
Submitted:
01 October 2023
Posted:
02 October 2023
You are already at the latest version
Abstract
Understanding the genetic architecture of a disease is crucial for development of valid diagnostic and therapeutic interventions. The analysis of genomic variations associated with pathological conditions is the starting point for uncovering disease-causing pathways (candidate processes). However, the complexity of intergenic and genetic-environmental interactions hinders the identification of pathogenic values of genomic changes. Furthermore, heredity, epigenetics and somatic mosaicism make the interpretation of genomic data even more sophisticated. To succeed, a variety of bioinformatic techniques are applied. Here, reviewing own and literature data, knowledge-based prioritization of genomic variations is described. Theoretical basis of the knowledge-based prioritization is given with a special regard to gene ontology, heuristics, hermeneutics (genomic hermeneutics) and analytics. Practical and methodological issues of prioritization using ontology- or pathway-based systems analysis are considered in the light of optimistic and realistic scenarios of cumulative phenotypic effects of the variome (the whole set of genomic variations in an individual or specific set of genomic variations for a phenotypic outcome). In the present communication, copy number variants (CNVs) in children with neurodevelopmental diseases are used as a practical foundation for the prioritization, inasmuch as these genome variations are systematically overlooked in the so-called NGS era. Nonetheless, it is highly likely that the prioritization is applicable to almost all types of genomic variations (e.g. chromosome abnormalities, gene mutations, functional synonymous variants etc.). The present methodology seems to be a valuable addition to current biomedical science widening the opportunities for medical genomics and genetics.
Keywords:
candidate processes
; disease
; genomic variations
; knowledge
; medical genomics
; ontology
; pathways
; prioritization
; systems analysis
; variome
1. Introduction
Knowledge is not a usual term for designating elements of data analysis in medical genomics. Taking into account suggested complexity of epistemological aspects of genomic data analysis as well as extreme variability of genomic ontologies [1,2], it is not surprising that researchers prefer to focus on specific tasks such as comparative analysis of genomic variations in different cohorts, selection of single causative mutations or evaluating functional consequences of detected sequence variants [3,4,5]. On the other hand, results of numerous studies dedicated to uncovering genomic variations associated with diseases and massive sets of data on gene ontologies [2,6,7,8,9] appear to be useful for developing knowledge-based approaches to determine the consequences of genomic changes.
Variome (the whole set of genomic variations of an individual or a set of genomic variations associated with specific pathogenic condition or phenotypic trait) seems to be a promising target for knowledge-based analysis. However, probably due to the complexity, targeting variomes by high-resolution bioinformatics technologies is rare [7,10]. Moreover, the generation of biomedical knowledge remains largely enigmatic [11,12]. Alternatively, several efforts in application of variomic, pathway-based and interpretational analyses have been successful in studying neurodevelopmental disorders (NDD) [13,14,15,16]. Pathway-based classification of NDD and cumulative effects of individual genomic variations have been reported in studies of association between copy number variants (CNVs) and brain disorders [12,17,18,19,20]. An intriguing addition to canonical evaluation of causative CNVs has referred to CNV and gene prioritization analyzing CNVariome or the whole set of individual CNVs, in which an effect of genomic variation saturation has been observed (i.e. pathway is unaltered by single CNVs, whereas CNVs affecting several genes implicated in the pathway alter the processes; for more details, see [7]). Unfortunately, the quest for pathway-based classification of diseases (phenotypic traits) starting from knowledge generation and application accompanied by relevant analyses of variome (i.e. evaluation of pathway-oriented saturation by genomic variations) has not been systematically presented.
The present communication describes theoretical aspects of knowledge generation, knowledge-based prioritization of genomic variations (CNVs in NDD) and practical issues of knowledge-oriented studies in medical genomics. Using previous data, it was possible to systemize processes related to biomedical knowledge generation and genome analysis for basic and diagnostic research. Finally, two scenarios (optimistic and realistic) of knowledge-based prioritization of genomic variations are proposed.
2. Theory
As it has been occasionally noted, genomic research has met epistemological difficulties. These are suggested to occur due to problems in defining the nature of biomedical knowledge, increasing amount of data generated by a wide spectrum of genomic (or, more precisely, OMICs) studies, and intrinsic (natural) limitations in human intellectual capacity [1,21]. Since problems of human intellectual capacity are anthropologic and are unrelated to the topic of this communication, the theoretical part is basically focused on the nature of biomedical knowledge and the relationship with genomic data, which represent a major epistemological substrate for current biomedicine.
The whole set of OMICs technologies has formed the firm (epistemological) foundation for uncovering the meaning of genomic, transcriptomic, proteomic, and metabolomics variations. Using sophisticated statistics and bioinformatic technologies of biomedical data analysis, it becomes possible to go beyond direct associations between single/specific mutations and phenotypes [22,23]. Moreover, data sets of genomic variations (i.e. variomes) are hardly processable without additional bioinformatic analyses, which are performed using gene ontologies and/or algorithms of pathway-based classification. Hence, it becomes possible to modulate or to estimate the effects of specific genomic variations at the pathway level or, in other words, to model abnormal genome, transcriptome, proteome and metabolome behavior altered because of genomic change (from single nucleotide polymorphisms (SNP) and gene mutations to CNVs and chromosome imbalances) [2,9,11,12,13,23,24,25]. Pathway-based analysis or classification of genomic data represents, thereby, an appreciable step forward in understanding gene ontologies and genetic architecture of diseases [11,12,13], challenging and changing the conceptualization and causation of genetic diseases in the widest sense [12,26]. Alternatively, the use of these approaches to address the outcomes of genomic changes provides for developing complex (holistic) models of genetic diseases, which seem to explain the sophistication of pathological genome behavior [12,27]. Finally, construction and analysis of gene networks representing molecular and cellular pathways to pathological processes offer numerous opportunities for prioritization of pathogenic intracellular processes and unraveling genetic architecture of human diseases [25,28,29]. In total, one may suggest that accumulation of empirical genomic data is the important part (starting point) of analysis/prioritization of genomic variations in human disease genetics, requiring, however, an appreciable amount of additional bioinformatic efforts for epistemological transition from information to knowledge.
