Submitted:
04 October 2023
Posted:
09 October 2023
You are already at the latest version
Abstract
The enormous influence in terms of bioactivity, affinity, and selectivity represented by the replacement of (L)-2,6-dimethyl tyrosine (Dmt) instead of Phenylalanine (Phe) into Nociceptin/orphanin (N/OFQ) neuropeptide analogues, has been well documented in literature. More recently, the non-natural amino acid (L)-2-methyl tyrosine (Mmt), with steric hindrance among Tyr and Dmt, has been studied because of the modulation of steric effects in opioid peptide chains. Here we report a new synthetic strategy to obtain Mmt based on the well-known Pd-catalyzed ortho-C(sp2)–H activation approach, since there is a paucity of other synthetic routes in literature, to achieve it. The aim of this work was to force only the mono ortho-methylation process over the double ortho-methylation one. In this regard we are pleased to report that the introduction of the dibenzylamine moiety on Tyr aromatic nucleus is a convenient and traceless solution to achieve such a goal. Interestingly, our method provided the aimed Mmt either as N-Boc or N-Fmoc derivatives ready to be inserted into peptide chains through solid phase peptide synthesis (SPPS). Importantly, the introduction of Mmt in place of Phe1 in the sequence of N/OFQ(1-13)-NH2 was very well tolerated in terms of pharmacological profile and bioactivity.
Keywords:
monomethyltyrosine
; dimethyltyrosine
; catalysis
; peptides
1. Introduction
(L)-2,6-dimethyl tyrosine (Dmt) and the corresponding monomethyl tyrosine (Mmt), have been widely used as tyrosine analogues introducing steric encumbrance in bioactive peptides. In particular, Dmt amino acid residue significantly increased bioactivity, affinity and selectivity of synthetic opioid peptides containing it in place of native Tyr or Phe amino acids. [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20] At the current state, while a series of synthetic pathways have been developed for the synthesis of Dmt [21,22,23,24,25,26,27,28], there are only few methods for the obtainment of Mmt. Thus, in 1991 McDonald et al. reported the synthesis of racemic Mmt by the alkylation of the anion of diethyl acetamido malonate with 4-bromomethyl-3-methyl anisole.[29] Later on, Lazarus and Okada [30] achieved 2-monomethyl tyrosine through palladium-catalyzed coupling of a suitably trisubstituted iodo-phenol with 2-acetamido acrylate. In the subsequent step, the resulting sterically hindered dehydroamino acid was subjected to enantioselective hydrogenation in order to establish the desired C-alpha configuration.[25] An elegant synthetic route to Mmt was proposed by Majer et al.[31] who conceived to obtain a regioselective monomethylation of Tyr via hydrogenolysis of tetrahydroisoquinoline-3-carboxylic acid prepared from Tyr, through a Pictet-Spengler cyclization. Unfortunately, the drastic conditions required in the reductive step led to extensive racemization leading to the obtainment of Mmt in racemic form (see Figure 1).
In this work, we describe the successful preparation of the targeted non-natural (L)-amino acid Mmt through a synthetic pathway inspired by our recently reported strategy for the synthesis of a small library of 4-substituted Dmt-like amino acids (Figure 2).[32] In our previous work, indeed, we established that the dibenzylamine functional group (Bn2N) was well tolerated in the pivotal Pd-catalyzed ortho-C(sp2)–H activation-methylation sequence carried out in accordance with Zhang and Ma.[28] In addition, Bn2N showed to be a convenient precursor for a list of functional groups that were subsequently introduced by classical ipso-substitution reaction of the corresponding diazonium salt. As anticipated, by using picolinamide (PA) as a directing group and methyl iodide as the alkylating reagent it was not possible to kinetically differentiate methylation at C-2 from the one at C-6 so that a double ortho-dimethylation of the aryl propionic acid derivative was the only recorded event. We were confident that by performing the Pd-catalyzed ortho-activation-methylation sequence, on a sterically constrained aryl propionic acid derivative, the methylation reaction would occur exclusively at the most accessible aromatic ortho-position. The previously acquired knowledge suggested us to turn to the Bn2N group as a traceless regioselective inductor since it is undoubtedly sterically demanding, chemically tolerant, and easily removable.
Although not yet tested as extensively as Dmt, the Mmt analogue appears to be an interesting amino acid residue to be inserted in peptide chains because of some promising biological evaluations already reported for MOP selective agonists. In particular, [Mmt1]DALDA (H-Tyr-D-Arg-Phe-Lys-NH2)[6] and [Mmt1]EM-2 (endomorphin-2)[30] have shown increased MOP receptor binding affinity and MOP agonist activity compared to the corresponding parent peptides. Furthermore, similar receptor selectivity has been confirmed for Mmt1 and Dmt1 peptide derivatives with the latter being more potent. Herein we also report some preliminary results on [Mmt1]N/OFQ(1-13)-NH2, the shortest active fragment of N/OFQ (i.e. N/OFQ(1-13)-NH2) in which the synthesized Mmt has been inserted at the N-terminal amino acid residue. Effectively, in vitro pharmacological evaluation displayed highly interesting results confirming that Mmt represents an intriguing target in terms of innovative neuropeptides.
2. Discussion and Results
2.1. Chemistry
As reported by Ma and Zhang [28] the picolinamide functional group acts as a suitable ortho-directing group in the key step of Pd-catalyzed C(sp2)–H activation-methylation. Consequently, our synthesis (Figure 3) started with the methyl esterification of the commercially available 3-nitro-L-tyrosine 1 in order to allow its binding to picolinic acid via the HATU-Dipea condensation system. The resulting N-protected tyrosine derivative 2 was transformed to the corresponding TBDMS ether 3 before the conversion of the nitro group to the aimed Bn2N group. To this end, compound 3 was submitted to catalytic hydrogenation and the resulting primary arylamine 4 was N-alkylated with benzyl bromide to give compound 5. The latter was reacted with MeI in the presence of Pd(OAc)2 according to the already tested conditions.[28],[32] Gratifyingly, the expected mono ortho-methylated compound 6 was isolated in 90% yield, confirming that the Bn2N moiety really prevents the methylation reaction from taking place at the adjacent ortho-position. The subsequent hydrogenolytic step removed both benzyl groups from the arylamine 6 affording compound 7 from which Boc-Mmt 10 or Fmoc-Mmt 11 could be promptly obtained in moderate to good yields through the sequence of proto-deamination, hydrolytic protective groups removal and classical N-protection. In details, the reaction of compound 7 with isopentyl nitrite-fluoroboric acid system gave the corresponding aryl diazonium salt 8 which was protodeaminated to 9 by treatment with FeSO4 in DMF. The following harsh acidic hydrolytic conditions yielded Mmt hydrochloride salt, once treated with Boc2O afforded 10 or, alternatively treated with Fmoc-Cl afforded compound 11. (L)-Mmt 10 has been used for the synthesis of [Mmt1]N/OFQ(1-13)-NH2, through SPPS, following a previously described protocol.[33]The reported synthetic strategy to Mmt widen the scope of the original stereo-conservative Pd-catalyzed ortho-C(sp2)–H activation-methylation of Tyr, already described to prepare Dmt.[28]
It has been verified that the Bn2N moiety judiciously introduced on the Tyr aromatic nucleus is a convenient traceless functional group to obtain a single Pd-catalyzed ortho-methylation, over the uncontrolled ortho dimethylation previously reported, see Figure 4. The overall process allowed the preparation of chiral non-racemic Mmt suitably N-protected for use in SPPS.
2.2. Pharmacology
Here, it has been reported the concentration-response curves of N/OFQ, N/OFQ(1-13)-NH2, [Mmt1]N/OFQ(1-13)-NH2, and [Dmt1]N/OFQ(1-13)-NH2 at the calcium mobilization assay; by taking advantage of CHOhNOP cells expressing the chimeric G protein Gαqi5, which allows the coupling of the otherwise Gi/o coupling NOP receptor to Gq-calcium. In this assay, all compounds evoked a concentration-dependent stimulation of calcium release. N/OFQ (1-13)-NH2 shows similar potency and maximal effects (pEC50 = 9.82; Emax = 358) as the natural peptide N/OFQ (pEC50 = 9.56; Emax = 335). The substitution of Phe1 with Dmt in [Dmt1]N/OFQ(1-13)-NH2 produced a pEC50 of 8.35, showing a 30-fold reduction of potency compared to the parental peptide. Importantly, [Mmt1]N/OFQ(1-13)-NH2 mimicked the effects of the standard, with a pEC50 of 9.47. Of note, the statistical comparison of N/OFQ (1-13)-NH2 and the other ligands’ maximal effects indicated no significant alteration.
The concentration-response curves obtained with these ligands in CHOhNOP cells are displayed in Figure 5 and their values of potency and maximal effects are summarized in Table 1.
