
Submitted:
18 August 2023
Posted:
21 August 2023
You are already at the latest version
Abstract
Intracortical microelectrode arrays (MEAs) can be used in a range of applications, from basic neuroscience research to providing an intimate interface with the brain as part of a Brain-Computer Interface (BCI) system aimed to restore function for people living with neurological disorders or injuries. Unfortunately, MEAs tend to fail prematurely, leading to a loss in functionality for many applications. An important contributing factor in MEA failure is oxidative stress resulting from chronically inflammatory-activated microglia and macrophages releasing reactive oxygen species (ROS) around the implant site. Antioxidants offer a means for mitigating oxidative stress and improving tissue health and MEA performance. Here, we investigate using the clinically available antioxidant dimethyl fumarate (DMF) to reduce the neuroinflammatory response and improve MEA performance in a rat MEA model. Daily treatment of DMF for 16 weeks resulted in a significant improvement in the recording capabilities of MEA devices during the sub-chronic (Weeks 5-11) phase (42% active electrode yield vs. 35% for control). However, these sub-chronic improvements were lost in the chronic implantation phase, as a more exacerbated neuroinflammatory response occurs in DMF-treated animals by 16 weeks post-implantation. Yet, neuroinflammation was indiscriminate between treatment and control groups during the sub-chronic phase. Although worse for chronic use, a temporary improvement (<12 weeks) in MEA performance is meaningful. Providing short-term improvement to MEA devices using DMF can allow for improved use for limited-duration studies. Further efforts should be taken to explore the mechanism behind a worsened neuroinflammatory response at the 16-week time point for DMF-treated animals to assess its usefulness to specific applications.
Keywords:
Intracortical Microelectrode Arrays
; Neuroinflammation
; Dimethyl fumarate
; antioxidant
; neural engineering
; brain-machine interface
; neural interface
1. Introduction
Intracortical microelectrode arrays (MEAs) are an integral part of BCI systems that allow for motor restoration in people with spinal cord injury, can offer an approach to reduce the burden of treatments for various neurological disorders, and are used to understand further the complexity of brain functions [1,2,3,4,5]. Unfortunately, MEAs tend to fail over weeks to months following implantation, limiting their utility in many applications [6,7,8,9,10]. Consequently, there is a need for new designs and treatments to prolong the function of MEAs. Specifically, new developments need to be made to address the biological response from the body triggered by an implanted MEA.
Upon implantation, MEAs can rupture blood vessels causing damage to the blood-brain barrier (BBB) and damaging nearby neurons [11,12,13,14]. Within minutes [8,15,16], microglia in the brain and infiltrating macrophages surround the MEA device and become activated via innate immunity pathways [17,18,19,20,21,22]. For the duration of the implant, microglia and macrophages are activated, presumably producing pro-inflammatory cytokines and chemokines and releasing reactive oxygen species (ROS) around the implant site that further damage the surrounding tissue and the device [23,24,25,26,27,28]. Over weeks, astrocytes activate and migrate around the MEAs to form a glial scar to isolate the device from the rest of the brain [29,30]. As a result, the biological response can contribute to MEA failure consisting of a loss of recording function.
One factor contributing to the perpetual biological response is the constant presence of ROS and subsequent production from immune cells. As a byproduct of immune cell metabolism, ROS are released into the surrounding tissue when generation in microglia and macrophages exceeds the ability for natural removal [31,32,33]. ROS are highly reactive molecules that may trigger oxidative damage to the MEA, cellular damage, and death to neurons and tissue surrounding the implant [34,35]. Limiting the amounts of ROS after implantation offers a potential solution to reducing cellular and device damage.
Our team has focused on improving MEA function and tissue health using antioxidant therapeutics to reduce ROS accumulation around the implant site. Systemic and local delivery of antioxidants resveratrol and curcumin have shown promise in reducing neuroinflammation and improving MEA performance [24,26,36]. Additionally, we have developed an antioxidant coating of the superoxide dismutase mimetic Mn(III)tetrakis(4- benzoic acid)porphyrin (MnTBAP) that shows reductions in oxidative stress and improved MEA performance [37,38].
Here, we aimed to further the understanding of novel antioxidant therapeutics for improving MEA performance using dimethyl fumarate (DMF). DMF (Tecfidera) is FDA-approved for treating multiple sclerosis due to its neuroprotective and antioxidant effects [39,40]. It has been shown to mitigate damage in rodent models of neurological disorders, including traumatic brain injury [41,42,43,44,45,46,47]. As MEA implantation may be considered a form of focal traumatic brain injury, we hypothesized that DMF could alleviate and improve the biological response after device implantation. DMF increases Nrf2 pathway activity, a transcription factor known as a master regulator of antioxidant abilities [48,49,50]. Furthermore, DMF and its biologically active metabolite, monomethyl fumarate (MMF), can cross the blood-brain barrier (BBB) in pharmacologically relevant levels as indicated by direct effects on tissue and cells for inducing antioxidant effects [51,52,53]. Here, we investigate the effect of DMF on the neuroinflammatory and oxidative stress response at both a sub-chronic (7 weeks) and a chronic time point (16 weeks post-implantation) and MEA recording performance across acute (1-5 weeks), sub-chronic (6-11 weeks), and chronic (12-16 weeks) neuroinflammatory phases.
2. Materials and Methods
2.1. Intracortical Microelectrode Array Preparation
Before implantation, the 16-channel (electrode), single-shank intracortical microelectrode array (A1x16-3mm-100-177-Z16, iridium electrode sites, NeuroNexus Technologies, Ann Arbor, MI, USA) quality was verified via electrochemical impedance spectroscopy (EIS) testing. In a Faraday cage, each MEA to be implanted underwent EIS testing with a Gamry Interface 1010E Potentiostat (Gamry Instruments, Warminster, PA, USA) consisting of each electrode site as the working electrode, a platinum wire as a counter electrode, and an Ag|AgCl reference electrode for measurements. EIS was performed in 1x PBS (pH = 7.4) over 1 to 106 Hz (12 points per decade) with an AC voltage of 50 mV rms. Device verification was determined by measuring the impedance value at 1 kHz frequency. If a device was above 1 MΏ or below 100 kΏ, it was not used for implantation. Following EIS verification, MEAs were cleaned via dipping in 70% ethanol and DI water to remove any residual 1x PBS and optically imaged using a Keyence Optical Microscope (Keyence Corporation, Osaka, Japan) at a magnification of 150x for visual inspection.
2.2. Animals and Surgical Implantation
The Louis Stokes Cleveland Department of Veteran Affairs Medical Center Institutional Animal Care and Use Committee approved all animal work outlined. Surgical procedures follow those of previously established protocols [38]. Briefly, 35 Sprague Dawley rats (Charles River Labs, Wilmington, MA, USA) aged 8-10 weeks old were anesthetized in an isoflurane chamber (3.5% in 1.5L/min O2). Of the 35 rats, 22 were implanted with functional devices for the 16-week recording study (11 for control and 11 for DMF treatment), and 13 non-functional “dummy” probes for the 7-week genomic assessment of the neuroinflammatory response (7 for control and 6 for DMF treatment). For all surgeries, anesthesia level was monitored by paw-pinch reflex and assessing drowsiness by tracking the animal’s movement. Eye ointment was applied to keep the eyes from drying. The incision site on the animal’s head was shaved clean, and nails clipped to prevent ripping out of sutures during recovery. The rat was then mounted onto a stereotaxic frame (David Kopf Instruments, Tujunga, CA, USA), and anesthesia was maintained via isoflurane delivery at 0.5 – 2.0% and 1.5L/min O2 via inhalation through a nose cone. The animal’s vital signs were monitored and maintained via a blood-oxygenation and heart rate measurement system with a warming pad (PhysioSuite® for Mice & Rats, Kent Scientific Corporation, Torrington, CT, USA). A single injection of Marcaine (0.25% - 0.15mL of 1% in H2O) was administered subcutaneously to the incision site. A subcutaneous injection of the analgesic Buprenorphine (0.05 mg/kg body weight) was administered by the scruff of the neck to help with pain and recovery. The surgical area was scrubbed with alternating chlorohexidine gluconate and 70% isopropanol swabs. A one-inch incision was made along the scalp midline, and underlying connective tissue or muscle was removed to expose the skull. Sterile alligator clips were used to pull back any excess skin. A swab of hydrogen peroxide was used on the skull to dry out the surface and make visible the cranial sutures, including the bregma and lambda points. A thin coat of Vetbond tissue adhesive (Catalog #70200742529, 3M, Saint Paul, MN, USA) was applied to the clean skull to prime the surface for later cement application. For the functional-implanted rats, craniotomies for three stainless steel bone screws (Stoelting Co., Wood Dale, IL, USA) were hand-drilled into the skull using a 1.35 mm drill bit [54,55]. A ground screw (-1.5 mm lateral to midline and -1.5 mm posterior to bregma), a reference screw (-1.5 mm lateral, -4.5 mm posterior to bregma), and an anchoring screw (1.5 mm lateral, -3 mm posterior to bregma) were inserted into the skull to act as ground and reference for electrophysiological recordings (only in the animals that received recording electrodes). A final craniotomy for the implant site was drilled using a 1.75 mm drill bit located at 2 mm lateral and 2 mm anterior to bregma, targeting the primary motor cortex (M1). For the non-functional “dummy” probes, a single craniotomy was drilled using a 1.35 mm drill bit at 2 mm lateral to bregma and 2 mm anterior. In both cohorts, the implant was ready for insertion once the dura was removed and the brain was clear of any dura or debris. The intracortical microelectrode was positioned above the craniotomy to avoid blood vessels. For the functional implant cohort, reference and ground wires for the device were wrapped around their bone screws before implantation. Once positioned, the MEA was implanted into the motor cortex to a depth of 2 mm via a hydraulic inserter (2650 Micropositioner, David Kopf Instruments, Tujunga, CA, USA). For the non-functional “dummy” probe cohort, the NeuralGlider (Actuated Medical, Bellefonte, PA, USA) inserter was used to insert the probe to a depth of 1.3 mm. Non-functional probes were not inserted to a depth of 2 mm due to the length of the probe. For both cohorts, a dural graft (Biodesign Dural Graft, Cook Medical, Bloomington, IN, USA) was placed around the craniotomy to promote healing, followed by a layer of Kwik-Sil silicone adhesive (World Precision Instruments, Sarasota, FL, USA) to seal the craniotomy site. Teets Cold Cure dental cement (A-M Systems, Sequim, WA, USA) was then applied around all the craniotomy sites to build up an anchored dental cement head cap to secure the implant into place. Following surgery, 5-0 monofilament polypropylene sutures were used to close the surgical site for proper healing while leaving enough skin not sutured to keep the electrode connector exposed. A one-time dose of antibiotic Cefazolin (5 mg/kg, subcutaneously) was given immediately after surgery, followed by a continuous dosing of antibiotic (Trimethoprim-Sulfamethoxazole oral suspension, 53 mg/kg/24 hours) in the animal’s drinking water for up to 7 days. Twice daily, animals received buprenorphine (0.05 mg/kg, subcutaneously) for up to 72 hours to treat pain.