Taking into account the aforementioned epistemological difficulties in genomics [1,21,22,26,27,28], it appears that there are two more essential elements in processing genomic data (additional to acquiring empirical data), which are interpretation and analysis. These both are the theoretical basis for bioinformatic analysis of genome variability (variome) and, as a result, underlie generation of genomic (biomedical) knowledge. However, genome research occasionally considers analysis of genomic data as a basic/epistemological discipline, whereas the discipline of interpretation of genomic (variomic) data is generally left aside in current biomedical research. To rebrand and to re-introduce these two elements of generating biomedical knowledge, a tradition of using ancient Greek/philosophic terms (similarly to as it has been done in [12]) has been followed. Interpretational efforts in social and much more rarely natural sciences are designated as hermeneutics [30]. The latter term has been already used for computational big data analysis [31]. In medical sciences, hermeneutic studies are generally of social nature [32,33]. However, hermeneutic approaches to acquiring knowledge and data are applicable in biomedical purposes [34]. Actually, in silico analyses of genomic data at subchromosomal and chromosomal levels, which are targeted at interpretation of genome variation and chromosome (genome) instability, have systematically demonstrated the efficiency for uncovering the meaning in the disease and biodiversity context [7,35,36,37]. All these interpretational efforts in the field of medical genomics corresponds to hermeneutic analysis in the all the senses [30]. In this light, it is to remember that hermeneutics means initially textual interpretation (i.e. interpretation of ancient texts) [30]. Ironically, the genome of Homo sapiens may be considered as a rather ancient text aged ~200 000 years [38]. Therefore, the existence of genomic hermeneutics is likely to be justified.
Systems analysis (systems theory) has become an integral part of medical genomics (genomic medicine) showing an unprecedented value for unraveling molecular or genetic architecture of human disease [39,40,41]. Still, accumulation of biomedical knowledge periodically suffers from the lack of network and systems analyses [42]. On the other hand, systems analysis for interpretation using OMICs is able to process large variome data or a large genomic change such as structural chromosome imbalance and aneuploidy (gains/losses of whole chromosomes) [25,35]. A thorough systems analysis of discoveries built on the Human Genome Project in numbers has demonstrated enormous albeit irregular (some genes/gene ontologies are preferentially analyzed) progress in medical genomics [43]. Moreover, this is also appropriate for single-cell OMICs studies [44]. Thus, genomic analytics or systems analysis of genome data appears to be an important source of knowledge in current biomedical research. The description of empirical, hermeneutic and analytical genome analysis has allowed to propose two models of biomedical knowledge generation for prioritization of genomic variations. The first one is basic, which establishes hierarchy between empirics (data collecting), hermeneutics (interpretation) and analytics (systems analysis). The second one is dynamic; it describes processing of empirical data using hermeneutic and analytical methodology to generate knowledge. Figure 1 summarizes theoretical issues concerning knowledge-based prioritization (knowledge generation) using the aforementioned models.
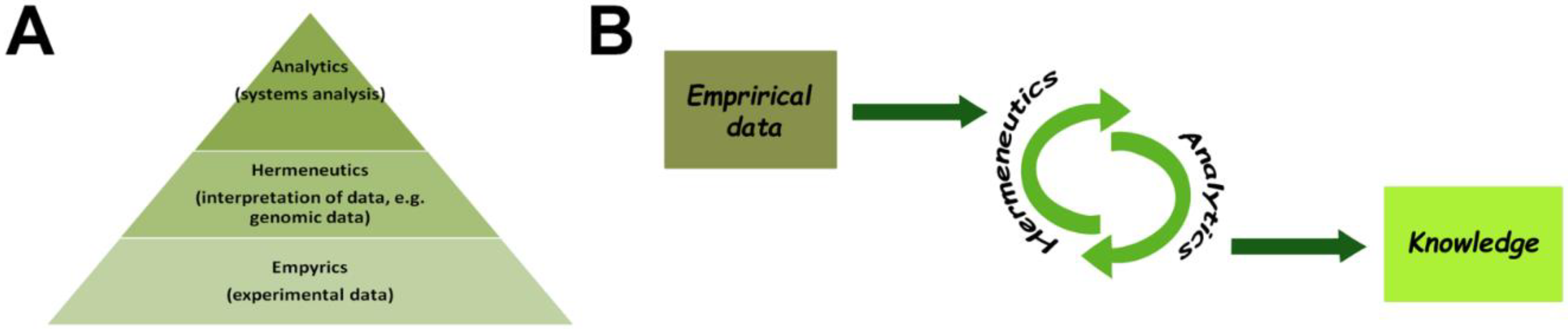
Figure 1.
Schematic representation of biomedical knowledge generation for knowledge-based prioritization of genomic variations using two models. (A) Basic model: empirics or acquiring experimental data is the foundation of knowledge generation followed by the interpretation of data (i.e. hermeneutics or genomic hermeneutics), which forms the basis for following systems analysis (analytics). (B) Dynamic model: empirical data is processed by hermeneutics and analytics (the transition from hermeneutics to analytics is hardly distinguishable during genomic data processing) being the main process for knowledge generation.
Figure 1.
Schematic representation of biomedical knowledge generation for knowledge-based prioritization of genomic variations using two models. (A) Basic model: empirics or acquiring experimental data is the foundation of knowledge generation followed by the interpretation of data (i.e. hermeneutics or genomic hermeneutics), which forms the basis for following systems analysis (analytics). (B) Dynamic model: empirical data is processed by hermeneutics and analytics (the transition from hermeneutics to analytics is hardly distinguishable during genomic data processing) being the main process for knowledge generation.
