
Submitted:
15 October 2023
Posted:
16 October 2023
You are already at the latest version
Abstract
Polyarteritis nodosa (PAN), also known as panarteritis nodosa, represents a form of necrotizing vasculitis that predominantly affects medium-sized vessels, although it is not restricted to them and can also involve smaller vessels. The clinical presentation is heterogeneous and characterized by a significant number of patients exhibiting general symptoms including asthenia, fever, and unintended weight loss. Although PAN can involve virtually any organ, it preferentially affects the skin, nervous system, and the gastrointestinal tract. Orchitis is a rare but specific manifestation of PAN. The absence of granulomas, glomerulonephritis and ANCA serves to distinguish PAN from other type of vasculitis. Major complications consist of hemorrhagic and thrombotic events occurring in mesenteric, cardiac, cerebral, and renal systems. Historically, PAN was frequently linked to hepatitis B virus (HBV) infection, but this association has dramatically changed in recent years due to declining HBV prevalence. Current epidemiological research often identifies connection between PAN and genetic syndromes as well as neoplasia. This article provides a comprehensive review of PAN, specifically focusing on the progression of its clinical manifestations over time.
Keywords:
PAN
; polyarteritis nodosa
; panarteritis nodosa
; VEXAS
; DADA2
1. Introduction
PAN was first described in 1866 by Kussmaul and Maier. They reported an "intermittent nodular appearance affecting arteries throughout the body sparing large vessels (the aorta and its branches), small vessels (arterioles, capillaries and venules) and pulmonary vessels" [1]. The distinct “pearl necklace” pattern led to the naming of this condition as polyarteritis nodosa. In 1952, pathologist Pearl Zeek laid out the first classification of PAN, distinguishing between hypersensitivity vasculitis, allergic vasculitis, PAN, and temporal arteritis, now respectively known as urticarial vasculitis, eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome), PAN, and giant cell arteritis (Horton's disease) [2]. A pivotal development came in 1982 when autoantibodies directed against neutrophil cytoplasm antigens (ANCA) were identified in 8 patients with clinical characteristic of vasculitis, introducing ANCA-associated vasculitis (AAV) as a distinct category of vasculitis separate from PAN [3]. The most recent definition of PAN came from the 2012 Chapel Hill Consensus Conference (CHCC) where the disease is described as "necrotizing arteritis of the medium or small arteries without glomerulonephritis or vasculitis of arterioles, capillaries or venules and without ANCA" [4,5,6]. Over the past two decades, the medical community’s understanding of PAN has significantly evolved. While initially described as a either primary vasculitis or Hepatitis B virus (HBV)-related, current understanding recognizes PAN as a disease often secondary to genetic syndromes and malignant hematologic disorders [7].
2. Epidemiology
Due to the lack of serum markers, heterogeneity in classification criteria, and numerous predisposing causes, estimating the overall prevalence of PAN is challenging. The prevalence of PAN varies greatly across different countries, ranging from 2 to 31 per million inhabitants in Europe [8,9]. There is notable North-South and seasonal gradient in the occurrence of the disease. Historically, PAN was frequently linked to Hepatitis B Virus (HBV) infection. With the advent of the HBV vaccine, the incidence of PAN has seen a significant decline, turning it from one of the most common vasculitis in the 1990s to one of the least common today [8,10]. In addition, the improvement of laboratory techniques for the detection of ANCA in the 1980s led to the reclassification of certain vasculitis that were initially diagnosed as PAN [11].
In the cohort from Pagnoux et al., which included 348 patients enrolled between 1963 and 2005, with a mean age of 51 ± 17 years old and a male to female ratio of 1.7 [12]. More recently, Rohmer et al. described 197 patients who were diagnosed with PAN between 2005 and 2019 and who had a mean age of 53.6 ± 18 years and a gender distribution with a male predominance of 1.5 [13].
3. Physiopathology
PAN is typically characterized by a segmental, necrotizing, and transmural inflammation, predominantly involving small to medium-sized arteries, although any arterial size could theoretically be susceptible. The disease most commonly impacts the visceral and muscular arteries, including their branches. In patient biopsies, it is common to observe co-existing lesions of diverse stages of inflammation and scarring within a single sample [14]. Because of the arterial inflammatory process, fibrinoid necrosis may develop, leading to the formation of microaneurysms. Over time, these complications can progress to chronic stages, characterized by fibrous scarring and vascular aneurysms, which can rupture and lead to severe bleeding [15,16]. During the acute phase, the cellular infiltrate, composed of macrophages, T lymphocytes, neutrophils, and eosinophils, is generally observed in the tunica media, but can also invade the tunica interna and tunica externa [17]. One distinctive feature of PAN, compared to other vasculitis is the absence of granulomas. This lack of granuloma formation provides a contrasting characteristic that aids in distinguishing PAN from other forms of vasculitis. This disease is also characterized by the coexistence of different stages of vascular inflammations at the same time.
The pathophysiology of PAN, not yet fully understood, may vary depending on the disease’s specific etiology. Serum cytokine profile analysis in PAN patients has revealed an elevation of interferon-alpha (IFN-⍺), interleukine-2 (IL-2), Tumor necrosis factor- (TNF⍺) and IL-1-ß compared to healthy individuals and those with Granulomatosis with Polyangiitis (GPA) [18]. Immunohistochemical studies of muscle and nerve biopsies from patients showed the presence of macrophages (41%) and mostly CD4+ T lymphocytes (41%) [19]. Most of these studies, however, primarily focuses on PAN associated with HBV.
Viral infections remain a common a trigger of PAN and should be excluded in all cases. With PAN associated with HBV, the HBs antigen is responsible for the formation of immune complexes [20,21], as suggested by animal models of hepatitis B antigen-associated PAN, which show an accumulation of immune complexes in blood vessels [22,23]. Hepatitis C virus (HCV) has also been linked to PAN, with HCV-associated PAN tending to present more severe and acute symptoms [24,25]. However, this only concerns 5% of patients with PAN and the distinction with cryoglobulinemic vasculitis can sometimes be challenging [26]. HIV infection has been associated with PAN, though HIV-associated PAN is generally less aggressive than HBV-associated PAN. The classical manifestation is mononeuritis multiplex and can occur at any stage of HIV infection [27]. Although parvovirus B19 has been associated with PAN, a study using PCR tests found no higher prevalence of this infection in people with PAN compared to those without [28,29,30,31].
More recently, vasculitis has been associated with SARS-CoV-2 infection, but to date, no cases of PAN have been reported [32,33]. Covid-19 vaccines have been associated with PAN manifestations [34,35,36]. Similarly to other vasculitis, PAN can be induced by the use of certain drugs, such as minocycline [37]. The association between PAN and neoplasia is well established, especially for hematological malignancies, such as hairy T cell leukemia or, more recently, myelodysplastic syndrome (MDS) [38,39,40,41,42]. In a study by Roupie et al., out of 70 patients with MDS and vasculitis, 9% presented with PAN. MDS is associated with a pro-inflammatory state in 10–30%, which also tend to present with autoimmune and inflammatory disorders. MDS and certain chronic inflammatory diseases share common genetic markers (such as HLA-B27) and polymorphisms (such as IL-1) [43,44].
More recently, genetic forms of PAN have been described. In the early 2000s, cases of PAN-like vasculitis were described in patients with Familial Mediterranean Fever (FMF) [45,46]. In a nationwide study in Turkey, PAN prevalence in patients with FMF was 0.9% [47]. FMF is caused by mutations in the MEFV gene that encodes for pyrin/marenostrin, which result in unregulated production of IL-1, leading to recurring inflammation, fever, and sometimes, autoimmune manifestations [48]. Patients with PAN associated to FMF present a higher incidence of perirenal hemorrhages and elevated levels of inflammation [49,50,51,52]. Another condition related to genetic forms of PAN is STING-associated vasculopathy, with onset in infancy (SAVI), which is a type I interferonopathy. This condition is caused by mutations in TMEM173 gene that induce inflammation of endothelial cells in children. It often presents PAN-like symptoms in affected children [53]. A monogenic syndrome resulting from a deficiency in Adenosine Deaminase 2 (DAD2) has been described in familial cases of necrotizing vasculitis that resemble PAN [54]. Since 2014, over 60 bi-allelic loss-of-function mutations in the ADA2 gene have been documented [55,56]. Vascular inflammation in DAD2 patients is believed to be cause by an imbalance in macrophages, favoring the M1 type over M2 type. To date, more than 200 cases of this condition have been recorded [57,58]. In 2020, Beck et al. published a cohort of 25 men exhibiting a somatic mutation affecting methionine-41 (p.Met41) in the UBA1 gene. Located on the X chromosome, this gene encodes for a critical enzyme involved in the initiation of ubiquitination. The syndrome associated with this mutation is known as VEXAS, an acronym for Vacuoles, E1 enzyme, X-linked, Autoinflammatory and Somatic. Although PAN-like features were initially reported in 12% of these patients, more recent studies suggest a lower incidence [59,60] (Figure 1).
