
Submitted:
11 September 2023
Posted:
13 September 2023
You are already at the latest version
Abstract
Many of the potential immune therapeutic targets are similarly affected in adult-onset neurodegenerative diseases such as Alzheimer’s (AD) disease, Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), and frontotemporal dementia (FTD), but also in a seemingly distinct Niemann-Pick type C disease with primarily juvenile-onset. This strongly argues for an overlap in pathogenic mechanisms. The commonly researched immune targets include various immune cell subsets such as microglia, peripheral macrophages, or regulatory T cells (Tregs), the complement system, and other soluble factors. In this review, we will compare these neurodegenerative diseases from a clinical point of view and point out the common pathways and mechanisms of protein aggregation, neurodegeneration and/or neuroinflammation that could potentially lead to shared treatment strategies. We also describe the common therapeutic approaches in treating the immune dysfunctions in these disorders, moving from immunization to microbiome regulation and stem cell treatment.
Keywords:
Alzheimer’s disease
; Parkinson’s disease
; Niemann-Pick type C disease
; neurodegeneration
; neuroinflammation
; immunomodulatory therapies
; rare diseases
1. Introduction
Neurodegenerative diseases (NDDs) share several common mechanisms, most prominent of which is neuroinflammation [1,2]. Neuroinflammation, evident as activation of microglia and astrocytes, which results in increased proinflammatory cytokine and reactive oxygen species generation, is one of the main mechanisms that causes neuronal death. Although it was traditionally considered to be almost exclusively a late step in disease pathogenesis, multiple lines of evidence have recently shown that it could be an early step as well {reviewed in [3,4]. This review will focus on overlapping neuroimmune mechanisms and potential shared therapeutic targets in several key NDDs. In a recent review article, we have already discussed the proposed immune imbalance underpinning amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) [5], which we will here extend to two other late-onset NDDs - Alzheimer’s disease (AD) and Parkinson’s disease (PD), and an early onset Niemann-Pick type C disease (NPC), due to its extensive and intriguing overlap with AD. We aim to depict common targets that could lead to faster translation and shared therapies.
1.1. Brief overview of distinct and overlapping clinical features in neurodegenerative diseases
AD is a progressive degenerative disease of the brain and the most common cause of dementia among elderly people, accounting for at least two-thirds of all dementia cases [6]. AD is defined as a progressive decline in cognitive functions, typically beginning with memory impairment, and a characteristic change in personality and executive functions. FTD represents a group of disorders considered to be clinically and pathologically distinct from AD, although FTD may be mistaken for AD in the early clinical stages [7,8]. The FTD clinically presents as either behavioural or aphasic variants, reflecting the topography of the underlying synaptic and neuronal loss [7,9,10]. The most common, behavioural or frontal variant of FTD is associated with disinhibition, impulsivity, apathy, and loss of insight that disturbs social component and typically is accompanied by marked frontal lobe atrophy. The aphasic variant is further divided into two subtypes: the non-fluent form (primary progressive aphasia), with hesitant diminished speech output for which left frontotemporal lobe involvement is characteristic; and the fluent form (semantic dementia) with severe naming and word comprehension and visual recognition deficit (agnosia) for faces and objects that involves bilateral anterior temporal lobes. Therefore, the clinical phenotype of FTD may overlap with AD in memory and executive dysfunction, but is distinct in terms of behavioural problems and language difficulties. ALS and FTD are related clinical phenotypes, which are characterized by decline in motor, cognitive and behavioural function, and short survival. ALS is the most common adult-onset motor neuron disease, characterized by the progressive, irreversible motor neuron loss leading to denervation atrophy of muscles and death by respiratory failure. However, it is increasingly recognized that ALS is a multisystem disorder in which other non-motor (cognitive and behavioural) impairments can be observed, whereas, on the other side, FTD can be associated to signs of motor neuron disease (FTD-MND) [11].
Niemann-Pick type C disease (NPC) is an autosomal recessive neurovisceral lipid storage disorder, characterized by liver dysfunction and progressive neurodegeneration [12,13]. It is characterized by a highly heterogenous and variable clinical phenotype, from a rapidly progressing neonatal form to an adult-onset chronic neurodegenerative condition. The neuropathological features of NPC disease include loss of Purkinje neurons in the cerebellum, hyperphosphorylation of tau and widespread occurrence of neurofibrillary tangles (NFTs), the presence of dendritic and axonal abnormalities, and profound neuroinflammation (activated astrocytes and microglia) [14,15,16,17]. While hepatosplenomegaly together with motor problems is initially observed in neonatal and infantile forms along with other symptoms, the juvenile and adult forms of NPC are typically manifested by a variety of progressive neurological and/or psychiatric symptoms including ataxia, dystonia, hearing loss, epileptic seizures, dysarthria, dysphagia, cognitive impairment, and dementia. Indeed, these NPC patients display an apolipoprotein E ε4-dependent accumulation of amyloid-ß peptides (Aß) into diffuse Aß plaques as well as widespread occurrence of NFTs in their brains [14,15,16] the two characteristic features of AD. For this reason, NPC disease is often called juvenile AD.
PD is the second most common NDD after AD, where the cardinal motor features include akinesia/bradykinesia, postural instability, and resting tremor. Here, it should be noted that while many of the motor symptoms arise from the loss of dopamine neurons in the substantia nigra, neuropathology occurs systemically and elsewhere in the brain, resulting in an array of additional motor and nonmotor symptoms; notably prominent are constipation (damage to enteric nervous system), mental health effects, and REM behaviour disorder, and loss of cognitive functions (can be especially prominent at the late disease stages) [18,19]. The affected functions vary from defects in performing executive tasks, visual perception, attention, memory loss and dementia. The cognitive impairment has been reported in up to 90% of PD cases, with dementia cases comprising up to 30% of cases, thus making cognitive manifestation one of the most important non-motor aspects of the disease. Overall, it has become clear that NDDs often show a broadly mixed pathologies and that many patients are now considered to belong to a disease spectrum rather than to a discrete NDD.
The importance of all of these overlapping features of NDDs should be more appreciated especially in cohort characterization for clinical studies, development of the future therapies as well as in biomarker design and its monitoring, with the ultimate goal towards precision/personal medicine.
2. Overlapping pathogenic mechanisms in neurodegenerative diseases
The pathogenic mechanisms implicated in NDDs are not linked to individual clinical entities. In contrast, NDD pathogenesis shows a considerable overlap in protein misfolding and aggregation, defects in endosomal-lysosomal network and clearance of damaged proteins by autophagy or proteasomes, mitochondrial dysfunction and oxidative stress, cellular calcium imbalance, impaired axonal, membrane or nucleocytoplasmic trafficking, DNA damage response and synaptic dysfunction, many of which crosstalk, and are directly or indirectly linked to neuroinflammation and even systemic immune imbalance [19,20,21,22,23,24,25,26,27]. Most of these mechanisms are also affected by ageing, the most prominent risk factor for adult-onset NDDs, which is in the immune system linked to immunosenescence, with increased activated adaptive immune cells and decreased repertoire of naïve cells, and chronic low-grade inflammation [28,29]. In this chapter we will focus on overlapping proteinopathies and immune imbalance in NDDs, the two features that have recently been tackled for designing and monitoring efficient therapies.
2.1. Overlapping proteinopathies
Thanks to our increased ability of detecting pathological protein species in the brains of patients affected by NDDs, it has been clear since the last decade that overlapping proteinopathies exist throughout the entire spectrum of most NDDs [30]. Notably, Aβ aggregates are commonly present in AD and NPC, tau in AD, NPC and FTD, TDP-43 in ALS and FTD, and α-synuclein (α–syn) in PD [14,31,32,33]. Less commonly, but by no means as an exception, TDP-43 pathology is found in AD and PD, Aβ in PD and α-syn in AD and FTD. Just to provide some indication of how frequent comorbidities could be in neurodegeneration - it has been estimated that only 20% of all AD cases that occur after the age of 70 can be defined as “pure” AD cases [34]. In most of these cases, the comorbidity occurs between tau pathology and another major actor in NDDs represented by the TDP-43 protein that plays a major role in ALS and FTLD-TDP, as recently reviewed by Riku et al. [35]. The occurrence of comorbidities has substantially changed our view of neurodegeneration processes: from considering AD, PD, ALS, FTD, etc. as distinct and well-defined pathologies to viewing them as a potentially interconnected spectrum of neurodegeneration, where comorbidities may often influence the main pathology and affect both disease progression and duration. When trying to address this new finding, another important question to answer is whether the overlapping pathologies have the same site-specific characteristics compared when they are alone. For both questions, some answers have already been provided with regards to prominent comorbidities. For example, TDP-43 pathology in the AD brains, considerably differs from the primary motor-cortex involvement that is characteristic of this protein in ALS. Rather, TDP-43 pathology in AD brains starts in the amygdala and goes through several stages to finally reach the basal ganglia and middle frontal cortex [36].
Another type of comorbidity that has been studied in the past especially at the mechanistic level is represented by α-syn in brain inclusions of AD patients, that has been recently reviewed by [37,38]. At the mechanistic level, α-syn and tau have been shown to be connected in several ways, with α-syn fibrils being able to promote tau aggregation [39,40]. Taken together, these observations suggest that protein aggregation comorbidities can play an important role in NDDs, and that their study could be prioritized in future studies to better understand their pathological and clinical connections.
In addition, a careful identification of comorbidities could also be pivotal for the interpretation of clinical trials results. In fact, through the development of appropriate biomarkers, it would be greatly advantageous to start stratifying treated subjects by presence and types of accompanying comorbidities. This action might be able to uncover clinical response variability in some groups as compared to others, potentially “rescuing” treatments that might otherwise fail completely if this factor is not taken into account. For this reason, accurate in vivo comorbidities detection methods are urgently needed for the future of therapeutic research.
2.2. Overlapping immune imbalance
As mentioned above, many pathogenic mechanisms in NDDs are intricately linked to neuroinflammation and systemic immune imbalance. At a cellular level, microglia, the resident macrophages in the brain and spinal cord, are the primary immunocompetent cells in the CNS that have a pleiotropic neurotrophic function [41,42]. In response to protein aggregation and various other neurotoxic conditions, microglia dynamically react in different ways, which range from neuroprotection to neurotoxicity. Similarly, astrocytes, the most abundant CNS glial cells, modify their functional status in pathological conditions [43]. When activated, microglia and astrocytes promote the release of immune mediators, including chemokines and interleukins, that, if persistently elevated, promote the onset and progression of NDDs, becoming potential therapeutic targets [44,45].
Several genes directly affecting immune functions have been linked to ALS and/or FTD, such as C9ORF72, TBK1, OPTN, CYLD, GRN (reviewed in [3,4,5,46]). Moreover, as detailed below, genes enriched in microglia have been increasingly implicated in AD pathogenesis over the last decade. It is interesting though, that there is only moderate genetic overlap between NDDs. A recent large GWAS study found evidence for only eleven shared loci in AD, PD and/or ALS, which were potentially linked to genes affecting lysosomal or autophagic functions, neuroinflammation, DNA damage response, and oxidative stress [47]. However, despite such comparably small overlap in individual genes, functions defective across the broad spectrum of NDDs are linked to phagocytosis, lysosomal functions, autophagy, inflammatory signalling, activation of complement and others [48,49,50,51,52]. Many of these functions, including activation of complement, inflammatory signalling, and phagocytosis are beneficial only in a narrow window, and if uncontrolled can lead to various harmful effects, including extensive synaptic pruning and bystander cytotoxicity [51,52,53,54,55]. The harmful effects of these factors are not limited to the CNS, and many lead to systemic immune imbalance. Higher neutrophil counts and blood proinflammatory factors are linked to a higher risk of developing ALS, AD and PD [56,57,58,59].
Given that genetics can explain only a small fraction of NDD cases and that most have a complex environmental component, many environmental factors have been researched, such as infectious diseases, microbiome composition, toxins (pesticides and other), most of which affect immune responses. The proposed link between viral infection and NDD has been studied since the Spanish flu of 1918, which was caused by influenza A virus subtype H1N1 [60]. Since then, multiple viruses proposed to increase the risk such as herpes simplex virus 1 (HSV-1) for AD, retroviruses for ALS, and others [61,62,63,64] Most recently, similar risk has been found for SARS-Cov2 and is predicted to have a great impact given the large number of affected individuals in the corona virus 19 (COVID-19) pandemic [65]. In perhaps the most comprehensive study thus far, exposure to 45 viruses was linked to NDDs in a Finnish cohort, 22 of which were replicated in an UK cohort [66]. While the large risk effects such as those reported between viral encephalitis and AD were rare, moderately increased risk was very common. Notably, severe cases of influenza and pneumonia were significantly associated with five NDDs (AD, PD, ALS, vascularized and general dementia). Most of the associations were more strongly linked to NDD one year prior to their diagnosis, but some exposures affected the risk up to 15 years prior to the diagnosis. Fittingly, associations with neurotropic viruses were the most common (>80%), and none of the viruses seemed to confer neuroprotection. Similar associations have been reported for severe systemic bacterial infections in AD, as further discussed below [67]. Overall, this strongly argues that CNS and systemic inflammation is linked to NDDs and that preventive vaccines should be pursued more aggressively, not only to avoid infections, but also their long-term aftermath like NDDs.
