cell niche and cell signaling
Acute inflammation kill Lgr5+ stem cells in both the small intestine and the colon [104]. Infections caused by bacteria, viruses, or parasites can damage significant regions of the gut, such as a plethora of crypt-villus units [105]. In addition, radiation, chemotherapeutic drugs, and antibiotics all cause intestinal injury in the crypts and villi [106]. Since the developed cells at the villi show a short life cycle of a few days, removing the stem or its progenitor cells in the crypts impairs the recovery of epithelial cells in the wound-associated lesion [107,108]. Since the cells do not perform all of the necessary intestinal tasks, this is only a temporary solution. The cells are then replaced within a week by creating functioning crypts from scratch. The fission of freshly generated crypts is necessary to compensate for lost crypts on a massive scale [109,110], a process slowed by crypt fusion [111].
Intestinal stem cells interact physically with those having epithelial and mesenchymal nature. The intestinal stem cell niche is made up of these cells and their interactions. Damage can be generated in several ways to examine the regeneration response. High-dose radiation exhausts Lgr5+ cells [112], which has been experimentally used to explore the regeneration response [113]. DSS, an experimental agent used to induce both acute and chronic colitis [114], also elicits crypt loss [115]. Using an animal model, the targeted knock-in of Lgr5 has been shown to provide a sophisticated strategy for understanding stem cell biology [116]. Interestingly, Lgr5+ specific deletion did not bring any visual changes in intestine architecture [116,117].
Stem cells inside the niche are subdivided and compete for limited niche space [118,119]. As a result, cells towards the niche’s boundary are more likely to be pushed out of the niche [119,120]. Consequently, stem cell-promoting stimuli facilitate cell differentiation to progenitors, which move towards the transit-amplifying zone at crypt compartments. They thereafter undergo many cycles of cell division. The most common lineage option involves choosing between the secretory and absorptive lineages [121]. In the in vivo experiments, the intestinal epithelium sustains after Paneth cells depletion, supportive of the alternate cascade activation [122,123]. Thus, a decrease in Paneth cells corresponds to a decrease in stem cells; their retention is required in the activity of in vitro stem cells [30]. WNT3, EGF, and DLL4 are produced in epithelial Paneth cells [30], which assists stem cell metabolism by providing lactate as a substrate for oxidative phosphorylation [124].
Enteroendocrine and tuft cells may substitute for deleted PCs and provide a juxtapose origin of Notch signals, even though the mesenchyme adjoining the epithelium releases adequate concentrations of Wnt ligands [125,126]. In the gut, the mesenchymal compartment comprises fibroblasts creating extracellular matrix components and myofibroblasts [127]. Many subpopulations have been shown to assist stem cells. Gli1+ cells express Wnt2b, whereas CD34+ cells express Rspo1 and Wnt2b [128]. Foxl1+ cells that express Wnt2b and Rspo3 and Pdgfra+ myofibroblasts are examples [129,130]. Mesenchymal cells thereby contribute to the activity of intestinal stem cells by establishing a healthy gradient of BMP signaling [131]. According to McCarthy et al. (2020), Pdgfra1low mesenchymal cells secrete gremlin 1. On the other hand, mesenchymal telocytes Pdgfra1high are found in the villus, and after their stimulation, they promote BMP signaling [132]. Apart from Paneth cells, deep crypt cells in the colon activate Notch [133]. Because canonical Wnt ligands are not generated in the epithelium, they must be obtained from the adjacent mesenchyme. As a result, when Wnt-secreting Gli1+ mesenchymal cells in the colon are diminished, the colonic architecture collapses [134].
2.6. Role of extracellular matrix in intestinal regeneration
Healthy cells do not proliferate in fluid, implying that they require anchoring to a solid matrix [135]. Thus, the characteristics of the extracellular matrix (ECM) have a substantial impact on the cells. Cells can assess ECM stiffness via receptors such as integrins and adjust their intracellular significance as per requirements. The stiffness is a critical parameter for stem cell differentiation [136]. Consequently, the ECM regulates cellular characteristics such as differentiation.
YAP/TAZ is considered the primary effectors of stiffness sensing, contributing to intestinal regeneration and stem cell proliferation. In an in vitro model, matrix stiffening leads to activation of YAP [137]. Under the umbrella of matrix stiffness, YAP/TAZ modulates the transition to proliferation in mammary epithelial cells [138]. The results of in vitro experiments reveal that intestinal stem cells demand a rigid matrix for optimal growth [139]. Gα12/13-coupled receptors block the Hippo pathway kinases (i.e., Lats1/2), triggering YAP and TAZ transcription coactivators [140].
It has been shown that DSS treatment increases the levels of collagen 1 and integrin [141]. In addition, DSS treatment inhibits FAK and hence compromises intestinal damage. FAK inhibition leads to decreased levels of YAP [141]. So, FAK plays a role in intestinal regeneration in association with YAP required for detecting ECM [141].
3. Dietary Lipids and IBD progression
The incidence of IBD is increasing concurrently with the rise in overweight and obesity rates. Contrary to traditional belief, a notable portion of IBD patients (31.5%) are obese, and this could potentially be linked to the development and progression of IBD [142]. Cystic fibrosis, another example of a disease characterized by extensive intestinal damage due to a genetic deficiency in CFTR, decrease fluid production in epithelial cells [143]. CF patients having increased inflammation and damage have a more static mucus layer that ineffectively protects against bacterial infections [144].
Experimental animal models fed with high-fat diets have been systematically studied, and scientific evidence shows that consumption of a western diet, which is high in fat, is directly linked to increased inflammation in the large intestine. Recent experimental studies have shown that mice fed a western diet were protected from colonic inflammation compared to those fed a normal diet [27]. However, it remains unclear whether fat consumption triggers defensive cascades against DSS-induced inflammatory pathways or impedes the well-known DSS-induced colitis.
It should be noted that the metabolic functions of fatty acids exhibit distinct characteristics in the function of intestinal epithelial cells. Although there is uncertainty surrounding the cellular activity of fatty acid oxidation, fatty acid synthesis has been shown to have an impact on intestinal epithelial cells. Studies suggest that acetyl-CoA-carboxylase 1-mediated FAS contributes to the maintenance of Lgr5+ stem cell function. As a result, FAS promotes the production of organoids and the differentiation of crypt structures by maintaining PPARδ/β-catenin [145]. Inhibition of the FAS pathway in intestinal epithelial cells led to a reduction in epithelial crypt structures and decreased Lgr5+ intestinal epithelial stem cells (Figure 4).
On the contrary, excess fat intake disturbs the phospholipid membrane structure of the epithelial cells. Intestines absorb lipids from the intestinal mucosa. In fasting conditions, the small intestine is efficient in using plasma fatty acid for oxidation and esterification and increases the uptake capacity of triacylglycerols absorption up to six-fold [146]. Triacylglycerol hydrolysis by pancreatic lipase then yields 2-monoacyglycerol (2-MAG), which is engulfed by the intestinal enterocytes [147], whereas esterified cholesterol hydrolyzed by means of cholesterol esterase generates cholesterol and fatty acid. The resulting cholesterol is taken up into micelles, which mainly contain bile acids, along with lower levels of phospholipids, FFAs, and 2-MAG [147,148]. The micelles are absorbed into enterocytes via the brush border, where they secrete fatty acid and 2-MAG and are absorbed, where it takes part in synthesizing chylomicrons. However, dietary and biliary lipids produce lysophosphatidylcholine and free fatty acids under the action of pancreatic phospholipase A2 [149].
Primary bile acids (PBA) production occurs in the liver via two cascades (i.e., classical and alternative pathways) (Figure 5). Cholic acid (CA) and chenodeoxycholic acid (CDCA) are prevalently abundant PBAs in humans. The classical pathway produces approximately 90% of the bile acid [150]. The 7α-hydroxyl group interacts with cholesterol to produce 7α-hydroxycholesterol in the presence of cytochrome P450s (CYPs). The production of 7α-hydroxycholesterol is a rate-limiting step catalyzed by CYP7A1 [151]. The CYP7A1 gene cipher an enzyme named cholesterol 7α-hydroxylase, responsible for cholesterol breakdown and bile acid synthesis [152]. Consistently, the results from animal studies show that homozygous deletion mutation in CYP7A1 resulted in hyperlipidemia [152]. It has also been shown that bile acid production is elevated in DSS-induced IBD due to a compensatory increase in CYP7A1 [153].
BA is transported into the small intestine via the ampulla of Vater in the second portion of the small intestine and accelerates the reabsorption of lipid molecules in the jejunum [154]. As BA is unable to get absorbed by the small intestine, it leads to the release of a significant portion (more than 90%) of BA through the small intestine (Figure 5), which is resorbed by the hepatic portal vein and named enterohepatic circulation of BAs [154]. The remnants of bile acids within the intestine undergo a series of chemical transformations, including deconjugation, desulfation, dehydrogenation, dehydroxylation, and isomerization, facilitated by colonic bacteria. [155,156].
PBAs dehydoxylation at carbon-7 leads to deconjugation and produces secondary bile acids (SBAs) such as lithocholic acid (LCA), deoxycholic acid (DCA), and ursodeoxycholic acid [157]. The recent mechanistic flow of converting PBAs, i.e., CA and CDCA into SBAs, LCA, and DCA, may be explained by the group of 7α-dehydroxylating bile-acid-induced (bai) operon enzymes naming BaiB, BaiCD, BaiA2, BaiE, BaiF, and BaiH found within Clostridium cluster XIVa species including Lachnospiraceae and Ruminococcaceae families, and also Eubacterium species [158].
In general, seven species are involved in bacterial/microbial clade, while Subdoligranulum, Gemmiger, and Faecalibacterium genera hold close links as they are involved in the butyrate production, which is found to have beneficial effects on IBD. However, clade production is decreased by the physiological and immunological reactions, consequently aggravating the IBD [159]. A group of Subdoligranulum species is involved in forming new clades and has been found to reduce IBD and IBD-linked metabolites such as bile acids and polyunsaturated fatty acids [160]. Summarizing the strain-level reporting of interlinked microorganisms with host epithelium reveals the organ-specific microbial species accountable for the IBD-allied surge of primary unconjugated bile acids and diminution of SBAs [159]. Post-cholecystectomy patients’ fecal BAs and mucosal microbiome analysis showed elevated immuno-regulatory activity and SBA negatively associated with peripheral monocyte levels [161]. The study’s results, including 14 healthy control patients and 39 CD patients, show that there were significantly low levels of SBA, LCA, and DCA observed in the serum and fecal of CD patients. Moreover, Enterobacteriaceae and Lachnospiraceae were robustly found in patients with CD, resulting in psychological comorbidity by disturbing their bile acids metabolism [162].
Conjugated bile acids make micelles with lipids containing phosphatidylcholine and cholesterol, while in the stomach, a gastric enzyme lipase acts on the dietary lipids and converts them into diacylglycerol and fatty acids [163]. Gastric lipase is different from pancreatic lipase but has a close resemblance. Newborn infants have low levels of pancreatic lipase, which puts emphasis on the alternative mechanism for fat digestion fulfilled by extra-pancreatic lipases (i.e., gastric lipase and ligual lipase in humans and rats) to meet the physiological demands [164,165]. The fatty acids in the stomach assist in the emulsification of lipids, followed by their movement towards the small intestine, where they are further emulsified via bile acids, strengthening the lipolytic activity of pancreatic lipases. Alternatively, the small intestine promotes the production of microbes that either promote lipid absorption or inhibit lipid intake [165,166]. These events may facilitate epithelial cell turnover, if in excess, injuries, potentially aggravating IBD progression. Hence, it remains to be established what the exact roles of appropriate amounts of dietary fat supplies and types of lipids are for the prevention of injury and promotion of stem cell regeneration.