The present study was carried out with the aim of evaluating the in vitro pharmacological activity of the new peptide [Mmt1]N/OFQ (1-13)-NH2, for its propensity of activating the human NOP receptor. The effects of [Mmt1]N/OFQ (1-13)-NH2, the previously reported [Dmt1]N/OFQ(1-13)-NH2,[10] the parental peptide N/OFQ(1-13)-NH2 and the endogenous neuropeptide N/OFQ were characterized via the calcium mobilization assay in the presence of chimeric G proteins. Intriguingly, in this assay, [Mmt1]N/OFQ (1-13)-NH2 potency was higher than that of the Dmt derivative and similar to that of N/OFQ(1-13)-NH2 and N/OFQ. The design of NOP opioid peptides was based on seminal structure-activity relationship (SAR) studies on N/OFQ [33], which demonstrated that: i) the amidation of the C-terminal protects the peptide from enzymatic degradation; ii) N/OFQ(1–13)-NH2 is the minimal bioactive sequence, which has been subsequently used as a chemical template for further SAR studies. In particular, a large series of N/OFQ(1–13)-NH2 derivatives were generated to investigate the importance of Phe1 for ligand efficacy and selectivity: Phe1 has an important role in the interaction with the receptor and its substitution leads to changes in affinity [10]; indeed the substitution with Phe1 in N/OFQ(1–13)-NH2 sequences causes a reduction in selectivity for NOP over mu-opioid receptors; [33,34] the substitution of Tyr1 with Dmt1 displayed a slight decrease in NOP potency associated with an increase in potency at mu opioid receptor.[10] Even though Mmt has not been tested as extensively as Dmt in position 1, previous reports demonstrate that Mmt is an interesting non proteinogenic amino acid. In particular, the work of Tingyou Li and colleagues [30] evaluated the opioid receptor binding affinities of EM-2 (endomorphin-2) analogs, and [Mmt1]EM-2 have shown increased biological activity compared to the corresponding parent peptides when compared to [Dmt1]EM-2. In the present experiments, the peptides were evaluated for their capability to stimulate the release of intracellular calcium. To do that, CHO cells stably co-expressing the NOP opioid receptors and the Gαqi5 protein were used. The chimeric G protein is able to activate the PLC-IP-Ca2+ pathway, thus increasing intracellular calcium concentrations upon activation of an otherwise Gi/o-coupling GPCR. [35] The calcium mobilization assay used for screening the NOP receptor ligands has been validated in previous studies. In particular, the pharmacological profile of the human NOP receptor coupled with calcium signaling has been assessed with large panels of well-known [36] and novel [10,37,38,39,40,41,42,43,44,45] ligands. In brief, the pharmacological profile obtained at the NOP receptor with this assay is in line with that of other more classical paradigms. Here, N/OFQ(1-13)-NH2, N/OFQ, and [Dmt1]N/OFQ(1-13)-NH2 effects were in line with those reported in the literature, with N/OFQ(1-13)-NH2 and N/OFQ sharing similar values of potency, and [Dmt1]N/OFQ(1-13)-NH2 being approximately 30-fold less potent. [10,42] Very importantly, the introduction of Mmt in place of Phe1 in the N/OFQ(1-13)-NH2 sequence, was very well tolerated (equipotent to N/OFQ) and, if compared to the Dmt modification, the occurring [Mmt1]N/OFQ (1-13)-NH2 was 10-fold more potent. The results obtained for [Mmt1]N/OFQ (1-13)-NH2 might be enlightening for a spatial disposition within the NOP receptor binding pocket close to that of the endogenous peptide. Further molecular dynamics studies could be of interest in validating this hypothesis. This has been now made possible by the recently reported high resolution active structure of the human NOP receptor in complex with N/OFQ and Gi. [46]
3. Conclusions
In conclusion, it was possible to obtain in good to moderate yield the (L)-2-methyl tyrosine, (L)-Mmt, through a novel pathway, different from the ones reported in literature, getting rid of possible byproducts originated by uncontrolled alkylation.
Moreover, the synthesis of this product turned out to be an affordable and low-cost synthesis, since it didn’t require complicated steps or expensive reagents.
Even if the presence of a meta substituted aromatic ring is widely known to represent a smart strategy to control the palladium-catalyzed alkylation leading to the only mono substituted product, the choice of an easily and selectively removable bulky functional group, permissive for the Pd-catalyzed C(sp2)–H activation reaction, was not obvious.
It has been possible to synthesize few N/OFQ related peptides, comparing the presence of Mmt and Dmt in place of the natural Phe1, which is present in N/OFQ and in the shorter fragment N/OFQ(1–13)-NH2. The four peptides have been tested on calcium mobilization assay in CHOhNOP cells expressing the Gαqi5 protein and showed good results in terms of potency, the occurring [Mmt1]N/OFQ(1-13)-NH2 was 10-fold more potent than the Dmt derivative and similar to N/OFQ(1-13)-NH2 and N/OFQ.
Finally, the present study describes the design, synthesis, and in vitro pharmacological evaluation of [Mmt1]N/OFQ(1–13)-NH2 which, thanks to our results, represents an intriguing possibility in terms of further exploration of highly potent synthetic peptides acting as NOP receptor agonists.
4. Material and methods
4.1. Chemistry
All commercial materials were purchased from Fluorochem and Sigma-Aldrich, and used as received unless otherwise noted. Pd(Oac)2 (>98%, Fluorochem) was used in the Pd-catalyzed reactions. Instruments: Analytical RP-HPLC analyses were performed on a Xbridge® C18 column (4.6 × 150 mm, 5 μm particle size) with a flow rate of 0.5 mL/min using a linear gradient of acetonitrile (0.1% TFA) in water (0.1% TFA) from 0% to 100% in 25 minutes. Retention times (Tr) from analytical RP-HPLC are reported in minutes. When necessary, compounds were purified on a reverse-phase Waters Prep 600 HPLC system equipped with a Jupiter column C18 (250 x 30 mm, 300 Å, 15 µm spherical particle size). Gradients used consisted of A (H2O + 0.1% TFA) and B (40% H2O in CH3CN + 0.1% TFA) at a flow rate of 20 mL/min. UV detection wavelength for semi-preparative HPLC was 220 nm. All final products showed a degree of purity >95% at 220 and 254 nm. The mass spectra were recorded with a MICROMASS ZMD 2000. TLC were performed on pre-coated plates of silica gel F254 (Merck, Darmstadt, Germany). 1H NMR and 13C, DEPT NMR analysis were obtained at ambient temperature using a Varian 400 MHz spectrometer and were referenced to residual 1H signals of the deuterated solvents respectively (δ 1H 7.26 for CDCl3, δ 1H 2.50 for DMSO-d6, δ 1H 3.31, 4.87 for CD3OD); the following abbreviations were used to describe the shape of the peaks: s: singlet; d: doublet; dd: double doublet; t: triplet; m: multiplet. Optical rotations were obtained on a Jasco P-2000 Polarimeter instrument with a path length of 1 dm (589 nm), and reported as follows: (c = g/100 mL, solvent). The infrared analyses were performed with a spectroscopy FT-IR spectrum 100 (Perkin Elmer Inc., Waltham, Massachusetts, USA). Hydrogenation reaction in AcOEt was performed under continuous-flow conditions in an H-Cube Pro™ setup (Thalesnano, Hungary) equipped with a module for automatic control of operational parameters (reaction temperature in °C and pressure in bar, flow rates of liquid feed in mL/min and hydrogen). Reactions with microwave assistance were performed using a Biotage Initiator TM 2.0 apparatus (Biotage Sweden).
4.2. Pharmacology
Drugs and reagents: the reagents used were purchased from Sigma Aldrich (St. Louis, MO, USA) and Thermo Fisher Scientific (Waltham, US). Peptides N/OFQ, N/OFQ(1-13)-NH2, [Mmt1] N/OFQ(1-13)-NH2, and [Dmt1]N/OFQ(1-13)-NH2 were synthesized at the Department of Chemical, Pharmaceutical and Agricultural Sciences of the University of Ferrara by the research group of prof. Remo Guerrini. Concentrated solutions of ligands were made in distilled water (1 mM) and kept at – 20°C until use. Successive dilutions of ligands were made in Hank’s Balanced Salt Solution (HBSS) and HEPES buffer (containing 0.005% Bovine Serum Albumine (BSA)). Cell Culture: Chinese Hamster Ovary (CHO) cells stably co-expressing the NOP opioid receptors and the Gαqi5 protein were used. Cells were generated as described by Camarda and co-workers. [37] They were cultured in Dulbecco’s Medium (DMEM) and Ham’s F-12 (1:1) supplemented with 10% fetal bovine serum (FBS), streptomycin (100 μg/ml), penicillin (100 IU/ml), l-glutamine (2 mmol/L), geneticin (G418; 200 μg/ml), fungizone (1 μg/ml) and hygromycin B (100 μg/ml). Cell cultures were kept at 37°C in 5% CO2-humidified air. When confluence was reached, cells were detached by trypsinization, and 50,000 cells/well were seeded into 96-well black, clear-bottom plates 24 h before the test. The following day, after 24 h, the cells were used for testing. Calcium Mobilization Assay: calcium mobilization studies were performed using the automated fluorometer microplate reader FlexStation II (Molecular Devices, Sunnyvale, CA), which was employed to detect changes in fluorescence intensity. At the assay time, cells were pre-incubated for 30 min at 37 °C protected from light with a loading solution consisting of HBSS supplemented with 2.5 mM probenecid, 3 μM Fluo-4 AM, and 0.01% pluronic acid. After that, the loading solution was discarded and 100 μL/well of Brillant Black solution (consisting of HBSS with 20 mM HEPES, 2.5 mM probenecid, and 500 μM Brilliant Black (Sigma-Aldrich, St. Louis, United States)) was added and incubated for an additional 10 min into the fluorometer. After placing both plates (cell culture and compounds plate) into the fluorometric imaging plate reader FlexStation II, fluorescence changes were measured. Serial dilutions of ligands were prepared in HBSS buffer with 20mM HEPES and 0.02% of BSA. Automated addition of peptides or vehicle solutions were automatically added in a volume of 50 μL/well. The present studies were performed at 37 °C to facilitate drug diffusion into the wells. Maximum change in fluorescence, expressed as percent over the baseline fluorescence, was used to determine agonist response. Data analysis and terminology: all data were analyzed using Graph Pad Prism 6.0 (La Jolla, CA, USA). Concentration-response curves were fitted using the four parameters log-logistic equation. Data are expressed as mean ± sem of 5 experiments performed in duplicate. Agonist effects were expressed as the maximum change in percent over the baseline fluorescence. Baseline fluorescence was measured in wells treated with vehicles. Agonist potency was expressed as pEC50, which is the negative logarithm to base 10 of the agonist molar concentration that produces 50% of the maximal possible effect of that agonist.
