Submitted:
22 September 2023
Posted:
26 September 2023
You are already at the latest version
Abstract
A series of novel substituted isoxazolidine derivatives was synthesized through 1,3-dipolar cycloaddition reactions of α-aryl-N-methyl nitrones with various ethylene moieties, resulting in the formation of isoxazolidine derivatives. The structures of the synthesized compounds were characterized using spectroscopic methods. The [3+2] cycloaddition (32CA) reactions of α-aryl-N-methyl nitrones with diverse ethylene substituents were computed employing Density Functional Theory (DFT) at the B3LYP/6–31G(d) level, upgrading bond parameters. An investigation of the electron localization function (ELF) unveiled the zwitterionic nature of α-aryl-N-methyl nitrones, devoid of pseudoradical or carbenoid centers. Utilization of CDFT indices enabled the prediction of global electronic flux from the strongly nucleophilic α-aryl-N-methyl nitrones to the varied electrophilic ethylene functionalities. Molecular docking and molecular dynamics simulations were conducted on the isoxazolidine derivatives against the EGFR receptor (PDB: 4ZAU). In silico assessment of the synthesized isoxazolidines involved molecular docking, wherein the compound with the highest docking score was subjected to molecular dynamics simulations. Compound 11a, out of the thirty isoxazolidine derivatives, displayed the most substantial binding energy, yielding a g score of -6.57 kcal/mol. These observations highlighted the functional role of amide NH2 and NO2 in the binding process with the target protein, influencing its activity. Furthermore, prediction of ADME drug-likeness properties was performed to identify a suitable candidate.
Keywords:
isoxazolidine derivatives
; 1
; 3-dipolar cycloaddition
; α-aryl-N-methyl nitrones
; Density Functional Theory (DFT)
; Electron Localization Function (ELF)
; CDFT indices
; docking
; molecular dynamics
; EGFR
; binding energy
1. Introduction
Heterocyclic compounds represent the predominant category of cyclic organic compounds and are characterized by the presence of at least one heteroatom, with nitrogen, oxygen, and sulfur being the most common. Additionally, heterocyclic rings containing diverse heteroatoms have been documented. Heterocycles hold significance in pharmaceutical and industrial domains. The notion of drug-like entities is widely employed in medicinal chemistry, with the use of molecular descriptors from existing pharmaceuticals to aid in the design of new molecules, a recognized strategy for enhancing clinical viability[1, 2].
Nitrones have been extensively employed as precursors for the synthesis of biologically active organic compounds, including heterocyclic isoxazolines [3], isoxazolidines[4, 5], oxazines[6] derivatives, and various natural products [7] . In a similar vein, the isoxazolidine functional group, formed through the 1,3-dipolar cycloaddition of a nitrone and an alkene, assumes a pivotal role in several chemical transformations due to the labile nature of the N-O bond [8].
Isoxazolidines represent a class of potent heterocycles widely utilized as precursors for 1,3-amino-alcohols and a diverse array of natural compounds and derivatives [7], encompassing amino acids [9, 10], alkaloids [11], and amino sugars[12]. Isoxazolidines exhibit intriguing biological properties, including anticancer [13], antioxidant [5, 14], antibacterial [15], antifungal [16], antiretroviral [17], and antimycobacterial [18] activities. Among the valuable reactions for synthesizing heterocyclic isoxazolidine derivatives, the 1,3-dipolar cycloaddition of nitrones and olefins stands out [19].
Lung cancer constitutes the primary cause of cancer-related fatalities globally and boasts a substantial mortality rate. Recent concepts in lung cancer research involve identifying genetic determinants within patient subgroups that can serve as predictive biomarkers and molecular targets for targeted cancer therapy, such as EGFR somatic mutations [20-23]. Subsequent randomized clinical trials have demonstrated remarkable therapeutic responses to EGFR tyrosine kinase inhibitors in specific patients with these mutations [24-26].
A diverse range of EGFR gene mutations, activating the EGFR kinase and triggering oncogenic transformation, are now linked to lung cancer development, especially in non-smokers or former smokers [27]. These mutations also confer sensitivity to tyrosine kinase inhibition. The most prevalent mutations are exon 19 deletions encompassing the LREA motif and the L858R point mutation in exon 21, both responsive to the ATP-competitive first-generation EGFR inhibitors erlotinib and gefitinib [21, 28, 29].
Molecular docking is a pivotal technique in computer-assisted drug design, providing valuable insights into binding modes, ligand-protein interactions, inhibitory characteristics, and interaction energies. Molecular docking is particularly effective in assessing the recognition level between test compounds and protein active site amino acids. Ligand molecules retain flexibility during virtual screening, while macromolecules remain inflexible [30].
This study encompasses the synthesis of thirty novel isoxazolidines and presents an extensive theoretical analysis of the 32CA reaction involving α-aryl-N-methyl nitrones and diverse ethylene derivatives. Additionally, docking studies and molecular dynamics simulations were conducted on the binding pocket of wild-type EGFR to explore the potential of the synthesized compounds.
2. Results
2.1. Chemistry
Quantitative yield of the nitrones (3a-e) was achieved by reacting the benzaldehyde derivatives (1a-e) with N-phenylhydroxylamine hydrochloride (2) in a mixture of ethanol and water (Scheme 1). Spectroscopic methods were employed to characterize the resulting compound. The 1H NMR spectrum of 3a, for instance, exhibited singlets at δ 3.96 and δ 7.54 ppm, corresponding to -NCH3 and CH=N protons, respectively, alongside resonances in the aromatic region. Furthermore, additional resonances appeared at 55.12 (NCH3) and 136.05 (CH=N) in the 13C NMR spectrum.
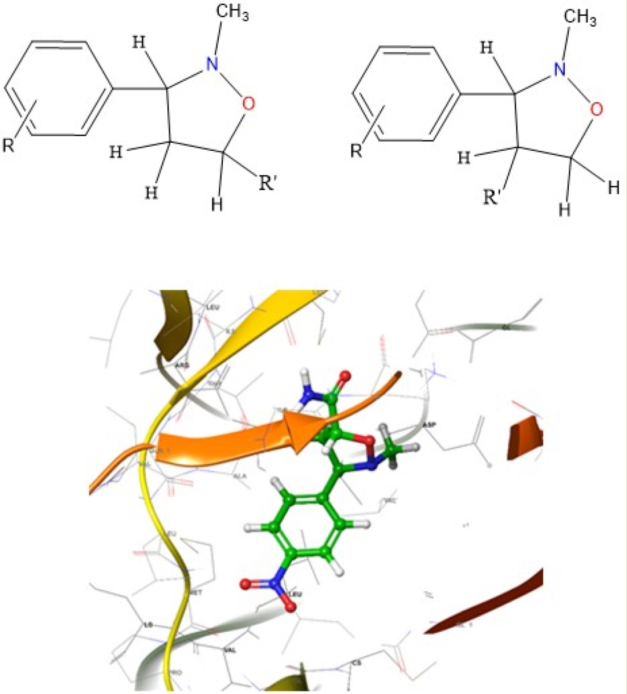
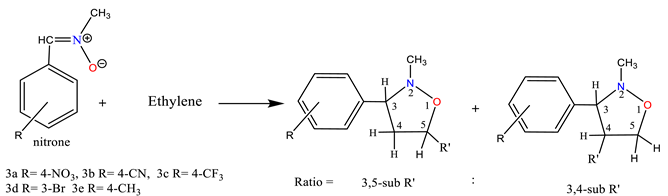
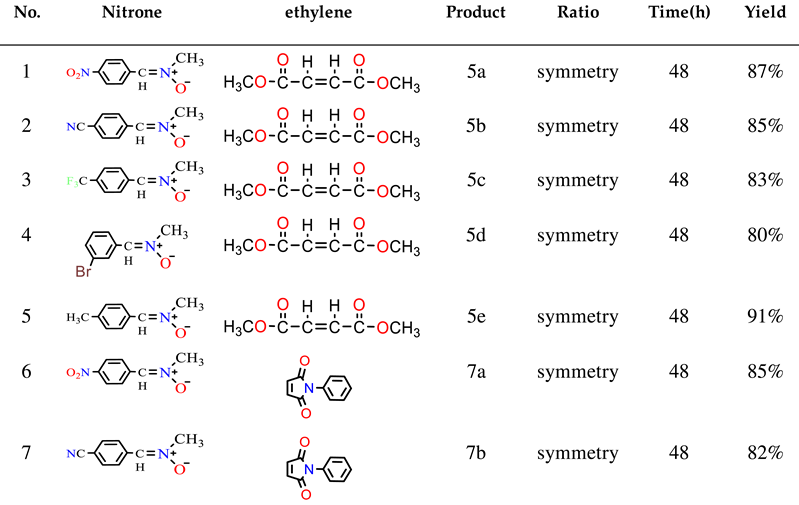
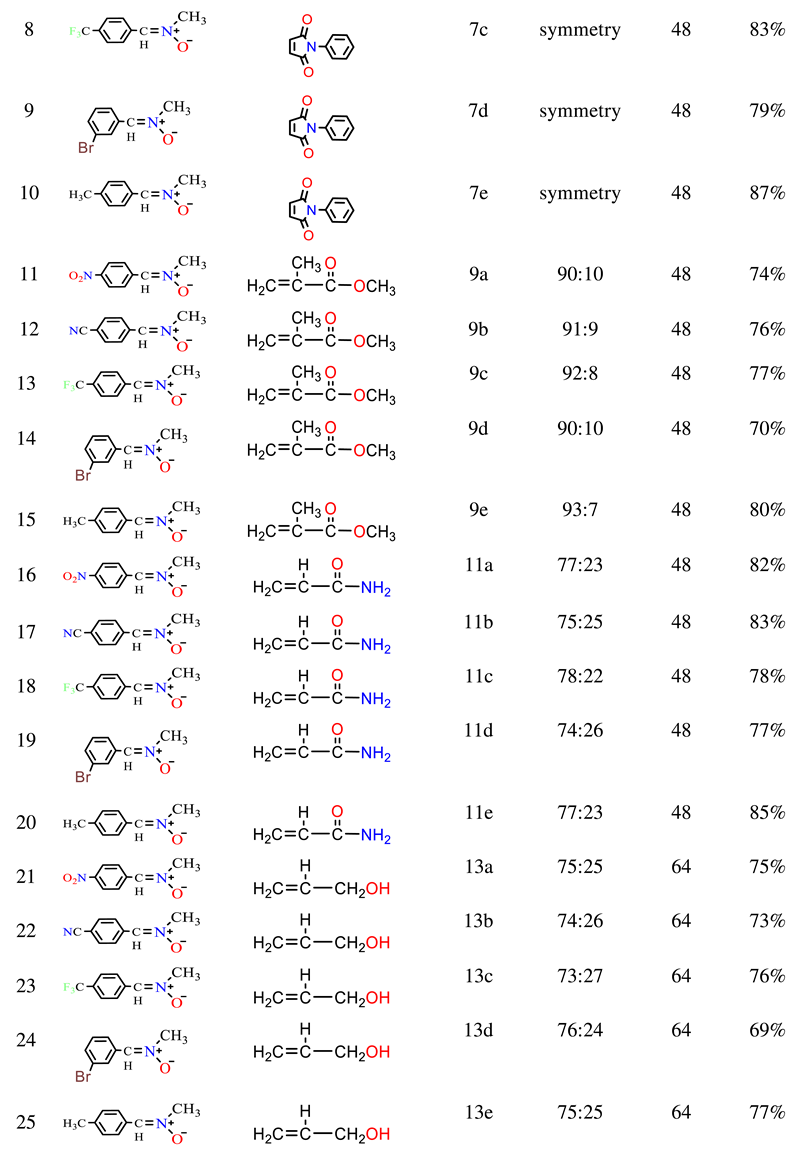
The cycloadditions of nitrones 3a–e with dimethyl maleate/ N-phenyl maleimide /methyl methacrylate /acrylamide / allyl alcohol / acrylonitrile 4, 6, 8, 10, 12, and 14 were carried out by refluxing equimolar solutions in benzene for 48–72 hours. Completion of the reaction (monitored by TLC) led to residues recovered by solvent removal under vacuum, followed by column chromatography over silica gel. This process yielded isoxazolidines 5a–e, 7a–e, 9a-e, 11a-e, 13a-e, and 15a-e, which underwent spectroscopic characterization (1H NMR, 13C NMR, and IR). A summary of the results is provided in Scheme 2 and Table 1.
2.1.1. A single product (5a-e) was obtained from the reaction of nitrone (3a-e) with symmetrical dimethyl maleate (4). The resulting cycloadduct (scheme 2 & table 1) was identified based on spectroscopic comparisons. In the 1H NMR spectrum of compound 5a, a singlet was observed for three protons each at δ 2.71, 3.68, and δ 3.80 ppm, attributed to -NCH3 and 2OCH3. The C4-H resonance appeared as a triplet at δ 3.71 (J = 9.1 and 9.7 Hz) ppm, the C3-H resonance as a doublet at δ 4.11 (J = 9.24 Hz), and the C5-H resonance as a doublet at δ 4.93 (J = 9.0 Hz) ppm, forming doublets at 7.63 and 8.23 (J = 9.16 and 8.88 Hz) ppm corresponding to four protons in the aromatic compounds. The 13C NMR spectrum exhibited coupled resonances for the newly formed isoxazolidine rings C4, C3, and C5 at 60.14, 74.3, and 76.9 ppm, respectively. Signals at 43.2, 52.5, 52.6, 168.9, and 169.6 ppm corresponded to NCH3, 2OCH3, and 2C=O carbons, while aromatic carbons were evident at 123.8 ppm, 128.7 ppm, 143.9 ppm, and 148.5 ppm.
2.1.2. The reaction between nitrone 3a-e and N-phenylmaleimide 6 yielded a single product. Alongside resonances in the aromatic region, the 1H NMR spectrum of 7b displayed a singlet at δ 2.70 attributed to NCH3, a triplet at δ 3.90 (t, J = 8.08 and 8.08 Hz) ppm, a doublet at δ 4.02 (J = 8.64 Hz) ppm, and a doublet at δ 5.06 (d, J = 7.48 Hz) ppm for C4-H, C3-H, and C5-H, respectively. The 13C NMR spectrum revealed methine carbons of the isoxazolidine ring at δ 54.4 (C4), δ 74.7 (C3), and δ 76.3 (C5), in addition to -NCH3 at δ 42.6 and carbonyl carbon resonances at δ 171.8 and δ 174.2 ppm.
