Submitted:
16 August 2023
Posted:
18 August 2023
You are already at the latest version
Abstract
The human T-cell leukemia virus type 1 (HTLV-1) is the only known human oncogenic retrovirus. HTLV-1 can cause a cancer called adult T-cell leukemia/lymphoma (ATL). The virus is transmitted through body fluids of infected individuals, primarily breast milk, blood, and semen. At least 5–10 million people in the world are infected with HTLV-1. In addition to ATL, HTLV-1 infection can also cause HTLV-I-associated myelopathy (HAM /TSP). ATL is characterized by low viral ex-pression and poor prognosis. The oncogenesis triggered by HTLV-1 is extremely complex and the molecular mechanisms are not fully understood. However, viral regulatory proteins Tax and HTLV-1 bZIP factor (HBZ) have been shown to play key roles in the transformation of HTLV-1-infected T cells. Moreover, several studies have shown that the final fate of HTLV-1-infected transformed T cell clones is the result of a complex interplay of HTLV-1 oncogenic protein expression with cellular transcription factors that subvert the cell cycle and disrupt reg-ulated cell death, thereby exerting their transforming effects. This review provides updated in-formation on the mechanisms underlying the transforming action of HTLV-1 and the potential therapeutic targets to combat ATL.
Keywords:
HTLV-1
; adult T-cell leukemia
; viral oncogenesis
; Tax
; HBZ
; apoptosis
1. Introduction
Human T-cell leukemia virus type 1 (HTLV-1), also known as human T-lymphotropic virus type 1, was the first human retrovirus discovered. It belongs to subtype Delta of the subfamily Orthoretrovirinae and is endemic to the southwestern part of Japan, South America, the Caribbean, the Middle East, Australo-Melanesia [1], and western and sub-Saharan Africa [2]. HTLV-1 has been recognized as the causative agent of adult T-cell leukemia/lymphoma (ATL), HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM /TSP), and other inflammatory diseases. ATL is an aggressive malignancy that usually occurs in adults, 30-50 years after HTLV-1 infection and in approximately 5% of infected individuals. The prognosis of ATL is very poor, with median survival ranging from 6 to 24 months. It is estimated that 5-10 million people are infected with HTLV-1 worldwide, although this number is most likely underestimated due to the lack of broader epidemiological studies. In vivo human-to-human transmission occurs through breastfeeding, blood transfusions, needle sharing, and sexual intercourse [3,4], and rapid spread of HTLV-1 has been demonstrated in transplant recipients [5]. To design a scenario of HTLV-1 infection/transformation occurring in vivo, several noteworthy questions need to be addressed. Clarification of these questions may represent the complex interplay between HTLV-1 and host gene expression that characterizes leukemogenesis and viral persistence in ATL [6]. This review will provide detailed information on the current knowledge of the interplay between the oncogenic viral proteins and the cellular factors underlying the transforming action of HTLV-1 and provide a brief overview of the potential therapeutic targets arising from this interplay and possible future therapeutic approaches.
2. HTLV-1 genome and virus transmission and spread
The HTLV-1 genome consists of a small positive ss(+) RNA with a size of approximately 9 kB. After infection, HTLV-1 is integrated into the host DNA as a provirus, similar to HIV-1. Both the sense strand and the anti-sense strand of the integrated provirus can be transcribed [7]. The viral genome is flanked by long terminal repeats (LTRs) at both the 5’ and 3’ ends. These direct repeats consist of three regions: the unique 3 (U3), the repeated (R), and the unique 5 (U5) region. The LTRs also contain important elements necessary for viral transcription, polyadenylation, and integration [8]. The HTLV-1 genome includes the standard structural and enzyme genes gag, pro, pol and env, which encode proteins essential for viral replication [9], as well as the coding potential for the accessory/regulatory proteins Tax, Rex, p12, p13 and p30 [10-13]. All these proteins are encoded by the sense strand, whereas the regulatory protein HTLV-1 bZIP factor (HBZ) is encoded by the anti-sense strand. HTLV-1 is a suitable model to study infection in vitro. Because HTLV-1 is strongly cell-associated, unlike HIV-1, cell-free transmission is very limited in favor of transmission by cell-to-cell contact. Cell-to-cell transmission of HTLV-1 is facilitated by several transport strategies that allow the virus to spread bypassing the host immune response. These strategies include transcytosis through epithelial barriers [14], and induction of membrane structures such as virological synapses [15], cellular conduits [16], and extracellular vesicles [17], which have been defined. Viral infection in vitro usually requires cocultivation between recipient lymphocytes and irradiated infected cells in the presence of IL -2 as a growth factor. After several passages in vitro, the number of which varies depending on the different cocultures, the newly infected cells can be immortalized, as indicated by the fact that they no longer require growth factor. After months in culture, they are then transformed [18]. During the period between infection and immortalization in vitro, different cells in the infected cultures undergo different and opposite fates simultaneously: some of them undergo strong proliferation and others undergo apoptosis. This might depend on the balance at the cellular level between the differential expression of viral and cellular proteins and the influence on the survival/death pathways induced by viral gene expression in the infected cells [4], as detailed in the next paragraph. Although this process has been described in detail in vitro, it is reasonable to assume that a similar pattern could occur in vivo. The spread of the virus in vivo, similar to all retroviruses, occurs through two distinct pathways: the infectious pathway, from cell to cell, and the mitotic pathway, once the HTLV-1 genome is integrated into the host cell genome [19]. It has been clarified that infectious spread persists in parallel with mitotic spread even during the chronic phase of HTLV-1 infection [20]. HTLV-1 primarily infects CD4+ T cells, the population that is preferentially selected after in vitro infection. Clonal expansion of HTLV-1-infected cells is influenced by the host immune response [21], and fluctuating levels of the virus LTR in vivo seem to indicate that the virus actively replicates during chronic infection [22]. Studies aimed at comparing the clone abundance distribution of the provirus in CD4+ and CD8+ T cell subpopulations in HTLV-1 infected individuals have shown that CD8+ T cells carried only 5% of the provirus load, whereas the provirus was present in a greater number of CD4+ clones [23]. Nevertheless, CD8+ T cells expand strongly in vivo, but their role has not been fully elucidated [23]. The mechanisms of cell-to-cell transmission of HTLV-1 are summarized in Figure 1. Essentially, ATL is the end result of selecting an infected clone, in most cases CD4+, from the many infected clones. The selected clone has acquired the characteristics of transformed cells over many years. The mechanisms underlying this long and complex process have been unraveled in recent years, thanks largely to advances in genomics and single-cell technology. See subsequent sections and reference [24].
3. ATL and HTLV-1-driven transformation: generalities
ATL develops in 5% of individuals infected with HTLV-1 and is characterized by lymphadenopathy, skin lesions, hypercalcemia, and severe involvement of organs such as the central nervous system, lungs, liver, spleen, and bone marrow. Survival prognosis is poor and may be limited to 6 months. ATL patients respond poorly to traditional anti-leukemia chemotherapy. Rather, more favorable responses have been achieved by combined treatment with zidovudine (AZT) plus interferon (IFN) [25] or allogeneic hematopoietic stem cell transplantation [26]. In addition, biologic therapy has relied on mogamulizumab, which targets chemokine receptor 4 [27], and the immunomodulator and antitumoral lenalinomide [28], a human CD30-directed chimeric antibody bound to the microtubule-disrupting agent [29], the EZH1 and EZH2 dual inhibitor valemetostat, which disrupts the hypermethylation of histone H3 lysine 27 (H3K27) and allows the re-expression of repressed genes [30], and histone deacetylase (HDAC) inhibitors [31,32]. The results of the above studies, although not informative, encourage finding potential new biological targets in the mechanism of viral transformation (see Section 6 and Section 7). Several studies have shown that the final fate of transformed T cell clones infected with HTLV-1 is the result of a fine-tuned regulation of viral gene expression. In particular, the balance between the two major oncogenic viral proteins Tax and HBZ determines the fate of HTLV-1 infection [33]. Therefore, a regulated interplay between cell survival and cell death plays a key role in the selection of malignant clones. One of the most important factors in the transformation and pathogenesis of HTLV-1 is the multifunctional viral protein Tax. This protein has the ability to interact directly with a variety of cellular proteins, including transcription factors, cell signaling proteins, cell cycle regulators, apoptotic proteins, and DNA damage response factors. The sustained activation of the cellular transcription factor Nuclear Factor-κB (NF-κB) by Tax [34] plays an important role in its multifunctionality towards cellular targets (see section 4). In addition, Tax is known to play a role as a transactivator and regulator of transcription of HTLV-1 structural and enzymatic proteins and its own transcription through interaction with the promoter within the 5’ LTR. Tax-mediated transactivation requires binding to a repeating sequence of21 nucleotides rich in G/C, representing the Tax-responsive element (TRE) located in the U3 region of the 5’ LTR [35]. Tax does not bind directly to DNA. Instead, it recruits the cAMP response element-binding protein (CREB)/activating transcription factor (ATF) to the cyclic AMP response elements (CREs) [36]. This leads to the formation of a nucleoprotein complex that recruits other CREB-binding proteins (CBP) and p300 [37]. This multiprotein complex is a potent activator of the 5’ LTR promoter and viral mRNA transcription. On the other hand, Tax can also upregulate viral transcription by directly binding to an epigenetic repressor, histone deacetylase 1 (HDAC1). Therefore, Tax inhibits or dissociates the binding of HDAC1 to the HTLV-1 promoter, thereby regulating viral protein transcription [38].