4.3. Experimental Protocols
4.3.1. Synthesis of methyl (S)-3-(4-hydroxy-3-nitrophenyl)-2-(picolinamido)propanoate (2)
To a solution of 4-Nitro-L-phenylalanine (1) (13.26 mmol, 3.00 g) in anhydrous MeOH (100 mL) was added SOCl2 (14.58 mmol, 1.06 mL) in a dropwise manner. The reaction mixture was heated at reflux overnight. The solvent was removed under vacuum to give the crude methyl-4-hydroxy-3-nitro phenylalanine hydrochloride, which was washed with 50 mL of saturated sodium bicarbonate aqueous solution (to pH ~ 8) and extracted with DCM. The organic layers were combined and evaporated under vacuum to give the corresponding ester as yellowish solid, which was used directly for the next step. A mixture of the previous crude amino product, picolinic acid (15.39 mmol, 1.89 g), HATU (15.39 mmol, 5.85 g) and DIPEA (32.07 mmol, 5.59 mL) in 150 mL of DCM was stirred at room temperature overnight. Then, the reaction was quenched with a saturated NH4Cl aqueous solution and the two layers were separated. The aqueous layer was extracted with DCM (3 times), and the organic layers were combined, dried over Na2SO4, filtered and concentrated in vacuum. The residue was purified by flash chromatography (1:1 petroleum ether/AcOEt) to afford compound 2 (3.54 g) as an orange oil. Yield: 80% overall. MS (ESI): m/z [M+H]+ 346,31 1H NMR (400 MHz, Chloroform-d) δ 10.47 (d, J = 0.4 Hz, 1H), 8.61 – 8.44 (m, 2H), 8.14 (dt, J = 7.8, 1.1 Hz, 1H), 7.91 (d, J = 2.2 Hz, 1H), 7.85 (td, J = 7.7, 1.7 Hz, 1H), 7.50 – 7.36 (m, 2H), 7.06 (d, J = 8.6 Hz, 1H), 5.06 (dt, J = 8.3, 6.0 Hz, 1H), 3.78 (s, 3H), 3.33 – 3.14 (m, 2H). 13C NMR (101 MHz, Chloroform-d) δ 171.39, 164.18, 154.31, 149.13, 148.52, 138.82, 137.53, 133.48, 128.74, 126.73, 125.42, 122.42, 120.34, 53.30, 52.77, 37.26.
4.3.2. Synthesis of methyl (S)-3-(4-((tert-butyldimethylsilyl)oxy)-3-nitrophenyl)-2-(picolinamido)propanoate (3)
To a solution of 2 (10.25 mmol, 3.54g) in DCM (150 mL), were added imidazole (11.27 mmol, 0.76g) and TBSCl (11.27 mmol, 1.69 g). The reaction mixture was stirred at room temperature overnight before the reaction was quenched by adding saturated NaHCO3. The organic layer was separated and the aqueous layer was extracted (3 times) with DCM. The reunited organic layers were dried over Na2SO4, filtered and concentrated under vacuum to give the TBS-ether as a yellowish oil, the crude was purified by flash chromatography (3:2 petroleum ether/AcOEt) to afford compound 3 (3.72 g) as a yellowish oil. Yield: 79% MS (ESI): m/z [M+H]+ 460,57 1H NMR (400 MHz, Chloroform-d) δ 8.56 (ddd, J = 4.8, 1.8, 1.0 Hz, 2H), 8.16 (dt, J = 7.8, 1.1 Hz, 1H), 7.86 (td, J = 7.7, 1.7 Hz, 1H), 7.62 (d, J = 2.3 Hz, 1H), 7.46 (ddd, J = 7.6, 4.8, 1.2 Hz, 1H), 7.31 – 7.23 (m, 1H), 6.88 (d, J = 8.5 Hz, 1H), 5.04 (dt, J = 8.3, 6.1 Hz, 1H), 3.76 (s, 3H), 3.31 – 3.14 (m, 2H), 0.98 (s, 9H), 0.22 (d, J = 1.3 Hz, 6H).13C NMR (101 MHz, Chloroform-d) δ 171.42, 163.97, 149.01, 148.44, 148.28, 141.75, 137.76, 134.79, 129.46, 126.75, 126.27, 122.59, 122.40, 53.39, 52.71, 37.21, 25.64, 18.30, -4.26.
4.3.3. Synthesis of methyl (S)-3-(3-amino-4-((tert-butyldimethylsilyl)oxy)phenyl)-2-(picolinamido)propanoate (4)
The compound 3 (8.09 mmol, 3.72g) was dissolved in AcOEt (53.96 mL, 0.15 M) and set up in continuous-flow hydrogenator reactor H-Cube Pro Thales-Nano at 55 °C, 20 bar, in 0.5 mL/min flow, with Pd/C (10 mol%) as catalyst. When the reaction was completed, monitored via mass spectrometry, the solvent was concentrated in vacuum to obtain the crude product 4 as red-orange oil which was purified by flash chromatography (1:1 petroleum ether/AcOEt) to afford compound 4 (3.02 g) as an orange oil. Yield: 87% MS (ESI): m/z [M+H]+ 430,59 1H NMR (400 MHz, Chloroform-d) δ 8.54 (ddd, J = 4.8, 1.7, 0.9 Hz, 1H), 8.45 (d, J = 8.4 Hz, 1H), 8.15 (dt, J = 7.9, 1.1 Hz, 1H), 7.82 (td, J = 7.7, 1.7 Hz, 1H), 7.41 (ddd, J = 7.6, 4.8, 1.2 Hz, 1H), 6.64 (d, J = 8.0 Hz, 1H), 6.59 (d, J = 2.2 Hz, 1H), 6.45 (dd, J = 8.1, 2.2 Hz, 1H), 4.98 (dt, J = 8.3, 6.2 Hz, 1H), 3.71 (s, 3H), 3.32 (s, 2H), 3.08 (d, J = 6.3 Hz, 2H), 1.05 – 0.95 (m, 9H), 0.21 (s, 6H). 13C NMR (101 MHz, Chloroform-d) δ 172.15, 164.11, 149.51, 148.40, 142.26, 137.85, 137.35, 129.48, 126.42, 122.38, 119.54, 118.55, 116.74, 53.65, 52.39, 37.87, 25.95, 18.36, -4.13.
4.3.4. Synthesis of methyl (S)-3-(4-((tert-butyldimethylsilyl)oxy)-3-(dibenzylamino)phenyl)-2-(picolinamido)propanoate (5)
To a solution of the aniline derivative 4 (7.03 mmol, 3.02 g) in CH3CN (100 mL) was added benzyl bromide (17.57 mmol, 2.09 mL) and potassium carbonate (14.06 mmol, 1.94 g). The mixture was stirred at 80°C overnight. The crude mixture was purified by flash chromatography (1:1 AcOEt/petroleum ether) to afford 5 as an orange oil (3.04 g). Yield: 71% MS (ESI): m/z [M+H]+ 610,84 1H NMR (400 MHz, Chloroform-d) δ 8.44 (d, J = 8.4 Hz, 1H), 8.38 (d, J = 4.8 Hz, 1H), 8.14 (dt, J = 7.8, 1.1 Hz, 1H), 7.80 (td, J = 7.7, 1.7 Hz, 1H), 7.34 (t, J = 6.3 Hz, 1H), 7.26 – 7.14 (m, 6H), 7.09 (d, J = 6.4 Hz, 4H), 6.78 (d, J = 8.0 Hz, 1H), 6.68 (d, J = 8.2 Hz, 1H), 6.56 (s, 1H), 4.93 (q, J = 6.5 Hz, 1H), 4.18 (d, J = 14.3 Hz, 2H), 4.08 (d, J = 14.3 Hz, 2H), 3.62 (s, 3H), 3.16 – 2.91 (m, 2H), 1.04 (s, 9H), 0.25 (s, 6H). 13C NMR (101 MHz, Chloroform-d) δ 171.90, 163.98, 149.53, 148.38, 147.88, 142.63, 138.24, 137.31, 129.21, 128.78, 128.17, 126.99, 126.38, 123.09, 122.34, 120.19, 54.19, 53.57, 52.31, 37.85, 26.11, 18.52, -3.89.