2.1.3. Similarly, the reaction between nitrone 3a-e and methyl methacrylate 8 yielded the predominant cycloadduct 9a-e. Regiochemistry followed the typical pattern, favoring the 3,5-isomer over the 3,4-derivative [31]. The cycloaddition occurred between nitrone and methyl methacrylate, with the N-oxygen atom attacking the germinal carbon of the carbonyl group. Extensive spectroscopic research supported the structure determination of 9a. The 1H NMR spectrum displayed a singlet for CH3, NCH3, and OCH3 protons at δ 1.44, 2.53, and δ 3.70 ppm, respectively. The C4-H resonance appeared as separate doublets at δ 2.23, 2.27 (J = 9.28, 8.24 Hz, 1H, H4b), and as a doublet at δ 3.15, 3.18 (J = 6.88, 6.60 Hz, 1H, H4a), while C3-H appeared as a triplet at δ 4.04 (J = 7.64 and 6.44 Hz). The 13C NMR spectrum reflected different isoxazolidine carbons: C4 at δ 48.99 ppm, indicating no attachment to a heteroatom, and C3, C5 at δ 71.15 and δ 82.13, respectively. -CH3, -NCH3, -OCH3, and C=O resonances appeared at δ 24.99, δ 44.03, δ 52.88, and δ 174.2, respectively.
2.1.4 The investigation of the reactions between nitrone (3a-e) and acrylamide 10 was carried out under analogous conditions (Scheme 2 & Table 1). Reacting the nitrone with acrylamide 10 led to the formation of the major product cycloadducts 11a-e (77-85%) upon separation from the mixture. By using chromatography on silica gel, it was possible to separate this compound from the mixture of isomers [32, 33]. The regiochemistry of addition in 11a was assigned based on its 1H NMR spectrum. The chemical shift of the C5-H methylene group appeared as a doublet-doublet at δ 4.59, 4.61 (J = 4.16, 4.76 Hz). The downfield-shifted position in the 1H NMR indicated the attachment of the methylene carbon to oxygen in compounds 11a. Assignments of 13C NMR chemical shifts and observed proton couplings provided further support for these conclusions. Notably, the vicinal relation of C4-H to both C3-H and C5-H was evident, corroborated by the 13C chemical shifts of various isoxazolidine moiety carbons. For example, the 13C NMR resonance of methylene carbon (C4) in 11a at δ 43.3 indicated its detachment from oxygen. The resonances of C3, C5 and C=O are shown at δ 72.21, 75.79 and δ 176.08 ppm, respectively.
2.1.5. Similarly, the reaction of nitrone 3a-e with allyl alcohol 12 was studied. Following separation from the isomeric mixture via chromatography on silica gel, only the major products 13a-e were obtained in the case of nitrone addition to allyl-alcohol (Scheme 2 & Table 1). The 1H NMR spectrum of 13e identified multiple resonances, including –CH2–O– and C5-H protons at δ 3.65-3.77 (3H, H5 & CH2OH), a C3–H resonance as a double doublet at δ 2.67, 2.69 (J=8.28, 8.28 Hz 1H, H3), an NCH3 resonance as a singlet at δ 2.48 ppm, and C4–H as multiple resonances at δ 2.10 - 2.22 (2H, H4). The 13C NMR spectrum featured three methine (CH–) resonances: δ 42.71 (C4), δ 73.63 (C3), and δ 77.21 (C5). Another resonance corresponding to an oxygen-linked carbon (CH2) appeared at δ 65.15 (-CH2-OH).
Steric factors and secondary interactions might contribute to determining the stereoisomeric outcome. In the case of allyl alcohol addition, a proposed [34, 35] secondary interaction involving allylic oxygen could lead to the formation of an endo adduct as the major product, with an exo isomer constituting the minor product.
2.1.6. Similarly, the reactions of nitrone 3a-e with acrylonitrile 14 were analyzed (Scheme 2 and Table 1). The compounds resulting from these reactions could be isolated from the isomeric mixture using silica gel chromatography. The reactions of nitrones with 14 produced major products 15a-e (65-72%) and minor (traces) cycloadducts, with 15a being a case in point. The 1H NMR spectrum of 15a distinctly indicated coupling of the methylene hydrogens (C4-H) with both C3-H and C5-H, manifesting as a doublet-doublet at 3.83, 3.87 (J= 10.48, 8.92). C3-H and C5-H resonances were observed at 4.20 and 4.44 respectively. The 13C chemical shifts of several isoxazolidine moiety carbons supported these findings, with the 13C NMR resonance of methylene carbon (C4) in 15a appearing at δ 42.96, indicating its detachment from oxygen. Resonances of C3, C5, and CN were evident at δ 69.02, 72.82, and 117.35 respectively.
2.2. Computational Investigations
2.2.1. ELF Analysis of Electronic Structures
ELF [36] analysis of the electronic structures of nitrones 3a, 3b, 3c, 3d, and 3e in conjunction with the olefins 4, 6, 8, 10, 12, and 14 was performed (Fig. 1) to establish connections between electronic structures and reactivity [37]. In the ELF analysis of nitrone 3a, a disynaptic basin, V(N2, C3), integrating 4.00 e, corresponding to the N2-C3 double bond, and another disynaptic basin, V(N2,O1), integrating 1.36 e, linked to the depopulated N2–O1 single bond, were identified. Two monosynaptic basins, V(O1) and V′(O1), integrating 5.83e in total, were associated with the non-bonding electron density of oxygen O1. The ELF of the nitrones 3b, 3c, 3d, and 3e is very similar to that of nitrone 3a. The absence of pseudoradical and carbenoid centers in nitrones 3a, 3b, 3c, 3d, and 3e indicates the participation of zwitterionic TACs in zw-type 32CA reactions [38].
ELF topological analyses of ethylenes 4, 6, 8, 10, 12, and 14 revealed the presence of two disynaptic basins, V(C4, C5) and V′(C4, C5), integrating a total charge of 3.46e, 3.40e, 3.52e, 3.48e, 3.54e, and 3.36e, respectively, associated with the C4-C5 double bond. Incorporating the electronic drawing group CN in the olefin resulted in decreased electron density in the C4–C5 bonding region.
Figure 1.
B3LYP/6–31G(d) ELF localization domains represented at an isosurface value of ELF = 0.82, ELF basin attractor positions together with the most relevant valence basin populations, and ELF-based Lewis-like structures together with natural atomic charges, of nitrones 3a, 3b, 3c, 3d, 3e, and ethylene 4, 6, 8, 10, and 12. Valence basin populations and natural atomic charges are given in average number of electrons, e. Negative charges are colored in red, positive charges in blue, and negligible charges in green.
Figure 1.
B3LYP/6–31G(d) ELF localization domains represented at an isosurface value of ELF = 0.82, ELF basin attractor positions together with the most relevant valence basin populations, and ELF-based Lewis-like structures together with natural atomic charges, of nitrones 3a, 3b, 3c, 3d, 3e, and ethylene 4, 6, 8, 10, and 12. Valence basin populations and natural atomic charges are given in average number of electrons, e. Negative charges are colored in red, positive charges in blue, and negligible charges in green.
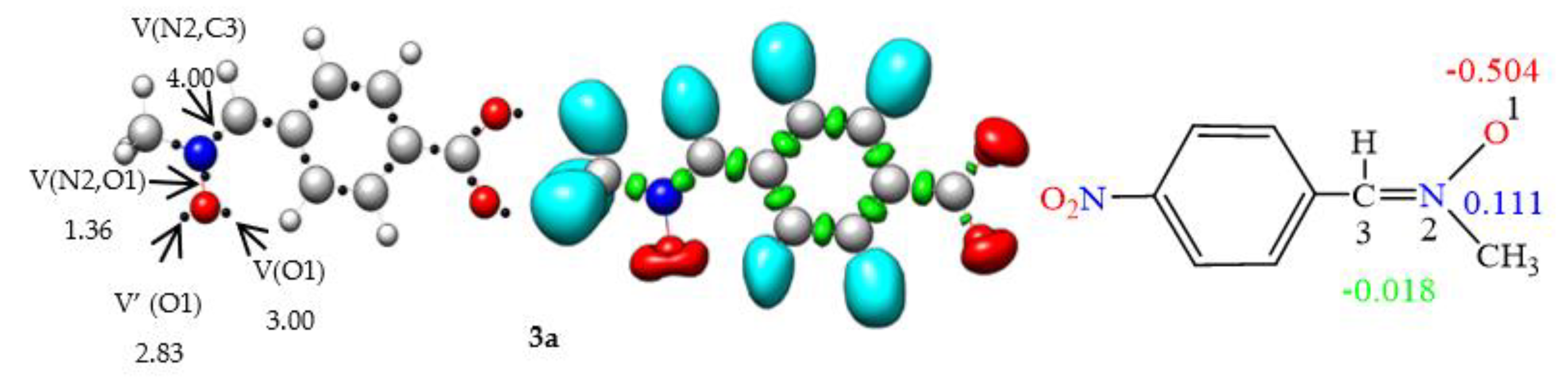
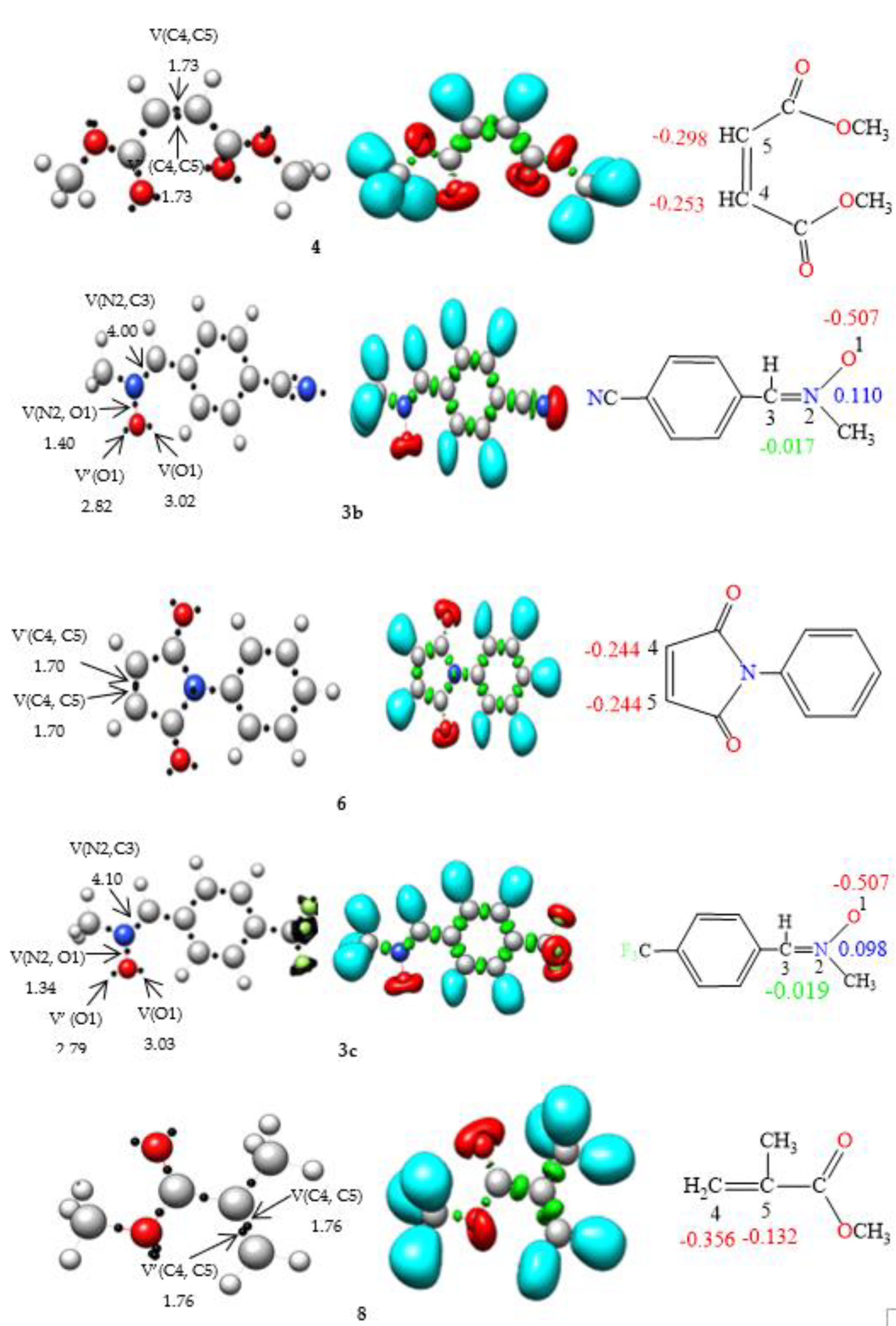
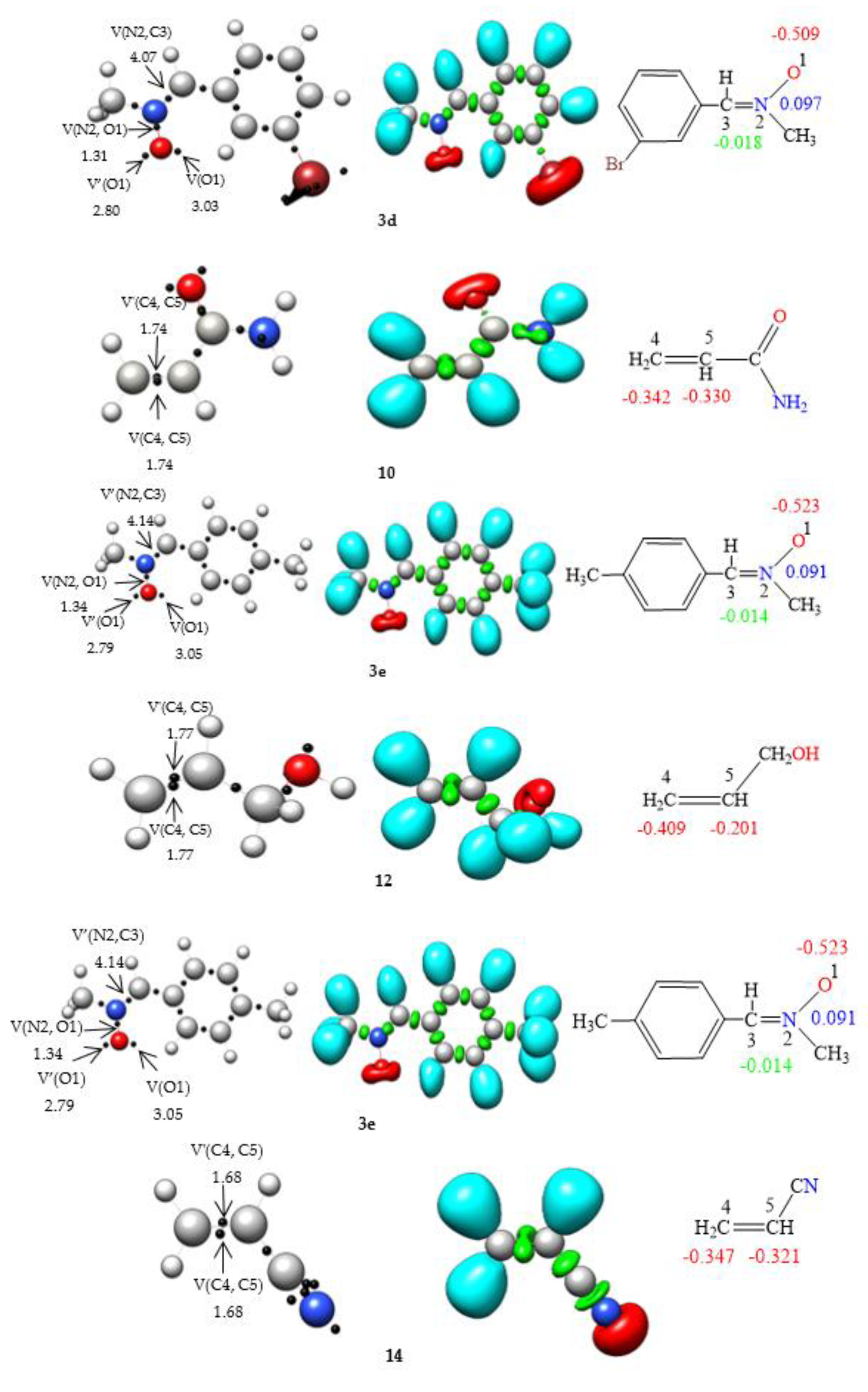
The natural atomic charges of these compounds, derived through natural population analysis [39, 40] (NPA), were analysed (see Fig. 1). In nitrones 3a, 3b, 3c, 3d and 3e, while carbon C3 has a negligible charge, oxygen O1 has a negative charge between -0.504e and -0.523. N2 atoms are positively charged between 0.111e and 0.091e. On the other hand, in ethylenes 4, 6, 8, 10, 12 and 14, carbon C4 has a negative charge of -0.253e, -0.244e, -0.356, -0.342, -0.409e and -0.347e, respectively, while C5 has a negative charge of -0.298e, -0.244e, -0.132e, -0.330, -0.201e and -0321, respectively.