4. Therapeutic Approaches and New Candidates
The available treatments for IBD, including filgotinib, tofacitinib, infliximab, and adalimumab, along with others, are being practiced by physicians to relief the symptoms of IBD (Table 2).
In addition, new drug candidates for IBD are described below:
- It has been shown that IL-6 was raised in mice treated with 5% DSS-induced acute colitis, while IL-6 was reduced after treatment with SM934 (artemisinin analog) and ameliorated experimental colitis [179]. The result of the tocilizumab trial on 36 patients with CD has been reported with clinical significance [180]
- In experimental models, it has been observed that deficiency of monoacylglycerol acyltransferase (MGAT) 2 provides protection against obesity. Moreover, the specific deletion of MGAT2 deters fat accumulation in the intestine [181]. In another study, monoacylglycerol lipase (MAGL) inhibition enhances the 2-arachidonoglycerol levels and results in decreased macroscopic and histological colon alterations, lowering cytokine levels [182]. MGAT2 deficiency in the intestine safeguards mice from metabolic disorders induced by high-fat feeding [183]. JTP-103237, currently in the preclinical stage, is an inhibitor of MGAT2 and impairs the absorption of luminal lipids in mice [184]. TNBS-induced murine colitis was reversed by the potent MAGL inhibitor JZL184 [182], and another MAGL inhibitor URB602 significantly repressed whole gut transient [185].
- Recent research suggests that ketogenic diets (KD) can increase the levels of circulating ketone bodies and have an anti-inflammatory effect [186]. However, the effects of this particular diet on colitis are still not well-understood. Animal studies have been conducted using KD, a low-carbohydrate diet, and a normal diet [186]. Following colitis, KD was found to protect intestinal barrier function and reduce inflammatory cytokines. Thus, KD may alleviate colitis by modifying microbiota.
- IBD frequently leads to liver injury. Milk fat globule membrane (MFGM) has been shown to mitigate colitis and liver injury [187]. Prophylactic MFGM therapy was found to be effective against colitis, improving weight loss, disease activity index, and pathological scores. Moreover, MFGM reduced levels of inflammatory mediators with an increase in IL-10 levels. MFGM thus alleviated DSS-induced injury, enhancing the mucosal barrier. It appears that MFGM may decrease oxidative stress in the liver [187].
- Signaling agents, including Wnt, EGF, Notch, and BMP ligands, promote the proliferation of Lgr5+ stem cells [188].
- Sphingosine-1-phosphate (S1P) is a signaling molecule involved in physiological processes. In IBD, the excessive infiltration of immune cells into the intestinal tissue is a significant contributor to the pathogenesis of the disease. Studies have shown that targeting S1P receptors could be a viable therapeutic strategy for IBD. Monoclonal antibodies directed to S1P have been tested in preclinical models of prostate and kidney cancer, but no studies have been done in IBD [189,190,191]. However, S1P receptor modulators have shown promise in preclinical studies [192] and are currently being evaluated in clinical trials for inflammatory disorders. These agents work downstream of S1P receptors to limit lymphocyte recruitment to inflammatory areas, reducing immune cell infiltration and mitigating inflammation in the intestine.
- Therapeutic agents that enhance insulin sensitivity, such as GLP-1, SGLT-2, and PPAR-γ ligands, have shown benefits for IBD patients by improving insulin-sensitized supplies of fuel and building block sources [193]. However, the potential impact of obesity on IBD treatment efficacy is still not well understood. Studies on various autoimmune diseases suggest that obesity can significantly affect therapeutic efficacy, leading to suboptimal treatment outcomes due to rapid clearance and decreased trough concentrations of medications. Therefore, further investigation is needed to better understand the interplay between obesity and IBD treatment outcomes.
5. Perspectives
- IBD results from the dysregulated immune system and the release of inflammatory mediators and lipotoxicity. Since inflammatory cytokines and lipotoxicity contribute to insulin resistance generation, the patients with IBD and those with metabolic disorders have common characteristics in the context of proinflammatory cytokines and oxidative stress.
- Inhibition of de novo FAS affects intestinal stem cell function and regeneration capacity, so intake of dietary lipids should be carefully interpreted to understand epithelial tissue repair and regeneration for IBD patients.
- Anti-inflammatory agents and insulin-sensitizing drugs are therapeutically beneficial to patients with IBD due to the inhibition of inflammatory injury, efficient cellular fuel oxidation, and increased tissue regeneration capacity.
Author Contributions
Conception, design, Kim S.G; Literature Review Kwon S.J. and Khan M.S. Original draft preparation, Kwon S.J. and Khan M.S; writing-review and editing, supervision, Kim S.G. All authors have read and agreed to the final draft of the manuscript.
Funding
This study was supported by the National Research Foundation (NRF) grant funded by the Korea government (NRF-2021R1A2B5B03086265).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
This manuscript has no associated data.
Acknowledgments
We would like to thank lab seniors Jihoon Tak, Sang Gil Lee, and Yun Seok Kim for their helpful discussions.
Conflicts of Interest
Authors share no conflict of interest.
Abbreviations
| IBD | Inflammatory bowel diseases |
| Lgr5 | Leucine-rich repeat-containing G-protein coupled receptor 5 |
| IEC | Intestinal epithelial cell |
| UC | Ulcerative colitis |
| CD | Crohn’s disease |
| NAFLD | Non-alcoholic fatty liver disease |
| MAFLD | Metabolic dysfunction-associated fatty liver disease |
| T2D | Type 2 diabetes |
| HFD | High-fat diet |
| Tslp | Thymic stromal lymphopoietin |
| TNF | Tumor necrosis factor |
| TRADD | Tumor necrosis factor receptor type 1-associated death domain protein |
| TRAF2 | TNF receptor-associated factor 2 |
| RIPK1 | Receptor-interacting serine/threonine-protein kinase 1 |
| cIAP | Calf intestinal alkaline phosphatase |
| IFN | Interferon |
| TH17 | T helper 17 cells |
| JAK | Janus kinase |
| STAT | Signal transducer and activator of transcription proteins |
| PI3K | Phosphoinositide 3-kinases |
| AKT | Protein kinase B (PKB), also known as Akt |
| mTOR | Mammalian target of rapamycin |
| RAS | Renin–angiotensin system |
| RAF | Rapidly accelerated fibrosarcoma |
| MEK | MAPK/ERK kinase |
| ERK | Extracellular signal-regulated kinases |
| ADAM | A Disintegrin and metalloproteinase domain-containing protein |
| SOCS3 | Suppressors of cytokine signaling 3 |
| AP-1 | Activator protein 1 |
| NLRP3 | NLR family pyrin domain containing 3 |
| DSS | Dextran sodium sulfate |
| TNBS | 2,4,6-Trinitrobenzene sulfonic acid |
| RORγt | RAR-related orphan receptor gamma |
| MAPK | Mitogen-activated protein kinases |
| NOD | Nucleotide-binding oligomerization domain-containing protein |
| TLR2 | Toll-like receptor 2 |
| PMNs | Polymorphonuclear leukocyte |
| ICAM1 | Intercellular adhesion molecule 1 |
| S1Ps | Sphingosine-1-phosphate |
| WNT | Wingless-related integration site |
| EGF | Epidermal growth factor |
| DLL4 | Delta like canonical notch ligand 4 |
| PCs | Paneth cells |
| Gli1 | Glioma-associated oncogene family zinc finger 1 |
| Rspo1 | R-Spondin 1 |
| Pdgfra | Platelet derived growth factor receptor alpha |
| BMP | Bone morphogenetic protein |
| ECM | Extracellular matrix |
| YAP | Yes-associated protein |
| TAZ | Transcriptional coactivator with PDZ-binding motif |
| FAK | Focal adhesion kinase |
| CF | Cystic fibrosis |
| FAS | Fatty acid synthesis |
| 2-MAG | 2-Monoacyglycerol |
| FFA | Free fatty acids |
| PBA | Primary bile acids |
| CA | Cholic acid |
| CDCA | Chenodeoxycholic acid |
| LCA | Lithocholic acid |
| DCA | Deoxycholic acid |
| SBA | Secondary bile acids |
| MAGL | Monoacylglycerol lipase |
| MFGM | Milk fat globule membrane |
| S1P | Sphingosine-1-phosphate |
| GLP-1 | Glucagon-Like Peptide 1 |
| SGLT-2 | Sodium-Glucose Cotransporter 2 |
References
- Yilmaz, B.; Juillerat, P.; Øyås, O.; Ramon, C.; Bravo, F.D.; Franc, Y.; Fournier, N.; Michetti, P.; Mueller, C.; et al. Microbial network disturbances in relapsing refractory Crohn’s disease. Nat Med 2019, 25, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Cleynen, I.; Boucher, G.; Jostins, L.; Schumm, L.P.; Zeissig, S.; Ahmad, T.; Andersen, V.; Andrews, J.M.; Annese, V.; et al. Inherited determinants of Crohn’s disease and ulcerative colitis phenotypes: a genetic association study. The Lancet 2016, 387, 156–167. [Google Scholar] [CrossRef]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Philip Schumm, L.; Sharma, Y.; et al. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, C.; Salleron, J.; Savoye, G.; Fumery, M.; Merle, V.; Laberenne, J.-E.; Vasseur, F.; Dupas, J.-L.; Cortot, A.; et al. Natural history of elderly-onset inflammatory bowel disease: a population-based cohort study. Gut 2014, 63, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Kappelman, M.D.; Rifas–Shiman, S.L.; Kleinman, K.; Ollendorf, D.; Bousvaros, A.; Grand, R.J.; Finkelstein, J.A. The prevalence and geographic distribution of Crohn’s disease and ulcerative colitis in the United States. Clin Gastroenterol Hepatol 2007, 5, 1424–1429. [Google Scholar] [CrossRef] [PubMed]