4.3.5. Synthesis of methyl (S)-3-(4-((tert-butyldimethylsilyl)oxy)-5-(dibenzylamino)-2-methylphenyl)-2-(picolinamido)propanoate (6)
To a solution of compound 5 (4.98 mmol, 3.04 g) in toluene (90 mL) were added K2CO3 (14.95 mmol, 2.06 g), CH3I (24.92 mmol, 1.55 mL), and Pd(OAc)2 (0.49 mmol, 0.11 g). The mixture was stirred at 120 °C overnight. After 24 hours the reaction was cooled to r.t. and filtered through celite pad, washed with AcOEt (50 mL). The filtrate was concentrated under vacuum to obtain the crude product. The crude product was purified by flash chromatography (3:7 AcOEt/petroleum ether), to afford 6 (2.83 g) as a yellowish solid. Yield: 91% MS (ESI): m/z [M+H]+ 624,87 1H NMR (400 MHz, Chloroform-d) δ 8.43 (s, 1H), 8.39 (s, 1H), 8.12 (dt, J = 7.8, 1.1 Hz, 1H), 7.78 (td, J = 7.7, 1.7 Hz, 1H), 7.38 – 7.28 (m, 1H), 7.20 (ddd, J = 12.6, 7.7, 5.9 Hz, 6H), 7.13 – 7.04 (m, 4H), 6.65 (s, 1H), 6.51 (s, 1H), 4.87 (q, J = 7.2 Hz, 1H), 4.10 (d, J = 10.8 Hz, 4H), 3.59 (s, 3H), 3.09 (dd, J = 14.1, 6.4 Hz, 1H), 3.00 (dd, J = 14.1, 7.3 Hz, 1H), 2.25 (s, 3H), 1.03 (s, 9H), 0.25 (d, J = 2.3 Hz, 6H). 13C NMR (101 MHz, Chloroform-d) δ 172.36, 164.01, 149.41, 148.33, 147.66, 140.28, 138.40, 137.33, 130.56, 129.48, 129.20, 128.31, 128.10, 126.91, 126.82, 126.39, 123.93, 122.34, 54.36, 52.96, 52.34, 35.34, 26.11, 18.95, -3.88.
4.3.6. Synthesis of methyl (S)-3-(5-amino-4-((tert-butyldimethylsilyl)oxy)-2-methylphenyl)-2-(picolinamido)propanoate (7)
The compound 6 (4.53 mmol, 2,83g) was dissolved in AcOEt (100 mL) and the reductive hydrogenation was performed by the addition of Pd/C (10 mol%) under H2 atmosphere. Once the reaction was completed, monitored via mass spectrometry, the solvent was concentrated in vacuum to afford the crude product, which was purified by flash chromatography (1:1 AcOEt/petroleum ether) to obtain 7 (1.69 g) as a yellowish oil. Yield: 84% MS (ESI): m/z [M+H]+ 444,62 1H NMR (400 MHz, Chloroform-d) δ 8.54 (ddd, J = 4.8, 1.7, 0.9 Hz, 1H), 8.45 (d, J = 8.3 Hz, 1H), 8.12 (dt, J = 7.8, 1.1 Hz, 1H), 7.81 (td, J = 7.7, 1.7 Hz, 1H), 7.40 (ddd, J = 7.6, 4.7, 1.2 Hz, 1H), 6.57 (s, 1H), 6.56 – 6.48 (m, 1H), 4.93 (dt, J = 8.3, 7.0 Hz, 1H), 3.69 (s, 3H), 3.63 (d, J = 20.4 Hz, 2H), 3.10 (dd, J = 14.1, 6.9 Hz, 1H), 3.04 (dd, J = 14.0, 7.3 Hz, 1H), 2.21 (s, 3H), 0.99 (s, 9H), 0.21 (s, 6H). 13C NMR (101 MHz, Chloroform-d) δ 172.52, 164.10, 149.43, 148.34, 142.21, 137.34, 135.25, 127.49, 127.02, 126.41, 122.36, 120.72, 117.76, 53.04, 52.34, 35.47, 25.91, 18.84, 18.33, -4.13.
4.3.7. Synthesis of (S)-2-((tert-butyldimethylsilyl)oxy)-5-(3-methoxy-3-oxo-2-(picolinamido)propyl)-4-methylbenzenediazonium (8)
To a solution of compound 7 (3.81 mmol, 1.69 g) dissolved in anhydrous THF (30 mL), cooled to -10 °C, iso-pentyl nitrite (7.61 mmol, 1.02 mL) and HBF4 (15.23 mmol, 0.95 mL) were added. The reaction was stirred for 4 hours at -10 °C and a orange precipitate was formed, which was directly used as wet crude for the next step. It was performed a diazocopulation assay on the crude 8, with positive result.
4.3.8. Synthesis of methyl (S)-3-(4-((tert-butyldimethylsilyl)oxy)-2-methylphenyl)-2-(picolinamido)propanoate (9)
To a solution of Fe2SO4 (3.81 mmol, 0.57 g) in 15 mL of DMF was added dropwise to the crude compound 8 (3.71 mmol, 1.73 g) solubilized in DMF (5.00 mL). The reaction was stirred overnight, at r.t. The solvent was removed under vacuum and the residue was dissolved in DCM. The organic layer was washed with water, dried over Na2SO4, filtered and concentrated in vacuum. The crude orange oil was purified by flash chromatography (3:7 AcOEt/petroleum ether) obtaining the compound 9 as a yellow solid (0.55 g). Yield: 34% MS (ESI): m/z [M+H]+ 429,60 1H NMR (400 MHz, Chloroform-d) δ 8.56 (ddd, J = 4.8, 1.9, 0.9 Hz, 1H), 8.49 (d, J = 8.4 Hz, 1H), 8.14 (dt, J = 7.8, 1.2 Hz, 1H), 7.89 – 7.77 (m, 1H), 7.48 – 7.38 (m, 1H), 6.96 (d, J = 8.3 Hz, 1H), 6.65 (d, J = 2.6 Hz, 1H), 6.57 (dd, J = 8.2, 2.6 Hz, 1H), 4.97 (dt, J = 8.5, 7.2 Hz, 1H), 3.68 (s, 3H), 3.18 (dd, J = 14.1, 6.9 Hz, 1H), 3.12 (dd, J = 14.1, 7.3 Hz, 1H), 2.31 (s, 3H), 0.96 (s, 9H), 0.16 (s, 6H). 13C NMR (101 MHz, Chloroform-d) δ 172.28, 163.63, 154.50, 149.02, 147.87, 137.98, 137.64, 130.80, 127.17, 126.41, 122.52, 122.10, 117.45, 52.89, 52.21, 35.38, 25.66, 19.49, 18.17, -4.44.
4.3.9. Synthesis of (S)-2-((tert-butoxycarbonyl)amino)-3-(4-hydroxy-2-methylphenyl)propanoic acid (10)
Once purified compound 9 was dissolved in HCl 6N aqueous solution (21.81 mmol, 1.81 mL) and heated at 110 °C for 24 hours. The obtained hydrolyzed crude product was concentrated under vacuum to reduce the volume and the crude solution was used directly for the subsequent protection step. The HCl salt just synthesized was directly used as crude, and diluted in water/dioxane (1:2) solution (0.2 M). The mixture was basified with NaOH 2N aqueous solution until reachment of pH value of 10/11 at 0 °C. Boc2O (1.53 mmol, 0.33 g) was added and the reaction was left stirring at r.t. for 12 hours. The completion of the reaction was monitored per ESI mass spectrometry and TLC. The dioxane was removed under vacuum and HCl 1N aqueous solution was added at 0 °C to pH 1. The mixture was extracted with ethyl acetate (3 times) and the organic phases combined were dried over Na2SO4 and concentrated under vacuum. The crude was purified by flash chromatography (7:3 AcOEt/petroleum ether/1% acetic acid) and crystallized with 2:1 Et2O/petroleum ether, obtaining a white solid as final product, 10. Yield: 45% (0,17g white solid) MS (ESI): m/z [M+H]+ 296,34 1H NMR (400 MHz, DMSO-d6) δ 12.47 (s, 1H), 9.06 (s, 1H), 7.06 (d, J = 8.4 Hz, 1H), 6.95 (d, J = 8.2 Hz, 1H), 6.54 (d, J = 2.6 Hz, 1H), 6.47 (dd, J = 8.2, 2.6 Hz, 1H), 3.99 (ddd, J = 10.0, 8.4, 4.6 Hz, 1H), 2.91 (dd, J = 14.2, 4.7 Hz, 1H), 2.67 (dd, J = 14.2, 10.0 Hz, 1H), 2.19 (s, 3H), 1.32 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 174.37, 156.17, 155.93, 137.49, 131.17, 126.78, 117.22, 112.79, 78.42, 54.59, 33.78, 28.62, 19.47.
