
Submitted:
12 October 2023
Posted:
17 October 2023
You are already at the latest version
Abstract
Immune checkpoint inhibitors (ICIs), including anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and anti-programmed death-1 (PD-1) antibodies, have initiated a new era in the treatment of malignant melanoma. ICIs can be used in various settings, including as first-line, adjuvant, and neoadjuvant therapy. In the scope of this review, we examined clinical studies utilizing ICIs in the context of treating oral mucosal melanoma, a rare disease albeit with an extremely poor prognosis, with a specific focus on unraveling the intricate web of resistance mechanisms. The absence of a comprehensive review focusing on ICIs in oral mucosal melanoma is notable. Therefore, this review seeks to address this deficiency by offering a novel and thorough analysis of the current status, potential resistance mechanisms, and future prospects of applying ICIs specifically to oral malignant melanoma. Clarifying and thoroughly understanding these mechanisms will facilitate the advancement of effective therapeutic approaches and enhance the prospects for patients suffering from oral mucosal melanoma.
Keywords:
immune checkpoint inhibitor
; resistance mechanism
; melanoma
; oral mucosal melanoma
; immunotherapy
; immune checkpoint blockade
; anti-PD-1
; anti-CTLA-4
1. Introduction
Oral melanoma is an uncommon cancer that is characterized by aggressive progression. It accounts for only 0.2–8% of all melanomas and 1–2% of all oral carcinomas [1]. The cause of oral melanoma and the risk factors contributing to the malignant transformation of cells remain unclear. Mechanical irritation due to prostheses, tobacco use, and infection have been proposed as potential contributors to the onset of oral mucosal melanoma [2].
Oral melanoma holds significant clinical relevance because it is correlated with a higher mortality rate than cutaneous melanoma. This is due to the fact that it is often not diagnosed or misdiagnosed in the early stages, and when diagnosed, the disease has often already invaded the surrounding tissues [3,4,5]. Consequently, the anatomical features limit surgical intervention, making it more complicated [6]. This underscores the significance of exploring and implementing effective systemic therapies while encouraging continued research in this field. Furthermore, compared with skin melanoma, fewer treatment options are available for oral melanoma. Therefore, the development of efficient immunotherapy for oral melanoma is vital for improving patient outcomes.
Surgical resection is the standard therapy for treating patients with melanoma. In situations where the lymph node status has the potential to influence treatment planning or the ability to participate in clinical trials, it is advisable to consider sentinel lymph node (SN) biopsy for accessible sinonasal or oral mucosal melanomas [7]. However, it is not desirable that routine complete lymph node dissection, i.e., completion neck dissection, be performed for patients with SN-positive oral melanoma [7,8].
Radiotherapy received after the surgery reduces the possibility of local recurrence [9]. Although certain guidelines suggest photon-based intensity-modulated radiotherapy after surgical operation in patients with head and neck mucosal melanoma, its effectiveness in addressing distant metastasis is limited [7,9]. Thus, achieving systemic disease control is of paramount importance, particularly for patients with a heightened likelihood of metastasis.
Dacarbazine has held a notable place on cancer treatment, serving as a commonly used initial chemotherapy option in the management of metastatic melanoma. It exhibits an overall response rate of 13.4%, and the median survival duration varies from 5.6 to 11 months [10]. Nonetheless, owing to its limited efficacy, researchers have been driven to investigate more effective treatment modalities, particularly for cases with unresectable and metastatic melanoma where surgery is not a viable option. For many years, dacarbazine has held the status of the standard of therapy, but since 2011, the Food and Drug Administration (FDA)-approved use of immune checkpoint inhibitors (ICIs) and small-molecule inhibitors has brought about substantial changes in the standard treatment approach [11].
The discovery of the BRAFV600E mutation was a milestone in the development of targeted and more personalized approaches to melanoma [12]. As a first-line treatment option, targeted therapies such as BRAF and MEK inhibitors are highly efficient, especially in combination [13].
Despite the great potential of targeted therapies for melanoma treatment, they have some limitations—only patients with targetable gene mutations are suitable candidates for therapy. Moreover, melanoma treatment that targets a single mutation tends to result in resistance [14,15]. Therefore, additional therapy is required.
Remodeling the immune system to leverage the host’s immune defenses against cancer cells has been an appealing concept for years, and curated knowledge of the immune system has created opportunities for the development of various immunotherapies.
Interleukin-2 (IL-2) administration represents the first effective immunotherapy for patients with melanoma [16]. A high dose of IL-2 induces a durable anti-tumor response, especially in advanced renal cell carcinoma and melanoma, but severe toxicity is the main obstacle to successful treatment [17]. However, immunotherapy did not meet expectations until the breakthrough discovery of immune checkpoint blockade. The groundbreaking research conducted by Honjo et al. led to the discovery of programmed death-1′s (PD-1) role in immune regulation in 1992 [18]. PD-1 is predominantly found on T-cell surfaces, whereas programmed death ligand-1 (PD-L1) is present on many cell types, including cancer cells [19]. When PD-1 on T cells binds to PD-L1 on cancer cells, it can lead to the suppression of the immune response, enabling cancer cells to avoid recognition and elimination by the immune system [19]. Thus, blocking PD-1 or PD-L1 has taken center stage in the realm of immunotherapy [20]. Targeting PD-1 and PD-L1 showed good anti-tumor activity along with less toxicity than IL-2 therapy [21]. PD-1 blockade also has a long-term therapeutic effect compared with other cancer therapies that target cancer cells, as it targets T cells instead of cancer cells, which constitute a heterogenous population [22].
Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) is another immune checkpoint protein first discovered by Brunet et al. in 1987 [23]. To ensure an efficient and well-regulated immune response, complete T cell activation necessitates both TCR engagement and the presence of co-stimulatory signals. Among these signals, CD28-mediated co-stimulation, whereby the CD28 on T cells binds to CD80/CD86 ligands on antigen-presenting cells (APCs), is important. However, CTLA-4 on T cells engages in competition with CD28 to bind to CD80/CD86, resulting in the inhibition of T cell activation. Therefore, blocking CTLA-4 activation has surfaced as an innovative approach within the field of immunotherapy [24]. After several clinical trials proved the effectiveness of ipilimumab, a monoclonal antibody that targets CTLA-4, the FDA authorized the inclusion of this antibody in the treatment of patients with unresectable or metastatic melanoma [25,26,27].
Currently, ICIs and small-molecule inhibitors play significant roles in melanoma treatment. The American Society of Clinical Oncology (ASCO) guidelines recommend nivolumab or pembrolizumab as adjuvant systemic therapy for patients with resected stage IIIA/B/C/D melanoma harboring wild-type BRAF. For BRAF-mutant patients, in addition to these options, dabrafenib plus trametinib is also recommended. Based on the Checkmate 238 trial, nivolumab has been suggested as adjuvant therapy for patients with resected stage IV melanoma.
For patients with unresectable or metastatic melanoma harboring wild-type BRAF, ipilimumab plus nivolumab followed by nivolumab or pembrolizumab or nivolumab, are recommended. For patients with BRAF mutations, a combination of BRAF/MEK inhibitor therapy is recommended. If the disease continues to progress after first-line anti-PD1 therapy, ipilimumab-containing regimens or therapeutic approaches that combine BRAF and MEK inhibitors are recommended based on the mutation status. Although primarily intended for patients with cutaneous melanoma, the guidelines state that these treatment regimens can also be applied to unresectable or metastatic mucosal melanoma [28]. Despite its reduced effectiveness in mucosal melanomas compared to cutaneous melanomas, immune checkpoint blockade remains a valuable treatment option [29].
The United Kingdom National Guidelines for Head and Neck Melanoma recommend anti-PD1 and anti-CTLA4 combination therapy for advanced and metastatic melanoma. If the patient is unsuitable for combination treatment, either nivolumab or pembrolizumab monotherapy is suggested. Depending on the mutation status, either BRAF or c-KIT inhibitors are recommended. In the case that immunotherapy and targeted therapy are not viable choices or resistance occurs, then chemotherapy may be a suitable alternative according to these guidelines [7].
Despite being a groundbreaking treatment approach in melanoma therapy, ICIs exhibit significant limitations, including immune-related adverse events (irAEs) and, most importantly, therapy resistance, which continues to stand as a crucial barrier to achieving successful outcomes. However, enhancing our comprehension of resistance mechanisms makes it possible to design treatments that optimize the benefits of ICIs. In the landscape of immunotherapy research, numerous studies have explored the application of ICIs across various malignancies. However, a notable gap exists in assessing their efficacy and potential in the context of oral malignant melanoma (OMM). To the best of our knowledge, the current literature also lacks a comprehensive review of the status of ICIs in OMM. Therefore, this review gives a comprehensive overview of the current status of ICIs in the treatment of oral mucosal melanoma as well as the molecular mechanisms of resistance to ICIs, which is the major obstacle to their effectiveness in melanoma treatment.
2. ICI therapy for OMM
In 1980, Umeda and Shimada proposed a successful treatment regimen for stage 1 and 2 oral melanomas. This protocol involves: 1) performing intraoral surgery to excise the primary lesion; 2) therapeutic radical neck dissection in cases with neck lymph node metastases; and 3) DAV and OK-432 as adjuvant therapy [30]. This approach, incorporating surgical treatment and dacarbazine-based chemotherapy, is considered the standard therapy for patients with stage 1 or 2 oral melanoma.
Another adjuvant therapy is high-dose interferon-α2b (HDI). In clinical trials, while HDI treatment group showed a prolonged relapse-free survival rate (RFS), a significant difference was not detected in terms of overall survival (OS) between patients diagnosed with stage-III oral mucosal melanoma who were treated with chemotherapy and those who received HDI after chemotherapy as adjuvant therapy. Nevertheless, it has been suggested that for patients with OMM, especially those in stage IVa who do not respond to chemotherapy, HDI can serve as an effective adjuvant therapy [31]. However, routine use of HDI is not within the standard adjuvant therapy recommendations according to ASCO guideline [28].
Clinical trials have been carried out to evaluate the effectiveness of specific small-molecule inhibitors in an adjuvant setting for melanoma. Patients with completely resected stage III cutaneous melanoma harboring BRAF V600E or V600K mutations showed improved RFS and OS, along with a decreased chance of relapse, when they received dabrafenib plus trametinib as adjuvant treatment [32]. Following the outcomes of the COMBI-AD clinical trial (NCT01682083), dabrafenib plus trametinib received FDA approval in 2018 for adjuvant treatment of melanoma patients harboring BRAF V600E or V600K mutations [33]. However, although 50–60% of patients with cutaneous melanoma have a BRAF mutation, this rate was found to be only 3.5% among 57 patients with OMM in a previous study [34].
Conversely, KIT mutations have been identified as more prevalent than BRAF mutations in OMM [35]. To date, there have been no clinical trials exploring the use of KIT inhibitors as adjuvant therapy for OMM. If such trials are to be conducted, it would be essential to consider that the potential benefits of imatinib, a KIT inhibitor, could vary depending on the specific exon in which the mutation is present [14]. Careful examination and understanding of the mutation location are crucial for determining the likelihood of patients gaining benefits from this treatment approach.
The favorable outcomes achieved with anti-PD-1 and PD-L1 treatment in patients with metastatic and unresectable melanoma have led to consideration of their potential use as adjuvant therapy. Pembrolizumab was approved by the FDA for use as adjuvant treatment for stage IIIA (>1 mm lymph node metastasis), IIIB, or IIIC cutaneous melanoma after surgery in 2019 based on the KEYNOTE-054 pivotal trial [36,37]. Furthermore, in 2021, it received FDA approval for the adjuvant treatment for patients aged 12 years and older with stage IIB or IIC melanoma following complete resection, based on the KEYNOTE-716 (NCT03553836) trial [38]. However, pembrolizumab, nivolumab, or combination therapy with dabrafenib and trametinib are not recommended as adjuvant therapy for stage II melanoma patients in the ASCO guidelines [28].
Studies have shown that patients with nodular-type oral mucosal melanoma who were treated with chemotherapy—dacarbazine and cisplatin—plus anti-PD-1 agents as adjuvant therapy showed improved 2-year OS and progression-free survival (PFS), along with less cytotoxic effects, while decreasing the likelihood of melanoma recurrence in the oral and distant regions compared to patients who received chemotherapy alone or chemotherapy plus high-dose interferon-α2b(HDI) [39].
In a double-blind phase III trial (EORTC 18071) involving patients with stage III cutaneous melanoma, researchers found that adjuvant treatment with intravenous infusions of ipilimumab at a dosage of 10 mg/kg, given every 3 weeks for four doses initially, followed by its administration every 3 months for a maximum duration of 3 years after complete resection led to a noteworthy improvement in RFS. In light of the findings from this study, the FDA authorized the use of ipilimumab as adjuvant treatment for high-risk stage III melanoma after complete resection. However, a noteworthy percentage of the patient cohort (245 out of 471) experienced side effects that resulted in the discontinuation of treatment [40,41]. Due to the high cost and severe toxicity associated with the use of ipilimumab as adjuvant therapy, its use is not recommended in adjuvant settings [42,43].
In a clinical trial comparing adjuvant nivolumab and ipilimumab in patients with resected stage III or IV melanoma, the 18-month RFS in patients treated with nivolumab was 66.4%, whereas that in patients treated with ipilimumab alone was 52.7%. Additionally, the ipilimumab treatment group demonstrated a drug-related death rate of 0.4%, whereas there were no recorded fatalities attributed to drug-related issues in the nivolumab alone group. Moreover, drug-related grade 3 or 4 side effects were observed in 45.9% of patients in the ipilimumab group, whereas only 14.4% of patients in the nivolumab group experienced such side effects. These results indicated that nivolumab was safer than ipilimumab [44]. However, the scarcity of mucosal melanoma cases, particularly OMM, has resulted in a restricted number of clinical trials exploring the utilization of ICIs in adjuvant therapy.
When surgery is not indicated, such as in patients with unresectable or metastatic melanoma, targeted therapy or immunotherapy have emerged as the preferred initial treatment options because the efficacy of chemotherapy in terms of OS is notably limited [45]. BRAF inhibitors, particularly in combination with MEK inhibitors, have proven to be highly efficient in patients with cutaneous melanoma. However, these mutations are seldom found in mucosal melanoma, making them infrequently considered a viable treatment for patients with mucosal melanoma [46]. In cases where a targetable mutation exists in the patient, targeted therapy may be a potential candidate as the first-line treatment.