4. Clinical manifestations
Signs and symptoms of PAN result from damage to the vascular walls, potentially affecting all organs. This section provides an overview of organ systems that can be impacted in PAN patients. Unless otherwise stated, the percentages and specifics of the manifestations come from the cohorts shown in Table 1.
4.1. General symptoms (85-93%)
General, non-specific symptoms such as asthenia, fever, weight loss, myalgia and arthralgia are frequently the initial symptoms of PAN. They are present in over 9 out of 10 patients.
4.2. Neurologic (59-79%)
Neurologic manifestations occur in more than two-thirds of patients, most commonly as motor and sensory mononeuritis multiplex of the peripheral nerves [61,62]. Peripheral neuropathy is typically distal, asymmetric, and can be rapid onset, often associated with localized skin edema. Of note, deep sensation is rarely affected. Foot drop, an important and disabling complication, may be the initial presentation [63]. Cranial nerves are affected in less than 1% of patients. Central manifestations such as strokes occur in 2 to 10% of patients, typically in the later stages of the disease [61,64,65]. Compared with classic PAN, DADA2 patients more frequently present with central neurologic manifestations, particularly stroke, usually at a young age.
4.3. Cutaneous (50-59%)
Skin lesions, including nodules, purpura, necrotic ulcers, and livedo reticularis are present in half of the patients [66]. In case of cutaneous manifestations suggestive of vasculitis, a skin biopsy is recommended. The biopsy should be deep enough to include the dermal layer where medium-sized arteries are located. A subset of PAN, known as cutaneous PAN (CPAN), is confined to the skin and requires different management [67,68].
4.4. Renal (15-75%)
Kidney involvement is typically characterized by stenosis and aneurysm, primarily affecting renal and interlobar arteries, less frequently the smaller arcuate and interlobular arteries [69,70]. This can manifest as hypertension (up to 35% of patients), micro or macro hematuria, mild proteinuria, and renal infarct [62,70,71]. Despite up to 75% of patients experiencing renal involvement, renal insufficiency occurs in only 15% of cases [62,72]. Glomerular involvement is infrequent [73]. Severe complication such as ruptured aneurysm and spontaneous perirenal hemorrhage, which require embolization or nephrectomy, are infrequent but can be life-threatening [74,75,76,77].
4.5. Gastrointestinal (22 to 38%)
Gastrointestinal manifestations of PAN are common, affecting up to 50% of patients. Abdominal pain is reported by one out of three patients [78,79]. Vascular inflammation in mesenteric arteries can be severe, leading to intestinal ischemia, perforation, and hemorrhage [80,81]. Reports have also shown gallbladder involvement, malabsorption with loss of weight and pancreatitis in patients with PAN [82,83]. In rare but severe instances, hepatic aneurysm can occur, potentially triggering acute liver failure and resulting in high mortality [84,85,86,87,88]. Gastrointestinal manifestations are associated with a poor prognosis; the mortality rate is around 25% for patients with such involvement [62,80]. This association with high mortality is supported by a retrospective study from 1988, which found that digestive complications contributed to the death of 16 % of PAN patients [89,90]. Diagnosing mesenteric arterial involvement in PAN can be challenging, and conventional angiography may be valuable, especially in patients with minimal clinical evidence of extraintestinal manifestations.
4.6. Genital (15 to 17%)
Manifestations such as testicular pain, with or without orchitis, have been described as potential symptoms specific to PAN, although similar symptoms have been noted in Behcet’s patients. Testicular biopsy can be useful for diagnosis [91]. Interestingly, there are reports of ovarian artery dissection associated with PAN in women [92].
4.7. Cardiovascular (7 to 78%)
Cardiac involvement predominantly affects the myocardium due to coronary artery vasculitis. The left anterior descending, circumflex branches and right coronary arteries are most affected. Pericarditis are relatively rare, often resulting from pre-existing myocardial involvement [93,94,95]. Myocardial infarction due to coronary infarction is also unusual [96]. Celiac artery involvement and new-onset hypertension are potential risk factors for coronary involvement [97]. Heart failure often presents during initial stages of the disease. Hypertrophic cardiomyopathy, which may be a result of uncontrolled hypertension, could trigger serious condition like ventricular tachycardia and syncope. Mild diffuse interstitial myocarditis can be caused by focal necrosis [98]. Among pediatric patients with PAN, cases of hemopericardium have been described [99,100,101]. In terms of vascular manifestations, large vessels can be affected due to necrosis of the vasa vasorum [102]. Symptoms of arterial claudication can be indicative of stenosis or ischemia in the lower extremities [103,104,105].
4.8. Other manifestations
Ophthalmic complications, including retinal vasculitis, are observed in PAN [106,107,108]. Unlike other forms of vasculitis such as granulomatosis with polyangiitis (GPA), eosinophilic granulomatosis with polyangiitis (EGPA) or microscopic polyangiitis (MPA), pulmonary lesions are notably absent in PAN. However, an autopsy study revealed bronchial artery damage in 7 out of 10 patients, despite the absence of symptoms [109]. Muscular manifestations in PAN can vary from nonspecific myalgia to paresis, and muscle biopsy shows inflammation related to PAN in up to 50% of cases [110].
VEXAS Syndrome presents general symptoms like fever or weight loss in 96% of patients, skin manifestations such as neutrophilic dermatosis or tender plaques (84%), pulmonary infiltrates (49%), chondritis (36%), and deep vein thrombosis (35%) [111]. Hematologic manifestations include macrocytic anemia (96%) and vacuoles in bone marrow myeloid and erythroid cells found all patients [60]. The vascular manifestations of VEXAS mimic small to large vessels vasculitis [60,112]. In the inaugural study of 25 VEXAS patients, 12% were diagnosed with PAN [60]. A recent literature review showed that among 9 cases of medium vessel vasculitis found, all were men with macrocytic anemia and skin lesions, 6 of whom had passed away prior to the article’s publication [113]. Patients with DADA2 exhibit vasculitis, immunodeficiency, and hematological manifestations. Vasculitis appears as mucocutaneous manifestations in 75% of cases, including livedo reticularis (50%), PAN-like skin lesions with non-granulomatous necrotizing inflammation of medium-sized arteries (34%), digital necrosis (22%), nodules (14%), Raynaud phenomenon (8 %) and aphthous ulcers (7%). Neurological manifestations occur in 51% of cases and may include ischemic strokes (27%), cranial nerve palsy (27 %), hemorrhagic strokes (12%) and polyneuropathy (9%). General symptoms such as fever elevated erythrocyte sedimentation rate or CRP are present in half of the patients. Immunodeficiency manifests as hypogammaglobulinemia (22%), low IgM (18%) or IgA (12%), and infections (20%). Viral infections (11%) are more frequent than bacterial ones (7%). Hematological diseases manifest as anemia (13%), neutropenia (7%) and thrombocytopenia (6%) due to bone marrow failure or autoimmune cytopenia. Lymphoproliferative symptoms (32%) including splenomegaly and lymphadenopathy are common in patients with DADA2. Most symptoms and signs appear (85%) occur before the age of 12 [114,115] (see Table 1).
5. Treatments
The treatment recommendations for PAN are primarily based on weak empirical evidence and are often drawn from recommendations for other forms of vasculitis, with modifications according to the disease severity. Mild PAN, characterized by non-life or organ-threatening manifestations like constitutional symptoms, arthritis, or skin lesions, is differentiated from moderate to severe PAN, which involves more severe complications, such as arterial stenosis—particularly those involving the renal arteries and aorta—and ischemic complications that affect the heart, peripheral nervous system, and gastrointestinal system [116]. To aid in risk stratification, the 1996 version of the Five-Factor Score (FFS) can be used. This score assigns +1 point for each: proteinuria greater >1 g/day, serum creatinine >140 µmol/L, cardiomyopathy, severe gastrointestinal involvement, and CNS involvement [117].