4.3.10. Synthesis of (S)-2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(4-hydroxy-2-methylphenyl)propanoic acid (11)
Once purified the compound 9 was dissolved in HCl 6N aqueous solution (21.93 mmol, 1.82 mL) and heated at 110 °C for 24 hours. The obtained hydrolyzed crude product was concentrated under vacuum to reduce the volume and the crude solution was used directly for the subsequent protection step. The HCl salt synthesized was used as crude. It was diluted in water/dioxane (1:2) solution (0.2 M) and basified with Na2CO3 (3,88 mmol, 0,41 g) aqueous solution until pH 10/11 at 0 °C. FmocCl (1,16 mmol, 0,30 g) was added and the reaction was left stirring at r.t. for 2 hours. The reaction was monitored by ESI mass spectrometry and TLC. The dioxane was then removed under vacuum and HCl 1N aqueous solution was added at 0 °C to pH 1. The mixture was extracted with ethyl acetate (3 times) and the organic phases combined were dried over Na2SO4 and concentrated under vacuum. The crude was purified by flash chromatography (1:1 AcOEt/petroleum ether/1% acetic acid) and crystallized with 2:1 Et2O/petroleum ether. Yield: 62% (0,34 g white solid) MS (ESI): m/z [M+H]+ 418,46 1H NMR (400 MHz, DMSO-d6) δ 9.09 (s, 1H), 7.95 – 7.84 (m, 2H), 7.74 – 7.59 (m, 3H), 7.46 – 7.36 (m, 2H), 7.36 – 7.24 (m, 2H), 7.00 (d, J = 8.2 Hz, 1H), 6.55 (d, J = 2.6 Hz, 1H), 6.48 (dd, J = 8.2, 2.6 Hz, 1H), 4.18 (q, J = 4.7 Hz, 3H), 4.13 – 3.99 (m, 1H), 3.00 (dd, J = 14.2, 4.5 Hz, 1H), 2.73 (dd, J = 14.2, 10.2 Hz, 1H), 2.21 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 173.68, 155.97, 155.77, 143.79, 140.67, 137.01, 130.79, 127.62, 127.29, 127.06, 125.26, 120.10, 116.84, 112.41, 65.62, 54.55, 46.58, 33.42, 19.05.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, 1H and 13C spectra of final compounds and application to SPPS.
Author Contributions
Conceptualization, D.I. and A.F.; methodology, D.I.; formal analysis, D.I., C.S.; investigation, V.A.; writing—original draft preparation, D.I., S.S.; writing—review and editing, A.F., V.Z., C.S., D.M.; visualization, C.T., R.G.; supervision, A.F.; funding acquisition, C.T., A.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Recovery and Resilience Plan (NRRP), Mission 04 Component 2 Investment 1.5—NextGenerationEU, Call for tender n. 3277 dated 30 December 2021. Award Number: 0001052 dated 23 June 2022.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article and Supplementary Materials.
Acknowledgments
We thank Erika Marzola for HPLC analysis and Paolo Formaglio for NMR analysis.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dygos, J.H.; Yonan, E.E.; Scaros, M.G.; Goodmonson, O.J.; Getman, D.P.; Periana, R.A.; Beck, G.R. A Convenient Asymmetric Synthesis of the Unnatural Amino Acid 2,6-Dimethyl-L-tyrosine. Synthesis 1992, 8, 741–743. [Google Scholar] [CrossRef]
- Balboni, G.; Marzola, E.; Sasaki, Y.; Ambo, A.; Marczak, E.D.; Lazarus, L.H.; Salvadori, S. Role of 2′,6′-dimethyl-l-tyrosine (Dmt) in some opioid lead compounds. Bioorg. Med. Chem. 2010, 18, 6024–6030. [Google Scholar] [CrossRef] [PubMed]
- Salvadori, S.; Attila, M.; Balboni, G.; Bianchi, C.; Bryant, S.D.; Crescenzi, O.; Guerrini, R.; Picone, D.; Tancredi, T.; Temussi, P.A.; Lazarus, L.H. Delta opioidmimetic antagonists: prototypes for designing a new generation of ultraselective opioid peptides. Mol. Med. 1995, 1, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Bryant, S.D.; Jinsmaa, Y.; Okada, Y.; Lazarus, L.H.; Salvadori, S. Dmt and opioid peptides: a potent alliance. Biopolymers 2003, 71, 86–102. [Google Scholar] [CrossRef]
- Guerrini, R.; Capasso, A.; Sorrentino, L.; Anacardio, R.; Bryant, S.D.; Lazarus, L.H.; Attila, M.; Salvadori, S. Opioid receptor selectivity alteration by single residue replacement: synthesis and activity profile of [Dmt1]deltorphin B. Eur. J.Pharm. 1996, 302, 37–42. [Google Scholar] [CrossRef]
- Schiller, P.W.; Nguyen, T.M.-D.; Berezowska, I.; Dupuis, S.; Weltrowska, G.; Chung, N.N.; Lemieux, C. Synthesis and in vitro opioid activity profiles of DALDA analogues. Eur. J. Med. Chem. 2000, 35, 895–901. [Google Scholar] [CrossRef]
- Mallareddy, J.R.; Borics, A.; Keresztes, A.; Toth, G. Design, Synthesis, Pharmacological Evaluation, and Structure-Activity Study of Novel Endomorphin Analogues with Multiple Structural Modifications. J. Med. Chem.; 2011, 54, 1462–1472. [Google Scholar] [CrossRef]
- Schiller, P.W.; Nguyen, T.M.-D.; Chung, N.N.; Lemieux, C. Dermorphin analogues carrying an increased positive net charge in their "message" domain display extremely high mu opioid receptor selectivity. J. Med. Chem.; 1989, 3, 698–703. [Google Scholar] [CrossRef]
- Schiller, P.W.; Fundytus, M.E.; Merovitz, L.; Weltrowska, G.; Nguyen T., M.-D.; Lemieux, C.; Chung N., N.; Coderre, T.J. J. Med. Chem. 1999, 42, 3520–3526. [CrossRef]
- Molinari, S.; Camarda, V.; Rizzi, A.; Marzola, G.; Salvadori, S.; Marzola, E.; Molinari, P.; McDonald, J.; Ko, M.C.; Lambert, D.G.; Calò, G.; Guerrini, R. [Dmt1]N/OFQ(1–13)-NH2: a potent nociceptin/orphanin FQ and opioid receptor universal agonist. Br. J. Pharmacol. 2013, 168, 151–162. [Google Scholar] [CrossRef]
- Pacifico, S.; Albanese, V.; Illuminati, D.; Marzola, E.; Fabbri, M.; Ferrari, F.; Holanda V. A., D.; Sturaro, C.; Malfacini, D.; Ruzza, C.; Trapella, C.; Preti, D.; Lo Cascio, E.; Arcovito, A.; Della Longa, S.; Marangoni, M.; Fattori, D.; Nassini, R.; Calò, G.; Guerrini, R. Novel Mixed NOP/Opioid Receptor Peptide Agonists. J. Med. Chem. 2021, 64, 6656–6669. [Google Scholar] [CrossRef]
- Ambo, A.; Murase, H.; Niizuma, H. , Ouchi H.; Yamamoto Y.; Sasaki A. Dermorphin and deltorphin heptapeptide analogues: replacement of Phe residue by Dmp greatly improves opioid receptor affinity and selectivity. Bioorg. Med. Chem. Lett. 2002, 12, 879–881. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y. , Sasaki A., Ariizumi T.; igari Y.; Sato K.; Kohara H.; Nizuma H.; Ambo A. 2′,6′-Dimethylphenylalanine (Dmp) Can Mimic the N-Terminal Tyr in Opioid Peptides. Biol. Pharm. Bull. 2004, 27, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Sasaki, A.; Niizuma, H.; Goto, H.; Ambo, A. Endomorphin 2 Analogues Containing Dmp Residue as an Aromatic Amino Acid Surrogate with High μ-Opioid Receptor Affinity and Selectivity. Bioorg. Med. Chem. Lett. 2003, 11, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Hirabuki, M.; Ambo, A.; Ouchi, H.; Yamamoto, Y. Enkephalin Analogues with 2’,6’-Dimethylphenylalanine Replacing Phenylalanine in Position 4. Bioorg. Med. Chem. Lett.; 2001, 11, 327–329. [Google Scholar] [CrossRef]
- Toll, L.; Bruchas, M.R.; Calo, G.; Cox, B.M.; Zaveri, N.T. Nociceptin/Orphanin FQ Receptor Structure, Signaling, Ligands, Functions, and Interactions with Opioid Systems. Pharmacol. Rev. 2016, 68, 419–457. [Google Scholar] [CrossRef]
- Lin, A.P.; Ko, M.C. The Therapeutic Potential of Nociceptin/Orphanin FQ Receptor Agonists as Analgesics without Abuse Liability. ACS Chem. Neurosci. 2013, 4, 214–224. [Google Scholar] [CrossRef]
- Günther, T.; Dasgupta, P.; Mann, A.; Miess, E.; Kliewer, A.; Fritzwanker, S.; Steinborn, R.; Schulz, S. Targeting multiple opioid receptors - improved analgesics with reduced side effects? Br. J.Pharmacol. 2018, 175, 2857–2868. [Google Scholar] [CrossRef]
- Pacifico, S. , Albanese V., Illuminati D., Fantinati A., Marzola E., Ferrari F., Neto J.A., Sturaro C., Ruzza C., Calò G., Preti D., Guerrini R. Tetrabranched Hetero-Conjugated Peptides as Bifunctional Agonists of the NOP and Mu Opioid Receptors. Bioconj. Chem. 2019, 30, 2444–2451. [Google Scholar] [CrossRef]
- Sasaki, Y.; Ambo, A. 2’, 6’-dimethylphenylalanine: a useful aromatic amino acid surrogate for Tyr or Phe residue in opioid peptides. Int. J. Med. Chem. 2012. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Tang, X.; Hruby, V.J. Large-scale asymmetric synthesis of novel sterically constrained 2′,6′-dimethyl- and α,2′,6′-trimethyltyrosine and -phenylalanine derivatives via alkylation of chiral equivalents of nucleophilic glycine and alanine. Tetrahedron 2001, 57, 6375–6382. [Google Scholar] [CrossRef]
- Balducci, D.; Contaldi, S.; Lazzari, I.; Porzi, G. A highly efficient stereocontrolled synthesis of (S)-2′,6′-dimethyltyrosine [(S)-DMT] Tetrahedron: Asymmetry 2009, 20, 1398–1401.