2.3. Drug Preparation and Delivery
The vehicle for drug delivery was chosen based on past literature [41,42,56] and formulated as 8% Methocel solution (Methocel® A15 LV, Catalog # 64605, Millipore Sigma, Burlington, MA, USA) in DI water. Methocel acts as a suitable suspending agent for orally delivering DMF. Methocel vehicle was aliquoted into 20 mL containers for either control or treatment preparation. For treatment, 97% Dimethyl Fumarate (DMF, Catalog #242926, Millipore Sigma, Burlington, MA, USA) was prepared by grinding into a fine powder via mortar and pestle. Powdered DMF was then measured at a concentration of 60 mg DMF per 1 mL of 8% Methocel solution. Following sufficient suspension using a planetary mixer, the DMF / Methocel solution was then drawn up in 1mL syringes to be frozen and stored in a -80°C freezer until the day of treatment. Another batch was prepared consisting of only 8% Methocel with no DMF to act as the vehicle treatment for the control group and frozen until the day of treatment. Each week for 16 weeks, every rat was weighed to determine the necessary DMF delivery to match a 60 mg/kg body weight dosing. During the day of treatment, DMF and control treatment syringes were taken from the freezer and allowed to thaw to room temperature before oral gavage treatment occurred. The control groups were given 8% Methocel at the same volume as if they were given a 60 mg/kg body weight dose to keep consistent. Due to the fast metabolism of DMF, a 1x daily oral gavage delivery of either DMF or vehicle control was required to maintain clinically relevant levels of DMF. Oral gavage provides accurate dosing to ensure the total dose of DMF is obtained by each animal and matches that of clinical trials in which clinically available DMF is provided for oral use in pill form.
2.4. Neurophysiological Recordings
Electrophysiological recordings were acquired from the MEA devices twice weekly for the 16-week study, including a recording taken on the same day as surgery once the animal had awoken (Day 0). Animals were placed under isoflurane anesthesia (3.5% in 1.5 L/min O2 ) for each recording session until unresponsive [57,58,59]. Animals were then placed in an acrylic box inside a Faraday cage with recording equipment to shield them from outside electromagnetic interference. The MEA connector was clipped into a 16-channel ZIF-Clip Head stage (Tucker-Davis Technologies Inc., Alachua, FL, USA) that was part of a 32-channel motorized commutator system (Catalog #ACO32, Tucker-Davis Technologies Inc) to allow for the animal to roam freely without getting caught on the cord [18,21]. The commutator was connected to a RA16PA 16-channel Medusa preamp (Tucker-Davis Technologies Inc) and from the preamp to an RZ5 Bioamp Processor for signal processing (Tucker-Davis Technologies Inc). Once the rat was awake and moving, recordings were taken for 10 minutes at a sampling rate of 25 kHz using the commercially available Synapse Software (Tucker-Davis Technologies Inc) with a built-in 300-3000 Hz bandpass filter. Using Plexon Offline Sorter (Plexon Inc, Dallas, TX, USA), data were analyzed to detect single unit activity and assess MEA performance. Data were digitally referenced to the common average across electrodes to remove artifacts because of animal movement, biological processes, and any other common mode signal. Individual spikes were detected using a -4σ standard deviation cutoff from the mean. Any existing artifacts were removed by setting cutoffs for amplitude (+/- 300 µV) and by removing spikes that occurred on more than five consecutive electrodes. Signals concurrently presented on over five electrodes can usually be identified as motion artifacts from the animal moving or grooming during recording. Motion and other artifacts identified were removed. Automated K-means scanning was done to sort and cluster spikes into individual single units and manually verified [38].
2.5. Neurophysiological Analysis
Additional calculations were done using MATLAB R2021a (Mathworks, Natick, MA, USA). Peak-to-peak voltage (Vpp) was defined as the voltage range from peak to trough of each waveform. The noise was calculated as the root-mean-square of the electrodes after removing spikes. The signal-to-noise ratio (SNR) was calculated by dividing the Vpp of the ensemble unit waveform by the Noise for each unit. The spike rate was defined as the inverse of the median interspike interval per unit as recorded in the Plexon Offline Sorter. Recording data from individual MEA devices that registered multiple active electrodes were averaged together to determine average values for Vpp, Noise, SNR, and Spike Rate for single recording sessions. Recording data were binned into three distinct intervals as based on previously defined by the normal progression of the foreign body response [58]: acute neuroinflammation up to 5 weeks post-implantation, the sub-chronic response between weeks 6 and 11, and finally, the chronic neuroinflammatory response during week 12 and onwards. For grouping Vpp, Noise, SNR, and Spike Rate, the values of each individual electrode were averaged together within the time phase and group (DMF vs control). For example, all Vpp values for electrode 1 of the DMF animals in the acute phase were averaged together to get a singular value. This was repeated for each electrode and each time phase between groups. To calculate the number of units per active electrode, the sum of units for each electrode of a device in that time phase was recorded and divided by the number of weeks in that specific time phase. This results in an average number of units per active electrode per device for all weeks in a specific time phase in each group. For all recording metrics, the sample size for the number of units per electrode is 125 for acute DMF and 121 for acute control, 141 for sub-chronic DMF and 137 for sub-chronic control, and 124 for chronic DMF and 144 for chronic control. To determine the proportion of active electrodes in each group, the total number of active electrodes (electrodes recording at least one unit) was summed and divided by the total possible electrodes (16 electrodes per device x 11 animals per group x 1 week) on a week-by-week basis. The active and total electrodes were summed across weeks, making up each time phase when grouped into acute, sub-chronic, and chronic phases. Doing so leaves a total sample size of 880 for acute (11 animals x 16 electrodes x 5 weeks), 1056 for sub-chronic (11 animals x 16 electrodes x 6 weeks), and 880 (11 animals x 16 electrodes x 5 weeks) for chronic. When considering individual week proportional comparisons, the sample size is determined by the total number of electrodes multiplied by the number of animals (n = 176 per week for both DMF and control).
2.6. Histological Tissue Processing
After reaching the 16-week endpoint for the study, each animal received an IP anesthetic injection of ketamine (160 mg/kg) and xylazine (20 mg/kg). Sufficient anesthetic depth was determined via toe pinch and achieved when there was no associated animal reaction following the pinch. After full anesthetization, a vertical incision was made just below the xiphoid process up the abdomen to expose the abdominal cavity. A second lateral incision was made across the abdominal cavity, followed by a vertical incision up each side of the rib cage. The rib cage was held open to expose the pleural cavity and the heart, and the diaphragm was cut with scissors. Afterward, the sternum was clamped with a hemostat to keep the heart and major blood vessels exposed, and a small incision was made in the left ventricle of the heart. A gavage was inserted into the aorta through the left ventricle and clamped to stay in place. To allow blood and perfused liquid to flow out, the right atrium was snipped, and the rat was perfused with 400-500 mL of 1x PBS (1X PBS, Invitrogen, Carlsbad USA) at 75-100 mL/min until the liver was flushed beige and the perfused liquid flowing out of the heart was clear. Directly after, 400-500 mL of 10% buffered formalin (Fisher Chemical, Waltham, MA) was perfused throughout the animal, and once complete, the brain was extracted following decapitation. After extraction, the brain was placed in refrigerated 10% buffered formalin for 24 hours, followed by 10% sucrose solution (Sucrose from Millipore Sigma, Burlington, MA) in 1x PBS for 24 hours, 20% sucrose for another 24 hours, 30% sucrose for 48 hours, and then a final fresh solution of 30% sucrose until the brains were frozen in Optimal Cutting Temperature compound (OCT, Sakura Finetek USA Inc, Torrance, CA, USA, #25608-930) for sectioning. Following freezing, to prepare for immunohistochemistry, each brain was sliced at 20 μm section thickness on a cryostat before being placed on microscope slides (SuperFrost Plus, FisherBrand, Hampton, NH).
2.7. Immunohistochemical Staining
Immunohistochemical protocols were followed utilizing the Bond RX automated staining system (Leica Biosystems, Wetzlar, Germany) and our current standard laboratory procedures [18,60,61,62]. Tissue was stained to analyze levels of astrocyte activity, microglia/macrophage activity, neuron levels, and BBB permeability. Brain tissue was thawed in a humidity chamber at room temperature for one hour, followed by a brief rehydration in 1x PBS to remove any residual OCT compound. The tissue was then loaded into the Bond RX system for automated staining. Proprietary Bond RX detergent wash buffer was applied to tissue to permeabilize cells and tissue for staining cells and molecules. A 10-minute heat-induced epitope retrieval step (HIER) was done on each slide using a proprietary sodium citrate-based solution at 80°C. After detergent washing and HIER, one-half of the tissue was incubated for 30 minutes with rabbit anti-immunoglobulin G (IgG, 1:100, Bio-Rad, Hercules, CA, USA, Catalog #618501) to visualize BBB permeability. Immediately after, the same set of tissue was then rewashed with detergent before incubation with mouse anti-neuronal nuclei (NeuN 1:250, Millipore Sigma, Burlington, MA, USA, Catalog #MAB3477) for neuron level analysis. The other half of tissue was incubated for 30 minutes with a combination solution containing rabbit anti-glial fibrillary acidic protein (GFAP, 1:500, Agilent Dako, Santa Clara, CA, USA, Catalog #Z0334429-2) to visualize astrocytes and mouse anti-CD68 (CD68, 1:100, Millipore Sigma, Burlington MA, USA, Catalog #MAB1435) to visualize activated microglia and macrophages. After incubation, both tissue sets were removed from the Bond RX automated stainer and placed back in a humidity chamber. Subsequently, the tissue was incubated at room temperature for two hours in a secondary antibody solution. Buffer was made up of fluorescent AlexaFluor secondary antibodies 488/594 (1:1000, Invitrogen, Waltham, MA, USA, Catalog #PIA32723, and #A11037, respectively) to visualize markers along with 4′6-diamidino-2-phenylindole (DAPI) (1:3600, 10.9 mM, Invitrogen, Waltham, MA, USA, Catalog #D3571) to visualize cell nuclei. Lastly, tissue was washed with dH2O, and excess dH2O was wiped off before slides were mounted with coverslips using a mounting medium (Fluoromount-G, Southern Biotech, Birmingham, AL, USA, Catalog #010001). For each group there is a sample size between 4 and 7. Variation in sample size was due to defective microscope slides leading to a loss in brain tissue for staining.
2.8. Imaging and Analysis
Stained tissues on slides were scanned under the 20x objective of the Axioscan.Z1 microscope (Zeiss Inc., Oberkochen, Germany) and saved as .CZI files. The exposure time was optimized for individual fluorescent markers and was kept consistent throughout the study. In each .CZI file, regions around the implantation hole (9500 × 9500 pixels) were cropped and converted into 16-bit .TIFF images for intensity analysis. A set of .PNG images containing NeuN and DAPI channels were also exported from the .CZI files for neuron counting. For analysis of stain intensity, a custom MATLAB (Mathworks, Natick, MA, US) program, SECOND, was used to outline the implant hole and mask artifacts from the subsequent analysis [63]. Using a custom Python script, the intensities of fluorescent markers for GFAP, CD68, and IgG in the unmasked parts of .TIFF images were quantified and binned into 50 µm intervals based on distance from the implantation site. All pixels within 700 µm from the implantation site were included in the analysis. Intensities in all bins were normalized to the average intensity in the 650–700 µm bin, which was defined as the background. Due to the regular activities of astrocytes in the brain, the normalization constant for GFAP was adjusted to 1. Given that we do not expect IgG and CD68 staining in healthy tissue, the background intensity level for these markers was adjusted to 0. To count neurons stained by NeuN, the Cellpose algorithm segmented the neurons with DAPI as the reference nuclei channel [64]. The cyto2 model for neuron counting was pre-trained on an independent set of images with default parameters. Another custom Python program was used to calculate the neuron density from the raw outputs of Cellpose. Using the same program, all density data were binned into 50 µm intervals, up to 500 µm away from the implantation hole. For each sample, the density of each bin was normalized to the value of 400–450 µm bin, which was defined as the baseline neuron density.