A patient with OMM harboring a KIT mutation underwent targeted therapy for this mutation with a KIT inhibitor, and no signs of recurrence were detected within a 41-month period [47]. Moreover, in patients with metastatic OMM with the KIT mutation, it was reported that treatment with imatinib extended the OS compared to conventional chemotherapy. However, of the 12 patients, five died due to treatment resistance [14]. Thus, the inherent heterogeneity of melanoma necessitates a multifaceted approach, targeting not only the individual mutations but also various mutation patterns or pathways in combination to enhance the response rate to treatment.
ICIs are of paramount importance in melanoma treatment, especially when patients lack a targetable mutation. In a pooled data analysis, patients who received nivolumab alone experienced a median PFS of 3.0 months for mucosal melanoma and 6.2 months for cutaneous melanoma, with objective response rates (ORR) of 23.3% and 40.9%, respectively. However, the most significant outcome was observed in patients who received nivolumab and ipilimumab in combination, where the median PFS increased to 5.9 months for mucosal melanoma and 11.7 months for cutaneous melanoma, with increased ORRs of 37.1% and 60.4%, respectively [48]. This suggests that combination therapy has more significant and favorable outcomes in terms of response rates and PFS in patients with either type of melanoma. However, it should be noted that the combination treatment also resulted in higher levels of toxicity.
Oral amelanotic melanoma, a subtype of oral melanoma without pigmentation, is an infrequent type of melanoma; however, it constitutes 75% of oral melanoma cases [49]. The absence of typical melanin pigmentation can make the diagnosis more challenging, further delaying timely and appropriate treatment [50]. Thus, oral amelanotic melanomas have a poorer prognosis than melanomas with pigmentation.
In a patient with metastatic oral amelanotic melanoma stage IVc with negative PD-1 levels, it was observed that combination therapy with ipilimumab at 3 mg/kg and nivolumab at 1 mg/kg administered every three weeks for four cycles visibly decreased the size of the oral melanoma lesion along with shrinkage of the metastatic lesions. However, following the administration of the second immunotherapy dose, the patient suffered from severe adverse side effects, such as myocarditis, hypophysitis, and neuritis, and the patient died due to cardiac arrest [51].
A post-hoc analysis of the KEYNOTE-001, 002, and 006 studies evaluated the effectiveness of pembrolizumab in advanced mucosal melanoma cases; the overall ORR in patients with mucosal melanoma was 19%, with a median PFS of 2.8 months and a median OS of 11.3 months. Notably, the responses were comparable between ipilimumab-naïve and ipilimumab-treated patients, indicating that pembrolizumab showed promising efficacy regardless of previous ipilimumab exposure [52].
In a patient with extensive advanced oral melanoma, treatment with ipilimumab followed by pembrolizumab showed a favorable response. This response was so effective that it eliminated the need for surgery as long as there was no tumor progression or recurrence [53]. However, to observe the long-term, reliable effects of pembrolizumab on mucosal melanoma, larger patient cohorts and longer-duration clinical studies are needed.
Numerous clinical trials have been conducted and are planned for advanced melanoma (Table 1.). However, due to its rarity, there are fewer clinical trials for mucosal melanoma compared to cutaneous melanoma. Particularly, patients with oral mucosal melanoma should be encouraged to participate in clinical trials, as it is crucial due to the condition’s rarity, limited treatment options, potential for personalized therapies, access to advancements, and the contribution to scientific progress.
3. Immunotherapy resistance
Resistance to immunotherapy can be clinically classified into two main categories. Patients who do not experience any therapeutic benefits from the initial attempt are considered to have primary resistance to the therapy. In contrast, some patients may initially benefit from the therapy, but over time, the tumor cells can acquire resistance, known as an acquired resistance, through adaptive changes, leading to tumor regrowth [54]. This resistance can be triggered intrinsically or extrinsically.
The discussion of resistance mechanisms primarily relies on cutaneous melanoma data, as it serves as a well-studied and more prevalent model in the field. This data offers insights into the broader understanding of melanoma resistance, even though the article’s main focus is on oral mucosal melanoma. However, it is crucial to recognize the unique characteristics of mucosal melanoma and its potential differences in resistance mechanisms to advance treatments for this less common subtype.
3.1. Mechanism of intrinsic resistance to immune checkpoint blockade therapy in melanoma
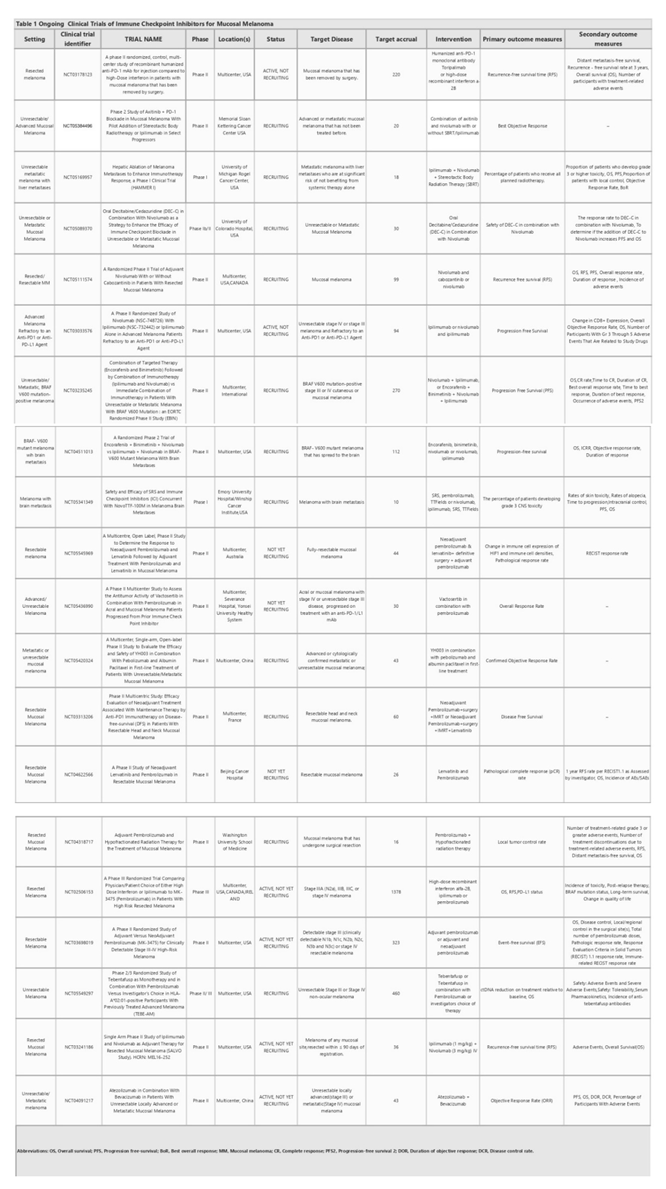
Low antigen expression is one of the primary contributors to the resistance against immunotherapy in cancer cells. Additionally, impairments in the system responsible for antigen processing and presentation, upregulation of constitutive PDL-1, absence of tumor-specific antigens and antigenic mutations, perturbations in the signaling pathways, genetic exclusion of T cells, and modifications in immune evasion mechanisms may significantly contribute to escaping immune responses, as elucidated by Sharma et al. in their comprehensive review [54]. In addition, regulatory networks dependent on TCF4/BRD4, MYC, the cytoprotective enzyme heme oxygenase-1 (HO-1), the loss of Kelch-like ECH-associated protein 1 (KEAP1), the loss of E-cadherin, and endogenous opioids are considered resistance-driver factors against immune checkpoint blockade in melanoma patients (Figure 1.).
3.1.1. Impairments in the antigen processing-presenting machinery
Therapies targeting CTLA-4 and PD-L1 promote T cell-driven immune enhancement against cancer. For these therapies to be effective, T cells present in the environment must recognize the cancer cells. Antigen presentation stimulates T cells to recognize the pathological cells; thus, any defect in the components of the tumor antigen-presenting machinery that prevents T cells from recognizing cancer cells may facilitate the evasion of immune defenses by tumor cells.
Human leukocyte antigen class I (HLA-I) is a noteworthy component of this machinery. Analysis of transcriptomic data from melanoma biopsies of immune checkpoint blockade responders and non-responders showed that responders had high levels of HLA-I in comparison to non-responders, suggesting that suppression of HLA-I antigen processing and presentation machinery plays a significant role in primary resistance to anti-CTLA-4 and anti-PD-1 therapy. Alternatively, the induction of Retinoic acid-inducible gene I (RIG-I) has been demonstrated to reverse HLA-I suppression in patients with melanoma, suggesting that it can be targeted to overcome resistance to immune checkpoint blockade [55]. Of note, Paulson et al. emphasized the importance of distinguishing between the two types of immunotherapy escape mechanisms, genetic HLA loss and transcriptional HLA loss, as genetic HLA loss demands the generation of novel T cell responses to target alternate HLAs to overcome immunotherapy resistance, while transcriptional HLA loss has the potential to be reversed through drug-based therapies to restore HLA expression [56]. This study underscores the importance of recognizing these distinct mechanisms to better understand and develop effective strategies for overcoming immunotherapy resistance.
Beta-2-microglobulin (B2M) is another indispensable element of APM, which participates in MHC Class I antigen presentation; its loss, particularly through loss of heterozygosity, may lead to the subsequent loss of MHC Class 1 and the proper presentation of tumor antigens, which hampers an effective anti-tumor response and contributes to immune evasion and resistance to therapy. The presence of B2M defects in patients is significantly correlated with non-responsiveness to anti-CTLA-4 therapy and anti-PDL-1 therapy. Notably, the fact that B2M defects are predominantly detected in samples taken before treatment from non-responders as well as in post-progression samples from patients with an initial response to immune checkpoint blockade suggests that B2M alterations may be involved in both acquired and primary resistance in metastatic melanoma [57].
IFN-γ displays a critical role in the antigen processing and presentation machinery, predominantly by upregulating MHC-1 and MHC-2 [58]. This becomes significant, especially within the scope of anti-PD-1 therapy, given that the initial response to such treatment is linked to preexisting immune activation mediated by IFN-γ, which is primarily manifested through the increase in expression of MHC-2 in metastatic melanoma [59]. IFN-γ also has a role in inducing PDL-1 expression [64]. Downregulation of this cytokine leads to downregulation of PDL-1, which causes insensitivity to anti-PD-1 immunotherapy and has been identified as an adaptive resistance mechanism [60].
Studies have indicated that impairments in the IFN-γ signal pathway in melanoma can also lead to a reduced response to anti-CTLA-4 therapy. Conversely, patients treated with ipilimumab showed an elevated level of IFN-γ and a better immune response [61]. In contrast, prolonged IFN-γ exposure causes resistance to radiotherapy plus anti-CTLA-4 treatment in melanoma cells. This is because IFN-γ increases PDL-1 expression, which suppresses T cells and results in adaptive resistance. Furthermore, extended IFN-γ exposure induces adaptive resistance through the STAT1 pathway, independent of its impact on PDL-1 expression [62]. This indicates that IFN-γ has a multifaceted role in inducing resistance to immune checkpoint blockade therapy, contributing to primary, adaptive, and acquired resistance to immune checkpoint blockade therapy in melanoma.
3.1.2. Alterations in signaling pathways
One of the resistance driver pathways is the MAPK pathway, whose upregulation has a negative effect on anti-tumor activity by regulating the production of VEGF and IL-8. The upregulation of these cytokines results in defective T-cell infiltration [54]. Jiang et al. showed that the MAPK pathway has the potential to stimulate PD-L1 expression in melanoma cells that have developed resistance to BRAF inhibition, suggesting that targeting the MAPK pathway can enhance the tumor response to immunotherapy [63]. Furthermore, mutations in this pathway increase CD73 expression. It has been suggested that these enzymes are upregulated in some patients undergoing immune checkpoint blockade therapy, and melanoma cells acquire a mesenchymal phenotype through the release of adenosine, ultimately causing adaptive resistance to therapy [64].
T-cell exclusion is another critical factor that leads to resistance to ICI therapy. It has been observed that the activation of the β-catenin pathway in melanoma cells leads to T cell exclusion and primary resistance to T cell-based cancer treatments such as ICIs due to the insufficient number of pre-existing T cells [65]. Tumor-intrinsic β-catenin signaling causes defective recruitment of CD103+ dendritic cells, which are crucial for CD8+ T cell function and immune infiltration [65].
PTEN is another determinant of T cell exclusion. Zhao et al. examined 66 patients with glioblastoma multiforme before and after PD-1 therapy to investigate the determinants of the therapeutic response. They detected an enrichment of PTEN mutations in non-responders, indicating that PTEN loss is connected to the development of resistance to anti-PD-1 therapy [66].
Loss of PTEN contributes to resistance to immunotherapy in melanoma, mainly by activating the PI3K pathway and decreasing the level of CD8 T-cells in tumors through the secretion of inhibitory cytokines, such as CCL2 and VEGF, as well as by inhibiting autophagy [67].
In contrast, ZEB1, a transcription factor that promotes the transition from epithelial to mesenchymal states may also contribute to immune escape by decreasing CD8+ T-cell accumulation in melanoma. ZEB1 causes a decrease in CD8+ T-cell infiltration into tumors through the downregulation of CD8+ T-cell-attracting chemokines and cytokines, such as CXCL10, CCL3, CCL4, IFN-γ, and TNF-α. Thus, ZEB-1 depletion is a positive regulator of anti-PD1 therapy [68].
3.1.3. Absence of tumor antigens and lack of antigenic mutation
Tumor neoantigens produced by tumor cells are distinct antigens formed by specific genetic mutations in cancerous cells. These mutations make them recognizable as foreign cells by the immune system and initiate an immune reaction against the tumors. However, tumor cells may sometimes lack these tumor antigens or may have some limitations in presenting them on the cell surface. This leads to an ineffective T-cell response and, in turn, resistance to T-cell-based immunotherapies such as anti-PDL-1 and anti-CTLA-4 in melanoma.
However, chronic stimulation with tumor antigens also promotes T-cell dysfunction and unresponsiveness [69]. Thus, antigenic mutations or newly emerged neoantigens are required to boost an effective anti-tumor response. Newly emerged neoantigens can serve as crucial factors in inducing durable T-cell responses and overcoming resistance. Studies have shown that the synergistic action of newly emerged neoantigen-induced CD8+ T cells and anti-PD-L1 therapy contributes to tumor elimination in a murine model of malignant melanoma [69]. The isolation of neoantigen-specific TCRs from metastatic melanoma tumor samples indicated the existence of neoantigen-specific T cell responses [70]. Tumors with a high abundance of clonal neoantigens are more responsive to immune checkpoint blockade in patients with melanoma [71]. Nevertheless, it is important to highlight that even melanomas with a low abundance of neoantigens can exhibit positive responses to immune checkpoint therapy [72]. Thus, understanding the role of clonal neoantigens in immune checkpoint blockade resistance is crucial for developing strategies to overcome resistance and improve treatment outcomes. Efforts are underway to identify and characterize the neoantigens in melanoma and to develop personalized immunotherapies that target these specific antigens, potentially enhancing the effectiveness of immune checkpoint blockade in resistant tumors.
