
Submitted:
10 October 2023
Posted:
10 October 2023
You are already at the latest version
Abstract
Introduction: Desmoid tumor (DT) is a rare benign neoplasm rising from muscle aponeurosis, having been associated mostly with trauma or pregnancy. DT has an infiltrative and locally aggressive growth pattern and usually does not metastasize. However, it has a high recurrence and complication rate. When they occur in pregnancy, are all pregnancies and deliveries taken as individual case for optimal management by the physicians and midwifes, who need to be cautious in finding the optimal delivery mode for patients, which depends on tumor size, location, behavior and past history. Study Objectives: We present a case report of a large desmoid tumor of the anterior abdominal wall in a 29-year-old woman in pregnancy, presenting a delivery problem, which resolved in surgical management. Moreover, we are bringing a systematic review of the literature to provide an overview on pathomechanism, symptoms, diagnostics and treatment management of the pregnant women affected by DT. Results: The authors report a case of pregnant women who underwent systemic oncological treatment for abdominal wall desmoid tumor and have been pregnant afterwards. The observational conservative management was chosen with an elective cesarean section at the 38+4 pregnancy week with uncomplicated postpartum follow-up. Conclusion: Pregnancy-associated desmoid tumors are very rare and optimal management of this tumor is not well established, despite some guidelines for non-pregnant patients. The authors reviewed the literature focusing on the actual modern management of desmoid tumors at all, including patients during the pregnancy, as well.
Keywords:
benign neoplasm
; desmoid tumors
; chemotherapy
; high-risk pregnancy
; delivery
; uncommon complications
; abdominal discomfort
; management
Introduction
Desmoid fibromatosis (DF) or sporadicall named as desmoid tumors (DT) represents rare type of tumors, accounting for 0.03% of all neoplasms and less than 3% of tumors arrising from soft tissue (1). DF according to WHO (2020) is defined as a locally aggressive but non-metastasizing deep-seated (myo)fibroblastic monoclonal neoplasm with infiltrative growth pattern and tendency for local recurrence (2). In the International Classification of Diseases (ICD) it is classified as D48.1 diagnosis (3).
Incidence of DF is 2.4 to 5 per one million population per year. DTs predominantly affect young females, mostly between age of 30 to 40 years and occurs more than twice as often in female than male patients. However, in older patients, the incidence do not depend on gender (4-6). Reflecting the age it can be described as follow: the "juvenile" type of DT, which ocusrs predominantly as extra-abdominal desmoid tumor mostly in women; "fertile" DT, that is exclusively abdominal localized DT in women in fertile age; "menopausal" DT, a predominantly abdominal tumor where the sex ratio approaches 1:1; and "senescent" type of DT, where abdominal and extra-abdominal varieties are equally frequent among female and male population (5).
Reflecting their location can be desmoid tumors classified as extra-abdominal and abdominal. Abdominal DTs are either superficial (abdominal wall) or intra-abdominal. These tumors shows a high recurrence rate even if there are og benign feature on microscopic histological assessment. Their biologic behavior often indicates patterns of the "malignant" disease because severe progression and local infiltration to vital organs leads to their impairment and sometimes to the patients death. Intraabdominal DTs can affect surrounding organs and vessels, what may greatly complicate their surgical treatment (7,8). As for the topical anatomic location are DT present in extra-abdominal locations (49%), abdominal wall (40%), and intra-abdominal areas including retroperitoneal space (8%) (6).
The extra-abdominal lesions mostly occur in the neck, shoulder, chest wall, breast, back, arm, buttock, thigh and leg. Multicentric extra-abdominal desmoids are very rare and thay have specific clinical presentation (9,10).
Historically, these tumors were first described by MacFarland in 1832. Etymologically, the word “desmoid” was first time used by professor Johannes Müller (1801–1858, chairman of the Department of Anatomy, Physiology and Pathology at the University of Berlin) in his monograph on cancer (11), published in Berlin in 1838, where he done the detailed analysis of the microscopic features of benign and malignant neoplasms in humans. The name is derived from the Greek word «Desmos», which means band or tendon-like (12). In 1951, Gardner first described the often presence of desmoid tumors in patients with Familial adenomatous polyposis (FAP).
The detailed pathogenesis and factors affecting clinical behavior of DTs are still uncertain. However, most cases of DTs were described in patients with previous abdominal trauma or surgery (either laparoscopic, robotic or open) (13-16).
DT is histologically arising from connective tissues of muscle (the result of abnormal monoclonal, fibroblastic proliferation or proliferation of myofibroblasts), the fascia or the aponeurosis. Recently, studies showed the important role of impairments in the Wnt/β-catenin signaling pathway or mutations in APC and CTNNB1 genes as a 'key' trigger for the development of desmoid tumors (17,18).
Although histology is the gold standard to sett the diagnosis, imaging modalities represents the basic tool in the diagnostic process of these tumors. To establish an adequate therapeutic approach, the proper diagnosis is necessary. Multimodal imaging tools, including computed tomography (CT), ultrasound (US) and Magnetic Resonance Imaging (MRI), are helpful tools in the assessment process. These techniques can be also used to guide the minimally invasive interventions and monitor their effectiveness in the treatment (19).
The management is multidisciplinary and often repetitive as DTs growth locally invasive and prone to the high local recurrence after resection.
Since their clinical behavior is heterogeneous and unpredictable where outcome is impacted by anatomic area, nearness to crucial organs, affiliation with FAP, and natural behavior, the treatment ought be individualized in aim to decrease the risk for treatment failure with subsequent tumor recurrence and achieving the acceptable morbidity with highets possible quality of life (20). Numerous issues with respect to ideal treatment of desmoids stay disputable. In any case, wide surgical extraction remains the treatment of choice, except when it is mutilating or is associated with significant organ function impairment and morbidity. Involvement of surgical margins is likely related with an increased risk of local recurrence. In this case, surgical re-resection, adjuvant radiation, systemic therapy or close clinical follow-up might all be appropriate alternatives (21).
Apart of this approach, there is in the last years increasing evidence proving significant progression in the systemic, biological or focused technological treatment possibilities for these patients.
In this paper, we report a case of young female who was diagnosed with desmoid tumor before pregnancy, underwent oncological treatment and has been pregnant with a residual desmoid tumor in anterior abdominal wall, thus leading to a special obstetric problem requiring individual management. Moreover, we are presenting an overview of the current local or systemic therapeutic possibilities for patients affected by desmoid fibromatosis with special attention to DTs in pregnancy.
Case report
A 29-year old primiparous woman with physiological pregnancy was refered to a primary care hospital for follow-up during pregnancy and planning of delivery with the first check in 12+4 week of pregnancy. She has underwent one legal abortion in the past history.
She was non-smoker, denied drinking alcohol or using illicit drugs, her pregravid body mass index was 20.2, and her past medical history was significant for FAP for which she underwent laparoscopic total proctocolectomy with ileoanal reservoir, with transient right-sided relieving colostomy at the age of nineteen. This due to papillous tubulovillious adenoma with low grade intraepithelial (LB-IEN) neoplasia. Subsequently regullarly (regularly) followed by colonoscopic examinations finding sporadic tubular adenomas with LG-IEN on excisional biopsies. Colostomy was closed back 8 months after the primary surgery.
When the patient turned 26 (6 years after the surgery) she developed a desmoid tumor sized 12 x 4.6 x 6.6 cm on abdominal ultrasound scan (Figure 1) and 8.8 x 4.8 x 15.3 cm on MRI scan (Figure 2) in anterior abdominal wall on the right side close to previous troacar point of incision after laparoscopic surgery for FAP and transient relieving stomy. Primarily suspected from sarcoma or desmoid tumor. A core-cutt biopsy was performed confirming the diagnosis desmoid fibromatos tumor, estrogene receptor positive (ER+). The tumor-board considered the situation as stable, with the possibility for partial spontaneous regression. As the risk for recurrence was assessed on to be 50%, no surgery was performed and she was referred for follow-up with new MRI control in about 3 months. During this follow up period she got pregnant and underwent a provoked medical legal abortion in week 6.
The 3-months MRI control (Figure 3) showed stable tumor size, however MRI check after six months showed significant tumor growth progression (Figure 4), sized 10.5 x 6.2 x 17.5 cm in cranio-caudal diameter located in musculus rectus abdominis on the right side. She was recommended to cut-of oral contraceptives and preventive devices (IUD) containing estrogene and/or progesterone and was referred to the National Oncology Center for further treatment. Here, the local Tumor-board concluded with the administration of chemotherapy, where treatment started with 3 cycles of Caelyx (doxorubicin hydrochloride), 40mg/m2 every 4th week with MRI control afterwards. The chemotherapy was administred at our local hospital. The patient suffered side effects in form of skin rashes, allergic respiratory problems and mucositis. She received every cyclus 60 mg of Caelyx i.v. Control MRI scan showed no effect of the treatment, furthermore slightly progression in the cranio-caudal tumor size (12 x 6.6 x 20 cm), (Figure 5). As the oncologist awaited the late response on chemotherapy, the further plan was to continue in the same treatment options (Caelyx, however in reduced dosage, set to 30mg/m2) due to previous side effects. Patient underwent 3 further cycles with total dose of 47 mg doxorubicin at each cycle. MRI scan after 6 cycles showed partial regression of the tumor, now sized 11.2 x 5.4 x 19.1 cm (Figure 6). She was continuously monitored by surgeon and radiologist with MR scans every 4 months and the 12 month-s scan after chemotherapy the tumor showed significant regress, sized only 1.2 x 3.7 x 8.7 cm (Figure 7). Five weeks later the patient became pregnant and was referred from the midwife for gynecological controls, starting on week 12+4.
All the prenatal controls showed no respiratory or bowels conditions worsening even in the tumor size/growth. All medical checks showed normal fetal growth charts and physiologic findings in maternal-fetal flow circulation, placenta morphology and amniotic fluid indexes out over the pregnancy.
Due to the previous total colectomy for FAP, chemotherapy and location of the tumor with size progression, the conclusion for an elective cesarean section (CS) were done and she delivered at 38+4 week of pregnancy through uncomplicated CS with Pfannenstiel incision and low transverse corporal incision on the uterus. She delivered the healthy baby weighing 3435 g, length 48 cm with Apgar score 10-10-10. Normal peroperative expulsion of placenta, bleeding estimated for 450 ml. The postpartum period followed uncomplicatedly, the patient was treated for anemia by i.v. iron substitution (Ferinject - ferric carboxymaltose; 500 IU), and was discharged home on 4th day postpartum without any clinical problems, advised to avoid hard physical activity in 4 weeks post partum.
Follow-up correspondence was done at 4 months post partum and the patient had recovered well, with a normal physical activity in life, DT is stable in size and the patient is followed-up by regular checks by gastric surgeons for desmoid tumor and FAP with regular colonoscopies and MR scans.
Discussion
The studies of patomechanism in desmoid tumors showed that 90% of them are sporadic, and the remaining 5-10% display a genetic link to FAP (Gardner syndrome). Patients with this syndrome have a 800-1000 times greater risk of developing DTs than in the general population (18,22). Sporadic DTs create most commonly in extra-abdominal areas, but those related with FAP are generally found within the small bowel mesentery and/or in the abdominal wall, predominantly arising in soft tissue (23-26) causing bowel obstruction or ulceration and ureter stenosis. Patients with FAP often develop intra-abdominal tumors after abdominal trauma or surgery (2). Moreover, the risk of DTs in FAP is increased by also certain pathologies in APC variants (27).