2.2.2. CDFT Reactivity Indices Analysis
The CDFT reactivity indices, including electronic chemical potential (μ), chemical hardness (η), electrophilicity (ω), and nucleophilicity (N), were calculated at the B3LYP/6-31G(d) level to define electrophilicity and nucleophilicity scales [41, 42]. These indices for nitrones 3a, 3b, 3c, 3d, 3e, and ethylenes 4, 6, 8, 10, 12, and 14 are presented in Table 2.
The electronic chemical potential μ [43] of nitrones 3a, 3b, 3c, 3d, and 3e (μ = -4.46 eV, -4.11 eV, -3.84 eV, -3.67 eV, and -3.27 eV) is higher compared to ethylene 4, 6, 8, 10, 12 and 14 (μ = -4.49 eV, -4.56 eV, -4.47 eV, -4.80 eV, -0.464 eV, and -4.70 eV). This disparity indicates an electronic flux from nitrones 3a, 3b, 3c, 3d, and 3e toward ethylene 4, 6, 8, 10, 12, and 14.
Comparing chemical hardness (η), nitrones 3a, 3b, 3c, 3d, and 3e exhibit lower values (η = 5.40 eV, 3.92 eV, 4.15 eV, 4.21 eV, and 4.19 eV) than ethylene 4, 6, 8, 10, 12, and 14 (η = 5.44 eV, 5.49 eV, 6.24 eV, 5.92 eV, 6.55 eV, and 6.34). Consequently, the TAC nitrones 3a, 3b, 3c, 3d, and 3e demonstrate reduced forcefulness and greater susceptibility to electron density deformation when contrasted with ethylene 4, 6, 8, 10, 12, and 14.
The electrophilicity ω [44, 45] indices of nitrones 3a, 3b, 3c, 3d, and 3e are 1.84, 2.15, 1.78, 1.60, and 1.28 eV, classifying them as strong electrophiles (1.51 eV < ω < 8.25 eV). Furthermore, the nucleophilicity N [46] indices for nitrones 3a are 2.32, indicative of a moderate nucleophile (2.00 eV < N < 3.00 eV). For nitrones 3b, 3c, 3d, and 3e, the corresponding values are 3.42, 3.57, 3.72, and 4.12, placing them in the category of strong nucleophiles (3.03 eV < N < 4.28 eV). This suggests their active participation in polar 32CA reactions involving potent electrophilic ethylene. Notably, electron-withdrawing substituents NO2, CN, and CF3 in 3a, 3b, and 3c lead to decreased nucleophilicity, heightened electrophilicity, and reduced electronic chemical potential. Conversely, electron-donating methyl (-CH3) groups in 3e considerably enhance nucleophilicity, diminish electrophilicity, and elevate electronic chemical potential, illustrating the substantial impact of substituents on the electronic behavior of nitrones.
For ethylene 4, 6, 8, and 10, electrophilicity ω indices are 1.85, 2.98, 1.39, and 1.22 eV, classifying them as strong electrophiles, while nucleophilicity N indices are 2.27, 2.97, 2.20, and 2.73 eV, categorizing them as moderate nucleophiles (2.03 eV < N < 3.00 eV) within standard electrophilicity [45] and nucleophilicity[47, 48] scales. Ethylene 12 displays an electrophilicity ω of 1.01, designating it as a moderate electrophile, and corresponding nucleophilicity indices are 2.57 eV, indicating moderate nucleophilicity. Electrophilicity ω for ethylene 14 is 1.74, labeling it as a strong electrophile, and the associated nucleophilicity index is 1.62 eV, classifying it as a marginal nucleophile. As the series of electrophilic ethylene 4, 6, 8, 10, 12, and 14 exhibits increasing electrophilicity in the order: allyl alcohol 12 < acrylamide 10 < methyl methacrylate 8 < acrylonitrile 14 < dimethyl maleate 4 < N-phenyl maleimide 6, it can be inferred that the reaction rate will follow the same sequence of escalation.
2.3. Molecular docking
In this study, isoxazolidine derivatives were subjected to molecular docking analysis to understand the various modes of binding to the receptor. The chosen ligands (30 different isoxazolidine derivatives) were docked to wild-type EGFR (PDB: 4ZAU). Figure 2 illustrates the interactions between the most effective isoxazolidine derivatives and the EGFR receptor. In the case of compounds 7a, 11a and 13b, the molecular docking study indicates the existence of strong hydrogen bonds between the electron-withdrawing groups, such as the NO2 group, and the amino acid residues GLU 762, LYS 745 (for 7a) and MET 793 (for 11a). In addition, the amide group of 11a has hydrogen bonds with the amino acid residues LEU 788, ALA 743 and the carbonyl group of 7a with CYS 797. Similarly, the electron-donating groups, such as the OH group, of 13b form strong hydrogen bonds with PRO 794. Figure 2 shows the 2D and 3D interactions of the primary isoxazolidine derivatives 7a, 11a and 13b.
Analyzing the presented isoxazolidine derivatives, 11a and 11e demonstrate hydrogen bonding between the amide group and the LEU 788 and ALA 743 residues. Notably, these compounds exhibit the highest Glide scores (-6.573 and -6.523 kcal/mol, respectively) within the series. Furthermore, these results underscored the role of hydrophobic interactions that contribute to the binding affinity. Table 3 summarizes the lowest binding energies observed for the ligand-EGFR interaction and highlights hydrogen bonding occurrences.
3.4. Oral drug-likeness
Lipinski rule parameters: molecular mass, number of hydrogen bond acceptors (Acceptor HB), number of hydrogen bond donors (Donor HB), and octanol-water partition coefficient, as well as the topological polar surface area (TPSA) have been calculated for all the compounds and are detailed in Table 4. Notably, none of the molecules exhibit more than ten hydrogen bond acceptors or five hydrogen bond donors, and the molecular masses of all compounds remain below 500 Daltons. The compounds under investigation conform to the predefined drug-like properties in this study, highlighting their potential for oral activity. For further comparison with documented compounds [49], additional pharmacophore models can be employed.
3.5. Molecular dynamics
For the investigation of complex conformational changes responsible for antagonistic behavior, a 200 ns molecular dynamics study on the natural 4ZAU and compound 11a bound complex structure was conducted.
The stability of the complex system was assessed through the examination of the RMSD plot throughout the entire 200 ns simulation period. The RMSD plot pertaining to the compound 11a complex system, in relation to its initial reference structure, is depicted in Figure 3. The reference structure corresponds to the complex's initial configuration. The analysis of the plot (shown on the left Y-axis of Fig. 3) demonstrates that the protein exhibits fluctuations until 51 ns, after which it maintains relative stability, with variations within the acceptable range of 0–3 Å for the entire simulation duration [50]. To gain insights into the diverse allosteric signals provoked by the ligand during the simulation, it becomes essential to examine the ligand's relative mean square deviation (RMSD). The RMSD of the ligand's heavy atoms was computed by aligning the complex structure with the protein backbone of the reference frame. The plot (illustrated on the right Y-axis of Fig. 3) makes it evident that the ligand undergoes greater fluctuations and eventually reaches a stable conformation around 130 ns. This analysis underscores that the ligand assumes an entirely distinct orientation compared to its initial binding site.
3. Materials and Methods
3.1. General Information
All chemicals and solvents utilized were of reagent grade. Monitoring of reactions was performed using analytical thin-layer chromatography (TLC) on Silica Gel 60 F254 (Merck), and detection was carried out using UV light (254 nm) and a solution of 1% potassium permanganate in water containing 1% NaOH. IR spectra were recorded employing a Shimadzu 1800 FT-IR spectrophotometer on KBr pellets. For Nuclear Magnetic Resonance spectra, Bruker DPX-300FT, 400 MHz for 1H-NMR and 100 MHz for 13C-NMR spectrometers were used. The recording was conducted using deuterium solvents such as CDCl3 and DMSO-d6.
3.2. Synthesis
3.2.1. General Procedure for the Synthesis of Nitrone Derivatives.
Benzaldehyde derivative (1 mmol), N-methylhydroxylamine hydrochloride (1 mmol), and NaHCO3 (1 mmol) were dissolved in EtOH/H2O 1:1 (20 ml) at room temperature. The mixture was stirred until the reactant disappeared, as indicated by thin-layer chromatography (TLC) (1:1, petroleum ether: ethyl acetate). The solvent was mostly removed under reduced pressure, and the residue was extracted with dichloromethane (3 x 20 mL).
3.2.2. General Procedure for the Synthesis of Isoxazolidine Derivatives.
The appropriate nitrone (0.01 mol) was dissolved in benzene (10 mL) and the corresponding olefin (0.01 mol) was added. The mixture was heated at 70°C for 48-72 h using an autoclave. The synthesis of the product was confirmed by thin-layer chromatography (TLC) (1:1, petroleum ether: ethyl acetate). Concentration of the solution under vacuum yielded the crude product, which was then subjected to chromatography to isolate the pure isoxazolidine derivative as an isolated isomer.
3.3. Characterization
3.3.1. N-methyl-C-4-nitrophenyl-nitrone (3a)
Yield 93%, yellow solid, m.p. 202-205 °C. IR (KBr): ν 1165 (N-O), 1338, 1508 (NO2), 1597 (C=N)) cm-1; 1H-NMR (400 MHz, CDCl3) : δ 3.96 (s, 3H, -NCH3), 7.54 (s, 1H, -N=CH), 8.26, 8.38 ppm (dd, J=8.84 and 8.40 Hz, 4H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 55.12 (N-CH3), 123.93 (Car), 128.58 (Car), 132.93 (Car), 136.05 (CH=N), 147.57 (Car) ppm.
3.3.2. N-methyl-C-4-cyanophenyl-nitrone (3b)
Yield 96%, pale yellow solid, m.p. 183-185 °C. IR (KBr): ν 1165 (N-O), 1581 (C=N)), 2222 (C≡N) cm-1; 1H-NMR (400 MHz, CDCl3): δ 3.96 (s, 3H, -NCH3), 7.46 (s, 1H, -N=CH), 7.68, 8.30 ppm (dd, J=6.52 and 6.32 Hz, 4H, Ar-H).
3.3.3. N-methyl-C-4-trifluoromethylphenyl-nitrone (3c)
Yield 95%, white solid, m.p. 103-105 °C. IR (KBr): ν 1172 (N-O), 1415 (C=N)) cm-1; 1H-NMR (400 MHz, CDCl3): δ 3.97 (s, 3H, -NCH3), 7.46 (s, 1H, -N=CH), 7.68, 8.33 ppm (dd, J=6.28 and 6.4 Hz, 4H, Ar-H).
3.3.4. N-methyl-C-3-bromophenyl-nitrone (3d)
Yield 91%, white solid, m.p. 47-49 °C. IR (KBr): ν 1176 (N-O), 1589 (C=N)) cm-1; 1H-NMR (400 MHz, CDCl3): δ 3.88 (s, 3H, -NCH3), 7.35 (s, 1H, -N=CH), 7.25-8.5 ppm (m, 4H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 54.62 (-NCH3), 122.57 (Car), 126.90 (Car), 129.69 (Car), 131.10 (Car), 132.25 (Car), 133.23 (Car), 133.85 (CH=N) ppm.
3.3.5. N-methyl-C-4-methylphenyl-nitrone (3e)
Yield 98%, white solid, m.p. 115-117 °C. IR (KBr): ν 1165 (N-O), 1585 (C=N)) cm-1; 1H-NMR (400 MHz, CDCl3): δ 2.37 (s, 3H, -CH3Ph), 3.85 (s, 3H, -NCH3), 7.33 (s, 1H, -N=CH), 7.23, 8.11 ppm (dd, J=7.44 and 8.36 Hz, 4H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 21.69 (s, 3H, -CH3Ph), 54.21 (-NCH3), 127.92 (Car), 128.48 (Car), 129.20 (Car), 135.33 (CH=N), 140.91 ppm (Car).
3.3.6. Dimethyl 3-(4-nitrophenyl)-2-methylisoxazolidine-4,5-dicarboxylate (5a)
Yield: 87%, white solid, m.p. 140-141°C. IR (KBr): ν 1219 (N-O), 1350, 1523 (NO2), 1732 (C=O), 2854, 2877, 2997 (C-H), 3070 cm-1 (Ar-H); 1H-NMR (400 MHz, CDCl3): δ 2.71 (s, 3H, -NCH3), 3.68 (s, 3H, -OCH3), 3.71 (t, J = 9.1 and 9.7 Hz, 1H, -C4H), 3.80 (s, 3H, -OCH3), 4.11 (d, J = 9.24 Hz, 1H, -C3H), 4.93 (d, J =9.0 Hz, 1H, -C5H), 7.63-8.23 ppm (dd, J=9.16 and 8.88 Hz, 4H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 43.2 (N-CH3), 52.5, 52.6 (-2OCH3), 60.14 (C4), 74.3 (C3), 76.9 (C5), 123.8 (Car), 128.7 (Car), 143.9 (Car), 148.5 (Car), 168.9, 169.6 (C=O) ppm.