4. Tax and cell signalling: role of the transcription factor nuclear NF-κB
In addition to regulating viral transcription, Tax regulates the expression of several signaling molecules involved in the processes of oncogenesis, proliferation, immune response, and apoptosis. The nuclear transcription factor NF-κB plays a central role in coordinating the various cellular signals. The prototypical NF-κB complex corresponds to a heterodimer of the p50 (NFKB1) and RelA (p65) members of the NF-κB/Rel family of transcription factors [39]. Tax induces phosphorylation and degradation of both IκBα and IκBβ, suggesting that this HTLV-1 regulatory protein can induce nuclear translocation of NF-κB by acting upstream of or at the level of IκB phosphorylation [40,41]. Activation of NF-κB is involved in a number of events involving cytokine production in an inflammatory context. Evidence that Tax regulates the expression of multiple cytokines is that its inhibition is dependent on the inhibition of NF-κB in HTLV-1-infected cells [42]. Sustained activation of NF-κB has a remarkable impact on immortalization and transformation of HTLV-I infected cells. Reduction of Tax in primary ATL cells rapidly abrogates NF-κB activation, leading to induction of apoptosis [43]. One of the features that characterize immortalized HTLV-1 cells is the strong expression of anti-apoptotic genes. A Tax-mediated increase in the expression of Bfl-1, a gene that belongs to the Bcl-2 family and is strongly expressed in HTLV-1-infected cells T cell lines, has been reported [44]. Direct evidence for the role of Bcl-2 family genes was provided by the fact that silencing of Bfl-1 and Bcl-xL decreased the survival of HTLV-1 cells [44]. In vitro studies have shown that the anti-apoptotic survivin promoter was also transactivated by Tax via activation of NF-κB [45]. Nevertheless, Tax has not been exclusively associated with the inhibition of apoptosis, but also with its activation (see Section 5).
5. Latency and leukemogenesis: role of Tax, HBZ and apoptosis
HTLV-1 infection can remain latent for years before full disease onset, suggesting that there are sophisticated mechanisms regulating the on/off switching of viral protein expression, but also an increase in function of genes related to the T cell receptor/NF-kB and signalling related to immune surveillance such as HLA and FAS [46]. The outcome and progression of infection is regulated by Tax, whose expression is also critical for stimulating the cytotoxic T-lymphocytes immune response which is thought to play a role in the viability of ATL long-term survivors [47]. Loss or reduction of Tax expression in immortalised cells protects them from immune response attack and renders them vulnerable to survival, expansion, and proliferation through a process of continuous transient expression [48]. However, Tax is not the only responsible for the various changes observed in viral expression and cell fate during infection. As already indicated in Section 3, HBZ also plays an important role by counteracting Tax to some extent and, on the other hand, integrating the process of virus-induced leukemogenesis [49]. In fact, HBZ does not directly affect HTLV-1-mediated immortalization: rather, it regulates the establishment and maintenance of chronic infection [50]. Expression of hbz has been shown to upregulate JunD abundance and HBZ heterodimerizes with JunD, which recruits the transcription factor Sp1 to the 3’ LTR of the provirus to enhance its activity [51]. Moreover, HBZ is indirectly involved in leukemogenesis by increasing the expression of two oncogenic miRNA, miR-17 and miR-21. These miRNA in turn downregulate the expression of the single-stranded DNA-binding protein hSSB2, thus promoting genomic instability [52]. Another indication of the possible involvement of HBZ in neoplastic transformation is the detection of its translocation to the nucleus in cells from ATL patients [53]. Other accessory proteins play a selective role in HTLV-1 leukemogenesis. The p30 counteracts viral transformation by inhibiting the export of tax/rex mRNA from the nucleus [54], while p13 is involved in the transformation by increasing the intracellular content of reactive oxygen species [55]. On the other hand, p12 enhances STAT5 activation in transduced peripheral blood mononuclear cells, allowing them to proliferate even in the presence of a low IL -2 concentration [56]. Thus, a number of different mechanisms underlie HTLV-1 latency/leukemogenesis involving both viral and cellular signals, although the major players are Tax and HBZ proteins. Apparently, Tax is involved in the initiation of immortalization/transformation, whereas HBZ is required for the maintenance of the transformed stage. Nevertheless, the differential low expression of Tax and HBZ at different stages of infection appears to favour the establishment of a dynamic, rather than static, state of latency and persistence. HBZ has been shown to inhibit viral sense transcription and favour the entry of HTLV-1 provirus into the latency phase [57]. Ex vivo experiments have shown that after HTLV-1 provirus integration into the cell, its replication essentially consists of a series of mechanisms based on successive phases of burst and reactivation alternating with viral latency and controlled by Tax, with the expression of pro- and anti-apoptotic genes playing a central role over cell cycle gene expression [58]. In addition, the role of Tax in the switch between life and death leading to the selection of immortalized clones as the final phase of the HTLV-1 transformation process has been investigated by experimental in vitro models (see below). However, an important point in this context is that seemingly conflicting results suggest that the role of Tax in controlling apoptosis is not clear. Indeed, several studies have supported a preventive effect of HTLV-1 gene expression against apoptosis in infected cells and have shown a reduced susceptibility to apoptosis induced by anti-Fas [59] and TNFα [60]. This resistance to apoptosis has been associated with activation of NF-κB via the Tax protein in HTLV-1 infected cells [45], or with Tax-induced repression of pro-apoptotic genes [61], or expression of anti-apoptotic genes [62]. However, despite the evidence for the prevention of cell death during HTLV-1 infection, other studies reported that the viral protein Tax not only exerts some anti-apoptotic effects but is also responsible for promoting apoptosis in HTLV-1-infected cells under certain experimental conditions, as also mentioned in the previous paragraph. Clear evidence for this phenomenon was first provided by a study reporting that the expression of Tax in an inducible system induced rather than inhibited cell death [63]. The same and other authors even provided mechanistic details on apoptosis induction by Tax expression, showing that Tax-triggered death was dependent on ICEprotease [64], related to upregulation of the Fas ligand (FasL) gene [65], and presumably dependent on nuclear expression of the CREB-binding protein (CBP)/p300-binding domain of the HTLV-1transactivator Tax [66]. In another study, Tax was shown to induce cell death through NF-κB-mediated activation of tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) [67]. Taken together, these data fit the apparent paradox of a dual role of Tax for apoptotic cell death in the early phase preceding the completion of the transformation process triggered by HTLV-1. To investigate this apparent paradox, some of us pioneered the experimental model of HTLV-1 infection in vitro several years ago. Our model consisted of long-term culture of PBMCs from healthy donors after exposure to irradiated HTLV-1-infected cells as viral cell donor [68]. We felt that such a model was the best strategy to recapitulate in vitro what occurs naturally in vivo in HTLV-1-infected patients during the long period of immortalization and transformation that could lead to ATL. Indeed, the results of this study have shown that in vitro HTLV-1 infection of mononuclear cells induces a high input of cell proliferation during the first weeks with concomitant high susceptibility to regulated cell death (RCD) through apoptosis and a progressive decrease in cell death rates during the phase of long-term culture towards immortalization [68]. Moreover, in situ hybridization showed that the cells undergoing apoptosis were indeed infected and had HTLV-1 proviral DNA, rather than residual uninfected cells [68]. Although this event was observed in a mass population, we hypothesized that some infected cells died by apoptosis because pro-apoptotic signals prevailed in response to infection triggered by viral gene expression, whereas the infected cells that survived were protected by the prevalent expression of anti-apoptotic genes, which were themselves triggered by viral proteins. Moreover, based on our results, we suggested for the first time that induction of massive apoptosis in response to infection might act as a selection pressure for the emergence of anti-apoptotic, well-endowed infected clones prone to immortalization [68]. A more recent study performed at the single cell level in the MT -1 ATL cell line attempted to further elucidate the same paradox of Tax's opposing effects on cell death by demonstrating a situation in which Tax expression affects susceptibility to apoptosis. The results showed that a balance between antiapoptotic and proapoptotic genes depended on the on/off of Tax expression in the cells used in this study. High expression of Tax was preferentially associated with an antiapoptotic gene expression scenario, whereas low or absent expression of Tax was associated with greater susceptibility to apoptosis. This switch was continuous in culture and due to the coexistence of different expanding clonesnt expanding clones [69]. To explain the dual role of Tax in ATL, in which cells progress in the cell cycle despite low Tax expression, the results of another study hypothesized that ATL cells acquire genetic/epigenetic alterations during the transformation process that can bypass the Tax/NF-kB-dependent induction of senescence [70]. However, it should be considered that the above studies were performed on ATL cells in which the HTLV-1 transformation process had already been completed. Thus, the role of Tax in these cells could explain what happens in the cells of ATL patients in an advanced phase of the disease to maintain the leukemic state and not describe the role of Tax in the long premalignant phase of HTLV-1 infection, characterized by oligo-/polyclonal expansion of non-malignant HTLV-1-infected cells, which precedes overt ATL in patients and actually involves HTLV-1-driven oncogenesis. A recent study investigated the association between early transcription of the positive viral strand, viral burst,and expression of pro- and anti-apoptotic genes in two naturally infected T cell clones from patients transduced with a Tax-responsive timer protein [58]. The results showed that anti-apoptotic genes were expressed during the early positive-strand virus burst, followed by a phase in which proapoptotic genes outlasted the virus burst [58]. Another study of naturally HTLV-1-infected patient Tax-expressing T cell clones showed that high Tax expression occurred during the burst phase, immediately followed by a phase of Tax expression heterogeneity associated with poor proliferation, slow cell cycle, and high susceptibility to apoptosis [71]. Conversely, Tax-expressing clones showed long-term increases in proliferation and decreases in apoptosis [71]. Interestingly, apoptosis may play a role in the awakening of the virus from latency. It has been provocatively hypothesized that in cells that become apoptotic RCD in response to infection, apoptosis itself may awaken viral latency and promote viral replication through direct upregulation of caspase 9, which, in addition to its proapoptotic effect, may form a complex with Sp1-p53 and activate viral LTR [72]. On the other hand, viral reactivation from latency could also be triggered by metabolic changes such as hypoxia, which has been shown to increase transcription of the HTLV-1 proviral plus strand; conversely, inhibition of glycolysis and mitochondrial electron transport chain hinders transcription of the proviral plus strand [73]. Interestingly, the results of these recent detailed studies appear to be essentially consistent with the observations reported above. Overall, the results of these studies suggest that both the apoptotic RCD signaling pathway and the metabolic pathway in HTLV-1 infection may represent potential targets for the development of molecules to reverse latency.