Etiology
The etiology of desmoid fibromatosis is multifactorial and incorporates hereditary components (most commonly sporadic somatic CTNNB1 mutations and less frequently germline APC mutations in Gardner syndrome), physical factors (surgery, injury), and hormonal factors as pregnancy or contraception (28-29). Surgery and trauma increases the risk of tumour development via its recovery process where proliferative phase of wound healing exhibit high β-catenin expression (30).
Desmoid tumors are associated with germline variations in APC involving codons 1310–2011 within the mid- to C-terminal parcel of the encoded protein (31). FAP-associated desmoid tumours are caused by APC rather than CTNNB1 variant, the latter being more common in sporadic counterparts; nevertheless, they also show nuclear beta-catenin immunoreactivity secondary to Wnt pathway activation (32).
Pathogenesis
In the pathomechanism of DTs, two mutually exclusive mechanisms have been described. The majority (85-90%) of sporadic DTs results from somatic mutation pathway of CTNNB1 gene. It act on three different point mutations in codons 41 and 45 of exon 3 of the CTNNB1 gene that encodes β-catenin, a protooncogene: p.Thr41Ala, p.Ser45Phe and p.Ser45Pro (33-37). The p.Thr41Ala (55%) and p.Ser45Phe (35%) are the most frequent mutations giving p.Ser45Phe only approximately 10% frequency (33,34,36,38).
A smaller percentage (10-15% of cases) of desmoid tumours (germline APC mutation associated pathway) emerge within the Gardner syndrome. These patients harbor germline mutations in the APC tumour suppressor gene, specifically are changes located at or beyond codon 1444, what lead to loss of heterozygosity of the wildtype allele, however, rare cases of DTs with sporadic APC mutations have been described, as well (39-46). The activating mutations in CTNNB1 or inactivating mutations in APC (where a truncated APC protein with low affinity to β-catenin is created) interfere with β-catenin proteasomal degradation. These processes results in abnormal accumulation of β-catenin within cell nuclei (47,48).
Because β-catenin functions are a part of the transcription apparatus in the nucleus, this increased activation of the WNT/β-catenin pathway is thought to drive tumorigenesis of DT through advance cell transcription, proliferation, adhesion and survival (49-51).
When simplified, this complex integration can be described as a process where DT pathogenesis is strongly connected to Wnt/ β-catenin cascade, and where β-catenin dysregulation plays a crucial part through binding to transducin β-like protein 1 (TBL1/TBLR1), and this complex stimulates the expression of several Wnt/APC/β-catenin pathway target genes, including some proliferative components, such as S100A4 or CTHRC1 (52). The APC genes plays a central part within the phosphorylation and proteosomal degradation of β-catenin and the Wnt pathway, in turn, restrains APC-dependent phosphorylation (53,54).
On grow macroscopy DT resemble scar tissue with firm consistence and gray or whitish color. Under the light microscopy they are displayed by a heterogeneous, poorly characterized and uniform proliferation of spindle cells that resembles myofibroblasts wrapped inside a stroma of abundant collagen and a vascular network missing capsule. No necrosis, cellular atypia or increased mitosis is noted. Inside the nuclei there may be detected euchromatin or heterochromatin. There is no histological difference between sporadic DT and FAP-related, however their molecular profile can be different.
On immunohistochemistry are DTs characterized by nuclear positivity for smooth muscle actin, muscle spocific actin, vimentin, β-catenin, PDGFRb, Cox-2, androgen and β-estrogen receptors. They are negative for S-100, h-caldesmon, desmin, CKIT and CD34 (2,18,55).
Prognosis, prediction and clinical behavior
The biological course of desmoid fibromatosis in certain patients varies a lot. Presentation may vary from asymptomatic lesions to impairing tumors with unpredictable growth, stabilization, and ev. regression. The variety of symptoms is directly associated with the size, location and progression speed of DTs. The intra-abdominal DTs grow asymptomatic until they reach large dimension, leading to intestinal, urinary, vessel obstruction, tissue ischemic dammage with possible perforations or bleeding (56-57). Although tumour-related deaths are rare, they are more common reported in patients with FAP (58). In spite of the fact that up-front surgery with negative surgical margins is considered as the standard of care, local recurrence may not absolutely correlate with status of tumor margins (32,59). Roughly 25-60% of patients show recurrence after resection; extensive surgery can be morbid causing significant loss of function or disabling chronic symptoms (18). Moreover, a subset of tumours may exhibit spontaneous regression (60). Rarely is complete remission seen also despite the recurrent feature of DTs after only simple observation. (61).
When considering surgery, it is extra-abdominal location, younger age, larger tumour size, and mutation status of DT what gives higher rates of local recurrence in patients (59,62,63). The most affecting mutation giving a high risk for local recurrence is p.Ser45Phe in CTNNB1 gene (34,36,64,65), linking this mutation with unfavorable prognostic factors but not a prognostic factor for event-free survival (progression, relapse, or death) (66).
For all these reasons, a watchful waiting approach with a individual period of initial observation has been refered for asymptomatic patients (28).
Point by point physical examination, imaging by ultrasound, CT and MRI, and if not previously done, biopsy should be performed in accordance to the recommendations for soft tissue sarcomas as it was adapted at consensus meeting in Milan (Italy, June 2018) (3). Following this consensus, the current strategy for management of DT advocates for an «active surveillance» period. This approach does not show up to impact the efficacy of ensuing treatment when required, but permits the clinician to arrange the following step in the therapeutic management. Hence, being cautious and maintaining a strategic distance from potential hurt is considered the best option in the first management approaches after setting the diagnosis in many patients. In this period, neither surgery nor other treatment forms are proposed. Any aggressive attempts for total eradication of the tumor may be more awful than the illness itself. It is important to comprehend that even a recurrent desmoid tumor can undergo spontaneous regression (61). Considering the disease biology with unpredictable course, active treatment should be indicated in cases with continuously growth progression of DTs. Tumor size progression at a single evaluation, particularly in the absence of severe clinical symptoms and in non-critical anatomic areas, should not be considered as an sign to begin an active treatment quickly. The active surveillance implies that patients ought to be checked with first MRI or CT inside 1-2 months and afterward in 3-6 months intervals until the fifth year, and yearly afterwards. The later occurrence of severe tumor progression with symptom burden is considered as a indication for active treatment after initial postponed decision for that (3).
Here, when active approach for DT is required, clinician should consider surgery as an up-front therapy. The achievement of R0 margins is the primary aim, but sporadic presence of positive (R1) microscopic margins can be also accepted. In these cases, the following radiation therapy or systemic treatment can be offered to the patients. Thus, a multidisciplinary approach in DT patients therapy is clearly required (3).
The last evidence for surgical management of abdominal desmoid tumours in terms of success, recurrence and morbidity was brought by Moore et. al. (2023). Authors have conducted a large systematic mata-analytical search for 20-yrs period, since November 2000, and concluded that the surgical resection for abdominal desmoids remains a valid treatment option in highly selective cases where negative margins can be obtained, with low morbidity (4.4%) and/or mortality (2% within 30 days after operation) (67).
As DTs shows localy infiltrative growth and may often exhibit relapses, most of R1 cases were treated with subsequent radiotherapy. Moreover, evolution in therapeutic options brought the shift from primary surgeries to less invasive approaches. The wide attention has gained the image-guided ablations with low morbidity as e.g. cryoablation and high-intensity focused ultrasound.
Adjuvant radiotherapy (RT) known for its significant reduced risk of local recurrence rate when applied in patients with incomplete surgical resection or in recurrent tumours is ofently indicated as second step in the manegament (68). However, due to side effects, it can be applied only in patients where anatomic conditions restrict complete resection and therapy is not giving high toxicity. Risk most common factors for local treatment failure include young age, large tumor size, recurrent disease feature, limb, girdle or intra-abdominal location, positive surgical margins, omission of RT, radiation dose < 50 Gy and inappropriate radiation field extention. The recommended dose is 50-56 Gy in 28 once-daily fractions of 2 Gy (69,70). The doses > 56 Gy have failed to demonstrate improvement in local disease control and are associated with greater toxicity including fibrosis, soft tissue necrosis, edema, pathological fractures or vascular complications as well as radio-induced neoplasms (71).
To the others local treatment approaches belongs a noninvasive high-intensity focused ultrasound (HIFU). Here, the low-power cumulative HIFU therapy may lead to significant efficacy and long-term control mostly in recurrent DTs. Zhong et all. (2022) has reported the mean ablation proportion of the HIFU treatment at 69.5%, the objective response rate 47.3% and the 5-year progression-free survival (PFS) rate at 69.3% (72).
The promising results for local approaches showed also study from Schmitz et al. (2016) pointing on the percutaneous cryoablation of extraabdominal DTs (73), which were recently confirmed by Vora et al. (2021). Authors in this meta-analysis study showed that the proportion of stable disease rate was 85.8%, and the progression free survival rate at 1 year was 84.5% and 78.0% at 3 years, with only 4.2% of major/minor complications. Moreover, the 37.5% to 96.9% of patients showed partial or complete symptom relief (74).
In patients that have anatomic barriers to effective surgery, radiotherapy, HIFU or cryoablation, the systemic treatment may be indicated. This option currently include nonsteroidal anti-inflammatory drugs (NSAID), anti-hormonal agents, tyrosine kinase inhibitors (TKI), «low-dose» chemotherapeutic regiments, and conventional chemotherapy as anthracycline-based regimens, mostly liposomal doxorubicin.
NSAID have shown the ability to block the Wnt/β-catetin signaling pathway mediated by COX-2 or prostaglandines which induces an objective size response and lowers pain perception in patients (75,76).
Anti-hormonal therapy is based on often ER+ immunohistochemical status of DTs and its linkage to pregnancy. The most used agents are tamoxifen and toremifene (77,78). They are used alone or in combination with NSAIDs. The therapeutic effect of these two drugs is thought to be involved in the affection of transforming growth factor-β (TGF-β) and its receptors, known as an important pathway regulating fibroblasts proliferation (75,76).
One of the first reports on tamoxifen use in DTs was a study from Sportiello et al. (1991) who reported a case of recurrent retroperitoneal desmoid tumor successfully treated with tamoxifen (Nolvadex tablets) in patient presented late in her second pregnancy with a large retroperitoneal pelvic DT what resulted in a complete tumor regression remaining stable for 27 months (79).
Despite the initial encouragement, later studies have shown that hormonal agents and nonsteroidal anti-inflammatory drugs have limited efficacy. Thus, they did not gain the wide clinical acceptance and clinicians have turned the focus on standard and low-dose chemotherapy. Here, the most often used are anthracycline-based therapies given for 6-8 cycles achieving the tumor shrinking or disease stabilization in 80% of the cases, and a lasting response in 45-50 % of patients (80,81). However, clinical usage is limited by the risk of cardiomyopathy and secondary malignancies. Nevertheless, pegylated liposomal doxorubicin has been reported to have significant effect with quite well tolerated toxicity profile for its lower cardiac toxicity than conventional doxorubicin (82). In trying to overcome the toxicity of antracyclines, the low-dose chemotherapy regiments based on the metotrexate and a vinca alkaloid (vinblastine or vinorelbine) have been also investigated and represent a preferable choice to full-dose chemotherapy (83).