3. Practice
Since the idea that practice is the criterion of truth remains widely acceptable in the materialistic world, knowledge generation and knowledge-based prioritization are to be considered in the light of basic and/or diagnostic practice (research). To pass from theory to practice, CNVs in NDD have been selected as a target. Apart from a relative ease of the management (amount of CNVs per individual genome is much more less than quantity of sequence variants detected in an individual), the selection is also produced by the intention to raise decreasing focus on CNVs in the so-called NGS era using appreciable data sets on the contribution to NDD pathogenesis. Analyzing a large set of own [7,15,24,35,44,45,46] and literature data [8,9,13,16,18,20,47] clearly demonstrates that gene ontologies and molecular pathways are key elements in a complex system determining disease mechanism, which gathers genomic, epigenomic, proteomic and metabolomic data. In other words, the interpretation of genomic/variomic data (e.g. CNVariome) or genomic hermeneutics is effective when based on systems analysis (analytics) and vice versa [7,25,35,41,42]. In addition, it has been demonstrated that heuristic algorithms appear to be the most applicable for this type of the prioritization inasmuch as the amount of biomedical data (especially, data on gene ontology) gradually increases as well as the results of these studies cannot be ultimately proven [46]. It also becomes evident that CNVs may interact with each other, i.e. multiple CNVs may affect molecular processes (alter ontologies) changing the dosage and structures of transcripts involved in disease candidate process. Accordingly, a saturation effect of pathway-altering genomic variations in an individual variome (CNVariome) certainly exits [7]. Therefore, the saturation is a promising target for assays applied to unravel genomic mechanisms of a disease. In total, prioritization of causative CNVs (genomic variations) using knowledge generated through the interpretation and (systems) analysis of genomic data appears to be attractive.
Despite the success of the aforementioned genomic studies [7,8,9,15,16,18,20,24,35,44,45,46,47], some practical limitations of assays for uncovering molecular disease mechanisms using knowledge-based prioritization of genome variations exist. Postgenomic knowledge (i.e. knowledge acquired from combining genomic, epigenomic, proteomic/interactomic and metabolomics data/information) and gene ontologies are naturally limited due to impossibility of synchronous processing of data on all tissues/cells of a human organism [28,42,48]. This becomes even more sophisticated in cases of CNVs and chromosomal imbalances (cytopostgenomic aspects), which are featured by simultaneous involvement of multiple genes [46,48,49]. Still, there are technological solutions for processing large genomic data sets [8,10,28,48]. Another source of the limitation of knowledge-based prioritization is genetic-environmental interactions, which add variability to functional consequences of genomic variations [50,51]. Genome or chromosome instability and somatic mosaicism (presence of cellular populations differing with respect to their genomic variations in an organism) are probably the best examples of related problems being the result of alterations to numerous molecular/cellular pathways coupled or produced by environmental effects; additionally, these phenomena affect phenotypic outcomes of genomic variation per se (for more details, see [50,51,52,53,54,55]. Complex systems of the human orgasm (i.e. brain or central nervous system) are the most promising, albeit uneasy, targets for sophisticated systems analysis when these difficulties are not left aside [56]. Fortunately, a number of models (e.g. variome or variome saturation model) may be used for solving the problems for uncovering genetic architecture of complex disorders [7,57]. Nonetheless, although developments in genomic bioinformatics have long been proved to be powerful in discovering causative mutations in humans [9,58], there is still a need for consistent improvements in available methodology. This is the particular case of knowledge-based prioritization and evaluation of variomic saturation.
4. Methodology
The methodological description requires several definitions. A genomic change (e.g. CNV) is an event. CNVs may be classified as pathogenic or non-pathogenic according to the occurrence (rare versus common) [20]. The variome as the set of all the genomic variants (or CNVs) represents a data set or information. The extraction of ‘pathogenic’ variants or, more accurately, pathway-altering genomic changes (CNVs) from a variome would produce a set of epistemological elements, which interact to form a system that represents a disease mechanism (pathogenesis) and is eventually knowledge. To develop a knowledge-based prioritization of genomic variations (a quest from empirics through hermeneutics-analytics to knowledge or transition from information to knowledge), there is a need for defining the interplay between events and their attributes or, in functional terms, probability. Consequently, probability is essential for developing the knowledge-based methodology.
Pathway-based definition of CNV pathogenic values among individuals with NDD is generally made through assessing ontologies of genes [2] affected by CNVs and involved in brain-specific molecular and cellular pathways [13,14,19,24,35,46]. In this instance, there are effective methods for determining the probability of pathogenicity (p-value) of each CNV (for theoretical and technical details, see [47,59,60,61]). Thus, this part of methodology has been already developed without requirements for further efforts. However, there are no similar methodology for multiple causative CNVs from an individual variome or for description the effect of genomic variation saturation, i.e. knowledge-based prioritization per se. Here, an evaluation of cumulative probability of pathogenicity of genomic variations appears to be required. Due to the heuristic nature of knowledge-based prioritization of CNVs [46], two scenarios for cumulative phenotypic effects of CNVariome through alterations of molecular pathways — optimistic and realistic — have been proposed.
4.1. Optimistic Scenario
The most optimistic situation in this methodology corresponds to an extremely simplified concept “one CNV alters one pathway”. This functionally corresponds to P(N)=N, where P — probability of specific pathway altering (pathogenicity) and N — number of CNV. P is determined for each candidate pathway (process). Certainly, the level of optimism seems to require a decrease. To do so, Pi (probability of pathway altering of each single CNV) is introduced. Consequently, the optimistic scenario may be functionally described as follows: P(N)=Pi∙N. Pi may be determined as previously [59,60,61]. Linear dependence between CNVs (number of events) and probability (presumed value or p-value of pathogenicity defined using a number of attributes [61]) suggests that optimistic scenario is also the simplest one. Graphically, the optimistic scenario may be shown as a series of linear graphs (P(N)= Pi∙N) differing with respect to Pi (Figure 2). The scenario is truly optimistic as one may note that even a moderate meaning of Pi (p-value equals to 0.5) corresponds to the case, in which 2 CNVs alter a brain-specific pathway and lead to NDD.