Latency could possibly be counteracted by activating the expression of viral antigens that are likely to be recognized by the immune system. This would allow the recruitment of effector cells capable of knocking out the virus. Recently, a treatment to reverse latency has been proposed by using the histone deacetylase inhibitors (HDACi) panobinostat and romidepsin [74]. These inhibitors were shown to repress the transcription of Tax and of Tax target genes, although only slightly. Simplified schematics of the complex and as yet not fully elucidated mechanisms leading to immortalization and transformation processes induced in HTLV-1-infected cells are summarized in Figure 2.
6. Proposals for HTLV-1/ATL-targeted therapy
Although the mechanisms of HTLV-1 immortalization/transformation have been extensively studied, it has not been possible to find a unique, suitable candidate treatment. This may be due to the large number of targets that could potentially be hit. The disease starts after a specific HTLV-1 infection of CD4+ cells. Therefore, hypothetically, a wide range of potential targets could be found within the viral genome/proteins as well as among host cell genes transcribed during infection that are highlysubject to virus-induced dysregulation. In particular, the particular cell signalling activated by HTLV-1 in infected cells includes multifunctional factors/complexes that could serve as targets for pharmacological intervention. Accordingly, several inhibitors of host cell signalling have recently been proposed in in vitro/ex vivo studies as potential anti-ATL therapeutics. These include apigenin as an inhibitor of the aryl hydrocarbon receptor and transcription factor that enhances the cytotoxicity of antiretroviral drugs, and dimethyl fumarate as an inhibitor of the so-called CBM complex [75,76]. Another potential cellular target studied for several years is phosphatidylinositol 3-kinases (PI3K). PI3K is involved in several processes of oncogenesis and is preferentially expressed in hematopoietic cells [77,78]. It has been demonstrated that the process of multiple nucleation and cell proliferation in ATL strictly depends on changes in the PI3K cascade [79]. Recently, a PI3K-δ/AKT inhibitor, idelalisib, was shown to specifically decrease proliferation of ATL cells in vitro [80]. In addition, a dual PI3K and HDAC inhibitor, CUDC-907, previously used in patients with hematologic malignancies, has been considered as a potential candidate for ATL treatment. It was found to induce a cytotoxic effect in HTLV-1-infected cells by inhibiting the phosphorylation of downstream PI3K targets such as AKT, REL A, and p70 S6K and decreasing HSP90 chaperone activity in ATL cells in vitro. Moreover, in HTLV-1-infected cells, CUDC-907 induced upregulation of caspases, concomitant downregulation of anti-apoptotic gene expression, and suppression of NF-κB activation by inhibiting IKKα/ß phosphorylation [81]. Suppression of NF-κB signalling proved to be one of the most important means to counteract the growth of HTLV-1-infected cells. This was demonstrated in HTLV-1-infected and ATL cells subjected to mono-treatment with butein, a polyphenol that possesses pro-apoptotic and anti-proliferative properties by down-regulating AKT, AP1 and NF-κB activation [82]. Given the signalling mediated by NF-κB, a suitable approach may be to combine an inhibitor of NF-κB signalling with a chemotherapeutic or antiviral drug to promote susceptibility to cell death. The results of a recent study conducted by some of us have shown how pharmacological inhibition of IκBα could enhance the pro-apoptotic effect of AZT in chronically infected HTLV-1 cell lines [83]. sing a similar approach, it was reported that combination treatment was the most fruitful approach for ATL treatment, even in the case of co-treatment with biologic drugs. The combination of the JAK /STAT inhibitor ruxolitinib and the Bcl-2/Bcl-xL inhibitor navitoclax showed antitumor efficacy in an additive/synergistic manner in IL -2–dependent ATL cell lines and ex vivo in lymphocytes from ATL patients [84]. Recently, a triple combination of an inhibitor of the oncogenic driver bromine and extra-terminal domain (BET) motif family involved in down-regulation of MYC transcription was synergistically used with NF-κB and PI3K inhibitors to achieve an antiproliferative effect in ATL cells in vitro and ex vivo [85]. To evaluate the response of ATL patients to treatment in vivo, it may be critical to determine the efficacy of therapy on viral replication in addition to its effects on cell survival. A recent study in ATL patients subjected to long-term therapy with AZT and alpha-interferon demonstrated that the combination treatment resulted in complete inhibition of the activity of RT, a reduction in two other virologic parameters, and a dramatic change in clonality pattern, as observed in short-term cultures of PBMCs from the patients who responded to therapy [86]. Studies conducted in ATL patients have shown that an important goal is to induce cells to undergo apoptosis, or generally move toward RCD, in response to therapy. Among the various ways to undergo RCD, interaction with the autophagy pathway should be considered. Autophagy may actually play a dual role in RCD either initiating cell death or maintaining it [87]. The autophagy protein Beclin-1 appears to be involved in maintaining activation of the factors NF-κB and STAT3 in HTLV-1 transformed cell lines [88]. Therefore, in the case of HTLV-1 infection, it seems that suppression of autophagy may be an appropriate approach to treat ATL. Indeed, autophagy appears to be a “self-feeding” mechanism in tumour progression that supports cancer cell growth. Recently, chloroquine/hydroxychloroquine was shown to inhibit autophagic flux in ATL cells ex vivo. The mechanism involved the accumulation of p47 together with the autophagic protein LC3II, and the accumulation of p47 led to the accumulation of IkBα, resulting in inhibition of NF-κB activation and susceptibility to apoptosis [89]. On the other hand, small molecule inhibitors of sirtuin 2, the nicotinamide adenine dinucleotide-dependent deacylase involved in the control of cell cycle modulation, inhibited the growth of patient ATL cell lines by not only inducing caspase-dependent and caspase-independent cell death, but also increasing autophagosome accumulation and inhibiting autophagosome degradation [90]. Therefore, in this case, upregulation of autophagic flux in ATL cells appears to be associated with mechanisms that induce cell death. These seemingly contradictory results encourage further studies on the role of autophagy in HTLV-1 infection and on a possible pharmacological modulation of autophagy as a novel strategy to target ATL. Potential targets could be identified not only in genes directly involved in cell signalling but also among regulators of gene expression. Recently, thanks to a bioinformatics approach, 12 miRNA associated with the regulation of key cell signalling genes in ATL cases have been identified [91]. Based on the seemingly conflicting results obtained in defining a satisfactory treatment for ATL, it seems plausible that combination treatments may be the most appropriate approach to inhibit a complex network of cell signalling as induced by HTLV-1 infection. Indeed, such a therapeutic strategy is likely to be the best weapon to avoid possible feedback control aimed at restoring leukemic cell survival. A summary of the suggestions for HTLV-1-targeted therapy is provided in Table 1.
7. Potential of gene editing technology in the eradication of persistent HTLV-1 infection and ATL therapy
CRISPR/Cas9 genome editing is a novel technology that uses a guide RNA (gRNA) to precisely cleave double-stranded DNA at a specific site. After cleavage, DNA double-strand breaks in human cells are normally repaired by the error-prone non-homologous end joining pathway [92]. This can lead to insertions and deletions that alter gene reading frames, disrupt DNA regulatory motifs, or damage RNA structures [93]. CRISPR technology has the potential to be a therapeutic strategy for HTLV-1 disease, as demonstrated by the use of zinc finger nucleases (ZFNs) to disrupt LTR promoter function [94] and inhibit proliferation of HTLV-1-positive cell lines [95]. However, CRISPR/Cas9 has advantages over ZFNs and other gene editing approaches, such as simplicity, cost-effectiveness, and efficiency, and has been shown to reduce ATL cell proliferation in vitro by targeting Hb [96]. Since the advent of gene editing, several studies have focused on targeting the HIV-1 provirus. This work may shed light on the efficacy, safety, and limitations of these approaches for targeting HTLV-1 [97].
The goal of gene editing approaches against HIV-1 is primarily to remove proviral DNA from the host genome. This can be achieved by targeting both LTRs and causing their disruption, followed by excision of the proviral DNA from the host genome. However, effective delivery and low off-target effects are critical for successful application in clinical trials [98,99]. In this sense, CRISPR technology is a promising gene editing tool, but it has some drawbacks that need to be addressed before it can be effectively used for antiretroviral therapy. In vitro, CRISPR/Cas9 can remove the integrated HIV-1 genome from cellular DNA, but in vivo, off-target activity, gene rearrangements, target selection limitations, and a limited number of effective transporters complicate the process. To overcome these problems, multiple RNA guide structures should be introduced into a single cell to ensure cleavage of the provirus. However, the use of multiple guide structures may increase off-target activity and unpredictable DNA rearrangements. A recent study examined the fate of HIV-1 provirus and cellular repair mechanisms triggered by CRISPR/Cas9. The study was conducted in two parts: The first part examined the fate of HIV-1 provirus in 293T cells, and the second part confirmed the results in a human T-cell leukemia line latently transduced with HIV-1-GFP and in T-cell leukemia cells infected with a clinical lymphotropic isolate of HIV-1. The study found that after CRISPR-mediated LTR ablation, the excised HIV-1 provirus remains in the cells for an extended period of time and can circulate as single molecules or concatemers that remain as episomes in the infected cells [100]. Non-integrated HIV-1 is abundant in resting, non-proliferating CD4+ T cells and leads to de novo virus production after cytokine exposure of resting cells. Although transducing particle production was at the limit of detection, the results of this study raise concerns about the persistence of CRISPR/Cas9-excised proviral DNA in the absence of antiretroviral therapy [100].