The last and very promising systemic therapy is a biological treatment where the therapeutic agents targets concrete signaling pathways in DTs patomechanism. Here, the tyrosine kinase inhibitors (imatinib, nilotinib, sorafenib, sunitinib, and pazopanib) are well studied (84), and sorafenib is now one of the most utilized therapies, though limited by its side effect profile. Among patients with refractory, progressive or symptomatic DTs, use of sorafenib (400-mg tablet once daily) led to significant prolongation in progression-free survival and initiated durable responses, where the 2-year PFS rate was 81% (85).
The promising effect of targeted biological therapy started the wide spectra of ongoing clinical trials focusing on the Notch pathway by using of gamma-secretase inhibitors (Nirogacestat, PF-0308401) showed efficacy in phase 1 and 2/3 trials (86,87), selective inhibitor of nuclear β-catenin acting through binding TBL-1 (Tegavivint, BC-2059) (88), or immune checkpoints inhibitors (PD-L1) (89), tumor microenvironment (matrix metalloproteinases), platelet-derived growth factor receptor (PDGFR), angiogenesis (vascular endhothelial growth factor, VEGF), cell-cycle regulatory proteins (RB1, CDKN2A and TP53), cell-cell adhesions (N-cadherin and α-catenin) or cell proliferation/survival by blocking the mTOR pathway (90) which can present multiple potential novel therapeutic targets (76).
The serious advancement showed the use of Nirogacestat in adults with growing desmoid tumors according to the Response Evaluation Criteria in Solid Tumors (RECIST). In a study from Gounder et al. (2023) DTs patients received the oral γ-secretase inhibitor nirogacestat (150 mg) showed significant progression-free survival benefit at 2 years at 76%. Nirogacestat was associated with significant benefits with respect to PFS, objective response, symptom burden, role and physical functioning, pain, and health-related quality of life in adults with progressing desmoid tumors. Adverse events with nirogacestat were frequent but mostly (95%) of low grade 1 or 2 (91).
There have been reported around 100-120 cases of DTs in pregnancy worldwide so far published on Pubmed, however only a few cases with systemic therapy for DT. The common sign of these reports was strong biological variability of DTs depending on hormonal background and mostly watch-&-see management with close follow-up and operative delivery. Moreover, the reports describing DTs in pregnancy showed that clinical picture of DTs can be unpredictable, especially when they develop before delivery or in post-partum period. Most of these cases initially diagnosed in pregnancy were bioptised (core biopsy) and if malignity was excluded, they were managed conservatively by follow-up with ev. precise MRI scans for assessment of the DT size and location where pregnancy was ended by CS with DT resection during CS or in the postpartum period, (Table 1) (57,61,92-102).
For decades, DF has been thought as a conceivable hormone-dependent malignancy based on these arguments: estrogen receptors positivity on imunnohistological assessment, the predominance of female patients with high incidence in the fertile age, studies reporting diagnosis or relapse of desmoid tumor in and/or after pregnancy, and watched tumor shrinkage with the administration of anti-estrogen drugs or in post-partum period.
One of the first multicentric study focusing on DTs behavior and pregnancy was published from Italy by Fiore et al., (2014) on ninety-two women with DT who analyzed long-term data linked to disease presentation during and after pregnancy. They showed that the initial treatment of DT in pregnant women was resection (52%), watchful waiting (43%) with later progression at 63% and only 4% of patients received medical therapy. Only 13% of patients relapsed after surgery. After pregnancy, 46% underwent treatment of DT, whereas 54% were managed with watchful waiting. Only 17% showed further progression after treatment. Spontaneous regression occurred in 14%. After further pregnancies, only 27% of patients progressed. The only advers obstetric event was a higher rate of cesarean sections (60). The linkage to pregnancy and postpartum events was even more strengthened by the report from Hanna et al. (2016), who reported a case of rapidly progressing DT in the post-partum period (95), and recently also confirmed by Debaudringhien et al. (2022) who showed that history of pregnancy was associated with an increased risk of progression/relapse in patients with newly diagnosed DF, whereas hormonal contraception did not showed an connestion with disease progression or relapse (29).
The important knowledge in the management of DTs in pregnancy was brought by Cates et al. (2015), who found that pregnancy itself does not increase the risk for local recurrence after surgical resection of desmoid tumors (103). Based on these findings, it seems that pregnancy can be taken as a risk factor for promoting growth and progression of desmoid-type fibromatosis in ongoing pregnancy and post-partum because of the estrogenenic stimulation of desmoid growth, however, it has of low impact on recurrence of DTs after its surgical resection. Thus, subsequent pregnancy should not be discouraged for women in fertile age after surgical resection of desmoid tumors.
Conclusion
Pregnancy-associated DTs are rare and optimal management of these tumors is yet to be established. Nowadays, controversy points on the follow-up approach or proper timing of surgical resection, which may be influenced by the increased potential for tumor growth and the negative reactions of a gravid uterus. Surgical resection of these tumors has been performed successfully both during and early after delivery, where the postpartum radiotherapy, chemotherapy and other medical intervention showed its effectiveness. Risk for disease progression during pregnancy is high, but it can be safely managed. Pregnancy-related DF has good outcomes. As desmoid tumors does not significantly increase obstetric risk, thay can not be a contraindication to future pregnancies.
The management of desmoid tumors, especially when diagnosed during pregnancy is complex and requires multidisciplinary expertise by an experienced team where treatment has to be individualized. We must identificate the reliable prognostic/predictive factors is they are the key for assessing the efficacy of local or systemic treatment. The better understanding of genetics and molecular alterations in signaling pathways enriched this attitude and led to the development of the tailored interventions for DT, which could revolutionize its therapy and management strategies.
Disclosure and Contributorship
The article or part of it has not been published in any other journal. PZ, KK, JV participated in writting of the article, EC, PZ and KOL was involved in clinical management of the patient, CMH, MEØ, PZ contributed in manuscript preparation and review, KOL and PZ participated in the final reviewing of the manuscript and post-surgical clinical management of the patient.
Funding and financial support
None.
Acknowledgments
Authors thank to Petra Zuborova for technical help with managing this paper and to the pasient who approved the content of the paper and signed the consent for the publishing of this case.
Conflict o finterests
The authors declare no conflicts o finterests regarding the publication of this manuscript.
References
- Sakorafas GH, Nissotakis C, Peros G. Abdominal desmoid tumors. Surg Oncol. 2007 Aug;16(2):131-42. [CrossRef]
- Hornick JL, Fritchle KJ, Matt van de Rijn. Desmoid fibromatosis. In: WHO Classification of Tumours Editorial Board. Soft tissue and bone tumours. Lyon (France): International Agency for Research on Cancer; 2020. (WHO classification of tumours series, 5th ed.; vol. 3). Available from: https://tumourclassification.iarc.who.int/chapters/33.
- Desmoid Tumor Working Group. The management of desmoid tumours: A joint global consensus-based guideline approach for adult and paediatric patients. Eur J Cancer. 2020 Mar;127:96-107. [CrossRef]
- Penel N, Coindre JM, Bonvalot S, Italiano A, Neuville A, Le Cesne A, Terrier P, Ray-Coquard I, Ranchere-Vince D, Robin YM, Isambert N, Ferron G, Duffaud F, Bertucci F, Rios M, Stoeckle E, Le Pechoux C, Guillemet C, Courreges JB, Blay JY. Management of desmoid tumours: A nationwide survey of labelled reference centre networks in France. Eur J Cancer. 2016 May;58:90-6. [CrossRef]
- Reitamo JJ, Häyry P, Nykyri E, Saxén E. The desmoid tumor. I. Incidence, sex-, age- and anatomical distribution in the Finnish population. Am J Clin Pathol. 1982 Jun;77(6):665-73. [CrossRef]
- Anneberg M, Svane HML, Fryzek J, Nicholson G, White JB, Edris B, Smith LM, Hooda N, Petersen MM, Baad-Hansen T, Keller JØ, Jørgensen PH, Pedersen AB. The epidemiology of desmoid tumors in Denmark. Cancer Epidemiol. 2022 Apr;77:102114. [CrossRef]
- Alfentoukh M, Salih A, Hassan ME, Alghamdi O, Alkhawaja KA, Ibrahim MA, Sabir EI. Desmoid Tumor of the Rectus Abdominis with Urinary Bladder Involvement: A Case Report and Review of Literature. Gulf J Oncolog. 2023 Jan;1(41):100-106.
- Mundada AV, Tote D, Zade A. Desmoid tumor of Meckel's diverticulum presenting as intestinal obstruction: A rare case report with literature review. J Cancer Res Ther. 2022 Jul-Sep;18(4):880-884. [CrossRef]
- Minami Y, Matsumoto S, Ae K, Tanizawa T, Hayakawa K, Saito M, Kurosawa N. The Clinical Features of Multicentric Extra-abdominal Desmoid Tumors. Cancer Diagn Progn. 2021 Jul 3;1(4):339-343. [CrossRef]
- Tzur R, Silberstein E, Krieger Y, Shoham Y, Rafaeli Y, Bogdanov-Berezovsky A. Desmoid Tumor and Silicone Breast Implant Surgery: Is There Really a Connection? A Literature Review. Aesthetic Plast Surg. 2018 Feb;42(1):59-63. [CrossRef]
- Müller, J. Müller J. Handbuch der Physiologie des Menschen. Hölscher, Coblenz, 1833–1838.
- https://www.wikidoc.org/index.php/Desmoid_tumor_historical_perspective.