3.1.4. Expression of PD-L1 and other contributing factors to resistance
The relationship between PDL-1 expression and resistance to immune checkpoint therapy is complex and needs to be clearly understood to select suitable patients for therapy and improve their prognosis.
PDL-1 can be either constitutively expressed or induced. Constitutive expression of PDL-1 is triggered by intrinsic oncogenic signaling pathways, whereas inducible PDL-1 is expressed as a response to inflammatory cytokines in the tumor microenvironment (TME) [73].
Intrinsic PDL-1 has a pro-tumoral effect, and this can lead to responsiveness to PD-1/PD-L1 inhibitor therapy [74]. Recent findings indicate that melanoma cells displaying increased constitutive PDL-1 expression have a diverse transcriptomic profile characterized by de-differentiation and active tumor necrosis factor (TNF) and interferon(IFN) signaling pathways, potentially contributing to resistance [75].
The presence of enhancers in patients before treatment or their acquisition during treatment can also lead to innate or adaptive resistance to ICI therapy in melanoma by activating several pathways that cause resistance [76].
In a recent study, a regulatory network dependent on TCF4/BRD4 was linked to the development of resistance to both targeted and immune checkpoint therapies in melanoma. This network supports the maintenance of a mesenchymal-like phenotype while inhibiting gene expression associated with antigen presentation, interferon signaling, and activation of leukocytes [77]. MYC appears to have a substantial influence on immunotherapy resistance by negatively affecting Janus Kinase 2(JAK2) expression and the responsiveness of melanoma cells to IFNγ [78].
Fructose consumption has also been shown to upregulate the cytoprotective enzyme heme oxygenase-1 (HO-1), which can contribute to resistance to checkpoint blockade in melanoma [79]. This indicates that dietary factors can influence the TME and potentially hinder the effectiveness of ICIs.
Furthermore, the loss of KEAP1 in melanoma has been identified as a factor leading to resistance against anti-PD-1 therapy. Patients with low KEAP1 expression, when treated with an anti-PD-1 antibody, exhibited worse OS [80]. This suggests that the status of KEAP1 expression may serve as a predictive biomarker for patient responses to anti-PD-1 therapy.
In addition, resistance to immune checkpoint blockade is connected to the loss of E-cadherin, which is indicative of mesenchymal transition [81]. E-cadherin loss in tumor cells may inhibit CD103 anti-tumor activity and diminish the effectiveness of immune checkpoint blockade [81]. Another study identified endogenous opioids as the potential drivers of T-cell dysfunction and resistance to immune checkpoint blockade in melanoma [82]. These observations highlight the intricate interplay of the factors contributing to resistance to immune checkpoint blockade therapy in melanoma and emphasize the need for more precise and targeted approaches.
3.2. Role of the extrinsic tumor resistance mechanism
Crosstalk between TME components such as immunosuppressive cells, inhibitory receptors, exosomes, and cancer cells allows cancer cells to gain invasive properties and escape immune attacks [83]. The abstract of the extrinsic mechanism of resistance to immune checkpoint blockade is shown (Figure 2.).
Tregs represent an important subgroup of immunosuppressive cells that infiltrate the melanoma microenvironment [84]. Treg cells employ their ability to dampen the immune response by producing cytokines like IL-10 and IL-35. Aside from their ability to suppress effector T cells, they induce tumor-infiltrating macrophages to generate B7-H molecules, and the interaction of these molecules with their ligands contributes to immune tolerance by dampening the T cell response [85]. Additionally, Tregs can communicate with other immunosuppressive cells in the TME by secreting various cytokines, which in turn enhance the immunosuppressive microenvironment [86]. Nevertheless, even after their death, Treg cells may still persist in exerting their immunosuppressive effects [87].
A study revealed that Tregs undergo programmed cell death within the TME, and contrary to expectations, apoptotic Tregs were more effective in suppressing T-cell activation, mainly by producing high levels of ATP and converting it into immunosuppressive adenosine using specific enzymes. It was also shown in mouse cancer models that apoptotic Tregs affect anti-PDL-1 therapy adversely. This study proposed that hypoxia-induced Treg apoptosis may serve as a novel immune evasion mechanism in the TME, potentially leading to immune checkpoint blockade resistance [88].
Myeloid-derived suppressor cells (MDSCs) are a diverse group of myeloid cells with immunosuppressive capabilities. The most important mechanism underlying their immunosuppressive activity is the increased expression of nitric oxide (NO) and activation of arginase (Arg)-1. Arg-1 causes L-arginine depletion, which is required for T-cell function, whereas NO expression inhibits T-cell proliferation. Through these mechanisms, MDSCs lead to T cell dysfunction and a reduced response to immunotherapies such as immune checkpoint blockade [85,89].
High MDSC levels in melanoma patients who received ipilimumab indicate an unfavorable prognosis[90]. Conversely, patients with advanced melanoma who had reduced levels of CD33+CD11b+HLA-DR-MDSCs prior to treatment with ipilimumab exhibited extended survival and an objective clinical response [91]. Studies have also provided evidence regarding the association between an increased MDSC population and a lack of functional T cells, which can limit the efficacy of ICIs [92].
Cell-to-cell interactions also have a notable impact on shaping the TME. Cells in the TME may have the ability to impact the characteristics of cancer cells and the expression of other cells in the TME. Tirosh et al. examined 471 tumors from The Cancer Genome Atlas dataset. They found that a TME in which cancer-associated fibroblasts (CAFs) were highly abundant was associated with an invasive phenotype (microphthalmia-associated transcription factor (MITF)-low/AXL-high) of melanoma cells [93]. Phenotype switching is the term used to describe the ability of melanoma cells to transition between various cell states [94]. Resistance to PD-1 inhibitors, a common type of ICIs, is often associated with melanoma de-differentiation [95]. Investigation of melanoma tumor tissue samples taken before the administration of anti-PD1 therapy revealed that patients who responded to the therapy showed differentiated gene signatures characterized by high proliferation and low invasiveness. In contrast, non-responders are characterized by highly invasive and de-differentiated gene expression signatures [96]. These findings emphasize that by undergoing a phenotypic switch from proliferative MITF-high differentiated subpopulations to invasive MITF-low de-differentiated subpopulations, melanoma cells potentially become resistant to ICB therapy.
IDO, mainly present in tumor and host immune cells, is an enzyme that degrades tryptophan and negatively regulates the immune response to immune checkpoint blockade by inducing T-cell exhaustion and Treg proliferation in the TME [97,98]. IDO-deficient mice show elevated intratumoral ratios of effector T cells to T regs, which is a positive indicator of prognosis, after treatment with CTLA-4 [99]. Its deficiency was also shown to improve the prognosis after anti-PD-1/PDL-1 treatment [99].
Thus, a combination therapy of IDO inhibitors and ICIs emerged to overcome resistance and enhance the efficacy of the treatment. Although Phase I/II trials of the IDO inhibitor plus pembrolizumab in solid tumors, including melanoma, showed promising results, a phase III trial in patients with unresectable or metastatic melanoma failed due to not meeting the primary end point [100,101] and no significant difference between the combination treatment and pembrolizumab alone groups was observed [101].
However, some studies showed that, while elevated IDO expression in surgically treated patients is correlated with shorter PFS or OS, treatment with PD-1 inhibitor showed longer PFS in patients with acral and mucosal melanoma who have elevated IDO levels. Iga et al. have stated that the reason for this is that the immune-suppressive environment caused by IDO can be reversed with immunotherapy, but the immune-suppressive environment created by IDO in patients treated with surgery alone cannot be reversed [102]. In essence, the conflicting findings suggest that IDO may have a dual role in cancer. In patients treated with surgery alone, IDO’s immune-suppressive effects may worsen outcomes. Still, in patients receiving immunotherapy, the therapy may counteract these effects, leading to better responses.
TAMs are another cell component present in TME. TAMs exhibit various phenotypes, encompassing M1-like and M2-like characteristics, with the latter being associated with immunosuppressive functions. In an experimental mouse model of melanoma, using a monoclonal antibody against MARCO, a scavenger receptor on TAMs, reduced the presence of M2 TAMs and improved the effectiveness of anti-CTLA-4 antibody therapy [103]. Several mechanisms have been proposed to explain how TAMs promote resistance to immune checkpoint therapy. One mechanism is through the secretion of immunosuppressive molecules such as TGF-β and PGE2. These molecules inhibit the activity of cytotoxic T cells and promote the expansion of Tregs, which dampens anti-tumor immune responses. Additionally, TAMs have the capability to produce immune checkpoint proteins like PD-L1, which can directly suppress the activity of T cells. The presence of PD-L1 in TAMs is correlated with resistance to PD-1/PD-L1 blockade therapy [104].
Additionally, hypoxia is a significant factor in influencing how patients with melanoma respond to ICIs and develop resistance to them. Hypoxia reduces the expression of MITF, a crucial gene involved in melanocyte differentiation. This decrease in MITF level is controlled by hypoxia-inducible factor 1α (HIF-1α). Consequently, melanoma cells adopt a more invasive phenotype, leading to resistance against immune checkpoint therapy [105]. As with chronic antigen stimulation, hypoxia can also affect multiple facets of T-cell function and contribute to therapeutic resistance by causing T-cell dysfunction. Moreover, lactate induced by hypoxia encourages Tregs to maintain their immunosuppressive properties and regulates the expression of PD-1 and PDL-1 [106,107]. In addition, cells exposed to hypoxia can undergo a hypermetabolic transformation marked by elevated glycolytic activity, leading to resistance [108].
In the case of an excessive immune response, a regulatory mechanism comes into effect to avoid excessive stimulation of T cells that could cause autoimmune diseases and tissue damage. In response, cells release inhibitory molecules such as CTLA-4, PD-1/PDL-1, T-cell immunoglobulin and mucin domain 3 (TIM-3) and lymphocyte-activation gene 3(LAG-3) for immune modulation [109]. The effectiveness of ICIs can be hindered in melanoma because of the increased expression of immune checkpoints such as TIM-3 and V-domain I g suppressor of T cell activation(VISTA) [110]. Preclinical studies have also correlated TIM-3 upregulation with resistance to anti-PD-1 therapy, and elevated VISTA expression has been observed in patients with melanoma who underwent disease progression while receiving anti-PD-1 inhibitor therapy [110]. T-cell immunoreceptor with Ig and ITIM domains(TIGIT), a recently identified immune checkpoint receptor, interacts with its ligand CD155, transmitting inhibitory signals and thus functioning in a manner similar to other immune checkpoints such as PD-1 and CTLA-4 [111]. Therefore, therapies aimed at blocking these co-inhibitory receptors to counteract immune resistance by reversing the negative effects they exert on T cells are in development.
4. Conclusion and future directions
The discovery of ICIs has driven significant improvements in immunotherapy. ICIs are effective in both adjuvant and primary treatment settings, particularly for melanoma, which has high immunogenicity. In addition to being used as a first-line or adjuvant treatment, recent research findings indicate that ICIs can enhance the effects of other conventional therapies such as radiation and chemotherapy.
The term “abscopal effect” refers to a rare phenomenon in which radiation therapy applied to one area has an anti-tumor effect on a distant tumor. This effect was observed in a patient with OMM who underwent maxillary resection and bilateral neck dissection, followed by adjuvant treatment with nivolumab. After the occurrence of brain, spleen, and liver metastases, the patient received radiation therapy for the brain tumor, and as a result of the abscopal effect, a regression was seen in the liver and spleen metastases. It was suggested that nivolumab played a role in exerting this effect [112].
ICIs can also improve the effectiveness of subsequent chemotherapy. In patients with malignant melanoma who developed resistance to PD-1 blockade, the overall response to chemotherapy administered with PD-1 inhibitor was higher than that in patients who received chemotherapy alone [113]. Recent studies have investigated the use of ICIs as a conversion therapy. Conversion therapy involves shrinking initially unresectable cancerous lesions through various treatment approaches, such as chemotherapy, immunotherapy, or radiotherapy, to make surgical operations feasible and safe. Zhang et al. demonstrated the effectiveness of combining PD-1 inhibitors with tyrosine kinase inhibitors as a conversion treatment for advanced hepatocellular carcinoma, showing a RFS rate of 75% at 12 months after surgery [114]. However, clinical studies on the use of ICIs as conversion therapy for melanoma and other types of cancer are limited. Especially in OMM, where a late diagnosis can make surgery extremely challenging, the application of ICIs as a conversion therapy holds promise, and further research in this field is needed.
Despite playing a significant role in various stages of cancer treatment, a significant number of patients acquire resistance to ICIs and fail to benefit from treatment. Therefore, to achieve more effective treatment outcomes, it is essential to elucidate the mechanisms that contribute to drug resistance to ICIs. Combination therapies targeting specific pathways or proteins have come to the forefront for overcoming immune checkpoint blockade resistance and enhancing the treatment response. Combination therapy using oncolytic viruses and ICIs is one of the most promising approaches.
An oncolytic virus refers to a virus, whether modified through genetic engineering or existing naturally, that specifically targets and reproduces within cancer cells, thus triggering a specific anti-tumor immunity, which leads to the death of cancer cells [115]. Oncolytic viruses assist in overcoming the resistance to immune checkpoint blockade therapy by infecting cancer cells and inducing their lysis, resulting in cell death. They also stimulate anti-tumor immunity, converting the cold TME into a hot TME [116,117].
In patients with advanced melanoma, combination therapy with pembrolizumab and talimogene laherparepvec (T-Vec), a double-mutated, second-generation oncolytic herpes simplex virus type 1 (HSV-1), resulted in a notable ORR of 62% and a complete response rate of 33%. In patients who demonstrated treatment efficacy, combination therapy was observed to modify the TME by enhancing various cell populations, primarily CD8+ cells [118].
When combined with anti-CTLA-4, G47Δ, a triple-mutated oncolytic HSV-1, was noticed to increase the influx of effector T cells into the TME in various cancer mouse models, including melanoma. This combination therapy augmented the effectiveness of systemic treatment, while also promoting the conversion of initially immune-resistant TME to a TME that may respond favorably to the immune response [119].
It has also been observed that G47Δ is highly effective in mouse models of oral squamous cell carcinoma, preventing cervical lymph node metastasis via viral traffics from primary lesion to the lymph node [120]. Especially in cases of oral melanoma where the risk of metastasis is considerably high due to late diagnosis, the combination of G47Δ with ICIs holds considerable potential.