2.9. Bulk Gene Analysis
A second cohort of 13 animals (7 Control and 6 DMF) were implanted with a non-functional “dummy” silicon shank matching the size and shape of the functional implant to evaluate the neuroinflammatory response at a sub-chronic timepoint (7 weeks) within the range of increased recording performance. Surgical differences are outlined in Section 2.3. After seven weeks, the rats were perfused with 1x PBS followed by 30% sucrose in 1x PBS, and brains were frozen in OCT after extraction.
Each frozen brain was sectioned in a cryostat at 150 µm thickness to isolate brain tissue around the implant site. Next, a 500 µm radius biopsy punch was centered over the implant site to isolate tissue radial to the implant. Biopsy punches were taken throughout the depth of the implant (~8 punches for a total of 1200 µm depth). Brain tissue from the biopsy punch was placed in sterile, nuclease-free tubes containing homogenization beads (#SKU 19-627, Omni International, Kennesaw, GA, USA). Following tissue isolation, samples were homogenized, and RNA was extracted using the Case Western Reserve University Translational Shared Resource core facility. Once RNA was extracted, samples were run on the NanoString nCounter for bulk gene analysis using a customized panel of 152 genes (Table 1). The custom panel of genes was selected based on available oxidative stress markers, previously analyzed genes upregulated after MEA implantation, and previously identified housekeeping genes for rat brain tissue (Table 1) [27,65,66,67,68]. Isolated RNA was hybridized with NanoString capture and reporter probe sets, which bind to the 152 RNA sequences of interest using the complementary sequences. These hybridized samples were then suspended in a clear cartridge. The capture probes secure the RNA in a stable position to align the reporter probes. The reporter probes consist of a fluorescent sequence unique to each gene of interest, which is then scanned by the nCounter. The NanoString nCounter then outputs raw counts of all 152 RNA sequences scanned within each sample.
2.10. Statistical Analysis
Excel (Microsoft Corporation, Redmon, WA, US), R Studio 2022.7.1+554 (RStudio, PBC, Boston, MA, US), and GraphPad Prism (Dotmatics, Boston, MA, US) were used to conduct data analysis, graphing, and statistical measurements. Outliers were identified based on 3 median absolute deviations away from the median and removed using the UnivOut1 package in R studio. A one-tailed test of proportions z-test was used for calculating statistical differences of the proportion of active electrodes between groups for the acute, sub-chronic, and chronic phases. Additional recording metrics were compared within and across acute, sub-chronic, and chronic neuroinflammatory phases using a Kruskal-Wallis test followed by a Benjamini-Krieger-Yekutieli test for multiple comparisons for non-normal distributions to increase statistical power and reduce type I errors. A standard Student’s t-test was performed for each distance interval for immunohistochemical analysis. In all cases, statistical significance was defined at p<0.05. Bar plots using error bars with the standard error of the mean (SEM). For recording data box plots, whiskers represent minimum and maximum values, the box represents the first and third quartiles of the data, and the horizontal line indicates the median. All recording numerical data is represented in the text as the mean ±SD. Bulk RNA expression data was analyzed using the NanoString nSolver software as previously described by our lab [65,67,68]. The raw expression counts were normalized with positive and negative control probe counts to account for assay efficiency and housekeeping genes that normalize to the amount of RNA collected per sample. Following normalization, a differential expression analysis was performed to determine how gene expression changes with DMF treatment. Any gene with less than 20 counts in 85% of the samples was removed. The expression ratio was plotted on a log2 scale and called the Log2FoldChange. Expression of Log2FoldChange >1 indicates a two-fold increase, whereas Log2FoldChange <-1 indicates a two-fold decrease. A 2-tailed, unequal variance t-test was performed for each gene. A Benjamini-Hochberg correction using a false discovery rate of 0.05 filtered out random significance due to the many genes tested.
3. Results
3.1. Effect of DMF on MEA performance and single-unit recordings
Figure 1A illustrates the proportion of active electrodes in acute (Weeks 1-5), sub-chronic (Weeks 6-11), and chronic (Weeks 12-16) phases. At the acute phase, DMF (32% active electrode yield ) showed no significant difference compared to controls (31% active electrodes yield). Within groups, DMF-treated animals (N = 11) demonstrated a significant but temporary increase in the active electrode yield with higher recording performance at the sub-chronic phase, only to decline in the chronic phase (Figure 1B). Control animals (N = 11) also improved from acute to sub-chronic to chronic phase (Figure 1C). However, the active electrode yield recovered in the chronic phase for control animals (Figure 1C). The steady increase in the proportion of active electrodes in the control animals may result from stabilization after the wound healing response in the acute phase and BBB stabilization during the late sub-chronic phase. The differential trends within treatment modalities led to differences in the comparison between treatment groups at both the sub-chronic and chronic phases (Figure 1A). Specifically, during the sub-chronic phase, DMF treatment (42% active electrodes) shows a significant improvement in the proportion of active electrodes (p = 0.0005) compared to control animals (35% active electrodes). The sub-chronic improvement in active electrode yield for DMF-treated animals was lost during the chronic phase, as DMF-treated animals declined in performance (37% active electrodes) while control animals continued to improve to a statistically higher proportional yield (p = 0.043; 41% active electrodes; Figure 1A).
Units per electrodes were compared within and between groups across neuroinflammatory phases for both DMF-treated and control animals. Despite changes in the active electrode yield between and within treatment groups, the average number of units per electrode site remained indistinguishable across all inflammatory phases (Figure 1D, no significant differences, p > 0.05). The week-by-week comparison (Figure 1E) shows the improvement of DMF compared to control for the acute phase at only week 4 and improvements in the sub-chronic phase in weeks 6, 7, and 9. We observe significant declines at weeks 12 and 16 during the chronic phase. Week 7 specifically showed the largest improvement between DMF and control of 11.9%.
As shown in Figure 2A, Vpp was comparable between DMF-treated animals and control groups across acute (44.8 ± 17.2 µV for DMF, 42.5 ± 20.2 µV for control), sub-chronic (34.6 ± 14.9 µV for DMF, 38.0 ± 19.8 µV for control), and chronic (32.7 ± 12.9 µV for DMF, 33.9 ± 19.1 µV for control) phases. DMF treatment resulted in a significant decline in Vpp from acute to sub-chronic (p < 0.0001) and acute to chronic (p < 0.0001) phases (Figure 2A). Control animals showed a similar decline from acute to chronic (p = 0.0003, Figure 2A).
Comparison of noise levels showed no significant differences between treatment groups for acute (7.5 ± 2.6 µV for DMF, 7.0 ± 3.0 µV for control), sub-chronic (6.1 ± 2.5 µV for DMF, 6.3 ± 3.1 µV for control), and chronic (5.7 ± 2.3 µV for DMF, 5.4 ± 2.7 µV for control) neuroinflammatory phases (Figure 2B). DMF-treated animals showed a significant decline in noise from acute to sub-chronic (p < 0.0001) and from acute to chronic (p < 0.0001) phases (Figure 2B). Control animals also showed a significant decline in noise from acute to sub-chronic (p = 0.0343), acute to chronic (p < 0.0001), and sub-chronic to chronic (p = 0.0097, Figure 2B). Due to a lack of differences in Vpp and Noise levels, the SNR (Figure 2C) for both treatment groups were not statistically different (acute: 6.2 ± 2.0 for DMF and 6.5 ± 2.9 for control, sub-chronic: 6.2 ± 2.4 for DMF and 6.6 ± 2.9 for control, chronic: 6.3 ± 2.6 for DMF and 7.0 ± 3.7 for control). There were no significant changes in SNR between phases, most likely due to similar declines in Vpp and Noise levels from acute to sub-chronic to chronic phases (Figure 2C). Spike rate (Figure 2D) showed no significant changes between control and DMF-treated within a given phase: acute (2.2 ± 1.5 for DMF and 2.7 ± 2.1 for control), sub-chronic (1.7 ± 0.9 for DMF and 2.0 ± 1.3 for control), chronic (2.2 ± 1.5 for DMF and 3.4 ± 3.6 for control). Additionally, no significant changes were observed across phases for a given treatment group.
3.2. Effect of DMF on tissue health and neuroinflammation
3.2.1. Chronic Inflammation and blood-brain barrier permeability
Immediately after implantation, microglia and macrophages migrate to the implant site, activate to clear debris, and combat the foreign body presence of the MEA [6]. Chronic activation results in the over-production of ROS and perpetual neuroinflammation, contributing to the failure of the MEA [33,69,70]. Figure 3 shows the neuroinflammatory response to implanted MEAs in DMF-treated and control animals following 16 weeks of implantation. DMF-treated animals have significantly (p<0.05) higher expressions of active microglia/macrophage markers (CD68; Figure 3A) within 150 µm of the implant compared to controls. Increased CD68 expression is indicative of an elevated neuroinflammatory response [14]. Furthermore, DMF-treated animals demonstrated significantly higher immunoglobulin (IgG; Figure 3B) levels within 250 µm of the implant site, indicating higher BBB permeability. Such results suggest chronic DMF usage may weaken the BBB, allowing more macrophages to infiltrate the implant site and further perpetuate the neuroinflammatory response. Staining for glial-fibrillary acidic protein (GFAP; Figure 3C) showed no statistical difference within the immediate recording area (within 150 µm) from the implant site, indicating no difference in glial scarring at 16 weeks post-implantation. Significance observed at 250-300 µm suggests that the DMF group has higher astrocytic activity in that region. However, the difference in intensity (1.19 for DMF vs. 1.12 for control) is minimal, suggesting that significance is simply due to low variance within animals. At 250-300 µm, we do not usually expect a biological difference due to implant unless the closer regions also display significant differences in expression.
3.2.2. Neural health and viability
Following MEA implantation, neuronal populations demonstrate a trend of reduced density, typically measured with neuronal nuclei (NeuN) staining, in the 200-250 µm closest to the implant surface [6]. Due to the antioxidant and neuroprotective effects of DMF previously demonstrated in the literature [41,42], neuronal density was hypothesized to improve over 16 weeks compared to control animals, leading to improvements in MEA recordings. However, no significant change was seen in normalized neuronal nuclei (NeuN; Figure 3D), indicating a relatively stable population of neurons compared to controls. Such results show that regular DMF treatments did not increase the ability to prevent neuron loss 16 weeks post-implantation of MEAs.