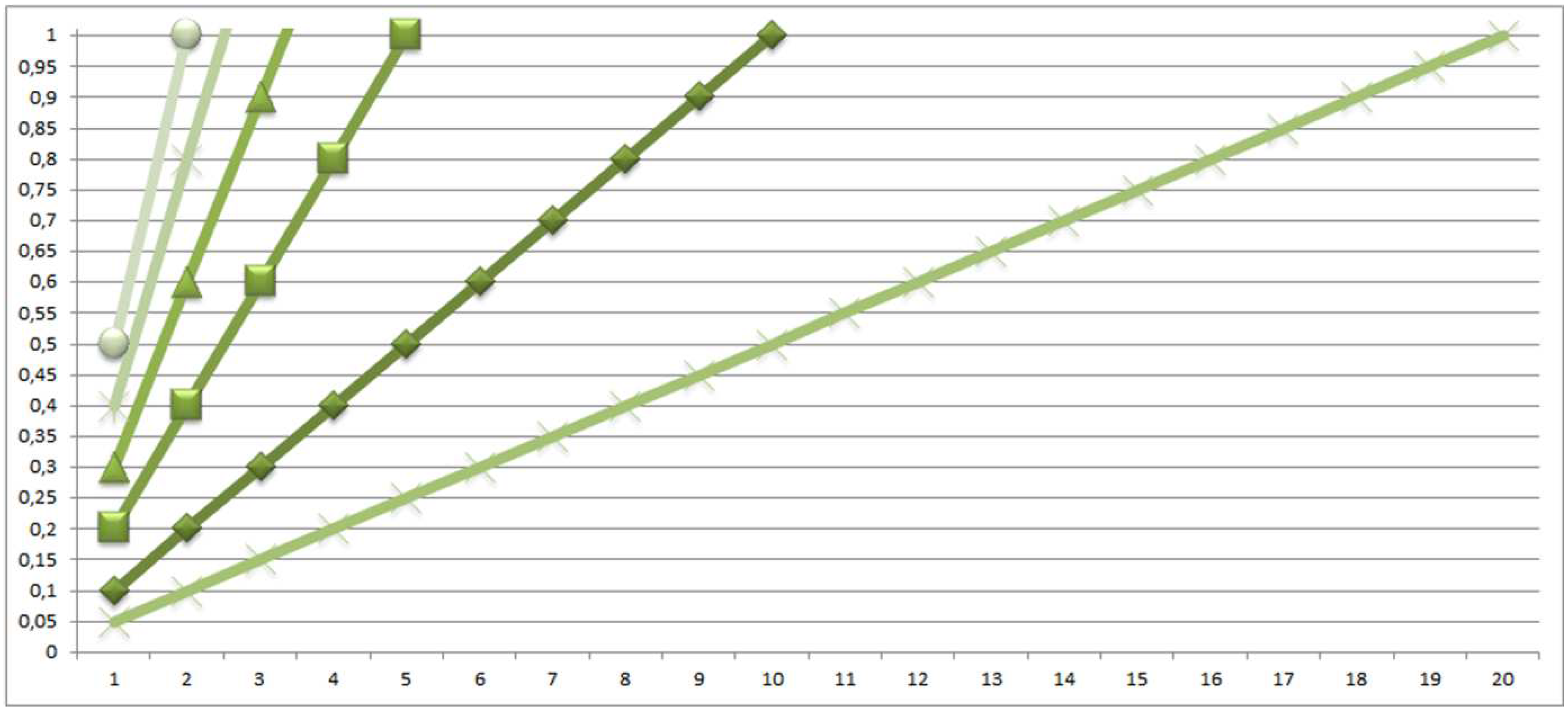
Figure 2.
Graphical depiction of the optimistic scenario, which is based on linear dependence between number of CNVs affecting genes implicated in an altered pathway (abscissa; N) and probability of the alteration (ordinate; P) or P(N)=Pi∙N; from left to right: Pi = 0.5, 0.4, 0.3, 0.2, 0.1 and 0.05 (note that when Pi>0.3 just four CNVs may produce the effect of CNVariome saturation in the context of specific pathway).
Figure 2.
Graphical depiction of the optimistic scenario, which is based on linear dependence between number of CNVs affecting genes implicated in an altered pathway (abscissa; N) and probability of the alteration (ordinate; P) or P(N)=Pi∙N; from left to right: Pi = 0.5, 0.4, 0.3, 0.2, 0.1 and 0.05 (note that when Pi>0.3 just four CNVs may produce the effect of CNVariome saturation in the context of specific pathway).
4.2. Realistic Scenario
Complicating the scenario by introducing the effect of genomic variation saturation on pathway dysfunctions (variome concept) [7] appears to be appropriate for transition from optimism to realism. Following idea originating from an immanent feature of materialistic knowledge (epistemology), which seems to be always relative to previous achievements, underlies the scenario: “we cannot be absolutely sure about 100% adverse effect of a CNV on a specific pathway”. Thus, any amount of CNVs affecting brain-specific pathway would produce a trend of P→1, but P=1 always remains unachievable. Apparently, the pathway-oriented saturation of genomic variations is describable by logarithmic dependence. Additional realism may be achieved by taking into account the fact that each CNV possesses own p-value (Pi of the optimistic scenario). Accordingly, a rank has been introduced to the function P(N) representing an average effect of all CNVs affecting specific pathway in relation to their number.
Hence, the realistic scenario may be mathematically defined as follows: P(N)=, where P — probability of pathogenicity or specific pathway altering, N — number of CNV, and R — rank determined as (Pi is determined as in the case of optimistic scenario and varies between 0 and 0.9). Figure 3 graphically demonstrates the basic variant of the scenario.
The realistic scenario is likely to be the most suitable candidate for becoming mathematical basis of knowledge-based prioritization of genomic variations. Firstly, it is based on an important epistemological feature (see below). Secondly, it links genomic hermeneutics and analytics (system analysis), if average P is established per pathway cluster or a set of candidate processes. Thirdly and finally, the logarithmic law used for definition of the scenario reflects the heuristic nature of bioinformatic interpretational analysis of genome data.
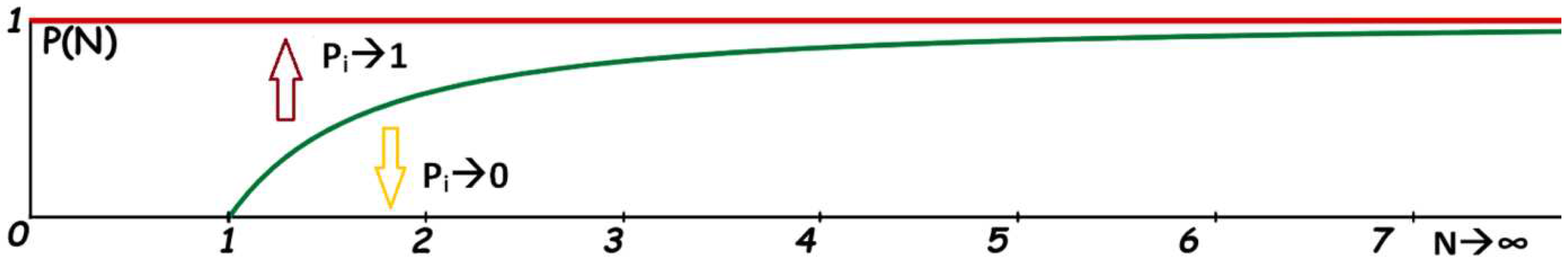
Figure 3.