Targeting TGF-β also holds promise in overcoming resistance and enhancing immunotherapy responses. TGF-β has a complex role in tumor immunity, hampering the ability of T cells to infiltrate tumors, reducing their effector functions, and promoting the differentiation of peripheral CD4+ T cells into Tregs, potentially limiting the host’s natural defense against cancer [121]. To counteract these effects and boost the efficacy of anti-PD-L1 treatment, researchers have proposed blocking TGF-β as a strategy [122]. One promising avenue in this endeavor involves the development of Fc fusion proteins that specifically target TGF-β. These fusion proteins have shown the potential to counteract TGF-β-induced mesenchymal properties in malignant melanoma cells and TβRI-TβRII-Fc chimeric receptor inhibited B16 melanoma tumor growth in vivo. Considering the ability of E-cadherin loss leading resistance to immune checkpoint blockade, it would provide insight into how immunotherapeutic approaches that block TGF-β could enhance responses and overcome resistance to immunotherapy. [123].
In this paper, we have included many studies that have examined resistance mechanisms in melanoma. However, these studies have primarily focused on cutaneous melanoma. Mucosal melanoma including OMM is relatively rare, comprising a smaller percentage of melanoma diagnoses, which results in limited available data for study. Despite this, cutaneous melanoma, being more extensively studied, may offer valuable insights and serve as reference points for understanding resistance in oral mucosal melanoma. Thus, our inclusion of such studies aims to bridge the knowledge gap and provide a broader perspective on immunotherapy resistance in melanoma.
Due to their shared origin as melanomas, it is possible that some resistance mechanisms may also be shared between cutaneous and mucosal melanoma. However, it’s important to note that OMM exhibits unique clinical and molecular characteristics that could give rise to resistance mechanisms not observed in cutaneous melanoma. Overall, it is important to thoroughly elucidate the factors contributing to resistance to ICI therapy and develop effective combination treatments aimed at overcoming these obstacles.
Author Contributions
Conceptualization: T.U., S.K., T.I..; Investigation: S.Z.U., T.U., S.K., K.K., T.S., A.S., S.S.; Data curation: S.Z.U., T.U.; Writing – original draft: S.Z.U., T.U.; Writing – review & editing: S.Z.U., T.U., S.S..; Supervision: S.T.; Project administration: T.U.
Funding
This research received no external funding.
Data Availability Statement
The data presented in this study are available in this article.
References
- Zito, P. M.; Brizuela, M.; Mazzoni, T. Oral Melanoma. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2023.
- Warszawik-Hendzel, O.; Słowińska, M.; Olszewska, M.; Rudnicka, L. Melanoma of the Oral Cavity: Pathogenesis, Dermoscopy, Clinical Features, Staging and Management. J. Dermatol. Case Rep. 2014, 8 (3), 60–66. https://doi.org/10.3315/jdcr.2014.1175. [CrossRef]
- Rapidis, A. D.; Apostolidis, C.; Vilos, G.; Valsamis, S. Primary Malignant Melanoma of the Oral Mucosa. J. Oral Maxillofac. Surg. 2003, 61 (10), 1132–1139. https://doi.org/10.1016/S0278-2391(03)00670-0. [CrossRef]
- Mascitti, M.; Santarelli, A.; Sartini, D.; Rubini, C.; Colella, G.; Salvolini, E.; Ganzetti, G.; Offidani, A.; Emanuelli, M. Analysis of Nicotinamide N-Methyltransferase in Oral Malignant Melanoma and Potential Prognostic Significance. Melanoma Res. 2019, 29 (2), 151–156. https://doi.org/10.1097/CMR.0000000000000548. [CrossRef]
- Hicks, M. J.; Flaitz, C. M. Oral Mucosal Melanoma: Epidemiology and Pathobiology. Oral Oncol. 2000, 36 (2), 152–169. https://doi.org/10.1016/S1368-8375(99)00085-8. [CrossRef]
- Meleti, M.; Leemans, C. R.; Mooi, W. J.; Vescovi, P.; van der Waal, I. Oral Malignant Melanoma: A Review of the Literature. Oral Oncol. 2007, 43 (2), 116–121. https://doi.org/10.1016/j.oraloncology.2006.04.001. [CrossRef]
- Nenclares, P.; Ap Dafydd, D.; Bagwan, I.; Begg, D.; Kerawala, C.; King, E.; Lingley, K.; Paleri, V.; Paterson, G.; Payne, M.; Silva, P.; Steven, N.; Turnbull, N.; Yip, K.; Harrington, K. J. Head and Neck Mucosal Melanoma: The United Kingdom National Guidelines. Eur. J. Cancer Oxf. Engl. 1990 2020, 138, 11–18. https://doi.org/10.1016/j.ejca.2020.07.017. [CrossRef]
- Testori, A. A. E.; Blankenstein, S. A.; van Akkooi, A. C. J. Surgery for Metastatic Melanoma: An Evolving Concept. Curr. Oncol. Rep. 2019, 21 (11), 98. https://doi.org/10.1007/s11912-019-0847-6. [CrossRef]
- Yao, J.-J.; Zhang, F.; Zhang, G.-S.; Deng, X.-W.; Zhang, W.-J.; Lawrence, W. R.; Zou, L.; Zhang, X.-S.; Lu, L.-X. Efficacy and Safety of Primary Surgery with Postoperative Radiotherapy in Head and Neck Mucosal Melanoma: A Single-Arm Phase II Study. Cancer Manag. Res. 2018, 10, 6985–6996. https://doi.org/10.2147/CMAR.S185017. [CrossRef]
- Yang, A. S.; Chapman, P. B. The History and Future of Chemotherapy for Melanoma. Hematol. Oncol. Clin. North Am. 2009, 23 (3), 583–x. https://doi.org/10.1016/j.hoc.2009.03.006. [CrossRef]
- Luke, J. J.; Flaherty, K. T.; Ribas, A.; Long, G. V. Targeted Agents and Immunotherapies: Optimizing Outcomes in Melanoma. Nat. Rev. Clin. Oncol. 2017, 14 (8), 463–482. https://doi.org/10.1038/nrclinonc.2017.43. [CrossRef]
- Vasudevan, S.; Flashner-Abramson, E.; Alkhatib, H.; Roy Chowdhury, S.; Adejumobi, I. A.; Vilenski, D.; Stefansky, S.; Rubinstein, A. M.; Kravchenko-Balasha, N. Overcoming Resistance to BRAFV600E Inhibition in Melanoma by Deciphering and Targeting Personalized Protein Network Alterations. Npj Precis. Oncol. 2021, 5 (1), 1–13. https://doi.org/10.1038/s41698-021-00190-3. [CrossRef]
- Flaherty, K. T.; Infante, J. R.; Daud, A.; Gonzalez, R.; Kefford, R. F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; Kudchadkar, R.; Burris, H. A.; Falchook, G.; Algazi, A.; Lewis, K.; Long, G. V.; Puzanov, I.; Lebowitz, P.; Singh, A.; Little, S.; Sun, P.; Allred, A.; Ouellet, D.; Kim, K. B.; Patel, K.; Weber, J. Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations. N. Engl. J. Med. 2012, 367 (18), 1694–1703. https://doi.org/10.1056/NEJMoa1210093. [CrossRef]
- Ma, X.; Wu, Y.; Zhang, T.; Song, H.; Jv, H.; Guo, W.; Ren, G. The Clinical Significance of C-Kit Mutations in Metastatic Oral Mucosal Melanoma in China. Oncotarget 2017, 8 (47), 82661–82673. https://doi.org/10.18632/oncotarget.19746. [CrossRef]
- Wong, D. J. L.; Ribas, A. Targeted Therapy for Melanoma. Cancer Treat. Res. 2016, 167, 251–262. https://doi.org/10.1007/978-3-319-22539-5_10. [CrossRef]
- Rosenberg, S. A. IL-2: The First Effective Immunotherapy for Human Cancer. J. Immunol. Baltim. Md 1950 2014, 192 (12), 5451–5458. https://doi.org/10.4049/jimmunol.1490019. [CrossRef]
- Atkins, M. B.; Lotze, M. T.; Dutcher, J. P.; Fisher, R. I.; Weiss, G.; Margolin, K.; Abrams, J.; Sznol, M.; Parkinson, D.; Hawkins, M.; Paradise, C.; Kunkel, L.; Rosenberg, S. A. High-Dose Recombinant Interleukin 2 Therapy for Patients with Metastatic Melanoma: Analysis of 270 Patients Treated between 1985 and 1993. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1999, 17 (7), 2105–2116. https://doi.org/10.1200/JCO.1999.17.7.2105. [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced Expression of PD-1, a Novel Member of the Immunoglobulin Gene Superfamily, upon Programmed Cell Death. EMBO J. 1992, 11 (11), 3887–3895. https://doi.org/10.1002/j.1460-2075.1992.tb05481.x. [CrossRef]
- Hamanishi, J.; Mandai, M.; Matsumura, N.; Abiko, K.; Baba, T.; Konishi, I. PD-1/PD-L1 Blockade in Cancer Treatment: Perspectives and Issues. Int J Clin Oncol 2016, 21 (3), 462–473. https://doi.org/10.1007/s10147-016-0959-z. [CrossRef]
- Ribas, A. Tumor Immunotherapy Directed at PD-1. N. Engl. J. Med. 2012, 366 (26), 2517–2519. https://doi.org/10.1056/NEJMe1205943. [CrossRef]
- Mahoney, K. M.; Freeman, G. J.; McDermott, D. F. The Next Immune-Checkpoint Inhibitors: PD-1/PD-L1 Blockade in Melanoma. Clin. Ther. 2015, 37 (4), 764–782. https://doi.org/10.1016/j.clinthera.2015.02.018. [CrossRef]
- Iwai, Y.; Hamanishi, J.; Chamoto, K.; Honjo, T. Cancer Immunotherapies Targeting the PD-1 Signaling Pathway. J. Biomed. Sci. 2017, 24 (1), 26. https://doi.org/10.1186/s12929-017-0329-9. [CrossRef]
- Brunet, J.-F.; Denizot, F.; Luciani, M.-F.; Roux-Dosseto, M.; Suzan, M.; Mattei, M.-G.; Golstein, P. A New Member of the Immunoglobulin Superfamily—CTLA-4. Nature 1987, 328 (6127), 267–270. https://doi.org/10.1038/328267a0. [CrossRef]
- Leach, D. R.; Krummel, M. F.; Allison, J. P. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science 1996, 271 (5256), 1734–1736. https://doi.org/10.1126/science.271.5256.1734. [CrossRef]
- Wolchok, J. D.; Hodi, F. S.; Weber, J. S.; Allison, J. P.; Urba, W. J.; Robert, C.; O’Day, S. J.; Hoos, A.; Humphrey, R.; Berman, D. M.; Lonberg, N.; Korman, A. J. Development of Ipilimumab: A Novel Immunotherapeutic Approach for the Treatment of Advanced Melanoma. Ann. N. Y. Acad. Sci. 2013, 1291 (1), 1–13. https://doi.org/10.1111/nyas.12180. [CrossRef]
- Camacho, L. H. CTLA-4 Blockade with Ipilimumab: Biology, Safety, Efficacy, and Future Considerations. Cancer Med. 2015, 4 (5), 661–672. https://doi.org/10.1002/cam4.371. [CrossRef]
- Hodi, F. S.; O’Day, S. J.; McDermott, D. F.; Weber, R. W.; Sosman, J. A.; Haanen, J. B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J. C.; Akerley, W.; van den Eertwegh, A. J. M.; Lutzky, J.; Lorigan, P.; Vaubel, J. M.; Linette, G. P.; Hogg, D.; Ottensmeier, C. H.; Lebbé, C.; Peschel, C.; Quirt, I.; Clark, J. I.; Wolchok, J. D.; Weber, J. S.; Tian, J.; Yellin, M. J.; Nichol, G. M.; Hoos, A.; Urba, W. J. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363 (8), 711–723. https://doi.org/10.1056/NEJMoa1003466. [CrossRef]
- Seth, R.; Messersmith, H.; Kaur, V.; Kirkwood, J. M.; Kudchadkar, R.; McQuade, J. L.; Provenzano, A.; Swami, U.; Weber, J.; Alluri, K. C.; Agarwala, S.; Ascierto, P. A.; Atkins, M. B.; Davis, N.; Ernstoff, M. S.; Faries, M. B.; Gold, J. S.; Guild, S.; Gyorki, D. E.; Khushalani, N. I.; Meyers, M. O.; Robert, C.; Santinami, M.; Sehdev, A.; Sondak, V. K.; Spurrier, G.; Tsai, K. K.; van Akkooi, A.; Funchain, P. Systemic Therapy for Melanoma: ASCO Guideline. J. Clin. Oncol. 2020, 38 (33), 3947–3970. https://doi.org/10.1200/JCO.20.00198. [CrossRef]
- Buchbinder, E. I.; Weirather, J. L.; Manos, M.; Quattrochi, B. J.; Sholl, L. M.; Brennick, R. C.; Bowling, P.; Bailey, N.; Magarace, L.; Ott, P. A.; Haq, R.; Izar, B.; Giobbie-Hurder, A.; Hodi, F. S. Characterization of Genetics in Patients with Mucosal Melanoma Treated with Immune Checkpoint Blockade. Cancer Med. 2021, 10 (8), 2627–2635. https://doi.org/10.1002/cam4.3789. [CrossRef]
- Umeda, M.; Shimada, K. Primary Malignant Melanoma of the Oral Cavity—Its Histological Classification and Treatment. Br. J. Oral Maxillofac. Surg. 1994, 32 (1), 39–47. https://doi.org/10.1016/0266-4356(94)90172-4. [CrossRef]
- Wang, R.; Jing, G.; Lv, J.; Song, H.; Li, C.; Wang, X.; Xia, W.; Wu, Y.; Ren, G.; Guo, W. Interferon-α-2b as an Adjuvant Therapy Prolongs Survival of Patients with Previously Resected Oral Muscosal Melanoma. Genet. Mol. Res. GMR 2015, 14 (4), 11944–11954. https://doi.org/10.4238/2015.October.5.8. [CrossRef]
- Long, G. V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; Robert, C.; Mortier, L.; Schachter, J.; Schadendorf, D.; Lesimple, T.; Plummer, R.; Ji, R.; Zhang, P.; Mookerjee, B.; Legos, J.; Kefford, R.; Dummer, R.; Kirkwood, J. M. Adjuvant Dabrafenib plus Trametinib in Stage III BRAF-Mutated Melanoma. N. Engl. J. Med. 2017, 377 (19), 1813–1823. https://doi.org/10.1056/NEJMoa1708539. [CrossRef]
- Research, C. for D. E. and. FDA Approves Dabrafenib plus Trametinib for Adjuvant Treatment of Melanoma with BRAF V600E or V600K Mutations. FDA 2019.