3.3. Bulk Gene Expression Analysis
To further understand the neuroinflammatory and oxidative stress response in the brain, we performed experiments with an additional cohort of animals. In this additional cohort, animals were given treatment for seven weeks. Week 7 was chosen due to observing the most significant margin of improvement observed in MEA recording performance in the functional implant cohort (Figure 1E). In this second cohort, six rats were treated with DMF, and seven were treated with the control vehicle. Rather than using 4-6 histological markers and performing immunohistochemistry again, we developed a custom gene expression panel with 152 genes associated with neuroinflammatory and oxidative stress pathways (Table 1). Here, our results from bulk gene analysis based on log2(fold change) of DMF-treated animals compared to control animals suggest no significant differences between groups after correction (Figure 4).
4. Discussion
In previous studies, we observed acute average electrode yield in control animals that were higher than those displayed for our controls here. It is important to note that the control animals here were administered a daily diluent gavage. While not treated with antioxidants, they were treated and handled more regularly than our historical control animals, which could account for lower active electrode yield than traditional control animals [38]. At sub-chronic time points (weeks 5-11), DMF-treated rats displayed a significantly improved recording performance compared to vehicle control (Figures 1 & 2). Specifically, week 7 showed the most significant margin of improvement in DMF compared to controls (Figure 1E). However, DMF-treated animals began to decline at chronic time points (12+ weeks) and were significantly worse than control. To understand why such a reversal of expected performance occurred, we measured the histological outcomes of the brain at 16 weeks (Figure 3) and genetic analysis of 152 neuroinflammatory and oxidative stress-related genes at seven weeks (Figure 4).
Immunohistochemical evaluation of the brain tissue adjacent to the MEA at 16 weeks post-implantation showed a significant increase in IgG and CD68 levels immediately around the implant (0-200 µm) for DMF-treated animals compared to control animals. IgG is naturally found in high abundance circulating in the bloodstream but not in the brain [71]. CD68 expression is a marker for increased microglia and macrophage activation to an inflammatory phenotype, indicating an elevated neuroinflammatory response [14]. Therefore, the abundance of IgG found at the MEA-tissue interface at 16 weeks indicates less stability in BBB integrity at some point post-implantation. There is debate in the literature if IgG expression is a static or dynamic measure of BBB permeability regarding if IgG is cleared or accumulates from/at the implant site. Additionally, higher levels of CD68 expression surrounding the implants in the DMF-treated group indicate increased microglia and macrophage activation (Figure 3A-B).
BBB integrity and CD68 expression may follow similar trends due to the prevalence of infiltrating macrophages coming from the bloodstream into the implant site [72,73,74]. Several labs have indicated a self-perpetuating relationship between activated microglia and macrophages and BBB integrity, partly due to the secretion of reactive oxygen species by activated microglia and macrophages, which compromises BBB integrity [6,11,26,75]. We have previously shown that blood-derived macrophages are responsible for a large portion of mouse models' neuroinflammatory response to MEAs [17,18]. Additionally, multiple studies have linked IgG expression to MEA recording performance [7,76,77].
Therefore, poor BBB stability and resulting macrophage infiltration may explain the reversal of sub-chronic improvements in recording performance and the subsequent lack of chronic improvements in neurophysiological recordings for DMF-treated animals. Elevated neuroinflammation during the chronic phase could mask the initial improvements in recording performance for DMF-treated animals.
To understand why sub-chronic recording improvements occurred, we set up additional animals to be sacrificed at the 7-week time point, where the greatest weekly difference in active electrode yield was seen (Figure 1E). Bulk gene analysis of oxidative and neuroinflammatory genes at seven weeks post-implant did not reveal any significant changes between DMF and control groups (Figure 4). It was unexpected to see a lack of differences given DMF’s antioxidant capabilities and the recording improvements observed at that time. However, changes to gene expression may be a predictive indicator for functional experiments like microelectrode recording differences. A future study could look at gene expression in preceding weeks (for example, Weeks 5 and 6 here) to observe if molecular level changes in the neuroinflammatory process take time to impact recording observations and MEA performance.
It is essential to discuss that DMF treatments designed to assess the impact on neurological disorders in rodent models have shown neuroprotective and antioxidant effects throughout the study. However, it is worth noting that the referenced studies were designed to end before our chronic phase time point of 16 weeks, with most studies lasting either less than a few weeks (acute) [41,42,45] or not beyond ten weeks (sub-chronic) [47,56]. While we did not uncover the apparent side effects of DMF treatment, as we have noted with daily IP injections of Resveratrol [24], there may be undetected side effects masking the beneficial effects during the chronic phase. For example, regular oral gavage treatments have been linked to adverse health effects due to changes in the gut microbiome and plasma metabolome [78]. While there is growing evidence suggesting that altering gut microbiome composition can alter brain and neuron health through the gut-brain axis [79,80,81,82,83,84,85], in the context of this study, we did not evaluate potential changes in gut microbiome for either control or DMF-treated animals, compared to naïve sham animals. To that end, it is essential to point out that both control and DMF-treated animals demonstrated a lower active electrode yield than we have seen in other studies in our labs [38,57,58]. It is possible that decreased recording performance in both groups was a result of unnoticed complications such as elevated stress levels due to regular oral gavage [86,87].
Regarding the known mechanism of action for the antioxidative effects of DFM treatment, DMF has been shown to exert neuroprotective and antioxidant effects through its ability to activate the Nrf2 pathway [47,52,56]. Nrf2 is a transcription factor that, when activated, provides anti-inflammatory, cytoprotective, and antioxidant effects to the brain [49,88]. Although DMF has been shown to act through the Nrf2 pathway, the complete mechanism of DMF activity remains to be determined. DMF has been shown to impact many aspects of the innate and adaptive immune systems, including reductions in absolute lymphocyte counts [52,89,90]. Declines in lymphocyte count are one reason why chronic use of DMF has been linked to an elevated risk of infection [91,92]. No signs of chronic infections were seen in this study. However, with such a broad range of effects and a need for a more precise understanding, DMF may trigger a response in which the BBB needs to heal appropriately over time due to persistent MEA presence, leading to poor chronic results. To that end, we have shown that over-attenuating the innate immune response to MEAs can negatively impact recording performance compared to partial inhibition of the same target [18,21]. Therefore, a smaller or less frequent dose of DMF could facilitate a longer-lasting improvement in MEA performance. Future studies may explore alternative means and dosing regimens for improvements in chronic MEA performance.
Although the current delivery and dose for DMF suggested a lack of recording improvement in the chronic period of implantation, there may still be utility in using DMF for temporary improvements in MEA performance on the sub-chronic timescale (<12 weeks). Clinically, MEA devices are designed to be “lifetime” solutions for brain-computer interfacing systems. However, there are still many short-term uses for MEAs [93]. Outside clinical use, sub-chronic improvements in recording performance can also benefit basic science and hypothesis-driven neuroscience studies. Poor recording performance is continuously an issue in signal processing for decoding brain activity into peripheral motor action [6,94,95]. Improving recordings in the sub-chronic phase can aid in the signal decoding process. Additionally, DMF may be one part of a treatment cocktail that utilizes a multipronged approach to mitigate several aspects of the neuroinflammatory response robustly and temporally to implanted MEAs.
5. Conclusions
DMF is a widely used therapy for treating relapsing multiple sclerosis due to its neuroprotective and antioxidant therapeutic effects. We show that DMF treatment produces a statistically significant improvement in active electrode yield within a sub-chronic neuroinflammatory phase (5-11 weeks). Though the current formulation and dosing did not provide an indefinite long-term improvement beyond ten weeks post-implantation to declining MEA performance, temporary improvements to neurophysiological recordings are still crucial for short-term brain-machine interface uses and basic science studies. Further optimization of the dosing of DMF or the inclusion of DMF in combination therapies could result in a more robust and comprehensive approach to mitigate MEA failure.
Author Contributions
Conceptualization, G.F.H, J.J.P., and J.R.C.; methodology, G.F.H, L.N.D., B.S., J.D., A.G. H-R, S.F.C., J.J.P, J.R.C.; validation, G.F.H, J.D., B.S., A.G. H-R; formal analysis, G.F.H., T.B., H.O., J.Z., L.N.D., B.S., M.S., P.B., J.D., A.G. H-R.; investigation, G.F.H., T.B., H.O., J.Z., M.S., P.B., J.D.; resources, S.F.G., J.J.P., and J.R.C.; data curation, G.F.P.; writing—original draft preparation, G.F.H. and J.R.C; writing—review and editing, all.; supervision, J.R.C.; funding acquisition, S.F.G., J.J.P., and J.R.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by the National Institutes of Health, National Institute for Neurological Disorders and Stroke (R01NS110823, Capadona/Pancrazio) diversity supplement (Hernandez-Reynoso), and the National Institute for Biomedical Imaging and Bioengineering, (T32EB004314, Capadona/Kirsch). Additional support was provided by Merit Review Award (I01RX002611, Capadona) and a Senior Research Career Scientist Award (I02RX003077 and IK6RX003077, Capadona) from the United States (US) Department of Veterans Affairs Rehabilitation Research and Development Service.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest and Disclosures