- Arima K, Komohara Y, Uchihara T, Yamashita K, Uemura S, Hanada N, Baba H. A Case of Mesenteric Desmoid Tumor Causing Bowel Obstruction After Laparoscopic Surgery. Anticancer Res. 2022 Jan;42(1):381-384. [CrossRef]
- Takada M, Okuyama T, Yoshioka R, Noie T, Takeshita E, Sameshima S, Oya M. A case with mesenteric desmoid tumor after laparoscopic resection of stage I sigmoid colon cancer. Surg Case Rep. 2019 Feb 28;5(1):38. [CrossRef]
- Fukuhara S, Yoshimitsu M, Yano T, Chogahara I, Yamasaki R, Ebara S, Okajima M. Mesenteric desmoid tumor after robot-assisted laparoscopic cystectomy with bladder replacement: a case report. J Surg Case Rep. 2022 Feb 15;2022(2):rjab529. [CrossRef]
- Wanjeri JK, Opeya CJ. A massive abdominal wall desmoid tumor occurring in a laparotomy scar: a case report. World J Surg Oncol. 2011 Mar 22;9:35. [CrossRef]
- Howard JH, Pollock RE. Intra-Abdominal and Abdominal Wall Desmoid Fibromatosis. Oncol Ther. 2016;4(1):57-72. [CrossRef]
- Garcia-Ortega DY, Martín-Tellez KS, Cuellar-Hubbe M, Martínez-Said H, Álvarez-Cano A, Brener-Chaoul M, Alegría-Baños JA, Martínez-Tlahuel JL. Desmoid-Type Fibromatosis. Cancers (Basel). 2020 Jul 9;12(7):1851. [CrossRef]
- Simonetti I, Bruno F, Fusco R, Cutolo C, Setola SV, Patrone R, Masciocchi C, Palumbo P, Arrigoni F, Picone C, Belli A, Grassi R, Grassi F, Barile A, Izzo F, Petrillo A, Granata V. Multimodality Imaging Assessment of Desmoid Tumors: The Great Mime in the Era of Multidisciplinary Teams. J Pers Med. 2022 Jul 16;12(7):1153. [CrossRef]
- Schut AW, Lidington E, Timbergen MJM, Younger E, van der Graaf WTA, van Houdt WJ, Bonenkamp JJ, Jones RL, Grünhagen DJ, Sleijfer S, Verhoef C, Gennatas S, Husson O. Unraveling Desmoid-Type Fibromatosis-Specific Health-Related Quality of Life: Who Is at Risk for Poor Outcomes. Cancers (Basel). 2022 Jun 16;14(12):2979. [CrossRef]
- Melis M, Zager JS, Sondak VK. Multimodality management of desmoid tumors: how important is a negative surgical margin? J Surg Oncol. 2008 Dec 15;98(8):594-602. [CrossRef]
- Napolitano A, Mazzocca A, Spalato Ceruso M, Minelli A, Baldo F, Badalamenti G, Silletta M, Santini D, Tonini G, Incorvaia L, Vincenzi B. Recent Advances in Desmoid Tumor Therapy. Cancers (Basel). 2020 Aug 1;12(8):2135. [CrossRef]
- Dehner LP, Askin FB. Tumors of fibrous tissue origin in childhood. A clinicopathologic study of cutaneous and soft tissue neoplasms in 66 children. Cancer. 1976 Aug;38(2):888-900. [CrossRef]
- Batsakis JG, Raslan W. Extra-abdominal desmoid fibromatosis. Ann Otol Rhinol Laryngol. 1994 Apr;103(4 Pt 1):331-4. [CrossRef]
- Nieuwenhuis MH, Casparie M, Mathus-Vliegen LM, Dekkers OM, Hogendoorn PC, Vasen HF. A nation-wide study comparing sporadic and familial adenomatous polyposis-related desmoid-type fibromatoses. Int J Cancer. 2011 Jul 1;129(1):256-61. [CrossRef]
- Tayeb Tayeb C, Parc Y, Andre T, Lopez-Trabada Ataz D. Familial adenomatous polyposis, desmoid tumors and Gardner syndrome. Bull Cancer. 2020 Mar;107(3):352-358. [CrossRef]
- Saito Y, Hinoi T, Ueno H, Kobayashi H, Konishi T, Ishida F, Yamaguchi T, Inoue Y, Kanemitsu Y, Tomita N, Matsubara N, Komori K, Kotake K, Nagasaka T, Hasegawa H, Koyama M, Ohdan H, Watanabe T, Sugihara K, Ishida H. Risk Factors for the Development of Desmoid Tumor After Colectomy in Patients with Familial Adenomatous Polyposis: Multicenter Retrospective Cohort Study in Japan. Ann Surg Oncol. 2016 Aug;23(Suppl 4):559-565. [CrossRef]
- Kasper B, Baumgarten C, Garcia J, Bonvalot S, Haas R, Haller F, Hohenberger P, Penel N, Messiou C, van der Graaf WT, Gronchi A; Desmoid Working Group. An update on the management of sporadic desmoid-type fibromatosis: a European Consensus Initiative between Sarcoma PAtients EuroNet (SPAEN) and European Organization for Research and Treatment of Cancer (EORTC)/Soft Tissue and Bone Sarcoma Group (STBSG). Ann Oncol. 2017 Oct 1;28(10):2399-2408. [CrossRef]
- Debaudringhien M, Blay JY, Bimbai AM, Bonvalot S, Italiano A, Rousset-Jablonski C, Corradini N, Piperno-Neumann S, Chevreau C, Kurtz JE, Guillemet C, Bompas E, Collard O, Salas S, Le Cesne A, Orbach D, Thery J, Le Deley MC, Mir O, Penel N. Association between recent pregnancy or hormonal contraceptive exposure and outcome of desmoid-type fibromatosis. ESMO Open. 2022 Oct;7(5):100578. [CrossRef]
- Gounder MM, Thomas DM, Tap WD. Locally Aggressive Connective Tissue Tumors. J Clin Oncol. 2018 Jan 10;36(2):202-209. [CrossRef]
- Slowik V, Attard T, Dai H, Shah R, Septer S. Desmoid tumors complicating Familial Adenomatous Polyposis: a meta-analysis mutation spectrum of affected individuals. BMC Gastroenterol. 2015 Jul 16;15:84. [CrossRef]
- Colombo C, Foo WC, Whiting D, Young ED, Lusby K, Pollock RE, Lazar AJ, Lev D. FAP-related desmoid tumors: a series of 44 patients evaluated in a cancer referral center. Histol Histopathol. 2012 May;27(5):641-9. [CrossRef]
- Amary MF, Pauwels P, Meulemans E, Roemen GM, Islam L, Idowu B, Bousdras K, Diss TC, O'Donnell P, Flanagan AM. Detection of beta-catenin mutations in paraffin-embedded sporadic desmoid-type fibromatosis by mutation-specific restriction enzyme digestion (MSRED): an ancillary diagnostic tool. Am J Surg Pathol. 2007 Sep;31(9):1299-309. [CrossRef]
- Lazar AJ, Tuvin D, Hajibashi S, Habeeb S, Bolshakov S, Mayordomo-Aranda E, Warneke CL, Lopez-Terrada D, Pollock RE, Lev D. Specific mutations in the beta-catenin gene (CTNNB1) correlate with local recurrence in sporadic desmoid tumors. Am J Pathol. 2008 Nov;173(5):1518-27. [CrossRef]
- Colombo C, Belfiore A, Paielli N, De Cecco L, Canevari S, Laurini E, Fermeglia M, Pricl S, Verderio P, Bottelli S, Fiore M, Stacchiotti S, Palassini E, Gronchi A, Pilotti S, Perrone F. β-Catenin in desmoid-type fibromatosis: deep insights into the role of T41A and S45F mutations on protein structure and gene expression. Mol Oncol. 2017 Nov;11(11):1495-1507. [CrossRef]
- Dômont J, Salas S, Lacroix L, Brouste V, Saulnier P, Terrier P, Ranchère D, Neuville A, Leroux A, Guillou L, Sciot R, Collin F, Dufresne A, Blay JY, Le Cesne A, Coindre JM, Bonvalot S, Bénard J. High frequency of beta-catenin heterozygous mutations in extra-abdominal fibromatosis: a potential molecular tool for disease management. Br J Cancer. 2010 Mar 16;102(6):1032-6. [CrossRef]
- Crago AM, Chmielecki J, Rosenberg M, O'Connor R, Byrne C, Wilder FG, Thorn K, Agius P, Kuk D, Socci ND, Qin LX, Meyerson M, Hameed M, Singer S. Near universal detection of alterations in CTNNB1 and Wnt pathway regulators in desmoid-type fibromatosis by whole-exome sequencing and genomic analysis. Genes Chromosomes Cancer. 2015 Oct;54(10):606-15. [CrossRef]
- Penel N, Chibon F, Salas S. Adult desmoid tumors: biology, management and ongoing trials. Curr Opin Oncol. 2017 Jul;29(4):268-274. [CrossRef]
- Giarola M, Stagi L, Presciuttini S, Mondini P, Radice MT, Sala P, Pierotti MA, Bertario L, Radice P. Screening for mutations of the APC gene in 66 Italian familial adenomatous polyposis patients: evidence for phenotypic differences in cases with and without identified mutation. Hum Mutat. 1999;13(2):116-23. [CrossRef]
- Bertario L, Russo A, Sala P, Eboli M, Giarola M, D'amico F, Gismondi V, Varesco L, Pierotti MA, Radice P; Hereditary Colorectal Tumours Registry. Genotype and phenotype factors as determinants of desmoid tumors in patients with familial adenomatous polyposis. Int J Cancer. 2001 Mar 20;95(2):102-7. [CrossRef]
- Gurbuz AK, Giardiello FM, Petersen GM, Krush AJ, Offerhaus GJ, Booker SV, Kerr MC, Hamilton SR. Desmoid tumours in familial adenomatous polyposis. Gut. 1994 Mar;35(3):377-81. [CrossRef]