The role of the gut microbiome in the pathogenesis of NDDs is of great interest since it has been shown that its composition differs in people afflicted with NDD when compared to the healthy population [68,69]. Changes in relative abundances of different microbial taxa have been shown to factor into neurodegeneration, going as far as to influence the severity of the symptoms of certain diseases, such as in the case of PD, where it has been shown that changes in the relative abundance of various taxa can be correlated, either positively or negatively, with motor and non-motor symptom severity [70]. It has been hypothesized that these effects are achieved, among others, through the gut-brain axis (GBA). GBA is a complex bidirectional system operating between the intestines and the brain, and it has been demonstrated in a number of studies that it could drive neurodegeneration in conditions such as AD and PD [71]. So far, several ways in which this system could operate have been described. These include retrograde axonal transport across the vagal nerve [72], microbial metabolites, such as short-chain fatty acids (SCFA) [72], serotonin-microbiota interaction [73], tryptophan-kynurenine metabolism [74] and immune signalling [75]. Direct evidence for the involvement of the immune system came also from studying C9ORF72, the most common genetic risk factor for both ALS and FTD, which acts in autophagy and endolysosomal pathways to suppress inflammation. Indeed, C9ORF72 has also been shown to suppress microbiota-induced inflammation in mouse models [76]. Therefore, the evidence that the immune signalling linked to microbiota can play an important part in disease pathogenesis is being gathered for many NDDs [77,78].
2.2.1. Immune imbalance in Alzheimer’s disease
Recent genome-wide association studies (GWAS) of the late-onset AD (LOAD) have identified risk factors in genes expressed by microglia (TREM2, CD33, CR1, INPP5D, SPI1, BIN1, PICALM, ABCA7, SORL1, CD2AP and the MS4A gene cluster) or crucial for microglial development and function (PU.1) [79,80,81,82,83], suggesting that immune dysfunction and neuroinflammation could drive neurodegeneration rather than being only considered as a (late) consequence of protein aggregation. Microglial dysfunction thus became considered as a major contributor to AD risk [57], challenging the long-standing Aβ hypothesis, which posits that Aβ is the initial disease trigger, while an excessive inflammatory response of microglia is secondary to accumulation of Aβ peptides and Aβ plaque formation [84,85]. Interestingly, several of the above-listed GWAS risk factors are functionally linked to microglial phagocytosis and Aβ clearance [86,87,88,89], placing dysfunction of the immune system in the CNS in the centre of AD pathobiology. Although the genesis of disease-associated microglia (DAMs) is a characteristic feature of AD, it is still a matter of debate whether DAMs are “good” or “bad”, i.e., whether microglial activation and neuroinflammation is beneficial (neuroprotective) or detrimental (neurotoxic) for disease progression. Notably, single nuclei RNA sequencing (snRNASeq) in AD brains recently showed that distinct microglia profiles are linked to Aβ and tau-associated pathology [90]. On the other hand, chronically altered microglia in AD could compromise not only phagocytosis, but also other physiological functions, including cytokine, chemokine, and growth factor secretion [91,92]. Moreover, APOE ε4 allele, a major genetic risk factor of LOAD [93], has been functionally linked to reduced Aβ clearance [94,95], implying its direct or indirect role in microglial function. Recent snRNA-seq analyses of the frozen AD patients’ brains and preclinical models revealed significantly upregulated expression of ApoE in DAMs and not exclusively in astrocytes, as previously assumed [96]supporting an emerging view that microglia are an important contributor to ApoE biology in the CNS [97,98,99,100]. In addition to genetic risk factors that link the genesis of LOAD with microglial dysfunction, ageing is another important risk factor that may influence microglial phagocytic capacity and neuroinflammation through epigenetic mechanisms. Indeed, age-dependent accumulation of Aβ in the LOAD patients seems to be associated with age-related decrease in microglial phagocytic capacity [101]. Furthermore, a study using an AD mouse model (APP/PS1) has shown that only young microglia from wild-type mice cleared Aβ plaques, and that exposure of old microglia to conditioned media of young microglia increased their proliferation and reduced Aβ plaque size [101]. This suggested that microglial dysfunction in AD could be reversible and that the phagocytic ability could be modulated to prevent and/or restrict Aβ accumulation. Lastly, it is now increasingly accepted that infectious diseases may be involved in the aetiology of AD, exemplified by the recent influence of COVID-19 on a spectrum of neurological manifestations [102,103,104]. Similarly, other severe infections requiring hospitalization have recently shown to increase the risk of dementia [105], further supporting an important role of immune system in the pathogenesis of AD.
2.2.2. Immune imbalance in Niemann-Pick type C disease
It is intriguing that a rare inherited lysosomal and lipid storage disorder Niemann-Pick type C disease (NPC) shares several key features with AD [106] (Figure 1). Among these, neuroinflammation seems to play an early and important role in disease progression, together with neurodegeneration. However, in contrast to the complex genetics of AD, NPC is a monogenic disease caused by mutations in NPC1 or NPC2 genes (95% and 5% of cases, respectively) [106]. These mutations result in dysfunction of cholesterol transport proteins NPC1 or NPC2, accumulation of unesterified cholesterol and other lipids (e.g., glycosphingolipid, sphingomyelin and sphingosine) in late endosomes/lysosomes and their dysfunction [107,108]. The molecular mechanism of neurodegeneration and neuroinflammation in NPC is currently unknown but, although peripheral organs such as liver and spleen are also affected, NPC1 expression restricted to CNS was capable to rescue both neurodegeneration and lethality in NPC1 null mice [109]. Notably, the restoration of NPC1 in neurons only does not fully rescue the phenotype, indicating that NPC1 is functionally important in other CNS cells as well [110,111,112,113]. Indeed, NPC1 is ubiquitously expressed throughout the brain with particularly high expression in microglia and oligodendrocytes [114]. It has been generally assumed that neuroinflammation in NPC is secondary to neuronal loss. However, recent findings in NPC1 mice and NPC patients’ blood-derived macrophages [114], suggest a possible causative rather than consequential role of neuroinflammation in NPC neuropathology. Notably, NPC microglia proteome changes precede neuronal loss and contribute to neuropathology in a cell autonomous manner. Importantly, lipid accumulation in NPC1-mouse microglia is a consequence of impaired lipid trafficking with a striking accumulation of multivesicular bodies, while lysosomal degradation function seems preserved. Among these, late endosomal/exosomal marker CD63 was the most significantly changed protein at the presymptomatic stage, suggesting that defects within endosomal/lysosomal trafficking and sorting may be among the earliest pathological alterations in NPC microglia. Recently, single cell transcriptomics of the NPC1 mouse cerebella identified the earliest gene expression changes in microglia cells together with endothelial cells [115], further supporting an important role of microglia dysfunction and neuroinflammation in the pathogenesis of NPC disease. Pathway analysis of differentially expressed genes revealed that activated microglia in NPC1 mice resemble those from AD mouse model, rather than those from an ALS mouse [115].
2.2.3. Immune imbalance in Parkinson’s disease
While specific neuronal populations (dopamine neurons in the substantia nigra) are affected in PD, there is also a considerable overlap to AD and ALS in pathogenic mechanisms [24,25,26,27]. In keeping with this, complex interactions have been revealed between α-syn, a neuronal protein associated with PD and other synucleinopathies, and microglia [116]. Like in ALS and AD, microglia are neuroprotective in the early stages of disease by clearing α-syn, whereas during the chronic disease stage they are considered to promote neurodegeneration by propagating the α-syn burden and creating an inflammatory environment [117]. In addition, viral infections, including COVID-19, have been associated with an increased risk of PD [118] suggesting that inflammation linked to infection could contribute not only to AD but also to PD pathogenesis (as detailed above). Importantly, these findings suggest that neuroinflammation may be a primary pathogenic event and that systemic neuroinflammatory insults may drive transsynaptic spread of PD pathology. The link with PD, microbiota and immunity is an important topic. Retrograde axonal transport of pathology across the vagal nerve has been hypothesised in 2003 by Heiko Braak, using the model of PD [118]. The idea was that α-syn accumulation actually begins in the gut, with pathologic forms of the protein migrating into the brain through the vagal nerve. Evidence of this has been shown in 2014, with microtubule-associated transport being a key mechanism [119]. More recently, the same enteric propagation was proposed for β-amyloid in AD. In a mouse model, it has been shown that intra-gastrointestinal application of Aβ plaques leads to a higher deposition of these plaques in various regions of the brain, with retrograde vagal transport being a key pathway behind it [119]. Microbiota metabolites have also been shown to have an effect on the CNS, with short-chain fatty acids (SCFA) being the most prominent products, affecting the regulation of enteric secretion and motility as well as gut-brain signalization [120]. In various models it has been shown that SCFA can have a neuroprotective role, such as in the case of Lactobacillus plantarum and its product butyrate, which has anti-inflammatory effects [120]. It also positively influences blood-brain-barrier (BBB) permeability [121]. On the other hand, in some cases SCFA can have a detrimental effect, such as in the case of propionic acid, where serum levels positively correlated with motor and non-motor symptom severity in PD[121]. Besides SCFA, microbiota has also been implicated in taurine metabolism [122] as well as magnetite and hydrogen sulphite production [122], both of which have been shown to have a role in PD. Serotonin is thought to be an important part of GBA signalling, with certain bacteria affecting both its colonic and serum levels, aiding in SCFA production, which in turn increases serotonin production [123]. Microbiota also have an effect on the serotonin precursor tryptophan. It has been shown in one study that a sex-dependent increase in hippocampal serotonin levels could be attributed to microbiota-related changes in tryptophan levels [124]. Furthermore, kynurenine, a metabolic product of tryptophan, has been shown to traverse the BBB and lead to neuroinflammation, as well as neurodegeneration [74]. Immune signalling linked to microbiota can play an important part in the development of neurodegenerative diseases [77,125]. Alterations of gut microbiota can lead to changes in the permeability of the intestinal mucus layer, leading to local immune activation and intestinal barrier dysfunction, eventually allowing various microbial triggers, such as lipopolysaccharide (endotoxin) and peptidoglycans to be released into the systemic circulation. This leads to a process known as metabolic endotoxemia, and it has been shown to trigger immune activation in various systems, including the central nervous system, most notably through microglia [126]. Macrophages have also been shown to be intrinsically linked to the gut microbiome. Antibiotics-induced changes in the microbiota have been shown to decrease macrophage levels and affect gastrointestinal motility [126]. With macrophages being essential responders to intestinal injury, this decrease could further enhance the neuroinflammatory effects of metabolic endotoxemia.
There are also direct examples of toxic insults that may influence both PD and ALS. For example, environmental exposures have been extensively studied as risk factors for PD [127]. The organophosphate pesticide, chlorpyrifos has been identified as a possible risk factor for PD in both human and animal studies, where in addition to inhibiting acetylcholinesterase, it also likely affects dopaminergic neurotransmission and produces oxidative stress [128,129,130,131,132,133,134]. While the role of environmental exposures in ALS is far less understood than in PD, pesticides have also been explored as risk factors, with some studies suggesting increased ALS risk with pesticide exposure, especially related to organophosphate pesticides such as chlorpyrifos [135]. Interestingly, mutations in genes specifically responsible for chlorpyrifos detoxification also seem to increase ALS risk, suggesting a gene-environment interaction in the aetiology [136,137]. With respect to genetic overlap, an example is optineurin, which has been extensively studied in ALS and glaucoma [138]; mutations may also be a risk factor in PD [139,140]. While optineurin has many roles (i.e., innate immunity, mitophagy), in vivo experimental PD models, alterations in mitophagy expression and localization suggest that these are likely important in both ALS and PD [141]. Taken together, there is therefore a clear pathogenic overlap between ALS and PD. Overall, there is mounting evidence that the immune system may be a primary target of PD-relevant exposure, versus just a downstream pathogenic pathway. For example, a recent line of research has advanced understating of how environmental agents interact with specific innate immune signaling pathways in microglia to stimulate conversion to a neurotoxic phenotype. Here, researchers showed that nuclear factor kappa B (NF-κB) signaling in microglia is critical to clearance of aberrant α-syn resulting from rotenone exposure, an important finding that identifies neurotoxin-immune system-phenotypic links [141].
3. Common therapeutic approaches in treating the immune (dys)functions in neurodegenerative diseases
The current treatments for NDDs are symptomatic and cannot affect and modify the underlying disease cause. However, recent understanding of multiple shared pathologies among NDDs, with considerable mechanistic overlap, has expanded to development of therapeutic targets. In fact, several emerging findings hold on the promising role of therapies able to act on the immune dysfunctions for slowing down the neurodegenerative process. As described above, many reports support the idea that molecules affecting the immune system pathways can have as a primary target the misfolded and aggregated proteins that accumulate in NDDs. The most investigated treatments include active and passive vaccinations, the molecules directly targeting the inflammatory mediators or pathways and the multimodal effects of stem cells, particularly of the mesenchymal ones (summarized in Figure 2).