Graphical depiction of the realistic scenario. The effect of the pathway-based variome saturation by CNVs (see [7] for details) or the dependence between number of CNVs affecting genes implicated in an altered pathway (abscissa; N) and probability of the alteration (ordinate; P) follows logarithmic law; green graph — P(N)=, where R=0 or, in other words, single CNV is unable to affect pathway. Red line (equals to the 100% probability) is used to depict the idea underlying scenario realism: the line is absolutely unachievable according to the corresponding logarithmic law. Finally, the variability of R (R is not always equals 0) may increase (red arrow) or decrease (yellow arrow) starting P values.
Figure 3.
Graphical depiction of the realistic scenario. The effect of the pathway-based variome saturation by CNVs (see [7] for details) or the dependence between number of CNVs affecting genes implicated in an altered pathway (abscissa; N) and probability of the alteration (ordinate; P) follows logarithmic law; green graph — P(N)=, where R=0 or, in other words, single CNV is unable to affect pathway. Red line (equals to the 100% probability) is used to depict the idea underlying scenario realism: the line is absolutely unachievable according to the corresponding logarithmic law. Finally, the variability of R (R is not always equals 0) may increase (red arrow) or decrease (yellow arrow) starting P values.
5. Conclusion
In the world of ultra-rapid accumulation of biomedical knowledge, there is a growing need of analytical methodology emerging theoretical and practical issues. The definition of genetic architecture of pathological conditions seems primarily to benefit from the existence of similar methodology. Since numerous practical/diagnostic efforts in the field of medical genomics lack firm theoretical background and bioinformatic algorithms are useless without testing in diagnostic/basic research practice (genomic/cytogenomic), a concept unifying theory and practice appears to be required. Variome concept appears to be one, inasmuch as it allows not only to unravel genomically altered pathways [7], but also to propose successful therapeutic interventions in presumably incurable genetic diseases (e.g. diseases caused by chromosomal abnormalities) [62,63]. Here, a large theoretical input to the concept and the practical issues are presented. Moreover, a model of biomedical knowledge generation for knowledge-based prioritization of genomic variations is proposed.
Attempts to determine entirely the genetic architecture of complex diseases (group of diseases) have been systematically limited by the available technologies [9,12,23,47,63,64]. Probably, separated theoretical and practical efforts in medical genomics hindered the development of an integrated view on the evaluation of genomic variation pathogenicity, especially in case of variomic analysis [7,64]. Combining the efforts, it was suggested that knowledge generated by empirical, hermeneutic and analytical analyses may be successfully used for prioritization of genomic variations by ontology- or pathway-based heuristic technologies. The methodology mathematically enforced by proposing realistic and optimistic scenarios of prioritization appears to have appreciable advantages comparing to pure empirical or isolated bioinformatic (theoretical) technologies. Fortunately, a number of previous studies have shown that similar approaches to the prioritization are effective for studying CNVs and chromosomal imbalances in clinical cohorts [6,8,13,14,15,16,17,18,19,20,25,35,46]. Finally, the aforementioned pathway-based analysis of saturation in genomic variations using the knowledge-based prioritization of specific variomes is not limited to CNVs and NDD. When CNVs are substituted by another type of genomic variation (e.g. single nucleotide polymorphisms/sequence variants or gene mutations, etc.), the knowledge-based prioritization seems to remain effective comparably to previous assays of bioinformatic interpretation of sequence variants [65,66,67] and causative gene mutations [68,69,70]. Finally, focusing on other pathways or candidate processes (process clusters) than those mentioned here before is able to be applicable for uncovering mechanisms of diseases and phenotypic features in the widest sense.
To this end, understanding of the generation and classification of biomedical knowledge and finding the routes to use it for diagnostic and basic research appears incomplete without systematic efforts in upgrading genomic theory and technology. Certainly, successful evidence-based molecular therapy of genetic diseases would be closely related to consistent efforts in improving methods for high-resolution processing of genomic data.
References
- Dougherty ER. On the epistemological crisis in genomics. Curr Genomics. 2008; 9(2):69-79. doi: 10.2174/138920208784139546. [CrossRef]
- The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019; 47(D1):D330-D338. doi: 10.1093/nar/gky1055. [CrossRef]
- Iourov IY, Yurov YB, Vorsanova SG. Chromosome-centric look at the genome. In: Iourov I, Vorsanova S, Yurov Y, editors. Human interphase chromosomes — Biomedical aspects. Springer; 2020. pp. 157–170.