TThe authors declare no conflicts of interest. The contents do not represent the views of the U.S. Department of Veterans Affairs, the National Institutes of Health, or the United States Government. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors. The funders had no role in the study’s design; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Simeral, J.D.; Kim, S.P.; Black, M.J.; Donoghue, J.P.; Hochberg, L.R. Neural control of cursor trajectory and click by a human with tetraplegia 1000 days after implant of an intracortical microelectrode array. J Neural Eng 2011, 8, 025027. [Google Scholar] [CrossRef] [PubMed]
- Vilela, M.; Hochberg, L.R. Applications of brain-computer interfaces to the control of robotic and prosthetic arms. Handb Clin Neurol 2020, 168, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Shih, J.J.; Krusienski, D.J.; Wolpaw, J.R. Brain-computer interfaces in medicine. Mayo Clin Proc 2012, 87, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Fatima, N.; Shuaib, A.; Saqqur, M. Intra-cortical brain-machine interfaces for controlling upper-limb powered muscle and robotic systems in spinal cord injury. Clinical Neurology and Neurosurgery 2020, 196, 106069. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Eguren, D.; Azorin, J.M.; Grossman, R.G.; Luu, T.P.; Contreras-Vidal, J.L. Brain-machine interfaces for controlling lower-limb powered robotic systems. J Neural Eng 2018, 15, 021004. [Google Scholar] [CrossRef]
- Jorfi, M.; Skousen, J.L.; Weder, C.; Capadona, J.R. Progress Towards Biocompatible Intracortical Microelectrodes for Neural Interfacing Applications. Journal of neural engineering 2015, 12, 011001. [Google Scholar] [CrossRef] [PubMed]
- Kozai, T.D.; Jaquins-Gerstl, A.S.; Vazquez, A.L.; Michael, A.C.; Cui, X.T. Brain tissue responses to neural implants impact signal sensitivity and intervention strategies. ACS Chem Neurosci 2015, 6, 48–67. [Google Scholar] [CrossRef]
- Wellman, S.M.; Li, L.; Yaxiaer, Y.; McNamara, I.; Kozai, T.D.Y. Revealing Spatial and Temporal Patterns of Cell Death, Glial Proliferation, and Blood-Brain Barrier Dysfunction Around Implanted Intracortical Neural Interfaces. Front Neurosci 2019, 13, 493. [Google Scholar] [CrossRef]
- Barrese, J.C.; Rao, N.; Paroo, K.; Triebwasser, C.; Vargas-Irwin, C.; Franquemont, L.; Donoghue, J.P. Failure mode analysis of silicon-based intracortical microelectrode arrays in non-human primates. J Neural Eng 2013, 10, 066014. [Google Scholar] [CrossRef]
- Freire, M.A.; Morya, E.; Faber, J.; Santos, J.R.; Guimaraes, J.S.; Lemos, N.A.; Sameshima, K.; Pereira, A.; Ribeiro, S.; Nicolelis, M.A. Comprehensive analysis of tissue preservation and recording quality from chronic multielectrode implants. PLoS One 2011, 6, e27554. [Google Scholar] [CrossRef]
- Bennett, C.; Mohammed, F.; Alvarez-Ciara, A.; Nguyen, M.A.; Dietrich, W.D.; Rajguru, S.M.; Streit, W.J.; Prasad, A. Neuroinflammation, oxidative stress, and blood-brain barrier (BBB) disruption in acute Utah electrode array implants and the effect of deferoxamine as an iron chelator on acute foreign body response. Biomaterials 2019, 188, 144–159. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.; Samikkannu, M.; Mohammed, F.; Dietrich, W.D.; Rajguru, S.M.; Prasad, A. Blood brain barrier (BBB)-disruption in intracortical silicon microelectrode implants. Biomaterials 2018, 164, 1–10. [Google Scholar] [CrossRef]
- Potter, K.A.; Buck, A.C.; Self, W.K.; Capadona, J.R. Stab injury and device implantation within the brain results in inversely multiphasic neuroinflammatory and neurodegenerative responses. J Neural Eng 2012, 9, 046020. [Google Scholar] [CrossRef]
- Polikov, V.; Tresco, P.; Reichert, W. Response of brain tissue to chronically implanted neural electrodes. J Neurosci Methods 2005, 148, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wellman, S.M.; Yaxiaer, Y.; Eles, J.R.; Kozai, T.D.Y. In vivo spatiotemporal patterns of oligodendrocyte and myelin damage at the neural electrode interface. Biomaterials 2021, 268, 120526–120526. [Google Scholar] [CrossRef]
- Kozai, T.D.Y.; Marzullo, T.C.; Hooi, F.; Langhals, N.B.; Majewska, A.K.; Brown, E.B.; Kipke, D.R. Reduction of neurovascular damage resulting from microelectrode insertion into the cerebral cortex using in vivo two-photon mapping. Journal of Neural Engineering 2010, 7, 046011. [Google Scholar] [CrossRef]
- Ravikumar, M.; Sunil, S.; Black, J.; Barkauskas, D.; Haung, A.Y.; Miller, R.H.; Selkirk, S.M.; Capadona, J.R. The Roles of Blood-derived Macrophages and Resident Microglia in the Neuroinflammatory Response to Implanted Intracortical Microelectrodes. Biomaterials 2014, S0142-9612, 8049–8064. [Google Scholar] [CrossRef]
- Bedell, H.W.; Hermann, J.K.; Ravikumar, M.; Lin, S.; Rein, A.; Li, X.; Molinich, E.; Smith, P.D.; Selkirk, S.M.; Miller, R.H. Targeting CD14 on blood derived cells improves intracortical microelectrode performance. Biomaterials 2018, 163, 163–173. [Google Scholar] [CrossRef]
- Bedell, H.W.; Song, S.; Li, X.; Molinich, E.; Lin, S.; Voit, W.E.; Pancrazio, J.J.; Capadona, J.R. Understanding the effects of both CD14-meditated innate immunity and device/tissue mechanical mismatch in the neuroinflammatory response to intracortical microelectrodes. Frontiers in Neuroscience 2018, 12, 772. [Google Scholar] [CrossRef] [PubMed]
- Bedell, H.W.; Capadona, J.R. Anti-inflammatory Approaches to Mitigate the Neuroinflammatory Response to Brain-Dwelling Intracortical Microelectrodes. J Immunol Sci 2018, 2, 15–21. [Google Scholar] [CrossRef]
- Hermann, J.K.; Ravikumar, M.; Shoffstall, A.J.; Ereifej, E.S.; Kovach, K.M.; Chang, J.; Soffer, A.; Wong, C.; Srivastava, V.; Smith, P.; et al. Inhibition of the cluster of differentiation 14 innate immunity pathway with IAXO-101 improves chronic microelectrode performance. J Neural Eng 2018, 15, 025002. [Google Scholar] [CrossRef] [PubMed]
- Hermann, J.K.; Lin, S.; Soffer, A.; Wong, C.; Srivastava, V.; Chang, J.; Sunil, S.; Sudhakar, S.; Tomaszewski, W.H.; Protasiewicz, G.; et al. The Role of Toll-Like Receptor 2 and 4 Innate Immunity Pathways in Intracortical Microelectrode-Induced Neuroinflammation. Front Bioeng Biotechnol 2018, 6, 113. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.M.; Rodriguez, A.; Chang, D.T. Foreign body reaction to biomaterials. Seminars in Immunology 2008, 20, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Potter-Baker, K.A.; Stewart, W.G.; Tomaszewski, W.H.; Wong, C.T.; Meador, W.D.; Ziats, N.P.; Capadona, J.R. Implications of chronic daily anti-oxidant administration on the inflammatory response to intracortical microelectrodes. J Neural Eng 2015, 12, 046002. [Google Scholar] [CrossRef]
- Potter, K.A.; Jorfi, M.; Householder, K.T.; Foster, E.J.; Weder, C.; Capadona, J.R. Curcumin-releasing mechanically-adaptive intracortical implants improve the proximal neuronal density and blood-brain barrier stability. Acta Biomater 2014, 10, 2209–2222. [Google Scholar] [CrossRef]
- Potter, K.A.; Buck, A.C.; Self, W.K.; Callanan, M.E.; Sunil, S.; Capadona, J.R. The effect of resveratrol on neurodegeneration and blood brain barrier stability surrounding intracortical microelectrodes. Biomaterials 2013, 34, 7001–7015. [Google Scholar] [CrossRef]
- Ereifej, E.S.; Rial, G.; Hermann, J.K.; Smith, C.S.; Meade, S.; Rayyan, J.; Chen, K.; Feng, H.; Capadona, J.R. Implantation of Neural Probes in the Brain Elicits Oxidative Stress. Frontiers in Bioengineering and Biotechnology 2018, Under Revision.
- Potter-Baker, K.A.; Capadona, J.R. Reducing the “Stress”: Antioxidative Therapeutic and Material Approaches May Prevent Intracortical Microelectrode Failure. ACS Macro Letters 2015, 275–279. [Google Scholar] [CrossRef]
- Salatino, J.W.; Ludwig, K.A.; Kozai, T.D.Y.; Purcell, E.K. Glial responses to implanted electrodes in the brain. Nature Biomedical Engineering 2017, 1, 862–877. [Google Scholar] [CrossRef]
- Spencer, K.C.; Sy, J.C.; Falcon-Banchs, R.; Cima, M.J. A three dimensional in vitro glial scar model to investigate the local strain effects from micromotion around neural implants. Lab Chip 2017, 17, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants (Basel) 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, S.; Guillemin, G.J.; Abiramasundari, R.S.; Essa, M.M.; Akbar, M.; Akbar, M.D. The Role of Reactive Oxygen Species in the Pathogenesis of Alzheimer's Disease, Parkinson's Disease, and Huntington's Disease: A Mini Review. Oxid Med Cell Longev 2016, 2016, 8590578. [Google Scholar] [CrossRef] [PubMed]
- Colton, C.A.; Chernyshev, O.N.; Gilbert, D.L.; Vitek, M.P. Microglial contribution to oxidative stress in Alzheimer's disease. Ann N Y Acad Sci 2000, 899, 292–307. [Google Scholar] [CrossRef]
- Han, N.; Ding, S.J.; Wu, T.; Zhu, Y.L. Correlation of free radical level and apoptosis after intracerebral hemorrhage in rats. Neurosci Bull 2008, 24, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Katsu, M.; Niizuma, K.; Yoshioka, H.; Okami, N.; Sakata, H.; Chan, P.H. Hemoglobin-induced oxidative stress contributes to matrix metalloproteinase activation and blood-brain barrier dysfunction in vivo. J Cereb Blood Flow Metab 2010, 30, 1939–1950. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Ereifej, E.S.; Schwartzman, W.E.; Meade, S.M.; Chen, K.; Rayyan, J.; Feng, H.; Aluri, V.; Mueller, N.N.; Bhambra, R.; et al. Investigation of the Feasibility of Ventricular Delivery of Resveratrol to the Microelectrode Tissue Interface. Micromachines 2021, 12. [Google Scholar] [CrossRef]
- Potter-Baker, K.A.; Nguyen, J.K.; Kovach, K.M.; Gitomer, M.M.; Srail, T.W.; Stewart, W.G.; Skousen, J.L.; Capadona, J.R. Development of Superoxide Dismutase Mimetic Surfaces to Reduce Accumulation of Reactive Oxygen Species Surrounding Intracortical Microelectrodes. Journal of Materials Chemistry B 2014, 2, 2248–2258. [Google Scholar] [CrossRef]
- Hernandez-Reynoso, A.G.; Krebs, O.K.; Sturgill, B.; Hoeferlin, G.F.; Radhakrishna, R.; Smith, T.J.; Zhang, J.; Mittal, G.; Thai, T.T.D.; Desai, M.S.; et al. The Effect of a Superoxide Dismutase Mimetic Coating on the Acute and Sub-Chronic Recording Performance of Planar Silicon Intracortical Microelectrode Arrays. Biomaterials 2023, Under Review.
- Idec, B. Tecfidera (dimethyl fumarate): US prescribing information. 2013.
- Administration, U.F.a.D. TECFIDERA™ prescribing information. 2013.