- Mollica, A.; Costante, R.; Mirzaie, S.; Carradori, S.; Macedonio, G.; Stefanucci, A.; Novellino, E. Preparation of Constrained Unnatural Aromatic Amino Acids via Unsaturated Diketopiperazine Intermediat. J. Heterocyclic Chem. 2016, 53, 2106–2110. [Google Scholar] [CrossRef]
- Abrash, H.I.; Niemann, C. Steric Hindrance in α-Chymotrypsin-catalyzed Reactions. Biochemistry 1963, 2, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Dygos, J.H.; Yonan, E.E.; Scaros, M.G.; Goodmonson, O.J.; Getman, D.P.; Periana, R.A.; Beck, G.R. A Convenient Asymmetric Synthesis of the Unnatural Amino Acid 2,6-Dimethyl-L-tyrosine. Synthesis 1992, 741–743. [Google Scholar] [CrossRef]
- Praquin, C.F.B.; de Koning, P.D.; Peach, P.J.; Howard, R.M.; Spencer, S.L. Development of an Asymmetric Hydrogenation Route to (S)-N-Boc-2,6-dimethyltyrosine. Org. Process Res. Dev. 2011, 15, 1124–1129. [Google Scholar] [CrossRef]
- Bender, A.M.; Griggs, N.W.; Gao, C.; Trask, T.J.; Traynor, J.R.; Mosberg, H.I. Rapid Synthesis of Boc-2′,6′-dimethyl-l-tyrosine and Derivatives and Incorporation into Opioid Peptidomimetics. ACS Med. Chem. Lett. 2015, 6, 1199–1203. [Google Scholar] [CrossRef]
- Wang, X.; Niu, S.; Xu, L.; Zhang, C.; Meng, L.; Zhang, X.; Ma, D. Pd-Catalyzed Dimethylation of Tyrosine-Derived Picolinamide for Synthesis of (S)-N-Boc-2,6-dimethyltyrosine and Its Analogues. Org. Lett. 2017, 19, 246–249. [Google Scholar] [CrossRef]
- McDonald, I.A.; Nice, P.L.; Jung, M.J.; Sabol, J.S. Syntheses of DL-2-fluoromethy-p-tyrosine and DL-2-difluoromethyl-p-tyrosine as potential inhibitors of tyrosine hydroxylase. Tetrahedron Lett.; 1991, 32, 887–890. [Google Scholar] [CrossRef]
- Li, T.; Fujita, Y.; Tsuda, Y.; Miyazaki, A.; Ambo, A.; Sasaki, Y.; Jinsmaa, Y.; Bryant, S.D.; Lazarus, L.H.; Okada, Y. Unique High-Affinity Synthetic μ-Opioid Receptor Agonists with Central- and Systemic-Mediated Analgesia. J. Med. Chem. 2005, 48, 586–592. [Google Scholar] [CrossRef]
- Majer, P.; Slaninova, J.; Lebl, M. Synthesis of methylated phenylalanines via hydrogenolysis of corresponding 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acids. In. J. Peptide Protein Res. 1994, 43, 62–68. [Google Scholar] [CrossRef]
- Illuminati, D.; Fantinati, A.; De Ventura, T.; Perrone, D.; Sturaro, C.; Albanese, V.; Marzola, E.; Cristofori, V.; Oble, J.; Poli, G.; Trapella, C. Synthesis of 2,6-Dimethyltyrosine-Like Amino Acids through Pinacolinamide-Enabled C–H Dimethylation of 4-Dibenzylamino Phenylalanine. J. Org. Chem. 2022, 87, 2580–2589. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, R.; Calò, G.; Rizzi, A.; Bianchi, C.; Lazarus, L.H.; Salvadori, S.; Temussi, P.A.; Regoli, D. Address and Message Sequences for the Nociceptin Receptor: A Structure−Activity Study of Nociceptin-(1−13)-peptide amide. J. Med. Chem. 1997, 40, 1789–1793. [Google Scholar] [CrossRef] [PubMed]
- Varani, K.; Rizzi, A.; Calò, G.; Bigoni, R.; Toth, G.; Guerrini, R.; Gessi, S.; Salvadori, S.; Borea, P.A.; Regoli, D. Pharmacology of [Tyr1]nociceptin analogs: receptor binding and bioassay studies. Naunyn Schmiedebergs Arch. Pharmacol. 1999, 360, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Coward, P.; Chan, S.D.H.; Wada, H.G.; Humphries, G.M.; Conklin, B.R. Chimeric G proteins allow a high-throughput signaling assay of Gi-coupled receptors. Anal. Biochem. 1999, 270, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Camarda, V.; Fischietti, C.; Anzellotti, N.; Molinari, P.; Ambrosio, C.; Kostenis, E.; Regoli, D.; Trapella, C.; Guerrini, R.; Salvadori, S.; Calò, G. Pharmacological profile of NOP receptors coupled with calcium signaling via the chimeric protein G alpha qi5. Naunyn Schmiedebergs Arch. Pharmacol. 2009, 379, 599–607. [Google Scholar] [CrossRef]
- Fischetti, C.; Camarda, V.; Rizzi, A.; Pelà, M.; Trapella, C.; Guerrini, R.; McDonald, J.; Lambert, D.G.; Salvadori, S.; Regoli, D.; Calo', G. Pharmacological characterization of the nociceptin/orphanin FQ receptor non peptide antagonist Compound 24. Eur. J. Pharmacol. 2009, 614, 50–57. [Google Scholar] [CrossRef]
- Bojnik, E.; Babos, F.; Fischetti, C.; Magyar, A.; Camarda, V.; Borsodi, A.; Bajusz, S.; Calo', G.; Benyhe, S. Comparative biochemical and pharmacological characterization of a novel, NOP receptor selective hexapeptide, Ac-RYYRIR-ol. Brain Res Bull. 2010, 81, 477–483. [Google Scholar] [CrossRef]
- Rizzi, A.; Malfacini, D.; Cerlesi, M.C.; Ruzza, C.; Marzola, E.; Bird, M.F.; Rowbotham, D.J.; Salvadori, S.; Guerrini, R.; Lambert, D.G.; Calo, G. In vitro and in vivo pharmacological characterization of nociceptin/orphanin FQ tetrabranched derivatives. Br. J. Pharmacol. 2014, 171, 4138–4153. [Google Scholar] [CrossRef]
- Guerrini, R.; Marzola, E.; Trapella, C.; Pacifico, S.; Cerlesi, M.C.; Malfacini, D.; Ferrari, F.; Bird, M.F.; Lambert, D.G.; Salvadori, S.; Calò, G. Structure activity studies of nociceptin/orphanin FQ(1–13)-NH2 derivatives modified in position 5. Bioorg. Med. Chem. 2015, 23, 1515–1520. [Google Scholar] [CrossRef]
- Ferrari, F.; Cerlesi, M.C.; Malfacini, D.; Asth, L.; Gavioli, E.C.; Journigan, B.V.; Kamakolanu, U.G.; Meyer, M.E.; Yasuda, D.; Polgar, W.E.; Rizzi, A.; Guerrini, R.; Ruzza, C.; Zaveri, N.T.; Calò, G. In vitro functional characterization of novel nociceptin/orphanin FQ receptor agonists in recombinant and native preparations. Eur. J. Pharmacol. 2016, 793, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cerlesi, M.C.; Ding, H.; Bird, M.F.; Kiguchi, N.; Ferrari, F.; Malfacini, D.; Rizzi, A.; Ruzza, C.; Lambert, D.G.; Ko, M.C.; Calo, G.; Guerrini, R. Pharmacological studies on the NOP and opioid receptor agonist PWT2-[Dmt1]N/OFQ(1-13). Eur. J. Pharmacol. 2017, 794, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, A.; Cerlesi M., C.; Ruzza, C.; Malfacini, D.; Ferrari, F.; Bianco, S.; Costa, T.; Guerrini, R.; Trapella, C.; Calo’, G. Pharmacological characterization of cebranopadol a novel analgesic acting as mixed nociceptin/orphanin FQ and opioid receptor agonist. Pharmacol. Res. Perspect. 2016, 4, e00247. [Google Scholar] [CrossRef]
- Pacifico, S.; Carotenuto, A.; Brancaccio, D.; Novellino, E.; Marzola, E.; Ferrari, F.; Cerlesi, M.C.; Trapella, C.; Preti, D.; Salvadori, S.; Calò, G.; Guerrini, R. Structure- and conformation-activity studies of nociceptin/orphanin FQ receptor dimeric ligands. Sci. Rep. 2017, 7, 45817. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, F.; Malfacini, D.; Journigan B., V.; Bird M., F.; Trapella, C.; Guerrini, R.; Lambert D., G.; Calo’, G.; Zaveri N., T. In vitro pharmacological characterization of a novel unbiased NOP receptor-selective nonpeptide agonist AT-403. Pharmacol. Res. Perspect. 2017, 5, 4–e00333. [Google Scholar] [CrossRef]
- Wang, Y.; Zhuang, Y.; DiBerto, J.F.; Zhou, X.E.; Schmitz, G.P.; Yuan, Q.; Jain, M.K.; Liu, W.; Melcher, K.; Jiang, Y.; Roth, B.L.; Xu, H.E. Structures of the entire human opioid receptor family. Cell. 2023, 186, 413–427. [Google Scholar] [CrossRef]
Figure 1.