- Liehr T. From human cytogenetics to human chromosomics. Int J Mol Sci. 2019; 20(4):826. doi: 10.3390/ijms20040826. [CrossRef]
- Liehr T. About classical molecular genetics, cytogenetic and molecular cytogenetic data not considered by Genome Reference Consortium and thus not included in genome browsers like UCSC, Ensembl or NCBI. Mol Cytogenet. 2021; 14(1):20. doi: 10.1186/s13039-021-00540-7. [CrossRef]
- Riggs ER, Ledbetter DH, Martin CL. Genomic variation: lessons learned from whole-genome CNV analysis. Curr Genet Med Rep. 2014; 2(3):146-150. doi: 10.1007/s40142-014-0048-4. [CrossRef]
- Iourov IY, Vorsanova SG, Yurov YB. The variome concept: focus on CNVariome. Mol Cytogenet. 2019; 12:52. doi: 10.1186/s13039-019-0467-8. [CrossRef]
- Wainschtein P, Jain D, Zheng Z; TOPMed Anthropometry Working Group; NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium; Cupples LA, Shadyab AH, McKnight B, Shoemaker BM, Mitchell BD, Psaty BM, Kooperberg C, Liu CT, Albert CM, Roden D, Chasman DI, Darbar D, Lloyd-Jones DM, Arnett DK, Regan EA, Boerwinkle E, Rotter JI, O’Connell JR, Yanek LR, de Andrade M, Allison MA, McDonald MN, Chung MK, Fornage M, Chami N, Smith NL, Ellinor PT, Vasan RS, Mathias RA, Loos RJF, Rich SS, Lubitz SA, Heckbert SR, Redline S, Guo X, Chen Y-I, Laurie CA, Hernandez RD, McGarvey ST, Goddard ME, Laurie CC, North KE, Lange LA, Weir BS, Yengo L, Yang J, Visscher PM. Assessing the contribution of rare variants to complex trait heritability from whole-genome sequence data. Nat Genet. 2022; 54(3):263-273. doi: 10.1038/s41588-021-00997-7. [CrossRef]
- Claussnitzer M, Cho JH, Collins R, Cox NJ, Dermitzakis ET, Hurles ME, Kathiresan S, Kenny EE, Lindgren CM, MacArthur DG, North KN, Plon SE, Rehm HL, Risch N, Rotimi CN, Shendure J, Soranzo N, McCarthy MI. A brief history of human disease genetics. Nature. 2020; 577(7789):179-189. doi: 10.1038/s41586-019-1879-7. [CrossRef]
- Burn J, Watson M. The Human Variome Project. Hum Mutat. 2016; 37(6):505-7. doi: 10.1002/humu.22986. [CrossRef]
- Manzoni C, Kia DA, Vandrovcova J, Hardy J, Wood NW, Lewis PA, Ferrari R. Genome, transcriptome and proteome: the rise of omics data and their integration in biomedical sciences. Brief Bioinform. 2018; 19(2):286-302. doi: 10.1093/bib/bbw114. [CrossRef]
- Iourov IY, Vorsanova SG, Yurov YB. Pathway-based classification of genetic diseases. Mol Cytogenet. 2019; 12:4. doi: 10.1186/s13039-019-0418-4. [CrossRef]
- Mullin AP, Gokhale A, Moreno-De-Luca A, Sanyal S, Waddington JL, Faundez V. Neurodevelopmental disorders: mechanisms and boundary definitions from genomes, interactomes and proteomes. Transl Psychiatry. 2013; 3(12):e329.
- Cardoso AR, Lopes-Marques M, Silva RM, Serrano C, Amorim A, Prata MJ, Azevedo L. Essential genetic findings in neurodevelopmental disorders. Hum Genomics. 2019; 13(1):31. doi: 10.1186/s40246-019-0216-4. [CrossRef]
- Zelenova MA, Yurov YB, Vorsanova SG, Iourov IY. Laundering CNV data for candidate process prioritization in brain disorders. Mol Cytogenet. 2019; 12:54. doi: 10.1186/s13039-019-0468-7. [CrossRef]
- Safizadeh Shabestari SA, Nassir N, Sopariwala S, Karimov I, Tambi R, Zehra B, Kosaji N, Akter H, Berdiev BK, Uddin M. Overlapping pathogenic de novo CNVs in neurodevelopmental disorders and congenital anomalies impacting constraint genes regulating early development. Hum Genet. 2023; 142(8):1201-1213. doi: 10.1007/s00439-022-02482-5. [CrossRef]
- Lupski JR. Brain copy number variants and neuropsychiatric traits. Biol Psychiatry. 2012; 72(8):617-9. doi: 10.1016/j.biopsych.2012.08.007. [CrossRef]
- Takumi T, Tamada K. CNV biology in neurodevelopmental disorders. Curr Opin Neurobiol. 2018; 48:183-192. doi: 10.1016/j.conb.2017.12.004. [CrossRef]
- Iourov IY, Vorsanova SG, Yurov YB, Zelenova MA, Kurinnaia OS, Vasin KS, Kutsev SI. The cytogenomic "theory of everything": chromohelkosis may underlie chromosomal instability and mosaicism in disease and aging. Int J Mol Sci. 2020; 21(21):8328. doi: 10.3390/ijms21218328. [CrossRef]
- Kopal J, Kumar K, Saltoun K, Modenato C, Moreau CA, Martin-Brevet S, Huguet G, Jean-Louis M, Martin CO, Saci Z, Younis N, Tamer P, Douard E, Maillard AM, Rodriguez-Herreros B, Pain A, Richetin S, Kushan L, Silva AI, van den Bree MBM, Linden DEJ, Owen MJ, Hall J, Lippé S, Draganski B, Sønderby IE, Andreassen OA, Glahn DC, Thompson PM, Bearden CE, Jacquemont S, Bzdok D. Rare CNVs and phenome-wide profiling highlight brain structural divergence and phenotypical convergence. Nat Hum Behav. 2023; 7(6):1001-1017. doi: 10.1038/s41562-023-01541-9. [CrossRef]
- Dougherty ER, Shmulevich I. On the limitations of biological knowledge. Curr Genomics. 2012; 13(7):574-87. doi: 10.2174/138920212803251445. [CrossRef]
- Mehta T, Tanik M, Allison DB. Towards sound epistemological foundations of statistical methods for high-dimensional biology. Nat Genet. 2004; 36(9):943-7. doi: 10.1038/ng1422. [CrossRef]
- Karczewski KJ, Snyder MP. Integrative omics for health and disease. Nat Rev Genet. 2018; 19(5):299-310. doi: 10.1038/nrg.2018.4. [CrossRef]
- Vorsanova SG, Yurov YB, Iourov IY. Neurogenomic pathway of autism spectrum disorders: linking germline and somatic mutations to genetic-environmental interactions. Curr Bioinform. 2017;12:19–26. doi: 10.2174/1574893611666160606164849. [CrossRef]
- Yurov YB, Vorsanova SG, Iourov IY. Network-based classification of molecular cytogenetic data. Curr Bioinform. 2017;12:27–33. doi: 10.2174/1574893611666160606165119. [CrossRef]
- Dekeuwer C. Conceptualization of genetic disease. In: Schramme T, Edwards S, editors. Handbook of the philosophy of medicine. Dordrecht: Springer; 2015. pp. 1–18.