- Lips DJ, Barker N, Clevers H, Hennipman A. The role of APC and beta-catenin in the aetiology of aggressive fibromatosis (desmoid tumors). Eur J Surg Oncol. 2009 Jan;35(1):3-10. [CrossRef]
- Nieuwenhuis MH, De Vos Tot Nederveen Cappel W, Botma A, Nagengast FM, Kleibeuker JH, Mathus-Vliegen EM, Dekker E, Dees J, Wijnen J, Vasen HF. Desmoid tumors in a dutch cohort of patients with familial adenomatous polyposis. Clin Gastroenterol Hepatol. 2008 Feb;6(2):215-9. [CrossRef]
- Järvinen HJ, Peltomäki P. The complex genotype-phenotype relationship in familial adenomatous polyposis. Eur J Gastroenterol Hepatol. 2004 Jan;16(1):5-8. [CrossRef]
- Nieuwenhuis MH, Vasen HF. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): a review of the literature. Crit Rev Oncol Hematol. 2007 Feb;61(2):153-61. [CrossRef]
- Wang WL, Nero C, Pappo A, Lev D, Lazar AJ, López-Terrada D. CTNNB1 genotyping and APC screening in pediatric desmoid tumors: a proposed algorithm. Pediatr Dev Pathol. 2012 Sep-Oct;15(5):361-7. [CrossRef]
- Barker, N. Barker N. The canonical Wnt/beta-catenin signalling pathway. Methods Mol Biol. 2008;468:5-15. [CrossRef]
- Brener-Chaoul M, Cervantes-Gutiérrez Ó, Padilla-Longoria R, Martín-Téllez KS. Desmoid tumors: diagnostic and therapeutic considerations. Gac Med Mex. 2020;156(5):439-445. [CrossRef]
- Lazar AJ, Hajibashi S, Lev D. Desmoid tumor: from surgical extirpation to molecular dissection. Curr Opin Oncol. 2009 Jul;21(4):352-9. [CrossRef]
- Nusse R, Clevers H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell. 2017 Jun 1;169(6):985-999. [CrossRef]
- Kotiligam D, Lazar AJ, Pollock RE, Lev D. Desmoid tumor: a disease opportune for molecular insights. Histol Histopathol. 2008 Jan;23(1):117-26. [CrossRef]
- Li J, Wang CY. TBL1-TBLR1 and beta-catenin recruit each other to Wnt target-gene promoter for transcription activation and oncogenesis. Nat Cell Biol. 2008 Feb;10(2):160-9. [CrossRef]
- Angers S, Moon RT. Proximal events in Wnt signal transduction. Nat Rev Mol Cell Biol. 2009 Jul;10(7):468-77. [CrossRef]
- Liu J, Xiao Q, Xiao J, Niu C, Li Y, Zhang X, Zhou Z, Shu G, Yin G. Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduct Target Ther. 2022 Jan 3;7(1):3. [CrossRef]
- Owens CL, Sharma R, Ali SZ. Deep fibromatosis (desmoid tumor): cytopathologic characteristics, clinicoradiologic features, and immunohistochemical findings on fine-needle aspiration. Cancer. 2007 Jun 25;111(3):166-72. [CrossRef]
- Devata S, Chugh R. Desmoid tumors: a comprehensive review of the evolving biology, unpredictable behavior, and myriad of management options. Hematol Oncol Clin North Am. 2013 Oct;27(5):989-1005. [CrossRef]
- Mohd Sulaiman N, Mohd Dali F, Mohd Hussain MSB, Ramli R. Abdominal wall desmoid tumour in pregnancy. BMJ Case Rep. 2022 Jun 16;15(6):e249966. [CrossRef]
- Desurmont T, Lefèvre JH, Shields C, Colas C, Tiret E, Parc Y. Desmoid tumour in familial adenomatous polyposis patients: responses to treatments. Fam Cancer. 2015 Mar;14(1):31-9. [CrossRef]
- Salas S, Dufresne A, Bui B, Blay JY, Terrier P, Ranchere-Vince D, Bonvalot S, Stoeckle E, Guillou L, Le Cesne A, Oberlin O, Brouste V, Coindre JM. Prognostic factors influencing progression-free survival determined from a series of sporadic desmoid tumors: a wait-and-see policy according to tumor presentation. J Clin Oncol. 2011 Sep 10;29(26):3553-8. [CrossRef]
- Fiore M, Coppola S, Cannell AJ, Colombo C, Bertagnolli MM, George S, Le Cesne A, Gladdy RA, Casali PG, Swallow CJ, Gronchi A, Bonvalot S, Raut CP. Desmoid-type fibromatosis and pregnancy: a multi-institutional analysis of recurrence and obstetric risk. Ann Surg. 2014 May;259(5):973-8. [CrossRef]
- Sueishi T, Arizono T, Nishida K, Hamada T, Inokuchi A. A Case of Spontaneous Regression of Recurrent Desmoid Tumor Originating From the Internal Obturator Muscle After Delivery. World J Oncol. 2016 Aug;7(4):75-80. [CrossRef]
- Crago AM, Denton B, Salas S, Dufresne A, Mezhir JJ, Hameed M, Gonen M, Singer S, Brennan MF. A prognostic nomogram for prediction of recurrence in desmoid fibromatosis. Ann Surg. 2013 Aug;258(2):347-53. [CrossRef]
- Lev D, Kotilingam D, Wei C, Ballo MT, Zagars GK, Pisters PW, Lazar AA, Patel SR, Benjamin RS, Pollock RE. Optimizing treatment of desmoid tumors. J Clin Oncol. 2007 May 1;25(13):1785-91. [CrossRef]
- Bo N, Wang D, Wu B, Chen L, Ruixue Ma. Analysis of β-catenin expression and exon 3 mutations in pediatric sporadic aggressive fibromatosis. Pediatr Dev Pathol. 2012 May-Jun;15(3):173-8. [CrossRef]
- Colombo C, Miceli R, Lazar AJ, Perrone F, Pollock RE, Le Cesne A, Hartgrink HH, Cleton-Jansen AM, Domont J, Bovée JV, Bonvalot S, Lev D, Gronchi A. CTNNB1 45F mutation is a molecular prognosticator of increased postoperative primary desmoid tumor recurrence: an independent, multicenter validation study. Cancer. 2013 Oct 15;119(20):3696-702. [CrossRef]
- Penel N, Bonvalot S, Bimbai AM, Meurgey A, Le Loarer F, Salas S, Piperno-Neumann S, Chevreau C, Boudou-Rouquette P, Dubray-Longeras P, Kurtz JE, Guillemet C, Bompas E, Italiano A, Le Cesne A, Orbach D, Thery J, Le Deley MC, Blay JY, Mir O. Lack of Prognostic Value of CTNNB1 Mutation Profile in Desmoid-Type Fibromatosis. Clin Cancer Res. 2022 Sep 15;28(18):4105-4111. [CrossRef]
- Moore D, Burns L, Creavin B, Ryan E, Conlon K, Kelly ME, Kavanagh D. Surgical management of abdominal desmoids: a systematic review and meta-analysis. Ir J Med Sci. 2023 Apr;192(2):549-560. [CrossRef]
- Janssen ML, van Broekhoven DL, Cates JM, Bramer WM, Nuyttens JJ, Gronchi A, Salas S, Bonvalot S, Grünhagen DJ, Verhoef C. Meta-analysis of the influence of surgical margin and adjuvant radiotherapy on local recurrence after resection of sporadic desmoid-type fibromatosis. Br J Surg. 2017 Mar;104(4):347-357. [CrossRef]
- de Bree E, Keus R, Melissas J, Tsiftsis D, van Coevorden F. Desmoid tumors: need for an individualized approach. Expert Rev Anticancer Ther. 2009 Apr;9(4):525-35. [CrossRef]
- Keus RB, Nout RA, Blay JY, de Jong JM, Hennig I, Saran F, Hartmann JT, Sunyach MP, Gwyther SJ, Ouali M, Kirkpatrick A, Poortmans PM, Hogendoorn PCW, van der Graaf WTA. Results of a phase II pilot study of moderate dose radiotherapy for inoperable desmoid-type fibromatosis--an EORTC STBSG and ROG study (EORTC 62991-22998). Ann Oncol. 2013 Oct;24(10):2672-2676. [CrossRef]
- Gronchi A, Colombo C, Le Péchoux C, Dei Tos AP, Le Cesne A, Marrari A, Penel N, Grignani G, Blay JY, Casali PG, Stoeckle E, Gherlinzoni F, Meeus P, Mussi C, Gouin F, Duffaud F, Fiore M, Bonvalot S; ISG and FSG. Sporadic desmoid-type fibromatosis: a stepwise approach to a non-metastasising neoplasm--a position paper from the Italian and the French Sarcoma Group. Ann Oncol. 2014 Mar;25(3):578-583. [CrossRef]
- Zhong X, Hu X, Zhao P, Wang Y, Fang XF, Shen J, Shen H, Yuan Y. The efficacy of low-power cumulative high-intensity focused ultrasound treatment for recurrent desmoid tumor. Cancer Med. 2022 May;11(10):2079-2084. [CrossRef]
- Schmitz JJ, Schmit GD, Atwell TD, Callstrom MR, Kurup AN, Weisbrod AJ, Morris JM. Percutaneous Cryoablation of Extraabdominal Desmoid Tumors: A 10-Year Experience. AJR Am J Roentgenol. 2016 Jul;207(1):190-5. [CrossRef]
- Vora BMK, Munk PL, Somasundaram N, Ouellette HA, Mallinson PI, Sheikh A, Abdul Kadir H, Tan TJ, Yan YY. Cryotherapy in extra-abdominal desmoid tumors: A systematic review and meta-analysis. PLoS One. 2021 Dec 23;16(12):e0261657. [CrossRef]
- Mignemi NA, Itani DM, Fasig JH, Keedy VL, Hande KR, Whited BW, Homlar KC, Correa H, Coffin CM, Black JO, Yi Y, Halpern JL, Holt GE, Schwartz HS, Schoenecker JG, Cates JM. Signal transduction pathway analysis in desmoid-type fibromatosis: transforming growth factor-β, COX2 and sex steroid receptors. Cancer Sci. 2012 Dec;103(12):2173-80. [CrossRef]