- Lyu, J.; Wu, Y.; Li, C.; Wang, R.; Song, H.; Ren, G.; Guo, W. Mutation Scanning of BRAF, NRAS, KIT, and GNAQ/GNA11 in Oral Mucosal Melanoma: A Study of 57 Cases. J. Oral Pathol. Med. 2016, 45 (4), 295–301. https://doi.org/10.1111/jop.12358. [CrossRef]
- Chen, F.; Zhang, Q.; Wang, Y.; Wang, S.; Feng, S.; Qi, L.; Li, X.; Ding, C. KIT, NRAS, BRAF and FMNL2 Mutations in Oral Mucosal Melanoma and a Systematic Review of the Literature. Oncol. Lett. 2018, 15 (6), 9786–9792. https://doi.org/10.3892/ol.2018.8558. [CrossRef]
- Nebhan, C. A.; Johnson, D. B. Pembrolizumab in the Adjuvant Treatment of Melanoma: Efficacy and Safety. Expert Rev. Anticancer Ther. 2021, 21 (6), 583–590. https://doi.org/10.1080/14737140.2021.1882856. [CrossRef]
- Eggermont, A. M. M.; Blank, C. U.; Mandala, M.; Long, G. V.; Atkinson, V.; Dalle, S.; Haydon, A.; Lichinitser, M.; Khattak, A.; Carlino, M. S.; Sandhu, S.; Larkin, J.; Puig, S.; Ascierto, P. A.; Rutkowski, P.; Schadendorf, D.; Koornstra, R.; Hernandez-Aya, L.; Maio, M.; van den Eertwegh, A. J. M.; Grob, J.-J.; Gutzmer, R.; Jamal, R.; Lorigan, P.; Ibrahim, N.; Marreaud, S.; van Akkooi, A. C. J.; Suciu, S.; Robert, C. Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma. N. Engl. J. Med. 2018, 378 (19), 1789–1801. https://doi.org/10.1056/NEJMoa1802357. [CrossRef]
- Research, C. for D. E. and. FDA Approves Pembrolizumab for Adjuvant Treatment of Stage IIB or IIC Melanoma. FDA 2021.
- Wu, Y.; Wei, D.; Ren, G.; Guo, W. Chemotherapy in Combination with Anti-PD-1 Agents as Adjuvant Therapy for High-Risk Oral Mucosal Melanoma. J. Cancer Res. Clin. Oncol. 2023, 149 (6), 2293–2300. https://doi.org/10.1007/s00432-022-04090-2. [CrossRef]
- Eggermont, A. M. M.; Chiarion-Sileni, V.; Grob, J.-J.; Dummer, R.; Wolchok, J. D.; Schmidt, H.; Hamid, O.; Robert, C.; Ascierto, P. A.; Richards, J. M.; Lebbé, C.; Ferraresi, V.; Smylie, M.; Weber, J. S.; Maio, M.; Konto, C.; Hoos, A.; de Pril, V.; Gurunath, R. K.; de Schaetzen, G.; Suciu, S.; Testori, A. Adjuvant Ipilimumab versus Placebo after Complete Resection of High-Risk Stage III Melanoma (EORTC 18071): A Randomised, Double-Blind, Phase 3 Trial. Lancet Oncol. 2015, 16 (5), 522–530. https://doi.org/10.1016/S1470-2045(15)70122-1. [CrossRef]
- Eggermont, A. M. M.; Chiarion-Sileni, V.; Grob, J.-J.; Dummer, R.; Wolchok, J. D.; Schmidt, H.; Hamid, O.; Robert, C.; Ascierto, P. A.; Richards, J. M.; Lebbé, C.; Ferraresi, V.; Smylie, M.; Weber, J. S.; Maio, M.; Bastholt, L.; Mortier, L.; Thomas, L.; Tahir, S.; Hauschild, A.; Hassel, J. C.; Hodi, F. S.; Taitt, C.; de Pril, V.; de Schaetzen, G.; Suciu, S.; Testori, A. Prolonged Survival in Stage III Melanoma with Ipilimumab Adjuvant Therapy. New England Journal of Medicine 2016, 375 (19), 1845–1855. https://doi.org/10.1056/NEJMoa1611299. [CrossRef]
- Petrella, T. M.; Fletcher, G. G.; Knight, G.; McWhirter, E.; Rajagopal, S.; Song, X.; Baetz, T. D. Systemic Adjuvant Therapy for Adult Patients at High Risk for Recurrent Cutaneous or Mucosal Melanoma: An Ontario Health (Cancer Care Ontario) Clinical Practice Guideline. Curr. Oncol. 2020, 27 (1), e43–e52. https://doi.org/10.3747/co.27.5933. [CrossRef]
- Goldstein, D. A. Adjuvant Ipilimumab for Melanoma—The $1.8 Million per Patient Regimen. JAMA Oncol. 2017, 3 (12), 1628–1629. https://doi.org/10.1001/jamaoncol.2017.3123. [CrossRef]
- Weber, J.; Mandala, M.; Del Vecchio, M.; Gogas, H. J.; Arance, A. M.; Cowey, C. L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Marquez-Rodas, I.; Grob, J.-J.; Butler, M. O.; Middleton, M. R.; Maio, M.; Atkinson, V.; Queirolo, P.; Gonzalez, R.; Kudchadkar, R. R.; Smylie, M.; Meyer, N.; Mortier, L.; Atkins, M. B.; Long, G. V.; Bhatia, S.; Lebbé, C.; Rutkowski, P.; Yokota, K.; Yamazaki, N.; Kim, T. M.; de Pril, V.; Sabater, J.; Qureshi, A.; Larkin, J.; Ascierto, P. A. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N. Engl. J. Med. 2017, 377 (19), 1824–1835. https://doi.org/10.1056/NEJMoa1709030. [CrossRef]
- Yi, J. H.; Yi, S. Y.; Lee, H. R.; Lee, S. I.; Lim, D. H.; Kim, J. H.; Park, K. W.; Lee, J. Dacarbazine-Based Chemotherapy as First-Line Treatment in Noncutaneous Metastatic Melanoma: Multicenter, Retrospective Analysis in Asia. Melanoma Res. 2011, 21 (3), 223. https://doi.org/10.1097/CMR.0b013e3283457743. [CrossRef]
- Tyrrell, H.; Payne, M. Combatting Mucosal Melanoma: Recent Advances and Future Perspectives. Melanoma Manag. 2018, 5 (3), MMT11. https://doi.org/10.2217/mmt-2018-0003. [CrossRef]
- Lyu, J.; Song, Z.; Chen, J.; Shepard, M. J.; Song, H.; Ren, G.; Li, Z.; Guo, W.; Zhuang, Z.; Shi, Y. Whole-Exome Sequencing of Oral Mucosal Melanoma Reveals Mutational Profile and Therapeutic Targets. J. Pathol. 2018, 244 (3), 358–366. https://doi.org/10.1002/path.5017. [CrossRef]
- D’Angelo, S. P.; Larkin, J.; Sosman, J. A.; Lebbé, C.; Brady, B.; Neyns, B.; Schmidt, H.; Hassel, J. C.; Hodi, F. S.; Lorigan, P.; Savage, K. J.; Miller, W. H.; Mohr, P.; Marquez-Rodas, I.; Charles, J.; Kaatz, M.; Sznol, M.; Weber, J. S.; Shoushtari, A. N.; Ruisi, M.; Jiang, J.; Wolchok, J. D. Efficacy and Safety of Nivolumab Alone or in Combination With Ipilimumab in Patients With Mucosal Melanoma: A Pooled Analysis. J. Clin. Oncol. 2017, 35 (2), 226–235. https://doi.org/10.1200/JCO.2016.67.9258. [CrossRef]
- Adisa, A. O.; Olawole, W. O.; Sigbeku, O. F. Oral Amelanotic Melanoma. Ann. Ib. Postgrad. Med. 2012, 10 (1), 6–8.
- Lamichhane, N. S.; An, J.; Liu, Q.; Zhang, W. Primary Malignant Mucosal Melanoma of the Upper Lip: A Case Report and Review of the Literature. BMC Res. Notes 2015, 8, 499. https://doi.org/10.1186/s13104-015-1459-3. [CrossRef]
- Khoury, Z. H.; Hausner, P. F.; Idzik-Starr, C. L.; Frykenberg, M. R. A.; Brooks, J. K.; Dyalram, D.; Basile, J. R.; Younis, R. H. Combination Nivolumab/Ipilimumab Immunotherapy For Melanoma With Subsequent Unexpected Cardiac Arrest: A Case Report and Review of Literature. J. Immunother. Hagerstown Md 1997 2019, 42 (8), 313–317. https://doi.org/10.1097/CJI.0000000000000282. [CrossRef]
- Hamid, O.; Robert, C.; Ribas, A.; Hodi, F. S.; Walpole, E.; Daud, A.; Arance, A. S.; Brown, E.; Hoeller, C.; Mortier, L.; Schachter, J.; Long, J.; Ebbinghaus, S.; Ibrahim, N.; Butler, M. Antitumour Activity of Pembrolizumab in Advanced Mucosal Melanoma: A Post-Hoc Analysis of KEYNOTE-001, 002, 006. Br. J. Cancer 2018, 119 (6), 670–674. https://doi.org/10.1038/s41416-018-0207-6. [CrossRef]
- Castaño, A.; Shah, S. S.; Cicero, G.; El Chaar, E. Primary Oral Melanoma - A Non-Surgical Approach to Treatment via Immunotherapy. Clin. Adv. Periodontics 2017, 7 (1), 9–17. https://doi.org/10.1902/cap.2016.160003. [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J. A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168 (4), 707–723. https://doi.org/10.1016/j.cell.2017.01.017. [CrossRef]
- Such, L.; Zhao, F.; Liu, D.; Thier, B.; Le-Trilling, V. T. K.; Sucker, A.; Coch, C.; Pieper, N.; Howe, S.; Bhat, H.; Kalkavan, H.; Ritter, C.; Brinkhaus, R.; Ugurel, S.; Köster, J.; Seifert, U.; Dittmer, U.; Schuler, M.; Lang, K. S.; Kufer, T. A.; Hartmann, G.; Becker, J. C.; Horn, S.; Ferrone, S.; Liu, D.; Van Allen, E. M.; Schadendorf, D.; Griewank, K.; Trilling, M.; Paschen, A. Targeting the Innate Immunoreceptor RIG-I Overcomes Melanoma-Intrinsic Resistance to T Cell Immunotherapy. J. Clin. Invest. 2020, 130 (8), 4266–4281. https://doi.org/10.1172/JCI131572. [CrossRef]
- Paulson, K. G.; Voillet, V.; McAfee, M. S.; Hunter, D. S.; Wagener, F. D.; Perdicchio, M.; Valente, W. J.; Koelle, S. J.; Church, C. D.; Vandeven, N.; Thomas, H.; Colunga, A. G.; Iyer, J. G.; Yee, C.; Kulikauskas, R.; Koelle, D. M.; Pierce, R. H.; Bielas, J. H.; Greenberg, P. D.; Bhatia, S.; Gottardo, R.; Nghiem, P.; Chapuis, A. G. Acquired Cancer Resistance to Combination Immunotherapy from Transcriptional Loss of Class I HLA. Nat Commun 2018, 9 (1), 3868. https://doi.org/10.1038/s41467-018-06300-3. [CrossRef]
- Sade-Feldman, M.; Jiao, Y. J.; Chen, J. H.; Rooney, M. S.; Barzily-Rokni, M.; Eliane, J.-P.; Bjorgaard, S. L.; Hammond, M. R.; Vitzthum, H.; Blackmon, S. M.; Frederick, D. T.; Hazar-Rethinam, M.; Nadres, B. A.; Van Seventer, E. E.; Shukla, S. A.; Yizhak, K.; Ray, J. P.; Rosebrock, D.; Livitz, D.; Adalsteinsson, V.; Getz, G.; Duncan, L. M.; Li, B.; Corcoran, R. B.; Lawrence, D. P.; Stemmer-Rachamimov, A.; Boland, G. M.; Landau, D. A.; Flaherty, K. T.; Sullivan, R. J.; Hacohen, N. Resistance to Checkpoint Blockade Therapy through Inactivation of Antigen Presentation. Nat. Commun. 2017, 8 (1), 1136. https://doi.org/10.1038/s41467-017-01062-w. [CrossRef]
- Castro, F.; Cardoso, A. P.; Gonçalves, R. M.; Serre, K.; Oliveira, M. J. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol. 2018, 9, 847. https://doi.org/10.3389/fimmu.2018.00847. [CrossRef]
- Rodig, S. J.; Gusenleitner, D.; Jackson, D. G.; Gjini, E.; Giobbie-Hurder, A.; Jin, C.; Chang, H.; Lovitch, S. B.; Horak, C.; Weber, J. S.; Weirather, J. L.; Wolchok, J. D.; Postow, M. A.; Pavlick, A. C.; Chesney, J.; Hodi, F. S. MHC Proteins Confer Differential Sensitivity to CTLA-4 and PD-1 Blockade in Untreated Metastatic Melanoma. Sci. Transl. Med. 2018, 10 (450), eaar3342. https://doi.org/10.1126/scitranslmed.aar3342. [CrossRef]
- Karachaliou, N.; Gonzalez-Cao, M.; Crespo, G.; Drozdowskyj, A.; Aldeguer, E.; Gimenez-Capitan, A.; Teixido, C.; Molina-Vila, M. A.; Viteri, S.; De Los Llanos Gil, M.; Algarra, S. M.; Perez-Ruiz, E.; Marquez-Rodas, I.; Rodriguez-Abreu, D.; Blanco, R.; Puertolas, T.; Royo, M. A.; Rosell, R. Interferon Gamma, an Important Marker of Response to Immune Checkpoint Blockade in Non-Small Cell Lung Cancer and Melanoma Patients. Ther. Adv. Med. Oncol. 2018, 10, 1758834017749748. https://doi.org/10.1177/1758834017749748. [CrossRef]
- Gao, J.; Shi, L. Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S. E.; Chen, P.-L.; Hwu, P.; Allison, J. P.; Futreal, A.; Wargo, J. A.; Sharma, P. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167 (2), 397-404.e9. https://doi.org/10.1016/j.cell.2016.08.069. [CrossRef]
- Benci, J. L.; Xu, B.; Qiu, Y.; Wu, T.; Dada, H.; Victor, C. T.-S.; Cucolo, L.; Lee, D. S. M.; Pauken, K. E.; Huang, A. C.; Gangadhar, T. C.; Amaravadi, R. K.; Schuchter, L. M.; Feldman, M. D.; Ishwaran, H.; Vonderheide, R. H.; Maity, A.; Wherry, E. J.; Minn, A. J. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167 (6), 1540-1554.e12. https://doi.org/10.1016/j.cell.2016.11.022. [CrossRef]
- Jiang, X.; Zhou, J.; Giobbie-Hurder, A.; Wargo, J.; Hodi, F. S. The Activation of MAPK in Melanoma Cells Resistant to BRAF Inhibition Promotes PD-L1 Expression That Is Reversible by MEK and PI3K Inhibition. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19 (3), 598–609. https://doi.org/10.1158/1078-0432.CCR-12-2731. [CrossRef]