- Hochstein E. Why one model is never enough: a defense of explanatory holism. Biol Philos. 2017;32(6):1105–1125.
- Iourov IY, Vorsanova SG, Yurov YB. Systems cytogenomics: are we ready yet? Curr Genomics. 2021; 22(2):75-78. doi: 10.2174/1389202922666210219112419. [CrossRef]
- Rosenthal SB, Wright SN, Liu S, Churas C, Chilin-Fuentes D, Chen CH, Fisch KM, Pratt D, Kreisberg JF, Ideker T. Mapping the common gene networks that underlie related diseases. Nat Protoc. 2023; 18(6):1745-1759. doi: 10.1038/s41596-022-00797-1. [CrossRef]
- Ricœur P. Le conflit des interprétation. Essais d’herméneutique 1. Paris: Le Seuil. 1969.
- Mohr JW, Wagner-Pacifici R, Breiger RL. Towards a computational hermeneutics. Big Data Soc. 2015; 2(2):2053951715613809.
- Vasileiou K, Barnett J, Thorpe S, Young T. Characterising and justifying sample size sufficiency in interview-based studies: systematic analysis of qualitative health research over a 15-year period. BMC Med Res Methodol. 2018; 18(1):148. doi: 10.1186/s12874-018-0594-7. [CrossRef]
- Miah SJ, Gammack J, Hasan N. Methodologies for designing healthcare analytics solutions: a literature analysis. Health Informatics J. 2020; 26(4):2300-2314. doi: 10.1177/1460458219895386. [CrossRef]
- Burgun A, Bodenreider O. Accessing and integrating data and knowledge for biomedical research. Yearb Med Inform. 2008:91-101.
- Iourov IY, Vorsanova SG, Yurov YB. In silico molecular cytogenetics: a bioinformatic approach to prioritization of candidate genes and copy number variations for basic and clinical genome research. Mol Cytogenet. 2014; 7(1):98. doi: 10.1186/s13039-014-0098-z. [CrossRef]
- Heng H.H. New data collection priority: focusing on genome-based bioinformation. Res Results Biomed. 2020;6(1):5–8. doi: 10.18413/2658-6533-2020-6-1-0-1. [CrossRef]
- Heng J, Heng HH. Karyotype coding: The creation and maintenance of system information for complexity and biodiversity. Biosystems. 2021; 208:104476. doi: 10.1016/j.biosystems.2021.104476. [CrossRef]
- Stringer C. The origin and evolution of Homo sapiens. Philos Trans R Soc Lond B Biol Sci. 2016; 371(1698):20150237. doi: 10.1098/rstb.2015.0237. [CrossRef]
- Auffray C, Chen Z, Hood L. Systems medicine: the future of medical genomics and healthcare. Genome Med. 2009; 1(1):2. doi: 10.1186/gm2. [CrossRef]
- Gustafsson M, Nestor CE, Zhang H, Barabási AL, Baranzini S, Brunak S, Chung KF, Federoff HJ, Gavin AC, Meehan RR, Picotti P, Pujana MÀ, Rajewsky N, Smith KG, Sterk PJ, Villoslada P, Benson M. Modules, networks and systems medicine for understanding disease and aiding diagnosis. Genome Med. 2014; 6(10):82. doi: 10.1186/s13073-014-0082-6. [CrossRef]
- Tanaka H, Kreisberg JF, Ideker T. Genetic dissection of complex traits using hierarchical biological knowledge. PLoS Comput Biol. 2021; 17(9):e1009373. doi: 10.1371/journal.pcbi.1009373. [CrossRef]
- Barabási DL, Bianconi G, Bullmore E, Burgess M, Chung S, Eliassi-Rad T, George D, Kovács IA, Makse H, Nichols TE, Papadimitriou C, Sporns O, Stachenfeld K, Toroczkai Z, Towlson EK, Zador AM, Zeng H, Barabási AL, Bernard A, Buzsáki G. Neuroscience needs network science. J Neurosci. 2023; 43(34):5989-5995. doi: 10.1523/JNEUROSCI.1014-23.2023. [CrossRef]
- Gates AJ, Gysi DM, Kellis M, Barabási AL. A wealth of discovery built on the Human Genome Project - by the numbers. Nature. 2021; 590(7845):212-215. doi: 10.1038/d41586-021-00314-6. [CrossRef]
- Iourov IY, Vorsanova SG, Yurov YB. Single cell genomics of the brain: focus on neuronal diversity and neuropsychiatric diseases. Curr Genomics. 2012; 13(6):477-88. doi: 10.2174/138920212802510439. [CrossRef]
- Iourov IY, Vorsanova SG, Korostelev SA, Zelenova MA, Yurov YB. Long contiguous stretches of homozygosity spanning shortly the imprinted loci are associated with intellectual disability, autism and/or epilepsy. Mol Cytogenet. 2015; 8:77. doi: 10.1186/s13039-015-0182-z. [CrossRef]
- Iourov IY, Vorsanova SG, Kurinnaia OS, Zelenova MA, Vasin KS, Demidova IA, Kolotii AD, Kravets VS, Iuditskaia ME, Iakushev NS, Soloviev IV, Yurov YB. Molecular cytogenetic and cytopostgenomic analysis of the human genome. Res Results Biomed. 2022; 8(4):412–423. doi: 10.18413/2658-6533-2022-8-4-0-1. [CrossRef]
- Lee C, Iafrate AJ, Brothman AR. Copy number variations and clinical cytogenetic diagnosis of constitutional disorders. Nat Genet. 2007; 39(7 Suppl):S48-54. doi: 10.1038/ng2092. [CrossRef]
- Iourov IY. Cytopostgenomics: what is it and how does it work? Curr Genomics. 2019; 20(2):77–78. doi: 10.2174/138920292002190422120524. [CrossRef]
- Liehr T. Cytogenomics. Cambridge: Academic Press; 2021.