3.1. Passive and active vaccination therapies targeting protein aggregates
In the last three decades, active vaccinations and monoclonal antibodies for passive vaccinations have been applied in several fields of medicine, including neoplastic and autoimmune diseases. More recently, this therapeutic strategy is also being evaluated for neurological diseases. In most NDDs, the primary underlying pathological mechanism is the abnormal accumulation of soluble proteins in the form of insoluble intracellular or extracellular aggregates, which can act directly as toxic aggregates or via precursors or mediators, and on which vaccination therapies can work [142]. More specifically, immunotherapeutic approaches include both passive and active immunization. The first one consists of an infusion with the monoclonal antibodies directed against the target molecules (e.g., the misfolded proteins); the second one provides specific antigens directed to a specific adaptive immune response [143], inducing the production of antibodies or modulating the inflammatory response. Certainly, the most relevant example is the removal of Aβ accumulation in AD. However, since the intracellular protein accumulation is a hallmark of most NDDs, the same approach was also used for intracellular proteins, α-syn and tau among them. We will summarize here the main clinical trials recently developed.
3.1.1. Clinical trials on Aβ immunization
Currently, passive immunization with the antibodies against Aβ is the most advanced immunotherapy under investigation. It started from solid preclinical data showing the ability of Aβ-targeting monoclonal antibodies to bind the Aβ-42 species, reducing the toxicity, preventing cell death, and restoring the plasticity at the hippocampal level in animal models [144,145,146]. Different Aβ-targeting monoclonal antibodies have been proposed in recent years for AD treatment. Due to different binding properties among them, we have aducanumab [147] and lecanemab [148]among the most promising. The aducanumab was approved after two large trials (EMERGE, 1638 patients and ENGAGE, 1647) with AD patients in the early stage of the disease [147]. The primary outcome was the changes over treatment in the global cognition (measured with the Clinical Dementia Rating Sum of Boxes test) (CDR-SB) [149], and it was met in the EMERGE trial but not in the ENGAGE. However, in both studies, promising results were obtained regarding biomarkers, confirming a dose-dependent reduction in AD markers pathophysiology. Based on these results, aducanumab (marketed as Aduhelm) was approved by the U.S. Food and Drug Administration in 2021 under the accelerated approval pathway. On the contrary, the European Medicines Agency withdrew its marketing authorization application for aducanumab to treat the early stages of AD. Also for the US patients, in July 2023, lecanemab, a humanized IgG1 monoclonal antibody that binds with high affinity to Aβ soluble protofibrils, was approved (marketed as Leqembi). In the phase III clinical trial, the drug obtained its primary outcome (the same as the aducanumab trial) and reported a significant reduction in brain amyloid burden. Clinical trials on active immunization for Aβ started in 2000 [150]. The first vaccine tested in humans, called AN1792 (Elan Pharmaceuticals), provided the inoculations of amyloid-β42 peptide with an adjuvant. Albeit positive effects were observed in post-mortem neuropathological studies[151], the vaccine had several severe side effects (i.e., meningoencephalitis) and the trial was stopped. Even if without clinical efficacy, in a long-term follow-up of survived patients (14 years) [152] all had only very sparse or undetectable plaques in all regions examined. Subsequent trials in this regard obtained better safety results, but no clinical and biological effects [153].
3.1.2. Clinical trials on tau immunization
Since intraneuronal aggregates of tau protein have been shown to directly correlate with cognitive decline in AD [154,155], they comprise a potentially even more interesting target than Aβ. In the last decades, several encouraging results were obtained from the experimental mouse models, where tau targeting resulted in a reduction in protein pathology, preservation of brain volume, and improvement of behavioural scores [156]. Among the most relevant of anti-tau vaccines is AADvac1, an active peptide vaccine targeting nonphosphorylated tau, which proved safe and immunogenic in AD patients [157], although there were no clinical effects in the whole cohort. Regarding passive immunization, three studies have been completed, but the results are still unpublished (BIIB092/Gosuranemab, a humanized monoclonal antibody that binds to N-terminal tau (NCT03352557), RO7105705/ Semorinemab, an anti-tau IgG4 antibody (NCT02820896), and LY3303560/Zagotenemab, a humanized anti-tau antibody derived from MCI-1 (NCT02754830). A trial with the JNJ-63733657, a humanized monoclonal anti-tau antibody that binds to phosphorylated tau (NCT04619420), is in a recruiting phase.
3.1.3. Clinical trials on α-syn immunization
Similar as for Aβ, passive immunization for synucleinopathies involves using different monoclonal antibodies against α-syn. In a mouse model of PD, Abs-α-syn significantly attenuated the cognitive and motor deficits, with a consequent reduction of α-syn aggregates and of pathological accumulations of the levels of soluble α-syn, total human α-syn, and α-syn oligomers [158]. Starting from results in animal models, large clinical trials started. However, unfortunately, no positive results were obtained from one large trial with prasinezumab [159], observing no meaningful effect in global and imaging measures of PD progression and a large percentage of infusion reactions (up to 34% of patients). A better safety profile has been obtained in the phase I trial with BIIB054 (cinpanemab) [160], but still without effects on clinical outcomes in the first year of the trial, leading to its discontinuation and premature termination (data not published). Clinical trials on α-syn active immunization were a continuation of several studies of animal models, which showed that antibodies to α-syn prevented pathogenic protein spread and promoted clearance of aggregates [161]. Several in-human studies have been proposed. One of the most promising involved the use of PD01A (AFFITOPE). The molecule is an eight amino acid peptide that mimics an epitope in the C-terminal region of human α-syn designed to stimulate B cell antibody responses bypassing the auto-reactive T cell mobilization. The first-in-human, randomized, phase 1 study of immunizations with PD01A [162], demonstrated that the repeated administrations of PD01A were safe and relatively well tolerated in a cohort of PD patients. From a biological point of view, a substantial humoral immune response was observed. However, to date, no other results have been published.
3.1.4. Immunization for ALS treatment
For ALS, monoclonal antibodies against intracellular proteins such as SOD1, C9orf72 and TDP-43 are under study or recently completed, although several improvements are needed to increase the efficiency in ALS patients and to obtain significant results [163]. Other antibody-based interventions investigated in ALS patients targeted other molecules involved in the neuroinflammatory pathways mainly localized at intracellular and extracellular level, including the neurite outgrowth inhibitor A (ozanezumab), the muscle specific kinase, the IL-6 receptor (tocilizumab) and other proteins (namely NRP-1, myostatin, CD40L, DR-6, IFN-γ, GD1a, CTGF and HMGB1). Also in this case, the results are only preliminary, contrasting[164,165], and mainly related to biological outcomes [164].
3.2. Targeting inflammatory mediators
Although there are interesting data on the crosstalk of aggregated proteins in different models of NDDs, to date, immunotherapies targeting aggregates alone have had a limited success. This leads us to assume that a more complex scenario exists where the pathological proteins are only one facet of a much more complex therapeutic challenge. As already described, one of the most common and relevant targets of neurodegeneration in NDDs is the alteration of CNS homeostasis mediated by the immune system, where microglia and astrocytes play a central role. In the past years, several clinical trials using anti-inflammatory drugs (e.g., aspirin, prednisone, naproxen, diclofenac, indomethacin) started in patients with NDDs, including AD, PD and ALS, but all failed to obtain clinical improvements [166,167]. This suggested that a complexity between microglia, astrocytes and neurodegeneration necessitates more precise targeting, and perhaps acting further upstream in the neuroinflammatory cascade, such as on inflammatory mediators (cytokines, chemokines and growth factors) or phagocytic functions. However, converging targets with great cytolytic potential such as the complement cascade, have also been targeted. For example, some potential targets are: 1) TNF-α; the use TNF-α inhibitors (antibodies and related fusion proteins) was promising in rodent models of AD; however, two previous studies in AD obtained contrasting results, mainly related to the difficulty of the drug (etanercept) to penetrate the CNS after perispinal injection [168]. Other routes of administration have been tried (e.g. subcutaneous), but with no relevant clinical results, probably related to its inability to penetrate the BBB [168]; 2) the interleukins: evidence reported that numerous interleukins (including IL-2, IL-17, IL-22) are associated with NDDs development and progression by activating glial cells and creating a pro-inflammatory environment. Preliminary evidence for using IL-2 in AD started from solid preclinical data [169], and some trials are currently ongoing (NCT05821153 and NCT05468073); 3) the GM-CSF; GM-CSF is an immunomodulatory growth factor that is clearly deregulated in NDDs. GM-CSF has shown a strong positive effect in mouse models showing a pleiotropic neuroprotective effect with the attenuation of neuroinflammation and cognitive decline by enhancing Aβ clearance by recruiting microglia to amyloid plaques [170]. The in-human results are also encouraging: a clinical trial published in 2021 on AD patients showed that the GM-CSF (sargramostim) treatment had no adverse events in patients, changed innate immune system markers, and significantly improved cognitive status [171] A larger trial is ongoing (NCT04902703). The same drug also showed encouraging effects in PD, even in a small pilot phase I clinical trial [172]: sargramostim showed, clinically, a modest improvement after treatment compared with placebo, and biologically, a Treg number increase.
Regarding ALS, some anti-inflammatory therapies targeting the immune system yield promising results. Very recently, the group of Professor Mandrioli published interesting results on rapamycin treatment in ALS patients [173]. Specifically, the trial observed a significant decrease in the mRNA relative expression of IL-18, plasmatic IL-18 protein, and increased monocytes and memory-switched B cells. Other promising results derived by an autologous infusion of expanded Tregs plus subcutaneous IL-2: Treg/IL-2 treatments promoted, from a biological point of view, a higher Treg suppressive function and, from a clinical point of view, a slowing in disease progression [174]. Lastly, the clinical trial with masitinib is underway (NCT03127267). Masitinib is a selective, oral tyrosine kinase inhibitor with neuroprotective effect preclinically demonstrated and with promising clinical effects in the ALS early trial stage. Interestingly, the same drug is in study also in mild to moderate AD patients (NCT05564169).
3.3. Targeting complement
As shown, the complement has a pathogenic relevance in NDDs, where the role of innate immune-driven inflammation is rapidly growing. Drug molecules that target players of the complement activation cascade can potentially stop the complement-mediated tissue damage, and some trials are ongoing in this regard in NDDs [175]. For example, in AD mouse models, positive results in terms of safety are obtained with ANX005 (a humanized immunoglobulin G4 recombinant antibody against C1q). No studies in humans are currently ongoing [176]. In ALS, similar results in mouse models were obtained using the PMX205, which could delay the grip decline and slow the disease progression [177]. Also, in ALS patients, two studies are ongoing targeting complement: the first one, a phase 2 trial with ANX005 (NCT04569435), and another phase 2 trial with Pegcetacoplan (APL-2, a complement C3 inhibitor) is ongoing (NCT04579666). Lastly, a phase 3 trial is recently terminated with Ravulizumab (a long-acting inhibitor of terminal complement protein C5) but without a positive outcome (the internal displacement monitoring Centre discontinued the study due to lack of efficacy). No trials in PD are ongoing.
3.4. Stem cell and related treatments
Due to their immunomodulatory, anti-inflammatory, and regenerative properties, stem cells and particularly mesenchymal stromal cell (MSC) treatments can have long-term effects on patients with NDDs, as recently demonstrated in ALS [178,179]. From a scientific point of view, this positive result lays the foundations for continuing research in this field, also applying it to other NDDs. MSC (Lomecel-B) have recently been preliminary tested in AD in a phase I clinical trial [180] with positive results, including improvement in cognitive function, hippocampal tropism and fluid biomarkers, providing mandatory information for larger phase II/III clinical trials. However, to date, there is no data about MSC transplantation in PD but from one preliminary report autologous bone marrow-MSCs injected into the subventricular zone appeared safe and well-tolerated, also with minimal motor function improvement [181,182].
Another important avenue in the treatment of NDDs are represented by exosomes. MSCs -derived exosomes demonstrated to exert more potent therapeutic effects over MSCs in NDDs, principally by delivering anti-neuroinflammatory processes [181,182]. Currently some clinical trials to test efficacy of exosomes in AD (NCT0438 8982) and PD (NCY01860118) are ongoing.
3.5. Targeting microbiota
With lot of the pathways behind gut-brain signalling still waiting to be discovered, the main incentive for further research should be potential therapeutic methods targeting the gut microbiota. Antibiotics have shown a positive effect on PD pathology in mice, either by altering microbiota composition [183] or inhibiting α-syn fibrillation [184]. Similarly, probiotics have been shown to prevent neuroinflammation and cognitive dysfunction also by modifying microbiome composition [185]. Specific diets are known to have a beneficial effect in neurodegenerative diseases such as AD and PD, most notably the Mediterranean diet, which is rich in Lactobacilli [186]. Certain clinical studies have shown promise, such as in the case of enema application [187,188] or fecal microbiota transplantation [189] In conclusion, since a great body of knowledge shows microbiome changes in NDDs, the gut, as an often-neglected organ in treating NDDs, should be taken into account. In time, microbiota regulation could prove to be a powerful tool for the prevention and management of neurodegenerative disorders.