- Reinhardt, J.; Landsberg, J.; Schmid-Burgk, J. L.; Ramis, B. B.; Bald, T.; Glodde, N.; Lopez-Ramos, D.; Young, A.; Ngiow, S. F.; Nettersheim, D.; Schorle, H.; Quast, T.; Kolanus, W.; Schadendorf, D.; Long, G. V.; Madore, J.; Scolyer, R. A.; Ribas, A.; Smyth, M. J.; Tumeh, P. C.; Tüting, T.; Hölzel, M. MAPK Signaling and Inflammation Link Melanoma Phenotype Switching to Induction of CD73 during Immunotherapy. Cancer Res. 2017, 77 (17), 4697–4709. https://doi.org/10.1158/0008-5472.CAN-17-0395. [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T. F. Melanoma-Intrinsic β-Catenin Signalling Prevents Anti-Tumour Immunity. Nature 2015, 523 (7559), 231–235. https://doi.org/10.1038/nature14404. [CrossRef]
- Zhao, J.; Chen, A. X.; Gartrell, R. D.; Silverman, A. M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; Filip, I.; Orenbuch, R.; Goetz, M.; Yamaguchi, J. T.; Cloney, M.; Horbinski, C.; Lukas, R. V.; Raizer, J.; Rae, A. I.; Yuan, J.; Canoll, P.; Bruce, J. N.; Saenger, Y. M.; Sims, P.; Iwamoto, F. M.; Sonabend, A. M.; Rabadan, R. Immune and Genomic Correlates of Response to Anti-PD-1 Immunotherapy in Glioblastoma. Nat. Med. 2019, 25 (3), 462–469. https://doi.org/10.1038/s41591-019-0349-y. [CrossRef]
- Peng, W.; Chen, J. Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M. T.; Xu, C.; McKenzie, J. A.; Zhang, C.; Liang, X.; Williams, L. J.; Deng, W.; Chen, G.; Mbofung, R.; Lazar, A. J.; Torres-Cabala, C. A.; Cooper, Z. A.; Chen, P.-L.; Tieu, T. N.; Spranger, S.; Yu, X.; Bernatchez, C.; Forget, M.-A.; Haymaker, C.; Amaria, R.; McQuade, J. L.; Glitza, I. C.; Cascone, T.; Li, H. S.; Kwong, L. N.; Heffernan, T. P.; Hu, J.; Bassett, R. L.; Bosenberg, M. W.; Woodman, S. E.; Overwijk, W. W.; Lizée, G.; Roszik, J.; Gajewski, T. F.; Wargo, J. A.; Gershenwald, J. E.; Radvanyi, L.; Davies, M. A.; Hwu, P. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6 (2), 202–216. https://doi.org/10.1158/2159-8290.CD-15-0283. [CrossRef]
- Plaschka, M.; Benboubker, V.; Grimont, M.; Berthet, J.; Tonon, L.; Lopez, J.; Le-Bouar, M.; Balme, B.; Tondeur, G.; de la Fouchardière, A.; Larue, L.; Puisieux, A.; Grinberg-Bleyer, Y.; Bendriss-Vermare, N.; Dubois, B.; Caux, C.; Dalle, S.; Caramel, J. ZEB1 Transcription Factor Promotes Immune Escape in Melanoma. J. Immunother. Cancer 2022, 10 (3), e003484. https://doi.org/10.1136/jitc-2021-003484. [CrossRef]
- Muramatsu, T.; Noguchi, T.; Sugiyama, D.; Kanada, Y.; Fujimaki, K.; Ito, S.; Gotoh, M.; Nishikawa, H. Newly Emerged Immunogenic Neoantigens in Established Tumors Enable Hosts to Regain Immunosurveillance in a T-Cell-Dependent Manner. Int. Immunol. 2021, 33 (1), 39–48. https://doi.org/10.1093/intimm/dxaa049. [CrossRef]
- Yc, L.; Z, Z.; Fj, L.; Jj, G.; Td, P.; Pf, R.; Sa, R. Direct Identification of Neoantigen-Specific TCRs from Tumor Specimens by High-Throughput Single-Cell Sequencing. J. Immunother. Cancer 2021, 9 (7). https://doi.org/10.1136/jitc-2021-002595. [CrossRef]
- Van Allen, E. M.; Miao, D.; Schilling, B.; Shukla, S. A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M. H. G.; Goldinger, S. M.; Utikal, J.; Hassel, J. C.; Weide, B.; Kaehler, K. C.; Loquai, C.; Mohr, P.; Gutzmer, R.; Dummer, R.; Gabriel, S.; Wu, C. J.; Schadendorf, D.; Garraway, L. A. Genomic Correlates of Response to CTLA-4 Blockade in Metastatic Melanoma. Science 2015, 350 (6257), 207–211. https://doi.org/10.1126/science.aad0095. [CrossRef]
- Liu, W. R.; Fisher, D. E. Epitope Spreading and the Efficacy of Immune Checkpoint Inhibition in Cancer. Int. J. Oncol. Res. 2021, 4 (1), 029. https://doi.org/10.23937/2643-4563/1710029. [CrossRef]
- Jenkins, R. W.; Barbie, D. A.; Flaherty, K. T. Mechanisms of Resistance to Immune Checkpoint Inhibitors. Br. J. Cancer 2018, 118 (1), 9–16. https://doi.org/10.1038/bjc.2017.434. [CrossRef]
- Yadollahi, P.; Jeon, Y.-K.; Ng, W. L.; Choi, I. Current Understanding of Cancer-Intrinsic PD-L1: Regulation of Expression and Its Protumoral Activity. BMB Rep. 2021, 54 (1), 12–20. https://doi.org/10.5483/BMBRep.2021.54.1.241. [CrossRef]
- Ahn, A.; Rodger, E. J.; Motwani, J.; Gimenez, G.; Stockwell, P. A.; Parry, M.; Hersey, P.; Chatterjee, A.; Eccles, M. R. Transcriptional Reprogramming and Constitutive PD-L1 Expression in Melanoma Are Associated with Dedifferentiation and Activation of Interferon and Tumour Necrosis Factor Signalling Pathways. Cancers 2021, 13 (17), 4250. https://doi.org/10.3390/cancers13174250. [CrossRef]
- Maitituoheti, M.; Shi, A.; Tang, M.; Ho, L.-L.; Terranova, C.; Galani, K.; Keung, E. Z.; Creasy, C. A.; Wu, M.; Chen, J.; Chen, N.; Singh, A. K.; Chaudhri, A.; Anvar, N. E.; Tarantino, G.; Yang, J.; Sarkar, S.; Jiang, S.; Malke, J.; Haydu, L.; Burton, E.; Davies, M. A.; Gershenwald, J. E.; Hwu, P.; Lazar, A.; Cheah, J. H.; Soule, C. K.; Levine, S. S.; Bernatchez, C.; Saladi, S. V.; Liu, D.; Wargo, J.; Boland, G. M.; Kellis, M.; Rai, K. Enhancer Reprogramming in Melanoma Immune Checkpoint Therapy Resistance. bioRxiv September 3, 2022, p 2022.08.31.506051. https://doi.org/10.1101/2022.08.31.506051. [CrossRef]
- Pozniak, J.; Pedri, D.; Landeloos, E.; Herck, Y. V.; Antoranz, A.; Karras, P.; Nowosad, A.; Makhzami, S.; Bervoets, G.; Dewaele, M.; Vanwynsberghe, L.; Cinque, S.; Kint, S.; Vandereyken, K.; Voet, T.; Vernaillen, F.; Annaert, W.; Lambrechts, D.; Boecxstaens, V.; Oord, J. van den; Bosisio, F.; Leucci, E.; Rambow, F.; Bechter, O.; Marine, J.-C. A TCF4/BRD4-Dependent Regulatory Network Confers Cross-Resistance to Targeted and Immune Checkpoint Therapy in Melanoma. bioRxiv August 13, 2022, p 2022.08.11.502598. https://doi.org/10.1101/2022.08.11.502598. [CrossRef]
- Markovits, E.; Harush, O.; Baruch, E. N.; Shulman, E. D.; Debby, A.; Itzhaki, O.; Anafi, L.; Danilevsky, A.; Shomron, N.; Ben-Betzalel, G.; Asher, N.; Shapira-Frommer, R.; Schachter, J.; Barshack, I.; Geiger, T.; Elkon, R.; Besser, M. J.; Markel, G. MYC Induces Immunotherapy and IFNγ Resistance Through Downregulation of JAK2. Cancer Immunol. Res. 2023, 11 (7), 909–924. https://doi.org/10.1158/2326-6066.CIR-22-0184. [CrossRef]
- Kuehm, L. M.; Khojandi, N.; Piening, A.; Klevorn, L. E.; Geraud, S. C.; McLaughlin, N. R.; Griffett, K.; Burris, T. P.; Pyles, K. D.; Nelson, A. M.; Preuss, M. L.; Bockerstett, K. A.; Donlin, M. J.; McCommis, K. S.; DiPaolo, R. J.; Teague, R. M. Fructose Promotes Cytoprotection in Melanoma Tumors and Resistance to Immunotherapy. Cancer Immunol. Res. 2021, 9 (2), 227–238. https://doi.org/10.1158/2326-6066.CIR-20-0396. [CrossRef]
- Fox, D. B.; Ebright, R. Y.; Hong, X.; Russell, H. C.; Guo, H.; LaSalle, T. J.; Wittner, B. S.; Poux, N.; Vuille, J. A.; Toner, M.; Hacohen, N.; Boland, G. M.; Sen, D. R.; Sullivan, R. J.; Maheswaran, S.; Haber, D. A. Downregulation of KEAP1 in Melanoma Promotes Resistance to Immune Checkpoint Blockade. Npj Precis. Oncol. 2023, 7 (1), 1–7. https://doi.org/10.1038/s41698-023-00362-3. [CrossRef]
- Shields, B. D.; Koss, B.; Taylor, E. M.; Storey, A. J.; West, K. L.; Byrum, S. D.; Mackintosh, S. G.; Edmondson, R.; Mahmoud, F.; Shalin, S. C.; Tackett, A. J. Loss of E-Cadherin Inhibits CD103 Antitumor Activity and Reduces Checkpoint Blockade Responsiveness in Melanoma. Cancer Res. 2019, 79 (6), 1113–1123. https://doi.org/10.1158/0008-5472.CAN-18-1722. [CrossRef]
- Mangani, D.; Huang, L.; Singer, M.; Li, R.; Barilla, R.; Escobar, G.; Tooley, K.; Cheng, H.; Delaney, C.; Newcomer, K.; Nyman, J.; Marjanovic, N.; Nevin, J.; Rozenblatt-Rosen, O.; Kuchroo, V.; Regev, A.; Anderson, A. 964 Dynamic Immune Landscapes during Melanoma Progression Reveal a Role for Endogenous Opioids in Driving T Cell Dysfunction. J. Immunother. Cancer 2022, 10 (Suppl 2). https://doi.org/10.1136/jitc-2022-SITC2022.0964. [CrossRef]
- Simiczyjew, A.; Dratkiewicz, E.; Mazurkiewicz, J.; Ziętek, M.; Matkowski, R.; Nowak, D. The Influence of Tumor Microenvironment on Immune Escape of Melanoma. Int. J. Mol. Sci. 2020, 21 (21), 8359. https://doi.org/10.3390/ijms21218359. [CrossRef]
- Jacobs, J. F. M.; Nierkens, S.; Figdor, C. G.; de Vries, I. J. M.; Adema, G. J. Regulatory T Cells in Melanoma: The Final Hurdle towards Effective Immunotherapy? Lancet Oncol. 2012, 13 (1), e32-42. https://doi.org/10.1016/S1470-2045(11)70155-3. [CrossRef]
- Fujimura, T.; Kambayashi, Y.; Aiba, S. Crosstalk between Regulatory T Cells (Tregs) and Myeloid Derived Suppressor Cells (MDSCs) during Melanoma Growth. Oncoimmunology 2012, 1 (8), 1433–1434. https://doi.org/10.4161/onci.21176. [CrossRef]
- Saleh, R.; Elkord, E. Treg-Mediated Acquired Resistance to Immune Checkpoint Inhibitors. Cancer Lett. 2019, 457, 168–179. https://doi.org/10.1016/j.canlet.2019.05.003. [CrossRef]
- Beier, U. H. Apoptotic Regulatory T Cells Retain Suppressive Function through Adenosine. Cell Metab. 2018, 27 (1), 5–7. https://doi.org/10.1016/j.cmet.2017.12.013. [CrossRef]
- Maj, T.; Wang, W.; Crespo, J.; Zhang, H.; Wang, W.; Wei, S.; Zhao, L.; Vatan, L.; Shao, I.; Szeliga, W.; Lyssiotis, C.; Liu, J. R.; Kryczek, I.; Zou, W. Oxidative Stress Controls Regulatory T Cell Apoptosis and Suppressor Activity and PD-L1-Blockade Resistance in Tumor. Nat. Immunol. 2017, 18 (12), 1332–1341. https://doi.org/10.1038/ni.3868. [CrossRef]
- Fujimura, T.; Mahnke, K.; Enk, A. H. Myeloid Derived Suppressor Cells and Their Role in Tolerance Induction in Cancer. Journal of Dermatological Science 2010, 59 (1), 1–6. https://doi.org/10.1016/j.jdermsci.2010.05.001. [CrossRef]
- Meyer, C.; Cagnon, L.; Costa-Nunes, C. M.; Baumgaertner, P.; Montandon, N.; Leyvraz, L.; Michielin, O.; Romano, E.; Speiser, D. E. Frequencies of Circulating MDSC Correlate with Clinical Outcome of Melanoma Patients Treated with Ipilimumab. Cancer Immunol. Immunother. 2014, 63 (3), 247–257. https://doi.org/10.1007/s00262-013-1508-5. [CrossRef]
- Sade-Feldman, M.; Kanterman, J.; Klieger, Y.; Ish-Shalom, E.; Olga, M.; Saragovi, A.; Shtainberg, H.; Lotem, M.; Baniyash, M. Clinical Significance of Circulating CD33+CD11b+HLA-DR− Myeloid Cells in Patients with Stage IV Melanoma Treated with Ipilimumab. Clin. Cancer Res. 2016, 22 (23), 5661–5672. https://doi.org/10.1158/1078-0432.CCR-15-3104. [CrossRef]
- Weide, B.; Martens, A.; Zelba, H.; Stutz, C.; Derhovanessian, E.; Di Giacomo, A. M.; Maio, M.; Sucker, A.; Schilling, B.; Schadendorf, D.; Büttner, P.; Garbe, C.; Pawelec, G. Myeloid-Derived Suppressor Cells Predict Survival of Patients with Advanced Melanoma: Comparison with Regulatory T Cells and NY-ESO-1- or Melan-A–Specific T Cells. Clin. Cancer Res. 2014, 20 (6), 1601–1609. https://doi.org/10.1158/1078-0432.CCR-13-2508. [CrossRef]