3.3.7. Dimethyl 3-(4-cyanomethylphenyl)-2-methylisoxazolidine-4,5-dicarboxylate (5b)
Yield: 85%, white solid, m.p. 115-117°C. IR (KBr): ν 1199 (N-O), 1739 (C=O), 2225 (C≡N), 2951 (C-H), 2997 cm-1 (Ar-H); 1H-NMR (400 MHz, CDCl3): δ 2.73 (s, 3H, -NCH3), 3.67 (s, 3H, -OCH3), 3.69 (t, J = 7.86 and 8.68 Hz, 1H, -C4H), 3.79 (s, 3H, -OCH3), 4.05 (d, J = 8.64 Hz, 1H, -C3H), 4.92 (d, J =9.44 Hz, 1H, -C5H), 7.55-7.68 ppm (dd, J=8.84 and 8.64 Hz, 4H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 43.2 (N-CH3), 52.5, 52.6 (-2OCH3), 59.5 (C4), 74.7 (C3), 76.9 (C5), 112.8 (CN-Car), 118.5 (Car), 128.7 (Car), 133.0 (Car), 142.2 (Car), 168.8, 169.5 (2C=O) ppm.
3.3.8. Dimethyl 3-(4-trifluoromethylphenyl)-2-methylisoxazolidine-4,5-dicarboxylate (5c)
Yield:83%, white solid, m.p. 65-67°C. IR (KBr): ν 1207 (N-O), 1732 (C=O), 2858, 2897, 2958 (C-H), 3005cm-1 (Ar-H); 1H-NMR (400 MHz, CDCl3): δ 2.72 (s, 3H, -NCH3), 3.67 (s, 3H, -OCH3), 3.72 (t, J = 9.24 and 9.12 Hz, 1H, -C4H), 3.79 (s, 3H, -OCH3), 4.03 (d, J = 9.24 Hz, 1H, -C3H), 4.93 (d, J =9.0 Hz, 1H, -C5H), 7.54-7.63 ppm (dd, J=8.12 and 8.12 Hz, 4H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 43.1 (-NCH3), 52.5, 52.6 (2OCH3), 59.8 (C4), 74.7 (C3), 76.9 (C5), 122.3 (-CF3Ph), 125.9 (Car), 128.7 (Car), 131.2 (Car), 140.5 (Car), 168.8, 169.7 (C=O) ppm.
3.3.9. Dimethyl 3-(3-bromomethylphenyl)-2-methylisoxazolidine-4,5-dicarboxylate (5d)
Yield:80%, colorless oil. IR (KBr): ν 1203 (N-O), 1735 (C=O), 2846, 2954, 2989, (C-H), 3050 cm-1 (Ar-H); 1H-NMR (400 MHz, DMSO): δ 2.53 (s, 3H, -NCH3), 3.57 (s, 3H, -OCH3), 3.59 (t, J = 9.56 and 9.00 Hz, 1H, -C4H), 3.66 (s, 3H, -OCH3), 3.92 (d, J = 9.00 Hz, 1H, -C3H), 4.99 (d, J =7.52 Hz, 1H, -C5H), 7.28-7.70 ppm (m, 4H, Ar-H); 13C-NMR (400 MHz, DMSO): δ 42.7 (-NCH3), 52.4, 52.6 (2OCH3), 59.1 (C4), 74.7 (C3), 77.2 (C5), 123.3 (Car), 127.9 (Car), 130.4 (Car), 131.1 (Car), 133.1 (Car), 139.8 (Car), 168.9, 170.7 (2C=O) ppm.
3.3.10. Dimethyl 3-(4-methylmethylphenyl)-2-methylisoxazolidine-4,5-dicarboxylate (5e)
Yield:91%, pale yellow solid, m.p. 59-62°C. IR (KBr): ν 1226 (N-O), 1728 (C=O), 2854, 2877,2958 (C-H), 2993 cm-1 (Ar-H); 1H-NMR (400 MHz, CDCl3): δ 2.32 (s, 3H, -CH3), 2.59 (s, 3H, -NCH3), 3.64 (s, 3H, -OCH3), 3.74 (t, J = 10.16 and 9.36 Hz, 1H, -C4H), 3.78 (s, 3H, -OCH3), 3.89 (d, J = 9.2 Hz, 1H, -C3H), 4.93 (d, J =9.32 Hz, 1H, -C5H), 7.17-7.27 ppm (dd, J=7.52 and 8.44 Hz, 4H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 21.1 (-CH3Ph), 42.9 (-NCH3), 52.3, 52.4 (2OCH3), 59.8 (C4), 75.7 (C3), 76.8 (C5), 127.8 (Car), 129.1 (Car), 133.0 (Car), 138.6 (Car), 169.1, 170.2 (2C=O) ppm.
3.3.11. 3-(4-nitrophenyl)-2-methyl-5-phenyl-3a,6a-dihydro-3H-pyrrolo[3,4-d] [1,2] oxazole-4,6-dione (7a)
Yield: 85%, yellow solid, m.p. 161-163°C. IR (KBr): ν 1207 (N-O), 1346, 1519 (NO2), 1716 (C=O), 2854, 2970, 2997 (C-H), 3074 cm-1 (Ar-H); 1H-NMR (400 MHz, CDCl3): δ 2.72 (s, 3H, -NCH3), 3.94 (t, J = 8.28 and 8.56 Hz, 1H, -C4H), 4.06 (d, J = 7.20 Hz, 1H, -C3H), 5.09 (d, J =8.56 Hz, 1H, -C5H), 7.14-8.30 ppm (m, 9H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 42.6 (-NCH3), 54.4 (C4), 74.5 (C3), 76.3 (C5), 124.1 (Car), 125.9 (Car), 126.3 (Car), 128.7 (Car), 129.4 (Car), 130.9 (Car), 141.3 (Car), 148.1 (Car), 171.8, 174.2 (2C=O) ppm.
3.3.12. 3-(4-cyanophenyl)-2-methyl-5-phenyl-3a,6a-dihydro-3H-pyrrolo[3,4-d] [1,2] oxazole-4,6-dione (7b)
Yield: 82%, white solid, m.p. 147-150 °C. IR (KBr): ν 1203 (N-O), 1716 (-C=O), 2225 (C≡N), 2877, 2924, 2966 (C-H), 3066 cm-1 (Ar-H); 1H-NMR (400 MHz, CDCl3): δ 2.70 (s, 3H, -NCH3), 3.90 (t, J = 8.08 and 8.08 Hz, 1H, -C4H), 4.02 (d, J = 8.64 Hz, 1H, -C3H), 5.06 (d, J =7.48 Hz, 1H, -C5H), 7.20-7.72 ppm (m, 9H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 42.6 (-NCH3), 54.4 (C4), 74.7 (C3), 76.3 (C5), 112.74 (-CNPh), 118.32 (Car), 125.9 (Car), 126.3 (Car), 128.6 (Car), 129.3 (Car), 132.6 (Car), 134.2 (Car), 139.2 (Car), 171.8, 174.2 (2C=O) ppm.
3.3.13. 3-(4-trifluoromethylphenyl)-2-methyl-5-phenyl-3a,6a-dihydro-3H-pyrrolo[3,4-d] [1,2] oxazole-4,6-dione (7c)
Yield: 83%, white solid, m.p. 131-133°C. IR (KBr): ν 1199 (N-O), 1716 (C=O), 2854, 2966 (C-H), 3070 cm-1 (Ar-H); 1H-NMR (400 MHz, CDCl3): δ 2.69 (s, 3H, -NCH3), 3.89 (t, J = 8.4 and 8.4 Hz, 1H, -C4H), 3.99 (d, J = 9.0 Hz, 1H, -C3H), 5.06 (d, J =8.56 Hz, 1H, -C5H), 7.22-7.68 ppm (m, 9H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 43.0 (-NCH3), 54.2 (C4), 75.11 (C3), 76.3 (C5), 122.50 (-CF3Ph), 125.90 (Car), 126.4 (Car), 128.2 (Car), 129.0 (Car), 129.3 (Car), 131.19 (Car), 134.2 (Car), 137.89 (Car), 172.19, 174.69 (2C=O) ppm.
3.3.14. 3-(3-bromophenyl)-2-methyl-5-phenyl-3a,6a-dihydro-3H-pyrrolo[3,4-d] [1,2] oxazole-4,6-dione (7d)
Yield: 79%, orange solid, m.p. 122-124°C. IR (KBr): ν 1199 (N-O), 1716 (C=O), 2870, 2962, 2985 (C-H), 3066 cm-1 (Ar-H); 1H-NMR (400 MHz, DMSO): δ 2.61 (s, 3H, -NCH3), 3.97 (dd, J = 3.48 and 3.52 Hz, 1H, -C4H), 4.08 (d, J = 5.84 Hz, 1H, -C3H), 5.22 (d, J = 5.44 Hz, 1H, -C5H), 7.20-7.71 ppm (m, 9H, Ar-H); 13C-NMR (400 MHz, DMSO): δ 43.07 (-NCH3), 55.2 (C4), 74.19 (C3), 77.62 (C5), 122.26 (Car), 127.14 (Car), 128.9 (Car), 129.5 (Car), 131.2 (Car), 132.2 (Car), 135.15 (Car), 138.09 (Car), 172.88, 175.73 (2C=O) ppm.
3.3.15. 3-(4-methylphenyl)-2-methyl-5-phenyl-3a,6a-dihydro-3H-pyrrolo [3,4-d] [1,2] oxazole-4,6-dione (7e)
Yield: 87%, orange solid, m.p. 122-124°C. IR (KBr): ν 1184 (N-O), 1716 (C=O), 2854, 2920, 2958 (C-H), 3066 cm-1 (Ar-H); 1H-NMR (400 MHz, DMSO): δ 2.32 (s, 3H, -CH3Ph), 2.67 (s, 3H, -NCH3), 3.80 (dd, J = 7.36 and 7.96 Hz, 1H, -C4H), 3.88 (d, J = 8.52 Hz, 1H, -C3H), 4.98 (d, J = 8.52 Hz, 1H, -C5H), 7.16-7.52 ppm (m, 9H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 21.33 (-CH3Ph), 42.46 (-NCH3), 54.59 (C4), 75.55 (C3), 76.49 (C5), 126.10 (Car), 126.44 (Car), 127.61 (Car), 128.65 (Car), 129.24 (Car), 129.70 (Car), 131.4 (Car), 138.7 (Car), 172.53, 175.04 (2C=O) ppm.
3.3.16. Methyl 5-methyl-3-(4-nitrophenyl)-2-methyl-1,2-oxazolidine-5-carboxylate (9a)
Yield: 74%, yellow solid, m.p. 78-80 °C. IR (KBr): ν 1203 (N-O), 1342, 1516 (NO2), 1747 (C=O), 2881, 2954, 2985 (C-H), 3062 cm-1 (Ar-H); 1H-NMR (400 MHz, DMSO): δ 1.44 (s, 3H, -CH3), 2.53 (s, 3H, -NCH3), 2.23, 2.27 (dd, J= 9.28, 8.24 Hz, 1H, H4b), 3.15, 3.18 (dd, J= 6.88, 6.60 Hz, 1H, H4a), 3.70 (s, 3H, -OCH3), 4.04 (t, J = 7.64 and 6.44 Hz, 1H, H3), 7.68-8.20 ppm (dd, J= 9.00 and 9.24 Hz, 4H, Ar-H); 13C-NMR (400 MHz, DMSO): δ 24.99 (-CH3), 44.03 (-NCH3), 48.99 (C4), 52.88 (-OCH3), 71.15 (C3), 82.13 (C5), 124.19 (Car), 129.3 (Car), 147.4 (Car), 147.5 (Car), 174.2 (C=O) ppm.
3.3.17. Methyl 5-methyl-3-(4-cyanophenyl)-2-methyl-1,2-oxazolidine-5-carboxylate (9b)
Yield: 76%, white solid, m.p. 65-68 °C. IR (KBr): ν 1203 (N-O), 1728 (C=O), 2225 (C≡N), 2862, 2962, 2989 (C-H), 3051 cm-1 (Ar-H); 1H-NMR (400 MHz, DMSO): δ 1.45 (s, 3H, -CH3), 2.53 (s, 3H, -NCH3), 2.23, 2.27 (dd, J= 8.88, 8.64 Hz, 1H, H4b), 3.13, 3.16 (dd, J= 7.24, 7.4 Hz, 1H, H4a), 3.76 (s, 3H, -OCH3), 3.96 (t, J = 7.44 and 7.32 Hz, 1H, H3), 7.62-7.84 ppm (dd, J= 8.44 and 8.20 Hz, 4H, Ar-H); 13C-NMR (400 MHz, DMSO): δ 25.37 (-CH3), 44.66 (-NCH3), 49.01 (C4), 52.44 (-OCH3), 71.37 (C3), 81.97 (C5), 110.88 (-CNPh), 119.27 (Car), 128.89 (Car), 132.93 (Car), 145.36 (Car), 173.66 (C=O) ppm.
3.3.18. Methyl 5-methyl-3-(4-trifluoromethylphenyl)-2-methyl-1,2 oxazolidine -5-carboxylate (9c)
Yield: 77%, colorless oil. IR (KBr): ν 1165 (N-O), 1735 (C=O), 2854, 2873, 2958 (C-H), 2989 cm-1 (Ar-H); 1H-NMR (400 MHz, DMSO): δ 1.49 (s, 3H, -CH3), 2.48 (s, 3H, -NCH3), 2.26, 2.29 (dd, J= 8.84, 8.84 Hz, 1H, H4b), 3.12, 3.15 (dd, J= 7.28, 7.28 Hz, 1H, H4a), 3.75 (s, 3H, -OCH3), 3.96 (t, J = 7.44 and 7.32 Hz, 1H, H3), 7.65-7.82 ppm (m, 4H, Ar-H); 13C-NMR (400 MHz, DMSO): δ 23.84 (-CH3), 43.07 (-NCH3), 49.32 (C4), 52.75 (-OCH3), 72.60 (C3), 82.27 (C5), 120.63 (-CF3Ph), 123.48 (Car), 125.92 (Car), 129.46 (Car), 144.63 (Car), 175.04 (C=O) ppm.
3.3.19. Methyl 5-methyl-3-(3-bromophenyl)-2-methyl-1,2-oxazolidine-5-carboxylate (9d)
Yield: 70%, pale yellow oil. IR (KBr): ν 1203 (N-O), 1735 (C=O), 2846, 2870, 2954 (C-H), 2989 cm-1 (Ar-H); 1H-NMR (400 MHz, CDCl3): δ 1.51 (s, 3H, -CH3), 2.57 (s, 3H, -NCH3), 2.39, 2.43 (dd, J= 8.36, 8.40 Hz, 1H, H4b), 2.98, 3.01 (dd, J= 8.92, 9.00 Hz, 1H, H4a), 3.44 (t, J = 8.28 and 8.28 Hz, 1H, H3), 3.72 (s, 3H, -OCH3), 7.23-7.46 ppm (m, 4H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 23.84 (-CH3), 43.38 (-NCH3), 49.32 (C4), 53.06 (-OCH3), 72.96 (C3), 81.66 (C5), 122.70 (Car), 126.16 (Car), 126.8 (Car), 130.26 (Car), 131.11 (Car), 140.51 (Car), 174.69 (C=O) ppm.
