
Submitted:
15 August 2023
Posted:
16 August 2023
You are already at the latest version
Abstract
Rotavirus is a major cause of diarrhea globally in animals and young children under 5 years. Here, molecular detection and genetic characterization of porcine rotavirus in smallholder and commercial pig farms in the Lusaka Province of Zambia were con-ducted. Screening of 148 stool samples by RT-PCR targeting the VP6 gene revealed a prevalence of 22.9 % (34/148). Further testing of VP6-positive samples with VP7-specific primers produced 12 positives, which were then Sanger-sequenced. BLASTn of the VP7 positives showed sequence similarity to porcine and human rota-virus strains with identities ranging from 87.5% to 97.1%. By next-generation se-quencing, the full-length genetic constellation of the representative strains RVA/pig-wt/ZMB/LSK0137 and RVA/pig-wt/ZMB/LSK0147 were determined. Geno-typing of these strains revealed a known Wa-like genetic backbone and their genetic constellations were G4-P[6]-I5-R1-C1-M1-A8-N1-T1-E1-H1 and G9-P[13]-I5-R1-C1-M1-A8-N1-T1-E1-H1, respectively. Phylogenetic analysis revealed that these two viruses might have their ancestral origin from pigs, though some of their gene segments were related to human strains. The study shows evidence of reas-sortment and possible interspecies transmission between pigs and humans in Zambia. Therefore, the “One Health” surveillance approach for rotavirus A in animals and humans is recommended to inform the design of effective control measures.
Keywords:
Rotavirus A
; Reassortment
; Interspecies transmission
; Genomic characterization
; Porcine
; Zambia
1. Introduction
Rotavirus is a segmented, non-enveloped dsRNA virus of the family Reoviridae, and it is among the major causes of acute gastroenteritis (AGEs) in young children and animals globally [1,2]. A yearly estimate ranging from 122,322-215,575 deaths in children under five years has been reported in humans before 2021 [3]. This is lower than the earlier estimate of more than 430,000 deaths per year before the introduction of rotavirus vaccines globally [4]. Africa alone accounts for about 50% of group A rotavirus (RVA)-associated deaths globally despite the introduction of RVA vaccines in the immunization programs in most countries [4]. In Zambia, diarrhea-related deaths are ranked third among the major causes of death in children younger than five years [4,5].
In the livestock sector, rotavirus-associated infections result in major economic losses to farmers through high treatment costs, stunted growth, and varying mortality rates [3,4]. Ultimately, this leads to low productivity. Besides economic losses to farmers animals are considered natural reservoirs for rotavirus [1,5]. The assumptions have been arrived at because RVA has been detected in asymptomatic animals like pigs, cattle, and avian [1,9]. Therefore, studying animal sources like pigs for zoonotic RVA is key in developing preventive strategies in both animals and humans.
The segmented nature of the RVA genome makes it to be highly prone to reassortment events [6,7]. Through these events, novel RVA strains emerge with the capacity to affect the effectiveness of current vaccines in pigs and humans [8,12,13]. Recent reports have shown that strains known to cause infections only in pigs are now being detected in humans with evidence of porcine-human reassortment [6,10,11,14]. Evidence also indicates that direct transmission of RVA from pigs to humans does occur[15,16]. For example, RVA strains of porcine origin have been detected in children with AGEs in Kenya, the Democratic Republic of Congo (DRC), Egypt, Thailand, Taiwan, China, and Argentina [17,18,19,20]. These data emphasize the need to understand the epidemiology of RVA of animal origin to assess their potential to spread in the human population.
In Zambia, like many developing countries, pigs are among the cheaper sources of protein. Therefore, the interaction of pigs and humans is unavoidable as they live in close proximity to humans, thus creating a public health risk. Hence, effective vaccines are the most effective way of preventing RVA-related infections in humans and animals [21]. Effective vaccines are strain-specific in animals, while in humans, they are not [22,23]. For example, currently approved vaccines for RVA in humans, like Rotarix are derived from the G1 genotype while RotaTeq is from genotype G1-4 and a bovine genotype G6 virus. These two vaccines have been reported to reduce RVA infections in the human population even from infections associated with non-vaccine strains [23]. However low efficacy of these vaccines has been reported in developing countries [24]. One of the proposed hypotheses for the observed low vaccine efficacy in these countries, besides nutrient deficiencies (Zinc, Vitamin A and D), breast milk antibodies, and environmental enteropathy, is the genetic diversity of RVA which is associated mainly with reassortment and interspecies transmission between animals and human[25]. Hence, understanding the epidemiology, evolution, ecology, and diversity of RVA in animals such as pigs is critical in generating valuable data that may be useful in explaining the low efficacy of these vaccines in developing countries, including informing vaccine design.