3.6. Potential future therapies
Preventive vaccination and/or boosting for influenza virus, HSV-1, and herpes zoster virus have already been conclusively linked to decreased risk for dementia [190,191]. Similar effects were reported for vaccines to prevent pneumococcal pneumonias. Although these vaccinations are still not a standard of care for adult populations in most countries, it is advisable that these notions are considered in future preventive measures for NDDs.
Regarding PD, the primary limitation from advancing beyond dopamine replacement therapies has been that most patients are diagnosed well after significant neuropathology has occurred. As earlier biomarkers are developed, the likelihood of successful bench to bedside translation for novel interventions would be expected to increase. As one promising example, a recent study found that mtDNA damage was increased in peripheral blood mononuclear cells derived from patients with idiopathic PD and those with the PD-associated leucine-rich repeat kinase 2 (LRRK2) G2019S mutation in comparison with age-matched controls. Importantly, mtDNA damage was elevated in non-disease-manifesting LRRK2 mutation carriers, suggesting that those with known risk factors, but not yet with a PD phenotype (not all will convert) could be identified prior to diagnosis. Of note, LRRK2 has a critical role in the central and systemic immune systems. These findings point to a broad future approach, where PD patients are identified earlier or may even be treated at prodromal stages with specific immune modulators.
Current treatments of NPC aim to lower the accumulation of free cholesterol and other GSLs in late endosomes/lysosomes – the primary feature of NPC disease. However, the use of miglustat or methyl-β-cyclodextrin, which inhibit cholesterol synthesis or reduce its accumulation, respectively, can only alleviate symptoms somewhat, but cannot sustainably halt the progression of the disease [192,193,194,195,196]. Given the overlap between AD and NPC pathology, including Aß plaque formation, tau accumulation, and early neuroinflammation (activation of microglia), future therapeutic options against NPC may benefit from those already tested in AD.
4. Conclusions
With their complex genetic and environmental aetiology and an ever-increasing incidence in modern societies, NDDs remain one of the leading medical challenges. Given an extensive overlap in many adult-onset neurodegenerative diseases, particularly AD, PD, ALS and FTD, it is encouraging that many of the potential immunosuppressive and immunomodulatory treatments directly targeting immune mediators have been studied across different NDDs. A prerequisite for moving forward were two paradigm shifts regarding immunity in NDDs, that is that immune mechanisms are not merely noxious but also protective, and that inflammation likely plays a role as a trigger not only a distal element but can contribute to early pathogenesis of the disease. The first resulted in a deeper understanding of mechanisms and designing more nuanced targeted therapies instead of using broadly acting immunosuppressants, whereas the second will in time perhaps allow us to focus more on prevention and vaccinations to mitigate the risk for NDDs. Common targets for immunomodulatory treatment are of specific interest, and it is encouraging that some have also shown promising results across the NDD spectrum (Tregs, complement, etc). In conclusion, given the multifactorial nature, the numerous disease mechanisms, and the overlapping proteinopathies in the different NDDs, combining treatments acting on different disease pathways may allow an integrated and synergic disease-management intervention, personalized on a single patient.
Author Contributions
Conceptualization: SH, LM, FDM and IM; original draft preparation and editing: FDM, LV, EP, VR, JN, IJ, BR, VV, RML, JRC, EB, LM, IM and SH. All authors have read and agreed to the published version of the manuscript.
Funding
I.M.: this work was supported by the Croatian Science Foundation IP-2018-01-8563 and the University of Rijeka grant 18-211-1369. F.D.M., L.M.: No dedicated funding. F.D.M., L.M.: This work was supported by the AGING Project for the Department of Excellence at the Department of Translational Medicine (DIMET), Università del Pie-monte Orientale, Novara, Italy. B.R. and J.N. were supported by the Slovenian Research Agency (grant numbers N3-0141, J3-9263, J3-4503, J3-3065, and P4-0127). S.H.: This work was supported by the Croatian Science Foundation project neuroNiPiC (grant number IP-2016-06-2799) and the Hubert Curien “COGITO” programme (grant number 57794).
Institutional Review Board Statement
N/A.
Informed Consent Statement
N/A.
Data Availability Statement
N/A.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Schwartz, M.; Deczkowska, A. Neurological Disease as a Failure of Brain–Immune Crosstalk: The Multiple Faces of Neuroinflammation. Trends Immunol. 2016, 37, 668–679. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Beers, D.R.; Appel, S.H. Immune dysregulation in amyotrophic lateral sclerosis: mechanisms and emerging therapies. Lancet Neurol. 2019, 18, 211–220. [Google Scholar] [CrossRef]
- Béland, L.-C.; Markovinovic, A.; Jakovac, H.; De Marchi, F.; Bilic, E.; Mazzini, L.; Kriz, J.; Munitic, I. Immunity in amyotrophic lateral sclerosis: blurred lines between excessive inflammation and inefficient immune responses. Brain Commun. 2020, 2, fcaa124. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, F.; Franjkic, T.; Schito, P.; Russo, T.; Nimac, J.; Chami, A.A.; Mele, A.; Vidatic, L.; Kriz, J.; Julien, J.-P.; et al. Emerging Trends in the Field of Inflammation and Proteinopathy in ALS/FTD Spectrum Disorder. Biomedicines 2023, 11, 1599. [Google Scholar] [CrossRef]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.F.; Maloney, J.; et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Liscic, R.M.; Storandt, M.; Cairns, N.J.; Morris, J.C. Clinical and Psychometric Distinction of Frontotemporal and Alzheimer Dementias. Arch. Neurol. 2007, 64, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.F.; Selwood, A.; Mastri, A.R.; Frey, W.H. 2d Pick’s Disease versus Alzheimer’s Disease: A Comparison of Clinical Characteristics. Neurology 1993, 43, 289. [Google Scholar] [CrossRef]
- Neumann, M.; Lee, E.B.; Mackenzie, I.R. Frontotemporal Lobar Degeneration TDP-43-Immunoreactive Pathological Subtypes: Clinical and Mechanistic Significance. Adv Exp Med Biol 2021, 1281, 201–217. [Google Scholar] [CrossRef]
- Filippi, M.; Agosta, F.; Ferraro, P.M. Charting Frontotemporal Dementia: From Genes to Networks. J. Neuroimaging 2015, 26, 16–27. [Google Scholar] [CrossRef]
- Liscic, R.M.; Alberici, A.; Cairns, N.J.; Romano, M.; Buratti, E. From basic research to the clinic: innovative therapies for ALS and FTD in the pipeline. Mol. Neurodegener. 2020, 15, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.J.; Patterson, M.C.; Dambrosia, J.M.; Pikus, A.T.; Pentchev, P.G.; Sato, S.; Brady, R.O.; Barton, N.W. A clinical staging classification for type C Niemann-Pick disease. Neurology 1992, 42, 2286–2286. [Google Scholar] [CrossRef] [PubMed]
- Vanier, M.T. Niemann–Pick Diseases. Handb Clin Neurol 2013, 113, 1717–1721. [Google Scholar] [PubMed]
- Saito, Y.; Suzuki, K.; Nanba, E.; Yamamoto, T.; Ohno, K.; Murayama, S. Niemann–Pick Type C Disease: Accelerated Neurofibrillary Tangle Formation and Amyloid β Deposition Associated with Apolipoprotein E Ε4 Homozygosity. Ann Neurol 2002, 52, 351–355. [Google Scholar] [CrossRef]
- Distl, R.; Treiber-Held, S.; Albert, F.; Meske, V.; Harzer, K.; Ohm, T.G. Cholesterol Storage and Tau Pathology in Niemann–Pick Type C Disease in the Brain. The Journal of Pathology: A Journal of the Pathological Society of Great Britain and Ireland 2003, 200, 104–111. [Google Scholar] [CrossRef]
- Treiber-Held, S.; Distl, R.; Meske, V.; Albert, F.; Ohm, T.G. Spatial and Temporal Distribution of Intracellular Free Cholesterol in Brains of a Niemann–Pick Type C Mouse Model Showing Hyperphosphorylated Tau Protein. Implications for Alzheimer’s Disease. The Journal of Pathology: A Journal of the Pathological Society of Great Britain and Ireland. 2003, 200, 95–103. [Google Scholar]
- Platt, N.; Speak, A.O.; Colaco, A.; Gray, J.; Smith, D.A.; Williams, I.M.; Wallom, K.; Platt, F.M. Immune dysfunction in Niemann-Pick disease type C. J. Neurochem. 2015, 136, 74–80. [Google Scholar] [CrossRef]
- Gratwicke, J.; Jahanshahi, M.; Foltynie, T. Parkinson’s disease dementia: a neural networks perspective. Brain 2015, 138, 1454–1476. [Google Scholar] [CrossRef]
- Dag, A.; Lucia, B.; Halliday, G.M.; Geurtsen, G.J.; Ballard, C.; Ray, C.K.; Weintraub, D. Parkinson Disease-Associated Cognitive Impairment (Primer). Nat Rev Dis Primers 2021, 7. [Google Scholar]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Mead, R.J.; Shan, N.; Reiser, H.J.; Marshall, F.; Shaw, P.J. Amyotrophic lateral sclerosis: a neurodegenerative disorder poised for successful therapeutic translation. Nat. Rev. Drug Discov. 2022, 22, 185–212. [Google Scholar] [CrossRef] [PubMed]
- Lutshumba, J.; Nikolajczyk, B.S.; Bachstetter, A.D. Dysregulation of Systemic Immunity in Aging and Dementia. Front. Cell. Neurosci. 2021, 15. [Google Scholar] [CrossRef] [PubMed]
- Grossman, M.; Seeley, W.W.; Boxer, A.L.; Hillis, A.E.; Knopman, D.S.; Ljubenov, P.A.; Miller, B.; Piguet, O.; Rademakers, R.; Whitwell, J.L. Frontotemporal Lobar Degeneration. Nat Rev Dis Primers 2023, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Öberg, M.; Fabrik, I.; Fabrikova, D.; Zehetner, N.; Härtlova, A. The role of innate immunity and inflammation in Parkinson´s disease. Scand. J. Immunol. 2021, 93, e13022. [Google Scholar] [CrossRef]
- Perry, V.H. Innate Inflammation in Parkinson’s Disease. Cold Spring Harb Perspect Med 2: A009373 2012.
- Phani, S.; Loike, J.D.; Przedborski, S. Neurodegeneration and Inflammation in Parkinson's disease. Park. Relat. Disord. 2012, 18, S207–S209. [Google Scholar] [CrossRef]
- Standaert, D.G.; Harms, A.S.; Childers, G.M.; Webster, J.M. Disease mechanisms as subtypes: Inflammation in Parkinson disease and related disorders. 2023, 95–106. [CrossRef]
- Henderson, R.D.; Kepp, K.P.; Eisen, A. ALS/FTD: Evolution, Aging, and Cellular Metabolic Exhaustion. Front. Neurol. 2022, 13, 890203. [Google Scholar] [CrossRef]
- Franceschi, C.; Zaikin, A.; Gordleeva, S.; Ivanchenko, M.; Bonifazi, F.; Storci, G.; Bonafè, M. Inflammaging 2018: An Update and a Model. In Proceedings of the Seminars in immunology; 2018; Vol. 40, pp. 1–5.