- Tirosh, I.; Izar, B.; Prakadan, S. M.; Wadsworth, M. H.; Treacy, D.; Trombetta, J. J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; Fallahi-Sichani, M.; Dutton-Regester, K.; Lin, J.-R.; Cohen, O.; Shah, P.; Lu, D.; Genshaft, A. S.; Hughes, T. K.; Ziegler, C. G. K.; Kazer, S. W.; Gaillard, A.; Kolb, K. E.; Villani, A.-C.; Johannessen, C. M.; Andreev, A. Y.; Van Allen, E. M.; Bertagnolli, M.; Sorger, P. K.; Sullivan, R. J.; Flaherty, K. T.; Frederick, D. T.; Jané-Valbuena, J.; Yoon, C. H.; Rozenblatt-Rosen, O.; Shalek, A. K.; Regev, A.; Garraway, L. A. Dissecting the Multicellular Ecosystem of Metastatic Melanoma by Single-Cell RNA-Seq. Science 2016, 352 (6282), 189–196. https://doi.org/10.1126/science.aad0501. [CrossRef]
- Diazzi, S.; Tartare-Deckert, S.; Deckert, M. The Mechanical Phenotypic Plasticity of Melanoma Cell: An Emerging Driver of Therapy Cross-Resistance. Oncogenesis 2023, 12 (1), 7. https://doi.org/10.1038/s41389-023-00452-8. [CrossRef]
- Lee, J. H.; Shklovskaya, E.; Lim, S. Y.; Carlino, M. S.; Menzies, A. M.; Stewart, A.; Pedersen, B.; Irvine, M.; Alavi, S.; Yang, J. Y. H.; Strbenac, D.; Saw, R. P. M.; Thompson, J. F.; Wilmott, J. S.; Scolyer, R. A.; Long, G. V.; Kefford, R. F.; Rizos, H. Transcriptional Downregulation of MHC Class I and Melanoma De- Differentiation in Resistance to PD-1 Inhibition. Nat. Commun. 2020, 11 (1), 1897. https://doi.org/10.1038/s41467-020-15726-7. [CrossRef]
- Hossain, S. M.; Gimenez, G.; Stockwell, P. A.; Tsai, P.; Print, C. G.; Rys, J.; Cybulska-Stopa, B.; Ratajska, M.; Harazin-Lechowska, A.; Almomani, S.; Jackson, C.; Chatterjee, A.; Eccles, M. R. Innate Immune Checkpoint Inhibitor Resistance Is Associated with Melanoma Sub-Types Exhibiting Invasive and de-Differentiated Gene Expression Signatures. 2022. https://doi.org/10.3389/fimmu.2022.955063. [CrossRef]
- Munn, D. H.; Mellor, A. L. Indoleamine 2,3-Dioxygenase and Tumor-Induced Tolerance. J. Clin. Invest. 2007, 117 (5), 1147–1154. https://doi.org/10.1172/JCI31178. [CrossRef]
- Uyttenhove, C.; Pilotte, L.; Théate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van den Eynde, B. J. Evidence for a Tumoral Immune Resistance Mechanism Based on Tryptophan Degradation by Indoleamine 2,3-Dioxygenase. Nat. Med. 2003, 9 (10), 1269–1274. https://doi.org/10.1038/nm934. [CrossRef]
- Holmgaard, R. B.; Zamarin, D.; Munn, D. H.; Wolchok, J. D.; Allison, J. P. Indoleamine 2,3-Dioxygenase Is a Critical Resistance Mechanism in Antitumor T Cell Immunotherapy Targeting CTLA-4. J. Exp. Med. 2013, 210 (7), 1389–1402. https://doi.org/10.1084/jem.20130066. [CrossRef]
- Mitchell, T. C.; Hamid, O.; Smith, D. C.; Bauer, T. M.; Wasser, J. S.; Olszanski, A. J.; Luke, J. J.; Balmanoukian, A. S.; Schmidt, E. V.; Zhao, Y.; Gong, X.; Maleski, J.; Leopold, L.; Gajewski, T. F. Epacadostat Plus Pembrolizumab in Patients With Advanced Solid Tumors: Phase I Results From a Multicenter, Open-Label Phase I/II Trial (ECHO-202/KEYNOTE-037). J. Clin. Oncol. 2018, 36 (32), 3223–3230. https://doi.org/10.1200/JCO.2018.78.9602. [CrossRef]
- Long, G. V.; Dummer, R.; Hamid, O.; Gajewski, T. F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M. S.; Grob, J.-J.; Kim, T. M.; Demidov, L.; Robert, C.; Larkin, J.; Anderson, J. R.; Maleski, J.; Jones, M.; Diede, S. J.; Mitchell, T. C. Epacadostat plus Pembrolizumab versus Placebo plus Pembrolizumab in Patients with Unresectable or Metastatic Melanoma (ECHO-301/KEYNOTE-252): A Phase 3, Randomised, Double-Blind Study. Lancet Oncol. 2019, 20 (8), 1083–1097. https://doi.org/10.1016/S1470-2045(19)30274-8. [CrossRef]
- Iga, N.; Otsuka, A.; Hirata, M.; Kataoka, T. R.; Irie, H.; Nakashima, C.; Matsushita, S.; Uchi, H.; Yamamoto, Y.; Funakoshi, T.; Fujisawa, Y.; Yoshino, K.; Fujimura, T.; Hata, H.; Ishida, Y.; Kabashima, K. Variable Indoleamine 2,3-Dioxygenase Expression in Acral/Mucosal Melanoma and Its Possible Link to Immunotherapy. Cancer Sci. 2019, 110 (11), 3434–3441. https://doi.org/10.1111/cas.14195. [CrossRef]
- Georgoudaki, A.-M.; Prokopec, K. E.; Boura, V. F.; Hellqvist, E.; Sohn, S.; Östling, J.; Dahan, R.; Harris, R. A.; Rantalainen, M.; Klevebring, D.; Sund, M.; Brage, S. E.; Fuxe, J.; Rolny, C.; Li, F.; Ravetch, J. V.; Karlsson, M. C. I. Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis. Cell Rep. 2016, 15 (9), 2000–2011. https://doi.org/10.1016/j.celrep.2016.04.084. [CrossRef]
- Pu, Y.; Ji, Q. Tumor-Associated Macrophages Regulate PD-1/PD-L1 Immunosuppression. Front. Immunol. 2022, 13, 874589. https://doi.org/10.3389/fimmu.2022.874589. [CrossRef]
- Arozarena, I.; Wellbrock, C. Phenotype Plasticity as Enabler of Melanoma Progression and Therapy Resistance. Nat Rev Cancer 2019, 19 (7), 377–391. https://doi.org/10.1038/s41568-019-0154-4. [CrossRef]
- Watson, M. J.; Vignali, P. D. A.; Mullett, S. J.; Overacre-Delgoffe, A. E.; Peralta, R. M.; Grebinoski, S.; Menk, A. V.; Rittenhouse, N. L.; DePeaux, K.; Whetstone, R. D.; Vignali, D. A. A.; Hand, T. W.; Poholek, A. C.; Morrison, B. M.; Rothstein, J. D.; Wendell, S. G.; Delgoffe, G. M. Metabolic Support of Tumour-Infiltrating Regulatory T Cells by Lactic Acid. Nature 2021, 591 (7851), 645–651. https://doi.org/10.1038/s41586-020-03045-2. [CrossRef]
- Daneshmandi, S.; Wegiel, B.; Seth, P. Blockade of Lactate Dehydrogenase-A (LDH-A) Improves Efficacy of Anti-Programmed Cell Death-1 (PD-1) Therapy in Melanoma. Cancers 2019, 11 (4), 450. https://doi.org/10.3390/cancers11040450. [CrossRef]
- Jaiswal, A. R.; Liu, A. J.; Pudakalakatti, S.; Dutta, P.; Jayaprakash, P.; Bartkowiak, T.; Ager, C. R.; Wang, Z.-Q.; Reuben, A.; Cooper, Z. A.; Ivan, C.; Ju, Z.; Nwajei, F.; Wang, J.; Davies, M. A.; Davis, R. E.; Wargo, J. A.; Bhattacharya, P. K.; Hong, D. S.; Curran, M. A. Melanoma Evolves Complete Immunotherapy Resistance through the Acquisition of a Hypermetabolic Phenotype. Cancer Immunol. Res. 2020, 8 (11), 1365–1380. https://doi.org/10.1158/2326-6066.CIR-19-0005. [CrossRef]
- Dyck, L.; Mills, K. H. G. Immune Checkpoints and Their Inhibition in Cancer and Infectious Diseases. European Journal of Immunology 2017, 47 (5), 765–779. https://doi.org/10.1002/eji.201646875. [CrossRef]
- Kato, S.; Okamura, R.; Kumaki, Y.; Ikeda, S.; Nikanjam, M.; Eskander, R.; Goodman, A.; Lee, S.; Glenn, S. T.; Dressman, D.; Papanicolau-Sengos, A.; Lenzo, F. L.; Morrison, C.; Kurzrock, R. Expression of TIM3/VISTA Checkpoints and the CD68 Macrophage-Associated Marker Correlates with Anti-PD1/PDL1 Resistance: Implications of Immunogram Heterogeneity. Oncoimmunology 2020, 9 (1), 1708065. https://doi.org/10.1080/2162402X.2019.1708065. [CrossRef]
- Kawashima, S.; Inozume, T.; Kawazu, M.; Ueno, T.; Nagasaki, J.; Tanji, E.; Honobe, A.; Ohnuma, T.; Kawamura, T.; Umeda, Y.; Nakamura, Y.; Kawasaki, T.; Kiniwa, Y.; Yamasaki, O.; Fukushima, S.; Ikehara, Y.; Mano, H.; Suzuki, Y.; Nishikawa, H.; Matsue, H.; Togashi, Y. TIGIT/CD155 Axis Mediates Resistance to Immunotherapy in Patients with Melanoma with the Inflamed Tumor Microenvironment. J. Immunother. Cancer 2021, 9 (11), e003134. https://doi.org/10.1136/jitc-2021-003134. [CrossRef]
- Igarashi, H.; Fukuda, M.; Konno, Y.; Takano, H. Abscopal Effect of Radiation Therapy after Nivolumab Monotherapy in a Patient with Oral Mucosal Melanoma: A Case Report. Oral Oncol. 2020, 108, 104919. https://doi.org/10.1016/j.oraloncology.2020.104919. [CrossRef]
- Vera Aguilera, J.; Paludo, J.; McWilliams, R. R.; Zhang, H.; Li, Y.; Kumar, A. B.; Failing, J.; Kottschade, L. A.; Block, M. S.; Markovic, S. N.; Dong, H.; Dronca, R. S.; Yan, Y. Chemo-Immunotherapy Combination after PD-1 Inhibitor Failure Improves Clinical Outcomes in Metastatic Melanoma Patients. Melanoma Res. 2020, 30 (4), 364–375. https://doi.org/10.1097/CMR.0000000000000669. [CrossRef]
- Zhang, W.; Hu, B.; Han, J.; Wang, Z.; Ma, G.; Ye, H.; Yuan, J.; Cao, J.; Zhang, Z.; Shi, J.; Chen, M.; Wang, X.; Xu, Y.; Cheng, Y.; Tian, L.; Wang, H.; Lu, S. Surgery After Conversion Therapy With PD-1 Inhibitors Plus Tyrosine Kinase Inhibitors Are Effective and Safe for Advanced Hepatocellular Carcinoma: A Pilot Study of Ten Patients. Front. Oncol. 2021, 11.
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic Virus Therapy: A New Era of Cancer Treatment at Dawn. Cancer Sci. 2016, 107 (10), 1373–1379. https://doi.org/10.1111/cas.13027. [CrossRef]
- Sivanandam, V.; LaRocca, C. J.; Chen, N. G.; Fong, Y.; Warner, S. G. Oncolytic Viruses and Immune Checkpoint Inhibition: The Best of Both Worlds. Molecular Therapy - Oncolytics 2019, 13, 93–106. https://doi.org/10.1016/j.omto.2019.04.003. [CrossRef]
- Yamada, T.; Tateishi, R.; Iwai, M.; Koike, K.; Todo, T. Neoadjuvant Use of Oncolytic Herpes Virus G47Δ Enhances the Antitumor Efficacy of Radiofrequency Ablation. Mol Ther Oncolytics 2020, 18, 535–545. https://doi.org/10.1016/j.omto.2020.08.010. [CrossRef]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R. H. I.; Michielin, O.; Olszanski, A. J.; Malvehy, J.; Cebon, J.; Fernandez, E.; Kirkwood, J. M.; Gajewski, T. F.; Chen, L.; Gorski, K. S.; Anderson, A. A.; Diede, S. J.; Lassman, M. E.; Gansert, J.; Hodi, F. S.; Long, G. V. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170 (6), 1109-1119.e10. https://doi.org/10.1016/j.cell.2017.08.027. [CrossRef]
- Sugawara, K.; Iwai, M.; Ito, H.; Tanaka, M.; Seto, Y.; Todo, T. Oncolytic Herpes Virus G47Δ Works Synergistically with CTLA-4 Inhibition via Dynamic Intratumoral Immune Modulation. Mol. Ther. - Oncolytics 2021, 22, 129–142. https://doi.org/10.1016/j.omto.2021.05.004. [CrossRef]
- Uchihashi, T.; Nakahara, H.; Fukuhara, H.; Iwai, M.; Ito, H.; Sugauchi, A.; Tanaka, M.; Kogo, M.; Todo, T. Oncolytic Herpes Virus G47Δ Injected into Tongue Cancer Swiftly Traffics in Lymphatics and Suppresses Metastasis. Mol. Ther. - Oncolytics 2021, 22, 388–398. https://doi.org/10.1016/j.omto.2021.06.008. [CrossRef]
- Batlle, E.; Massagué, J. Transforming Grown Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50 (4), 924–940. https://doi.org/10.1016/j.immuni.2019.03.024. [CrossRef]
- Mariathasan, S.; Turley, S. J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel III, E. E.; Koeppen, H.; Astarita, J. L.; Cubas, R.; Jhunjhunwala, S.; Banchereau, R.; Yang, Y.; Guan, Y.; Chalouni, C.; Ziai, J.; Şenbabaoğlu, Y.; Santoro, S.; Sheinson, D.; Hung, J.; Giltnane, J. M.; Pierce, A. A.; Mesh, K.; Lianoglou, S.; Riegler, J.; Carano, R. A. D.; Eriksson, P.; Höglund, M.; Somarriba, L.; Halligan, D. L.; van der Heijden, M. S.; Loriot, Y.; Rosenberg, J. E.; Fong, L.; Mellman, I.; Chen, D. S.; Green, M.; Derleth, C.; Fine, G. D.; Hegde, P. S.; Bourgon, R.; Powles, T. TGFβ Attenuates Tumour Response to PD-L1 Blockade by Contributing to Exclusion of T Cells. Nature 2018, 554 (7693), 544–548. https://doi.org/10.1038/nature25501. [CrossRef]
- Kodama, S.; Podyma-Inoue, K. A.; Uchihashi, T.; Kurioka, K.; Takahashi, H.; Sugauchi, A.; Takahashi, K.; Inubushi, T.; Kogo, M.; Tanaka, S.; Watabe, T. Progression of Melanoma Is Suppressed by Targeting All Transforming Growth Factor-β Isoforms with an Fc Chimeric Receptor. Oncol Rep 2021, 46 (3), 197. https://doi.org/10.3892/or.2021.8148. [CrossRef]
Figure 1.