Although retroviral proviruses are largely restricted to HIV-1, HTLV-1 offers more targeted gRNA targeting than HIV-1 due to its highly conserved viral genome with remarkable sequence homogeneity. Moreover, as shown in Figure 3, CRISPR/Cas9 can disable both latent and actively replicating HTLV-1 and abrogate the function or expression of viral Tax and HBZ, which are the main drivers of HTLV-1-mediated transformation and proliferation. Targeting viral LTRs involved in viral genome integration and gene expression may allow effective treatment of HTLV-1-infected individuals, asymptomatic carriers, and ATL and HAM / TSP patients. Careful design of gRNAs that disrupt two viral elements simultaneously can disrupt overlapping reading frames between HBZ and the 3'LTR and Tax and the 3' LTR [97].
8. Conclusions
Although some progress has been made in slowing progression, ATL is unfortunately still an incurable disease. However, in recent years, considerable progress has been made in defining the mechanisms at the molecular genetic level associated with the events underlying the complex viral/cellular system that leads to selection of the transformed clone that ultimately determines the onset of ATL in HTLV-1-infected patients. The definition of these mechanisms has already led to the identification of new therapeutic targets and corresponding agents that can act on them. Therefore, this seems to be an indication of how new therapeutic strategies may be found to counteract ATL in the near future. Nevertheless, a precise prediction of if and when it will actually be possible to use therapeutic treatments that can prevent or control ATL and potentially transform HTLV-1 infection into a chronic disease is currently not possible.
Author Contributions
All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gessain, A.; Cassar, O. Epidemiological aspects and world distribution of htlv-1 infection. Front Microbiol 2012, 3, 388. [Google Scholar] [CrossRef]
- Gessain, A.; Ramassamy, J.L.; Afonso, P.V.; Cassar, O. Geographic distribution, clinical epidemiology and genetic diversity of the human oncogenic retrovirus htlv-1 in africa, the world's largest endemic area. Front Immunol 2023, 14, 1043600. [Google Scholar] [CrossRef]
- Percher, F.; Jeannin, P.; Martin-Latil, S.; Gessain, A.; Afonso, P.V.; Vidy-Roche, A.; Ceccaldi, P.E. Mother-to-child transmission of htlv-1 epidemiological aspects, mechanisms and determinants of mother-to-child transmission. Viruses 2016, 8. [Google Scholar] [CrossRef]
- Lairmore, M.D.; Haines, R.; Anupam, R. Mechanisms of human t-lymphotropic virus type 1 transmission and disease. Curr Opin Virol 2012, 2, 474–481. [Google Scholar] [CrossRef]
- Cook, L.B.; Melamed, A.; Demontis, M.A.; Laydon, D.J.; Fox, J.M.; Tosswill, J.H.; de Freitas, D.; Price, A.D.; Medcalf, J.F.; Martin, F. , et al. Rapid dissemination of human t-lymphotropic virus type 1 during primary infection in transplant recipients. Retrovirology 2016, 13, 3. [Google Scholar] [CrossRef]
- Nasr, R.; El Hajj, H.; Kfoury, Y.; de The, H.; Hermine, O.; Bazarbachi, A. Controversies in targeted therapy of adult t cell leukemia/lymphoma: On target or off target effects? Viruses 2011, 3, 750–769. [Google Scholar] [CrossRef]
- Laverdure, S.; Polakowski, N.; Hoang, K.; Lemasson, I. Permissive sense and antisense transcription from the 5' and 3' long terminal repeats of human t-cell leukemia virus type 1. J Virol 2016, 90, 3600–3610. [Google Scholar] [CrossRef]
- Fujisawa, J.; Seiki, M.; Kiyokawa, T.; Yoshida, M. Functional activation of the long terminal repeat of human t-cell leukemia virus type i by a trans-acting factor. Proc Natl Acad Sci U S A 1985, 82, 2277–2281. [Google Scholar] [CrossRef]
- Boxus, M.; Willems, L. Mechanisms of htlv-1 persistence and transformation. Br J Cancer 2009, 101, 1497–1501. [Google Scholar] [CrossRef]
- Baydoun, H.; Duc-Dodon, M.; Lebrun, S.; Gazzolo, L.; Bex, F. Regulation of the human t-cell leukemia virus gene expression depends on the localization of regulatory proteins tax, rex and p30ii in specific nuclear subdomains. Gene 2007, 386, 191–201. [Google Scholar] [CrossRef]
- Sarkis, S.; Galli, V.; Moles, R.; Yurick, D.; Khoury, G.; Purcell, D.F.J.; Franchini, G.; Pise-Masison, C.A. Role of htlv-1 orf-i encoded proteins in viral transmission and persistence. Retrovirology 2019, 16, 43. [Google Scholar] [CrossRef] [PubMed]
- Silic-Benussi, M.; Marin, O.; Biasiotto, R.; D'Agostino, D.M.; Ciminale, V. Effects of human t-cell leukemia virus type 1 (htlv-1) p13 on mitochondrial k+ permeability: A new member of the viroporin family? Febs Lett 2010, 584, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- Younis, I.; Khair, L.; Dundr, M.; Lairmore, M.D.; Franchini, G.; Green, P.L. Repression of human t-cell leukemia virus type 1 and type 2 replication by a viral mrna-encoded posttranscriptional regulator. J Virol 2004, 78, 11077–11083. [Google Scholar] [CrossRef]
- Martin-Latil, S.; Gnadig, N.F.; Mallet, A.; Desdouits, M.; Guivel-Benhassine, F.; Jeannin, P.; Prevost, M.C.; Schwartz, O.; Gessain, A.; Ozden, S. , et al. Transcytosis of htlv-1 across a tight human epithelial barrier and infection of subepithelial dendritic cells. Blood 2012, 120, 572–580. [Google Scholar] [CrossRef]
- Mulherkar, T.H.; Gomez, D.J.; Sandel, G.; Jain, P. Co-infection and cancer: Host-pathogen interaction between dendritic cells and hiv-1, htlv-1, and other oncogenic viruses. Viruses 2022, 14. [Google Scholar] [CrossRef]
- Van Prooyen, N.; Gold, H.; Andresen, V.; Schwartz, O.; Jones, K.; Ruscetti, F.; Lockett, S.; Gudla, P.; Venzon, D.; Franchini, G. Human t-cell leukemia virus type 1 p8 protein increases cellular conduits and virus transmission. Proc Natl Acad Sci U S A 2010, 107, 20738–20743. [Google Scholar] [CrossRef]
- Pinto, D.O.; Al Sharif, S.; Mensah, G.; Cowen, M.; Khatkar, P.; Erickson, J.; Branscome, H.; Lattanze, T.; DeMarino, C.; Alem, F. , et al. Extracellular vesicles from htlv-1 infected cells modulate target cells and viral spread. Retrovirology 2021, 18, 6. [Google Scholar] [CrossRef]
- Mohanty, S.; Harhaj, E.W. Mechanisms of oncogenesis by htlv-1 tax. Pathogens 2020, 9. [Google Scholar] [CrossRef]
- Overbaugh, J.; Bangham, C.R. Selection forces and constraints on retroviral sequence variation. Science 2001, 292, 1106–1109. [Google Scholar] [CrossRef]
- Laydon, D.J.; Sunkara, V.; Boelen, L.; Bangham, C.R.M.; Asquith, B. The relative contributions of infectious and mitotic spread to htlv-1 persistence. PLoS Comput Biol 2020, 16, e1007470. [Google Scholar] [CrossRef]
- Izaki, M.; Yasunaga, J.I.; Nosaka, K.; Sugata, K.; Utsunomiya, H.; Suehiro, Y.; Shichijo, T.; Yamada, A.; Sugawara, Y.; Hibi, T. , et al. In vivo dynamics and adaptation of htlv-1-infected clones under different clinical conditions. PLoS Pathog 2021, 17, e1009271. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.M.; Hilburn, S.; Demontis, M.A.; Brighty, D.W.; Rios Grassi, M.F.; Galvao-Castro, B.; Taylor, G.P.; Martin, F. Long terminal repeat circular DNA as markers of active viral replication of human t lymphotropic virus-1 in vivo. Viruses 2016, 8, 80. [Google Scholar] [CrossRef]