- Iourov IY, Vorsanova SG, Yurov YB. Somatic cell genomics of brain disorders: a new opportunity to clarify genetic-environmental interactions. Cytogenet Genome Res. 2013; 139(3):181–188. doi: 10.1159/000347053. [CrossRef]
- Ye CJ, Sharpe Z, Heng HH. Origins and consequences of chromosomal instability: from cellular adaptation to genome chaos-mediated system survival. Genes (Basel) 2020; 11(10):1162. doi: 10.3390/genes11101162. [CrossRef]
- Iourov IY, Yurov YB, Vorsanova SG, Kutsev SI. Chromosome instability, aging and brain diseases. Cells. 2021; 10(5):1256. doi: 10.3390/cells10051256. [CrossRef]
- Iourov IY, Vorsanova SG, Yurov YB, Kutsev SI. Ontogenetic and pathogenetic views on somatic chromosomal mosaicism. Genes (Basel). 2019; 10(5):379. doi: 10.3390/genes10050379. [CrossRef]
- Vorsanova SG, Yurov YB, Iourov IY. Dynamic nature of somatic chromosomal mosaicism, genetic-environmental interactions and therapeutic opportunities in disease and aging. Mol Cytogenet. 2020; 13:16. doi: 10.1186/s13039-020-00488-0. [CrossRef]
- Costantino I, Nicodemus J, Chun J. Genomic mosaicism formed by somatic variation in the aging and diseased brain. Genes (Basel) 2021; 12(7):1071. doi: 10.3390/genes12071071. [CrossRef]
- Ji Z, Song Q, Su J. Editorial: Advanced computational systems biology approaches for accelerating comprehensive research of the human brain. Front Genet. 2023; 14:1143789. doi: 10.3389/fgene.2023.1143789. [CrossRef]
- Iourov IY, Vorsanova SG, Kurinnaia OS, Kutsev SI, Yurov YB. Somatic mosaicism in the diseased brain. Mol Cytogenet. 2022; 15(1):45. doi: 10.1186/s13039-022-00624-y. [CrossRef]
- Andrade MA, Sander C. Bioinformatics: from genome data to biological knowledge. Curr Opin Biotechnol. 1997; 8(6):675-83. doi: 10.1016/s0958-1669(97)80118-8. [CrossRef]
- Kirov G, Pocklington AJ, Holmans P, Ivanov D, Ikeda M, Ruderfer D, Moran J, Chambert K, Toncheva D, Georgieva L, Grozeva D, Fjodorova M, Wollerton R, Rees E, Nikolov I, van de Lagemaat LN, Bayés A, Fernandez E, Olason PI, Böttcher Y, Komiyama NH, Collins MO, Choudhary J, Stefansson K, Stefansson H, Grant SG, Purcell S, Sklar P, O’Donovan MC, Owen MJ. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry. 2012; 17(2):142-53. doi: 10.1038/mp.2011.154. [CrossRef]
- Vulto-van Silfhout AT, Hehir-Kwa JY, van Bon BW, Schuurs-Hoeijmakers JH, Meader S, Hellebrekers CJ, Thoonen IJ, de Brouwer AP, Brunner HG, Webber C, Pfundt R, de Leeuw N, de Vries BB. Clinical significance of de novo and inherited copy-number variation. Hum Mutat. 2013; 34(12):1679-87. doi: 10.1002/humu.22442. [CrossRef]
- Zhang L, Shi J, Ouyang J, Zhang R, Tao Y, Yuan D, Lv C, Wang R, Ning B, Roberts R, Tong W, Liu Z, Shi T. X-CNV: genome-wide prediction of the pathogenicity of copy number variations. Genome Med. 2021; 13(1):132. doi: 10.1186/s13073-021-00945-4. [CrossRef]
- Iourov IY, Vorsanova SG, Voinova VY, Yurov YB. 3p22.1p21.31 microdeletion identifies CCK as Asperger syndrome candidate gene and shows the way for therapeutic strategies in chromosome imbalances. Mol Cytogenet. 2015; 8:82. doi: 10.1186/s13039-015-0185-9. [CrossRef]
- Iourov IY. Cytogenomic bioinformatics: practical issues. Curr Bioinform. 2019; 14(5):372–3.
- Piñero J, Ramírez-Anguita JM, Saüch-Pitarch J, Ronzano F, Centeno E, Sanz F, Furlong LI. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2020; 48(D1):D845-D855. doi: 10.1093/nar/gkz1021. [CrossRef]
- Polonikov AV, Klyosova EYu, Azarova IE. Bioinformatic tools and internet resources for functional annotation of polymorphic loci detected by genome wide association studies of multifactorial diseases (review). Res Results Biomed. 2021; 7(1):15-31. DOI: 10.18413/2658-6533-2020-7-1-0-2. [CrossRef]
- Lin BC, Katneni U, Jankowska KI, Meyer D, Kimchi-Sarfaty C. In silico methods for predicting functional synonymous variants. Genome Biol. 2023; 24(1):126. doi: 10.1186/s13059-023-02966-1. [CrossRef]
- Mikhalitskaya EV, Vyalova NM, Ermakov EA, Levchuk LA, Simutkin GG, Bokhan NA, Ivanova SA. Association of single nucleotide polymorphisms of cytokine genes with depression, schizophrenia and bipolar disorder. Genes (Basel). 2023; 14(7):1460. doi: 10.3390/genes14071460. [CrossRef]
- Goldstein DB, Allen A, Keebler J, Margulies EH, Petrou S, Petrovski S, Sunyaev S. Sequencing studies in human genetics: design and interpretation. Nat Rev Genet. 2013; 14(7):460-70. doi: 10.1038/nrg3455. [CrossRef]
- Ceyhan-Birsoy O, Murry JB, Machini K, Lebo MS, Yu TW, Fayer S, Genetti CA, Schwartz TS, Agrawal PB, Parad RB, Holm IA, McGuire AL, Green RC, Rehm HL, Beggs AH; BabySeq Project Team. Interpretation of genomic sequencing results in healthy and ill newborns: results from the BabySeq Project. Am J Hum Genet. 2019; 104(1):76-93. doi: 10.1016/j.ajhg.2018.11.016. [CrossRef]
- Zhang J, Yao Y, He H, Shen J. Clinical interpretation of sequence variants. Curr Protoc Hum Genet. 2020; 106(1):e98. doi: 10.1002/cphg.98. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