- McLean TD, Duchi S, Di Bella C. Molecular Pathogenesis of Sporadic Desmoid Tumours and Its Implications for Novel Therapies: A Systematised Narrative Review. Target Oncol. 2022 May;17(3):223-252. [CrossRef]
- Ohashi T, Shigematsu N, Kameyama K, Kubo A. Tamoxifen for recurrent desmoid tumor of the chest wall. Int J Clin Oncol. 2006 Apr;11(2):150-2. [CrossRef]
- Fiore M, Colombo C, Radaelli S, Callegaro D, Palassini E, Barisella M, Morosi C, Baldi GG, Stacchiotti S, Casali PG, Gronchi A. Hormonal manipulation with toremifene in sporadic desmoid-type fibromatosis. Eur J Cancer. 2015 Dec;51(18):2800-7. [CrossRef]
- Sportiello DJ, Hoogerland DL. A recurrent pelvic desmoid tumor successfully treated with tamoxifen. Cancer. 1991 Mar 1;67(5):1443-6. [CrossRef]
- Janinis J, Patriki M, Vini L, Aravantinos G, Whelan JS. The pharmacological treatment of aggressive fibromatosis: a systematic review. Ann Oncol. 2003 Feb;14(2):181-90. [CrossRef]
- Garbay D, Le Cesne A, Penel N, Chevreau C, Marec-Berard P, Blay JY, Debled M, Isambert N, Thyss A, Bompas E, Collard O, Salas S, Coindre JM, Bui B, Italiano A. Chemotherapy in patients with desmoid tumors: a study from the French Sarcoma Group (FSG). Ann Oncol. 2012 Jan;23(1):182-186. [CrossRef]
- Constantinidou A, Jones RL, Scurr M, Al-Muderis O, Judson I. Pegylated liposomal doxorubicin, an effective, well-tolerated treatment for refractory aggressive fibromatosis. Eur J Cancer. 2009 Nov;45(17):2930-4. [CrossRef]
- Palassini E, Frezza AM, Mariani L, Lalli L, Colombo C, Fiore M, Messina A, Casale A, Morosi C, Collini P, Stacchiotti S, Casali PG, Gronchi A. Long-term Efficacy of Methotrexate Plus Vinblastine/Vinorelbine in a Large Series of Patients Affected by Desmoid-Type Fibromatosis. Cancer J. 2017 Mar/Apr;23(2):86-91. [CrossRef]
- Sparber-Sauer M, Orbach D, Navid F, Hettmer S, Skapek S, Corradini N, Casanova M, Weiss A, Schwab M, Ferrari A. Rationale for the use of tyrosine kinase inhibitors in the treatment of paediatric desmoid-type fibromatosis. Br J Cancer. 2021 May;124(10):1637-1646. [CrossRef]
- Gounder MM, Mahoney MR, Van Tine BA, Ravi V, Attia S, Deshpande HA, Gupta AA, Milhem MM, Conry RM, Movva S, Pishvaian MJ, Riedel RF, Sabagh T, Tap WD, Horvat N, Basch E, Schwartz LH, Maki RG, Agaram NP, Lefkowitz RA, Mazaheri Y, Yamashita R, Wright JJ, Dueck AC, Schwartz GK. Sorafenib for Advanced and Refractory Desmoid Tumors. N Engl J Med. 2018 Dec 20;379(25):2417-2428. [CrossRef]
- Zhou MY, Bui NQ, Charville GW, Ghanouni P, Ganjoo KN. Current management and recent progress in desmoid tumors. Cancer Treat Res Commun. 2022;31:100562. [CrossRef]
- Federman, N. Federman N. Molecular pathogenesis of desmoid tumor and the role of γ-secretase inhibition. NPJ Precis Oncol. 2022 Sep 6;6(1):62. [CrossRef]
- Braggio DA, Costas C de Faria F, Koller D, Jin F, Zewdu A, Lopez G, Batte K, Casadei L, Welliver M, Horrigan SK, Han R, Larson JL, Strohecker AM, Pollock RE. Preclinical efficacy of the Wnt/β-catenin pathway inhibitor BC2059 for the treatment of desmoid tumors. PLoS One. 2022 Oct 14;17(10):e0276047. [CrossRef]
- Siozopoulou V, Marcq E, Jacobs J, Zwaenepoel K, Hermans C, Brauns J, Pauwels S, Huysentruyt C, Lammens M, Somville J, Smits E, Pauwels P. Desmoid tumors display a strong immune infiltration at the tumor margins and no PD-L1-driven immune suppression. Cancer Immunol Immunother. 2019 Oct;68(10):1573-1583. [CrossRef]
- Weiss AR, Dry S, Maygar C, Cutler A, Lary CW, Khoo C, Fergione JE, Hounchell MM, Glick K, Browning M, Choo SH, Hawkins DS, Lagmay J, Manalang M, Skapek SX, Weigel B, Verwys S, Federman N. A pilot study evaluating the use of sirolimus in children and young adults with desmoid-type fibromatosis. Pediatr Blood Cancer. 2023 Jun 7:e30466. [CrossRef]
- Gounder M, Ratan R, Alcindor T, Schöffski P, van der Graaf WT, Wilky BA, Riedel RF, Lim A, Smith LM, Moody S, Attia S, Chawla S, D'Amato G, Federman N, Merriam P, Van Tine BA, Vincenzi B, Benson C, Bui NQ, Chugh R, Tinoco G, Charlson J, Dileo P, Hartner L, Lapeire L, Mazzeo F, Palmerini E, Reichardt P, Stacchiotti S, Bailey HH, Burgess MA, Cote GM, Davis LE, Deshpande H, Gelderblom H, Grignani G, Loggers E, Philip T, Pressey JG, Kummar S, Kasper B. Nirogacestat, a γ-Secretase Inhibitor for Desmoid Tumors. N Engl J Med. 2023 Mar 9;388(10):898-912. [CrossRef]
- Marsh-Armstrong B, Veerapong J, Taddonio M, Boles S, Sicklick JK, Binder P. Pregnancy-associated large pelvic desmoid tumor: A case report of fetal-protective strategies and fertility preservation. Gynecol Oncol Rep. 2021 Dec 14;39:100901. [CrossRef]
- Jin L, Tan Y, Su Z, Huang S, Pokhrel S, Shi H, Chen Y. Gardner syndrome with giant abdominal desmoid tumor during pregnancy: a case report. BMC Surg. 2020 Nov 12;20(1):282. [CrossRef]
- Palacios-Zertuche JT, Cardona-Huerta S, Juárez-García ML, Valdés-Flores E, Muñoz-Maldonado GE. Case report: Rapidly growing abdominal wall giant desmoid tumour during pregnancy. Cir Cir. 2017 Jul-Aug;85(4):339-343. [CrossRef]
- Hanna D, Magarakis M, Twaddell WS, Alexander HR, Kesmodel SB. Rapid progression of a pregnancy-associated intra-abdominal desmoid tumor in the post-partum period: A case report. Int J Surg Case Rep. 2016;29:30-33. [CrossRef]
- Awwad J, Hammoud N, Farra C, Fares F, Abi Saad G, Ghazeeri G. Abdominal Wall Desmoid during Pregnancy: Diagnostic Challenges. Case Rep Obstet Gynecol. 2013;2013:350894. [CrossRef]
- Choi SH, Lee JH, Seo BF, Kim SW, Rhie JW, Ahn ST. Desmoid tumor of the rectus abdominis muscle in a postpartum patient. Arch Plast Surg. 2012 Jul;39(4):439-41. [CrossRef]
- Durkin AJ, Korkolis DP, Al-Saif O, Zervos EE. Full-term gestation and transvaginal delivery after wide resection of an abdominal desmoid tumor during pregnancy. J Surg Oncol. 2005 Feb 1;89(2):86-90. [CrossRef]
- Michopoulou A, Germanos S, Kanakopoulos D, Milonas A, Orfanos N, Spyratou C, Markidis P. Management of a large abdominal wall desmoid tumor during pregnancy. Case report. Ann Ital Chir. 2010 Mar-Apr;81(2):153-6. PMID: 20726395.
- Viriyaroj V, Yingsakmongkol N, Pasukdee P, Rermluk N. A large abdominal desmoid tumor associated with pregnancy. J Med Assoc Thai. 2009 Jun;92 Suppl 3:S72-5. PMID: 19702071.
- Gurluler E, Gures N, Citil I, Kemik O, Berber I, Sumer A, Gurkan A. Desmoid tumor in puerperium period: a case report. Clin Med Insights Case Rep. 2014 Mar 16;7:29-32. [CrossRef]
- De Cian F, Delay E, Rudigoz RC, Ranchère D, Rivoire M. Desmoid tumor arising in a cesarean section scar during pregnancy: monitoring and management. Gynecol Oncol. 1999 Oct;75(1):145-8. [CrossRef]
- Cates, JM. Cates JM. Pregnancy does not increase the local recurrence rate after surgical resection of desmoid-type fibromatosis. Int J Clin Oncol. 2015 Jun;20(3):617-22. [CrossRef]
Figure 1.
Abdominal ultrasound (A – transverse view; B – longitudinal view) picture of desmoid tumor sized 12 x 4.6 x 6.6 cm on the first diagnosis.
Figure 1.
Abdominal ultrasound (A – transverse view; B – longitudinal view) picture of desmoid tumor sized 12 x 4.6 x 6.6 cm on the first diagnosis.
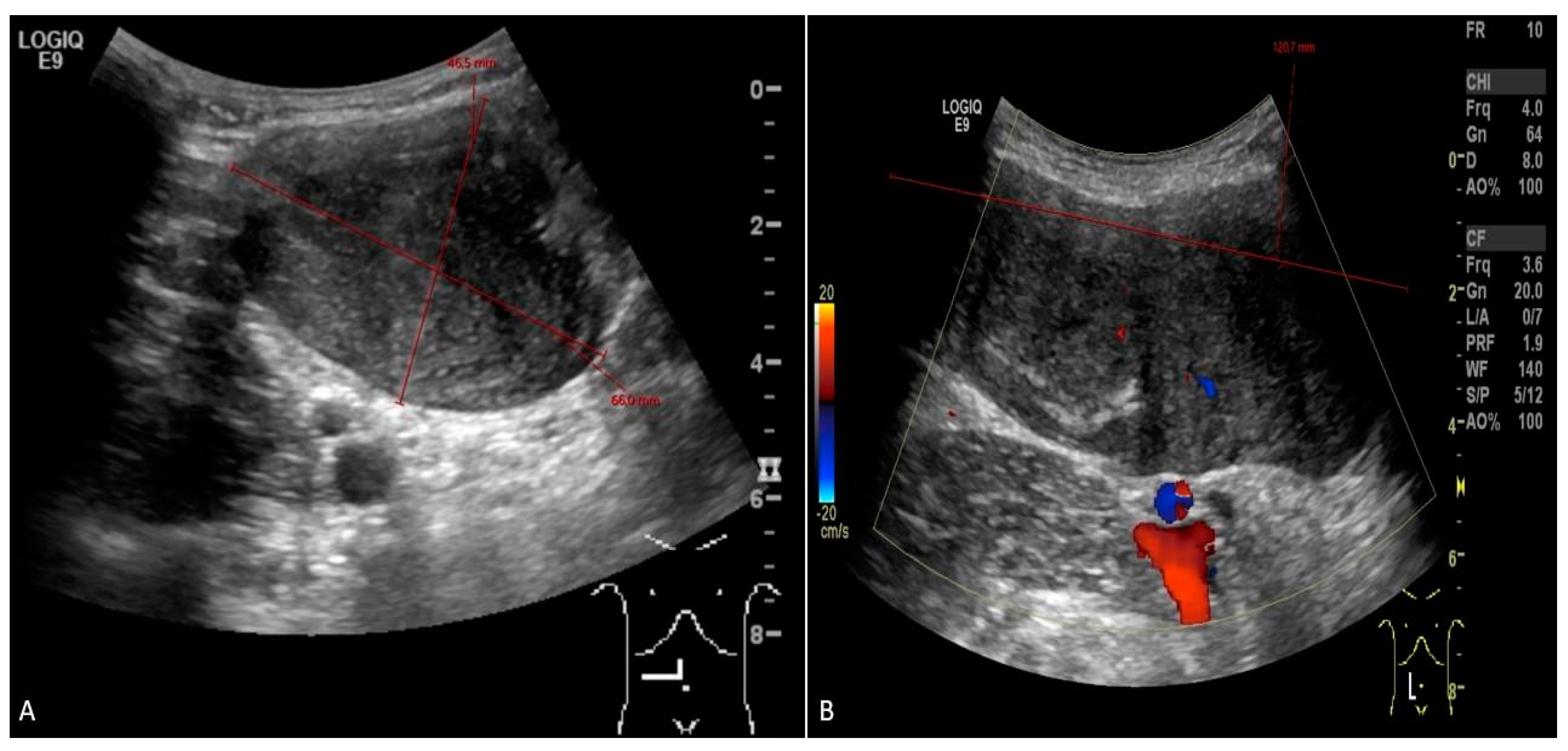
Figure 2.
MRI scan (A – axiall plane; B – sagital plane) asessment of fibromatosis lesion at the first diagnostic scan sizing 8.8 x 4.8 x 15.3 cm.
Figure 2.
MRI scan (A – axiall plane; B – sagital plane) asessment of fibromatosis lesion at the first diagnostic scan sizing 8.8 x 4.8 x 15.3 cm.
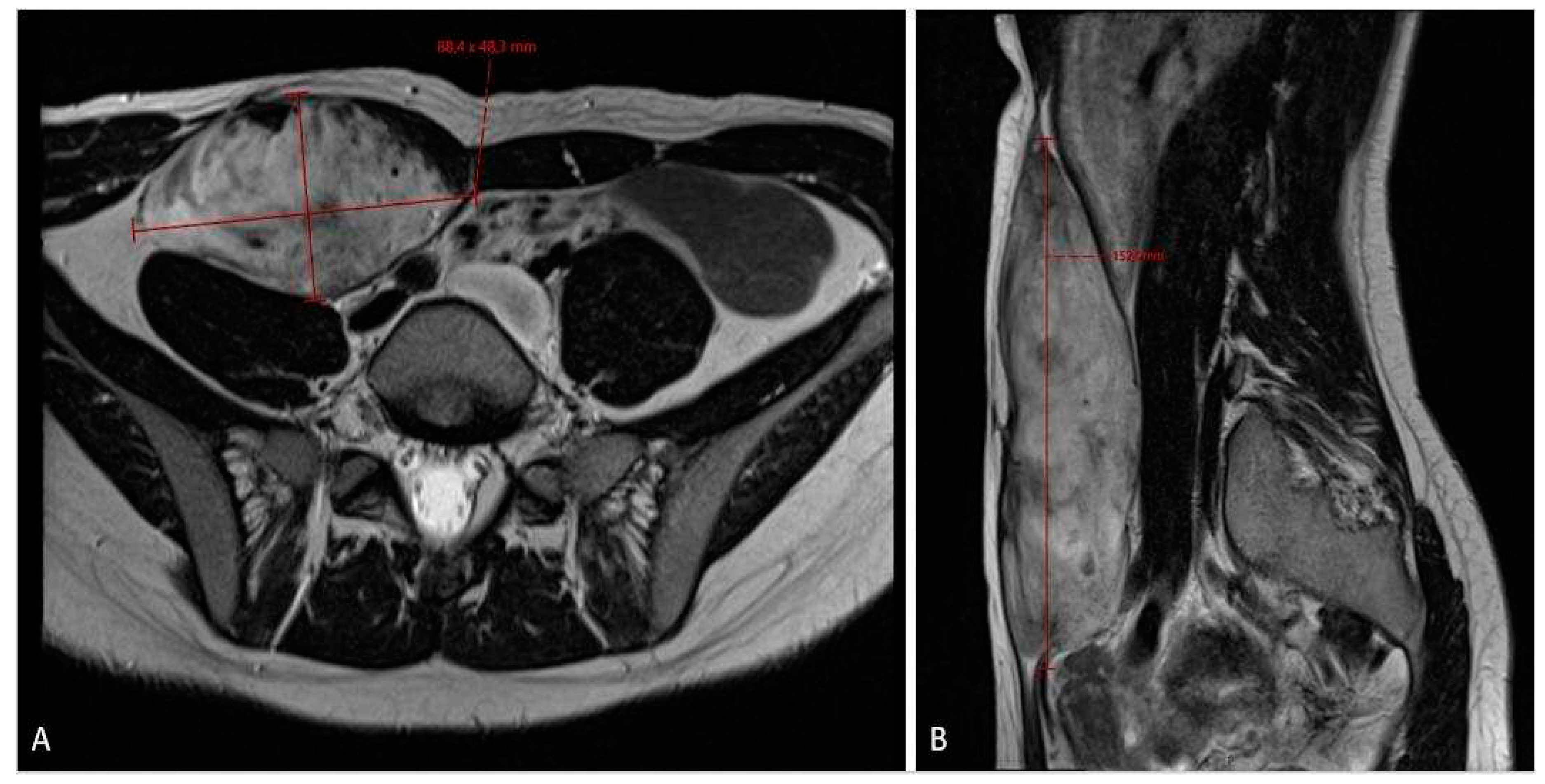
Figure 3.