- Kovacs, G.G. Are Comorbidities Compatible with a Molecular Pathological Classification of Neurodegenerative Diseases? Curr Opin Neurol 2019, 32, 279–291. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Distl, R.; Treiber-Held, S.; Albert, F.; Meske, V.; Harzer, K.; Ohm, T.G. Cholesterol Storage and Tau Pathology in Niemann–Pick Type C Disease in the Brain. The Journal of Pathology: A Journal of the Pathological Society of Great Britain and Ireland 2003, 200, 104–111. [Google Scholar] [CrossRef]
- Treiber-Held, S.; Distl, R.; Meske, V.; Albert, F.; Ohm, T.G. Spatial and Temporal Distribution of Intracellular Free Cholesterol in Brains of a Niemann–Pick Type C Mouse Model Showing Hyperphosphorylated Tau Protein. Implications for Alzheimer’s Disease. The Journal of Pathology: A Journal of the Pathological Society of Great Britain and Ireland 2003, 200, 95–103. 2003, 200, 95–103. [Google Scholar]
- Beach, T.G.; Malek-Ahmadi, M. Alzheimer’s Disease Neuropathological Comorbidities are Common in the Younger-Old. J. Alzheimer's Dis. 2021, 79, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Riku, Y.; Yoshida, M.; Iwasaki, Y.; Sobue, G.; Katsuno, M.; Ishigaki, S. TDP-43 Proteinopathy and Tauopathy: Do They Have Pathomechanistic Links? Int J Mol Sci 2022, 23, 15755. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.A.; Murray, M.E.; Whitwell, J.L.; Tosakulwong, N.; Weigand, S.D.; Petrucelli, L.; Liesinger, A.M.; Petersen, R.C.; Parisi, J.E.; Dickson, D.W. Updated TDP-43 in Alzheimer’s disease staging scheme. Acta Neuropathol. 2016, 131, 571–585. [Google Scholar] [CrossRef] [PubMed]
- Moussaud, S.; Jones, D.R.; Moussaud-Lamodière, E.L.; Delenclos, M.; Ross, O.A.; McLean, P.J. Alpha-Synuclein and Tau: Teammates in Neurodegeneration? Mol Neurodegener 2014, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Twohig, D.; Nielsen, H.M. α-Synuclein in the Pathophysiology of Alzheimer’s Disease. Mol Neurodegener 2019, 14, 1–19. [Google Scholar] [CrossRef]
- Waxman, E.A.; Giasson, B.I. Induction of Intracellular Tau Aggregation Is Promoted by α-Synuclein Seeds and Provides Novel Insights into the Hyperphosphorylation of Tau. J. Neurosci. 2011, 31, 7604–7618. [Google Scholar] [CrossRef]
- Oikawa, T.; Nonaka, T.; Terada, M.; Tamaoka, A.; Hisanaga, S.-I.; Hasegawa, M. α-Synuclein Fibrils Exhibit Gain of Toxic Function, Promoting Tau Aggregation and Inhibiting Microtubule Assembly. J. Biol. Chem. 2016, 291, 15046–15056. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.-E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef]
- Escartin, C.; Galea, E.; Lakatos, A.; O’callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Su, C.; Zhao, K.; Xia, H.; Xu, Y. Peripheral inflammatory biomarkers in Alzheimer's disease and mild cognitive impairment: a systematic review and meta-analysis. Psychogeriatrics 2019, 19, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Abdi, I.Y.; Ghanem, S.S.; El-Agnaf, O.M. Immune-related biomarkers for Parkinson's disease. Neurobiol. Dis. 2022, 170, 105771. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, F.; Tondo, G.; Corrado, L.; Menegon, F.; Aprile, D.; Anselmi, M.; D’alfonso, S.; Comi, C.; Mazzini, L. Neuroinflammatory Pathways in the ALS-FTD Continuum: A Focus on Genetic Variants. Genes 2023, 14, 1658. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, M.; Andrews, S.J.; Tripathy, S.J. Shared Genetic Risk Loci between Alzheimer’s Disease and Related Dementias, Parkinson’s Disease, and Amyotrophic Lateral Sclerosis. Alzheimers Res Ther 2023, 15, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Griciuc, A.; Patel, S.; Federico, A.N.; Choi, S.H.; Innes, B.J.; Oram, M.K.; Cereghetti, G.; McGinty, D.; Anselmo, A.; Sadreyev, R.I.; et al. TREM2 Acts Downstream of CD33 in Modulating Microglial Pathology in Alzheimer’s Disease. Neuron 2019, 103, 820–835. [Google Scholar] [CrossRef]
- Root, J.; Merino, P.; Nuckols, A.; Johnson, M.; Kukar, T. Lysosome dysfunction as a cause of neurodegenerative diseases: Lessons from frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol. Dis. 2021, 154, 105360–105360. [Google Scholar] [CrossRef]
- Gulen, M.F.; Samson, N.; Keller, A.; Schwabenland, M.; Liu, C.; Glück, S.; Thacker, V.V.; Favre, L.; Mangeat, B.; Kroese, L.J.; et al. cGAS–STING drives ageing-related inflammation and neurodegeneration. Nature 2023, 620, 374–380. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Alexopoulos, H.; Spaeth, P.J. Complement in neurological disorders and emerging complement-targeted therapeutics. Nat. Rev. Neurol. 2020, 16, 601–617. [Google Scholar] [CrossRef]
- Michailidou, I.; Fluiter, K.; Boziki, M.; Grigoriadis, N.; Baas, F. Editorial: Complement in nervous system disease. Front. Cell. Neurosci. 2023, 17, 1268023. [Google Scholar] [CrossRef]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Butler, C.A.; Popescu, A.S.; Kitchener, E.J.A.; Allendorf, D.H.; Puigdellívol, M.; Brown, G.C. Microglial Phagocytosis of Neurons in Neurodegeneration, and Its Regulation. J Neurochem 2021, 158, 621–639. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J. The role of peripheral immune cells in the CNS in steady state and disease. Nat. Neurosci. 2017, 20, 136–144. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and Microglial Activation in Alzheimer Disease: Where Do We Go from Here? Nat Rev Neurol 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Cao, W.; Cao, Z.; Tian, Y.; Zhang, L.; Wang, W.; Tang, L.; Xu, C.; Fan, D. Neutrophils are Associated with Higher Risk of Incident Amyotrophic Lateral Sclerosis in a BMI- and Age-dependent Manner. Ann. Neurol. 2023. [Google Scholar] [CrossRef]
- Tondo, G.; Aprile, D.; De Marchi, F.; Sarasso, B.; Serra, P.; Borasio, G.; Rojo, E.; Arenillas, J.F.; Comi, C. Investigating the Prognostic Role of Peripheral Inflammatory Markers in Mild Cognitive Impairment. J. Clin. Med. 2023, 12, 4298. [Google Scholar] [CrossRef]
- A Hoffman, L.; A Vilensky, J. Encephalitis lethargica: 100 years after the epidemic. Brain 2017, 140, 2246–2251. [Google Scholar] [CrossRef]
- Alfahad, T.; Nath, A. Retroviruses and amyotrophic lateral sclerosis. Antivir. Res. 2013, 99, 180–187. [Google Scholar] [CrossRef]
- Küry, P.; Nath, A.; Créange, A.; Dolei, A.; Marche, P.; Gold, J.; Giovannoni, G.; Hartung, H.-P.; Perron, H. Human Endogenous Retroviruses in Neurological Diseases. Trends Mol. Med. 2018, 24, 379–394. [Google Scholar] [CrossRef]
- Douville, R.; Liu, J.; Rothstein, J.; Nath, A. Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann. Neurol. 2011, 69, 141–151. [Google Scholar] [CrossRef]
- Marcocci, M.E.; Napoletani, G.; Protto, V.; Kolesova, O.; Piacentini, R.; Puma, D.D.L.; Lomonte, P.; Grassi, C.; Palamara, A.T.; De Chiara, G. Herpes Simplex Virus-1 in the Brain: The Dark Side of a Sneaky Infection. Trends Microbiol. 2020, 28, 808–820. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhou, Z. COVID-19 and the Risk of Alzheimer’s Disease, Amyotrophic Lateral Sclerosis, and Multiple Sclerosis. Ann Clin Transl Neurol 2022, 9, 1953–1961. [Google Scholar] [CrossRef] [PubMed]
- Levine, K.S.; Leonard, H.L.; Blauwendraat, C.; Iwaki, H.; Johnson, N.; Bandres-Ciga, S.; Ferrucci, L.; Faghri, F.; Singleton, A.B.; Nalls, M.A. Virus exposure and neurodegenerative disease risk across national biobanks. Neuron 2023, 111, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Sipilä, P.N.; Heikkilä, N.; Lindbohm, J. V; Hakulinen, C.; Vahtera, J.; Elovainio, M.; Suominen, S.; Väänänen, A.; Koskinen, A.; Nyberg, S.T. Hospital-Treated Infectious Diseases and the Risk of Dementia: A Large, Multicohort, Observational Study with a Replication Cohort. Lancet Infect Dis 2021, 21, 1557–1567. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, Y.; Wang, Z.; Xie, G.; Liu, M.; Yuan, B.; Chai, H.; Wang, W.; Cheng, P. Implications of Gut Microbiota in Neurodegenerative Diseases. Front. Immunol. 2022, 13, 785644. [Google Scholar] [CrossRef] [PubMed]
- Di Gioia, D.; Cionci, N.B.; Baffoni, L.; Amoruso, A.; Pane, M.; Mogna, L.; Gaggìa, F.; Lucenti, M.A.; Bersano, E.; Cantello, R.; et al. A prospective longitudinal study on the microbiota composition in amyotrophic lateral sclerosis. BMC Med. 2020, 18, 153. [Google Scholar] [CrossRef]
- Papić, E.; Rački, V.; Hero, M.; Tomić, Z.; Starčević-Čižmarević, N.; Kovanda, A.; Kapović, M.; Hauser, G.; Peterlin, B.; Vuletić, V. The effects of microbiota abundance on symptom severity in Parkinson’s disease: A systematic review. Front. Aging Neurosci. 2022, 14, 1020172. [Google Scholar] [CrossRef]
- Fülling, C.; Dinan, T.G.; Cryan, J.F. Gut Microbe to Brain Signaling: What Happens in Vagus…. Neuron 2019, 101, 998–1002. [Google Scholar] [CrossRef]
- Erny, D.; Hrabě de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef]
- Fung, T.C.; Vuong, H.E.; Luna, C.D.G.; Pronovost, G.N.; Aleksandrova, A.A.; Riley, N.G.; Vavilina, A.; McGinn, J.; Rendon, T.; Forrest, L.R.; et al. Intestinal serotonin and fluoxetine exposure modulate bacterial colonization in the gut. Nat. Microbiol. 2019, 4, 2064–2073. [Google Scholar] [CrossRef]
- Kennedy, P.J.; Cryan, J.F.; Dinan, T.G.; Clarke, G. Kynurenine pathway metabolism and the microbiota-gut-brain axis. Neuropharmacology 2017, 112, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Mazmanian, S.K. Innate immune recognition of the microbiota promotes host-microbial symbiosis. Nat. Immunol. 2013, 14, 668–675. [Google Scholar] [CrossRef]
- Burberry, A.; Wells, M.F.; Limone, F.; Couto, A.; Smith, K.S.; Keaney, J.; Gillet, G.; van Gastel, N.; Wang, J.-Y.; Pietilainen, O.; et al. C9orf72 suppresses systemic and neural inflammation induced by gut bacteria. Nature 2020, 582, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pardo, P.; Dodiya, H.B.; Engen, P.A.; Forsyth, C.B.; Huschens, A.M.; Shaikh, M.; Voigt, R.M.; Naqib, A.; Green, S.J.; Kordower, J.H.; et al. Role of TLR4 in the gut-brain axis in Parkinson’s disease: a translational study from men to mice. Gut 2019, 68, 829–843. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, F.; Munitic, I.; Amedei, A.; Berry, J.D.; Feldman, E.L.; Aronica, E.; Nardo, G.; Van Weehaeghe, D.; Niccolai, E.; Prtenjaca, N.; et al. Interplay between immunity and amyotrophic lateral sclerosis: Clinical impact. Neurosci. Biobehav. Rev. 2021, 127, 958–978. [Google Scholar] [CrossRef]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef]