Treatment for mild PAN (FFS of 0) may include glucocorticoids (GC) only. The clinical benefit of supplementing glucocorticoids with an immunosuppressive agent is not definitively established, but it could potentially offset the high 40% relapse rate and function as a steroid-sparing strategy [116,118]. Guidelines, however, show divergence in recommendations. The French protocol typically prescribes glucocorticoids as standalone treatment, introducing immunosuppressants such as methotrexate or azathioprine only in instances of resistance or intolerance. In contrast, the ACR's 2021 guidelines advocate for a combined approach right from the beginning, recommending the incorporation of azathioprine (administered orally at 2–3 mg/kg/day) or methotrexate (preferably given subcutaneously at 0.3 mg/kg/week) with glucocorticoids [110, 113, 116,118]. Moderate to severe PAN (FFS >0) is treated with intravenous (IV) GC in conjunction with an immunosuppressive agent, preferably cyclophosphamide [116,117]. The start of treatment marks the induction phase, lasting 3 to 6 months, aimed at achieving disease remission, defined by American College of Rheumatology (ACR), as a complete absence of clinical manifestations, with or without immunosuppressive treatment [116]. Initial treatment strategies recommend starting with at least 1 mg/kg/day of prednisone equivalent, capped at 60 mg/day. In patients with severe manifestations requiring rapid intervention, IV boluses of methylprednisolone are recommended. If remission is incomplete, the duration of cyclophosphamide therapy may be extended, although it is recommended not to exceed a period of 6 months given its potential toxicity [119].
Alternative therapies, including rituximab, mycophenolate mofetil, tocilizumab, anti-TNF-alpha, JAK inhibitors, IV immunoglobulins or plasma exchange, have not been well studied and their application is only reserved for certain refractory or relapsed patients [30,120,121,122,123,124,125,126]. A recent European retrospective study analyzed 42 patients treated for relapsed and/or refractory PAN. Tocilizumab, TNF inhibitors and rituximab achieved complete remission in 50%, 40% and 33% of cases, respectively, with a comparable safety profile. The induction phase is followed by the maintenance phase, with the objective of preventing relapse. Patients treated with cyclophosphamide with a complete remission may be switched to azathioprine or methotrexate for 12 to 18 months [117]. In secondary forms of PAN, the therapeutic approach focuses on the underlying etiology. In the context of HBV-associated PAN, antiviral therapy is used as the primary intervention [117]. When PAN is concomitant with MDS, interventions targeting the MDS are often effective in attenuating the vasculitic manifestations [39]. From this perspective, Mekinian et al. showed that azacytidine successfully treated autoimmune manifestations in 9 of 11 patients with MDS [127]. In the case of DADA2-associated PAN, numerous treatments have been explored (azathioprine, cyclosporine, tacrolimus, cyclophosphamide, methotrexate) with mitigated results. The ACR 2021 guidelines have now approved the use of steroids and anti-TNF-agents (etanercept, infliximab or adalimumab) following demonstration of their efficacy in PAN associated with DADA2 [116]. Hematopoietic stem cell transplantation (HSCT) has been reported to treat cytopenia [128]. Regarding VEXAS syndrome, several drugs have been tested with mixed results [129]. GC in combination with azacytidine (possibly supplemented by HSCT) seem most effective for patients with MDS features. For those without myelodysplasia, JAK inhibitors or tocilizumab may be suitable [130,131]. Finally, vasculitis associated with primary immunodeficiency can be managed with biotherapies, HSCT, or IV immunoglobulin therapy [48].
6. Conclusions
Over the past two decades, our understanding of PAN has undergone a significant evolution. While historically viewed through the binary lens of primary vs HBV-related PAN, the landscape has expanded. We now recognize a spectrum of PAN, from those linked to infections and paraneoplastic syndromes, to novel classifications associated with DADA2, interferonopathies, and the VEXAS syndrome. Interestingly, these auto-inflammatory related PAN variants present distinct clinical manifestations. For instance, VEXAS syndrome has heterogenous manifestations, with heightened general symptoms, cutaneous, ophthalmic and thromboembolic manifestations, unique chondritis, and frequent but mild pulmonary involvement. DADA2, which mainly affects children and young adults, shows a deviation from traditional PAN symptoms. It presents fewer general symptoms, lacks orchitis, and its neurological manifestations predominantly target the central nervous system. Identification of potential secondary causes of PAN is critical, as it can significantly influence treatment decisions. While conventional immunosuppressants such as cyclophosphamide are often the standard of care for primary PAN, secondary forms may benefit from more specific agents: anti-TNF-alpha for DADA2, JAK inhibitors for VEXAS, and targeted MDS treatment for PAN associated with MDS. Overall, despite the emergence of new forms of PAN, the important decrease in HBV prevalence due to vaccination has contributed to a decreased overall incidence of PAN.
Author Contributions
Conceptualization, L.W. and D.C.; methodology, L.W., A.H., L.M., M.P.K. and D.C..; writing—original draft preparation, L.W. and D.C..; writing—review and editing, M.P.K.. A.H. and L.M.; supervision, D.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Stone, J.H. Vasculitis: A Collection of Pearls and Myths. Rheum. Dis. Clin. North Am. 2007, 33, 691–739. [Google Scholar] [CrossRef] [PubMed]
- Zeek, P.M. Periarteritis Nodosa: A Critical Review. Am. J. Clin. Pathol. 1952, 22, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.J.; E Moran, J.; Niall, J.F.; Ryan, G.B. Segmental necrotising glomerulonephritis with antineutrophil antibody: possible arbovirus aetiology? BMJ 1982, 285, 606–606. [Google Scholar] [CrossRef] [PubMed]
- Karadag, O.; Jayne, D.J. Polyarteritis nodosa revisited: a review of historical approaches, subphenotypes and a research agenda. Clin Exp Rheumatol 2018, 135–142. [Google Scholar]
- Jennette, J.C.; Falk, R.J.; Bacon, P.A.; Basu, N.; Cid, M.C.; Ferrario, F.; Flores-Suarez, L.F.; Gross, W.L.; Guillevin, L.; Hagen, E.C.; et al. 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2012, 65, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Guillevin, L.; Mahr, A.; Cohen, P. Les vascularites nécrosantes systémiques : classification et stratégies actuelles de traitement. La Rev. de Médecine Interne 2003, 24, 172–182. [Google Scholar] [CrossRef]
- Rohmer, J.; Trefond, L.; Nguyen, Y.; Durel, C.; Lacout, C.; Maurier, F.; Rouzaud, D.; Cohen, P.; Lazaro, E.; Mekinian, A.; et al. Caractéristiques cliniques et évolution à long-terme des périartérite noueuses systémiques diagnostiquées depuis 2005. La Rev. de Médecine Interne 2020, 41, A33–A34. [Google Scholar] [CrossRef]
- Mahr, A.; Guillevin, L.; Poissonnet, M.; Aymé, S. Prevalences of polyarteritis nodosa, microscopic polyangiitis, Wegener's granulomatosis, and Churg-Strauss syndrome in a French urban multiethnic population in 2000: A capture–recapture estimate. Arthritis Care Res. 2004, 51, 92–99. [Google Scholar] [CrossRef]