3.3.20. Methyl 5-methyl-3-(4-methylphenyl)-2-methyl-1,2-oxazolidine-5-carboxylate (9e)
Yield: 80%, colorless oil. IR (KBr): ν 1203 (N-O), 1735 (C=O), 2846, 2866, 2954 (C-H), 2989 cm-1 (Ar-H); 1H-NMR (400 MHz, CDCl3): δ 1.56 (s, 3H, -CH3), 1.66 (s, 3H, -CH3Ph), 2.33 (s, 3H, -NCH3), 2.44, 2.49 (dd, J= 9.44, 11.72 Hz, 1H, H4b), 2.99, 3.04 (dd, J= 10.32, 10.24 Hz, 1H, H4a), 3.49 (t, J = 9.40 and 7.88 Hz, 1H, H3), 3.83 (s, 3H, -OCH3), 7.16-7.23 ppm (m, 4H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 21.02 (-CH3Ph), 23.84 (-CH3), 43.38 (-NCH3), 48.71 (C4), 53.06 (-OCH3), 73.88 (C3), 81.36 (C5), 127.97 (Car), 129.50 (Car), 134.83 (Car), 137.89 (Car), 175.50 (C=O) ppm.
3.3.21. 3-(4-nitrophenyl)-2-methylisoxazolidine-5-carboxamide (11a)
Yield: 82%, pale yellow solid m.p. 78-79 °C. IR (KBr): ν 1161 (N-O), 1346, 1519 (NO2), 1685 (C=O), 2854, 2920 (C-H), 2989 (Ar-H), 3452 cm-1 (NH2); 1H-NMR (400 MHz, CDCl3): δ 2.69 (s, 3H, -NCH3), 2.51, 2.56 (dd, J= 7.32, 8.64 Hz, 1H, H4b), 3.13, 3.17 (dd, J= 9.52, 8.76 Hz, 1H, H4a), 3.65 (t, J = 8.36 and 7.48 Hz, 1H, H3), 4.59, 4.61 (dd, J = 4.16, 4.76 Hz, 1H, H5),), 6.19, 6.89 (ss, 2H, NH2), 7.52, 8.2 ppm (dd, J= 9 and 9 Hz, 4H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 42.2 (-NCH3), 43.3 (C4), 72.21 (C3), 75.79 (C5), 123.83 (Car), 128.77 (Car), 145.33 (Car), 147.83 (Car), 176.08 (C=O) ppm.
3.3.22. 3-(4-cyanophenyl)-2-methylisoxazolidine-5-carboxamide (11b)
Yield: 83%, white solid m.p. 90-91°C. IR (KBr): ν 1107 (N-O),1685 (C=O), 2225 (C≡N), 2785, 2854, 2912 (C-H), 2997 (Ar-H), 3471 cm-1 (NH2); 1H-NMR (400 MHz, CDCl3): δ 2.63 (s, 3H, -NCH3), 2.47, 2.51 (dd, J= 11.32, 8.4 Hz, 1H, H4b), 3.10, 3.13 (dd, J= 10.04, 13.28 Hz, 1H, H4a), 3.60 (t, J = 8.08 and 8.08 Hz, 1H, H3), 4.56, 4.58 (dd, J = 4.68, 4.68 Hz, 1H, H5),), 6.22, 6.86 (ss, 2H, NH2), 7.45, 7.63 (dd, J= 7.64 and 7.76 Hz, 4H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 42.19 (-NCH3) , 43.33 (C4), 72.41 (C3), 75.65 (C5), 112.39 (-CNPh), 118.50 (Car), 128.63 (Car), 132.82 (Car), 143.63 (Car), 175.92 (C=O) ppm.
3.3.23. 3-(4-trifluoromethylphenyl)-2-methylisoxazolidine-5-carboxamide (11c)
Yield: 78%, white solid m.p. 90-91 °C. IR (KBr): ν 1114 (N-O), 1670 (C=O), 2854, 2881, 2958 (C-H), 3008 (Ar-H), 3383 cm-1 (NH2); 1H-NMR (400 MHz, CDCl3): δ 2.55 (s, 3H, -NCH3), 2.42, 2.45 (dd, J= 10.24, 7.48 Hz, 1H, H4b), 3.01, 3.04 (dd, J= 7.72, 7.72 Hz, 1H, H4a), 3.49 (t, J = 7.48 and 6.00 Hz, 1H, H3), 4.46, 4.50 (dd, J = 5.04, 5.78 Hz, 1H, H5),), 6.21, 6.83 (ss, 2H, NH2), 7.41, 7.55 (dd, J= 7.44 and 8.04 Hz, 4H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 42.15 (-NCH3), 42.51 (C4), 71.51 (C3), 74.70 (C5), 121.64 (-CF3Ph), 124.81 (Car), 127.10 (Car), 129.66 (Car), 141.06 (Car), 175.44 (C=O) ppm.
3.3.24. 3-(3-bromophenyl)-2-methylisoxazolidine-5-carboxamide (11d)
Yield: 77%, pale yellow solid m.p. 65-68°C. IR (KBr): ν 1103 (N-O),1666 (C=O), 2885, 2920, 2962 (C-H), 3059 (Ar-H), 3448 cm-1 (NH2); 1H-NMR (400 MHz, CDCl3): δ 2.60 (s, 3H, -NCH3), 2.39, 2.43 (dd, J= 11.52, 12.72 Hz, 1H, H4b), 2.99, 3.02 (dd, J= 9.16, 8.08 Hz, 1H, H4a), 3.38 (t, J = 8.92 and 6.96 Hz, 1H, H3), 4.45, 4.48 (dd, J = 7.44, 7.00 Hz, 1H, H5), 6.20, 6.86 (ss, 2H, NH2), 7.15 - 7.36 (m, 4H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 43.07 (-NCH3), 43.45 (C4), 72.48 (C3), 75.65 (C5), 122.83 (Car), 126.47 (Car), 130.42 (Car), 130.74 (Car), 131.38 (Car), 140.17 (Car), 176.27 (C=O) ppm.
3.3.25. 3-(4-methylphenyl)-2-methylisoxazolidine-5-carboxamide (11e)
Yield: 85%, pale yellow solid mp. 92-93°C. IR (KBr): ν 1076 (N-O), 1643 (C=O), 2850, 2931, 2954 (C-H), 3032 (Ar-H), 3406 cm-1 (NH2); 1H-NMR (400 MHz, CDCl3): δ 2.33 (s, 3H, -CH3Ph), 2.61 (s, 3H, -NCH3), 2.50, 2.53 (dd, J= 7.52, 6.44 Hz, 1H, H4b), 3.03, 3.08 (dd, J= 9.8, 8.4 Hz, 1H, H4a), 3.47 (t, J = 8.72 and 7.00 Hz, 1H, H3), 4.51, 4.56 (dd, J = 6.2, 7.28 Hz, 1H, H5), 6.41, 7.01 (ss, 2H, NH2), 7.15, 7.25 (dd, J = 6.52, 7.28 4H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 21.02 (-CH3Ph), 42.94 (-NCH3), 43.51 (C4), 73.31 (C3), 76.10 (C5), 127.65 (Car), 129.46 (Car), 134.40 (Car), 137.95 (Car), 176.78 (C=O) ppm.
3.3.26. 3-(4-nitrophenyl)-2-methyl-1,2-oxazolidine-5-methanol (13a)
Yield: 75%, pale yellow solid m.p. 66-69 °C. IR (KBr): ν 1111 (N-O), 1342, 1512 (NO2), 2866, 2920 (C-H), 3109 (Ar-H), 3468 cm-1 (OH); 1H-NMR (400 MHz, CDCl3): δ 2.21 (m, 2H, H4), 2.62 (s, 3H, -NCH3), 2.86, 2.90 (dd, J=7.8, 7.82 Hz, 1H, H3), 3.67-3.85 (m, 3H, H5 & CH2OH), 4.42 (s, 1H, OH), 7.60, 8.19 (dd, J= 8.20 and 9.84 Hz, 4H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 41.02 (-NCH3), 43.22 (C4), 65.39 (-CH2OH), 72.47 (C3), 77.65 (C5), 123.60 (Car), 128.50 (Car), 146.70 (Car), 147.58 (Car) ppm.
3.3.27. 3-(4-cyanophenyl)-2-methyl-1,2-oxazolidine-5-methanol (13b)
Yield: 73%, brown oil. IR (KBr): ν 1111 (N-O), 2229 (C≡N), 2858, 2958 (C-H), 3062 (Ar-H), 3502 cm-1 (OH); 1H-NMR (400 MHz, CDCl3): δ 2.19 (m, 2H, H4), 2.57 (s, 3H, -NCH3), 2.81, 2.89 (dd, J=8.2, 8.0 Hz, 1H, H3), 3.63-3.80 (m, 3H, H5 & CH2OH), 4.33 (s, 1H, OH), 7.52, 7.65 (dd, J= 8.16 and 8.20 Hz, 4H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 41.02 (-NCH3), 43.16 (C4), 65.49 (-CH2OH), 72.96 (C3), 77.01 (C5), 112.10 (-CNPh), 118.66 (Car), 128.63 (Car), 132.82 (Car), 144.66 (Car) ppm.
3.3.28. 3-(4-trifluoromethylphenyl)-2-methyl-1,2-oxazolidine-5-methanol (13c)
Yield: 76%, brown viscous oil. IR (KBr): ν 1122 (N-O), 2873, 2939 (C-H), 3070 (Ar-H), 3414cm-1 (OH); 1H-NMR (400 MHz, CDCl3): δ 2.24 (m, 2H, H4), 2.55 (s, 3H, -NCH3), 2.79, 2.81 (dd, J=7.72, 8.04 Hz, H, H3), 3.59-3.77 (m, 3H, H5 & CH2OH), 4.33 (s, 1H, OH), 7.50, 7.59 (dd, J= 8.72 and 8.76 Hz, 4H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 41.22 (-NCH3), 43.00 (C4), 65.72 (-CH2OH), 72.96 (C3), 76.71 (C5), 122.68 (-CF3Ph), 125.72 (Car), 128.23 (Car), 130.41 (Car), 143.04 (Car) ppm.
3.3.29. 3-(3-bromophenyl)-2-methyl-1,2-oxazolidine-5-methanol (13d)
Yield: 69%, brown viscous oil. IR (KBr): ν 1122 (N-O), 2873, 2920 (C-H), 3062 (Ar-H), 3417cm-1 (OH); 1H-NMR (400 MHz, CDCl3): δ 2.10 2.18 (m, 2H, H4), 2.51 (s, 3H, -NCH3), 2.68, 2.71 (dd, J=8.16, 8.24 Hz, 1H, H3), 3.64-3.73 (m, 3H, H5 & CH2OH), 4.28 (s, 1H, OH), 7.19-7.51 (m, 4H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 40.83 (-NCH3), 42.90 (C4), 65.78 (-CH2OH), 73.01 (C3), 77.36 (C5), 122.78 (Car), 126.31 (Car), 130.40 (Car), 130.72 (Car), 131.28 (Car), 140.73 (Car) ppm.
3.3.30. 3-(4-methylphenyl)-2-methyl-1,2-oxazolidine-5-methanol (13e)
Yield: 77%, brown viscous oil. IR (KBr): ν 1114 (N-O), 2866, 2924 (C-H), 2951 (Ar-H), 3441 cm-1 (OH); 1H-NMR (400 MHz, CDCl3): δ 2.10 - 2.22 (m, 2H, H4), 2.26 (s, 3H, -CH3Ph), 2.48 (s, 3H, -NCH3), 2.67, 2.69 (dd, J=8.28, 8.28 Hz 1H, H3), 3.65-3.77 (m, 3H, H5 & CH2OH), 4.29 (s, 1H, OH), 7.08, 7.21 (dd, J= 7.36 and 8.08 Hz, 4H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 21.15 (-CH3Ph), 40.73 (-NCH3), 42.71 (C4), 65.15 (-CH2OH), 73.63 (C3), 77.21 (C5), 127.65 (Car), 129.46 (Car), 134.75 (Car), 137.95 (Car) ppm.
3.3.31. 3-(4-nitrophenyl)-2-methyl-1,2-oxazolidine-5-carbonitrile (15a)
Yield: 72%, pale yellow solid m.p. 88-90 °C. IR (KBr): ν 1107 (N-O), 1346, 1516 (NO2), 2241 (C≡N), 2854, 2885, 2962 (C-H), 3066 (Ar-H), cm-1; 1H-NMR (400 MHz, CDCl3): δ 2.66 (s, 3H, -NCH3), 3.83, 3.87 (dd, J=10.48, 8.92, 2H, H4), 4.20 (t, J= 6.12, 5.0 1H, H3 ), 4.44 (t, J= 7.24, 7.92 1H, H5), 7.66, 8.28 (dd, J= 8.32 and 8.28 Hz, 4H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 40.48 (-NCH3), 42.96 (C4), 69.02 (C3), 72.82 (C5), 117.35 (CN-C5), 124.07 (Car), 129.46 (Car), 141.74 (Car), 148.01 (Car) ppm.
3.3.32. 3-(4-cyanophenyl)-2-methyl-1,2-oxazolidine-5-carbonitrile (15b)
Yield: 70%, pale yellow oil. IR (KBr): ν 1107 (N-O), 2225 (C≡N), 2881, 2916 (C-H), 2962 (Ar-H), cm-1; 1H-NMR (400 MHz, CDCl3): δ 2.59 (s, 3H, -NCH3), 3.49, 3.52 (dd, J=10.48, 8.92, 2H, H4), 3.52 (t, J= 7.46, 7.92, 1H, H3 ), 435 (t, J= 9.2, 7.64, 1H, H5), 7.52-7.60 (m, 4H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 42.33 (-NCH3), 43.84 (C4), 68.56 (C3), 71.24 (C5), 112.15 (CN-C5),118.45 (-CNPh), 128.12 (Car), 133.08 (Car), 141.43 (Car), 143.29 (Car) ppm.
3.3.33. 3-(4-trifluoromethylphenyl)-2-methyl-1,2-oxazolidine-5-carbonitrile (15c)
Yield: 66%, white oil. IR (KBr): ν 1126 (N-O), 2245 (C≡N), 2877, 2966 (C-H), 2997 (Ar-H), cm-1; 1H-NMR (400 MHz, CDCl3): δ 2.64 (s, 3H, -NCH3), 3.66, 3.82 (dd, J=8.6, 3.6 2H, H4), 4.18 (t, J= 7.08, 7.4 1H, H3), 4.40 (t, J= 7.4, 7.08 1H, H5), 7.57, 7.70 (dd, J= 7.76 and 8.92 Hz, 4H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 40.49 (-NCH3), 42.54 (C4), 69.02 (C3), 73.27 (C5), 117.53 (CN-C5), 126.02 (Car), 129.19 (Car), 131.40 (Car), 138.50 (Car) ppm.