- Magro, F.; Langner, C.; Driessen, A.; Ensari, A.; Geboes, K.; Mantzaris, G.; Villanacci, V.; Becheanu, G.; Nunes, P.B.; et al. European consensus on the histopathology of inflammatory bowel disease. J Crohns Colitis 2013, 7, 827–851. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Lu, D.; Zhuo, J.; Lin, Z.; Yang, M.; Xu, X. The gut-liver axis in immune remodeling: new insight into liver diseases. Int J Biol Sci 2020, 16, 2357. [Google Scholar] [CrossRef]
- Yoneyama, H.; Matsuno, K.; Zhang, Y.; Murai, M.; Itakura, M.; Ishikawa, S.; Hasegawa, G.; Naito, M.; Asakura, H.; et al. Regulation by chemokines of circulating dendritic cell precursors, and the formation of portal tract–associated lymphoid tissue, in a granulomatous liver disease. J Exp Med 2001, 193, 35–50. [Google Scholar] [CrossRef]
- Trivedi, P.J.; Adams, D.H. Gut–liver immunity. J Hepatol 2016, 64, 1187–1189. [Google Scholar] [CrossRef] [PubMed]
- Principi, M.; Iannone, A.; Losurdo, G.; Mangia, M.; Shahini, E.; Albano, F.; Rizzi, S.F.; La Fortezza, R.F.; Lovero, R.; et al. Nonalcoholic fatty liver disease in inflammatory bowel disease: prevalence and risk factors. Inflamm Bowel Dis 2018, 24, 1589–1596. [Google Scholar] [CrossRef]
- Shama, S.; Jang, H.; Wang, X.; Zhang, Y.; Shahin, N.N.; Motawi, T.K.; Kim, S.; Gawrieh, S.; Liu, W. Phosphatidylethanolamines Are Associated with Nonalcoholic Fatty Liver Disease (NAFLD) in Obese Adults and Induce Liver Cell Metabolic Perturbations and Hepatic Stellate Cell Activation. Int J Mol Sci 2023, 24, 1034. [Google Scholar] [CrossRef] [PubMed]
- Powell, E.E.; Wong, V.W.-S.; Rinella, M. Non-alcoholic fatty liver disease. The Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef]
- Khan, M.S.; Lee, C.; Kim, S.G. Non-alcoholic fatty liver disease and liver secretome. Arch Pharm Res 2022, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Ekstedt, M.; Hagström, H.; Nasr, P.; Fredrikson, M.; Stål, P.; Kechagias, S.; Hultcrantz, R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef] [PubMed]
- Moran, G.W.; Dubeau, M.-F.; Kaplan, G.G.; Panaccione, R.; Ghosh, S. The increasing weight of Crohn’s disease subjects in clinical trials: a hypothesis-generatings time-trend analysis. Inflamm Bowel Dis 2013, 19, 2949–2956. [Google Scholar] [CrossRef] [PubMed]
- Khalili, H.; Ananthakrishnan, A.N.; Konijeti, G.G.; Higuchi, L.M.; Fuchs, C.S.; Richter, J.M.; Chan, A.T. Measures of obesity and risk of Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis 2015, 21, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Harpsøe, M.C.; Basit, S.; Andersson, M.; Nielsen, N.M.; Frisch, M.; Wohlfahrt, J.; Nohr, E.A.; Linneberg, A.; Jess, T. Body mass index and risk of autoimmune diseases: a study within the Danish National Birth Cohort. Int J Epidemiol 2014, 43, 843–855. [Google Scholar] [CrossRef]
- Flores, A.; Burstein, E.; Cipher, D.J.; Feagins, L.A. Obesity in inflammatory bowel disease: a marker of less severe disease. Dig Dis Sci 2015, 60, 2436–2445. [Google Scholar] [CrossRef] [PubMed]
- Mendall, M.A.; Jensen, C.B.; Sørensen, T.I. A.; Ängquist, L.H.; Jess, T. Body mass index in young men and risk of inflammatory bowel disease through adult life: a population-based Danish cohort study. Sci Rep 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Chan, S.S. M.; Chen, Y.; Casey, K.; Olen, O.; Ludvigsson, J.F.; Carbonnel, F.; Oldenburg, B.; Gunter, M.J.; Tjønneland, A.; et al. Obesity is associated with increased risk of Crohn’s disease, but not ulcerative colitis: a pooled analysis of five prospective cohort studies. Clin Gastroenterol Hepatol 2022, 20, 1048–1058. [Google Scholar] [CrossRef] [PubMed]
- Zamani, M.; Alizadeh-Tabari, S.; Singh, S.; Loomba, R. Meta-analysis: prevalence of, and risk factors for, non-alcoholic fatty liver disease in patients with inflammatory bowel disease. Aliment Pharmacol Ther 2022, 55, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Lonardo, A.; Byrne, C.D. Nonalcoholic fatty liver disease and chronic vascular complications of diabetes mellitus. Nat Rev Endocrinol 2018, 14, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C. Ulceration of the colon with a much enlarged fatty liver. Trans Pathol Soc Phil 1873, 4, 87–88. [Google Scholar]
- Bargiggia, S.; Maconi, G.; Elli, M.; Molteni, P.; Ardizzone, S.; Parente, F.; Todaro, I.; Greco, S.; Manzionna, G.; et al. Sonographic prevalence of liver steatosis and biliary tract stones in patients with inflammatory bowel disease: study of 511 subjects at a single center. J Clin Gastroenterol 2003, 36, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Sourianarayanane, A.; Garg, G.; Smith, T.H.; Butt, M.I.; McCullough, A.J.; Shen, B. Risk factors of non-alcoholic fatty liver disease in patients with inflammatory bowel disease. J Crohns Colitis 2013, 7, e279–e285. [Google Scholar] [CrossRef] [PubMed]
- Blain, A.; Cattan, S.; Beaugerie, L.; Carbonnel, F.; Gendre, J.; Cosnes, J. Crohn’s disease clinical course and severity in obese patients. Clin Nutr 2002, 21, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Papoutsis, D.; da Rocha, S.D. C.; Herfindal, A.M.; Bøhn, S.K.; Carlsen, H. A high-fat western diet attenuates intestinal changes in mice with DSS-induced low-grade inflammation. J Nutr 2022, 152, 758–769. [Google Scholar] [CrossRef]
- Knoop, K.A.; Newberry, R.D. Goblet cells: multifaceted players in immunity at mucosal surfaces. Mucosal Immunol 2018, 11, 1551–1557. [Google Scholar] [CrossRef]
- Neutra, M.R.; O’Malley, L.J.; Specian, R.D. Regulation of intestinal goblet cell secretion. II. A survey of potential secretagogues. Am J Physiol Gastrointest 1982, 242, G380–G387. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Van Es, J.H.; Snippert, H.J.; Stange, D.E.; Vries, R.G.; Van Den Born, M.; Barker, N.; Shroyer, N.F.; Van De Wetering, M.; et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 2011, 469, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Worthington, J.J.; Reimann, F.; Gribble, F. Enteroendocrine cells-sensory sentinels of the intestinal environment and orchestrators of mucosal immunity. Mucosal Immunol 2018, 11, 3–20. [Google Scholar] [CrossRef]
- Piccand, J.; Vagne, C.; Blot, F.; Meunier, A.; Beucher, A.; Strasser, P.; Lund, M.L.; Ghimire, S.; Nivlet, L.; et al. Rfx6 promotes the differentiation of peptide-secreting enteroendocrine cells while repressing genetic programs controlling serotonin production. Mol Metab 2019, 29, 24–39. [Google Scholar] [CrossRef] [PubMed]
- Birchenough, G.M.; Nyström, E.E.; Johansson, M.E.; Hansson, G.C. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science 2016, 352, 1535–1542. [Google Scholar] [CrossRef] [PubMed]
- Haber, A.L.; Biton, M.; Rogel, N.; Herbst, R.H.; Shekhar, K.; Smillie, C.; Burgin, G.; Delorey, T.M.; Howitt, M.R.; et al. A single-cell survey of the small intestinal epithelium. Nature 2017, 551, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Petersson, J.; Schreiber, O.; Hansson, G.C.; Gendler, S.J.; Velcich, A.; Lundberg, J.O.; Roos, S.; Holm, L.; Phillipson, M. Importance and regulation of the colonic mucus barrier in a mouse model of colitis. Am J Physiol Gastrointest 2011, 300, G327–G333. [Google Scholar] [CrossRef]
- Johansson, M.E.; Gustafsson, J.K.; Sjöberg, K.E.; Petersson, J.; Holm, L.; Sjövall, H.; Hansson, G.C. Bacteria penetrate the inner mucus layer before inflammation in the dextran sulfate colitis model. PloS one 2010, 5, e12238. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, M.M.; Shen, L.; Rajapakse, H.; Raleigh, D.R.; Wang, Y.; Wang, Y.; Lingaraju, A.; Zha, J.; Abbott, E.; et al. Occludin OCEL-domain interactions are required for maintenance and regulation of the tight junction barrier to macromolecular flux. Mol Biol Cell 2013, 24, 3056–3068. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.M.; Holmes, J.; Bridges, A.; Gookin, J.L.; Coccaro, M.R.; Proctor, W.; Colegio, O.R.; Anderson, J.M. The density of small tight junction pores varies among cell types and is increased by expression of claudin-2. J Cell Sci 2008, 121, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Present, D.H.; Rutgeerts, P.; Targan, S.; Hanauer, S.B.; Mayer, L.; Van Hogezand, R.A.; Podolsky, D.K.; Sands, B.E.; Braakman, T.; et al. Infliximab for the treatment of fistulas in patients with Crohn’s disease. N Engl J Med 1999, 340, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Puimège, L.; Libert, C.; Van Hauwermeiren, F. Regulation and dysregulation of tumor necrosis factor receptor-1. Cytokine Growth Factor Rev 2014, 25, 285–300. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Nakabayashi, O.; Nakano, H. FLIP the Switch: Regulation of Apoptosis and Necroptosis by cFLIP. Int J Mol Sci 2015, 16, 30321–30341. [Google Scholar] [CrossRef] [PubMed]
- Targan, S.R.; Hanauer, S.B.; Van Deventer, S.J.; Mayer, L.; Present, D.H.; Braakman, T.; DeWoody, K.L.; Schaible, T.F.; Rutgeerts, P.J. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor α for Crohn’s disease. N Engl J Med 1997, 337, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Plevy, S.E.; Landers, C.J.; Prehn, J.; Carramanzana, N.M.; Deem, R.L.; Shealy, D.; Targan, S.R. A role for TNF-alpha and mucosal T helper-1 cytokines in the pathogenesis of Crohn’s disease. J Immunol 1997, 159, 6276–6282. [Google Scholar] [CrossRef]
- Colombel, J.-F.; Sands, B.E.; Rutgeerts, P.; Sandborn, W.; Danese, S.; D’Haens, G.; Panaccione, R.; Loftus, E.V.; Sankoh, S.; et al. The safety of vedolizumab for ulcerative colitis and Crohn’s disease. Gut 2017, 66, 839–851. [Google Scholar] [CrossRef]