- Lane, S.E.; Watts, R.; Scott, D.G.I. Epidemiology of systemic vasculitis. Curr. Rheumatol. Rep. 2005, 7, 270–275. [Google Scholar] [CrossRef]
- Sönmez, H.E.; Armağan, B.; Ayan, G.; Barut, K.; Batu, E.D.; Erden, A.; Ugurlu, S.; Bilginer, Y.; Kasapcopur, O.; Karadag, O.; et al. Polyarteritis nodosa: lessons from 25 years of experience. Clin Exp Rheumatol 2018, 52–56. [Google Scholar]
- Saadoun, D.; Vautier, M.; Cacoub, P. Medium- and Large-Vessel Vasculitis. Circ. 2021, 143, 267–282. [Google Scholar] [CrossRef]
- Pagnoux, C.; Seror, R.; Henegar, C.; Mahr, A.; Cohen, P.; Le Guern, V.; Bienvenu, B.; Mouthon, L.; Guillevin, L. Clinical features and outcomes in 348 patients with polyarteritis nodosa: A systematic retrospective study of patients diagnosed between 1963 and 2005 and entered into the French vasculitis study group database. Arthritis Rheum. 2010, 62, 616–626. [Google Scholar] [CrossRef]
- Rohmer, J.; Nguyen, Y.; Trefond, L.; Agard, C.; Allain, J.S.; Berezne, A.; Charles, P.; Cohen, P.; Gondran, G.; Groh, M.; et al. Clinical features and long-term outcomes of patients with systemic polyarteritis nodosa diagnosed since 2005: Data from 196 patients. J. Autoimmun. 2023, 139, 103093. [Google Scholar] [CrossRef] [PubMed]
- De Virgilio, A.; Greco, A.; Magliulo, G.; Gallo, A.; Ruoppolo, G.; Conte, M.; Martellucci, S.; de Vincentiis, M. Polyarteritis nodosa: A contemporary overview. Autoimmun. Rev. 2016, 15, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Kobayashi, S.; Ogishima, D.; Aoki, Y.; Sonoue, H.; Abe, H.; Fukumura, Y.; Nobukawa, B.; Kumasaka, T.; Mori, S.; et al. Isolated necrotizing arteritis (localized polyarteritis nodosa): examination of the histological process and disease entity based on the histological classification of stage and histological differences from polyarteritis nodosa. Cardiovasc. Pathol. 2007, 16, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Lie, J.T. Systemic and isolated vasculitis. A rational approach to classification and pathologic diagnosis. Pathol Annu 1989, 25–114. [Google Scholar]
- Kallenberg, C.G.; Brouwer, E.; Weening, J.J.; Tervaert, J.W.C. Anti-neutrophil cytoplasmic antibodies: Current diagnostic and pathophysiological potential. Kidney Int. 1994, 46, 1–15. [Google Scholar] [CrossRef] [PubMed]
- E Grau, G.; Roux-Lombard, P.; Gysler, C.; Lambert, C.; Lambert, P.H.; Dayer, J.M.; Guillevin, L. Serum cytokine changes in systemic vasculitis. Immunology 1989, 68, 196–8. [Google Scholar] [PubMed]
- Cid, M.; Grau, J.M.; Casademont, J.; Campo, E.; Coll-Vinent, B.; López-Soto, A.; Ingelmo, M.; Urbano-Márquez, A. Immunohistochemical characterization of inflammatory cells and immunologic activation markers in muscle and nerve biopsy specimens from patients with systemic polyarteritis nodosa. Arthritis Rheum. 1994, 37, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Guillevin, L.; Ronco, P.; Verroust, P. Circulating immune complexes in systemic necrotizing vasculitis of the polyarteritis nodosa group. Comparison of HBV-related polyarteritis nodosa and churg strauss angiitis. J. Autoimmun. 1990, 3, 789–792. [Google Scholar] [CrossRef]
- Prince, A.; Trepo, C. ROLE OF IMMUNE COMPLEXES INVOLVING SH ANTIGEN IN PATHOGENESIS OF CHRONIC ACTIVE HEPATITIS AND POLYARTERITIS NODOSA. Lancet 1971, 297, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Trepo, C.G.; Zuckerman, A.J.; Bird, R.C.; Prince, A.M. The role of circulating hepatitis B antigen/antibody immune complexes in the pathogenesis of vascular and hepatic manifestations in polyarteritis nodosa. J. Clin. Pathol. 1974, 27, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Fye, K.H.; Becker, M.J.; Theofilopoulos, A.N.; Moutsopoulos, H.; Feldman, J.-L.; Talal, N. Immune complexes in hepatitis B antigen-associated periarteritis nodosa. Am. J. Med. 1977, 62, 783–791. [Google Scholar] [CrossRef]
- Carson, C.W.; Conn, D.L.; Czaja, A.J.; Wright, T.L.; E Brecher, M. Frequency and significance of antibodies to hepatitis C virus in polyarteritis nodosa. J Rheumatol 1993, 20, 304–9. [Google Scholar] [PubMed]
- Saadoun, D.; Terrier, B.; Semoun, O.; Sene, D.; Maisonobe, T.; Musset, L.; Amoura, Z.; Rigon, M.R.; Cacoub, P. Hepatitis C virus–associated polyarteritis nodosa. Arthritis Care Res. 2010, 63, 427–435. [Google Scholar] [CrossRef]
- Cacoub, P.; Maisonobe, T.; Thibault, V.; Gatel, A.; Servan, J.; Musset, L.; Piette, J.C. Systemic vasculitis in patients with hepatitis C. J Rheumatol 2001, 28, 109–18. [Google Scholar]
- Patel, N.; Patel, N.; Khan, T.; Patel, N.; Espinoza, L.R. HIV Infection and Clinical Spectrum of Associated Vasculitides. Curr. Rheumatol. Rep. 2011, 13, 506–512. [Google Scholar] [CrossRef]
- Leruez-Ville, M.; Laugé, A.; Morinet, F.; Guillevin, L.; Dény, P. Polyarteritis riodosa and parvovirus B19. Lancet 1994, 344, 263–264. [Google Scholar] [CrossRef]
- Gherardi, R.; Belec, L.; Mhiri, C.; Gray, F.; Lescs, M.; Sobel, A.; Guillevin, L.; Wechsler, J. The spectrum of vasculitis in human immunodeficiency virus–infected patients. a clinicopathologic evaluation. Arthritis Rheum. 1993, 36, 1164–1174. [Google Scholar] [CrossRef]
- Viguier, M.; Guillevin, L.; Laroche, L. Treatment of Parvovirus B19–Associated Polyarteritis Nodosa with Intravenous Immune Globulin. New Engl. J. Med. 2001, 344, 1481–1482. [Google Scholar] [CrossRef]
- Finkel, T.; Leung, D.; Harbeck, R.; Gelfand, E.; Török, T.; Zaki, S.; Anderson, L.; Ferguson, P.; Saulsbury, F.; Durigon, E.; et al. Chronic parvovirus B19 infection and systemic necrotising vasculitis: opportunistic infection or aetiological agent? Lancet 1994, 343, 1255–1258. [Google Scholar] [CrossRef]
- Ramadan, S.M.; Kasfiki, E.V.; Kelly, C.W.; Ali, I. An interesting case of small vessel pathology following coronavirus infection. BMJ Case Rep. 2020, 13, e237407. [Google Scholar] [CrossRef]
- Vacchi, C.; Meschiari, M.; Milic, J.; Marietta, M.; Tonelli, R.; Alfano, G.; Volpi, S.; Faltoni, M.; Franceschi, G.; Ciusa, G.; et al. COVID-19-associated vasculitis and thrombotic complications: from pathological findings to multidisciplinary discussion. Rheumatology 2020, 59, e147–e150. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Hsu, H.; Chen, Y. Cutaneous polyarteritis nodosa following ChAdOx1 nCoV-19 vaccination. Int. J. Dermatol. 2022, 61, 630–631. [Google Scholar] [CrossRef] [PubMed]
- Ohkubo, Y.; Ohmura, S.-I.; Ishihara, R.; Miyamoto, T. Possible case of polyarteritis nodosa with epididymitis following COVID-19 vaccination: A case report and review of the literature. Mod. Rheumatol. Case Rep. 2022, 7, 172–176. [Google Scholar] [CrossRef]
- Ohmura, S.-I.; Ohkubo, Y.; Ishihara, R.; Otsuki, Y.; Miyamoto, T. Medium-vessel Vasculitis Presenting with Myalgia Following COVID-19 Moderna Vaccination. Intern. Med. 2022, 61, 3453–3457. [Google Scholar] [CrossRef]
- Kermani, T.A.; Ham, E.K.; Camilleri, M.J.; Warrington, K.J. Polyarteritis Nodosa-like Vasculitis in Association with Minocycline Use: A Single-Center Case Series. Semin. Arthritis Rheum. 2012, 42, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, M.T.; West, S.G. Polyarteritis nodosa in hairy cell leukemia: treatment with interferon-alpha. J Rheumatol 1994, 21, 1150–2. [Google Scholar] [PubMed]