3.3.34. 3-(3-bromophenyl)-2-methyl-1,2-oxazolidine-5-carbonitrile (15d)
Yield: 65%, brown oil. IR (KBr): ν 1107 (N-O), 2245 (C≡N), 2877, 2962 (C-H), 2993 (Ar-H), cm-1; 1H-NMR (400 MHz, CDCl3): δ 2.54 (s, 3H, -NCH3), 3.26, 3.29 (dd, J=9.12, 6.68 2H, H4), 4.14 (t, J= 5.44, 4.80 1H, H3), 4.75 (t, J= 4.28, 4.28 1H, H5), 7.20-7.51 (m 4H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 42.73 (-NCH3), 43.95 (C4), 68.51 (C3), 71.34 (C5), 119.27 (CN-C5), 122.92 (Car), 126.56 (Car), 130.69 (Car), 131.66 (Car), 138.19 (Car), 139.87 (Car) ppm.
3.3.35. 3-(4-methylphenyl)-2-methyl-1,2-oxazolidine-5-carbonitrile (15e)
Yield: 72%, white solid m.p. 62-63 °C. IR (KBr): ν 1111 (N-O), 2245 (C≡N), 2873, 2920 (C-H), 2993 (Ar-H), cm-1; 1H-NMR (400 MHz, CDCl3): δ 2.36 (s, 3H, CH3Ph), 2.54 (s, 3H, -NCH3), 3.58, 3.61 (dd, J=7.72, 9.24 2H, H4), 4.09 (t, J= 7.16, 7.16 1H, H3), 4.26 (t, J= 7.88, 7.88 1H, H5), 7.09, 7.29 (dd, J= 6.2 and 7.24 Hz, 4H, Ar-H); 13C-NMR (400 MHz, CDCl3): δ 21.77 (-CH3Ph), 40.62 (-NCH3), 42.77 (C4), 68.97 (C3), 73.81 (C5), 118.12 (CN-C5), 128.35 (Car), 129.88 (Car), 130.85 (Car), 138.99 (Car) ppm.
3.3. Computational methods
The optimization of the stationary points associated with the 32CA reactions of nitrones 3a, 3b, 3c, 3d, and 3e with ethylene 4, 6, 8, 10, 12, and 14 was carried out using the Berny analytical gradient optimization technique [51, 52] along with the B3LYP functional and the 6-31G(d) basis set[53]. Calculations of the Conceptual Density Functional Theory (CDFT) indices[41, 42] were performed according to the equations in reference [41]. All computations were executed using the Gaussian 16 suite of software[54]. The Electron Localization Function[36] (ELF) analyses were conducted utilizing the Multiwfn software [55], employing suitable B3LYP/6-31G(d) monodeterminant wave functions and assuming a high-quality grid. The representation of ELF localization regions was accomplished using the UCSF Chimera program at an isovalue of 0.82 a.u. [56].
3.4. Molecular docking
3.4.1. Protein preparation
The 3D crystal coordinates of the protein structure, namely the AZD9291 complex with wild-type EGFR (PDB ID: 4ZAU) containing 330 amino acids and with a resolution of 2.80 Å, were downloaded from the protein data bank website (http://www.rcsb.org/pdb/home/home.do). The protein was initially subjected to the "Protein preparation wizard" module [57] of Schrödinger maestro. Corrections were made to heavy atoms, water molecules, cofactors, and metal ions in the protein structure. Missing hydrogen atoms, side chains, and protons were also added. Using the modules EPIK [58] and IMPREF[59], additional refinement and minimization of the protein were conducted. The receptor grid generation module was utilized to further optimize the protein, generating a grid surrounding its ligand-bound active site. This enabled identification of an active site through formation of a centroid (cubic) shape over the workspace occupied by said ligand [59].
3.4.2. Docking
Ligands of isoxazolidine were developed by altering the functional group and substitutions, which were then prepared using LigPrep. The Schrödinger software package was employed to generate high-resolution 3D structures of these ligands, including 2D to 3D conversion, optimization, energy state reduction and other modifications [60]. The protein structure (PDB: 4ZAU), containing the AZD9291 complex with wild type EGFR was downloaded from the PDB database. Protein preparation wizard improved this protein structure [61]. Receptor grid generation in Schrodinger software suite 2018-1 [60, 62] was used for receptor grid generation The additional precision approach (SP) was used for docking. Glide score (Gscore) in kcal/mol was calculated based on a formula comprising van der Waals interactions ('vdW'), Coulombic interactions ('Coul'), lipophilic interaction ('Lipo'), hydrogen bonding('Hbond') , metal coordination('Metal' ), burial penalty term( 'BuryP' )and rotatable bond count( 'RotB') as well as site terms('Site').
G Score = a*vdW + b*Coul + Lipo+ Hbond +Metal + BuryP + RotB + Site
'Site' represents polar interactions at the active site.
Computational investigations were conducted on a Linux system [63].
3.4.3. Oral drug-likeness
Lipinski rule parameters of the compounds (5a-e, 7a-e, 9a-e, 11a-e, 13a-e, 15a-e) were calculated using the SwissADME, which predicts physically and pharmaceutically relevant properties of organic molecules [64].
3.4.4. Molecular dynamics MD
To investigate the stability of coupled 11a/4ZAU complex, a 200 ns Molecular Dynamics simulation study was performed using Desmond module of Schrödinger 2018-1 in an explicit solvent system with OPLS3 force field. Crystallographic water (TIP3P) molecules were added to solvate the molecular system with periodic orthorhombic boundary conditions for 10-buffer region; overlapping water molecules were eliminated while Na+ ions acted as counterions for neutralizing it. The system contained 39,270 atoms and 11,507 water molecules. A collection of Nose-Hoover thermostats (NPT) [65, 66] was employed to maintain a constant temperature (300 K) and pressure (1 bar). To achieve this goal, we used a hybrid energy minimization algorithm consisting of 1000 steps of steepest descent followed by conjugate gradient algorithms.
4. Conclusions
Various isoxazolidine derivatives were synthesized in moderate to high yields through a two-step process. All synthesized compounds were subjected to purification and characterization using IR, 1H NMR, and 13C NMR spectroscopy. DFT calculations at the B3LYP/6-31G(d) computation level were employed to investigate the reaction between reactant α-aryl-N-methyl nitrones and various ethylenes. Chemoisomeric reaction paths related to α-aryl-N-methyl nitrones and various ethylenes were identified and examined. The ground-state structures of α-aryl-N-methyl nitrones were subjected to ELF topological analysis, classifying them as zwitter-ionic TACs, enabling their participation in zw-type 32CA reactions that necessitate suitable electrophilic-nucleophilic interactions. Due to the higher electronic chemical potential and strong nucleophilicity of α-aryl-N-methyl nitrones compared to various ethylenes, a global electronic flux from α-aryl-N-methyl nitrones to various ethylenes is anticipated. Furthermore, molecular docking and dynamics simulations were conducted for isoxazolidine and wild-type EGFR (PDB ID: 4ZAU). The results were compared to explore potential interactions between compound 11a and 4ZAU. Significant interactions with residues ALA 743, LEU 788, and MET 793 were observed in both molecular docking and molecular dynamics simulations. Molecular dynamics simulations corroborated the results of molecular docking, affirming the binding affinities of the ligands and the accuracy of their interactions.
Author Contributions
Conceptualization and methodology, S. A. M. S., H. A. B. and H. A. M. S.; Organic synthesis, S. A. M. S., H. A. B. and H. A. M. S.; Supervision and Project Administration, J. V. de J.-O.; Original draft preparation, S. A. M. S., J. V. de J.-O. and H. A. M. S.; Final version, all authors. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data available upon request to the authors.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Taylor, R.D.; MacCoss, M.; Lawson, A.D. Rings in drugs: Miniperspective. Journal of medicinal chemistry 2014, 57, 5845–5859. [Google Scholar] [CrossRef]
- Mumtaz, A.; et al. Synthesis, molecular modelling and biological evaluation of tetrasubstituted thiazoles towards cholinesterase enzymes and cytotoxicity studies. Bioorganic chemistry 2018, 78, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; et al. Synthesis and biological evaluation of new piperazine substituted 3, 5-diarylisoxazolines. Current Organic Synthesis 2019, 16, 294–302. [Google Scholar] [CrossRef]
- Chiacchio, M.; et al. Isoxazolidines as biologically active compounds. Current Organic Synthesis 2016, 13, 726–749. [Google Scholar] [CrossRef]
- Ghannay, S.; et al. Design, synthesis, molecular properties and in vitro antioxidant and antibacterial potential of novel enantiopure isoxazolidine derivatives. Arabian Journal of Chemistry 2020, 13, 2121–2131. [Google Scholar] [CrossRef]
- Gaonkar, S.L.; Nagaraj, V.U.; Nayak, S. A review on current synthetic strategies of oxazines. Mini-Reviews in Organic Chemistry 2019, 16, 43–58. [Google Scholar] [CrossRef]
- Berthet, M.; et al. Isoxazolidine: a privileged scaffold for organic and medicinal chemistry. Chemical Reviews 2016, 116, 15235–15283. [Google Scholar] [CrossRef]
- Periyasami, G.; Arumugam, N.; Aldalbahi, A. Inexpensive ionic liquid mediated green synthetic approach of multi-functionalized regioselective β-lactam fused isoxazolidine heterocyclic hybrids. Tetrahedron 2017, 73, 322–330. [Google Scholar] [CrossRef]
- Aouadi, K.; et al. New synthetic routes toward enantiopure (2S, 3R, 4R)-4-hydroxyisoleucine by 1, 3-dipolar cycloaddition of a chiral nitrone to C4 alkenes. Synthesis 2007, 3399–3405. [Google Scholar]
- Aouadi, K.; et al. 1, 3-Dipolar cycloaddition of a chiral nitrone to (E)-1, 4-dichloro-2-butene: a new efficient synthesis of (2S, 3S, 4R)-4-hydroxyisoleucine. Tetrahedron Letters 2012, 53, 2817–2821. [Google Scholar] [CrossRef]
- Seerden, J.-P.G.; Boeren, M.M.; Scheeren, H.W. 1, 3-Dipolar cycloaddition reactions of nitrones with alkyl vinyl ethers catalyzed by chiral oxazaborolidines. Tetrahedron 1997, 53, 11843–11852. [Google Scholar] [CrossRef]
- Frederickson, M. Optically active isoxazolidines via asymmetric cycloaddition reactions of nitrones with alkenes: applications in organic synthesis. Tetrahedron 1997, 53, 403–425. [Google Scholar] [CrossRef]
- Li, W.-T.; et al. Synthesis and biological evaluation of N-heterocyclic indolyl glyoxylamides as orally active anticancer agents. Journal of medicinal chemistry 2003, 46, 1706–1715. [Google Scholar] [CrossRef]
- Brahmi, J.; et al. Unprecedented stereoselective synthesis of 3-methylisoxazolidine-5-aryl-1, 2, 4-oxadiazoles via 1, 3-dipolar cycloaddition and study of their in vitro antioxidant activity. Synthetic Communications 2016, 46, 2037–2044. [Google Scholar] [CrossRef]
- Ghannay, S.; et al. Stereoselective synthesis of enantiopure N-substituted pyrrolidin-2, 5-dione derivatives by 1, 3-dipolar cycloaddition and assessment of their in vitro antioxidant and antibacterial activities. Bioorganic & Medicinal Chemistry Letters 2017, 27, 2302–2307. [Google Scholar]
- Kumar, K.R.R.; Mallesha, H.; Rangappa, K.S. Synthesis of novel isoxazolidine derivatives and their antifungal and antibacterial properties. Archiv der Pharmazie: An International Journal Pharmaceutical and Medicinal Chemistry 2003, 336, 159–164. [Google Scholar] [CrossRef]
- Loh, B.; et al. Inhibition of HIV-1 Replication by Isoxazolidine and Isoxazole Sulfonamides. Chemical Biology & Drug Design 2010, 75, 461–474. [Google Scholar]
- Kumar, R.S.; et al. 1, 3-Dipolar cycloaddition of C-aryl-N-phenylnitrones to (R)-1-(1-phenylethyl)-3-[(E)-arylmethylidene] tetrahydro-4 (1H)-pyridinones: Synthesis and antimycobacterial evaluation of enantiomerically pure spiroisoxazolidines. European journal of medicinal chemistry 2010, 45, 124–133. [Google Scholar] [CrossRef]
- Nguyen, T.B.; et al. 1, 3-Dipolar cycloadditions of nitrones to heterosubstituted alkenes. Part 1: oxa and aza-substituted alkenes. Organic Preparations and Procedures International 2010, 42, 387–431. [Google Scholar] [CrossRef]
- Ding, L.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef]
- Huang, S.-F.; et al. High frequency of epidermal growth factor receptor mutations with complex patterns in non–small cell lung cancers related to gefitinib responsiveness in Taiwan. Clinical Cancer Research 2004, 10, 8195–8203. [Google Scholar] [CrossRef]
- Kosaka, T.; et al. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer research 2004, 64, 8919–8923. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small-cell lung cancer to gefitinib. New England Journal of Medicine 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Maemondo, M.; et al. Gefitinib or chemotherapy for non–small-cell lung cancer with mutated EGFR. New England Journal of Medicine 2010, 362, 2380–2388. [Google Scholar] [CrossRef]
- Mitsudomi, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. The lancet oncology 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Mok, T.S.; et al. Gefitinib or carboplatin–paclitaxel in pulmonary adenocarcinoma. New England Journal of Medicine 2009, 361, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Shigematsu, H.; et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. Journal of the National Cancer Institute 2005, 97, 339–346. [Google Scholar] [CrossRef]
- Carey, K.D.; et al. Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer research 2006, 66, 8163–8171. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T., D. Bell, and R. Sordella, S. Gurubhagavatu la, RA Okimoto, BW Brannigan, PL Harris, SM Haserlat, JG Supko, FG Haluska, et al. N Engl J Med 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Raji, V.; et al. Gold nanoparticles against respiratory diseases: oncogenic and viral pathogens review. Therapeutic Delivery 2020, 11, 521. [Google Scholar]
- Harwood, L.; Vickers, R. The Chemistry of Heterocyclic Compounds: Synthetic Applications of 1, 3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; Wiley & Sons: New York, NY, USA, 2002; pp. 169–252. [Google Scholar]
- Singh, G.; et al. Formation and reductive ring opening reactions of indolyl-isoxazolidines: access to novel natural product analogs and precursors. Tetrahedron 2016, 72, 900–911. [Google Scholar] [CrossRef]
- Wang, L.; et al. Dipolar cycloadditions of HMF-based nitrones: Stepwise and multicomponent reactions, stereochemical outcome and structural scope. Green Chemistry 2020, 22, 7907–7912. [Google Scholar] [CrossRef]
- Ali, S.A.; Wazeer, M. The 13-dipolar cycloaddition reactions of 3456-tetrahydro-2h-azepine 1-oxide. Tetrahedron 1990, 46, 7207–7218. [Google Scholar]
- Bernardi, L.; et al. First 1, 3-dipolar cycloaddition of Z-α-phenyl-N-methylnitrone with allylic fluorides: a stereoselective route to enantiopure fluorine-containing isoxazolidines and amino polyols. Tetrahedron: Asymmetry 2004, 15, 245–250. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. The Journal of chemical physics 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Ríos-Gutiérrez, M.; Domingo, L.R. Unravelling the mysteries of the [3+ 2] cycloaddition reactions. European Journal of Organic Chemistry 2019, 2019, 267–282. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. The Journal of Chemical Physics 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chemical Reviews 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef]
- Geerlings, P.; FProft, D.; Langenaeker, W. Conceptual density functional theory. Chemical reviews 2003, 103, 1793–1874. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Yang, W. Book density functional theory of atoms and molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Parr, R.G.; Szentpály, L.V.; Liu, S. Electrophilicity index. Journal of the American Chemical Society 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Domingo, L.R.; et al. Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels–Alder reactions. Tetrahedron 2002, 58, 4417–4423. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Perez, P. Understanding the high reactivity of the azomethine ylides in [3+ 2] cycloaddition reactions. Letters in Organic Chemistry 2010, 7, 432–439. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P. The nucleophilicity N index in organic chemistry. Organic & biomolecular chemistry 2011, 9, 7168–7175. [Google Scholar]
- Ríos-Gutiérrez, M.; Sousa, A.S.; Domingo, L.R. Electrophilicity and nucleophilicity scales at different DFT computational levels. Journal of Physical Organic Chemistry 2023, e4503. [Google Scholar] [CrossRef]
- Sahayarayan, J.J.; et al. In-silico protein-ligand docking studies against the estrogen protein of breast cancer using pharmacophore based virtual screening approaches. Saudi Journal of Biological Sciences 2021, 28, 400–407. [Google Scholar] [CrossRef]
- Feng, X.-Y.; et al. Identification of novel PPARα/γ dual agonists by pharmacophore screening, docking analysis, ADMET prediction and molecular dynamics simulations. Computational Biology and Chemistry 2019, 78, 178–189. [Google Scholar] [CrossRef]
- Schlegel, H.B. Optimization of equilibrium geometries and transition structures. Journal of computational chemistry 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Schlegel, H. Advanced series in physical chemistry. Modern Electronic Structure Theory 1994, 2. [Google Scholar]
- Hehre, W.; Radom, L.; Schleyer, P.V.R.; Pople, J.A. Ab initio molecular orbital theory 1986, 228-251.