- Glassman, C.R.; Mathiharan, Y.K.; Jude, K.M.; Su, L.; Panova, O.; Lupardus, P.J.; Spangler, J.B.; Ely, L.K.; Thomas, C.; et al. Structural basis for IL-12 and IL-23 receptor sharing reveals a gateway for shaping actions on T versus NK cells. Cell 2021, 184, 983–999. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F. IL-23 in inflammatory bowel diseases and colon cancer. Cytokine Growth Factor Rev 2019, 45, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wirtz, S.; Becker, C.; Blumberg, R.; Galle, P.R.; Neurath, M.F. Treatment of T cell-dependent experimental colitis in SCID mice by local administration of an adenovirus expressing IL-18 antisense mRNA. J Immunol 2002, 168, 411–420. [Google Scholar] [CrossRef]
- Dmitrieva-Posocco, O.; Dzutsev, A.; Posocco, D.F.; Hou, V.; Yuan, W.; Thovarai, V.; Mufazalov, I.A.; Gunzer, M.; Shilovskiy, I.P.; et al. Cell-type-specific responses to interleukin-1 control microbial invasion and tumor-elicited inflammation in colorectal cancer. Immunity 2019, 50, 166–180.e7. [Google Scholar] [CrossRef]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and related cytokines in the regulation of inflammation and immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Pohin, M.; Jackson, M.A.; Korsunsky, I.; Bullers, S.J.; Rue-Albrecht, K.; Christoforidou, Z.; Sathananthan, D.; Thomas, T.; et al. IL-1-driven stromal–neutrophil interactions define a subset of patients with inflammatory bowel disease that does not respond to therapies. Nat Med 2021, 27, 1970–1981. [Google Scholar] [CrossRef]
- Veenbergen, S.; Li, P.; Raatgeep, H.C.; Lindenbergh-Kortleve, D.J.; Simons-Oosterhuis, Y.; Farrel, A.; Costes, L.M. M.; Joosse, M.E.; van Berkel, L.A.; et al. IL-10 signaling in dendritic cells controls IL-1β-mediated IFNγ secretion by human CD4+ T cells: relevance to inflammatory bowel disease. Mucosal Immunol 2019, 12, 1201–1211. [Google Scholar] [CrossRef]
- Aschenbrenner, D.; Quaranta, M.; Banerjee, S.; Ilott, N.; Jansen, J.; Steere, B.; Chen, Y.-H.; Ho, S.; Cox, K.; et al. Deconvolution of monocyte responses in inflammatory bowel disease reveals an IL-1 cytokine network that regulates IL-23 in genetic and acquired IL-10 resistance. Gut 2021, 70, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Mossotto, E.; Gao, Y.; Haggarty, R.; Ashton, J.J.; Batra, A.; Stafford, I.S.; Beattie, R.M.; Williams, A.P.; et al. Immunological profiling of paediatric inflammatory bowel disease using unsupervised machine learning. J Pediatr Gastroenterol Nutr 2020, 70, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Li, M.-C.; He, S.-H. IL-10 and its related cytokines for treatment of inflammatory bowel disease. World J Gastroenterol 2004, 10, 620. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Baird, A.W.; Parsons, M.J.; Fan, K.; Skerrett-Byrne, D.A.; Nair, P.M.; Makanyengo, S.; Chen, J.; Neal, R.; et al. Platelet activating factor receptor acts to limit colitis-induced liver inflammation. FASEB J 2020, 34, 7718–7732. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Sun, M.; Wu, W.; Yang, W.; Huang, X.; Xiao, Y.; Ma, C.; Xu, L.; Yao, S.; et al. Microbiota metabolite butyrate differentially regulates Th1 and Th17 cells’ differentiation and function in induction of colitis. Inflamm Bowel Dis 2019, 25, 1450–1461. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yang, W.; Yu, T.; Yu, Y.; Cui, X.; Zhou, Z.; Yang, H.; Yu, Y.; Bilotta, A.J.; et al. Th17 cell-derived amphiregulin promotes colitis-associated intestinal fibrosis through activation of mTOR and MEK in intestinal myofibroblasts. Gastroenterology 2023, 164, 89–102. [Google Scholar] [CrossRef]
- Xu, Z.-S.; Zhang, H.-X.; Li, W.-W.; Ran, Y.; Liu, T.-T.; Xiong, M.-G.; Li, Q.-L.; Wang, S.-Y.; Wu, M.; et al. FAM64A positively regulates STAT3 activity to promote Th17 differentiation and colitis-associated carcinogenesis. Proc Natl Acad Sci USA 2019, 116, 10447–10452. [Google Scholar] [CrossRef]
- Coccia, M.; Harrison, O.J.; Schiering, C.; Asquith, M.J.; Becher, B.; Powrie, F.; Maloy, K.J. IL-1β mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4+ Th17 cells. J Exp Med 2012, 209, 1595–1609. [Google Scholar] [CrossRef] [PubMed]
- Namai, F.; Shigemori, S.; Ogita, T.; Sato, T.; Shimosato, T. Microbial therapeutics for acute colitis based on genetically modified Lactococcus lactis hypersecreting IL-1Ra in mice. Exp Mol Med 2020, 52, 1627–1636. [Google Scholar] [CrossRef] [PubMed]
- Wennerås, C.; Hagberg, L.; Andersson, R.; Hynsjö, L.; Lindahl, A.; Okroj, M.; Blom, A.M.; Johansson, P.; Andreasson, B.; et al. Distinct inflammatory mediator patterns characterize infectious and sterile systemic inflammation in febrile neutropenic hematology patients. PloS one 2014, 9, e92319. [Google Scholar] [CrossRef] [PubMed]
- Comini, L.; Pasini, E.; Bachetti, T.; Dreano, M.; Garotta, G.; Ferrari, R. Acute haemodynamic effects of IL-6 treatment in vivo: involvement of vagus nerve in NO-mediated negative inotropism. Cytokine 2005, 30, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Jones, S.A. IL-6 as a keystone cytokine in health and disease. Nat Immunol 2015, 16, 448–457. [Google Scholar] [CrossRef]
- Ohshima, S.; Saeki, Y.; Mima, T.; Sasai, M.; Nishioka, K.; Nomura, S.; Kopf, M.; Katada, Y.; Tanaka, T.; et al. Interleukin 6 plays a key role in the development of antigen-induced arthritis. Proc Natl Acad Sci USA 1998, 95, 8222–8226. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.-H.; Hsiao, H.-F.; Yeh, Y.-C.; Chen, T.-W.; Li, T.-K. Inflammatory interferon activates HIF-1α-mediated epithelial-to-mesenchymal transition via PI3K/AKT/mTOR pathway. J Exp Clin Cancer Res 2018, 37, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Baran, P.; Hansen, S.; Waetzig, G.H.; Akbarzadeh, M.; Lamertz, L.; Huber, H.J.; Ahmadian, M.R.; Moll, J.M.; Scheller, J. The balance of interleukin (IL)-6, IL-6· soluble IL-6 receptor (sIL-6R), and IL-6· sIL-6R· sgp130 complexes allows simultaneous classic and trans-signaling. J Biol Chem 2018, 293, 6762–6775. [Google Scholar] [CrossRef]
- Yan, I.; Schwarz, J.; Lücke, K.; Schumacher, N.; Schumacher, V.; Schmidt, S.; Rabe, B.; Saftig, P.; Donners, M.; et al. ADAM17 controls IL-6 signaling by cleavage of the murine IL-6Rα from the cell surface of leukocytes during inflammatory responses. J Leukoc Biol 2016, 99, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.I.; Alhayyani, S.; McLeod, L.; Yu, L.; Alanazi, M.; Deswaerte, V.; Tang, K.; Jarde, T.; Smith, J.A.; et al. ADAM 17 selectively activates the IL-6 trans-signaling/ERK MAPK axis in KRAS-addicted lung cancer. EMBO Mol Med 2019, 11, e9976. [Google Scholar] [CrossRef] [PubMed]
- Li, H.S.; Liu, C.; Xiao, Y.; Chu, F.; Liang, X.; Peng, W.; Hu, J.; Neelapu, S.S.; Sun, S.-C.; et al. Bypassing STAT3-mediated inhibition of the transcriptional regulator ID2 improves the antitumor efficacy of dendritic cells. Sci Signal 2016, 9, ra94–ra94. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Gadina, M.; Siegel, R.M. Cytokines and cytokine receptors. In Clinical immunology, Elsevier: 2019; pp 127-155.
- Guo, H.; Zhuang, K.; Ding, N.; Hua, R.; Tang, H.; Wu, Y.; Yuan, Z.; Li, T.; He, S. High-fat diet induced cyclophilin B enhances STAT3/lncRNA-PVT1 feedforward loop and promotes growth and metastasis in colorectal cancer. Cell Death Dis 2022, 13, 883. [Google Scholar] [CrossRef] [PubMed]
- Samavati, L.; Rastogi, R.; Du, W.; Hüttemann, M.; Fite, A.; Franchi, L. STAT3 tyrosine phosphorylation is critical for interleukin 1 beta and interleukin-6 production in response to lipopolysaccharide and live bacteria. Mol Immunol 2009, 46, (8–9), 1867-1877. [CrossRef]
- Gupta, S.C.; Phromnoi, K.; Aggarwal, B.B. Morin inhibits STAT3 tyrosine 705 phosphorylation in tumor cells through activation of protein tyrosine phosphatase SHP1. Biochem Pharmacol 2013, 85, 898–912. [Google Scholar] [CrossRef] [PubMed]
- Schust, J.; Sperl, B.; Hollis, A.; Mayer, T.U.; Berg, T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol 2006, 13, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Zeng, K.; Ma, X.; Song, F.; Jiang, Y.; Tu, P.; Wang, X. Resokaempferol-mediated anti-inflammatory effects on activated macrophages via the inhibition of JAK2/STAT3, NF-κB and JNK/p38 MAPK signaling pathways. Int Immunopharmacol 2016, 38, 104–114. [Google Scholar] [CrossRef]
- Garg, A.D.; Kaczmarek, A.; Krysko, O.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. ER stress-induced inflammation: does it aid or impede disease progression? Trends Mol Med 2012, 18, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, C.; Hirota, K.; Stieglitz, B.; Van Snick, J.; Tolaini, M.; Lahl, K.; Sparwasser, T.; Helmby, H.; Stockinger, B. An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nat Immunol 2011, 12, 1071–1077. [Google Scholar] [CrossRef]
- Nalleweg, N.; Chiriac, M.T.; Podstawa, E.; Lehmann, C.; Rau, T.T.; Atreya, R.; Krauss, E.; Hundorfean, G.; Fichtner-Feigl, S.; et al. IL-9 and its receptor are predominantly involved in the pathogenesis of UC. Gut 2015, 64, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Hong, W.; Li, S.; Chen, Z.; Zhou, W.; Dai, J.; Deng, X.; Zhou, H.; Li, B.; et al. Wood smoke particulate matter (WSPM2. 5) induces pyroptosis through both Caspase-1/IL-1β/IL-18 and ATP/P2Y-dependent mechanisms in human bronchial epithelial cells. Chemosphere 2022, 307, 135726. [Google Scholar] [CrossRef] [PubMed]
- Rauch, I.; Deets, K.A.; Ji, D.X.; von Moltke, J.; Tenthorey, J.L.; Lee, A.Y.; Philip, N.H.; Ayres, J.S.; Brodsky, I.E.; et al. NAIP-NLRC4 inflammasomes coordinate intestinal epithelial cell expulsion with eicosanoid and IL-18 release via activation of caspase-1 and-8. Immunity 2017, 46, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Ten Hove, T.; Corbaz, A.; Amitai, H.; Aloni, S.; Belzer, I.; Graber, P.; Drillenburg, P.; Van Deventer, S.J. H.; Chvatchko, Y.; et al. Blockade of endogenous IL-18 ameliorates TNBS-induced colitis by decreasing local TNF-α production in mice. Gastroenterology 2001, 121, 1372–1379. [Google Scholar] [CrossRef] [PubMed]