- Roupie, A.L.; Guedon, A.; Terrier, B.; Lahuna, C.; Jachiet, V.; Regent, A.; de Boysson, H.; Carrat, F.; Seguier, J.; Terriou, L.; et al. Vasculitis associated with myelodysplastic syndrome and chronic myelomonocytic leukemia: French multicenter case-control study. Semin. Arthritis Rheum. 2020, 50, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Veitch, D.; Tsai, T.; Watson, S.; Joshua, F. Paraneoplastic polyarteritis nodosa with cerebral masses: case report and literature review. Int. J. Rheum. Dis. 2014, 17, 805–809. [Google Scholar] [CrossRef]
- Hamidou, M.A.; Boumalassa, A.; Larroche, C.; El Kouri, D.; Blétry, O.; Grolleau, J.-Y. Systemic medium-sized vessel vasculitis associated with chronic myelomonocytic leukemia. Semin. Arthritis Rheum. 2001, 31, 119–126. [Google Scholar] [CrossRef]
- Fain, O.; Hamidou, M.; Cacoub, P.; Godeau, B.; Wechsler, B.; ParIès, J.; Stirnemann, J.; Morin, A.; Gatfosse, M.; Hanslik, T.; et al. Vasculitides associated with malignancies: Analysis of sixty patients. Arthritis Care Res. 2007, 57, 1473–1480. [Google Scholar] [CrossRef]
- Komrokji, R.S.; Kulasekararaj, A.; Al Ali, N.H.; Kordasti, S.; Bart-Smith, E.; Craig, B.M.; Padron, E.; Zhang, L.; Lancet, J.E.; Pinilla-Ibarz, J.; et al. Autoimmune diseases and myelodysplastic syndromes. Am. J. Hematol. 2016, 91, E280–E283. [Google Scholar] [CrossRef] [PubMed]
- Saif, M.W.; Hopkins, J.L.; Gore, S.D. Autoimmune Phenomena in Patients with Myelodysplastic Syndromes and Chronic Myelomonocytic Leukemia. Leuk. Lymphoma 2002, 43, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Balbir-Gurman, A.; Nahir, A.M.; Braun-Moscovici, Y. Vasculitis in siblings with familial Mediterranean fever: a report of three cases and review of the literature. Clin. Rheumatol. 2006, 26, 1183–1185. [Google Scholar] [CrossRef]
- Ozen, S.; Ben-Chetrit, E.; Bakkaloglu, A.; Gur, H.; Tinaztepe, K.; Calguneri, M.; Turgan, C.; Turkmen, A.; Akpolat, I.; Danaci, M.; et al. Polyarteritis nodosa in patients with Familial Mediterranean Fever (FMF): A concomitant disease or a feature of FMF? Semin. Arthritis Rheum. 2001, 30, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Tunca, M.; Akar, S.; Onen, F.; Ozdogan, H.; Kasapcopur, O.; Yalcinkaya, F.; Tutar, E.; Ozen, S.; Topaloglu, R.; Yilmaz, E.; et al. Familial Mediterranean Fever (FMF) in Turkey. Medicine 2005, 84, 1–11. [Google Scholar] [CrossRef]
- Ozen, S. The changing face of polyarteritis nodosa and necrotizing vasculitis. Nat. Rev. Rheumatol. 2017, 13, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Yalçınkaya, F.; Özçakar, Z.B.; Kasapçopur, O.; Öztürk, A.; Akar, N.; Bakkaloğlu, A.; Arısoy, N.; Ekı̇m, M.; Özen, S. Prevalence of the MEFV Gene Mutations in Childhood Polyarteritis Nodosa. J. Pediatr. 2007, 151, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Balbir-Gurman, A.; Nahir, A.M.; Braun-Moscovici, Y. Vasculitis in siblings with familial Mediterranean fever: a report of three cases and review of the literature. Clin. Rheumatol. 2006, 26, 1183–1185. [Google Scholar] [CrossRef]
- Ozen, S.; Ben-Chetrit, E.; Bakkaloglu, A.; Gur, H.; Tinaztepe, K.; Calguneri, M.; Turgan, C.; Turkmen, A.; Akpolat, I.; Danaci, M.; et al. Polyarteritis nodosa in patients with Familial Mediterranean Fever (FMF): A concomitant disease or a feature of FMF? Semin. Arthritis Rheum. 2001, 30, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Tunca, M.; Akar, S.; Onen, F.; Ozdogan, H.; Kasapcopur, O.; Yalcinkaya, F.; Tutar, E.; Ozen, S.; Topaloglu, R.; Yilmaz, E.; et al. Familial Mediterranean Fever (FMF) in Turkey. Medicine 2005, 84, 1–11. [Google Scholar] [CrossRef]
- Liu, Y.; Jesus, A.A.; Marrero, B.; Yang, D.; Ramsey, S.E.; Montealegre Sanchez, G.A.; Tenbrock, K.; Wittkowski, H.; Jones, O.Y.; Kuehn, H.S.; et al. Activated STING in a Vascular and Pulmonary Syndrome. N. Engl. J. Med. 2014, 371, 507–518. [Google Scholar] [CrossRef]
- Caratsch, L.; Schnider, C.; Moi, L.; Theodoropoulou, K.; Candotti, F.; Hofer, M. Déficit en adénosine désaminase 2 : une maladie aux présentations multiples. Rev Med Suisse. [CrossRef]
- Elkan, P.N.; Pierce, S.B.; Segel, R.; Walsh, T.; Barash, J.; Padeh, S.; Zlotogorski, A.; Berkun, Y.; Press, J.J.; Mukamel, M.; et al. Mutant Adenosine Deaminase 2 in a Polyarteritis Nodosa Vasculopathy. New Engl. J. Med. 2014, 370, 921–931. [Google Scholar] [CrossRef]
- Zhou, Q.; Yang, D.; Ombrello, A.K.; Zavialov, A.V.; Toro, C.; Zavialov, A.V.; Stone, D.L.; Chae, J.J.; Rosenzweig, S.D.; Bishop, K.; et al. Early-Onset Stroke and Vasculopathy Associated with Mutations in ADA2. New Engl. J. Med. 2014, 370, 911–920. [Google Scholar] [CrossRef]
- Kaljas, Y.; Liu, C.; Skaldin, M.; Wu, C.; Zhou, Q.; Lu, Y.; Aksentijevich, I.; Zavialov, A. Human adenosine deaminases ADA1 and ADA2 bind to different subsets of immune cells. Cell Mol. Life Sci. 2016, 74, 555–570. [Google Scholar] [CrossRef]
- Zavialov, A.V.; Gracia, E.; Glaichenhaus, N.; Franco, R.; Zavialov, A.V.; Lauvau, G. Human adenosine deaminase 2 induces differentiation of monocytes into macrophages and stimulates proliferation of T helper cells and macrophages. J. Leukoc. Biol. 2010, 88, 279–290. [Google Scholar] [CrossRef]
- Zakine, E.; Rodrigues, F.; Papageorgiou, L.; Georgin-Lavialle, S.; Mekinian, A.; Terrier, B.; Kosmider, O.; Hirsch, P.; Jachiet, M.; Bouaziz, J.; et al. Caractéristiques cliniques et histologiques des manifestations cutanées du syndrome VEXAS : une étude rétrospective centralisée de 59 cas. La Rev. de Médecine Interne 2022, 43, A350–A351. [Google Scholar] [CrossRef]
- Beck, D.B.; Ferrada, M.A.; Sikora, K.A.; Ombrello, A.K.; Collins, J.C.; Pei, W.; Balanda, N.; Ross, D.L.; Cardona, D.O.; Wu, Z.; et al. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. New Engl. J. Med. 2020, 383, 2628–2638. [Google Scholar] [CrossRef] [PubMed]
- Tervaert, J.W.C.; Kallenberg, C. NEUROLOGIC MANIFESTATIONS OF SYSTEMIC VASCULITIDES. Rheum. Dis. Clin. North Am. 1993, 19, 913–940. [Google Scholar] [CrossRef]
- Pagnoux, C.; Seror, R.; Henegar, C.; Mahr, A.; Cohen, P.; Le Guern, V.; Bienvenu, B.; Mouthon, L.; Guillevin, L. Clinical features and outcomes in 348 patients with polyarteritis nodosa: A systematic retrospective study of patients diagnosed between 1963 and 2005 and entered into the French vasculitis study group database. Arthritis Rheum. 2010, 62, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Neves, F.S.; Lin, K. Bilateral Foot Drop in Polyarteritis Nodosa. New Engl. J. Med. 2012, 367, e9. [Google Scholar] [CrossRef]
- de Boysson, H.; Guillevin, L. Polyarteritis Nodosa Neurologic Manifestations. Neurol. Clin. 2019, 37, 345–357. [Google Scholar] [CrossRef]
- Provenzale, J.M.; Allen, N.B. Neuroradiologic findings in polyarteritis nodosa. AJNR Am J Neuroradiol 1996, 17, 1119–1126. [Google Scholar] [PubMed]
- Morgan, A.J.; A Schwartz, R. Cutaneous polyarteritis nodosa: a comprehensive review. Int. J. Dermatol. 2010, 49, 750–756. [Google Scholar] [CrossRef]
- Stewart, M.; Lo, A.; Shojania, K.; Au, S.; Seidman, M.A.; Dutz, J.P.; Chan, J. Cutaneous polyarteritis nodosa diagnosis and treatment: A retrospective case series. J. Am. Acad. Dermatol. 2022, 87, 1370–1373. [Google Scholar] [CrossRef] [PubMed]
- Sunderkötter, C.H.; Zelger, B.; Chen, K.; Requena, L.; Piette, W.; Carlson, J.A.; Dutz, J.; Lamprecht, P.; Mahr, A.; Aberer, E.; et al. Nomenclature of Cutaneous Vasculitis. Arthritis Rheumatol. 2018, 70, 171–184. [Google Scholar] [CrossRef]
- El-Reshaid, K.; Kapoor, M.M.; El-Reshaid, W.; Madda, J.P.; Varro, J. The spectrum of renal disease associated with microscopic polyangiitis and classic polyarteritis nodosa in Kuwait. Nephrol. Dial. Transplant. 1997, 12, 1874–1882. [Google Scholar] [CrossRef]