- Frisch, M.E.; et al. Gaussian 16, revision C. 01; Gaussian, Inc.: Wallingford CT, USA, 2016. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. Journal of computational chemistry 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Pettersen, E.F.; et al. UCSF Chimera—a visualization system for exploratory research and analysis. Journal of computational chemistry 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Madhavi Sastry, G.; et al. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. Journal of computer-aided molecular design 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, J.R.; et al. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. Journal of computer-aided molecular design 2010, 24, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Kumar, T.A.; et al. Design, 3D QSAR modeling and docking of TGF-β type I inhibitors to target cancer. Computational biology and chemistry 2018, 76, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, L. Schrödinger Release 2022-3: LigPrep; Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- Impact, S. , LLC, New York, NY, 2016; Prime, Schrödinger, LLC, New York, NY, 2020. Google Scholar There is no corresponding record for this reference.
- Release, S. 1: Glide, Schrödinger, LLC, New York, NY, 2021. Schrödinger Release, 2021. 3.
- Maruthanila, V.; et al. In silico molecular modelling of selected natural ligands and their binding features with estrogen receptor alpha. Current computer-aided drug design 2019, 15, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Duffy, E.M. Prediction of drug solubility from structure. Advanced drug delivery reviews 2002, 54, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–Hoover chains: The canonical ensemble via continuous dynamics. The Journal of chemical physics 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. The Journal of chemical physics 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
Scheme 1.
Synthesis of phenyl nitrone derivatives.
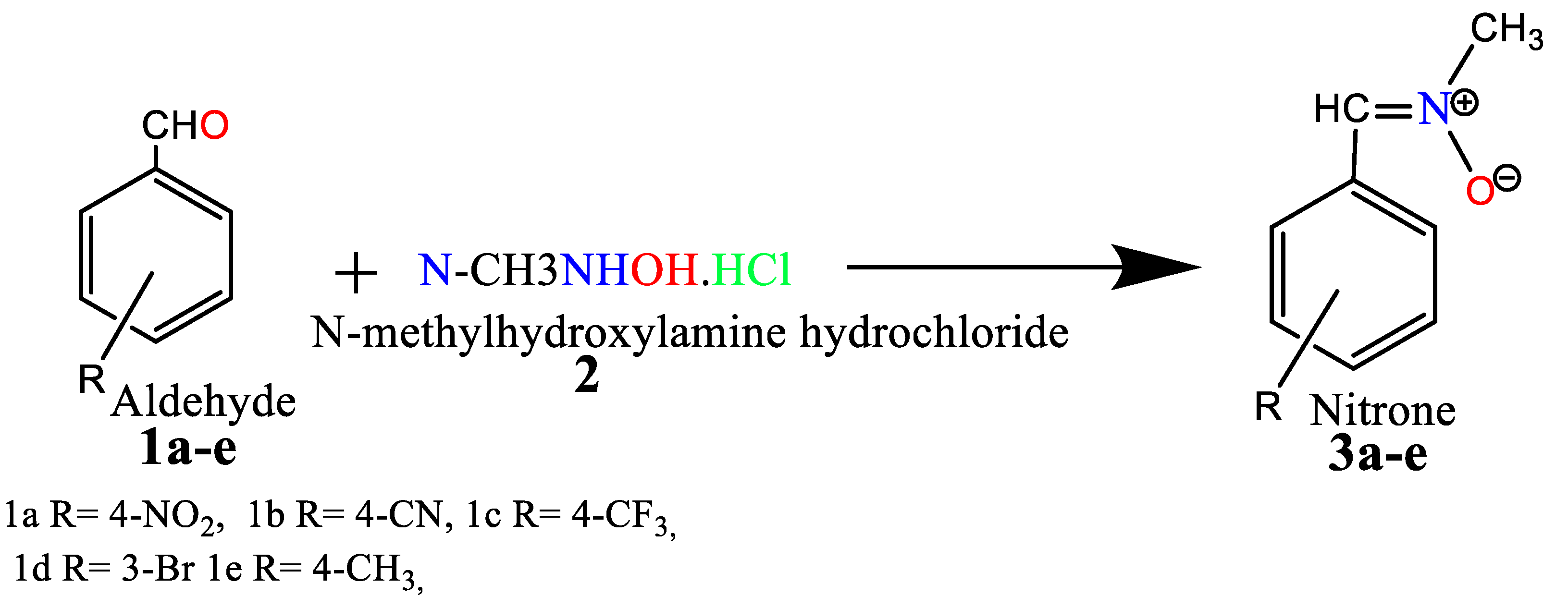
Scheme 2.
Synthesis of isoxazolidine derivatives from the 32CA reaction of nitrones and various ethylenes.
Scheme 2.
Synthesis of isoxazolidine derivatives from the 32CA reaction of nitrones and various ethylenes.
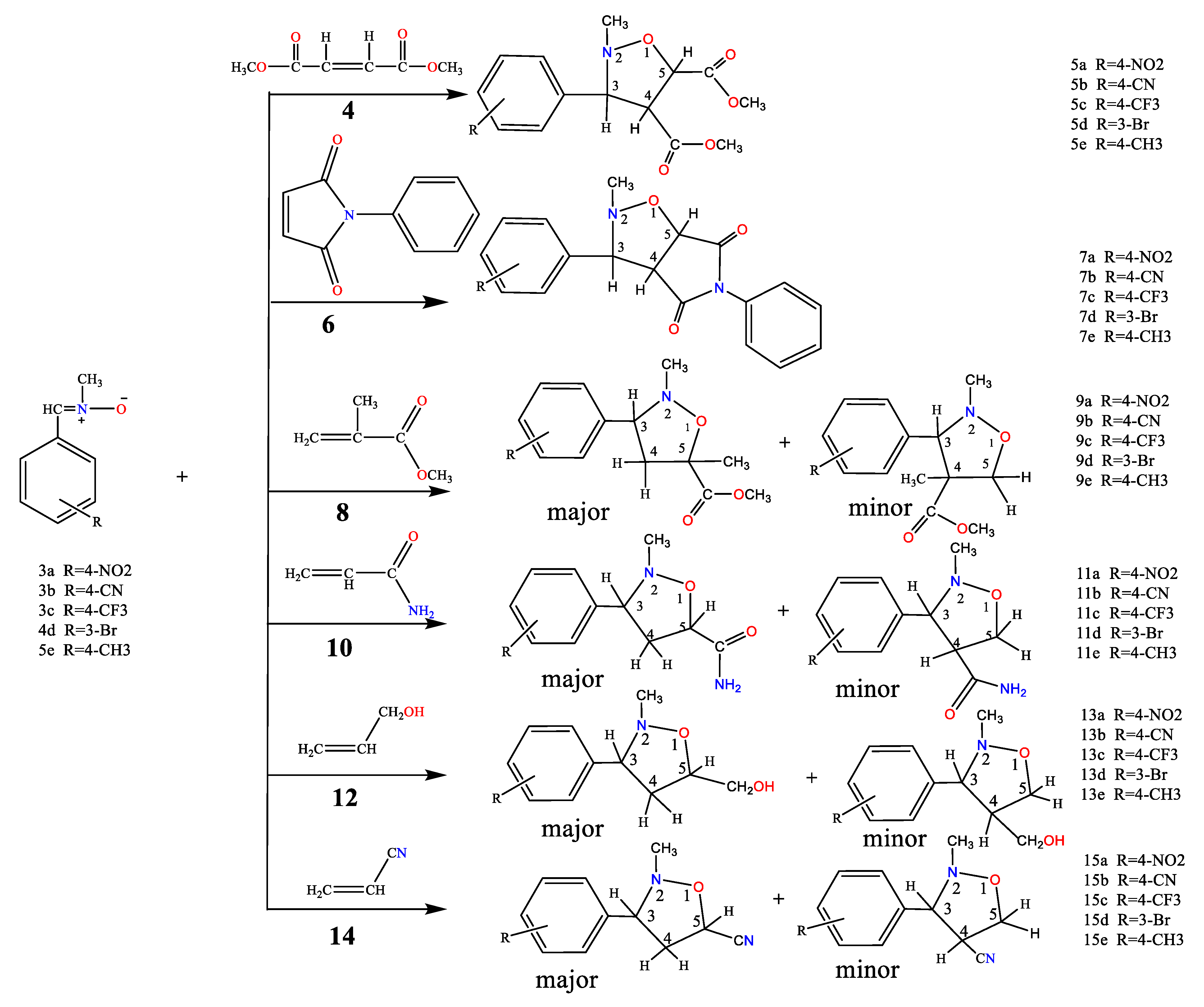
Figure 2.
2D and 3D interactions of the isoxazolidine derivatives, 7a, 11a and 13b.
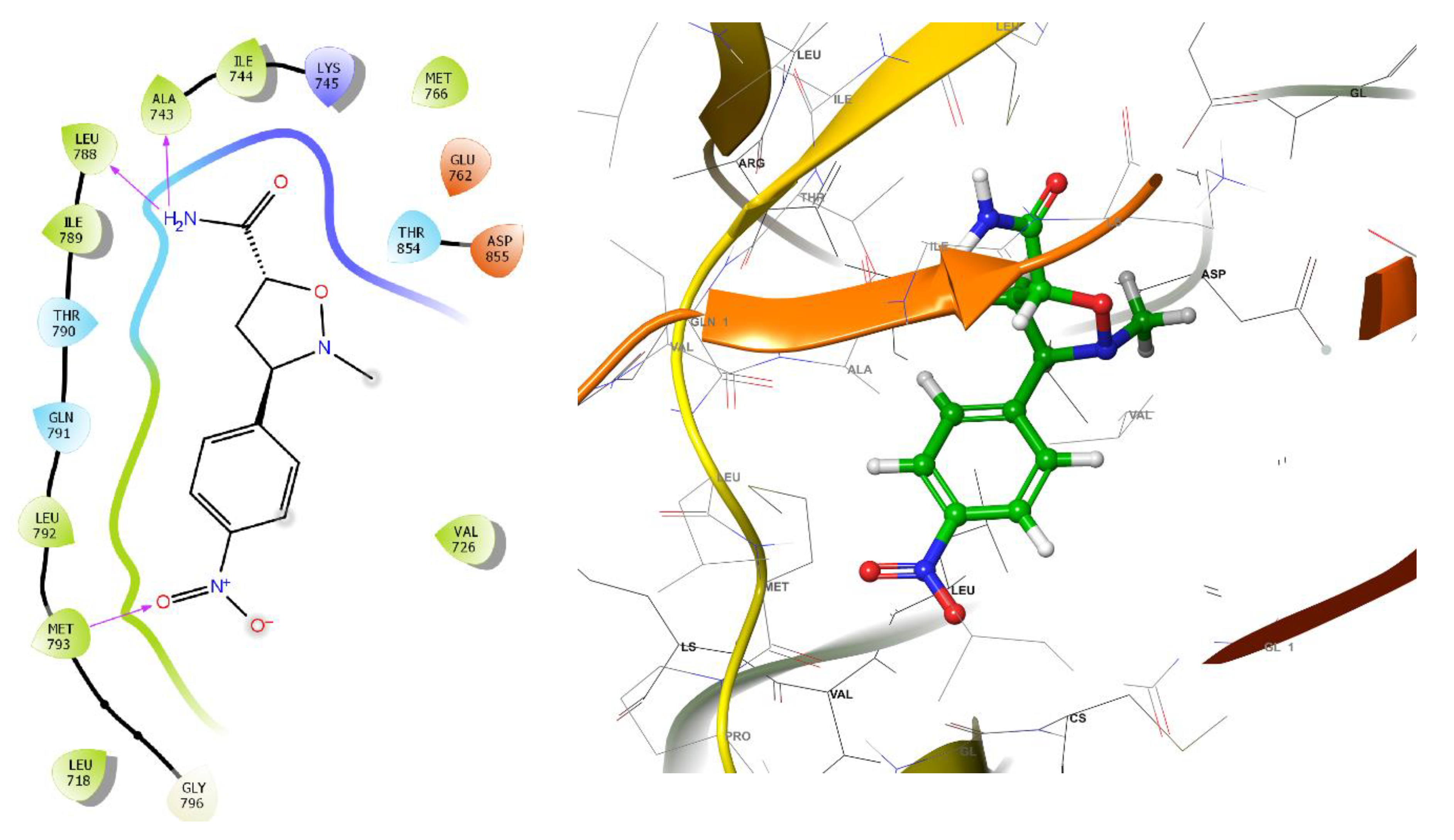
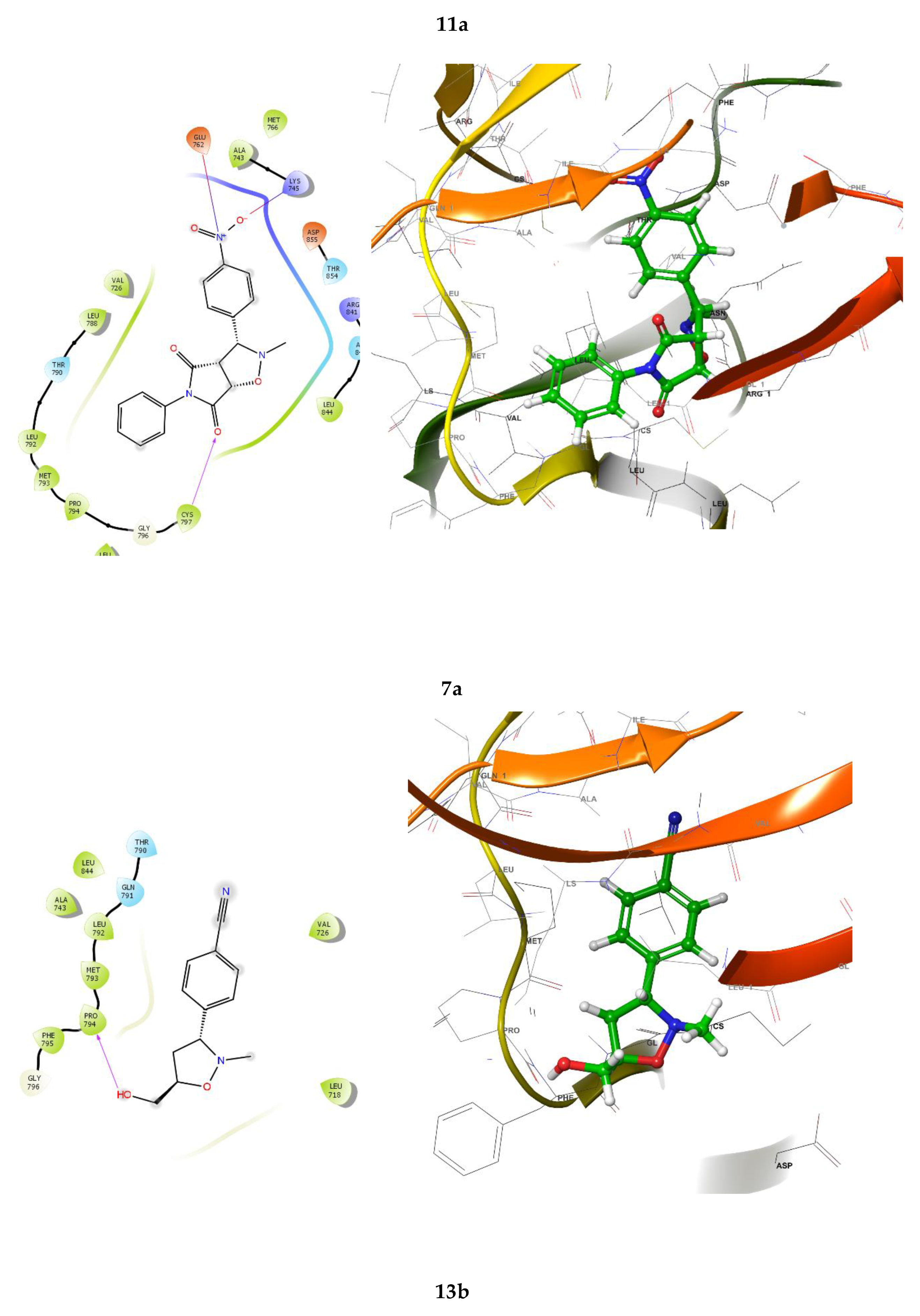
Figure 3.