- Melamed, A.; Laydon, D.J.; Al Khatib, H.; Rowan, A.G.; Taylor, G.P.; Bangham, C.R. Htlv-1 drives vigorous clonal expansion of infected cd8(+) t cells in natural infection. Retrovirology 2015, 12, 91. [Google Scholar] [CrossRef]
- Yasunaga, J. Viral, genetic, and immune factors in the oncogenesis of adult t-cell leukemia/lymphoma. International Journal of Hematology 2023, 117, 504–511. [Google Scholar] [CrossRef] [PubMed]
- El Hajj, H.; Tsukasaki, K.; Cheminant, M.; Bazarbachi, A.; Watanabe, T.; Hermine, O. Novel treatments of adult t cell leukemia lymphoma. Front Microbiol 2020, 11, 1062. [Google Scholar] [CrossRef]
- Cook, L.B.; Fuji, S.; Hermine, O.; Bazarbachi, A.; Ramos, J.C.; Ratner, L.; Horwitz, S.; Fields, P.; Tanase, A.; Bumbea, H. , et al. Revised adult t-cell leukemia-lymphoma international consensus meeting report. J Clin Oncol 2019, 37, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Joh, T.; Uike, N.; Yamamoto, K.; Utsunomiya, A.; Yoshida, S.; Saburi, Y.; Miyamoto, T.; Takemoto, S.; Suzushima, H. , et al. Defucosylated anti-ccr4 monoclonal antibody (kw-0761) for relapsed adult t-cell leukemia-lymphoma: A multicenter phase ii study. J Clin Oncol 2012, 30, 837–842. [Google Scholar] [CrossRef]
- Tanaka, T.; Inamoto, Y.; Ito, A.; Watanabe, M.; Takeda, W.; Aoki, J.; Kim, S.W.; Fukuda, T. Lenalidomide treatment for recurrent adult t-cell leukemia/lymphoma after allogeneic hematopoietic cell transplantation. Hematol Oncol 2022. [CrossRef] [PubMed]
- Baba, Y.; Sakai, H.; Kabasawa, N.; Harada, H. Successful treatment of an aggressive adult t-cell leukemia/lymphoma with strong cd30 expression using brentuximab vedotin as combination and maintenance therapy. Intern Med 2023, 62, 613–616. [Google Scholar] [CrossRef]
- Izutsu, K.; Makita, S.; Nosaka, K.; Yoshimitsu, M.; Utsunomiya, A.; Kusumoto, S.; Morishima, S.; Tsukasaki, K.; Kawamata, T.; Ono, T. , et al. An open-label, single-arm phase 2 trial of valemetostat for relapsed or refractory adult t-cell leukemia/lymphoma. Blood 2023, 141, 1159–1168. [Google Scholar]
- Utsunomiya, A.; Izutsu, K.; Jo, T.; Yoshida, S.; Tsukasaki, K.; Ando, K.; Choi, I.; Imaizumi, Y.; Kato, K.; Kurosawa, M. , et al. Oral histone deacetylase inhibitor tucidinostat (hbi-8000) in patients with relapsed or refractory adult t-cell leukemia/lymphoma: Phase iib results. Cancer Science 2022, 113, 2778–2787. [Google Scholar] [CrossRef]
- Katsuya, H. Current and emerging therapeutic strategies in adult t-cell leukemia-lymphoma. Int J Hematol 2023, 117, 512–522. [Google Scholar] [CrossRef]
- Zhi, H.J.; Yang, L.P.; Kuo, Y.L.; Ho, Y.K.; Shih, H.M.; Giam, C.Z. Nf-kappa b hyper-activation by htlv-1 tax induces cellular senescence, but can be alleviated by the viral anti-sense protein hbz. Plos Pathogens 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C.; Ballard, D.W. Persistent activation of nf-kappab by the tax transforming protein of htlv-1: Hijacking cellular ikappab kinases. Oncogene 1999, 18, 6948–6958. [Google Scholar] [CrossRef] [PubMed]
- Shimotohno, K.; Takano, M.; Teruuchi, T.; Miwa, M. Requirement of multiple copies of a 21-nucleotide sequence in the u3 regions of human t-cell leukemia virus type i and type ii long terminal repeats for trans-acting activation of transcription. Proc Natl Acad Sci U S A 1986, 83, 8112–8116. [Google Scholar] [CrossRef]
- Giam, C.Z.; Xu, Y.L. Htlv-i tax gene product activates transcription via pre-existing cellular factors and camp responsive element. J Biol Chem 1989, 264, 15236–15241. [Google Scholar] [CrossRef] [PubMed]
- Adya, N.; Giam, C.Z. Distinct regions in human t-cell lymphotropic virus type i tax mediate interactions with activator protein creb and basal transcription factors. J Virol 1995, 69, 1834–1841. [Google Scholar] [CrossRef]
- Lu, H.; Pise-Masison, C.A.; Linton, R.; Park, H.U.; Schiltz, R.L.; Sartorelli, V.; Brady, J.N. Tax relieves transcriptional repression by promoting histone deacetylase 1 release from the human t-cell leukemia virus type 1 long terminal repeat. J Virol 2004, 78, 6735–6743. [Google Scholar] [CrossRef]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the nfkappab system. Wiley Interdiscip Rev Syst Biol Med 2016, 8, 227–241. [Google Scholar] [CrossRef]
- Kanno, T.; Brown, K.; Franzoso, G.; Siebenlist, U. Kinetic analysis of human t-cell leukemia virus type i tax-mediated activation of nf-kappa b. Mol Cell Biol 1994, 14, 6443–6451. [Google Scholar] [CrossRef]
- Geleziunas, R.; Ferrell, S.; Lin, X.; Mu, Y.; Cunningham, E.T., Jr.; Grant, M.; Connelly, M.A.; Hambor, J.E.; Marcu, K.B.; Greene, W.C. Human t-cell leukemia virus type 1 tax induction of nf-kappab involves activation of the ikappab kinase alpha (ikkalpha) and ikkbeta cellular kinases. Mol Cell Biol 1998, 18, 5157–5165. [Google Scholar] [CrossRef] [PubMed]
- Fuggetta, M.P.; Bordignon, V.; Cottarelli, A.; Macchi, B.; Frezza, C.; Cordiali-Fei, P.; Ensoli, F.; Ciafre, S.; Marino-Merlo, F.; Mastino, A. , et al. Downregulation of proinflammatory cytokines in htlv-1-infected t cells by resveratrol. J Exp Clin Cancer Res 2016, 35, 118. [Google Scholar] [CrossRef]
- Hleihel, R.; Skayneh, H.; de The, H.; Hermine, O.; Bazarbachi, A. Primary cells from patients with adult t cell leukemia/lymphoma depend on htlv-1 tax expression for nf-kappab activation and survival. Blood Cancer J 2023, 13, 67. [Google Scholar] [CrossRef] [PubMed]
- Macaire, H.; Riquet, A.; Moncollin, V.; Biemont-Trescol, M.C.; Duc Dodon, M.; Hermine, O.; Debaud, A.L.; Mahieux, R.; Mesnard, J.M.; Pierre, M. , et al. Tax protein-induced expression of antiapoptotic bfl-1 protein contributes to survival of human t-cell leukemia virus type 1 (htlv-1)-infected t-cells. J Biol Chem 2012, 287, 21357–21370. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, H.; Tomita, M.; Matsuda, T.; Ohta, T.; Tanaka, Y.; Fujii, M.; Hatano, M.; Tokuhisa, T.; Mori, N. Transcriptional activation of survivin through the nf-kappab pathway by human t-cell leukemia virus type i tax. Int J Cancer 2005, 115, 967–974. [Google Scholar] [CrossRef]
- Kogure, Y.; Kataoka, K. Genetic alterations in adult t-cell leukemia/lymphoma. Cancer Sci 2017, 108, 1719–1725. [Google Scholar] [CrossRef]
- Jo, T.; Noguchi, K.; Sakai, T.; Kubota-Koketsu, R.; Irie, S.; Matsuo, M.; Taguchi, J.; Abe, K.; Shigematsu, K. Htlv-1 tax-specific memory cytotoxic t lymphocytes in long-term survivors of aggressive-type adult t-cell leukemia/lymphoma. Cancer Med 2022, 11, 3238–3250. [Google Scholar] [CrossRef] [PubMed]
- Yasunaga, J.I. Strategies of human t-cell leukemia virus type 1 for persistent infection: Implications for leukemogenesis of adult t-cell leukemia-lymphoma. Front Microbiol 2020, 11, 979. [Google Scholar] [CrossRef]
- Akkouche, A.; Moodad, S.; Hleihel, R.; Skayneh, H.; Chambeyron, S.; El Hajj, H.; Bazarbachi, A. In vivo antagonistic role of the human t-cell leukemia virus type 1 regulatory proteins tax and hbz. PLoS Pathog 2021, 17, e1009219. [Google Scholar] [CrossRef]
- Arnold, J.; Yamamoto, B.; Li, M.; Phipps, A.J.; Younis, I.; Lairmore, M.D.; Green, P.L. Enhancement of infectivity and persistence in vivo by hbz, a natural antisense coded protein of htlv-1. Blood 2006, 107, 3976–3982. [Google Scholar] [CrossRef]