- Maritati, F.; Iannuzzella, F.; Pavia, M.P.; Pasquali, S.; Vaglio, A. Kidney involvement in medium- and large-vessel vasculitis. J. Nephrol. 2016, 29, 495–505. [Google Scholar] [CrossRef]
- Pourafshar, N.; Sobel, E.; Segal, M. A case of isolated renal involvement of polyarteritis nodosa successfully treated with steroid monotherapy. BMJ Case Rep. 2016, 2016. [Google Scholar] [CrossRef]
- Kaur, J.S.; Goldberg, J.P.; Schrier, R.W. Acute Renal Failure Following Arteriography in a Patient With Polyarteritis Nodosa. JAMA 1982, 247, 833–834. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, S.; Yamamoto, Y.; Saita, M.; Hisanaga, S.; Morita, S.; Tanaka, K.; Sumiyoshi, A.; Koono, M. [Renal failure associated with polyarteritis nodosa]. Nihon Jinzo Gakkai Shi 1990, 32, 739–44. [Google Scholar] [PubMed]
- Launay, D.; Michon-Pasturel, U.; Boumbar, Y.; Dubrulle, F.; Bouroz-Joly, J.; Hachulla, E.; Lemaitre, L.; Devulder, B. Hématome périrénal spontané bilatéral: une complication rare de la panartérite noueuse. La Rev. de Médecine Interne 1998, 19, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Nandwani, G.M.; Musker, M.P.; Chaplin, B.J.; El Madhoun, I.; Akbani, H. Spontaneous perirenal haemorrhage in polyarteritis nodosa. J Coll Physicians Surg--Pak JCPSP 2013, 23, 445–7. [Google Scholar]
- Miyagawa, T.; Iwata, Y.; Oshima, M.; Ogura, H.; Sato, K.; Nakagawa, S.; Yamamura, Y.; Kitajima, S.; Toyama, T.; Hara, A.; et al. Polyarteritis nodosa with perirenal hematoma due to the rupture of a renal artery aneurysm. CEN Case Rep. 2021, 10, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.W.; Waybill, P.N.; Singh, H.; Brown, D.B. Polyarteritis Nodosa Presenting as Spontaneous Perirenal Hemorrhage: Angiographic Diagnosis and Treatment with Microcoil Embolization. J. Vasc. Interv. Radiol. 1999, 10, 1361–1363. [Google Scholar] [CrossRef]
- Levine, S.M.; Hellmann, D.B.; Stone, J.H. Gastrointestinal involvement in polyarteritis nodosa (1986–2000):: Presentation and outcomes in 24 patients. Am. J. Med. 2002, 112, 386–391. [Google Scholar] [CrossRef]
- Castelhano, R.; Win, K.M.; Carty, S. Celiac artery aneurysm causing an acute abdomen. BMJ Case Rep. 2021, 14, e240533. [Google Scholar] [CrossRef]
- Gendreau, S.; Porcher, R.; Thoreau, B.; Paule, R.; Maurier, F.; Goulenok, T.; Frumholtz, L.; Bernigaud, C.; Ingen-Housz-Oro, S.; Mekinian, A.; et al. Characteristics and risk factors for poor outcome in patients with systemic vasculitis involving the gastrointestinal tract. Semin. Arthritis Rheum. 2021, 51, 436–441. [Google Scholar] [CrossRef]
- Waisayarat, J.; Niyasom, C.; Vilaiyuk, S.; Molagool, S. Polyarteritis Nodosa with Cytomegalovirus Enteritis and Jejunoileal Perforation: Report of a Case with a Literature Review. Vasc. Heal. Risk Manag. 2022, ume 18, 595–601. [Google Scholar] [CrossRef]
- Dillard, B.M.; Black, W.C. Polyarteritis nodosa of the gallbladder and bile ducts. Am Surg 1970, 36, 423–7. [Google Scholar] [PubMed]
- Ito, M.; Sano, K.; Inaba, H.; Hotchi, M. Localized necrotizing arteritis. A report of two cases involving the gallbladder and pancreas. Arch Pathol Lab Med 1991, 115, 780–3. [Google Scholar]
- Empen, K.; Jung, M.-C.; Engelhardt, D.; Sackmann, M. Successful treatment of acute liver failure due to polyarteritis nodosa. Am. J. Med. 2002, 113, 349–351. [Google Scholar] [CrossRef] [PubMed]
- Herskowitz, M.M.; Flyer, M.A.; Sclafani, S.J.A. Percutaneous transhepatic coil embolization of a ruptured intrahepatic aneurysm in polyarteritis nodosa. Cardiovasc. Interv. Radiol. 1993, 16, 254–256. [Google Scholar] [CrossRef] [PubMed]
- Parent, B.A.; Cho, S.W.; Buck, D.G.; Nalesnik, M.A.; Gamblin, T.C. Spontaneous Rupture of Hepatic Artery Aneurysm Associated with Polyarteritis Nodosa. Am. Surg. 2010, 76, 1416–1419. [Google Scholar] [CrossRef] [PubMed]
- Roberto, M.; Meytes, V.; Liu, S. Ruptured hepatic aneurysm as first presenting symptom of polyarteritis nodosa. Oxf. Med Case Rep. 2018, 2018, 64–67. [Google Scholar] [CrossRef]
- Stambo, G.W.; Guiney, M.J.; Cannella, X.F.; Germain, B.F. Coil embolization of multiple hepatic artery aneurysms in a patient with undiagnosed polyarteritis nodosa. J. Vasc. Surg. 2004, 39, 1122–1124. [Google Scholar] [CrossRef]
- Guillevin, L.; Lhote, F.; Gallais, V.; Jarrousse, B.; Royer, I.; Gayraud, M.; Benichou, J. Gastrointestinal tract involvement in polyarteritis nodosa and Churg-Strauss syndrome. Ann Med Interne (Paris) 1995, 146, 260–7. [Google Scholar]
- Cacoub, P.; Guillevin, L.; Godeau, P. [Causes of death in systemic vasculitis of polyarteritis nodosa. Analysis of a series of 165 patients].. 1988, 139, 381–90. [Google Scholar]
- Teichman, J.M.; Mattrey, R.F.; Demby, A.M.; Schmidt, J.D. Polyarteritis Nodosa Presenting as Acute Orchitis: A Case Report and Review of the Literature. J. Urol. 1993, 149, 1139–1140. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Matsubara, S.; Yagi, K.; Kinoshita, K.; Fukunaga, T.; Yamamoto, A.; Uno, M. Intra-abdominal hemorrhage due to segmental arterial mediolysis of an ovarian artery pseudoaneurysm and concomitant aneurysmal subarachnoid hemorrhage: illustrative case. J. Neurosurgery: Case Lessons 2022, 4. [Google Scholar] [CrossRef]
- Kastner, D.; Gaffney, M.; Tak, T. Polyarteritis nodosa and myocardial infarction. Can J Cardiol 2000, 16, 515–8. [Google Scholar] [PubMed]
- Bae, Y.D.; Choi, H.J.; Lee, J.C.; Park, J.J.; Lee, Y.J.; Lee, E.B.; Song, Y.W. Clinical Features of Polyarteritis Nodosa in Korea. J. Korean Med Sci. 2006, 21, 591–595. [Google Scholar] [CrossRef]
- Schrader, M.L.; Hochman, J.S.; Bulkley, B.H. The heart in polyarteritis nodosa: A clinicopathologic study. Am. Hear. J. 1985, 109, 1353–1359. [Google Scholar] [CrossRef]
- Blétry, O.; Godeau, P.; Charpentier, G.; Guillevin, L.; Herreman, G. [Cardiac manifestations of periarteritis nodosa. Incidence of non-hypertensive cardiomyopathy].. 1980, 73, 1027–36. [Google Scholar]
- Lai, J.; Zhao, L.; Zhong, H.; Zhou, J.; Guo, X.; Xu, D.; Tian, X.; Zhang, S.; Zeng, X. Characteristics and Outcomes of Coronary Artery Involvement in Polyarteritis Nodosa. Can. J. Cardiol. 2020, 37, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, D.P.; Moyssakis, I.; Votteas, V.E. Polyarteritis nodosa and hypertrophic obstructive cardiomyopathy. A true association? Clin. Rheumatol. 2004, 23, 57–58. [Google Scholar] [CrossRef]
- Schafigh, M.; Bakhtiary, F.; Kolck, U.W.; Zimmer, S.; Greschus, S.; Silaschi, M. Coronary Artery Aneurysm Rupture in a Patient With Polyarteritis Nodosa. JACC: Case Rep. 2022, 4, 1522–1528. [Google Scholar] [CrossRef]
- Reimold, E.W.; Weinberg, A.G.; Fink, C.W.; Battles, N.D. Polyarteritis in Children. Arch. Pediatr. Adolesc. Med. 1976, 130, 534–541. [Google Scholar] [CrossRef]
- Holt, S.; Jackson, P. Ruptured coronary aneurysm and valvulitis in an infant with polyarthritis nodosa. J. Pathol. 1975, 117, 83–87. [Google Scholar] [CrossRef]
- Iino, T.; Eguchi, K.; Sakai, M.; Nagataki, S.; Ishijima, M.; Toriyama, K. Polyarteritis nodosa with aortic dissection: necrotizing vasculitis of the vasa vasorum. J Rheumatol 1992, 19, 1632–6. [Google Scholar] [PubMed]
- Shukla, A.; Aggarwal, A. Polyarteritis nodosa presenting as peripheral vascular disease and acute limb ischemia. J. Postgrad. Med. 2017, 63, 47–49. [Google Scholar] [CrossRef] [PubMed]