The 3-months MRI controll scan (A – axiall plane; B – sagital plane) after initial diagnosis showing stable tumor size.
Figure 3.
The 3-months MRI controll scan (A – axiall plane; B – sagital plane) after initial diagnosis showing stable tumor size.
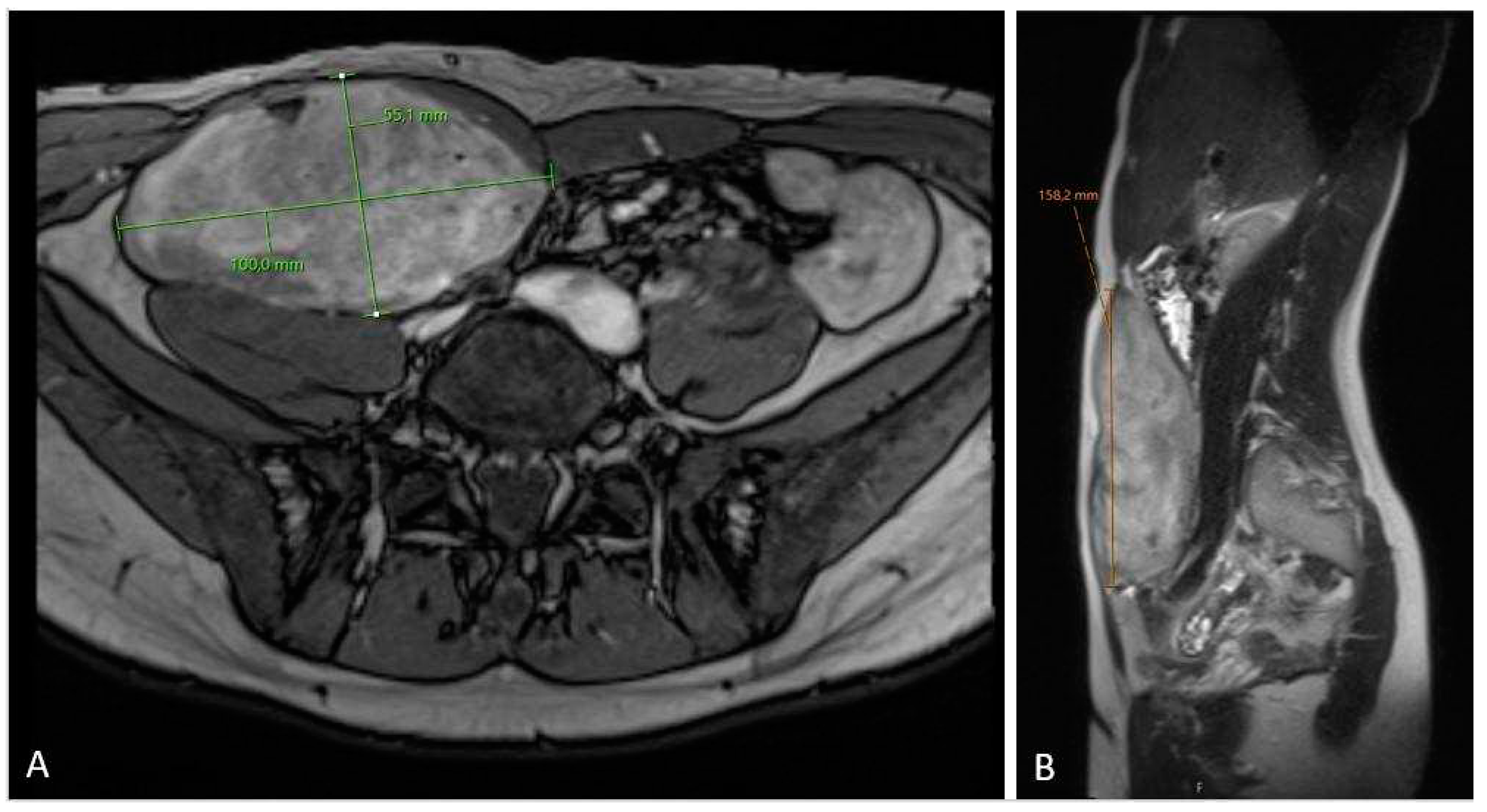
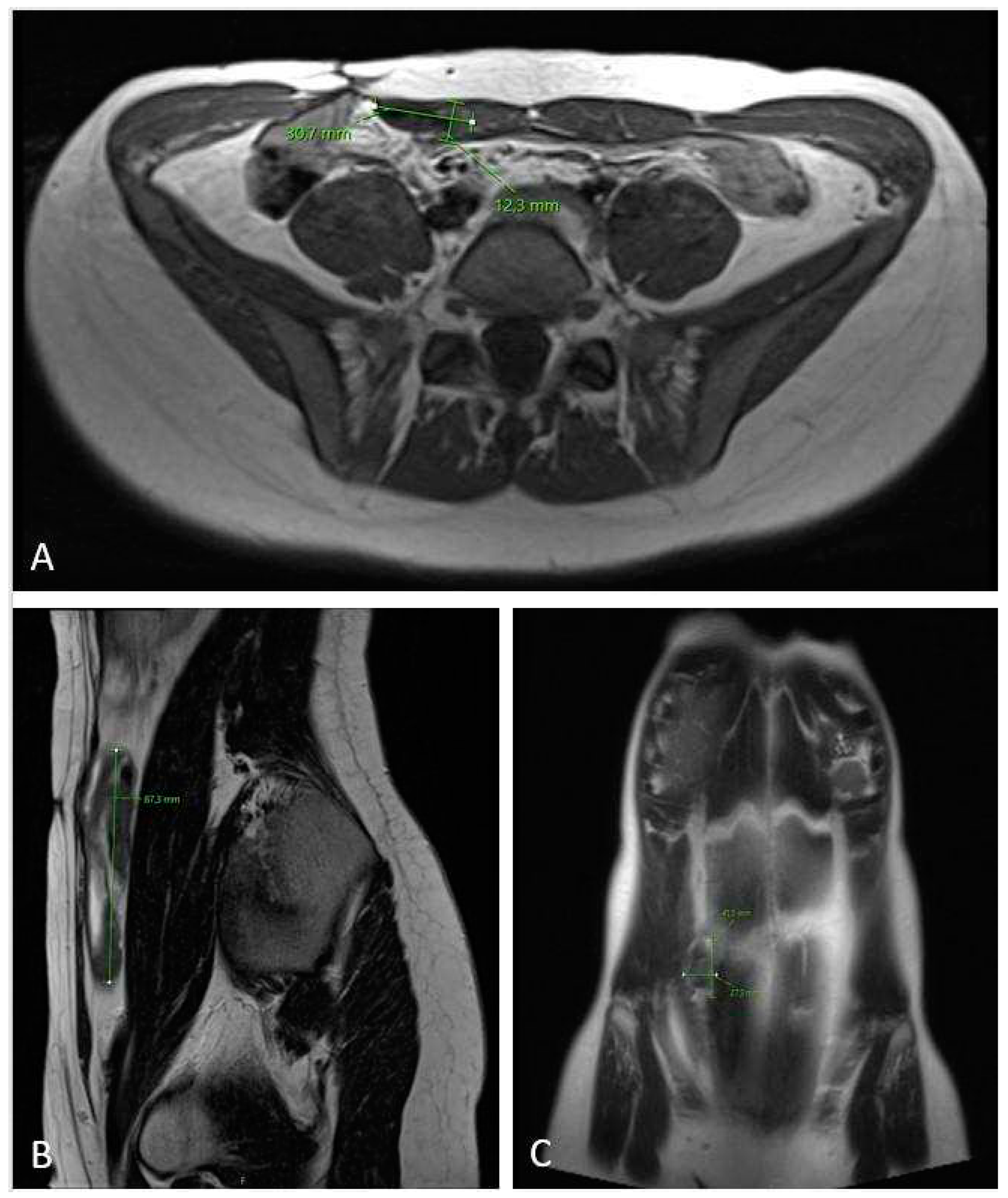
Figure 4.
MRI scan (A – axiall plane; B – sagital plane) on 6-month follow-up check showing significant tumor growth progression, sized 10.5 x 6.2 x 17.5 cm in cranio-caudal diameter located in musculus rectus abdominis on the right side.
Figure 4.
MRI scan (A – axiall plane; B – sagital plane) on 6-month follow-up check showing significant tumor growth progression, sized 10.5 x 6.2 x 17.5 cm in cranio-caudal diameter located in musculus rectus abdominis on the right side.