- Lambert, J.-C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; Jun, G.; DeStefano, A.L.; Bis, J.C.; Beecham, G.W. Meta-Analysis of 74,046 Individuals Identifies 11 New Susceptibility Loci for Alzheimer’s Disease. Nat Genet 2013, 45, 1452–1458. [Google Scholar] [CrossRef]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S. Alzheimer Genetic Analysis G (2013) TREM2 Variants in Alzheimer’s Disease. N Engl J Med 2013, 368, 117–127. [Google Scholar] [CrossRef]
- Jonsson, T.; Stefansson, K. TREM2 and Neurodegenerative Disease. N Engl J Med 2013, 369, 1568–1569. [Google Scholar]
- Huang, K.; Marcora, E.; Pimenova, A.A.; Di Narzo, A.F.; Kapoor, M.; Jin, S.C.; Harari, O.; Bertelsen, S.; Fairfax, B.P.; Czajkowski, J. A Common Haplotype Lowers PU. 1 Expression in Myeloid Cells and Delays Onset of Alzheimer’s Disease. Nat Neurosci 2017, 20, 1052–1061. [Google Scholar]
- Hardy, J.A.; Higgins, G.A. Alzheimer's disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s Disease Risk Gene CD33 Inhibits Microglial Uptake of Amyloid Beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Thambisetty, M.; Beason-Held, L.L.; An, Y.; Kraut, M.; Nalls, M.; Hernandez, D.G.; Singleton, A.B.; Zonderman, A.B.; Ferrucci, L.; Lovestone, S.; et al. Alzheimer Risk Variant CLU and Brain Function During Aging. Biol. Psychiatry 2013, 73, 399–405. [Google Scholar] [CrossRef]
- Kleinberger, G.; Yamanishi, Y.; Suárez-Calvet, M.; Czirr, E.; Lohmann, E.; Cuyvers, E.; Struyfs, H.; Pettkus, N.; Wenninger-Weinzierl, A.; Mazaheri, F.; et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 2014, 6, 243ra86–243ra86. [Google Scholar] [CrossRef]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 Lipid Sensing Sustains the Microglial Response in an Alzheimer’s Disease Model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef]
- Gerrits, E.; Brouwer, N.; Kooistra, S.M.; Woodbury, M.E.; Vermeiren, Y.; Lambourne, M.; Mulder, J.; Kummer, M.; Möller, T.; Biber, K.; et al. Distinct amyloid-β and tau-associated microglia profiles in Alzheimer’s disease. Acta Neuropathol. 2021, 141, 681–696. [Google Scholar] [CrossRef]
- Solito, E.; Sastre, M. Microglia Function in Alzheimer’s Disease. Front. Pharmacol. 2012, 3, 14. [Google Scholar] [CrossRef]
- Orre, M.; Kamphuis, W.; Osborn, L.M.; Melief, J.; Kooijman, L.; Huitinga, I.; Klooster, J.; Bossers, K.; Hol, E.M. Acute isolation and transcriptome characterization of cortical astrocytes and microglia from young and aged mice. Neurobiol. Aging 2014, 35, 1–14. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Deane, R.; Sagare, A.; Zlokovic, B. V The Role of the Cell Surface LRP and Soluble LRP in Blood-Brain Barrier Aβ Clearance in Alzheimer’s Disease. Curr Pharm Des 2008, 14, 1601–1605. [Google Scholar] [CrossRef] [PubMed]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human apoE Isoforms Differentially Regulate Brain Amyloid-β Peptide Clearance. Sci. Transl. Med. 2011, 3, 89ra57–89ra57. [Google Scholar] [CrossRef] [PubMed]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290. [Google Scholar] [CrossRef]
- Rujescu, D.; Jung, M.; S, K.; C, M.; R, C.; C, B.; N, C.; R, E.F.; L, B.; E, O.; et al. Faculty Opinions recommendation of The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. . 2020, 47. [Google Scholar] [CrossRef]
- Parhizkar, S.; Arzberger, T.; Brendel, M.; Kleinberger, G.; Deussing, M.; Focke, C.; Nuscher, B.; Xiong, M.; Ghasemigharagoz, A.; Katzmarski, N.; et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 2019, 22, 191–204. [Google Scholar] [CrossRef]
- Parhizkar, S.; Holtzman, D.M. APOE mediated neuroinflammation and neurodegeneration in Alzheimer’s disease. Semin. Immunol. 2022, 59, 101594–101594. [Google Scholar] [CrossRef]
- Daria, A.; Colombo, A.; Llovera, G.; Hampel, H.; Willem, M.; Liesz, A.; Haass, C.; Tahirovic, S. Young microglia restore amyloid plaque clearance of aged microglia. EMBO J. 2016, 36, 583–603. [Google Scholar] [CrossRef]
- Moir, R.D.; Lathe, R.; Tanzi, R.E. The antimicrobial protection hypothesis of Alzheimer's disease. Alzheimer's Dement. 2018, 14, 1602–1614. [Google Scholar] [CrossRef]
- Zubair, A.S.; McAlpine, L.S.; Gardin, T.; Farhadian, S.; Kuruvilla, D.E.; Spudich, S. Neuropathogenesis and Neurologic Manifestations of the Coronaviruses in the Age of Coronavirus Disease 2019. JAMA Neurol. 2020, 77, 1018–1027. [Google Scholar] [CrossRef]
- Rai, S.N.; Tiwari, N.; Singh, P.; Singh, A.K.; Mishra, D.; Imran, M.; Singh, S.; Hooshmandi, E.; Vamanu, E.; Singh, S.K.; et al. Exploring the Paradox of COVID-19 in Neurological Complications with Emphasis on Parkinson’s and Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2022, 2022, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sipilä, P.N.; Heikkilä, N.; Lindbohm, J. V; Hakulinen, C.; Vahtera, J.; Elovainio, M.; Suominen, S.; Väänänen, A.; Koskinen, A.; Nyberg, S.T. Hospital-Treated Infectious Diseases and the Risk of Dementia: A Large, Multicohort, Observational Study with a Replication Cohort. Lancet Infect Dis 2021, 21, 1557–1567. [Google Scholar] [CrossRef] [PubMed]
- Carstea, E.D.; Morris, J.A.; Coleman, K.G.; Loftus, S.K.; Zhang, D.; Cummings, C.; Gu, J.; Rosenfeld, M.A.; Pavan, W.J.; Krizman, D.B.; et al. Niemann-Pick C1 Disease Gene: Homology to Mediators of Cholesterol Homeostasis. Science 1997, 277, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Karten, B.; Peake, K.B.; Vance, J.E. Mechanisms and consequences of impaired lipid trafficking in Niemann–Pick type C1-deficient mammalian cells. Biochim. et Biophys. Acta (BBA) - Mol. Cell Biol. Lipids 2009, 1791, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Wu, X.; Du, X.; Yao, X.; Zhao, X.; Lee, J.; Yang, H.; Yan, N. Structural Basis of Low-PH-Dependent Lysosomal Cholesterol Egress by NPC1 and NPC2. Cell 2020, 182, 98–111. [Google Scholar] [CrossRef]
- Loftus, S.K.; Erickson, R.P.; Walkley, S.U.; Bryant, M.A.; Incao, A.; Heidenreich, R.A.; Pavan, W.J. Rescue of neurodegeneration in Niemann-Pick C mice by a prion-promoter-driven Npc1 cDNA transgene. Hum. Mol. Genet. 2002, 11, 3107–3114. [Google Scholar] [CrossRef]
- Chen, G.; Li, H.-M.; Chen, Y.-R.; Gu, X.-S.; Duan, S. Decreased estradiol release from astrocytes contributes to the neurodegeneration in a mouse model of Niemann-Pick disease type C. Glia 2007, 55, 1509–1518. [Google Scholar] [CrossRef]
- Lopez, M.E.; Klein, A.D.; Dimbil, U.J.; Scott, M.P. Anatomically Defined Neuron-Based Rescue of Neurodegenerative Niemann–Pick Type C Disorder. J. Neurosci. 2011, 31, 4367–4378. [Google Scholar] [CrossRef]
- Marshall, C.A.; Watkins-Chow, D.E.; Palladino, G.; Deutsch, G.; Chandran, K.; Pavan, W.J.; Erickson, R.P. In Niemann-Pick C1 mouse models, glial-only expression of the normal gene extends survival much further than do changes in genetic background or treatment with hydroxypropyl-beta-cyclodextrin. Gene 2018, 643, 117–123. [Google Scholar] [CrossRef]
- Zhang, Q.-G.; Wang, R.; Khan, M.; Mahesh, V.; Brann, D.W. Role of Dickkopf-1, an Antagonist of the Wnt/β-Catenin Signaling Pathway, in Estrogen-Induced Neuroprotection and Attenuation of Tau Phosphorylation. J. Neurosci. 2008, 28, 8430–8441. [Google Scholar] [CrossRef]
- Colombo, A.; Dinkel, L.; Müller, S.A.; Sebastian Monasor, L.; Schifferer, M.; Cantuti-Castelvetri, L.; König, J.; Vidatic, L.; Bremova-Ertl, T.; Lieberman, A.P. Loss of NPC1 Enhances Phagocytic Uptake and Impairs Lipid Trafficking in Microglia. Nat Commun 2021, 12, 1158. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290. [Google Scholar] [CrossRef] [PubMed]
- Heiden, D.L.; Monogue, B.; Ali, H.; Beckham, J.D. A functional role for alpha-synuclein in neuroimmune responses. J. Neuroimmunol. 2023, 376, 578047. [Google Scholar] [CrossRef] [PubMed]
- Deyell, J.S.; Sriparna, M.; Ying, M.; Mao, X. The Interplay between α-Synuclein and Microglia in α-Synucleinopathies. Int J Mol Sci 2023, 24, 2477. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; De Vos, R.A.I.; Steur, E.N.H.J.; Braak, E. Staging of Brain Pathology Related to Sporadic Parkinson’s Disease. Neurobiol Aging 2003, 24, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Sommerville, N.R.; Liu, J.Y.H.; Ngan, M.P.; Poon, D.; Ponomarev, E.D.; Lu, Z.; Kung, J.S.C.; Rudd, J.A. Intra-gastrointestinal Amyloid-β1–42 Oligomers Perturb Enteric Function and Induce Alzheimer’s Disease Pathology. J Physiol 2020, 598, 4209–4223. [Google Scholar] [CrossRef]
- Storelli, G.; Defaye, A.; Erkosar, B.; Hols, P.; Royet, J.; Leulier, F. Lactobacillus plantarum Promotes Drosophila Systemic Growth by Modulating Hormonal Signals through TOR-Dependent Nutrient Sensing. Cell Metab. 2011, 14, 403–414. [Google Scholar] [CrossRef]
- Wu, G.; Jiang, Z.; Pu, Y.; Chen, S.; Wang, T.; Wang, Y.; Xu, X.; Wang, S.; Jin, M.; Yao, Y.; et al. Serum short-chain fatty acids and its correlation with motor and non-motor symptoms in Parkinson’s disease patients. BMC Neurol. 2022, 22, 1–9. [Google Scholar] [CrossRef]
- Murros, K.E.; Huynh, V.A.; Takala, T.M.; Saris, P.E.J. Desulfovibrio Bacteria Are Associated With Parkinson’s Disease. Front. Cell. Infect. Microbiol. 2021, 11. [Google Scholar] [CrossRef]
- Reigstad, C.S.; Salmonson, C.E.; Rainey III, J.F.; Szurszewski, J.H.; Linden, D.R.; Sonnenburg, J.L.; Farrugia, G.; Kashyap, P.C. Gut Microbes Promote Colonic Serotonin Production through an Effect of Short-Chain Fatty Acids on Enterochromaffin Cells. The FASEB Journal 2015, 29, 1395. [Google Scholar] [CrossRef]