Brief comparison between previous Mmt syntheses and this work.
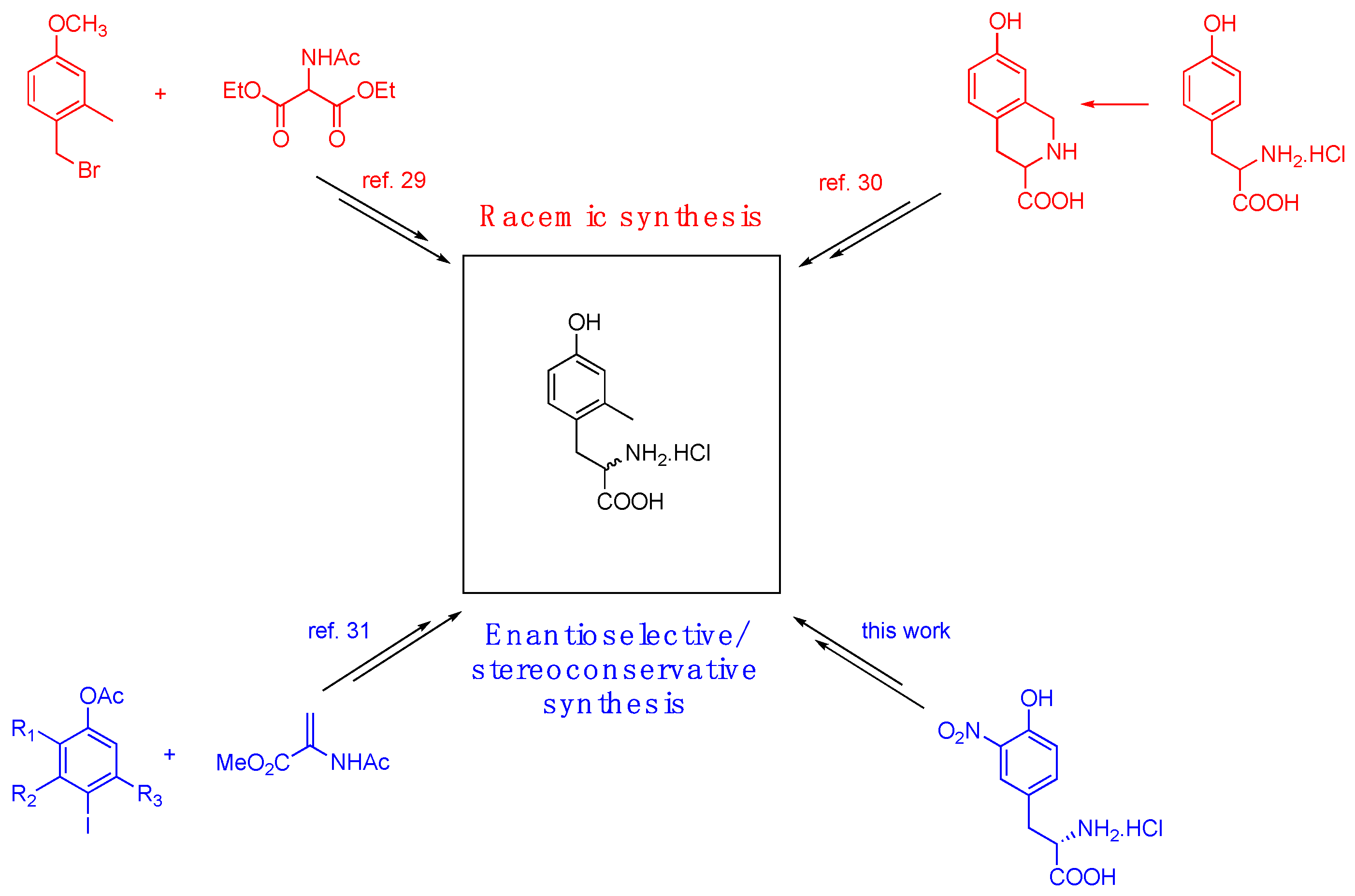
Figure 2.
Dmt synthesis via Pd-catalyzed C(sp2)–H, Trapella and Poli pathway.
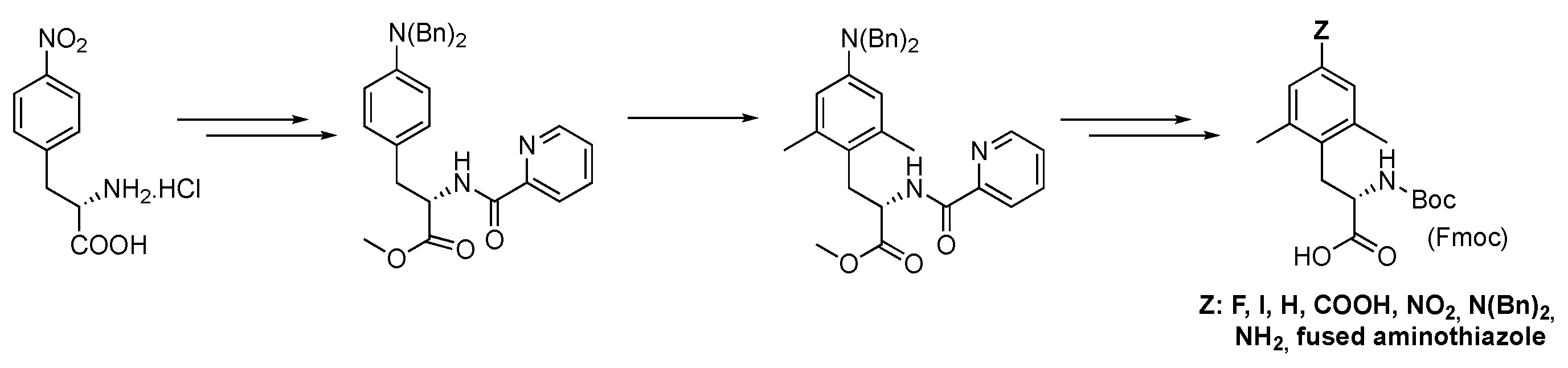
Figure 3.
Traceless bulky regioselective synthesis of Mmt via Pd-catalyzed C(sp2)–H activation.