- Holscher, C.M.; Stonko, D.P.; Weaver, M.L.; Reifsnyder, T. Successful bilateral popliteal-plantar bypasses for polyarteritis nodosa induced ischemia. J. Vasc. Surg. Cases Innov. Tech. 2020, 7, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Zahoor, S.; Siddique, S.; Mahboob, H.M. An unusual presentation of polyarteritis nodosa: A case report. Reumatol Clin 2022, 18, 124–126. [Google Scholar] [CrossRef] [PubMed]
- Akova, Y.A.; Jabbur, N.S.; Foster, C.S. Ocular Presentation of Polyarteritis Nodosa. Ophthalmology 1993, 100, 1775–1781. [Google Scholar] [CrossRef] [PubMed]
- Miloslavsky, E.; Unizony, S. The Heart in Vasculitis. Rheum. Dis. Clin. North Am. 2014, 40, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.T.; Kerrison, J.B.; Miller, N.R.; Goldberg, M.F. CHOROIDAL INFARCTION, ANTERIOR ISCHEMIC OPTIC NEUROPATHY, AND CENTRAL RETINAL ARTERY OCCLUSION FROM POLYARTERITIS NODOSA. Retina 2001, 21, 348–351. [Google Scholar] [CrossRef]
- Matsumoto, T.; Homma, S.; Okada, M.; Kuwabara, N.; Kira, S.; Hoshi, T.; Uekusa, T.; Saiki, S. The lung in polyarteritis nodosa: A pathologic study of 10 cases. Hum. Pathol. 1993, 24, 717–724. [Google Scholar] [CrossRef]
- Fort, J.G.; Griffin, R.; Tahmoush, A.; Abruzzo, J.L. Muscle involvement in polyarteritis nodosa: report of a patient presenting clinically as polymyositis and review of the literature. J Rheumatol 1994, 21, 945–8. [Google Scholar]
- Georgin-Lavialle, S.; Terrier, B.; Guedon, A.; Heiblig, M.; Comont, T.; Lazaro, E.; Lacombe, V.; Terriou, L.; Ardois, S.; Bouaziz, J.; et al. Further characterization of clinical and laboratory features in VEXAS syndrome: large-scale analysis of a multicentre case series of 116 French patients*. Br. J. Dermatol. 2021, 186, 564–574. [Google Scholar] [CrossRef]
- Grambow-Velilla, J.; Braun, T.; Pop, G.; Louzoun, A.; Soussan, M. Aortitis PET Imaging in VEXAS Syndrome. Clin. Nucl. Med. 2022, 48, e67–e68. [Google Scholar] [CrossRef]
- Watanabe, R.; Kiji, M.; Hashimoto, M. Vasculitis associated with VEXAS syndrome: A literature review. Front. Med. 2022, 9, 983939. [Google Scholar] [CrossRef]
- Meyts, I.; Aksentijevich, I. Deficiency of Adenosine Deaminase 2 (DADA2): Updates on the Phenotype, Genetics, Pathogenesis, and Treatment. J. Clin. Immunol. 2018, 38, 569–578. [Google Scholar] [CrossRef]
- Fayand, A.; Sarrabay, G.; Belot, A.; Hentgen, V.; Kone-Paut, I.; Grateau, G.; Melki, I.; Georgin-Lavialle, S. Les multiples facettes du déficit en ADA2, vascularite, maladie auto-inflammatoire et immunodéficit : mise au point à partir des 135 cas de la littérature. La Rev. de Médecine Interne 2018, 39, 297–306. [Google Scholar] [CrossRef]
- Chung, S.A.; Gorelik, M.; Langford, C.A.; Maz, M.; Abril, A.; Guyatt, G.; Archer, A.M.; Conn, D.L.; Full, K.A.; Grayson, P.C.; et al. 2021 American College of Rheumatology/Vasculitis Foundation Guideline for the Management of Polyarteritis Nodosa. Arthritis Rheumatol. 2021, 73, 1384–1393. [Google Scholar] [CrossRef] [PubMed]
- Terrier, B.; Darbon, R.; Durel, C.-A.; Hachulla, E.; Karras, A.; Maillard, H.; Papo, T.; Puechal, X.; Pugnet, G.; Quemeneur, T.; et al. French recommendations for the management of systemic necrotizing vasculitides (polyarteritis nodosa and ANCA-associated vasculitides). Orphanet J. Rare Dis. 2020, 15, 1–44. [Google Scholar] [CrossRef] [PubMed]
- Ribi, C.; Cohen, P.; Pagnoux, C.; Mahr, A.; Arène, J.; Puéchal, X.; Carli, P.; Kyndt, X.; Le Hello, C.; Letellier, P.; et al. Treatment of polyarteritis nodosa and microscopic polyangiitis without poor-prognosis factors: A prospective randomized study of one hundred twenty-four patients. Arthritis Rheum. 2010, 62, 1186–1197. [Google Scholar] [CrossRef]
- Samson, M.; Puéchal, X.; Mouthon, L.; Devilliers, H.; Cohen, P.; Bienvenu, B.; Ly, K.H.; Bruet, A.; Gilson, B.; Ruivard, M.; et al. Microscopic polyangiitis and non-HBV polyarteritis nodosa with poor-prognosis factors: 10-year results of the prospective CHUSPAN trial. Clin Exp Rheumatol 2017, 176–184. [Google Scholar]
- Hadjadj, J.; Canzian, A.; Karadag, O.; Contis, A.; Maurier, F.; Sanges, S.; Sartorelli, S.; Denis, L.; de Moreuil, C.; Durel, C.-A.; et al. Use of biologics to treat relapsing and/or refractory polyarteritis nodosa: data from a European collaborative study. Rheumatology 2022, 62, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Guillevin, L.; Fain, O.; Lhote, F.; Jarrousse, B.; Huong, D.L.T.; Bussel, A.; Leon, A. Lack of Superiority of Steroids Plus Plasma Exchange to Steroids Alone in the Treatment of Polyarteritis Nodosa and Churg-Strauss Syndrome. Arthritis Rheum. 1992, 35, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Machet, L.; Vincent, O.; Machet, M.C.; Barruet, K.; Vaillant, L.; Lorette, G. [Cutaneous periarteritis nodosa resistant to combined corticosteroids and immunosuppressive agents. Efficacy of treatment with intravenous immunoglobulins]. Ann Dermatol Venereol 1995, 122, 769–72. [Google Scholar] [PubMed]
- Seri, Y.; Shoda, H.; Hanata, N.; Nagafuchi, Y.; Sumitomo, S.; Fujio, K.; Yamamoto, K. A case of refractory polyarteritis nodosa successfully treated with rituximab. Mod. Rheumatol. 2015, 27, 696–698. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, E.; Cressend, T.; Duffau, P.; Grenouillet-Delacre, M.; Rouanet-Larivière, M.; Vital, A.; Longy-Boursier, M.; Mercié, P. Rituximab Efficacy during a Refractory Polyarteritis Nodosa Flare. Case Rep. Med. 2009, 2009, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Al-Homood, I.A.; Aljahlan, M.A. Successful use of combined corticosteroids and rituximab in a patient with refractory cutaneous polyarteritis nodosa. J. Dermatol. Dermatol. Surg. 2017, 21, 24–26. [Google Scholar] [CrossRef]
- Boistault, M.; Corbeto, M.L.; Quartier, P.; Arcobé, L.B.; Durall, A.C.; Aeschlimann, F.A. A young girl with severe polyarteritis nodosa successfully treated with tocilizumab: a case report. Pediatr. Rheumatol. 2021, 19, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Mekinian, A.; Grignano, E.; Braun, T.; Decaux, O.; Liozon, E.; Costedoat-Chalumeau, N.; Kahn, J.-E.; Hamidou, M.; Park, S.; Puéchal, X.; et al. Systemic inflammatory and autoimmune manifestations associated with myelodysplastic syndromes and chronic myelomonocytic leukaemia: a French multicentre retrospective study. Rheumatology 2015, 55, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.A.; Gorelik, M.; Langford, C.A.; Maz, M.; Abril, A.; Guyatt, G.; Archer, A.M.; Conn, D.L.; Full, K.A.; Grayson, P.C.; et al. 2021 American College of Rheumatology/Vasculitis Foundation Guideline for the Management of Polyarteritis Nodosa. Arthritis Rheumatol. 2021, 73, 1384–1393. [Google Scholar] [CrossRef]
- Boyadzhieva, Z.; Ruffer, N.; Kötter, I.; Krusche, M. How to treat VEXAS syndrome: a systematic review on effectiveness and safety of current treatment strategies. Rheumatology 2023, 62, 3518–3525. [Google Scholar] [CrossRef]
- Diarra A, Duployez N, Fournier E, Preudhomme C, Coiteux V, Magro L, Quesnel B, Heiblig M, Sujobert P, Barraco F, Balsat M, Scanvion Q, Hachulla E, Launay D, Yakoub-Agha I, Terriou L. Successful allogeneic hematopoietic stem cell transplantation in patients with VEXAS syndrome: a 2-center experience. Blood Adv. 2022 Feb 8;6(3):998-1003. [CrossRef]
- Comont, T.; Heiblig, M.; Rivière, E.; Terriou, L.; Rossignol, J.; Bouscary, D.; Rieu, V.; Le Guenno, G.; Mathian, A.; Aouba, A.; et al. Azacitidine for patients with Vacuoles, E1 Enzyme, X-linked, Autoinflammatory, Somatic syndrome (VEXAS) and myelodysplastic syndrome: data from the French VEXAS registry. Br. J. Haematol. 2021, 196, 969–974. [Google Scholar] [CrossRef]
Figure 1.