Rotavirus consists of 11 segments that encode six structural and six non-structural proteins, namely; VP1-VP4, VP6, VP7, and NSP1-NSP4, NSP5/NSP6 [26,27]. Currently, the rotavirus classification working group (RCWG) uses open reading frames to classify RVA based on the 11 segments as GX-P[x]-Ix-Rx-Cx-Mx-Ax-Nx-Tx-Ex-Hx [28]. Based on this, three major genotypes have been reported to cause infections in humans, namely, genotype 1, genotype 2, and genotype 3, referred to as Wa-like, DS-1-like, and AU-1-like, respectively [28]. The genetic backbone for genotype 1, genotype 2, and genotype 3 are I1-R1-C1-M1-A1-N1-T1-E1-H1, I2-R2-C2-M2-A2-N2-T2-E2-H2, and I3-R3-C3-M3-A3-N3-T3-E3-H3, respectively [29,30]. Interestingly, all these genotypes are believed to have an ancestral origin from animal sources (i.e., genotype 1; porcine, genotype 2; bovine and genotype 3; cats, dogs, and avian) [30]. To date, at least 42 G-types, 58 P-types, 32 I-types, 28 Rtypes,24 C-types, 23 M-types, 39 A-types, 28 N-types,28 T-types, 32 E-types, and 28 H-types have been detected (https://rega.kuleuven.be/cev/viralmetagenomics/virus-classification/newgenotypes) accessed on 25th June 2023. In addition, standardized naming criteria have been developed by the RCWG for RVA, which include the rotavirus group/species origin/country of identification/common name of the strain/year of detection /G/P types [28].
Host specificity for all the known RVA genotypes identified thus far has been described [31]. For instance, in humans, the predominant G-types are G1-G4, G9, and G12 in combination with the P-type P[4], P[6], and P[8], with G1P[8] and G9P[8] being the most common G-P combinations known to cause human infections [32]. In contrast, porcine G3-G5, G9 and G11, along with P[6], P[7] and P[8], with G5P[7] and G4P[8] are the most prevalent G-P combination [33]. In calves, the predominant genotype is G6 [34]. Recent studies from India, China, Argentina, Kenya, and the Democratic Republic of Congo have provided evidence of the new evolving genotype G4P[6] among the RVA strains causing severe diarrhea in young children globally [19,20,31,35,36,37]. Full genome analysis of the G4P[6] genotype from those studies revealed Wa-like and DS-1-like genetic backbone. These findings implied that the possible origin of G4P[6] would be porcine and bovine respectively [17,19]. Also, whole genome analysis of a human G12 from Zambia, South Africa, Ethiopia, and Cameroon revealed a porcine-human reassortment [14]. However, in Zambia, G2 was the most common genotype in humans post-rotavirus vaccine introduction and G1 and G9 were predominant during the pre-vaccination period [38]. Even though several genotypes have been detected in humans in Zambia, little is known about the origin of these genotypes. In animals, limited data is known about RVA in Zambia. To date, only three studies have been conducted, two in bats and one in rodents and from these studies, novel genotypes were reported [39,40]. This implies that the diversity of RVA in the animal population in Zambia is highly probable. Therefore, surveillance of RVA in possible natural reservoirs of RVA such as pigs is of great importance to understand the possible source of existing and new emerging strains of RVA. Therefore, in this study, we screened stool samples from smallholder and commercial pig farms for RVA and conducted sequence analyses of the detected viruses to improve our understanding of the epidemiology and genetic diversity of RVA circulating in domestic pigs in Zambia.