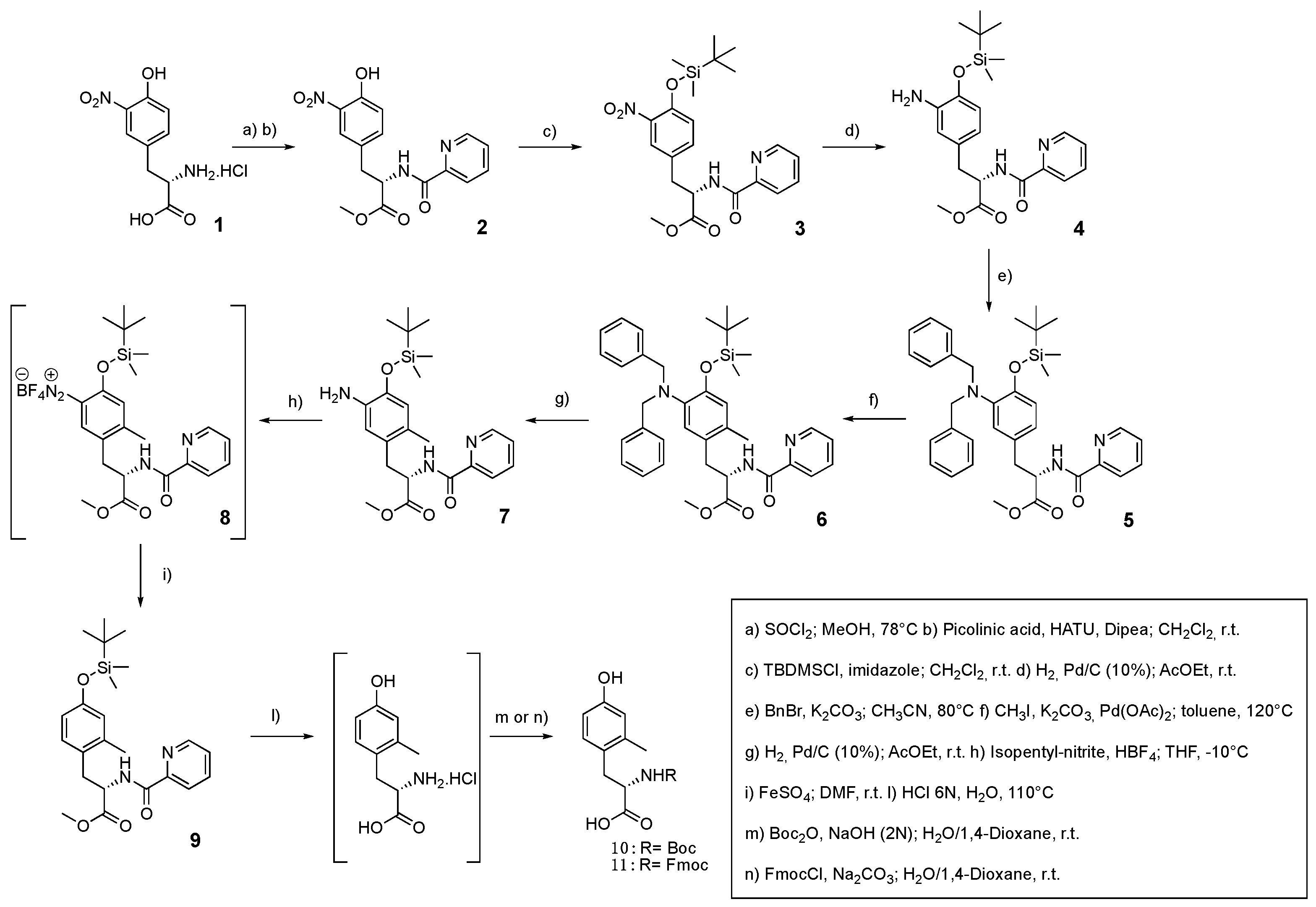
Figure 4.
Proposed regioselective Pd-catalyzed C(sp2)–H sterically constrained mono-methylation mechanism.
Figure 4.
Proposed regioselective Pd-catalyzed C(sp2)–H sterically constrained mono-methylation mechanism.

Figure 5.
Calcium mobilization assay in CHO cells stably expressing the NOP receptor and the Gαqi5 chimeric G protein. Concentration-response curves to N/OFQ, N/OFQ (1-13)-NH2, [Mmt1] N/OFQ (1-13)-NH2, [Dmt1] N/OFQ (1-13)-NH2. Data are mean ± SEM of at least 6 separate experiments made in duplicate.
Figure 5.
Calcium mobilization assay in CHO cells stably expressing the NOP receptor and the Gαqi5 chimeric G protein. Concentration-response curves to N/OFQ, N/OFQ (1-13)-NH2, [Mmt1] N/OFQ (1-13)-NH2, [Dmt1] N/OFQ (1-13)-NH2. Data are mean ± SEM of at least 6 separate experiments made in duplicate.
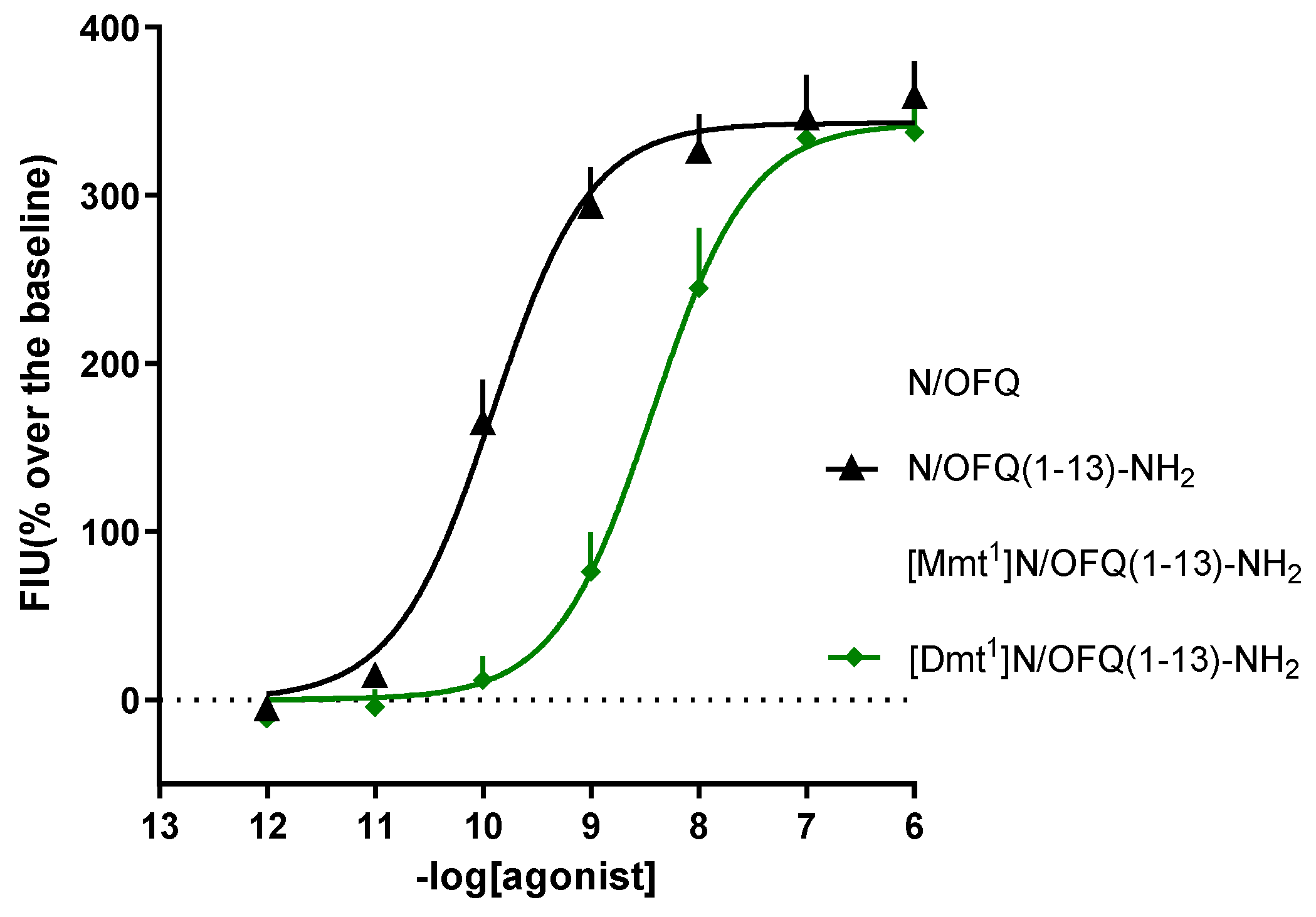

Table 1.
In Vitro Effects of the synthesized compounds in Calcium Mobilization studies performed on CHO cells co-expressing the NOP receptor and the Gαqi5 chimeric protein.
Table 1.
In Vitro Effects of the synthesized compounds in Calcium Mobilization studies performed on CHO cells co-expressing the NOP receptor and the Gαqi5 chimeric protein.
| Calcium mobilization in CHONOP + Gαqi5 cells | ||
|---|---|---|
| pEC50 (CL95%) | Emax + S.E.M. | |
| N/OFQ | 9.56 (9.02 – 10.09) |
335 + 33 |
| N/OFQ(1-13)-NH2 | 9.82 (9.45 – 10.18) |
358 + 22 |
| [Mmt1]N/OFQ(1-13)-NH2 | 9.47 (8.92 – 10.01) |
302 + 39 |
| [Dmt1]N/OFQ(1-13)-NH2 | 8.35 (7.94 – 8.77) |
337 + 26 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