- Casili, G.; Campolo, M.; Paterniti, I.; Lanza, M.; Filippone, A.; Cuzzocrea, S.; Esposito, E. Dimethyl Fumarate Attenuates Neuroinflammation and Neurobehavioral Deficits Induced by Experimental Traumatic Brain Injury. J Neurotrauma 2018, 35, 1437–1451. [Google Scholar] [CrossRef]
- Campolo, M.; Casili, G.; Biundo, F.; Crupi, R.; Cordaro, M.; Cuzzocrea, S.; Esposito, E. The Neuroprotective Effect of Dimethyl Fumarate in an MPTP-Mouse Model of Parkinson's Disease: Involvement of Reactive Oxygen Species/Nuclear Factor-kappaB/Nuclear Transcription Factor Related to NF-E2. Antioxid Redox Signal 2017, 27, 453–471. [Google Scholar] [CrossRef]
- Cerina, M.; Narayanan, V.; Delank, A.; Meuth, P.; Graebenitz, S.; Gobel, K.; Herrmann, A.M.; Albrecht, S.; Daldrup, T.; Seidenbecher, T.; et al. Protective potential of dimethyl fumarate in a mouse model of thalamocortical demyelination. Brain Struct Funct 2018, 223, 3091–3106. [Google Scholar] [CrossRef]
- Gafson, A.R.; Savva, C.; Thorne, T.; David, M.; Gomez-Romero, M.; Lewis, M.R.; Nicholas, R.; Heslegrave, A.; Zetterberg, H.; Matthews, P.M. Breaking the cycle: Reversal of flux in the tricarboxylic acid cycle by dimethyl fumarate. Neurol Neuroimmunol Neuroinflamm 2019, 6, e562. [Google Scholar] [CrossRef]
- Kunze, R.; Urrutia, A.; Hoffmann, A.; Liu, H.; Helluy, X.; Pham, M.; Reischl, S.; Korff, T.; Marti, H.H. Dimethyl fumarate attenuates cerebral edema formation by protecting the blood-brain barrier integrity. Experimental neurology 2015, 266, 99–111. [Google Scholar] [CrossRef]
- Ruggieri, S.; Tortorella, C.; Gasperini, C. Pharmacology and clinical efficacy of dimethyl fumarate (BG-12) for treatment of relapsing-remitting multiple sclerosis. Ther Clin Risk Manag 2014, 10, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Saha, L.; Kumari, P.; Singh, J.; Bhatia, A.; Banerjee, D.; Chakrabarti, A. Effect of dimethyl fumarate on neuroinflammation and apoptosis in pentylenetetrazol kindling model in rats. Brain Res Bull 2019, 144, 233–245. [Google Scholar] [CrossRef]
- Abbas, K.; Breton, J.; Planson, A.G.; Bouton, C.; Bignon, J.; Seguin, C.; Riquier, S.; Toledano, M.B.; Drapier, J.C. Nitric oxide activates an Nrf2/sulfiredoxin antioxidant pathway in macrophages. Free Radic Biol Med 2011, 51, 107–114. [Google Scholar] [CrossRef]
- Innamorato, N.G.; Rojo, A.I.; Garcıa-Yague, A.J.; Yamamoto, M.; Ceballos, M.L.d.; Cuadrado, A. The Transcription Factor Nrf2 Is a Therapeutic Target against Brain Inflammation. The Journal of Immunology 2008, 181, 680–389. [Google Scholar] [CrossRef]
- Yadav, S.K.; Soin, D.; Ito, K.; Dhib-Jalbut, S. Insight into the mechanism of action of dimethyl fumarate in multiple sclerosis. J Mol Med (Berl) 2019, 97, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Al-Jaderi, Z.; Maghazachi, A.A. Utilization of Dimethyl Fumarate and Related Molecules for Treatment of Multiple Sclerosis, Cancer, and Other Diseases. Front Immunol 2016, 7, 278. [Google Scholar] [CrossRef]
- Mills, E.A.; Ogrodnik, M.A.; Plave, A.; Mao-Draayer, Y. Emerging Understanding of the Mechanism of Action for Dimethyl Fumarate in the Treatment of Multiple Sclerosis. Front Neurol 2018, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Parodi, B.; Rossi, S.; Morando, S.; Cordano, C.; Bragoni, A.; Motta, C.; Usai, C.; Wipke, B.T.; Scannevin, R.H.; Mancardi, G.L.; et al. Fumarates modulate microglia activation through a novel HCAR2 signaling pathway and rescue synaptic dysregulation in inflamed CNS. Acta Neuropathol 2015, 130, 279–295. [Google Scholar] [CrossRef]
- Hoeferlin, G.F.; Menendez, D.M.; Krebs, O.K.; Capadona, J.R.; Shoffstall, A.J. Assessment of Thermal Damage from Robot-Drilled Craniotomy for Cranial Window Surgery in Mice. JoVE 2022, In Press.
- Shoffstall, A.J.; Paiz, J.E.; Miller, D.M.; Rial, G.M.; Willis, M.T.; Menendez, D.M.; Hostler, S.R.; Capadona, J.R. Potential for thermal damage to the blood-brain barrier during craniotomy: implications for intracortical recording microelectrodes. J Neural Eng 2018, 15, 034001. [Google Scholar] [CrossRef]
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Sturgill, B.; Radhakrishna, R.; Thai, T.T.; Patnaik, S.S.; Capadona, J.R.; Pancrazio, J.J. Characterization of Active Electrode Yield for Intracortical Arrays: Awake versus Anesthesia. Micromachines 2022, 13. [Google Scholar] [CrossRef]
- Usoro, J.O.; Dogra, K.; Abbott, J.R.; Radhakrishna, R.; Cogan, S.F.; Pancrazio, J.J.; Patnaik, S.S. Influence of Implantation Depth on the Performance of Intracortical Probe Recording Sites. Micromachines 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Stiller, A.M.; Usoro, J.; Frewin, C.L.; Danda, V.R.; Ecker, M.; Joshi-Imre, A.; Musselman, K.C.; Voit, W.; Modi, R.; Pancrazio, J.J.; et al. Chronic Intracortical Recording and Electrochemical Stability of Thiol-ene/Acrylate Shape Memory Polymer Electrode Arrays. Micromachines 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Ereifej, E.S.; Smith, C.S.; Meade, S.M.; Chen, K.; Feng, H.; Capadona, J.R. The neuroinflammatory response to nanopatterning parallel grooves into the surface structure of intracortical microelectrodes. Advanced Functional Materials 2018, 28, 1704420. [Google Scholar] [CrossRef]
- Nguyen, J.K.; Jorfi, M.; Buchanan, K.L.; Park, D.J.; Foster, E.J.; Tyler, D.J.; Rowan, S.J.; Weder, C.; Capadona, J.R. Influence of resveratrol release on the tissue response to mechanically adaptive cortical implants. Acta Biomater 2016, 29, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Potter, K.A.; Simon, J.S.; Velagapudi, B.; Capadona, J.R. Reduction of autofluorescence at the microelectrode-cortical tissue interface improves antibody detection. J Neurosci Methods 2012, 203, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Lindner, S.C.; Yu, M.; Capadona, J.R.; Shoffstall, A.J. A graphical user interface to assess the neuroinflammatory response to intracortical microelectrodes. Journal of Neuroscience Methods 2019, 317, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Stringer, C.; Wang, T.; Michaelos, M.; Pachitariu, M. Cellpose: a generalist algorithm for cellular segmentation. Nat Methods 2021, 18, 100–106. [Google Scholar] [CrossRef]
- Bedell, H.W.; Schaub, N.J.; Capadona, J.R.; Ereifej, E.S. Differential expression of genes involved in the acute innate immune response to intracortical microelectrodes. Acta Biomater 2020, 102, 205–219. [Google Scholar] [CrossRef]
- Ramhoj, L.; Axelstad, M.; Svingen, T. Validation of endogenous reference genes in rat cerebral cortex for RT-qPCR analyses in developmental toxicity studies. PeerJ 2019, 7, e7181. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Regan, B.; Ereifej, E.S.; Chan, E.R.; Capadona, J.R. Neuroinflammatory Gene Expression Analysis Reveals Pathways of Interest as Potential Targets to Improve the Recording Performance of Intracortical Microelectrodes. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- S. Song, L.N.D., H.W. Bedell, R. Chan, J.R. Capadona. Differential expression of genes involved in the chronic response to intracortical microelectrodes. Acta Biomaterialia 2023. (Accepted).
- Readnower, R.D.; Chavko, M.; Adeeb, S.; Conroy, M.D.; Pauly, J.R.; McCarron, R.M.; Sullivan, P.G. Increase in blood-brain barrier permeability, oxidative stress, and activated microglia in a rat model of blast-induced traumatic brain injury. Journal of Neuroscience Research 2010, 88, 3530–3539. [Google Scholar] [CrossRef] [PubMed]
- McConnell, G.C.; Rees, H.D.; Levey, A.I.; Gutekunst, C.A.; Gross, R.E.; Bellamkonda, R.V. Implanted neural electrodes cause chronic, local inflammation that is correlated with local neurodegeneration. J Neural Eng 2009, 6, 056003. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab 2012, 32, 1959–1972. [Google Scholar] [CrossRef]
- Hsieh, C.L.; Kim, C.C.; Ryba, B.E.; Niemi, E.C.; Bando, J.K.; Locksley, R.M.; Liu, J.; Nakamura, M.C.; Seaman, W.E. Traumatic brain injury induces macrophage subsets in the brain. Eur J Immunol 2013, 43, 2010–2022. [Google Scholar] [CrossRef]
- Kumar, A.; Alvarez-Croda, D.M.; Stoica, B.A.; Faden, A.I.; Loane, D.J. Microglial/Macrophage Polarization Dynamics following Traumatic Brain Injury. J Neurotrauma 2016, 33, 1732–1750. [Google Scholar] [CrossRef]
- Alam, A.; Thelin, E.P.; Tajsic, T.; Khan, D.Z.; Khellaf, A.; Patani, R.; Helmy, A. Cellular infiltration in traumatic brain injury. J Neuroinflammation 2020, 17, 328. [Google Scholar] [CrossRef] [PubMed]
- Takmakov, P.; Ruda, K.; Phillips, K.S.; Isayeva, I.S.; Krauthamer, V.; Welle, C.G. Rapid evaluation of the durability of cortical neural implants using accelerated aging with reactive oxygen species. Journal of neural engineering 2015, 12, 026003. [Google Scholar] [CrossRef]
- Nolta, N.F.; Christensen, M.B.; Crane, P.D.; Skousen, J.L.; Tresco, P.A. BBB leakage, astrogliosis, and tissue loss correlate with silicon microelectrode array recording performance. Biomaterials 2015, 53, 753–762. [Google Scholar] [CrossRef]
- Saxena, T.; Karumbaiah, L.; Gaupp, E.A.; Patkar, R.; Patil, K.; Betancur, M.; Stanley, G.B.; Bellamkonda, R.V. The impact of chronic blood–brain barrier breach on intracortical electrode function. Biomaterials 2013, 34, 4703–4713. [Google Scholar] [CrossRef]
- Landsiedel, R.; Hahn, D.; Ossig, R.; Ritz, S.; Sauer, L.; Buesen, R.; Rehm, S.; Wohlleben, W.; Groeters, S.; Strauss, V.; et al. Gut microbiome and plasma metabolome changes in rats after oral gavage of nanoparticles: sensitive indicators of possible adverse health effects. Particle and Fibre Toxicology 2022, 19, 1828. [Google Scholar] [CrossRef]
- Castaneda, M.; Smith, K.M.; Nixon, J.C.; Hernandez, C.J.; Rowan, S. Alterations to the gut microbiome impair bone tissue strength in aged mice. Bone Rep 2021, 14, 101065. [Google Scholar] [CrossRef] [PubMed]
- Galland, L. The gut microbiome and the brain. J Med Food 2014, 17, 1261–1272. [Google Scholar] [CrossRef]
- Zhu, S.; Jiang, Y.; Xu, K.; Cui, M.; Ye, W.; Zhao, G.; Jin, L.; Chen, X. The progress of gut microbiome research related to brain disorders. J Neuroinflammation 2020, 17, 25. [Google Scholar] [CrossRef] [PubMed]
- Gershon, M.D.; Margolis, K.G. The gut, its microbiome, and the brain: connections and communications. J Clin Invest 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Hanscom, M.; Loane, D.J.; Shea-Donohue, T. Brain-gut axis dysfunction in the pathogenesis of traumatic brain injury. J Clin Invest 2021, 131. [Google Scholar] [CrossRef]
- Dinan, T.G.; Cryan, J.F. Brain-Gut-Microbiota Axis and Mental Health. Psychosom Med 2017, 79, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Fulling, C.; Dinan, T.G.; Cryan, J.F. Gut Microbe to Brain Signaling: What Happens in Vagus. Neuron 2019, 101, 998–1002. [Google Scholar] [CrossRef]
- Brown, A.P.; Dinger, N.; Levine, B.S. Stress produced by gavage administration in the rat. Contemp Top Lab Anim Sci 2000, 39, 17–21. [Google Scholar]
- Dobrakovova, M.; Jurcovicova, J. Corticosterone and prolactin responses to repeated handling and transfer of male rats. Exp Clin Endocrinol 1984, 83, 21–27. [Google Scholar] [CrossRef]
- Sandberg, M.; Patil, J.; D'Angelo, B.; Weber, S.G.; Mallard, C. NRF2-regulation in brain health and disease: implication of cerebral inflammation. Neuropharmacology 2014, 79, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Zaro, B.W.; Vinogradova, E.V.; Lazar, D.C.; Blewett, M.M.; Suciu, R.M.; Takaya, J.; Studer, S.; de la Torre, J.C.; Casanova, J.L.; Cravatt, B.F.; et al. Dimethyl Fumarate Disrupts Human Innate Immune Signaling by Targeting the IRAK4-MyD88 Complex. J Immunol 2019, 202, 2737–2746. [Google Scholar] [CrossRef] [PubMed]
- Montes Diaz, G.; Fraussen, J.; Van Wijmeersch, B.; Hupperts, R.; Somers, V. Dimethyl fumarate induces a persistent change in the composition of the innate and adaptive immune system in multiple sclerosis patients. Sci Rep 2018, 8, 8194. [Google Scholar] [CrossRef]
- Winkelmann, A.; Loebermann, M.; Reisinger, E.C.; Hartung, H.P.; Zettl, U.K. Disease-modifying therapies and infectious risks in multiple sclerosis. Nat Rev Neurol 2016, 12, 217–233. [Google Scholar] [CrossRef]
- Longbrake, E.E.; Naismith, R.T.; Parks, B.J.; Wu, G.F.; Cross, A.H. Dimethyl fumarate-associated lymphopenia: Risk factors and clinical significance. Mult Scler J Exp Transl Clin 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Dobariya, A.; El Ahmadieh, T.Y.; Good, L.B.; Hernandez-Reynoso, A.G.; Jakkamsetti, V.; Brown, R.; Dunbar, M.; Ding, K.; Luna, J.; Kallem, R.R.; et al. Recording of pig neuronal activity in the comparative context of the awake human brain. Sci Rep 2022, 12, 15503. [Google Scholar] [CrossRef]
- Raspopovic, S.; Cimolato, A.; Panarese, A.; Vallone, F.; Del Valle, J.; Micera, S.; Navarro, X. Neural signal recording and processing in somatic neuroprosthetic applications. A review. J Neurosci Methods 2020, 337, 108653. [Google Scholar] [CrossRef]
- Carnicer-Lombarte, A.; Chen, S.T.; Malliaras, G.G.; Barone, D.G. Foreign Body Reaction to Implanted Biomaterials and Its Impact in Nerve Neuroprosthetics. Front Bioeng Biotechnol 2021, 9, 622524. [Google Scholar] [CrossRef]
Figure 1.