2. Materials and Methods
2.1. Study Design and Study Site
We employed a cross-sectional study in selected farms in three districts of Lusaka city in the Lusaka Province of Zambia, namely; Chilanga, Kafue, and Chongwe (Figure 1). Our study sites included both commercial and smallholder farms. Farms with more than 100 pigs were considered commercial, and those with less than 100 pigs were categorized as smallholder farms. Sample collections were conducted during all three main seasons of Zambia, namely, Wet Rainy from mid-November to April, Hot and Dry season from mid-August to mid-November, and Cold and Dry seasons from May to mid-August. We sampled piglets, weaners, and growers, regardless of whether they were symptomatic or asymptomatic for diarrhea. Pigs less than 4 weeks, 8 weeks, and greater than 8 weeks old were considered piglets, weaners, and growers, respectively. Random sampling was conducted from January 2018 to December 2018.
2.2. Sample Collection, Preparation, and RNA Extraction
Fecal samples were collected from piglets, weaners, and growers from selected farms in peri-urban areas of Lusaka Province. They were transported on a portable 4°C refrigerator to the Virology laboratory at the University of Zambia, School of Veterinary Medicine. A 10% suspension of each sample was prepared in phosphate-buffered saline (PBS) and was centrifuged at 6,000 x g for 10 minutes. Then 100 µL of the supernatant was used for RNA extraction using Trizol LS reagent according to the manufacturer’s instruction (Life Technologies Corporation, Carlsbad, USA). Total RNA was eluted in 50 µL of RNase-free water and stored at -80°C until use.
2.3. Genomic Screening for Group A Rotavirus
Nested polymerase chain reaction (PCR) was used for screening of RVA targeting the VP6 gene using primer pairs described previously [41]. A one-step reverse transcriptase PCR kit (Qiagen, Hilden, Germany) was used for the initial step under the following conditions 30 min at 50°C, 15 min at 95°C, 45 cycles of 20 s at 95°C, 30 s at 50°C, 1 min at 72°C, and a final extension step of 10 min at 72°C. The first-round PCR products were used as templates for the second-round PCR using the ExTaq DNA polymerase according to the manufacturer’s instructions (Takara Biotechnology Inc, Shiga, Japan). The following thermal cycling conditions were used: 98°C for 10 s, 35 cycles of 98°C for 30 s, 54°C for 30 s, 72°C for 1 min, and final extension at 72°C for 5 min. Visualization of the PCR products was achieved using gel electrophoresis with ethidium bromide staining. The positives obtained from the initial screening for VP6 were later used for genotyping using the major outer surface protein VP7 using primer pairs as described previously [39]. VP7 PCR products were purified for Sanger sequencing using Wizard® SV Gel and Clean-Up System (Promega, Madison, WI, USA) according to the manufacturer’s protocol. The purified DNA was then sequenced directly using a Big Dye terminator cycle sequencing ready reaction kit v3.1 on a 3500 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Sequences generated were assembled and edited using GENETYX version 12 (GENETYX Corporation, Tokyo, Japan).
2.4. Whole Genome Sequencing
Viral dsRNA in the positive fecal samples for the VP7 gene was isolated and enriched for whole genome sequencing. About 0.5-1.0 g of stool samples positive for the RVA genes were added to 10 mL of PBS and vortexed until well blended. The suspension was centrifuged at 10,000 x g for 3 min, and the supernatant was filtered through a 0.45 µm membrane to remove unpelleted bacterial-sized substances. The filtrate was concentrated using Amicon Ultra-15 centrifuge filters (Merck Millipore, Darmstadt, Germany) according to the manufacturer's instructions. The subsequent filtrate was used as material for RNA extraction with the QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions. Then, viral dsRNA in extracted total RNAs were purified using the ISOVIRUS II kit (Nippon Gene, Tokyo, Japan) according to the manufacturer’s instructions. The purified dsRNA was used for cDNA synthesis. Double-stranded cDNA was synthesized using a Prime Script Double Strand cDNA Synthesis Kit (Takara Biotechnology Inc, Shiga, Japan) according to the manufacturer’s instructions. The cDNA libraries were prepared using Nextera XT DNA Library Preparation Kit (Illumina, San Diego, CA, USA) according to the manufacturer’s instruction and subsequently subjected to whole-genome sequencing on the Illumina HiSeq X Ten (Illumina way, San Diego, USA).