- Clarke, M.T.M.; Brinkmalm, A.; Foiani, M.S.; Woollacott, I.O.C.; Heller, C.; Heslegrave, A.; Keshavan, A.; Fox, N.C.; Schott, J.M.; Warren, J.D.; et al. CSF synaptic protein concentrations are raised in those with atypical Alzheimer’s disease but not frontotemporal dementia. Alzheimer's Res. Ther. 2019, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-H.; Chen, C.-C.; Chiang, H.-L.; Liou, J.-M.; Chang, C.-M.; Lu, T.-P.; Chuang, E.Y.; Tai, Y.-C.; Cheng, C.; Lin, H.-Y.; et al. Altered gut microbiota and inflammatory cytokine responses in patients with Parkinson’s disease. J. Neuroinflammation 2019, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Koscsó, B.; Rajani, G.M.; Stevanovic, K.; Berres, M.-L.; Hashimoto, D.; Mortha, A.; Leboeuf, M.; Li, X.-M.; Mucida, D.; et al. Crosstalk between Muscularis Macrophages and Enteric Neurons Regulates Gastrointestinal Motility. Cell 2014, 158, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.R.; Greenamyre, J.T. The Role of Environmental Exposures in Neurodegeneration and Neurodegenerative Diseases. Toxicol. Sci. 2011, 124, 225–250. [Google Scholar] [CrossRef]
- Fitzmaurice, A.G.; Rhodes, S.L.; Cockburn, M.; Ritz, B.; Bronstein, J.M. Aldehyde dehydrogenase variation enhances effect of pesticides associated with Parkinson disease. Neurology 2014, 82, 419–426. [Google Scholar] [CrossRef]
- Fitzmaurice, A.G.; Rhodes, S.L.; Lulla, A.; Murphy, N.P.; Lam, H.A.; O'Donnell, K.C.; Barnhill, L.; Casida, J.E.; Cockburn, M.; Sagasti, A.; et al. Aldehyde dehydrogenase inhibition as a pathogenic mechanism in Parkinson disease. Proc. Natl. Acad. Sci. USA 2013, 110, 636–641. [Google Scholar] [CrossRef]
- Gatto, N.M.; Cockburn, M.; Bronstein, J.; Manthripragada, A.D.; Ritz, B. Well-Water Consumption and Parkinson’s Disease in Rural California. Environ. Heal. Perspect. 2009, 117, 1912–1918. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Tarbutton, G.L.; Levin, J.L.; Plotkin, G.M.; Lowry, L.K.; Nalbone, J.T.; Shepherd, S. Pesticide/Environmental Exposures and Parkinson's Disease in East Texas. J. Agromedicine 2008, 13, 37–48. [Google Scholar] [CrossRef]
- Manthripragada, A.D.; Costello, S.; Cockburn, M.G.; Bronstein, J.M.; Ritz, B. Paraoxonase 1, Agricultural Organophosphate Exposure, and Parkinson Disease. Epidemiology 2010, 21, 87–94. [Google Scholar] [CrossRef]
- Porras, G.; Ruiz, S.; Maestro, I.; Borrego-Hernández, D.; Redondo, A.G.; Martínez, A.; Martín-Requero. Functional Characterization of a Familial ALS-Associated Missense TBK1 (p-Arg573Gly) Mutation in Patient-Derived Lymphoblasts. Int. J. Mol. Sci. 2023, 24, 2847. [Google Scholar] [CrossRef]
- Voorhees, J.R.; Remy, M.T.; Erickson, C.M.; Dutca, L.M.; Brat, D.J.; Pieper, A.A. Occupational-like organophosphate exposure disrupts microglia and accelerates deficits in a rat model of Alzheimer’s disease. npj Aging Mech. Dis. 2019, 5, 3. [Google Scholar] [CrossRef]
- Andrew, A.; Zhou, J.; Gui, J.; Harrison, A.; Shi, X.; Li, M.; Guetti, B.; Nathan, R.; Tischbein, M.; Pioro, E.P.; et al. Pesticides applied to crops and amyotrophic lateral sclerosis risk in the U.S. NeuroToxicology 2021, 87, 128–135. [Google Scholar] [CrossRef]
- Landers, J.E.; Shi, L.; Cho, T.-J.; Glass, J.D.; Shaw, C.E.; Leigh, P.N.; Diekstra, F.; Polak, M.; Rodriguez-Leyva, I.; Niemann, S.; et al. A common haplotype within the PON1 promoter region is associated with sporadic ALS. Amyotroph. Lateral Scler. 2008, 9, 306–314. [Google Scholar] [CrossRef]
- Arnold, G.E.; Manchester, J.I.; Townsend, B.D.; Ornstein, R.L. Investigation of Domain Motions in Bacteriophage T4 Lysozyme. J. Biomol. Struct. Dyn. 1994, 12, 457–474. [Google Scholar] [CrossRef]
- Markovinovic, A.; Cimbro, R.; Ljutic, T.; Kriz, J.; Rogelj, B.; Munitic, I. Optineurin in amyotrophic lateral sclerosis: Multifunctional adaptor protein at the crossroads of different neuroprotective mechanisms. Prog. Neurobiol. 2017, 154, 1–20. [Google Scholar] [CrossRef]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; DeStefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef]
- Lill, C.M.; Roehr, J.T.; McQueen, M.B.; Kavvoura, F.K.; Bagade, S.; Schjeide, B.-M.M.; Schjeide, L.M.; Meissner, E.; Zauft, U.; Allen, N.C.; et al. Comprehensive Research Synopsis and Systematic Meta-Analyses in Parkinson’s Disease Genetics: The PDGene Database. PLoS Genet. 2012, 8, e1002548. [Google Scholar] [CrossRef]
- Rocha, S.M.; Kirkley, K.S.; Chatterjee, D.; Aboellail, T.A.; Smeyne, R.J.; Tjalkens, R.B. Microglia-specific Knock-out of NF-κB/IKK2 Increases the Accumulation of Misfolded A-synuclein through the Inhibition of P62/Sequestosome-1-dependent Autophagy in the Rotenone Model of Parkinson’s Disease. Glia 2023. [CrossRef]
- Sengupta, U.; Kayed, R. Amyloid β, Tau, and α-Synuclein Aggregates in the Pathogenesis, Prognosis, and Therapeutics for Neurodegenerative Diseases. Prog Neurobiol 2022, 214, 102270. [Google Scholar] [CrossRef]
- I Arevalo-Villalobos, J.; Rosales-Mendoza, S.; Zarazua, S. Immunotherapies for neurodegenerative diseases: current status and potential of plant-made biopharmaceuticals. Expert Rev. Vaccines 2016, 16, 151–159. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y. The Antibody Aducanumab Reduces Aβ Plaques in Alzheimer’s Disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Cheng, L.; Zhang, J.; Li, X.-Y.; Yuan, L.; Pan, Y.-F.; Chen, X.-R.; Gao, T.-M.; Qiao, J.-T.; Qi, J.-S. A novel antibody targeting sequence 31-35 in amyloid β protein attenuates Alzheimer's disease-related neuronal damage. Hippocampus 2016, 27, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Minami, S.S.; Sidahmed, E.; Aid, S.; Shimoji, M.; Niikura, T.; Mocchetti, I.; Rebeck, G.W.; Prendergast, J.S.; Dealwis, C.; Wetzel, R.; et al. Therapeutic versus neuroinflammatory effects of passive immunization is dependent on Aβ/amyloid burden in a transgenic mouse model of Alzheimer's disease. J. Neuroinflammation 2010, 7, 57–57. [Google Scholar] [CrossRef] [PubMed]
- Haeberlein, S.B.; Aisen, P.; Barkhof, F.; Chalkias, S.; Chen, T.; Cohen, S.; Dent, G.; Hansson, O.; Harrison, K.; von Hehn, C.; et al. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. J. Prev. Alzheimer's Dis. 2022, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S. Lecanemab in Early Alzheimer’s Disease. New England Journal of Medicine 2023, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.C. The Clinical Dementia Rating (CDR): Current Version and Scoring Rules. Neurology 1993. [CrossRef] [PubMed]
- Bayer, A.J.; Bullock, R.; Jones, R.W.; Wilkinson, D.; Paterson, K.R.; Jenkins, L.; Millais, S.B.; Donoghue, S. Evaluation of the Safety and Immunogenicity of Synthetic Aβ42 (AN1792) in Patients with AD. Neurology 2005, 64, 94–101. [Google Scholar] [CrossRef]
- Paquet, C.; Nicoll, J.A.R.; Love, S.; Mouton-Liger, F.; Holmes, C.; Hugon, J.; Boche, D. Downregulated Apoptosis and Autophagy after Anti-Aβ Immunotherapy in Alzheimer’s Disease. Brain Pathology 2018, 28, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, J.A.R.; Buckland, G.R.; Harrison, C.H.; Page, A.; Harris, S.; Love, S.; Neal, J.W.; Holmes, C.; Boche, D. Persistent neuropathological effects 14 years following amyloid-β immunization in Alzheimer’s disease. Brain 2019, 142, 2113–2126. [Google Scholar] [CrossRef]
- van Dyck, C.; Sadowsky, C.; Leterme, G.L.P.; Booth, K.; Peng, Y.; Marek, K.; Ketter, N.; Liu, E.; Wyman, B.; Jackson, N.; et al. VANUTIDE CRIDIFICAR (ACC-001) AND QS-21 ADJUVANT IN INDIVIDUALS WITH EARLY ALZHEIMER’S DISEASE: AMYLOID IMAGING POSITRON EMISSION TOMOGRAPHY AND SAFETY RESULTS FROM A PHASE 2 STUDY. J. Prev. Alzheimer's Dis. [CrossRef]
- Goedert, M.; Wischik, C.M.; A Crowther, R.; E Walker, J.; Klug, A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc. Natl. Acad. Sci. 1988, 85, 4051–4055. [Google Scholar] [CrossRef]
- Brier, M.R.; Gordon, B.; Friedrichsen, K.; McCarthy, J.; Stern, A.; Christensen, J.; Owen, C.; Aldea, P.; Su, Y.; Hassenstab, J. Tau and Aβ Imaging, CSF Measures, and Cognition in Alzheimer’s Disease. Sci Transl Med 2016, 8, 338ra66–338ra66. [Google Scholar] [CrossRef] [PubMed]
- Katsinelos, T.; Tuck, B.J.; Mukadam, A.S.; McEwan, W.A. The Role of Antibodies and Their Receptors in Protection Against Ordered Protein Assembly in Neurodegeneration. Front. Immunol. 2019, 10, 1139. [Google Scholar] [CrossRef] [PubMed]
- Novak, P.; Kovacech, B.; Katina, S.; Schmidt, R.; Scheltens, P.; Kontsekova, E.; Ropele, S.; Fialova, L.; Kramberger, M.; Paulenka-Ivanovova, N. ADAMANT: A Placebo-Controlled Randomized Phase 2 Study of AADvac1, an Active Immunotherapy against Pathological Tau in Alzheimer’s Disease. Nat Aging 2021, 1, 521–534. [Google Scholar] [CrossRef]
- Huang, Y.-R.; Xie, X.-X.; Ji, M.; Yu, X.-L.; Zhu, J.; Zhang, L.-X.; Liu, X.-G.; Wei, C.; Li, G.; Liu, R.-T. Naturally occurring autoantibodies against α-synuclein rescues memory and motor deficits and attenuates α-synuclein pathology in mouse model of Parkinson's disease. Neurobiol. Dis. 2018, 124, 202–217. [Google Scholar] [CrossRef]
- Pagano, G.; Taylor, K.I.; Anzures-Cabrera, J.; Marchesi, M.; Simuni, T.; Marek, K.; Postuma, R.B.; Pavese, N.; Stocchi, F.; Azulay, J.-P.; et al. Trial of Prasinezumab in Early-Stage Parkinson’s Disease. New Engl. J. Med. 2022, 387, 421–432. [Google Scholar] [CrossRef]
- Brys, M.; Fanning, L.; Hung, S.; Ellenbogen, A.; Penner, N.; Yang, M.; Welch, M.; Koenig, E.; David, E.; Fox, T. Randomized Phase I Clinical Trial of Anti–A-synuclein Antibody BIIB054. Movement Disorders 2019, 34, 1154–1163. [Google Scholar] [CrossRef]
- Bae, E.-J.; Lee, H.-J.; Rockenstein, E.; Ho, D.-H.; Park, E.-B.; Yang, N.-Y.; Desplats, P.; Masliah, E.; Lee, S.-J. Antibody-Aided Clearance of Extracellular α-Synuclein Prevents Cell-to-Cell Aggregate Transmission. J. Neurosci. 2012, 32, 13454–13469. [Google Scholar] [CrossRef]
- Investigators, H.S.G.R. Safety, Tolerability, and Efficacy of PBT2 in Huntington’s Disease: A Phase 2, Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Neurol 2015, 14, 39–47. [Google Scholar]
- Poulin-Brière, A.; Rezaei, E.; Pozzi, S. Antibody-Based Therapeutic Interventions for Amyotrophic Lateral Sclerosis: A Systematic Literature Review. Front. Neurosci. 2021, 15. [Google Scholar] [CrossRef]