- Gazon, H.; Lemasson, I.; Polakowski, N.; Cesaire, R.; Matsuoka, M.; Barbeau, B.; Mesnard, J.M.; Peloponese, J.M., Jr. Human t-cell leukemia virus type 1 (htlv-1) bzip factor requires cellular transcription factor jund to upregulate htlv-1 antisense transcription from the 3' long terminal repeat. J Virol 2012, 86, 9070–9078. [Google Scholar] [CrossRef]
- Vernin, C.; Thenoz, M.; Pinatel, C.; Gessain, A.; Gout, O.; Delfau-Larue, M.H.; Nazaret, N.; Legras-Lachuer, C.; Wattel, E.; Mortreux, F. Htlv-1 bzip factor hbz promotes cell proliferation and genetic instability by activating oncomirs. Cancer Res 2014, 74, 6082–6093. [Google Scholar] [CrossRef]
- Forlani, G.; Shallak, M.; Tedeschi, A.; Cavallari, I.; Marcais, A.; Hermine, O.; Accolla, R.S. Dual cytoplasmic and nuclear localization of htlv-1-encoded hbz protein is a unique feature of adult t-cell leukemia. Haematologica 2021, 106, 2076–2085. [Google Scholar] [CrossRef]
- Nicot, C.; Dundr, M.; Johnson, J.M.; Fullen, J.R.; Alonzo, N.; Fukumoto, R.; Princler, G.L.; Derse, D.; Misteli, T.; Franchini, G. Htlv-1-encoded p30ii is a post-transcriptional negative regulator of viral replication. Nat Med 2004, 10, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Silic-Benussi, M.; Cavallari, I.; Vajente, N.; Vidali, S.; Chieco-Bianchi, L.; Di Lisa, F.; Saggioro, D.; D'Agostino, D.M.; Ciminale, V. Redox regulation of t-cell turnover by the p13 protein of human t-cell leukemia virus type 1: Distinct effects in primary versus transformed cells. Blood 2010, 116, 54–62. [Google Scholar] [CrossRef]
- Nicot, C.; Mulloy, J.C.; Ferrari, M.G.; Johnson, J.M.; Fu, K.; Fukumoto, R.; Trovato, R.; Fullen, J.; Leonard, W.J.; Franchini, G. Htlv-1 p12(i) protein enhances stat5 activation and decreases the interleukin-2 requirement for proliferation of primary human peripheral blood mononuclear cells. Blood 2001, 98, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Gazon, H.; Chauhan, P.S.; Porquet, F.; Hoffmann, G.B.; Accolla, R.; Willems, L. Epigenetic silencing of htlv-1 expression by the hbz rna through interference with the basal transcription machinery. Blood Adv 2020, 4, 5574–5579. [Google Scholar] [CrossRef] [PubMed]
- Kiik, H.; Ramanayake, S.; Miura, M.; Tanaka, Y.; Melamed, A.; Bangham, C.R.M. Time-course of host cell transcription during the htlv-1 transcriptional burst. PLoS Pathog 2022, 18, e1010387. [Google Scholar] [CrossRef]
- Copeland, K.F.; Haaksma, A.G.; Goudsmit, J.; Krammer, P.H.; Heeney, J.L. Inhibition of apoptosis in t cells expressing human t cell leukemia virus type i tax. AIDS Res Hum Retroviruses 1994, 10, 1259–1268. [Google Scholar] [CrossRef]
- Yang, Y.C.; Hsu, T.Y.; Lin, R.H.; Su, I.J.; Chen, J.Y.; Yang, C.S. Resistance to tumor necrosis factor-alpha-induced apoptosis in human t-lymphotropic virus type i-infected t cell lines. AIDS Res Hum Retroviruses 2002, 18, 207–212. [Google Scholar] [CrossRef]
- Brauweiler, A.; Garrus, J.E.; Reed, J.C.; Nyborg, J.K. Repression of bax gene expression by the htlv-1 tax protein: Implications for suppression of apoptosis in virally infected cells. Virology 1997, 231, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Mori, N.; Fujii, M.; Cheng, G.; Ikeda, S.; Yamasaki, Y.; Yamada, Y.; Tomonaga, M.; Yamamoto, N. Human t-cell leukemia virus type i tax protein induces the expression of anti-apoptotic gene bcl-xl in human t-cells through nuclear factor-kappab and c-amp responsive element binding protein pathways. Virus Genes 2001, 22, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Chlichlia, K.; Moldenhauer, G.; Daniel, P.T.; Busslinger, M.; Gazzolo, L.; Schirrmacher, V.; Khazaie, K. Immediate effects of reversible htlv-1 tax function: T-cell activation and apoptosis. Oncogene 1995, 10, 269–277. [Google Scholar]
- Chlichlia, K.; Busslinger, M.; Peter, M.E.; Walczak, H.; Krammer, P.H.; Schirrmacher, V.; Khazaie, K. Ice-proteases mediate htlv-i tax-induced apoptotic t-cell death. Oncogene 1997, 14, 2265–2272. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zachar, V.; Zdravkovic, M.; Guo, M.; Ebbesen, P.; Liu, X. Role of the fas/fas ligand pathway in apoptotic cell death induced by the human t cell lymphotropic virus type i tax transactivator. J Gen Virol 1997, 78 ( Pt 12), 3277–3285. [Google Scholar] [CrossRef]
- Nicot, C.; Harrod, R. Distinct p300-responsive mechanisms promote caspase-dependent apoptosis by human t-cell lymphotropic virus type 1 tax protein. Mol Cell Biol 2000, 20, 8580–8589. [Google Scholar] [CrossRef]
- Rivera-Walsh, I.; Waterfield, M.; Xiao, G.; Fong, A.; Sun, S.C. Nf-kappab signaling pathway governs trail gene expression and human t-cell leukemia virus-i tax-induced t-cell death. J Biol Chem 2001, 276, 40385–40388. [Google Scholar] [CrossRef]
- Matteucci, C.; Balestrieri, E.; Macchi, B.; Mastino, A. Modulation of apoptosis during htlv-1-mediated immortalization process in vitro. J Med Virol 2004, 74, 473–483. [Google Scholar] [CrossRef]
- Mahgoub, M.; Yasunaga, J.I.; Iwami, S.; Nakaoka, S.; Koizumi, Y.; Shimura, K.; Matsuoka, M. Sporadic on/off switching of htlv-1 tax expression is crucial to maintain the whole population of virus-induced leukemic cells. Proc Natl Acad Sci U S A 2018, 115, E1269–E1278. [Google Scholar] [CrossRef]
- Shudofsky, A.M.D.; Giam, C.Z. Cells of adult t-cell leukemia evade htlv-1 tax/nf-kappab hyperactivation-induced senescence. Blood Adv 2019, 3, 564–569. [Google Scholar] [CrossRef]
- Ramanayake, S.; Moulding, D.A.; Tanaka, Y.; Singh, A.; Bangham, C.R.M. Dynamics and consequences of the htlv-1 proviral plus-strand burst. PLoS Pathog 2022, 18, e1010774. [Google Scholar] [CrossRef]
- Abou-Kandil, A.; Chamias, R.; Huleihel, M.; Godbey, W.T.; Aboud, M. Role of caspase 9 in activation of htlv-1 ltr expression by DNA damaging agents. Cell Cycle 2011, 10, 3337–3345. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Mateus, M.; Thinnes, C.C.; McCullagh, J.S.; Schofield, C.J.; Taylor, G.P.; Bangham, C.R.M. Glucose metabolism and oxygen availability govern reactivation of the latent human retrovirus htlv-1. Cell Chem Biol 2017, 24, 1377–+. [Google Scholar] [CrossRef] [PubMed]
- Schnell, A.P.; Kohrt, S.; Aristodemou, A.; Taylor, G.P.; Bangham, C.R.M.; Thoma-Kress, A.K. Hdac inhibitors panobinostat and romidepsin enhance tax transcription in htlv-1-infected cell lines and freshly isolated patients' t-cells. Front Immunol 2022, 13, 978800. [Google Scholar] [CrossRef] [PubMed]
- Sales, D.; Lin, E.; Stoffel, V.; Dickson, S.; Khan, Z.K.; Beld, J.; Jain, P. Apigenin improves cytotoxicity of antiretroviral drugs against htlv-1 infected cells through the modulation of ahr signaling. NeuroImmune Pharm Ther 2023, 2, 49–62. [Google Scholar] [CrossRef]
- Sato, T.; Maeta, T.; Ito, S. Dimethyl fumarate suppresses the proliferation of htlv-1-infected t cells by inhibiting cbm complex-triggered nf-kappa b signaling. Anticancer Res 2023, 43, 1901–1908. [Google Scholar] [CrossRef]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. Pi3k in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef]
- Hemmati, S.; Sinclair, T.; Tong, M.; Bartholdy, B.; Okabe, R.O.; Ames, K.; Ostrodka, L.; Haque, T.; Kaur, I.; Mills, T.S. , et al. Pi3k alpha and delta promote hematopoietic stem cell activation. Jci Insight 2019, 4. [Google Scholar]