Neurophysiological recording activity to evaluate the A) proportion of active electrodes (AEY %) on each recording device for comparison between DMF-treated animals and control animals. Additional direct comparisons from the same data set across neuroinflammatory phases for DMF-treated animals (B) or control animals (C). D) Evaluation of the number of units detected per active electrode compared between DMF and control. E) Week-by-Week comparison of MEA recording performance for each group. Significance denoted as p < 0.05 = *, p < 0.01 = **, p < 0.001 = ***, p < 0.0001 = ****. No symbol indicates a lack of statistical significance. Sample size for A, B, and C is determined by total number of electrodes multiplied by number of weeks in each phase and number of animals in each group. Sample size for D is determined by the number of active electrodes multiplied by the number of weeks in each time point and the number of animals in each group. Sample size for E is determined by total number of electrodes multiplied by number of animals for each group on a week-by-week basis.
Figure 1.
Neurophysiological recording activity to evaluate the A) proportion of active electrodes (AEY %) on each recording device for comparison between DMF-treated animals and control animals. Additional direct comparisons from the same data set across neuroinflammatory phases for DMF-treated animals (B) or control animals (C). D) Evaluation of the number of units detected per active electrode compared between DMF and control. E) Week-by-Week comparison of MEA recording performance for each group. Significance denoted as p < 0.05 = *, p < 0.01 = **, p < 0.001 = ***, p < 0.0001 = ****. No symbol indicates a lack of statistical significance. Sample size for A, B, and C is determined by total number of electrodes multiplied by number of weeks in each phase and number of animals in each group. Sample size for D is determined by the number of active electrodes multiplied by the number of weeks in each time point and the number of animals in each group. Sample size for E is determined by total number of electrodes multiplied by number of animals for each group on a week-by-week basis.
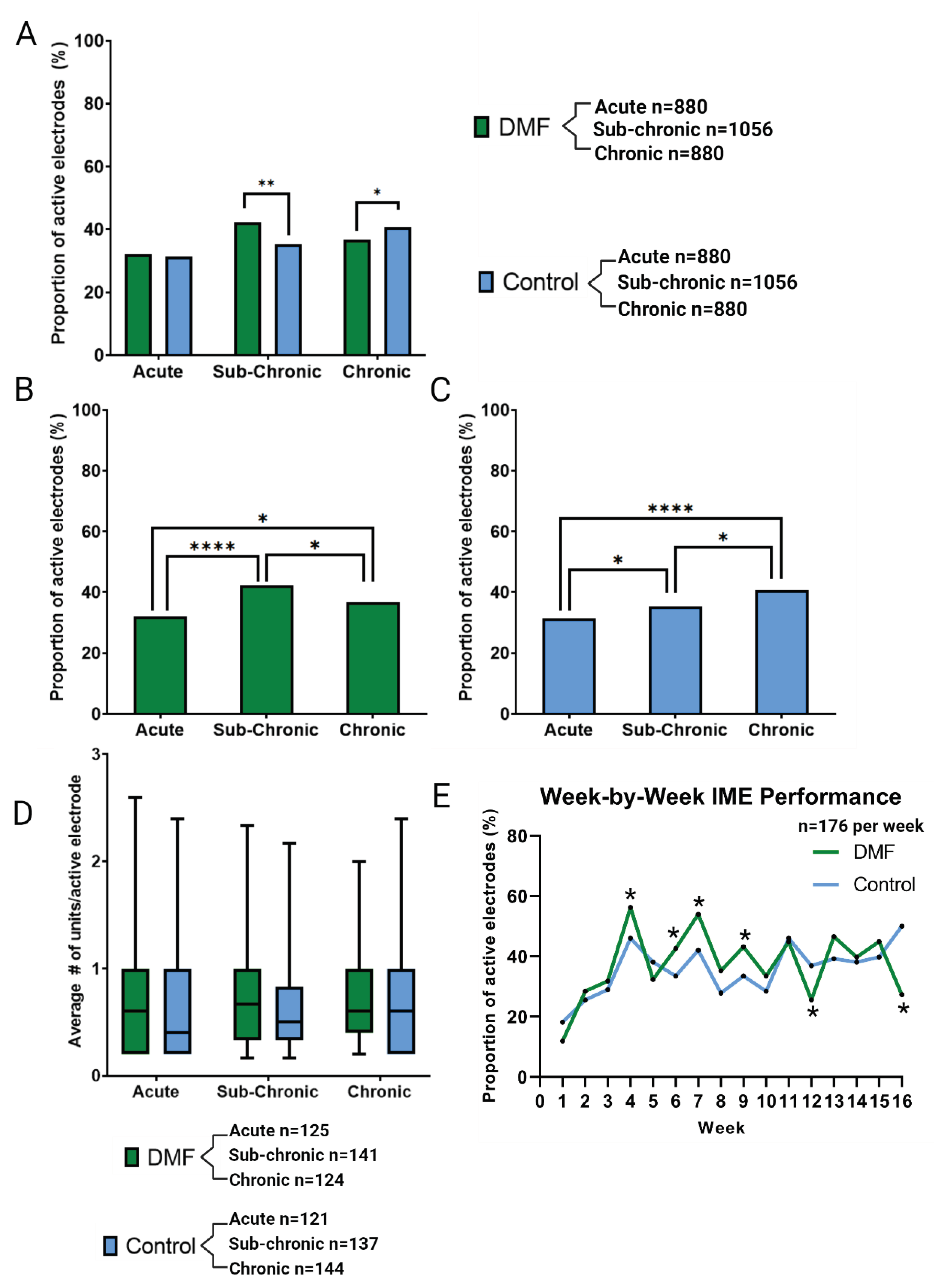
Figure 2.
Additional neurophysiological metrics to evaluate recordings performance, including A) Peak-to-peak voltage of signals (Vpp), B) Noise levels for each electrode, C) Signal-to-noise ratio, and D) the median spike rate for activity. Significance denoted as p < 0.05 = *, p < 0.01 = **, p < 0.001 = ***, p < 0.0001 = ****. No symbol indicates a lack of significance. Sample size is determined by the number of active electrodes multiplied by the number of weeks in each time point and the number of animals in each group.
Figure 2.
Additional neurophysiological metrics to evaluate recordings performance, including A) Peak-to-peak voltage of signals (Vpp), B) Noise levels for each electrode, C) Signal-to-noise ratio, and D) the median spike rate for activity. Significance denoted as p < 0.05 = *, p < 0.01 = **, p < 0.001 = ***, p < 0.0001 = ****. No symbol indicates a lack of significance. Sample size is determined by the number of active electrodes multiplied by the number of weeks in each time point and the number of animals in each group.