- Nowarski, R.; Jackson, R.; Gagliani, N.; de Zoete, M.R.; Palm, N.W.; Bailis, W.; Low, J.S.; Harman, C.C. D.; Graham, M.; et al. Epithelial IL-18 equilibrium controls barrier function in colitis. Cell 2015, 163, 1444–1456. [Google Scholar] [CrossRef] [PubMed]
- Scheibe, K.; Backert, I.; Wirtz, S.; Hueber, A.; Schett, G.; Vieth, M.; Probst, H.C.; Bopp, T.; Neurath, M.F.; et al. IL-36R signalling activates intestinal epithelial cells and fibroblasts and promotes mucosal healing in vivo. Gut 2017, 66, 823–838. [Google Scholar] [CrossRef] [PubMed]
- Ngo, V.L.; Abo, H.; Maxim, E.; Harusato, A.; Geem, D.; Medina-Contreras, O.; Merlin, D.; Gewirtz, A.T.; Nusrat, A.; et al. A cytokine network involving IL-36γ, IL-23, and IL-22 promotes antimicrobial defense and recovery from intestinal barrier damage. Proc Natl Acad Sci USA 2018, 115, E5076–E5085. [Google Scholar] [CrossRef] [PubMed]
- Scheibe, K.; Kersten, C.; Schmied, A.; Vieth, M.; Primbs, T.; Carlé, B.; Knieling, F.; Claussen, J.; Klimowicz, A.C.; et al. Inhibiting interleukin 36 receptor signaling reduces fibrosis in mice with chronic intestinal inflammation. Gastroenterology 2019, 156, 1082–1097. [Google Scholar] [CrossRef] [PubMed]
- Imaeda, H.; Takahashi, K.; Fujimoto, T.; Kasumi, E.; Ban, H.; Bamba, S.; Sonoda, H.; Shimizu, T.; Fujiyama, Y.; et al. Epithelial expression of interleukin-37b in inflammatory bowel disease. Clin Exp Immunol 2013, 172, 410–416. [Google Scholar] [CrossRef] [PubMed]
- McNamee, E.N.; Masterson, J.C.; Jedlicka, P.; McManus, M.; Grenz, A.; Collins, C.B.; Nold, M.F.; Nold-Petry, C.; Bufler, P.; et al. Interleukin 37 expression protects mice from colitis. Proc Natl Acad Sci USA 2011, 108, 16711–16716. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-q.; Dong, K.; Zhou, L.; Jiao, G.-h.; Zhu, C.-z.; Li, W.-w.; Yu, G.; Wu, W.-t.; Chen, S.; et al. IL-37b gene transfer enhances the therapeutic efficacy of mesenchumal stromal cells in DSS-induced colitis mice. Acta Pharmacol Sin 2015, 36, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Duchmann, R.; Kaiser, I.; Hermann, E.; Mayet, W.; Ewe, K.; BÜSCHENFELDE, K.H. M. Z. Tolerance exists towards resident intestinal flora but is broken in active inflammatory bowel disease (IBD). Clin Exp Immunol 1995, 102, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Mow, W.S.; Vasiliauskas, E.A.; Lin, Y.-C.; Fleshner, P.R.; Papadakis, K.A.; Taylor, K.D.; Landers, C.J.; Abreu-Martin, M.T.; Rotter, J.I.; et al. Association of antibody responses to microbial antigens and complications of small bowel Crohn’s disease. Gastroenterology 2004, 126, 414–424. [Google Scholar] [CrossRef]
- Mannon, P.J.; Fuss, I.J.; Mayer, L.; Elson, C.O.; Sandborn, W.J.; Present, D.; Dolin, B.; Goodman, N.; Groden, C.; et al. Anti–interleukin-12 antibody for active Crohn’s disease. N Engl J Med 2004, 351, 2069–2079. [Google Scholar] [CrossRef]
- Sawada, K.; Kusugami, K.; Suzuki, Y.; Bamba, T.; Munakata, A.; Hibi, T.; Shimoyama, T. Leukocytapheresis in ulcerative colitis: results of a multicenter double-blind prospective case-control study with sham apheresis as placebo treatment. Am J Gastroenterol 2005, 100, 1362–1369. [Google Scholar] [CrossRef]
- Lichtiger, S.; Present, D.H.; Kornbluth, A.; Gelernt, I.; Bauer, J.; Galler, G.; Michelassi, F.; Hanauer, S. Cyclosporine in severe ulcerative colitis refractory to steroid therapy. N Engl J Med 1994, 330, 1841–1845. [Google Scholar] [CrossRef] [PubMed]
- Sartor, R.B.; Hoentjen, F. Proinflammatory cytokines and signaling pathways in intestinal innate immune cells. Mucosal Immunol 2005, 30, 681–701. [Google Scholar] [CrossRef]
- Eshleman, E.M.; Shao, T.-Y.; Woo, V.; Rice, T.; Engleman, L.; Didriksen, B.J.; Whitt, J.; Haslam, D.B.; Way, S.S.; et al. Intestinal epithelial HDAC3 and MHC class II coordinate microbiota-specific immunity. J Clin Invest 2023. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Diao, J.; Liu, H.; Liu, S.; Liu, J.; Yuan, J.; Lin, J. The pathogenicity and synergistic action of Th1 and Th17 cells in inflammatory bowel diseases. Inflamm Bowel Dis 2022. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.; Di Giovangiulio, M.; Stolfi, C.; Franzè, E.; Fehling, H.-J.; Carsetti, R.; Giorda, E.; Colantoni, A.; Ortenzi, A.; et al. RORγt-Expressing Tregs Drive the Growth of Colitis-Associated Colorectal Cancer by Controlling IL6 in Dendritic CellsRORγt+ Tregs Drive CAC by Controlling IL6 in DCs. Cancer Immunol Res 2018, 6, 1082–1092. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, H.K.; Bording-Jorgensen, M.; Santer, D.M.; Zhang, Z.; Valcheva, R.; Rieger, A.M.; Kim, J.S.-H.; Dijk, S.I.; Mahmood, R.; et al. Unfermented β-fructan fibers fuel inflammation in select inflammatory bowel disease patients. Gastroenterology 2023, 164, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Fritz, J.H.; Ferrero, R.L.; Philpott, D.J.; Girardin, S.E. Nod-like proteins in immunity, inflammation and disease. Nat Immunol 2006, 7, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Abraham, C.; Cho, J.H. Functional consequences of NOD2 (CARD15) mutations. Inflamm Bowel Dis 2006, 12, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat Immunol 2004, 5, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Reaves, T.A.; Chin, A.C.; Parkos, C.A. Neutrophil transepithelial migration: role of toll-like receptors in mucosal inflammation. Mem Inst Oswaldo Cruz 2005, 100, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Smythies, L.E.; Sellers, M.; Clements, R.H.; Mosteller-Barnum, M.; Meng, G.; Benjamin, W.H.; Orenstein, J.M.; Smith, P.D. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J Clin Investig 2005, 115, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Davidson, L.A.; Goldsby, J.S.; Callaway, E.S.; Shah, M.S.; Barker, N.; Chapkin, R.S. Alteration of colonic stem cell gene signatures during the regenerative response to injury. Biochim Biophys Acta Mol Basis Dis 2012, 1822, 1600–1607. [Google Scholar] [CrossRef]
- Andersson-Rolf, A.; Zilbauer, M.; Koo, B.-K.; Clevers, H. Stem cells in repair of gastrointestinal epithelia. Physiology 2017, 32, 278–289. [Google Scholar] [CrossRef]
- Duncan, M.; Grant, G. Oral and intestinal mucositis—causes and possible treatments. Aliment Pharmacol Ther 2003, 18, 853–874. [Google Scholar] [CrossRef]
- Miyoshi, H.; Ajima, R.; Luo, C.T.; Yamaguchi, T.P.; Stappenbeck, T.S. Wnt5a potentiates TGF-β signaling to promote colonic crypt regeneration after tissue injury. Science 2012, 338, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Seno, H.; Miyoshi, H.; Brown, S.L.; Geske, M.J.; Colonna, M.; Stappenbeck, T.S. Efficient colonic mucosal wound repair requires Trem2 signaling. Proc Natl Acad Sci USA 2009, 106, 256–261. [Google Scholar] [CrossRef]
- Cairnie, A.B.; Millen, B.H. Fission of crypts in the small intestine of the irradiated mouse. Cell Prolif 1975, 8, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Dekaney, C.M.; Gulati, A.S.; Garrison, A.P.; Helmrath, M.A.; Henning, S.J. Regeneration of intestinal stem/progenitor cells following doxorubicin treatment of mice. Am J Physiol Gastrointest 2009, 297, G461–G470. [Google Scholar] [CrossRef] [PubMed]
- Bruens, L.; Ellenbroek, S.I. J.; van Rheenen, J.; Snippert, H.J. In vivo imaging reveals existence of crypt fission and fusion in adult mouse intestine. Gastroenterology 2017, 153, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Van Es, J.H.; Kuipers, J.; Kujala, P.; Van Den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef]
- Montgomery, R.K.; Carlone, D.L.; Richmond, C.A.; Farilla, L.; Kranendonk, M.E. G.; Henderson, D.E.; Baffour-Awuah, N.Y.; Ambruzs, D.M.; Fogli, L.K.; et al. Mouse telomerase reverse transcriptase (mTert) expression marks slowly cycling intestinal stem cells. Proc Natl Acad Sci USA 2011, 108, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Okayasu, I.; Hatakeyama, S.; Yamada, M.; Ohkusa, T.; Inagaki, Y.; Nakaya, R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990, 98, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Rakoff-Nahoum, S.; Paglino, J.; Eslami-Varzaneh, F.; Edberg, S.; Medzhitov, R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004, 118, 229–241. [Google Scholar] [CrossRef]
- Tian, H.; Biehs, B.; Warming, S.; Leong, K.G.; Rangell, L.; Klein, O.D.; De Sauvage, F.J. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature 2011, 478, 255–259. [Google Scholar] [CrossRef]
- Metcalfe, C.; Kljavin, N.M.; Ybarra, R.; de Sauvage, F.J. Lgr5+ stem cells are indispensable for radiation-induced intestinal regeneration. Cell Stem Cell 2014, 14, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Garcia, C.; Klein, A.M.; Simons, B.D.; Winton, D.J. Intestinal stem cell replacement follows a pattern of neutral drift. Science 2010, 330, 822–825. [Google Scholar] [CrossRef] [PubMed]
- Snippert, H.J.; Van Der Flier, L.G.; Sato, T.; Van Es, J.H.; Van Den Born, M.; Kroon-Veenboer, C.; Barker, N.; Klein, A.M.; Van Rheenen, J.; et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 2010, 143, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Ritsma, L.; Ellenbroek, S.I. J.; Zomer, A.; Snippert, H.J.; De Sauvage, F.J.; Simons, B.D.; Clevers, H.; Van Rheenen, J. Intestinal crypt homeostasis revealed at single-stem-cell level by in vivo live imaging. Nature 2014, 507, 362–365. [Google Scholar] [CrossRef]
- Koch, U.; Lehal, R.; Radtke, F. Stem cells living with a Notch. Development 2013, 140, 689–704. [Google Scholar] [CrossRef] [PubMed]