2.5. Sequence Data Analysis
Sequence data generated were produced at an average estimate depth of 37.7 from the Illumina HiSeq X ten and were analyzed using Geneious Prime® version 2020.0.2. The workflow for the analysis of the imported FastQ files in Geneious prime was constructed as follows: trim leads (index), error correct and normalization, and lastly, assembly, which was done by mapping the normalized, reads to RVA reference sequences downloaded from the GenBank. The consensus sequences generated were exported in Fasta files and genotyped using the Rotavirus Virus A Genotyping tool (https://www.rivm.nl/mpf/typingtool/rotavirusa) accessed on 21st June 2023. Phylogenetic trees of the 11 segments (VP1-VP4, VP6, VP7, NSP1-NSP5/6) of RVA were constructed using the maximum likelihood method in Molecular Evolutionary Genetics Analysis version 7 (MEGA 7) employing Gamma distributed with invariant sites model [42]. The trees generated included selected reference strains of RVA from pigs and humans with high nucleotide similarity to our study strains from Africa and the rest of the globe. The reference strains were identified using the Basic Local Alignment Search Tool (BLAST) (https://blast.ncbi.nlm.nih.gov/Blast.cgi). The nucleotide sequences that were shorter than the study strains were removed from the analysis. Nucleotide alignment was done using Multiple Alignment Fast Fourier Transform (MAFFT) (https://mafft.cbrc.jp/alignment/software/), accessed on 20th January 2023. A bootstrap of 1,000 replicates was used to determine the dependability of the branching order.
2.6. Statistical Analysis
Data collected regarding age, seasons, and health status (diarrhea) from pig farms were analysed using the R package dplyr v1.0.7 (RStudio, Boston, USA) and visualized using ggplot2 v3.3.5 (Houston, Texas, USA). Furthermore, data analysis was conducted in R, with statistical significance set at p ≤ 0.05. Specifically, univariate logistic regression was performed to select which variables to use in the multiple regression analysis, where variables with a p-value less than 0.2 qualified for analysis. Finally, the coefficients of the variables in the univariate and multivariate regression models were exponentiated to obtain odds ratios.
3. Results
3.1. Detection of Group A Rotaviruses
A total of 148 pigs consisting of 49 piglets, 89 weaners, and 10 growers from six farms were screened for the presence of RVA using nested PCR, targeting segment 6 encoding the VP6 gene. Of the 148 samples, 47 were obtained from asymptomatic pigs while 101 were obtained from pigs with diarrhoea. From the total number of samples screened, 34 were positive on PCR, comprising 6 from asymptomatic and 28 from symptomatic pigs, representing a prevalence of 22.9% (34/148). Using binary logistic regression, we modeled the dependence of RVA infections on different variables. First, we performed a univariate screening using p = 0.2 as our cut-off point (Table 1). The results revealed that all variables ( i.e., season, diarrhea, and age) had a p-value below the set threshold (i.e., p < 0.2 and thus qualified for inclusion in the multivariate regression analysis (Table 2). The result of the final model showed that only Season was significantly associated with RVA infections (Odds ratio (OR) = 3.13, 95% confidence interval (CI) = 1.37-7.47, p-value = 0.008). However, diarrhea was not significantly associated with RVA infections p > 0.05) (Table 2). Similarly, age (grower, piglets, and weaners) had no significant association with RVA infections (p > 0.05) (Table 2).
3.2. Genotyping of Group A Rotavirus Using VP7 Gene
Thirty-four stool samples positive for RVA on the VP6 gene were further screened for the VP7 gene by PCR to determine the G-genotypes of each positive sample. Out of these, 12 were positive for the VP7 gene and were all successfully Sanger-sequenced and the Biotechnology Information (NCBI) database. Out of the 12 positives, six had high nucleotide sequence similarity to porcine RVA strains and the other six to human RVA strains, with the percentage identity ranging from 87.5% to 97.1%. Notably, from the 12 positives for the VP7 gene, only one sample was recovered from asymptomatic pigs, and 11 were from symptomatic pigs. BLASTn also revealed three distinct G-genotypes that had high nucleotide sequence similarity to our study sequences, namely G5 (n = 5), G9 (n = 6), and G4 (n = 1) (Table 3). It was noted that some strains that showed high sequence similarity to our study samples in GenBank were isolated from pigs while others were detected in humans (Table 3).