Comparison of various etiologies of PAN across different cohorts over distinct time periods.
Figure 1.
Comparison of various etiologies of PAN across different cohorts over distinct time periods.
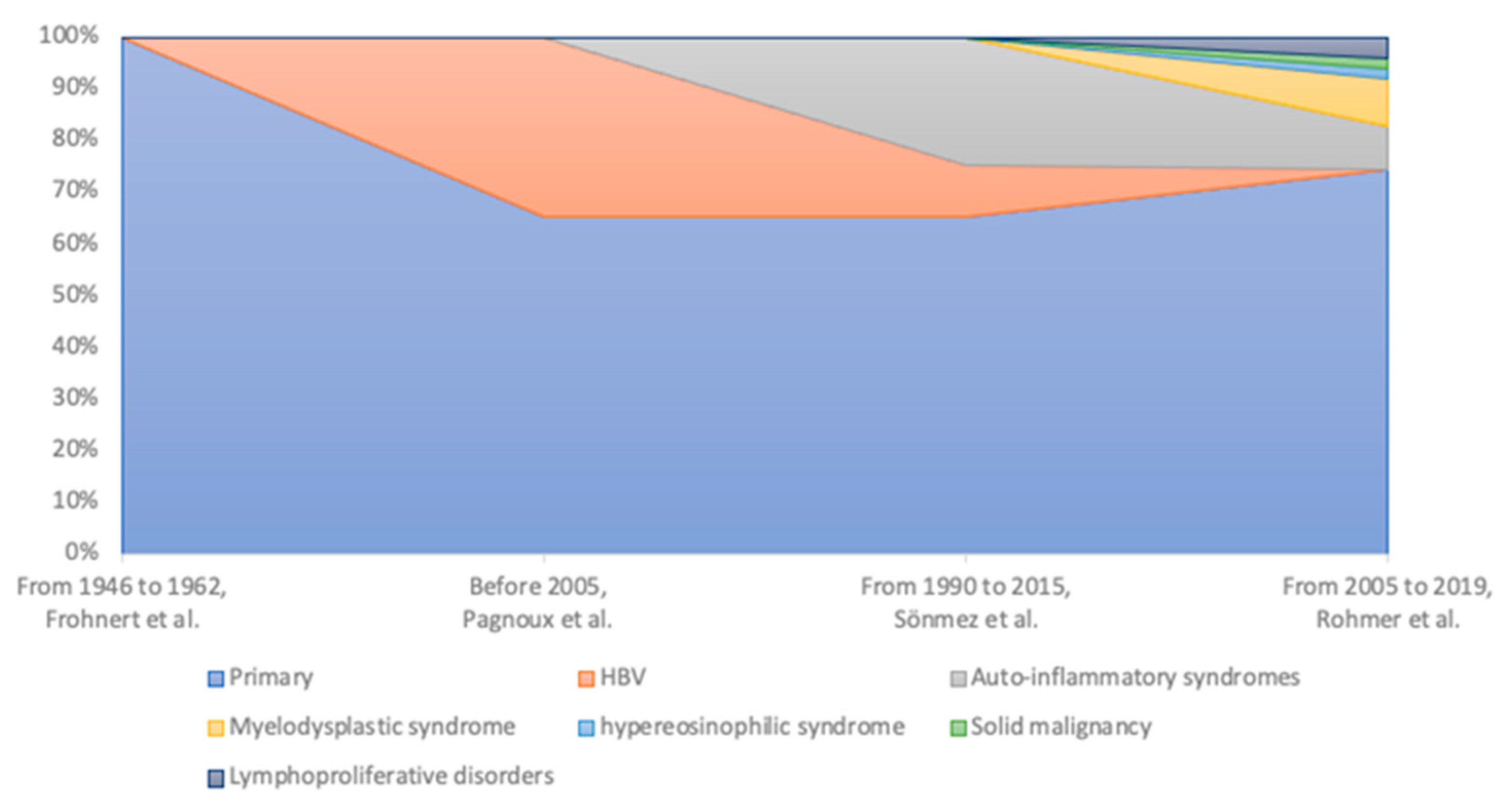

Table 1.
Characteristics of PAN patients reported in different cohorts (PNP: peripheral neuropathy, CNS : central nervous system, FFS : Five Factor Score, 1996). Results are expressed in percentage.
Table 1.
Characteristics of PAN patients reported in different cohorts (PNP: peripheral neuropathy, CNS : central nervous system, FFS : Five Factor Score, 1996). Results are expressed in percentage.
| Characteristics |
Pagnoux et al. (1963 to 2005) |
Sönmez et al (1990 to 2015) |
Rohmer et al. (2005 to 2019) |
Georgin- Lavialle et al. (VEXAS) |
Meyts et al. (ADA2) |
|---|---|---|---|---|---|
|
General symptoms Fever Loss of weight Myalgia |
93.1 63.8 69.5 58.6 |
53.7 53.7 46.2 |
85 54 50 50 |
95.7 64.6 54.5 |
50 |
|
All cutaneous Nodules Purpura Livedo Panniculitis |
49.7 17.2 22.1 16.7 |
67.2 17.9 |
59 7.5 |
83.6 12.9 |
75 14 50 |
|
Renal Hematuria Proteinuria Hypertension |
50.6 15.2 21.6 34.8 |
47.7 41.7 |
20 | 9.5 |
21 |
| Orchitis | 17 | 14.9 | 16 | 4 | |
|
Neurologic PNP Mononeuritis CNS |
79.0 74.1 70.7 4.6 |
43.2 | 59 |
5.2 2.6 |
9 53 |
|
Digestive Abdominal pain Bleeding Perforation |
37.9 35.6 3.4 4.3 |
22.3 37.3 |
28 | 13.8 8.6 0.9 0.9 |
33 12 2 |
|
Cardiovascular Pericarditis Distal necrosis Thrombo-embolism |
22.4 5.5 6.3 |
13.5 |
39 |
4.3 35.3 |
22 |
|
Ophtalmic Retinal vasculitis |
8.6 4.3 |
40.5 | |||
|
Pulmonary Cough Lung infiltrate Pleural effusion |
5.7 3.4 3.4 |
2.9 | 8 | 49.1 40.5 9.5 |
|
| Chondritis | 36.2 | ||||
|
Arthralgia Arthritis |
58.2 17.9 |
28.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