The intrinsic mechanism underlying the resistance to immune checkpoint blockade. This mechanism includes the following elements: 1. alterations in signaling pathways, transcription factors, and regulatory networks; 2. lack of antigenic mutations/neoantigens; and 3. defects in antigen processing and presenting machinery. Upregulation of the MAPK pathway can contribute to resistance by increasing PDL-1 expression and also through defective T cell infiltration and the acquisition of a mesenchymal phenotype by melanoma cells. Activation of the β-catenin pathway leads to T cell exclusion and resistance, as does the loss of Pten and the upregulation of the transcription factor Zeb1. The lack of tumor antigens or antigenic mutations poses a barrier to the immune system’s ability to recognize tumor cells. A regulatory network dependent on TCF4/BRD4 not only hinders antigen presentation, interferon signaling, and activation of leukocytes-associated gene expression but also supports the acquisition of a mesenchymal phenotype, leading to resistance. For an effective immune response, tumor antigens need to be presented to the immune system. Dysregulation of MHC-1, B2M, and TAP in the antigen processing and presenting machinery hinders proper antigen presentation, thereby impeding a robust immune response. Interferon-gamma plays a dual role in resistance mechanisms by upregulating both MHC-1 and MHC-2, contributing to antigen processing and presenting machinery, while also leading to PDL-1 expression. Although PDL-1 expression can lead to T cell dysfunction and resistance, it has been observed that cases with lower PDL-1 expression can result in insensitivity to anti-PDL-1 immunotherapy. Abbreviations: MHC, major histocompatibility complex; TAP, the transporter associated with antigen processing; B2M, beta-2-microglobulin; IFN-γ, interferon gamma; MAPK, mitogen-activated protein kinases; PTEN, phosphatase and tensin homolog; Zeb1, zinc finger E-box-binding homeobox 1; ER, endoplasmic reticulum.
Figure 1.
The intrinsic mechanism underlying the resistance to immune checkpoint blockade. This mechanism includes the following elements: 1. alterations in signaling pathways, transcription factors, and regulatory networks; 2. lack of antigenic mutations/neoantigens; and 3. defects in antigen processing and presenting machinery. Upregulation of the MAPK pathway can contribute to resistance by increasing PDL-1 expression and also through defective T cell infiltration and the acquisition of a mesenchymal phenotype by melanoma cells. Activation of the β-catenin pathway leads to T cell exclusion and resistance, as does the loss of Pten and the upregulation of the transcription factor Zeb1. The lack of tumor antigens or antigenic mutations poses a barrier to the immune system’s ability to recognize tumor cells. A regulatory network dependent on TCF4/BRD4 not only hinders antigen presentation, interferon signaling, and activation of leukocytes-associated gene expression but also supports the acquisition of a mesenchymal phenotype, leading to resistance. For an effective immune response, tumor antigens need to be presented to the immune system. Dysregulation of MHC-1, B2M, and TAP in the antigen processing and presenting machinery hinders proper antigen presentation, thereby impeding a robust immune response. Interferon-gamma plays a dual role in resistance mechanisms by upregulating both MHC-1 and MHC-2, contributing to antigen processing and presenting machinery, while also leading to PDL-1 expression. Although PDL-1 expression can lead to T cell dysfunction and resistance, it has been observed that cases with lower PDL-1 expression can result in insensitivity to anti-PDL-1 immunotherapy. Abbreviations: MHC, major histocompatibility complex; TAP, the transporter associated with antigen processing; B2M, beta-2-microglobulin; IFN-γ, interferon gamma; MAPK, mitogen-activated protein kinases; PTEN, phosphatase and tensin homolog; Zeb1, zinc finger E-box-binding homeobox 1; ER, endoplasmic reticulum.
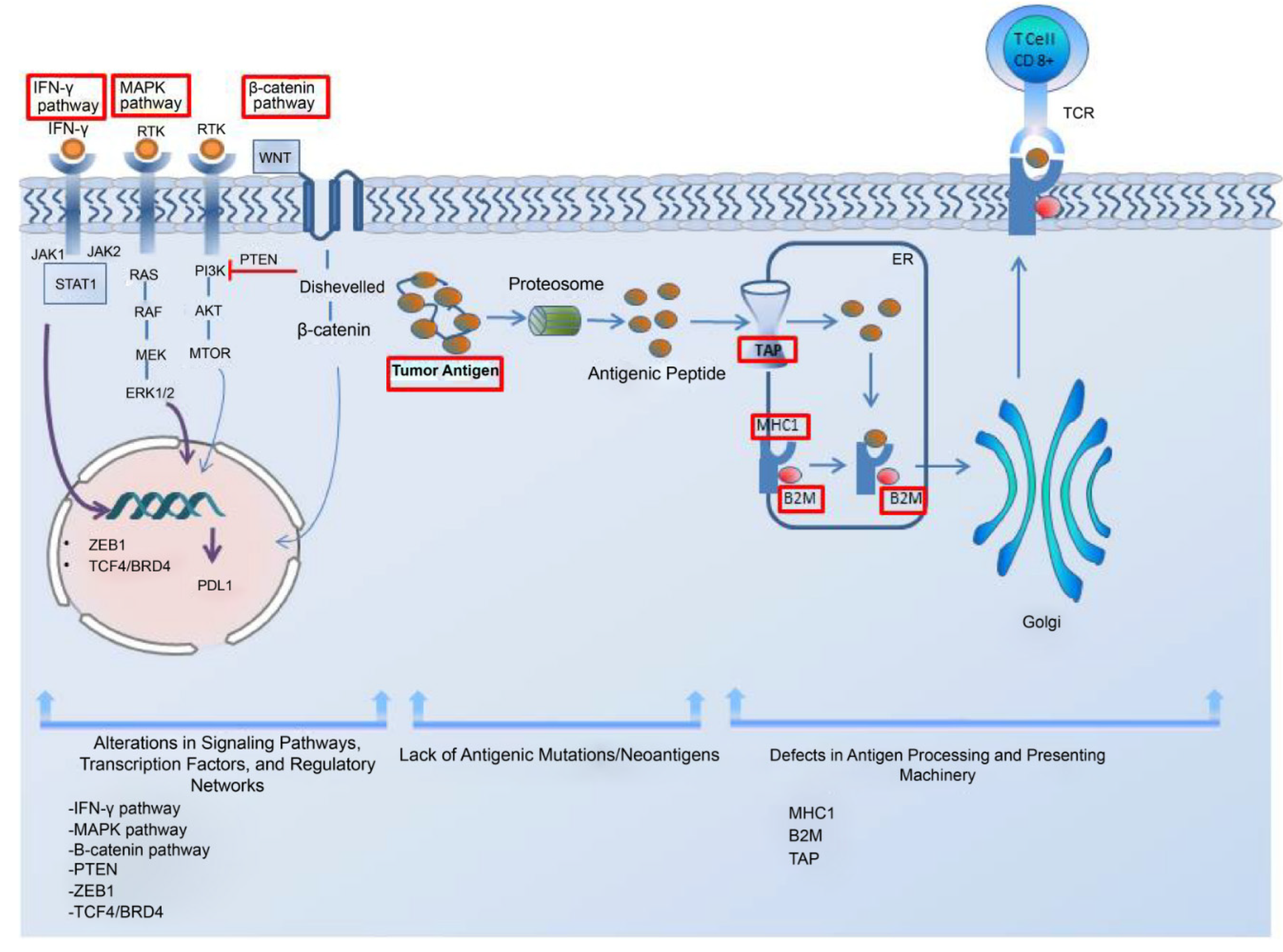
Figure 2.
The extrinsic mechanism underlying the resistance to immune checkpoint blockade.This mechanism of resistance to immune checkpoint blockade includes extrinsic (A) immune suppressive cell components such as regulatory T cells (Treg), tumor-associated macrophages (TAMs), regulatory/tolerogenic dendritic cells (DC), and myeloid-derived suppressor cells (MDSC), (B) various inhibitory receptors such as PDL-1, CTLA-4, LAG-3, TIMS-3, and VISTA, (C) hypoxic tumor microenvironment, and (D) phenotype switching. Abbreviations: LAG-3, lymphocyte-activation gene 3; PDL1, programmed death-ligand 1; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; TIM-3, T-cell immunoglobulin and mucin domain 3; VISTA, V-domain I g suppressor of T cell activation; HIF-1α, hypoxia-inducible factor 1-alpha; MITF, microphthalmia-associated transcription factor
Figure 2.
The extrinsic mechanism underlying the resistance to immune checkpoint blockade.This mechanism of resistance to immune checkpoint blockade includes extrinsic (A) immune suppressive cell components such as regulatory T cells (Treg), tumor-associated macrophages (TAMs), regulatory/tolerogenic dendritic cells (DC), and myeloid-derived suppressor cells (MDSC), (B) various inhibitory receptors such as PDL-1, CTLA-4, LAG-3, TIMS-3, and VISTA, (C) hypoxic tumor microenvironment, and (D) phenotype switching. Abbreviations: LAG-3, lymphocyte-activation gene 3; PDL1, programmed death-ligand 1; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; TIM-3, T-cell immunoglobulin and mucin domain 3; VISTA, V-domain I g suppressor of T cell activation; HIF-1α, hypoxia-inducible factor 1-alpha; MITF, microphthalmia-associated transcription factor
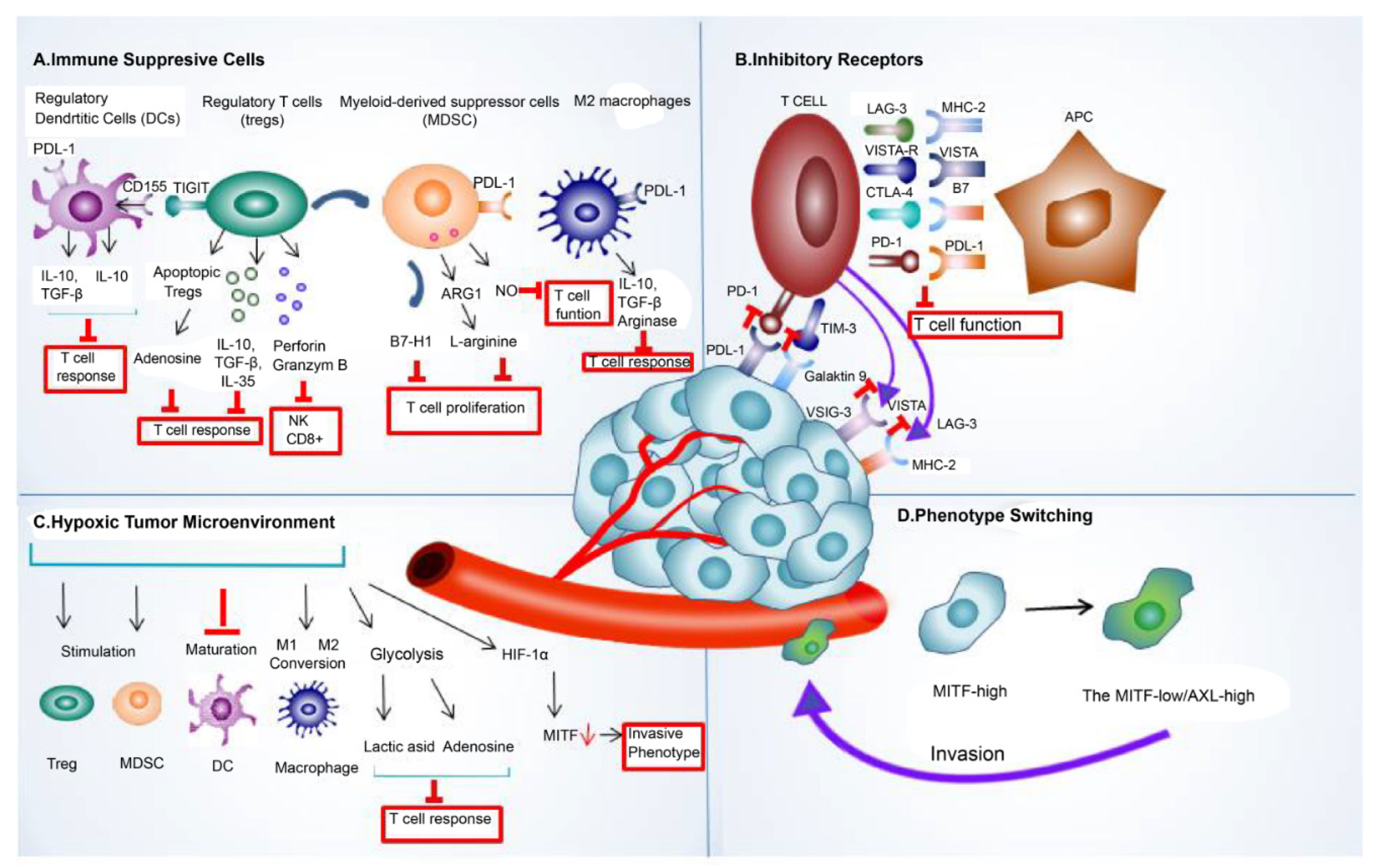
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