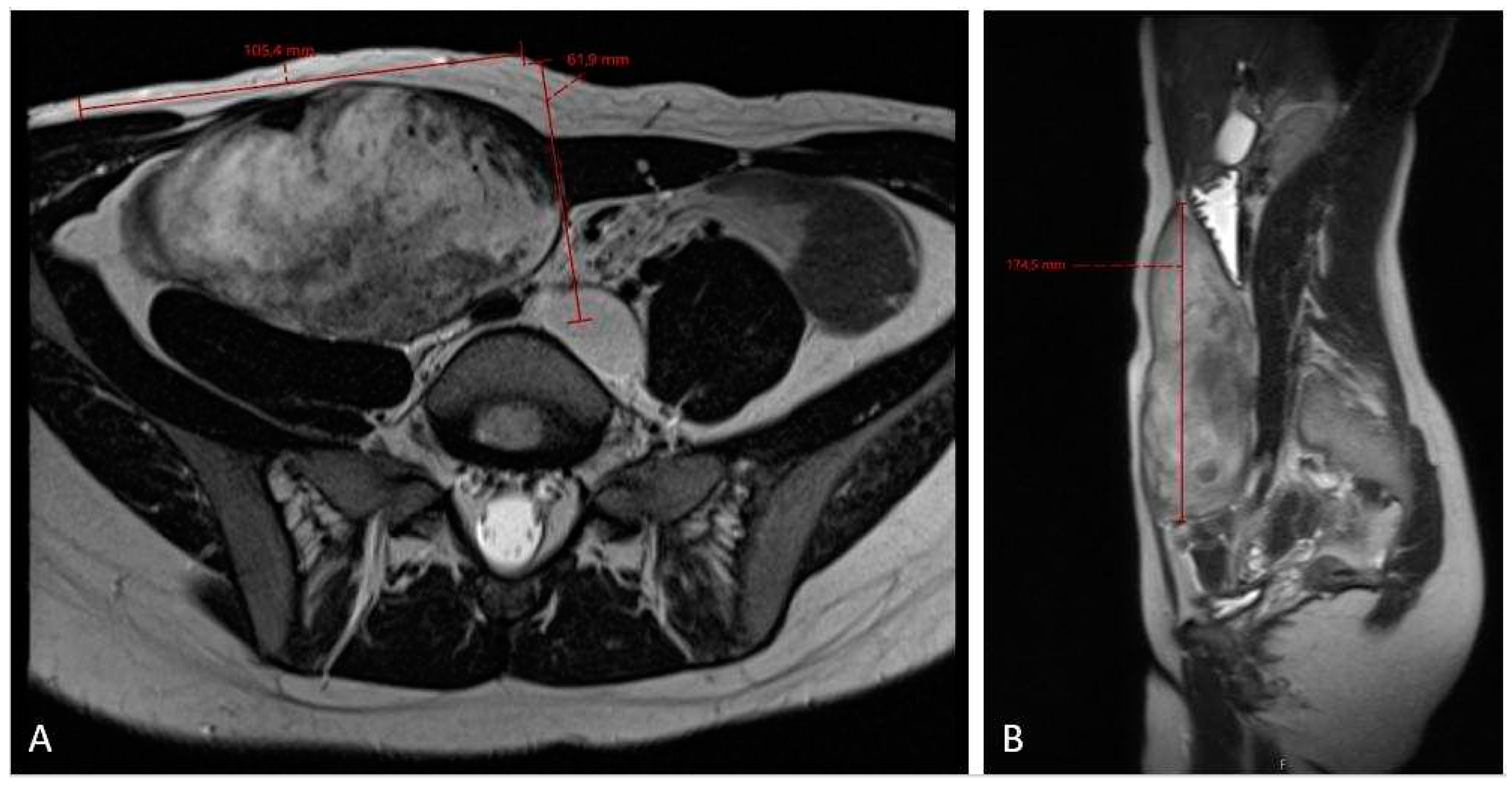
Figure 5.
MRI scan (A – axiall plane; B – sagital plane) after 3 cycles chemotherapy showing no effect of the treatment and slightly progression in the cranio-caudal tumor size (12 x 6.6 x 20 cm).
Figure 5.
MRI scan (A – axiall plane; B – sagital plane) after 3 cycles chemotherapy showing no effect of the treatment and slightly progression in the cranio-caudal tumor size (12 x 6.6 x 20 cm).
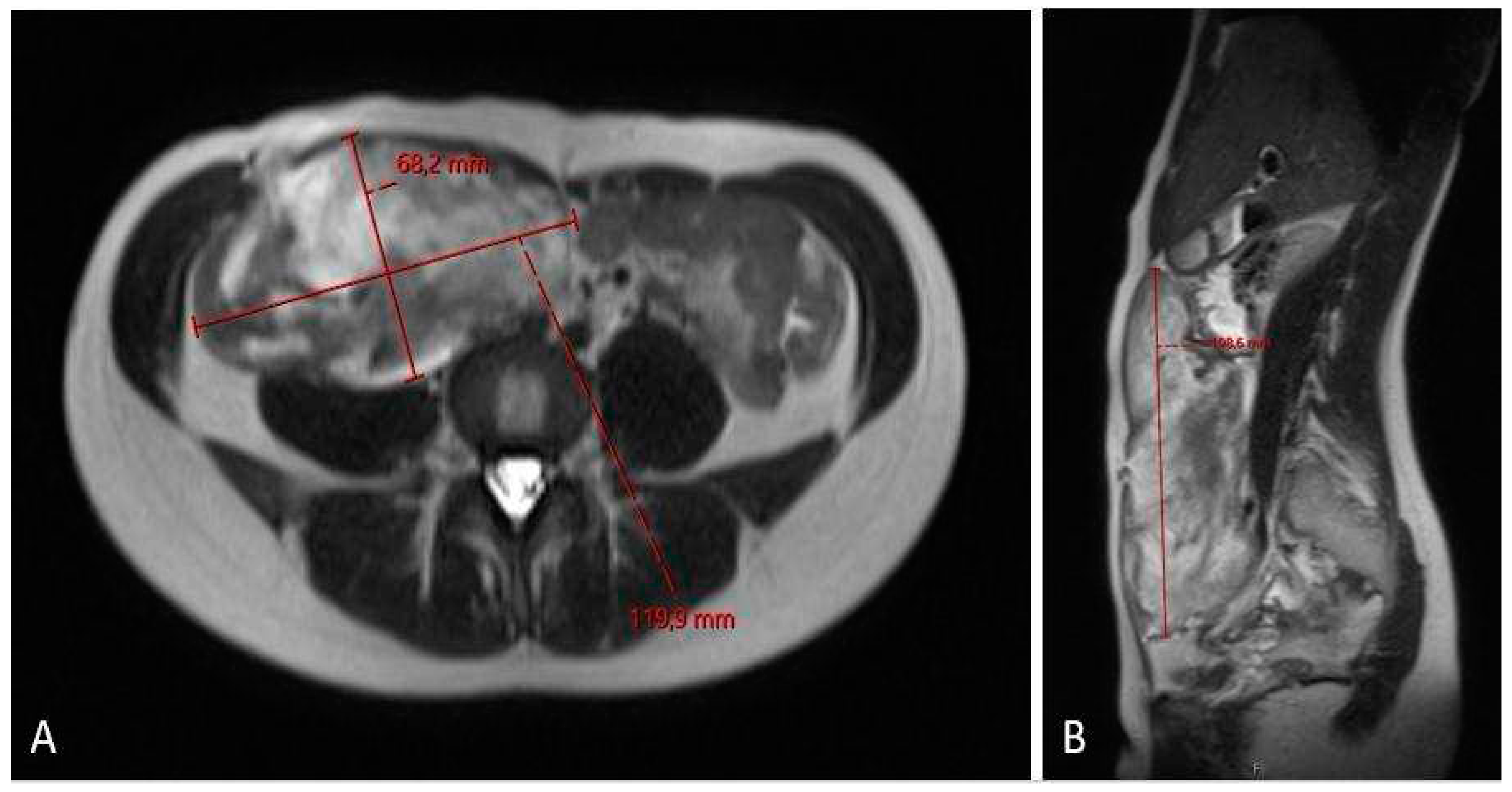
Figure 6.
MRI scan (A – axiall plane; B – sagital plane) after 6 cycles chemotherapy showing partial regression of the tumor, now sized 11.2 x 5.4 x 19.1 cm.
Figure 6.
MRI scan (A – axiall plane; B – sagital plane) after 6 cycles chemotherapy showing partial regression of the tumor, now sized 11.2 x 5.4 x 19.1 cm.
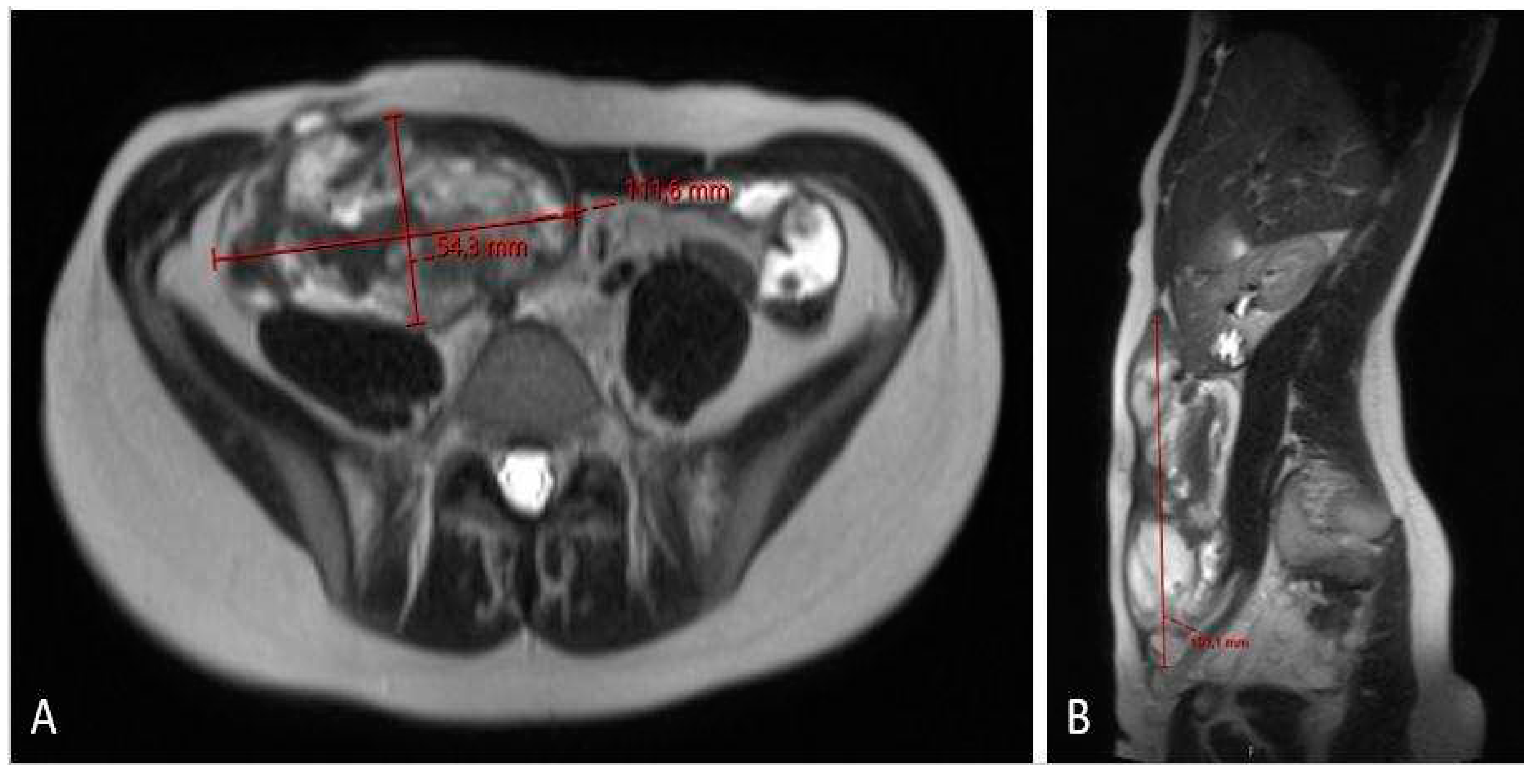
Figure 7.
MRI scan (A – axiall plane; B – sagital plane; C – coronal plane) 12 months after chemotherapy showing significant tumor regress, sized only 1.2 x 3.7 x 8.7 cm.
Figure 7.
MRI scan (A – axiall plane; B – sagital plane; C – coronal plane) 12 months after chemotherapy showing significant tumor regress, sized only 1.2 x 3.7 x 8.7 cm.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