- Fukuda, R.; Hayashi, A.; Utsunomiya, A.; Nukada, Y.; Fukui, R.; Itoh, K.; Tezuka, K.; Ohashi, K.; Mizuno, K.; Sakamoto, M. , et al. Alteration of phosphatidylinositol 3-kinase cascade in the multilobulated nuclear formation of adult t cell leukemia/lymphoma (atll). Proc Natl Acad Sci U S A 2005, 102, 15213–15218. [Google Scholar] [CrossRef]
- Katsuya, H.; Cook, L.B.M.; Rowan, A.G.; Satou, Y.; Taylor, G.P.; Bangham, C.R.M. Phosphatidylinositol 3-kinase-delta (pi3k-delta) is a potential therapeutic target in adult t-cell leukemia-lymphoma. Biomark Res 2018, 6, 24. [Google Scholar] [CrossRef]
- Ishikawa, C.; Mori, N. The role of cudc-907, a dual phosphoinositide-3 kinase and histone deacetylase inhibitor, in inhibiting proliferation of adult t-cell leukemia. Eur J Haematol 2020, 105, 763–772. [Google Scholar] [CrossRef]
- Ishikawa, C.; Senba, M.; Mori, N. Butein inhibits nf-kappab, ap-1 and akt activation in adult t-cell leukemia/lymphoma. Int J Oncol 2017, 51, 633–643. [Google Scholar] [CrossRef]
- Matteucci, C.; Marino-Merlo, F.; Minutolo, A.; Balestrieri, E.; Valletta, E.; Macchi, B.; Mastino, A.; Grelli, S. Inhibition of ikappabalpha phosphorylation potentiates regulated cell death induced by azidothymidine in htlv-1 infected cells. Cell Death Discov 2020, 6, 9. [Google Scholar] [CrossRef]
- Zhang, M.; Mathews Griner, L.A.; Ju, W.; Duveau, D.Y.; Guha, R.; Petrus, M.N.; Wen, B.; Maeda, M.; Shinn, P.; Ferrer, M. , et al. Selective targeting of jak/stat signaling is potentiated by bcl-xl blockade in il-2-dependent adult t-cell leukemia. Proc Natl Acad Sci U S A 2015, 112, 12480–12485. [Google Scholar] [CrossRef]
- Daenthanasanmak, A.; Bamford, R.N.; Yoshioka, M.; Yang, S.M.; Homan, P.; Karim, B.; Bryant, B.R.; Petrus, M.N.; Thomas, C.J.; Green, P.L. , et al. Triple combination of bet plus pi3k and nf-kappab inhibitors exhibit synergistic activity in adult t-cell leukemia/lymphoma. Blood Adv 2022, 6, 2346–2360. [Google Scholar] [CrossRef]
- Macchi, B.; Balestrieri, E.; Frezza, C.; Grelli, S.; Valletta, E.; Marcais, A.; Marino-Merlo, F.; Turpin, J.; Bangham, C.R.; Hermine, O. , et al. Quantification of htlv-1 reverse transcriptase activity in atl patients treated with zidovudine and interferon-alpha. Blood Adv 2017, 1, 748–752. [Google Scholar] [CrossRef]
- Zhang, T.; Yu, J.; Cheng, S.; Zhang, Y.; Zhou, C.H.; Qin, J.; Luo, H. Research progress on the anticancer molecular mechanism of targets regulating cell autophagy. Pharmacology 2023, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, D.; Zhang, Y.; Zhang, H.; Cheng, H. The autophagy molecule beclin 1 maintains persistent activity of nf-kappab and stat3 in htlv-1-transformed t lymphocytes. Biochem Biophys Res Commun 2015, 465, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Fauzi, Y.R.; Nakahata, S.; Chilmi, S.; Ichikawa, T.; Nueangphuet, P.; Yamaguchi, R.; Nakamura, T.; Shimoda, K.; Morishita, K. Antitumor effects of chloroquine/hydroxychloroquine mediated by inhibition of the nf-kappab signaling pathway through abrogation of autophagic p47 degradation in adult t-cell leukemia/lymphoma cells. PLoS One 2021, 16, e0256320. [Google Scholar] [CrossRef] [PubMed]
- Kozako, T.; Mellini, P.; Ohsugi, T.; Aikawa, A.; Uchida, Y.I.; Honda, S.I.; Suzuki, T. Novel small molecule sirt2 inhibitors induce cell death in leukemic cell lines. BMC Cancer 2018, 18, 791. [Google Scholar] [CrossRef]
- Machado, C.B.; da Cunha, L.S.; Maues, J.H.D.; Pessoa, F.M.C.D.; de Oliveira, M.B.; Ribeiro, R.M.; Lopes, G.S.; de Moraes, M.O.; de Moraes, M.E.A.; Khayat, A.S. , et al. Role of mirnas in human t cell leukemia virus type 1 induced t cell leukemia: A literature review and bioinformatics approach. International Journal of Molecular Sciences, 2022; 23. [Google Scholar]
- Huang, J.; Cook, D.E. The contribution of DNA repair pathways to genome editing and evolution in filamentous pathogens. FEMS Microbiol Rev 2022, 46. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Greene, E.C. DNA repair pathway choices in crispr-cas9-mediated genome editing. Trends Genet 2021, 37, 639–656. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Takeda, S.; Kariya, R.; Matsuda, K.; Urano, E.; Okada, S.; Komano, J. A novel therapeutic molecule against htlv-1 infection targeting provirus. Leukemia 2013, 27, 1621–1627. [Google Scholar] [CrossRef] [PubMed]
- Rojo-Romanos, T.; Karpinski, J.; Millen, S.; Beschorner, N.; Simon, F.; Paszkowski-Rogacz, M.; Lansing, F.; Schneider, P.M.; Sonntag, J.; Hauber, J. , et al. Precise excision of htlv-1 provirus with a designer-recombinase. Mol Ther 2023, 31, 2266–2285. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Shaffer, A.L., 3rd; Ceribelli, M.; Zhang, M.; Wright, G.; Huang, D.W.; Xiao, W.; Powell, J.; Petrus, M.N.; Yang, Y. , et al. Targeting the htlv-i-regulated batf3/irf4 transcriptional network in adult t cell leukemia/lymphoma. Cancer Cell 2018, 34, 286–297 e210. [Google Scholar] [CrossRef] [PubMed]
- Panfil, A.R.; Green, P.L.; Yoder, K.E. Crispr genome editing applied to the pathogenic retrovirus htlv-1. Front Cell Infect Microbiol 2020, 10, 580371. [Google Scholar] [CrossRef]
- Kaminski, R.; Chen, Y.; Fischer, T.; Tedaldi, E.; Napoli, A.; Zhang, Y.; Karn, J.; Hu, W.; Khalili, K. Corrigendum: Elimination of hiv-1 genomes from human t-lymphoid cells by crispr/cas9 gene editing. Sci Rep 2016, 6, 28213. [Google Scholar] [CrossRef]
- Xiao, Q.; Guo, D.; Chen, S. Application of crispr/cas9-based gene editing in hiv-1/aids therapy. Front Cell Infect Microbiol 2019, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.; Maori, E.; Quaranta, P.; Matteoli, G.; Maggi, F.; Sgarbanti, M.; Crucitta, S.; Pacini, S.; Turriziani, O.; Antonelli, G. , et al. Crispr/cas9 ablation of integrated hiv-1 accumulates proviral DNA circles with reformed long terminal repeats. J Virol 2021, 95, e0135821. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of HTLV-1 cell-to-cell transmission. VS = virological synapses. CC = cellular conduits. EV = extracellular vesicles. The mechanism of transcytosis was not reported due to difficulties in graphical representation.
Figure 1.
Schematic representation of HTLV-1 cell-to-cell transmission. VS = virological synapses. CC = cellular conduits. EV = extracellular vesicles. The mechanism of transcytosis was not reported due to difficulties in graphical representation.
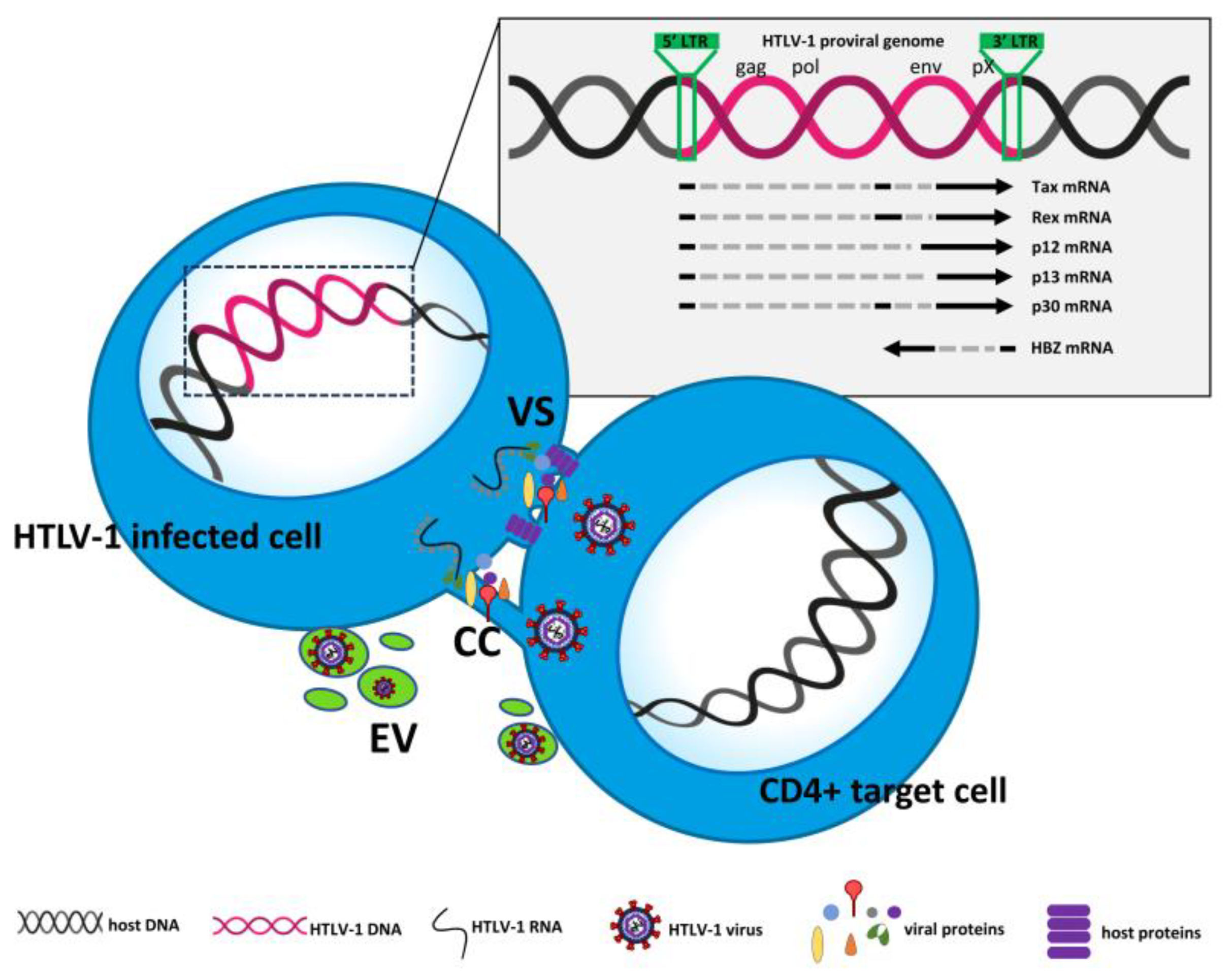
Figure 2.
Schematic representation of (a) the outcome of HTLV-1 infection in vitro leading to immortalization/transformation and (b) the essential processes of leukemogenesis leading to ATL in HTLV-1-infected cells. The fragmented lower edge of Tax expression at low levels and the small green arrow for Tax in ATL cells in (b) show that Tax can be sporadically turned on and off during leukemogenesis.
Figure 2.
Schematic representation of (a) the outcome of HTLV-1 infection in vitro leading to immortalization/transformation and (b) the essential processes of leukemogenesis leading to ATL in HTLV-1-infected cells. The fragmented lower edge of Tax expression at low levels and the small green arrow for Tax in ATL cells in (b) show that Tax can be sporadically turned on and off during leukemogenesis.