- Durand, A.; Donahue, B.; Peignon, G.; Letourneur, F.; Cagnard, N.; Slomianny, C.; Perret, C.; Shroyer, N.F.; Romagnolo, B. Functional intestinal stem cells after Paneth cell ablation induced by the loss of transcription factor Math1 (Atoh1). Proc Natl Acad Sci USA 2012, 109, 8965–8970. [Google Scholar] [CrossRef]
- Kim, T.-H.; Escudero, S.; Shivdasani, R.A. Intact function of Lgr5 receptor-expressing intestinal stem cells in the absence of Paneth cells. Proc Natl Acad Sci USA 2012, 109, 3932–3937. [Google Scholar] [CrossRef]
- Rodríguez-Colman, M.J.; Schewe, M.; Meerlo, M.; Stigter, E.; Gerrits, J.; Pras-Raves, M.; Sacchetti, A.; Hornsveld, M.; Oost, K.C.; et al. Interplay between metabolic identities in the intestinal crypt supports stem cell function. Nature 2017, 543, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Farin, H.F.; Van Es, J.H.; Clevers, H. Redundant sources of Wnt regulate intestinal stem cells and promote formation of Paneth cells. Gastroenterology 2012, 143, 1518–1529. [Google Scholar] [CrossRef] [PubMed]
- Kabiri, Z.; Greicius, G.; Madan, B.; Biechele, S.; Zhong, Z.; Zaribafzadeh, H.; Aliyev, J.; Wu, Y.; Bunte, R.; et al. Stroma provides an intestinal stem cell niche in the absence of epithelial Wnts. Development 2014, 141, 2206–2215. [Google Scholar] [CrossRef]
- Powell, D.W.; Pinchuk, I.V.; Saada, J.I.; Chen, X.; Mifflin, R.C. Mesenchymal cells of the intestinal lamina propria. Annual review of physiology 2011, 73, 213–237. [Google Scholar] [CrossRef] [PubMed]
- Stzepourginski, I.; Nigro, G.; Jacob, J.-M.; Dulauroy, S.; Sansonetti, P.J.; Eberl, G.; Peduto, L. CD34+ mesenchymal cells are a major component of the intestinal stem cells niche at homeostasis and after injury. Proc Natl Acad Sci USA 2017, 114, E506–E513. [Google Scholar] [CrossRef] [PubMed]
- Aoki, R.; Shoshkes-Carmel, M.; Gao, N.; Shin, S.; May, C.L.; Golson, M.L.; Zahm, A.M.; Ray, M.; Wiser, C.L.; et al. Foxl1-expressing mesenchymal cells constitute the intestinal stem cell niche. Cell Mol Gastroenterol Hepatol 2016, 2, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Shoshkes-Carmel, M.; Wang, Y.J.; Wangensteen, K.J.; Tóth, B.; Kondo, A.; Massasa, E.E.; Itzkovitz, S.; Kaestner, K.H. Subepithelial telocytes are an important source of Wnts that supports intestinal crypts. Nature 2018, 557, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Kosinski, C.; Li, V.S. W.; Chan, A.S. Y.; Zhang, J.; Ho, C.; Tsui, W.Y.; Chan, T.L.; Mifflin, R.C.; Powell, D.W.; et al. Gene expression patterns of human colon tops and basal crypts and BMP antagonists as intestinal stem cell niche factors. Proc Natl Acad Sci USA 2007, 104, 15418–15423. [Google Scholar] [CrossRef]
- McCarthy, N.; Manieri, E.; Storm, E.E.; Saadatpour, A.; Luoma, A.M.; Kapoor, V.N.; Madha, S.; Gaynor, L.T.; Cox, C.; et al. Distinct mesenchymal cell populations generate the essential intestinal BMP signaling gradient. Cell Stem Cell 2020, 26, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Sachs, N.; Wiebrands, K.; Ellenbroek, S.I. J.; Fumagalli, A.; Lyubimova, A.; Begthel, H.; van den Born, M.; van Es, J.H.; et al. Reg4+ deep crypt secretory cells function as epithelial niche for Lgr5+ stem cells in colon. Proc Natl Acad Sci USA 2016, 113, E5399–E5407. [Google Scholar] [CrossRef] [PubMed]
- Degirmenci, B.; Valenta, T.; Dimitrieva, S.; Hausmann, G.; Basler, K. GLI1-expressing mesenchymal cells form the essential Wnt-secreting niche for colon stem cells. Nature 2018, 558, 449–453. [Google Scholar] [CrossRef]
- Discher, D.E.; Janmey, P.; Wang, Y.-l. Tissue cells feel and respond to the stiffness of their substrate. Science 2005, 310, 1139–1143. [Google Scholar] [CrossRef] [PubMed]
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix elasticity directs stem cell lineage specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Aragona, M.; Panciera, T.; Manfrin, A.; Giulitti, S.; Michielin, F.; Elvassore, N.; Dupont, S.; Piccolo, S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 2013, 154, 1047–1059. [Google Scholar] [CrossRef]
- Gjorevski, N.; Sachs, N.; Manfrin, A.; Giger, S.; Bragina, M.E.; Ordóñez-Morán, P.; Clevers, H.; Lutolf, M.P. Designer matrices for intestinal stem cell and organoid culture. Nature 2016, 539, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.-X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Trappmann, B.; Gautrot, J.E.; Connelly, J.T.; Strange, D.G. T.; Li, Y.; Oyen, M.L.; Cohen Stuart, M.A.; Boehm, H.; Li, B.; et al. Extracellular-matrix tethering regulates stem-cell fate. Nat Mater 2012, 11, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Seminerio, J.L.; Koutroubakis, I.E.; Ramos-Rivers, C.; Hashash, J.G.; Dudekula, A.; Regueiro, M.; Baidoo, L.; Barrie, A.; Swoger, J.; et al. Impact of obesity on the management and clinical course of patients with inflammatory bowel disease. Inflamm Bowel Dis 2015, 21, 2857–2863. [Google Scholar] [CrossRef] [PubMed]
- De Lisle, R.C.; Borowitz, D. The cystic fibrosis intestine. Cold Spring Harb Perspect Med 2013, 3, a009753. [Google Scholar] [CrossRef] [PubMed]
- Smyth, R.L.; Croft, N.M.; O’Hea, U.; Marshall, T.G.; Ferguson, A. Intestinal inflammation in cystic fibrosis. Arch Dis Child 2000, 82, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Lu, C.-W.; Diem, E.C.; Li, W.; Guderian, M.; Lindenberg, M.; Kruse, F.; Buettner, M.; Floess, S.; et al. Acetyl-CoA-Carboxylase 1-mediated de novo fatty acid synthesis sustains Lgr5+ intestinal stem cell function. Nat Commun 2022, 13, 3998. [Google Scholar] [CrossRef] [PubMed]
- Shiau, Y.F.; Popper, D.A.; Reed, M.; Umstetter, C.; Capuzzi, D.; Levine, G.M. Intestinal triglycerides are derived from both endogenous and exogenous sources. Am J Physiol Gastrointest 1985, 248, G164–G169. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.-A. L. S.; Skinner, J.R.; Shew, T.M.; Abumrad, N.A.; Wolins, N.E. Monoacylglycerol disrupts Golgi structure and perilipin 2 association with lipid droplets. BioRxiv 2021, 2021–07. [Google Scholar] [CrossRef]
- Phan, C.T.; Tso, P. Intestinal lipid absorption and transport. Front Biosci 2001, 6, D299–D319. [Google Scholar] [CrossRef]
- Venuti, E.; Shishmarev, D.; Kuchel, P.W.; Dutt, S.; Blumenthal, C.S.; Gaskin, K.J. Bile salt stimulated lipase: Inhibition by phospholipids and relief by phospholipase A2. J Cyst Fibros 2017, 16, 763–770. [Google Scholar] [CrossRef]
- Fiorucci, S.; Distrutti, E.; Carino, A.; Zampella, A.; Biagioli, M. Bile acids and their receptors in metabolic disorders. Prog Lipid Res 2021, 82, 101094. [Google Scholar] [CrossRef] [PubMed]
- Gälman, C.; Arvidsson, I.; Angelin, B.; Rudling, M. Monitoring hepatic cholesterol 7α-hydroxylase activity by assay of the stable bile acid intermediate 7α-hydroxy-4-cholesten-3-one in peripheral blood. J Lipid Res 2003, 44, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Pullinger, C.R.; Eng, C.; Salen, G.; Shefer, S.; Batta, A.K.; Erickson, S.K.; Verhagen, A.; Rivera, C.R.; Mulvihill, S.J.; et al. Human cholesterol 7α-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J Clin Investig 2002, 110, 109–117. [Google Scholar] [CrossRef]
- Zhou, X.; Cao, L.; Jiang, C.; Xie, Y.; Cheng, X.; Krausz, K.W.; Qi, Y.; Sun, L.; Shah, Y.M.; et al. PPARα-UGT axis activation represses intestinal FXR-FGF15 feedback signalling and exacerbates experimental colitis. Nat Commun 2014, 5, 4573. [Google Scholar] [CrossRef] [PubMed]
- Krupa, Ł.; Staroń, R.; Dulko, D.; Łozińska, N.; Mackie, A.R.; Rigby, N.M.; Macierzanka, A.; Markiewicz, A.; Jungnickel, C. Importance of bile composition for diagnosis of biliary obstructions. Molecules 2021, 26, 7279. [Google Scholar] [CrossRef] [PubMed]
- Pavlidis, P.; Powell, N.; Vincent, R.P.; Ehrlich, D.; Bjarnason, I.; Hayee, B. Systematic review: bile acids and intestinal inflammation-luminal aggressors or regulators of mucosal defence? Aliment Pharmacol Ther 2015, 42, 802–817. [Google Scholar] [CrossRef] [PubMed]
- Almeida, J.I.; Tenreiro, M.F.; Martinez-Santamaria, L.; Guerrero-Aspizua, S.; Gisbert, J.P.; Alves, P.M.; Serra, M.; Baptista, P.M. Hallmarks of the human intestinal microbiome on liver maturation and function. J Hepatol 2022, 76, 694–725. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.R.; Haileselassie, Y.; Nguyen, L.P.; Tropini, C.; Wang, M.; Becker, L.S.; Sim, D.; Jarr, K.; Spear, E.T.; et al. Dysbiosis-induced secondary bile acid deficiency promotes intestinal inflammation. Cell Host Microbe 2020, 27, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Funabashi, M.; Grove, T.L.; Wang, M.; Varma, Y.; McFadden, M.E.; Brown, L.C.; Guo, C.; Higginbottom, S.; Almo, S.C.; et al. A metabolic pathway for bile acid dehydroxylation by the gut microbiome. Nature 2020, 582, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Pasolli, E.; Asnicar, F.; Manara, S.; Zolfo, M.; Karcher, N.; Armanini, F.; Beghini, F.; Manghi, P.; Tett, A.; et al. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 2019, 176, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, J.; Ren, X.; Zhang, Y.; Ke, Z.; Zhou, J.; Wang, Y.; Zhang, Y.; Liu, Y. Cholecystectomy-induced secondary bile acids accumulation ameliorates colitis through inhibiting monocyte/macrophage recruitment. Gut Microbes 2022, 14, 2107387. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Zhou, N.; Li, Z.; Fu, D.; Guo, Y.; Gao, X.; Liu, X. Co-occurrence of gut microbiota dysbiosis and bile acid metabolism alteration is associated with psychological disorders in Crohn’s disease. FASEB 2022, 36, e22100. [Google Scholar] [CrossRef] [PubMed]