The RMSD plot obtained for the complex between 11a and the 4ZAU structure.
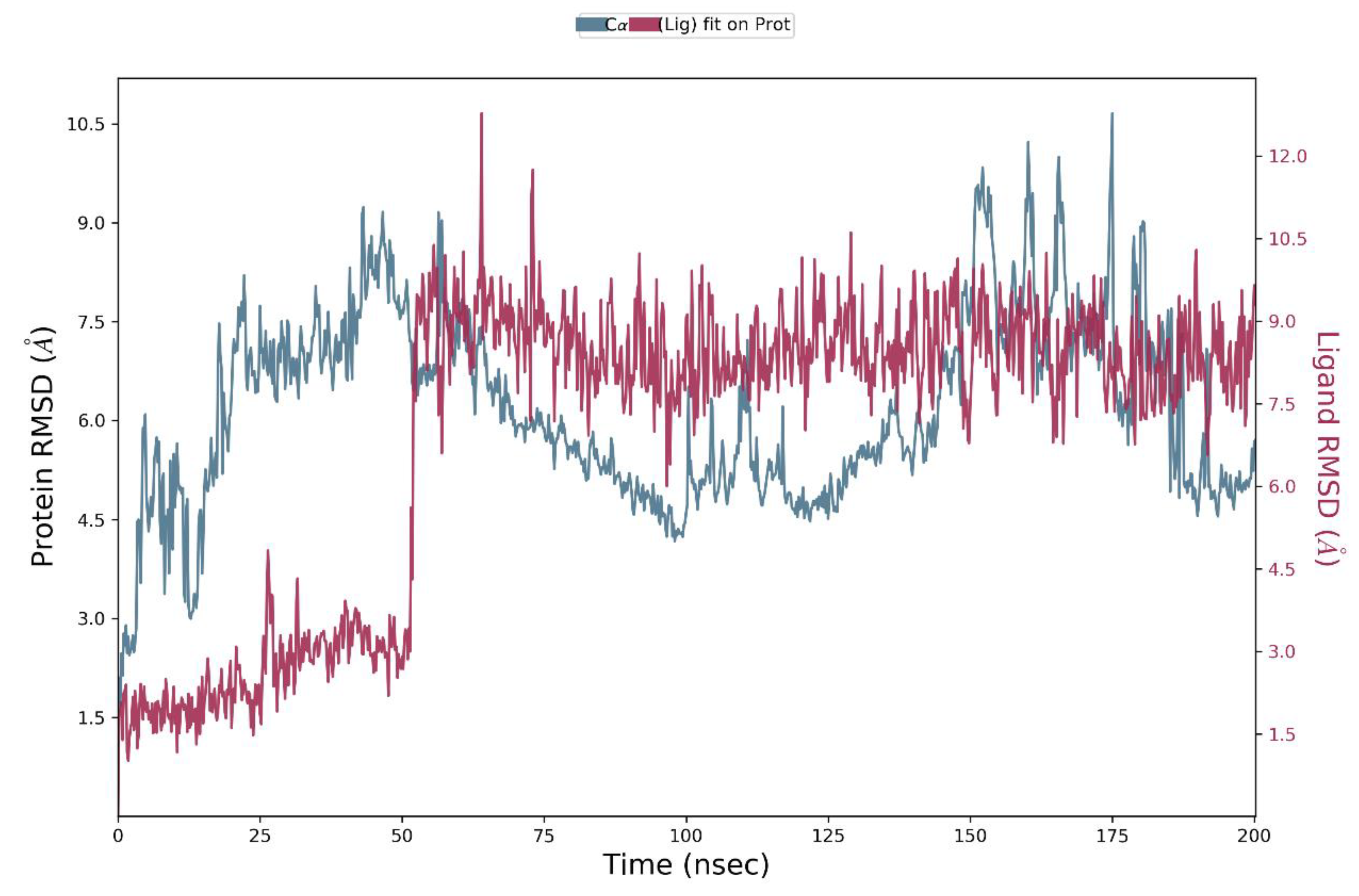

Table 1.
Structural scope of the cycloaddition of a derived nitrone and ethylene.
 | ||||||
 | ||||||
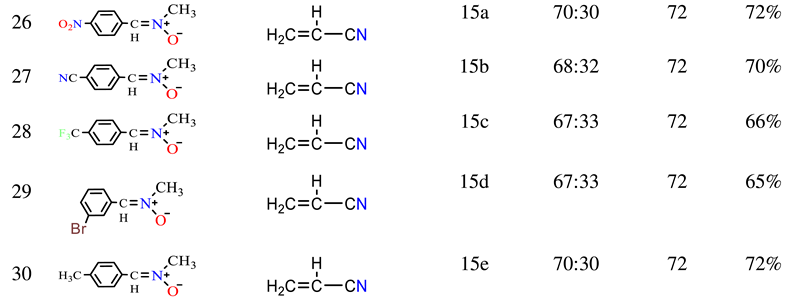 |
Table 2.
B3LYP/6-31G(d) Electronic Chemical Potential μ, Chemical Hardness η, Electrophilicity ω, and Nucleophilicity N Indices (in eV) of Nitrones and Ethylenes.
Table 2.
B3LYP/6-31G(d) Electronic Chemical Potential μ, Chemical Hardness η, Electrophilicity ω, and Nucleophilicity N Indices (in eV) of Nitrones and Ethylenes.
| Reactant | η | µ | ω | N |
| 3a | 5.40 | -4.46 | 1.84 | 2.32 |
| 3b | 3.92 | -4.11 | 2.15 | 3.42 |
| 3c | 4.15 | -3.84 | 1.78 | 3.57 |
| 3d | 4.21 | -3.67 | 1.60 | 3.72 |
| 3e | 4.19 | -3.27 | 1.28 | 4.12 |
| 4 | 5.44 | -4.49 | 1.85 | 2.27 |
| 6 | 5.49 | -4.56 | 2.98 | 2.97 |
| 8 | 6.24 | -4.47 | 1.39 | 2.20 |
| 10 | 5.92 | -4.80 | 1.22 | 2.73 |
| 12 | 6.55 | -4.64 | 1.01 | 2.57 |
| 14 | 6.34 | -4.70 | 1.74 | 1.62 |
Table 3.
Lowest binding energies for the ligand-EGFR interaction.
| Ligand | Affinity (kcal/mol) | H-Bonding | Interacting residues |
| 11a | -6.573 | 3 | ALA743, LEU788, MET793 |
| 11e | -6.523 | 2 | THR854, THR790 |
| 7c | -6.099 | 1 | CYC797 |
| 11d | -6.097 | 1 | PRO794 |
| 11 c | -6.835 | 2 | THR854, THR790 |
| 7 d | -6.08 | 1 | CYS797 |
| 13 b | -5.866 | 1 | PRO794 |
| 11 b | -5.803 | 2 | MET793, ASP800 |
| 9 d | -5.696 | 1 | CYS797 |
| 15 c | -5.606 | 0 | |
| 9 e | -5.536 | 1 | LYS745 |
| 7a | -5.506 | 3 | LYS745, GLU762, CYS797 |
| 13 e | -5.449 | 1 | PRO794 |
| 15 b | -5.437 | 0 | |
| 13 a | -5.392 | 1 | PHE795 |
| 15 a | -5.389 | 0 | |
| 7 b | -5.385 | 1 | CYS797 |
| 15 d | -5.37 | 0 | |
| 5 b | -5.362 | 2 | MET793, CYS797 |
| 13 c | -5.316 | 2 | MET793, MET793 |
| 5a | -5.234 | 1 | CYS797 |
| 15 e | -5.195 | 1 | LYS745 |
| 5 c | -5.129 | 1 | CYS797 |
| 7 e | -5.113 | 1 | CYS797 |
| 13 d | -5.114 | 1 | MET793 |
| 5 d | -5.105 | 1 | CYS797 |
| 9a | -5.022 | 1 | THR854 |
| 5 e | -4.798 | 1 | CYS797 |
| 9 c | -4.658 | 1 | CYS797 |
| 9 b | -4.493 | 1 | CYS797 |
Table 4.
Prediction of Lipinski Rule of five for the test compounds.
| Lig. | Formula | MW (>500) | Accept HB (<=10) | Donor HB (<=5) | TPSA |
iLOGP w (<5) |
| 5a | C14H16N2O7 | 324.29 | 8 | 0 | 110.89 | 2.61 |
| 5b | C15H16N2O5 | 304.3 | 7 | 0 | 88.86 | 2.77 |
| 5c | C15H16F3NO5 | 347.29 | 9 | 0 | 65.07 | 3.21 |
| 5d | C14H16BrNO5 | 358.18 | 6 | 0 | 65.07 | 3.27 |
| 5e | C15H19NO5 | 293.32 | 6 | 0 | 65.07 | 3.16 |
| 7a | C18H15N3O5 | 353.33 | 6 | 0 | 95.67 | 2.13 |
| 7b | C19H15N3O3 | 333.34 | 5 | 0 | 73.64 | 2.51 |
| 7c | C19H15F3N2O3 | 376.33 | 7 | 0 | 49.85 | 2.78 |
| 7d | C18H15BrN2O3 | 387.23 | 4 | 0 | 49.85 | 2.94 |
| 7e | C19H18N2O3 | 322.36 | 4 | 0 | 49.85 | 2.76 |
| 9a | C13H16N2O5 | 280.28 | 6 | 0 | 84.59 | 2.26 |
| 9b | C14H16N2O3 | 260.29 | 5 | 0 | 62.56 | 2.73 |
| 9c | C14H16F3NO3 | 303.28 | 7 | 0 | 38.77 | 2.84 |
| 9d | C13H16BrNO3 | 314.18 | 4 | 0 | 38.77 | 3.04 |
| 9e | C14H19NO3 | 249.31 | 4 | 0 | 38.77 | 2.91 |
| 11a | C11H13N3O4 | 251.24 | 5 | 1 | 101.38 | 1.19 |
| 11b | C12H13N3O2 | 231.25 | 4 | 1 | 79.35 | 1.29 |
| 11c | C12H13F3N2O2 | 274.24 | 6 | 1 | 55.56 | 1.78 |
| 11d | C11H13BrN2O2 | 285.14 | 3 | 1 | 55.56 | 1.97 |
| 11e | C12H16N2O2 | 220.27 | 3 | 1 | 55.56 | 1.76 |
| 13a | C11H14N2O4 | 238.24 | 5 | 1 | 78.52 | 1.63 |
| 13b | C12H14N2O2 | 218.25 | 4 | 1 | 56.49 | 2.04 |
| 13c | C12H14F3NO2 | 261.24 | 6 | 1 | 32.7 | 2.47 |
| 13d | C11H14BrNO2 | 272.14 | 3 | 1 | 32.7 | 2.51 |
| 13e | C12H17NO2 | 207.27 | 3 | 1 | 32.7 | 2.43 |
| 15a | C11H11N3O3 | 233.22 | 5 | 0 | 82.08 | 1.51 |
| 15b | C12H11N3O | 213.24 | 4 | 0 | 60.05 | 1.84 |
| 15c | C12H11F3N2O | 256.22 | 6 | 0 | 36.26 | 2.35 |
| 15d | C11H11BrN2O | 267.12 | 3 | 0 | 36.26 | 2.27 |
| 15e | C12H14N2O | 202.25 | 3 | 0 | 36.26 | 2.29 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