3.3. Whole Genome Sequence Analysis and Genetic Constellation Determination
From the 12 samples that were positive for the VP7 gene, six were selected for whole genome analysis based on PCR product intensity and the RNA concentration of the sample. Out of the six samples that were subjected to full genome sequencing, we obtained the two full-length genomic sequences of samples, LSK0137 and LSK0147, with good reads of 5.1 x 107 and 6.6 x 107, respectively. The raw sequence reads for the two samples were deposited in NCBI under the bioproject accession number PRJNA997783. The rest of the samples yielded few reads and had poor coverage. Therefore, we genotyped these two strains (LSK0137) and LSK0147) using the Rotavirus Virus A Genotypying tool (https://www.rivm.nl/mpf/typingtool/rotavirusa) accessed on 21st June 2023 and their genetic constellation was found to be G4-P[6]-I5-R1-C1-M1-A8-N1-T1-E1-H1 and G9-P[x]-I5-R1-C1-M1-Ax-N1-T1-E1-H1 respectively. However, for sample LSK0147 only nine segments were genotyped; the other two segments were short and did not meet the minimum % score for assigning genotypes (Table 4) [29].
Interestingly, out of the 11 segments of sample LSK0137, BLASTn assessment showed that nine had nucleotide sequence similarity to human RVA, and two were similar to porcine RVA with the percentage identity ranging from 95.2% to 98.8% (Table 4). Even though we did not genotype all the segments for LSK0147, all 11 segments were assessed on BLASTn. The results showed that out of 11 segments, six showed the highest nucleotide sequence similarity to human strains, and five were similar to porcine RVA, with the percentage identity ranging from 93.2% to 99.6%.
3.4. Phylogenetic Analysis of RVA Structural Proteins Genes
Phylogenetic analysis of segment 9 encoding the VP7 gene revealed that LSK0137 and LSK0147 belonged to G4 and G9 genotypes, respectively (Figure 2). Segment 9 of LSK0137 was closely related RVA strain from China and distantly related to a RVA strain detected in Sri Lanka, while LSK0147 grouped with viruses obtained from pigs in Taiwan (Figure 2). Segment 6 of LSK0137 clustered with porcine RVA which was detected in Vietnam while LSK0147 clustered separately. However, both belonged genotype I5 viruses (Figure 3). Phylogenetic analysis of segment 4 of LSK0137 revealed that it belonged to genotype P[6] and it was closely related to a human RVA which was detected in China in 2018 (Figure 4).
The sequences of segment 1 encoding the VP1 gene of LSK0137 and LSK0147 clustered with strains detected in pigs and humans from Africa and Asia. The VP1 sequence of LSK0137 was closely related to a porcine RVA strain from Mozambique, while that of LSK0147 was closely related to a human RVA strain detected in Thailand (Supplemental Figure S1). Phylogenetic analysis of the VP2 gene of LSK0137 showed that it belonged to a cluster of RVA strains detected from pigs and humans in China while the sequence of LSK0147 clustered distinctly (Supplemental Figure S2). The virus sequence of segment 3 encoding the VP3 gene of LSK0137 grouped distinctly, while that of LSK0147 clustered with RVA strains of porcine origin from Mozambique and Vietnam (Supplemental Figure S3).
3.4. Phylogenic Analysis of RVA Nonstructural Proteins Genes
Phylogenetic analysis of segment 5 encoding the NSP1 gene revealed that LSK0137 belonged to genotype A8 and clustered in a clade with human RVA strains detected in Thailand and one porcine RVA from Vietnum (Supplemental Figure S4). Analysis of the NSP2 gene sequence of LSK0137 and LSK0147 showed that they belonged to genotype N1, but were grouped in separate clusters. The NSP2 gene sequence of LSK0137 was closely related to human RVA strains from China, while that of LSK0147 was closely related to porcine RVA from China (Supplemental Figure S5). The analysis of the NSP3 gene virus sequences showed that both LSK0137 and LSK0147 belonged to genotype T1 and they were closely related to porcine RVA from China and Sri Lanka respectively (Figure 5). Analysis of the enterotoxin NSP4 gene of LSK0137 revealed that it belonged to genotype E1 and clustered separately (Figure 6). Phylogenetic analysis of the NSP5 gene showed that LSK0137 belonged to H1genotype (Supplemental Figure S6), and it clustered with RVA strains of pig and human origin from China.