- Bernbäck, S.; Bläckberg, L.; Hernell, O. Fatty acids generated by gastric lipase promote human milk triacylglycerol digestion by pancreatic colipase-dependent lipase. Biochim Biophys Acta Mol Cell Biol Lipids 1989, 1001, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Hamosh, M.; Scanlon, J.W.; Ganot, D.; Likel, M.; Scanlon, K.B.; Hamosh, P. Fat digestion in the newborn: characterization of lipase in gastric aspirates of premature and term infants. J Clin Investig 1981, 67, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Watkins, J.B. Lipid digestion and absorption. Pediatrics 1985, 75, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Guryn, K.; Hubert, N.; Frazier, K.; Urlass, S.; Musch, M.W.; Ojeda, P.; Pierre, J.F.; Miyoshi, J.; Sontag, T.J.; et al. Small intestine microbiota regulate host digestive and absorptive adaptive responses to dietary lipids. Cell Host Microbe 2018, 23, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Dal Buono, A.; Gabbiadini, R.; Solitano, V.; Vespa, E.; Parigi, T.L.; Repici, A.; Spinelli, A.; Armuzzi, A. Critical Appraisal of Filgotinib in the Treatment of Ulcerative Colitis: Current Evidence and Place in Therapy. Clin Exp Gastroenterol 2022, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Feagan, B.G.; Danese, S.; Loftus, E.V.; Vermeire, S.; Schreiber, S.; Ritter, T.; Fogel, R.; Mehta, R.; Nijhawan, S.; et al. Filgotinib as induction and maintenance therapy for ulcerative colitis (SELECTION): a phase 2b/3 double-blind, randomised, placebo-controlled trial. The Lancet 2021, 397, 2372–2384. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Grisham, M.; Hodge, J.; Telliez, J.-B. JAK inhibition using tofacitinib for inflammatory bowel disease treatment: a hub for multiple inflammatory cytokines. Am J Physiol Gastrointest Liver Physiol 2016, 310, G155–G162. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Su, C.; Sands, B.E.; D’Haens, G.R.; Vermeire, S.; Schreiber, S.; Danese, S.; Feagan, B.G.; Reinisch, W.; et al. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N Engl J Med 2017, 376, 1723–1736. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Peyrin-Biroulet, L.; Sharara, A.I.; Su, C.; Modesto, I.; Mundayat, R.; Gunay, L.M.; Salese, L.; Sands, B.E. Efficacy and safety of tofacitinib in ulcerative colitis based on prior tumor necrosis factor inhibitor failure status. Clin Gastroenterol Hepatol 2022, 20, 591–601.e8. [Google Scholar] [CrossRef] [PubMed]
- Kruis, W.; Bar–Meir, S.; Feher, J.; Mickisch, O.; Mlitz, H.; Faszczyk, M.; Chowers, Y.; Lengyele, G.; Kovacs, A.; et al. The optimal dose of 5-aminosalicylic acid in active ulcerative colitis: a dose-finding study with newly developed mesalamine. Clin Gastroenterol Hepatol 2003, 1, 36–43. [Google Scholar] [CrossRef]
- Wallis, R.S. Tumour necrosis factor antagonists: structure, function, and tuberculosis risks. Lancet Infect Dis 2008, 8, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, G.; Samaan, M.A.; Irving, P.M. Golimumab in the treatment of ulcerative colitis. Therap Adv Gastroenterol 2019, 12, 1756284818821266. [Google Scholar] [CrossRef] [PubMed]
- Arora, Z.; Shen, B. Biological therapy for ulcerative colitis. Gastroenterol Rep 2015, 3, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, O.H.; Steenholdt, C.; Juhl, C.B.; Rogler, G. Efficacy and safety of methotrexate in the management of inflammatory bowel disease: a systematic review and meta-analysis of randomized, controlled trials. EClinicalMedicine 2020, 20. [Google Scholar] [CrossRef]
- Pithadia, A.B.; Jain, S. Treatment of inflammatory bowel disease (IBD). Pharmacol Rep 2011, 63, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Burger, D.; Travis, S. Conventional medical management of inflammatory bowel disease. Gastroenterology 2011, 140, 1827–1837. [Google Scholar] [CrossRef]
- Yan, Y.-x.; Shao, M.-j.; Qi, Q.; Xu, Y.-s.; Yang, X.-q.; Zhu, F.-h.; He, S.-j.; He, P.-l.; Feng, C.-l.; et al. Artemisinin analogue SM934 ameliorates DSS-induced mouse ulcerative colitis via suppressing neutrophils and macrophages. Acta Pharmacol Sin 2018, 39, 1633–1644. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Takazoe, M.; Fukuda, Y.; Hibi, T.; Kusugami, K.; Andoh, A.; Matsumoto, T.; Yamamura, T.; Azuma, J.; et al. A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn’s disease. Gastroenterology 2004, 126, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.W.; Gao, Y.; Yen, M.-I.; Yen, C.-L. E. Intestine-specific deletion of acyl-CoA: monoacylglycerol acyltransferase (MGAT) 2 protects mice from diet-induced obesity and glucose intolerance. J Biol Chem 2014, 289, 17338–17349. [Google Scholar] [CrossRef] [PubMed]
- Alhouayek, M.; Lambert, D.M.; Delzenne, N.M.; Cani, P.D.; Muccioli, G.G. Increasing endogenous 2-arachidonoylglycerol levels counteracts colitis and related systemic inflammation. FASEB J 2011, 25, 2711–2721. [Google Scholar] [CrossRef]
- Yen, C.-L. E.; Cheong, M.-L.; Grueter, C.; Zhou, P.; Moriwaki, J.; Wong, J.S.; Hubbard, B.; Marmor, S.; Farese Jr, R.V. Deficiency of the intestinal enzyme acyl CoA: monoacylglycerol acyltransferase-2 protects mice from metabolic disorders induced by high-fat feeding. Nat Med 2009, 15, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Okuma, C.; Ohta, T.; Tadaki, H.; Hamada, H.; Oda, T.; Taniuchi, H.; Yamanaka, K.; Ishii, Y.; Ohe, Y.; et al. JTP-103237, a novel monoacylglycerol acyltransferase inhibitor, modulates fat absorption and prevents diet-induced obesity. Eur J Pharmacol 2015, 758, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.; Thomas, A.D.; Cluny, N.L.; Patel, A.; Patel, K.D.; Lutz, B.; Piomelli, D.; Alexander, S.P. H.; Sharkey, K.A. Distribution and function of monoacylglycerol lipase in the gastrointestinal tract. Am J Physiol Gastrointest 2008, 295, G1255–G1265. [Google Scholar] [CrossRef] [PubMed]
- Kong, C.; Yan, X.; Liu, Y.; Huang, L.; Zhu, Y.; He, J.; Gao, R.; Kalady, M.F.; Goel, A.; et al. Ketogenic diet alleviates colitis by reduction of colonic group 3 innate lymphoid cells through altering gut microbiome. Signal Transduct Target Ther 2021, 6, 154. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Liu, X.; Huang, S.; Li, T.; Zhang, X.; Pang, J.; Zhao, J.; Chen, L.; Zhang, B.; et al. Milk fat globule membrane attenuates acute colitis and secondary liver injury by improving the mucus barrier and regulating the gut microbiota. Front Immunol 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Gehart, H.; Clevers, H. Tales from the crypt: new insights into intestinal stem cells. Nat Rev Gastroenterol Hepatol 2019, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, X.; Bullock, A.J.; Callea, M.; Shah, H.; Song, J.; Moreno, K.; Visentin, B.; Deutschman, D.; et al. Anti-S1P Antibody as a Novel Therapeutic Strategy for VEGFR TKI-Resistant Renal CancerS1P Inhibition as a New Treatment for RCC. Clin Cancer Res 2015, 21, 1925–1934. [Google Scholar] [CrossRef] [PubMed]
- Cuvillier, O.; Ader, I.; Bouquerel, P.; Sabbadini, R.A.; Malavaud, B. Effect of a therapeutic sphingosine 1-phosphate antibody on intratumoral hypoxia and standard chemotherapy in a preclinical model of prostate cancer. J Clin Oncol 2012, 30, (5_suppl), 223-223. [CrossRef]
- Sabbadini, R.A. Sphingosine-1-phosphate antibodies as potential agents in the treatment of cancer and age-related macular degeneration. Br J Pharmacol 2011, 162, 1225–1238. [Google Scholar] [CrossRef] [PubMed]
- Mao-Draayer, Y.; Sarazin, J.; Fox, D.; Schiopu, E. The sphingosine-1-phosphate receptor: A novel therapeutic target for multiple sclerosis and other autoimmune diseases. Clin Immunol 2017, 175, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Zhang, Z.; Jing, D.; Huang, X.; Ren, D.; Shao, Z.; Zhang, Z. SGLT2 inhibitor activates the STING/IRF3/IFN-β pathway and induces immune infiltration in osteosarcoma. Cell Death Dis 2022, 13, 523. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A scheme showing TLR2 and interleukin-mediated activation of key transcription factors responsible for the activation of genes associated with IBD progression. CARD15 (formerly NOD2) and homologous intracellular receptors bind to diaminopimelic acid and muramyl dipeptide to activate NF-κB. Abbreviations: TLR2, toll-like receptor 2; IL-6R, CARD15, caspase activating recruitment domain 15; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells.
Figure 1.
A scheme showing TLR2 and interleukin-mediated activation of key transcription factors responsible for the activation of genes associated with IBD progression. CARD15 (formerly NOD2) and homologous intracellular receptors bind to diaminopimelic acid and muramyl dipeptide to activate NF-κB. Abbreviations: TLR2, toll-like receptor 2; IL-6R, CARD15, caspase activating recruitment domain 15; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells.
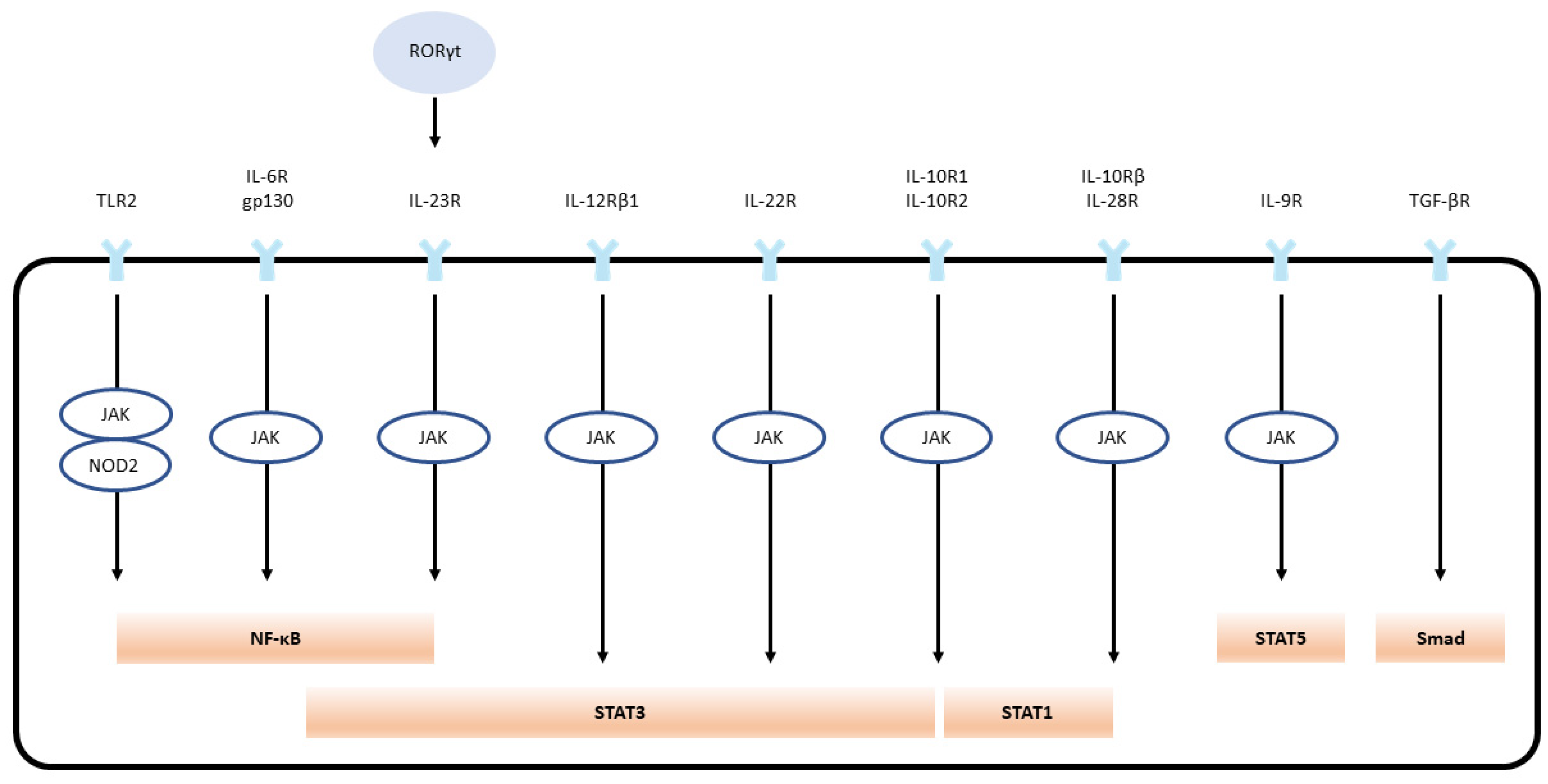
Figure 2.
The role of IL-6R interaction with gp130 for Stat3-mediated inflammatory target gene expression. Gp130 chain dimerization by the IL-6-IL6R complex stimulates the non-overlapping intracellular signaling pathway via phosphorylation of the cytoplasmic region of gp130 linked with the Janus kinase family.Abbreviations: IL-6, interlukin-6; IL-6R, interlukin-6 Receptor; gp130, glycoprotein 130.
Figure 2.
The role of IL-6R interaction with gp130 for Stat3-mediated inflammatory target gene expression. Gp130 chain dimerization by the IL-6-IL6R complex stimulates the non-overlapping intracellular signaling pathway via phosphorylation of the cytoplasmic region of gp130 linked with the Janus kinase family.Abbreviations: IL-6, interlukin-6; IL-6R, interlukin-6 Receptor; gp130, glycoprotein 130.