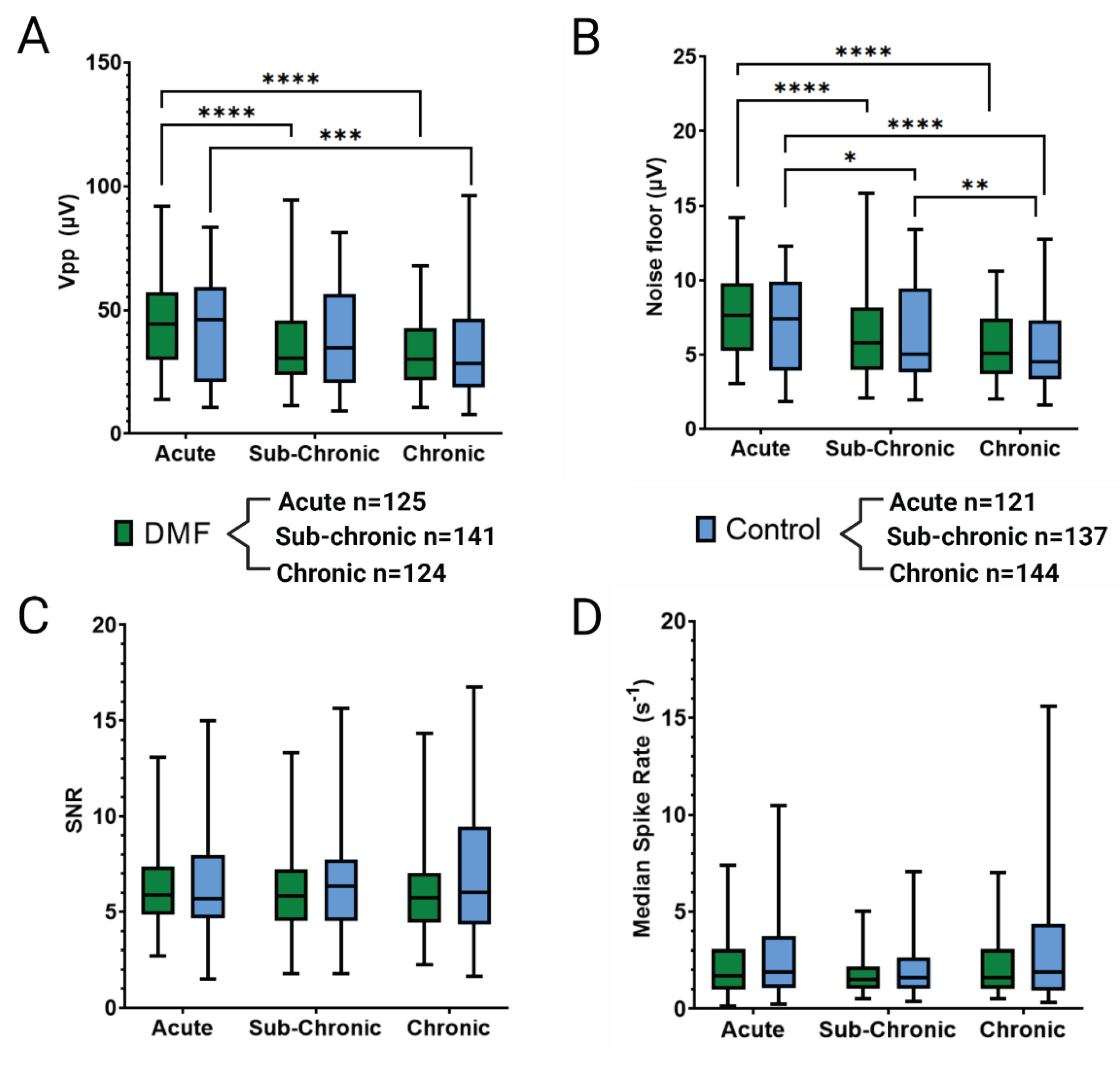
Figure 3.
Immunohistochemical evaluation of the implant site 16 weeks post-implantation. Representative images for each stain are included to the right of the quantification plots. A) CD68 expression indicating the presence of active microglia and macrophages, B) IgG expression indicating BBB stability, C) GFAP activity indicating the presence of hypertrophic astrocytes and glial scar formation, and D) Neuronal Nuclei (NeuN) expression to evaluate the density of neurons around the implant. Scale bars = 200 µm. Significance is denoted as p < 0.05 = *. No symbol indicates a lack of significance. Variation in sample size is a result of a set of defective microscope slides leading to a loss in brain tissue when staining.
Figure 3.
Immunohistochemical evaluation of the implant site 16 weeks post-implantation. Representative images for each stain are included to the right of the quantification plots. A) CD68 expression indicating the presence of active microglia and macrophages, B) IgG expression indicating BBB stability, C) GFAP activity indicating the presence of hypertrophic astrocytes and glial scar formation, and D) Neuronal Nuclei (NeuN) expression to evaluate the density of neurons around the implant. Scale bars = 200 µm. Significance is denoted as p < 0.05 = *. No symbol indicates a lack of significance. Variation in sample size is a result of a set of defective microscope slides leading to a loss in brain tissue when staining.
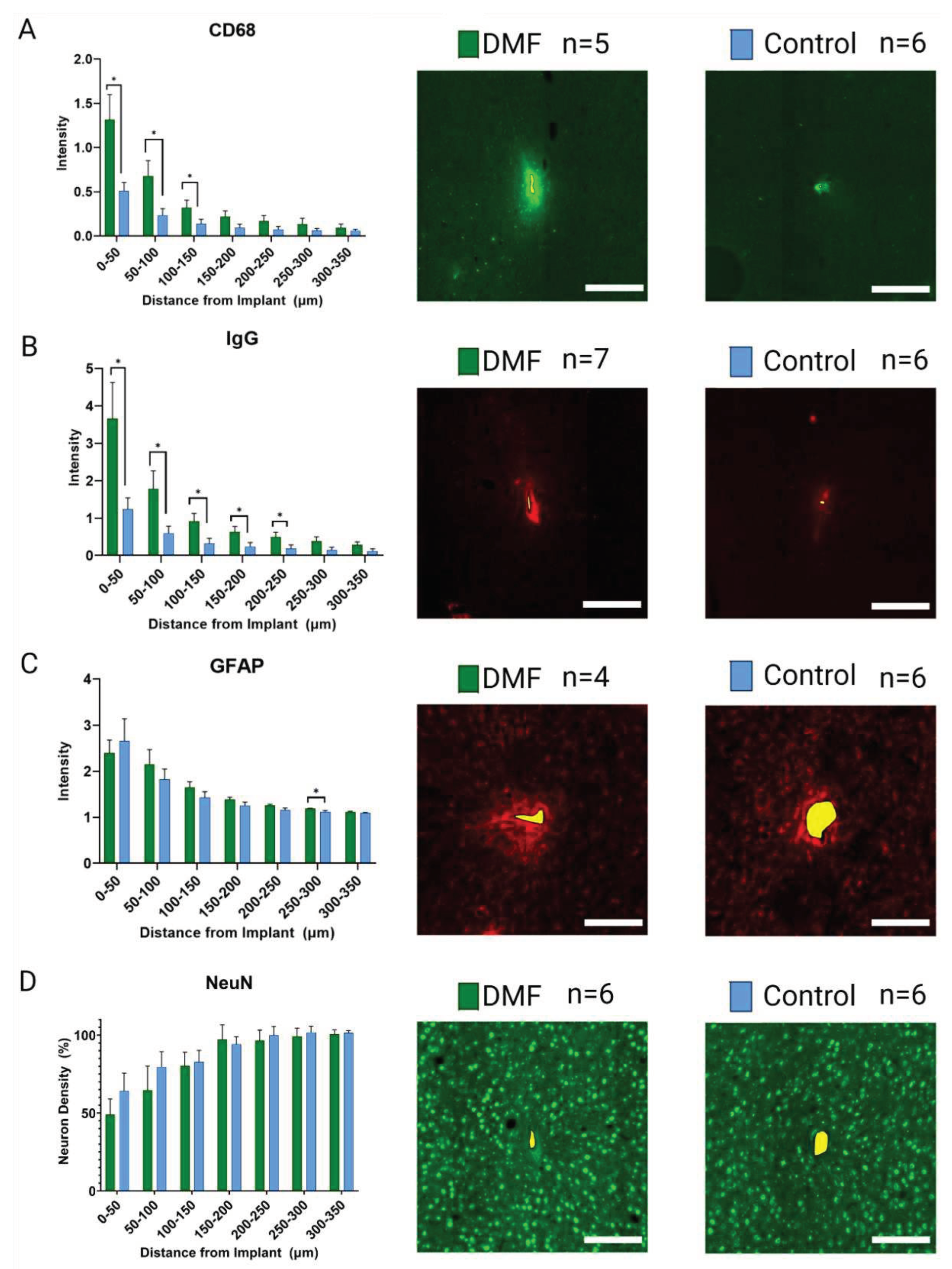
Figure 4.
Bulk gene analysis 7-weeks post implantation on 152 oxidative stress and neuroinflammatory genes associated with neural implants. Log2(fold change) is based on DMF values compared to the control using differential expression analysis with a 2-tailed unequal variance t-test for each gene. Grey points indicate no significant difference compared to control. N = 6 for DMF-treated; N = 7 for Control.
Figure 4.
Bulk gene analysis 7-weeks post implantation on 152 oxidative stress and neuroinflammatory genes associated with neural implants. Log2(fold change) is based on DMF values compared to the control using differential expression analysis with a 2-tailed unequal variance t-test for each gene. Grey points indicate no significant difference compared to control. N = 6 for DMF-treated; N = 7 for Control.

Table 1.
Complete list of neuroinflammatory and oxidative stress genes of interest utilized in the study. Here we list the 152 genes examined in rat brain tissue in this study using a combination of custom genes (shown in red with light grey shading) and preset genes from NanoString (shown in black). Housekeeping genes are highlighted in blue with dark grey shading.
Table 1.
Complete list of neuroinflammatory and oxidative stress genes of interest utilized in the study. Here we list the 152 genes examined in rat brain tissue in this study using a combination of custom genes (shown in red with light grey shading) and preset genes from NanoString (shown in black). Housekeeping genes are highlighted in blue with dark grey shading.
| Abl1 | C4A | Cycs | GSTA1 | Ins2 | Ngfg | Prnp | SPP1 |
| Ager | C5AR1 | Ddit3 | Gsta2 | Ipcef1 | Ngfr | Psen1 | Src |
| Aif1 | Casp3 | Dnm2 | GSTM2 | IRAK4 | Nme5 | PSMB8 | Srxn1 |
| AIM2 | CASP8 | DOCK2 | Gstp1 | IRF7 | Nol3 | Ptgs2 | Stx2 |
| Akt1 | CCL1 | Ehd2 | Gucy1b3 | ITGAM | Nos1 | PTPN6 | Tbp |
| Apoe | Ccl5 | Ep300 | H2-T23 | Jun | Nos3 | PTX3 | Tnf |
| App | Ccs | Ercc6 | Hdac2 | KEAP1 | Noxa1 | Rela | TNFRSF1A |
| ARC | CD14 | Fas | Hdac6 | LILRB4A | Nqo1 | Rpl13a | TNFRSF25 |
| Atf4 | CD36 | FCER1G | Hgf | Lpo | Nr2f6 | Rps18 | Tor1a |
| Atp13a2 | CD45 | FCGR2B | Hif1a | Lrrk2 | Nr4a2 | SCD1 | Tpm1 |
| Atp7a | CD68 | Fn1 | Hmox1 | Mapt | Osgin1 | Sdha | Trp53 |
| Atrn | CD74 | Fos | Hprt | Mgmt | OSMR | SERPINA3N | Trpm2 |
| Bad | CD84 | Fxn | Hspb1 | MMP12 | Oxr1 | Sirt1 | Txnl1 |
| Bcl2 | Cdk2 | GFAP | Htra2 | Mmp14 | Park7 | Sirt2 | Txnrd1 |
| Bdnf | CIM | Gnao1 | Idh1 | MPEG1 | Parp1 | Slc8a1 | TYROBP |
| BLNK | CLEC7A | Gpr37 | Il1b | Mutyh | Pdgfrb | Snca | Ubc |
| Bnip3 | Cln8 | Gsk3b | Il1r1 | Ncf1 | Pink1 | Sod1 | Ubqln1 |
| C3 | CTSS | Gsr | IL2RG | Nefh | Pla2g4a | Sod2 | Vegfa |
| C3AR1 | Cybb | Gss | Il6 | Nfe2l2 | Ppargc1a | Sod3 | Xbp1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