4. Discussion
In the present study, we report the detection and genetic characterization of RVA in domestic pigs in the Lusaka Province of Zambia. We screened a total of 148 samples, out of which 34 samples were positive for RVA, representing a prevalence of 22.9% (34/148), which is within the reported global prevalence of RVA in pigs of 20 to 60 % [33]. In Africa, the overall prevalence of porcine RVA infection is unknown due to limited studies [33]. However, the prevalence of RVA infection in pigs in some African countries has been reported. For example, Kenya and Uganda (26.2%), Tanzania (35.3%), Mozambique (11.8%), Ghana (10.4%), and South Africa (27.3%) [7,8,43]. These prevalence trends suggest that the overall prevalence of porcine RVA infections in Africa might be considerably high.
Our study also showed that the rainy season is one of the major risk factors associated with RVA infections. This may have been associated with poor drainage systems in most farms and low temperatures that characterized the rainy season[44]. However, more studies are needed to explore the association of RVA infections and seasonality in pigs in Zambia, and how this may affect RVA infection in humans. Nevertheless, our finding suggests that stringent measures should be employed during the rainy seasons to curtail the spread of RVA. Additionally, the insignificant association of RVA infections to diarrhea in pigs adds to the existing knowledge that pigs may be among the natural reservoirs of RVA and may explain why RVA is detected in asymptomatic pigs [45]. Hence, more surveillance is vital in pigs to understand the epidemiology and diversity of porcine RVA in Zambia.
Further, BLASTn of the VP7 gene produced a low overall percentage identity, with the reference strains from the Genbank, suggesting that these may be unique strains [6]. Interestingly, some of the identified genotypes were similar to human strains and others to porcine strains. Therefore, the results may suggest possible interspecies transmission of RVA between pigs and humans [1]. Moreover, the circulation of these genotypes in pigs may be the source of new strains in the human population and may affect the effectiveness of both pig and human vaccines [46]. Comprehensive genetic characterization of RVA in animals such as pigs is vital in detecting variants that may be involved in interspecies transmission of RVA. For example, strainse known to cause porcine infections have been detected in children with AGE in Egypt, Nigeria, and Cameroon [15,16,49]. This may suggest that reassortment events and interspecies transmission of RVA may be the source of these strains in the human population.
We obtained the whole genetic constellation of the LSK0137 strain and a near full-length genetic constellation of the LSK0147 strain, both with a Wa-like genetic backbone as G4-P[6]-I5-R1-C1-M1-A8-T1-E1-H1 and G9-P[x]-I5-R1-C1-M1-Ax-T1-E1-H1, respectively. Though we did not manage to get the full genome of LSK0147strain, the two missing segments VP4 and NSP1, had the high nucleotide similarity to P[13] and A8 with the reference strains. Thus the genotypes of VP4 and NSP1 of LSK0147 strains are most likely P[13] and A8 respectively. Hence, the most probable genetic constellation of the LSK0147 strain would be G9-P[13]-I5-R1-C1-M1-A8-T1-E1-H1. Therefore, the Wa-like genetic backbone of our study strain suggests that their origin may be from pigs. This is because the Wa-like genetic backbone is associated with porcine origin [30]. We propose the name for porcine RVA in Zambia for LSK0137 and LSK0137 according to RCWG as RVA/pig-wt/ZMB/2018/G4P6 and RVA/pig-wt/ZMB/2018/G9P[13] respectively [28]. Analysis of both strains revealed some segments with genotypes similar to human and pig strains on BLASTn within the same genetic constellation. This is suggestive of the possible occurrence of reassortment events in the study strains.
Notably, reassortment events are known to be among the primary contributors to new evolving strains of RVA in animals and humans [8]. Recent studies from animals in Zambia revealed novel RVA genotypes [40]. Therefore, the detection of reassortant strains in pigs reared in close proximity to humans emphasizes the importance of surveillance in animals like pigs to avoid spillover of such strains in the human population. For example, rare genotypes like G4 with features of reassortment within its genetic constellation have been detected in Kenya and the Democratic Republic of Congo, and their genetic characteristics were traced to porcine [3,6]. These reports indicate that reassortment might be playing a role in the evolvement of new strains of porcine RVA with the potential to infect humans. While our results suggest RVA interspecies transmission, viral isolation-based RNA screening may be recommended to fully assess the zoonotic potential of these reassortant RVAs circulating in the pig population [40].