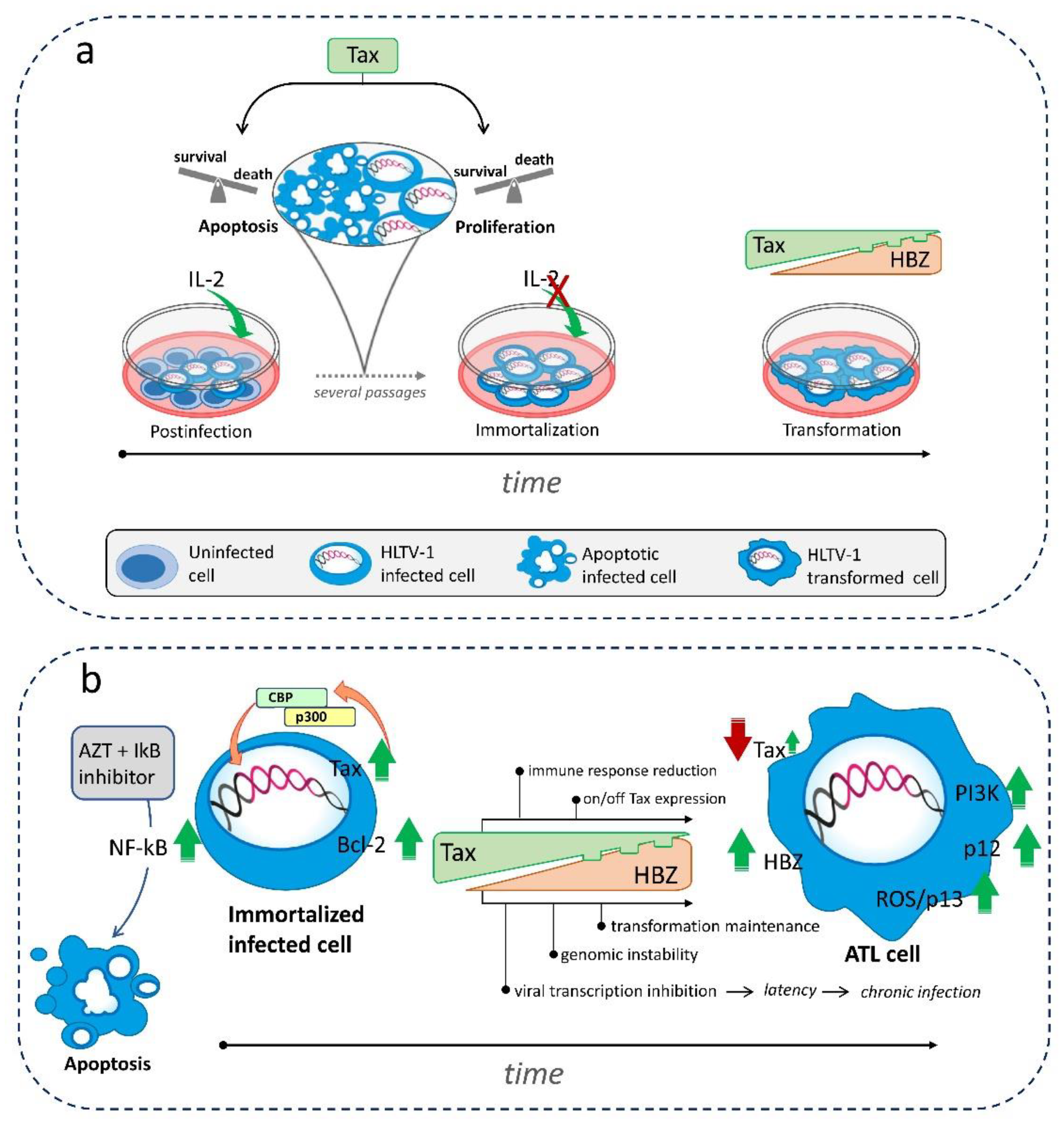
Figure 3.
Schematic representation of how CRISPR-Cas9 gene editing could affect HTLV-1 integration and cell transformation.
Figure 3.
Schematic representation of how CRISPR-Cas9 gene editing could affect HTLV-1 integration and cell transformation.
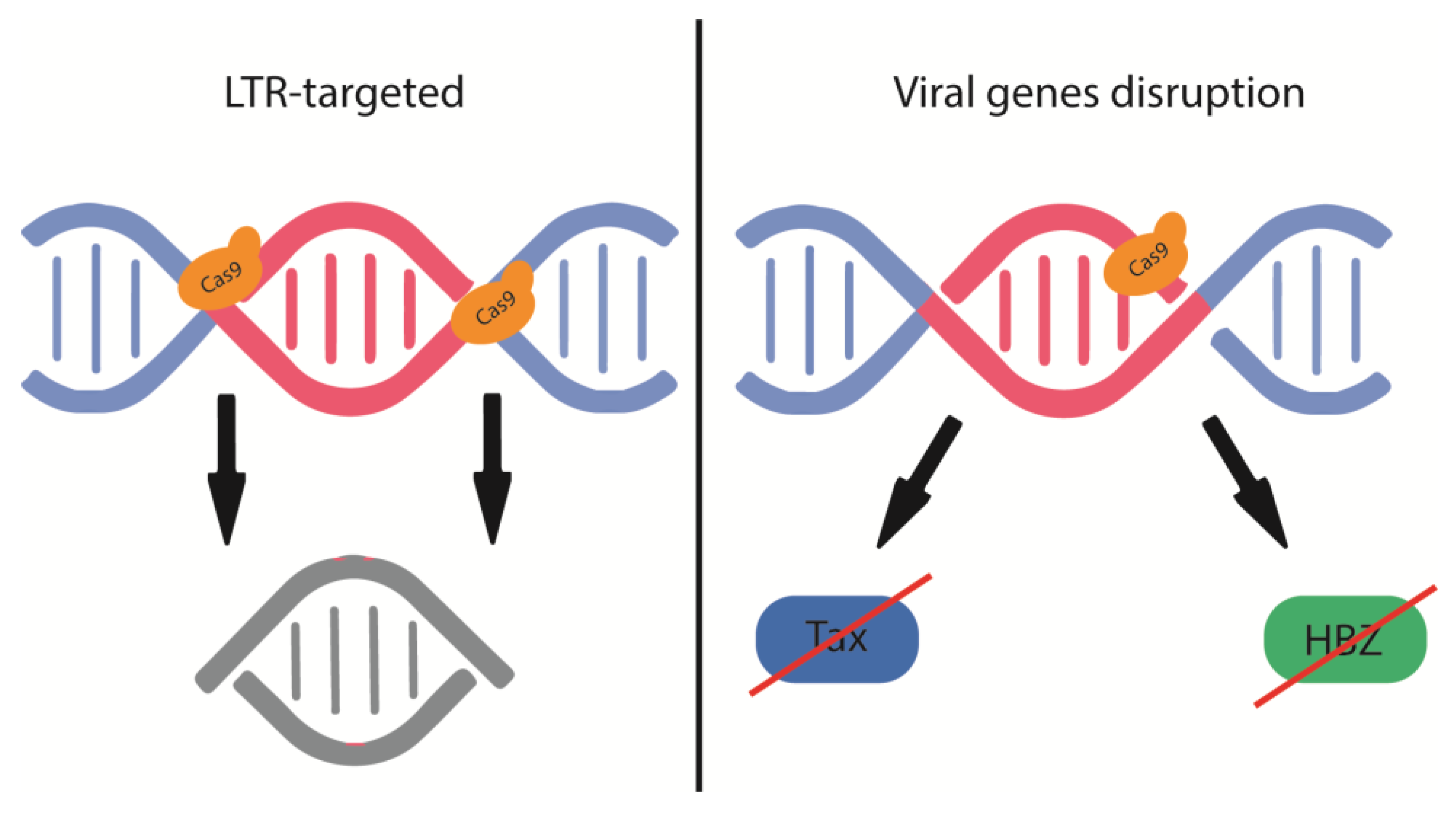

Table 1.
Proposals for HTLV-1/ATL-targeted therapy.
| Proposed therapeutic treatment |
Target | Available results | ||
|---|---|---|---|---|
| Viral | Cellular | in vitro/ex vivo | in vivo | |
| AZT+IFNα | RT | IFN-receptor other?1 |
Samples from patients a) complete inhibition of RT activity and b) reduction of virus parameters in resp. patients, c) dramatic change in the clonality pattern |
Prolonged survival with respect to untreated patients |
| Idelalisib | ? | PI3K-δ/AKT | inhibition of proliferation in ATL cells | no |
| CUDC-907 | ? | PI3K/HDAC | a) induction of cytotoxicity in HTLV-1-infected cells b) inhibition of HSP90 activity c) increased caspases activity, in ATL cells |
no |
| Butein | ? | AKT/AP1 NF-kB |
a) induction of apoptosis b) inhibition of proliferation in HTLV-1-infected and ATL cells |
no |
| AZT+ Bay 11-7085 |
RT | IκBα phosphorylation | a) increased apoptosis and b) up-reg. pro-apoptotic genes and down-reg. anti-apoptotic genes, in HTLV-1-infected/transformed cells |
no |
| Ruxolitinib+ navitoclax |
? | JAK/STAT Bcl-2/Bcl-xL |
increased cytotoxicity in IL-2–dependent ATL cell lines and ex vivo in lymphocytes from ATL patients | no |
| I-BET762+ Copanlisib+ bardoxolone methyl |
? | BET NF-κB PI3K |
Inhibition of proliferation in ATL cells in vitro and ex vivo samples from patients | Prolonged survival of ATL-bearing xenograft mice |
| Chloroquine/ Hydroxy chloroquine |
? | Autophagic flux |
Ex vivo, from ATL patients: a) inhibition of autophagy b) accumulation of p47 with LC3II, leading to inhibition of NF-κB activation c) proneness to apoptosis |
no |
| NCO-90/141 | ? | Sirtuin 2 | a) increased apoptosis and b) autophagy in ATL cells |
no |
| ? | ? | 12 miRNA | In silico analysis identified 12 miRNA deregulated in HTLV-1 samples predicted to interact with 90 genes | no |
1 ? = unknown/not investigated.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