- Milligan, C.; Atassi, N.; Babu, S.; Barohn, R.J.; Caress, J.B.; Cudkowicz, M.E.; Evora, A.; Hawkins, G.A.; Wosiski-Kuhn, M.; Macklin, E.A. Tocilizumab Is Safe and Tolerable and Reduces C-reactive Protein Concentrations in the Plasma and Cerebrospinal Fluid of ALS Patients. Muscle Nerve 2021, 64, 309–320. [Google Scholar] [CrossRef]
- Meininger, V.; Genge, A.; Berg, L.H.v.D.; Robberecht, W.; Ludolph, A.; Chio, A.; Kim, S.H.; Leigh, P.N.; Kiernan, M.C.; Shefner, J.M.; et al. Safety and efficacy of ozanezumab in patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol. 2017, 16, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Group, A.-F.R. Follow-up Evaluation of Cognitive Function in the Randomized Alzheimer’s Disease Anti-Inflammatory Prevention Trial and Its Follow-up Study. Alzheimer’s & Dementia 2015, 11, 216–225. [Google Scholar]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS Clinical Trials Review: 20 Years of Failure. Are We Any Closer to Registering a New Treatment? Front Aging Neurosci 2017, 9, 68. [Google Scholar] [PubMed]
- Butchart, J.; Brook, L.; Hopkins, V.; Teeling, J.; Püntener, U.; Culliford, D.; Sharples, R.; Sharif, S.; McFarlane, B.; Raybould, R.; et al. Etanercept in Alzheimer disease: A randomized, placebo-controlled, double-blind, phase 2 trial. Neurology 2015, 84, 2161–2168. [Google Scholar] [CrossRef]
- Alves, S.; Churlaud, G.; Audrain, M.; Michaelsen-Preusse, K.; Fol, R.; Souchet, B.; Braudeau, J.; Korte, M.; Klatzmann, D.; Cartier, N. Interleukin-2 Improves Amyloid Pathology, Synaptic Failure and Memory in Alzheimer’s Disease Mice. Brain 2017, 140, 826–842. [Google Scholar] [CrossRef]
- Kiyota, T.; Machhi, J.; Lu, Y.; Dyavarshetty, B.; Nemati, M.; Yokoyama, I.; Mosley, R.; Gendelman, H.E. Granulocyte-macrophage colony-stimulating factor neuroprotective activities in Alzheimer’s disease mice. J. Neuroimmunol. 2018, 319, 80–92. [Google Scholar] [CrossRef]
- Potter, H.; Woodcock, J.H.; Boyd, T.D.; Coughlan, C.M.; O'Shaughnessy, J.R.; Borges, M.T.; Thaker, A.A.; Raj, B.A.; Adamszuk, K.; Scott, D.; et al. Safety and efficacy of sargramostim (GM-CSF) in the treatment of Alzheimer's disease. Alzheimer's Dementia: Transl. Res. Clin. Interv. 2021, 7, e12158. [Google Scholar] [CrossRef]
- Gendelman, H.E.; Zhang, Y.; Santamaria, P.; Olson, K.E.; Schutt, C.R.; Bhatti, D.; Shetty, B.L.D.; Lu, Y.; Estes, K.A.; Standaert, D.G.; et al. Evaluation of the safety and immunomodulatory effects of sargramostim in a randomized, double-blind phase 1 clinical Parkinson’s disease trial. npj Park. Dis. 2017, 3, 1–12. [Google Scholar] [CrossRef]
- Mandrioli, J.; D’amico, R.; Zucchi, E.; De Biasi, S.; Banchelli, F.; Martinelli, I.; Simonini, C.; Tartaro, D.L.; Vicini, R.; Fini, N.; et al. Randomized, double-blind, placebo-controlled trial of rapamycin in amyotrophic lateral sclerosis. Nat. Commun. 2023, 14, 1–14. [Google Scholar] [CrossRef]
- Thonhoff, J.R.; Berry, J.D.; Macklin, E.A.; Beers, D.R.; Mendoza, P.A.; Zhao, W.; Thome, A.D.; Triolo, F.; Moon, J.J.; Paganoni, S.; et al. Combined Regulatory T-Lymphocyte and IL-2 Treatment Is Safe, Tolerable, and Biologically Active for 1 Year in Persons With Amyotrophic Lateral Sclerosis. Neurol. - Neuroimmunol. Neuroinflammation 2022, 9, e200019. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Alexopoulos, H.; Spaeth, P.J. Complement in neurological disorders and emerging complement-targeted therapeutics. Nat. Rev. Neurol. 2020, 16, 601–617. [Google Scholar] [CrossRef] [PubMed]
- Lansita, J.A.; Mease, K.M.; Qiu, H.; Yednock, T.; Sankaranarayanan, S.; Kramer, S. Nonclinical Development of ANX005: A Humanized Anti-C1q Antibody for Treatment of Autoimmune and Neurodegenerative Diseases. Int. J. Toxicol. 2017, 36, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.D.; Kumar, V.; Fung, J.N.T.; Ruitenberg, M.J.; Noakes, P.G.; Woodruff, T.M. Pharmacological inhibition of complement C5a-C5a1 receptor signalling ameliorates disease pathology in the hSOD1G93A mouse model of amyotrophic lateral sclerosis. Br. J. Pharmacol. 2017, 174, 689–699. [Google Scholar] [CrossRef]
- De Marchi, F.; Mareschi, K.; Ferrero, I.; Cantello, R.; Fagioli, F.; Mazzini, L. Effect of mesenchymal stromal cell transplantation on long-term survival in amyotrophic lateral sclerosis. Cytotherapy 2023, 25, 798–802. [Google Scholar] [CrossRef]
- Sironi, F.; De Marchi, F.; Mazzini, L.; Bendotti, C. Cell therapy in ALS: An update on preclinical and clinical studies. Brain Res. Bull. 2023, 194, 64–81. [Google Scholar] [CrossRef] [PubMed]
- Brody, M.; Agronin, M.; Herskowitz, B.J.; Bookheimer, S.Y.; Small, G.W.; Hitchinson, B.; Ramdas, K.; Wishard, T.; McInerney, K.F.; Vellas, B.; et al. Results and insights from a phase I clinical trial of Lomecel-B for Alzheimer's disease. Alzheimer's Dement. 2022, 19, 261–273. [Google Scholar] [CrossRef]
- Cai, J.; Wu, J.; Wang, J.; Li, Y.; Hu, X.; Luo, S.; Xiang, D. Extracellular vesicles derived from different sources of mesenchymal stem cells: therapeutic effects and translational potential. Cell Biosci. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Qian, X.; An, N.; Ren, Y.; Yang, C.; Zhang, X.; Li, L. Immunosuppressive Effects of Mesenchymal Stem Cells-derived Exosomes. Stem Cell Rev. Rep. 2020, 17, 411–427. [Google Scholar] [CrossRef]
- Pu, Y.; Chang, L.; Qu, Y.; Wang, S.; Zhang, K.; Hashimoto, K. Antibiotic-induced microbiome depletion protects against MPTP-induced dopaminergic neurotoxicity in the brain. Aging 2019, 11, 6915–6929. [Google Scholar] [CrossRef]
- Li, W.; Lesuisse, C.; Xu, Y.; Troncoso, J.C.; Price, D.L.; Lee, M.K. Stabilization of α-Synuclein Protein with Aging and Familial Parkinson's Disease-Linked A53T Mutation. J. Neurosci. 2004, 24, 7400–7409. [Google Scholar] [CrossRef]
- Ojha, S.; Patil, N.; Jain, M.; Kole, C.; Kaushik, P. Probiotics for Neurodegenerative Diseases: A Systemic Review. Microorganisms 2023, 11, 1083. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, R.N.; Gu, Y.; Mejia-Santana, H.; Cote, L.; Marder, K.S.; Scarmeas, N. The association between Mediterranean diet adherence and Parkinson's disease. Mov. Disord. 2012, 27, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Mandrioli, J.; Amedei, A.; Cammarota, G.; Niccolai, E.; Zucchi, E.; D'Amico, R.; Ricci, F.; Quaranta, G.; Spanu, T.; Masucci, L. FETR-ALS Study Protocol: A Randomized Clinical Trial of Fecal Microbiota Transplantation in Amyotrophic Lateral Sclerosis. Front. Neurol. 2019, 10, 1021. [Google Scholar] [CrossRef] [PubMed]
- Hegelmaier, T.; Lebbing, M.; Duscha, A.; Tomaske, L.; Tönges, L.; Holm, J.B.; Nielsen, H.B.; Gatermann, S.G.; Przuntek, H.; Haghikia, A. Interventional Influence of the Intestinal Microbiome Through Dietary Intervention and Bowel Cleansing Might Improve Motor Symptoms in Parkinson’s Disease. Cells 2020, 9, 376. [Google Scholar] [CrossRef]
- Xue, L.-J.; Yang, X.-Z.; Tong, Q.; Shen, P.; Ma, S.-J.; Wu, S.-N.; Zheng, J.-L.; Wang, H.-G. Fecal Microbiota Transplantation Therapy for Parkinson’s Disease: A Preliminary Study. Medicine 2020, 99. [Google Scholar] [CrossRef]
- Chia, R.; Sabir, M.S.; Bandres-Ciga, S.; Saez-Atienzar, S.; Reynolds, R.H.; Gustavsson, E.; Walton, R.L.; Ahmed, S.; Viollet, C.; Ding, J.; et al. Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nat. Genet. 2021, 53, 294–303. [Google Scholar] [CrossRef]
- Bukhbinder, A.S.; Ling, Y.; Hasan, O.; Jiang, X.; Kim, Y.; Phelps, K.N.; Schmandt, R.E.; Amran, A.; Coburn, R.; Ramesh, S.; et al. Risk of Alzheimer’s Disease Following Influenza Vaccination: A Claims-Based Cohort Study Using Propensity Score Matching. J. Alzheimer's Dis. 2022, 88, 1061–1074. [Google Scholar] [CrossRef]
- Bradbury, A.; Bagel, J.; Sampson, M.; Farhat, N.; Ding, W.; Swain, G.; Prociuk, M.; Odonnell, P.; Drobatz, K.; Gurda, B.; et al. Cerebrospinal Fluid Calbindin D Concentration as a Biomarker of Cerebellar Disease Progression in Niemann-Pick Type C1 Disease. Experiment 2016, 358, 254–261. [Google Scholar] [CrossRef]
- Davidson, C.D.; Ali, N.F.; Micsenyi, M.C.; Stephney, G.; Renault, S.; Dobrenis, K.; Ory, D.S.; Vanier, M.T.; Walkley, S.U. Chronic Cyclodextrin Treatment of Murine Niemann-Pick C Disease Ameliorates Neuronal Cholesterol and Glycosphingolipid Storage and Disease Progression. PLOS ONE 2009, 4, e6951. [Google Scholar] [CrossRef]
- Ory, D.S.; Ottinger, E.A.; Farhat, N.Y.; King, K.A.; Jiang, X.; Weissfeld, L.; Berry-Kravis, E.; Davidson, C.D.; Bianconi, S.; Keener, L.A. Intrathecal 2-Hydroxypropyl-β-Cyclodextrin Decreases Neurological Disease Progression in Niemann-Pick Disease, Type C1: A Non-Randomised, Open-Label, Phase 1–2 Trial. The Lancet 2017, 390, 1758–1768. [Google Scholar] [CrossRef]
- Heras, M.L.; Szenfeld, B.; Ballout, R.A.; Buratti, E.; Zanlungo, S.; Dardis, A.; Klein, A.D. Understanding the phenotypic variability in Niemann-Pick disease type C (NPC): a need for precision medicine. npj Genom. Med. 2023, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Santos-Lozano, A.; García, D.V.; Sanchis-Gomar, F.; Fiuza-Luces, C.; Pareja-Galeano, H.; Garatachea, N.; Gadea, G.N.; Lucia, A. Niemann-Pick disease treatment: a systematic review of clinical trials. Ann. Transl. Med. 2015, 3, 360. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Common features and differences between Alzheimer’s disease and Niemann-Pick type C disease. An especially prominent overlap has been observed between AD and NPC, two disorders with very distinct aetiology. Created by BioRender.com.
Figure 1.
Common features and differences between Alzheimer’s disease and Niemann-Pick type C disease. An especially prominent overlap has been observed between AD and NPC, two disorders with very distinct aetiology. Created by BioRender.com.

Figure 2.
Schematic representation of the main immunomodulatory mechanisms currently on study as a treatment for neurodegenerative diseases.
Figure 2.
Schematic representation of the main immunomodulatory mechanisms currently on study as a treatment for neurodegenerative diseases.
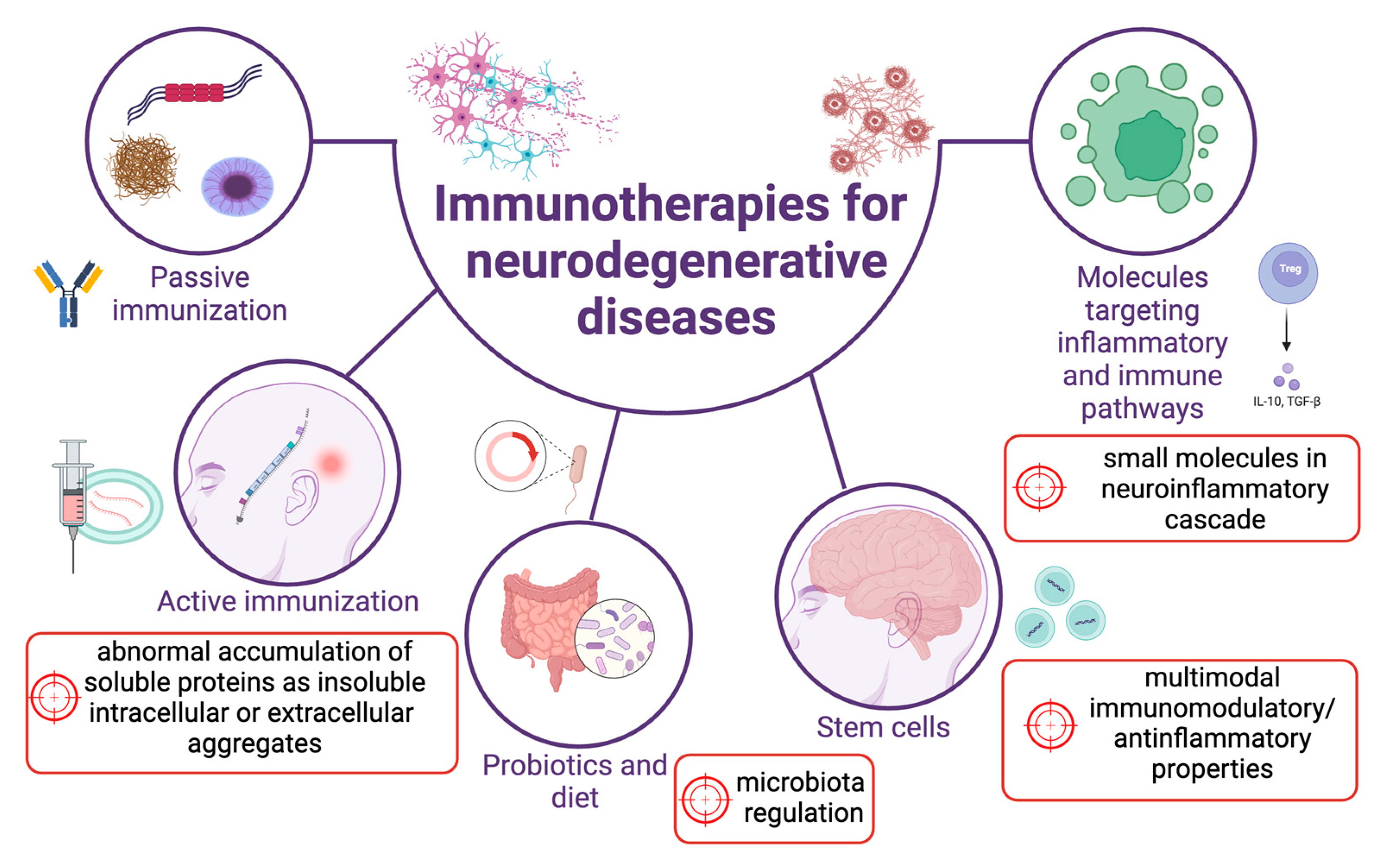
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