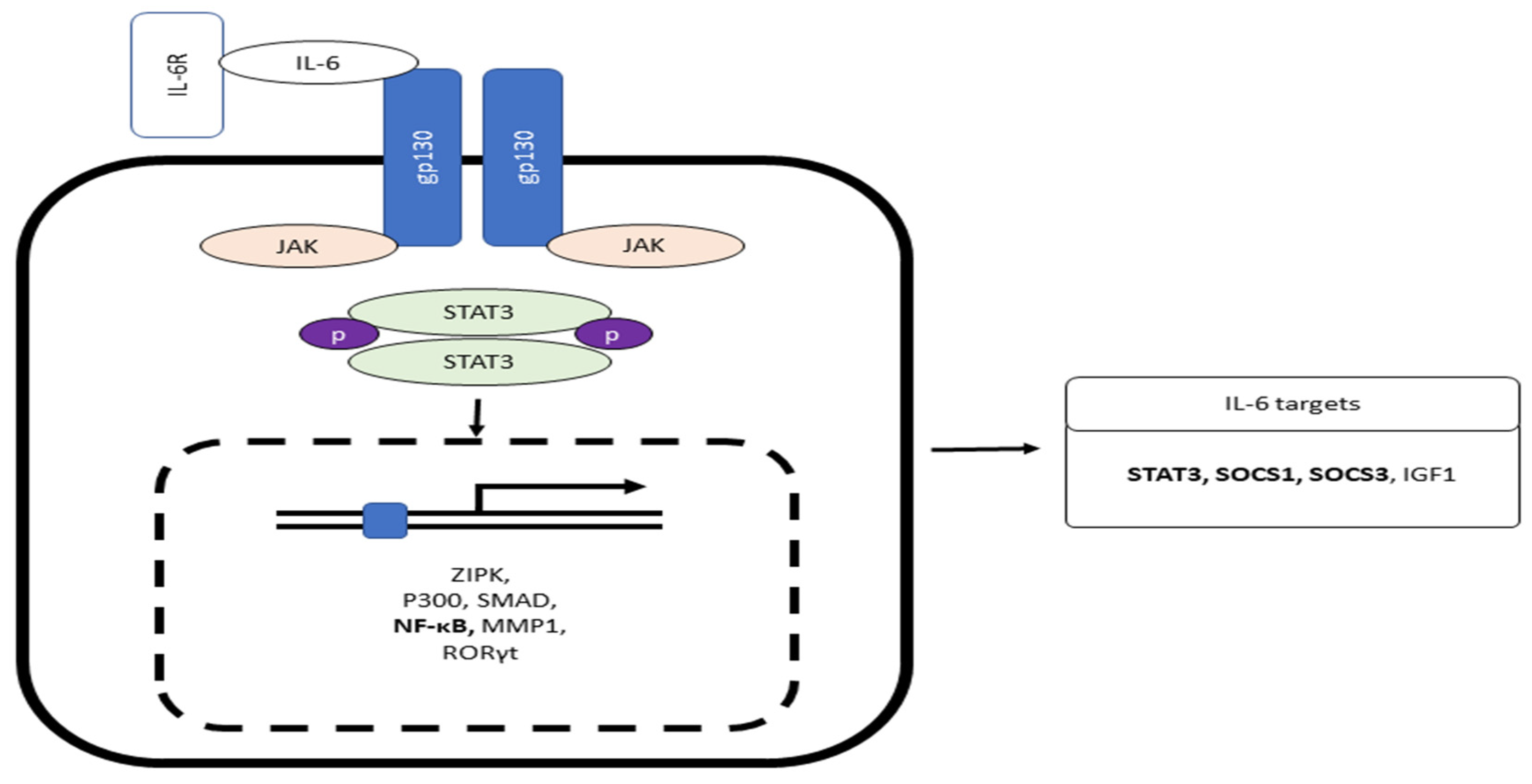
Figure 4.
A scheme showing the effects of dietary lipids on intestinal stem cell growth and differentiation. (A) MGAT2 deficiency inhibits fat accumulation in the intestine, protecting IECs in an animal model with diet-induced obesity and glucose intolerance. (B) Inhibition of de novo FAS in IECs results in a deficiency of epithelial crypt structures, which in turn leads to a reduction in Lgr5+ stem cells (ISCs). Abbreviations: MGAT2, monoacylglycerol acyltransferase (MGAT) 2; IECS, intestinal epithelial cells; FAS, fatty acid synthase, LGR5+, Leucine-rich repeat containing G protein-coupled receptor 5.
Figure 4.
A scheme showing the effects of dietary lipids on intestinal stem cell growth and differentiation. (A) MGAT2 deficiency inhibits fat accumulation in the intestine, protecting IECs in an animal model with diet-induced obesity and glucose intolerance. (B) Inhibition of de novo FAS in IECs results in a deficiency of epithelial crypt structures, which in turn leads to a reduction in Lgr5+ stem cells (ISCs). Abbreviations: MGAT2, monoacylglycerol acyltransferase (MGAT) 2; IECS, intestinal epithelial cells; FAS, fatty acid synthase, LGR5+, Leucine-rich repeat containing G protein-coupled receptor 5.
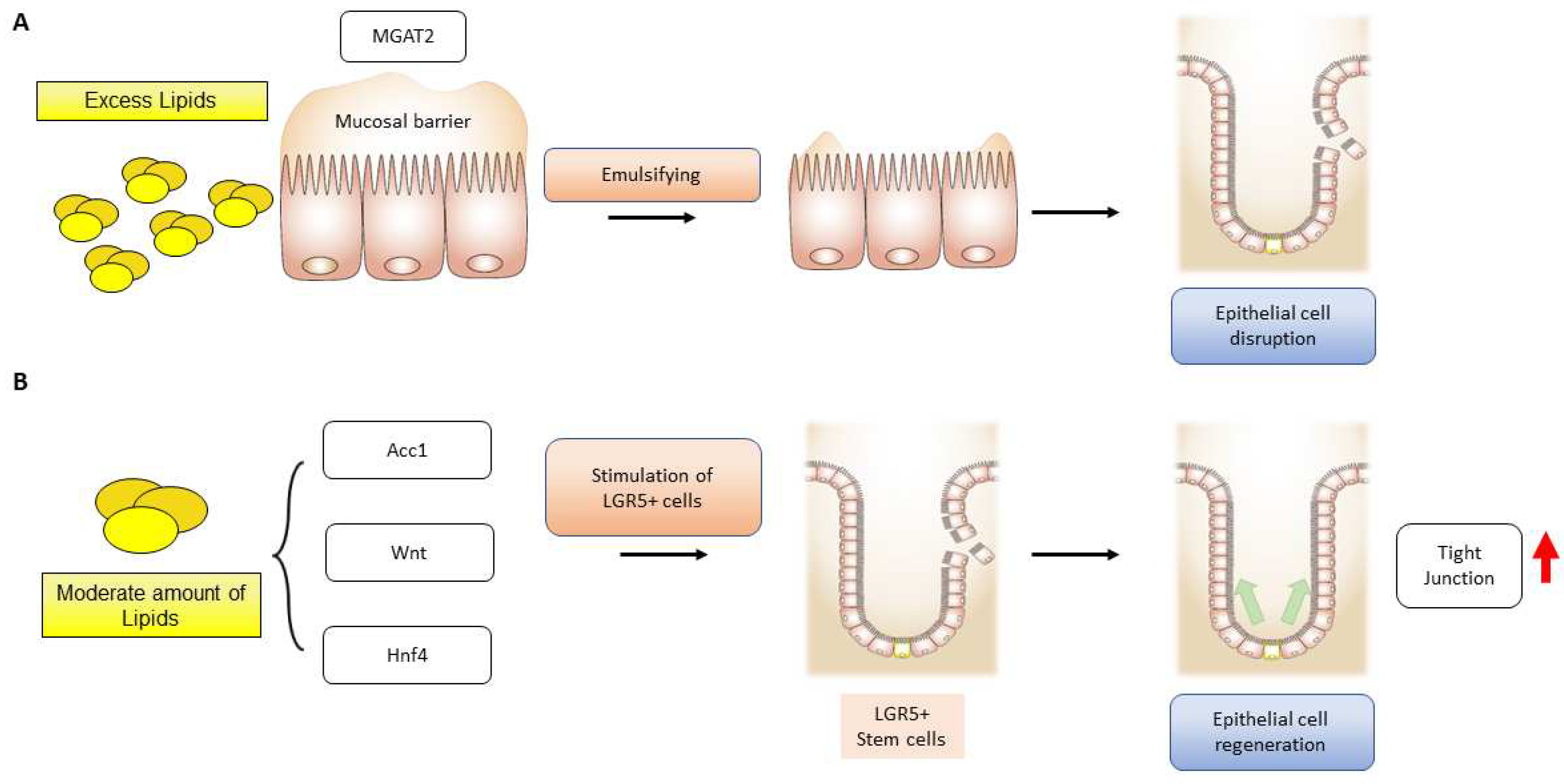
Figure 5.
A schematic flow of primary and secondary bile acids in gastrointestinal processes and microbial interactions. Abbreviations: PBAs, primary bile acids; SBAs, secondary bile acids; CA, cholic acid; CDCA, chenodeoxycholic acid.
Figure 5.
A schematic flow of primary and secondary bile acids in gastrointestinal processes and microbial interactions. Abbreviations: PBAs, primary bile acids; SBAs, secondary bile acids; CA, cholic acid; CDCA, chenodeoxycholic acid.


Table 1.
Association of IBD and obesity risk.
| Number of Patients and Characteristics | Results | References |
|---|---|---|
| 153 Crohn’s disease (CD) and 229 ulcerative colitis (UC) patients, respectively | The risk of developing CD is 2.3 times higher in obese women (18 years) and no significant association UC patients with obesity | [15] |
| CD (n = 138) and UC (n = 394) | The risk of CD was found to be increased by 1.9 times in obese non-pregnant women, but no significant association was reported for UC | [16] |
| CD (n = 75) and UC (n = 177) | According to the findings of the study, obesity alone is not enough to trigger the development of either CD or UC | [17] |
| CD (n = 297) and UC (n = 284), respectively | No statistically significant association was found between obesity and either UC or CD in the studied patient populations | [18] |
| 377,597 men with increased BMI have associative risk in developing CD and UC | The results of the COX regression analysis showed a positive correlation between BMI and CD in the group of 1523 patients, while an inverse correlation was observed in UC patients (n = 3323) | [19] |
| A pooled analysis of cohort studies inlcuding CD (n = 563) and UC (n = 1047) patients | The findings suggest that there is a significant association between CD and obese patients with BMI ≥30 kg/m2, while not significance relation between UC and obese patients | [20] |
| Systemic studies comprising 14,947 IBD subjects | Notably, 13.6% of IBD patients with NAFLD were found to have liver fibrosis | [21] |
Table 2.
Drugs available for IBD treatment.
| Drugs | Mechanisms of Action | Doses | References |
|---|---|---|---|
| Filgotinib | JAK1 inhibitor | 100 mg O.D. While 200 mg resulted in a primary embolism |
[167,168] |
| Tofacitinib | JAK1, JAK3 inhibitor | 5 or 10 mg B.I.D. for moderately to severe UC | [169,170,171] |
| Mesalazine (5-ASA) | Anti-inflammatory effect on colonic epithelial cells | 0.5 g and can be increased to 1 g 5-aminosalicylic acid T.I.D. against ulcerative colitis | [172] |
| Infliximab | Monoclonal antibody to TNF-α | Highest blood concentration via intravenous infusion 80-100 µg/ml and not less than 5µg/in 4-6 weeks |
[173] |
| Adalimumab | Monoclonal antibody to TNF-α | Subcutaneous injection 5-10 µg/ml leads to reduced TNF-α levels |
[173] |
| Golimumab | Monoclonal antibody to TNF-α | Initial starting with 200 mg and reduced to 100 mg after 2 weeks, and the dose is maintained by either 50 mg or 100 mg administered at intervals of 4 weeks for UC treatment | [174] |
| Vedolizumab | Anti-α4β7 integrin | 300 mg within 2 weeks | [175] |
| Methotrexate | inhibit enzymes responsible for nucleotide synthesis | 12.5–25 mg/week p.o or i.p. | [176] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