Phylogenetic analysis of the VP7 gene of the LSK0137 strain revealed that it belonged to the G4 genotype, and its origin may be from pigs as it formed a clustered with porcine and human-porcine reassortant RVA from China and Sri Lanka, respectively. The relatedness of the VP7 gene with reassortant strains may suggest that genotype G4 of LSK0137 may be zoonotic [48]. However, the VP7 gene of the LSK0147 strain grouped with porcine-origin strains from Taiwan belonging to the G9 genotype suggesting that its origin would be from pigs. Analysis of the VP6 gene of the LSK0137 and LSK0147 strains showed that they belonged to genotype I5, which is mostly associated with RVA of porcine origin. This implies that their ancestral origin would possibly be pigs. The analysis of segment 4 (VP4 gene) of the LSK0137 strain which belonged to the P6 genotype, showed that it was closely related to a human strain from China. This finding suggests possible interspecies transmission of RVA between pigs and humans. On the other hand, analysis of segments 1, 2, and 3 (VP1, VP2, and VP3) of the LSK0137 and LSK0147 strains revealed that these segments might be of porcine lineage as they mainly clustered with strains from pigs. However, the origin of the VP2 gene of the LSK0147 strain and the VP3 gene of the LSK0137 strain could not be ascertained as they clustered separately. This may imply that the genotypes of these segments may be new [9]. In addition, the relatedness of some of these segments with those from humans may imply that reassortment between pig and human RVAs does occur. Analysis of all the non-structural proteins (NSP1, NSP2, NSP3, NSP4, and NSP5/6) of the LSK0137 and LSK0147 strains showed a similar pattern with the structural proteins as they mainly clustered with strains from pigs, and some from humans. This suggests that the segments encoding the above-mentioned non-structural proteins may originate in porcine. The overall analysis of all 11 segments of RVA of the LSK0137 strain and some segments of the LSK0147 strain revealed that their likely origin would be porcine. However, the relatedness of some segments to human strains might suggest that interspecies transmission and reassortment may be occurring between pig and human RVAs in Zambia [36]. Therefore, the high sequence similarity and close phylogenetic relationships of some gene segments of viruses characterized in this study to those viruses detected in humans suggest that porcine might be among the sources of the unusual RVA emerging in humans like G4P[6] in Africa [3,12,16]. Thus, there is a need for continued surveillance of both pigs and humans to ascertain the possibility of interspecies transmission of RVA in Zambia.
5. Conclusions
This study has shown for the first-time evidence of the circulation of porcine RVA in Zambia. Nucleotide sequence and phylogenetic analysis suggest possible reassortment and interspecies transmission of RVA involving pigs and humans. Further, the insignificant statistical association of RVA infections with diarrhea supports the idea that pigs may be among the natural reservoirs of RVA in Zambia. Therefore, genomic surveillance of RVA in humans, pigs, and other animals is recommended for a better understanding of the epidemiology of RVA in Zambia.
Author Contributions
Conceptualization, J.N., H.H., and E.S.; data curation, J.N., H.H., H.M.C., and E.S.; formal analysis, J.N., H.H., H.M.C., A.H., W.M., M.K., and E.S.; funding acquisition, A.T, H.S., and E.S.; investigation, J.N., H.H., and E.S.; methodology, J.N., H.M.C, M.S., J.Y., H.H., and E.S.; project administration, J.N., and E.S.; resources, J.N., H.H., M.S., and E.S.; supervision, N.S., and E.S.; validation, J.N., H.H., N.S., and E.S; visualization, J.N., H.H., H.M.C., and E.S.; writing—original draft, J.N., H.H, A.K., M.S., and E.S.; writing—review & editing, J.N., H.H., M.S., N.S., H.S., M.K., A.T., and E.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the grant for the Japan Agency for Medical Research and Development (AMED) and Japan International Cooperation Agency (JICA) within the framework of the Science and Technology Research Partnership for Sustainable Development (SATREPS) (JP21jm0110019), the Japan Initiative for Global Research Network of Infectious Diseases from AMED (JP15fm0108008), the Japan Program for Infectious Diseases Research and Infrastructure from AMED (JP20wm0125008) and by AMED under Grants (JP223fa627005).
Ethics Statement
The study protocol was approved by the University of Zambia Health Science Research Ethics Committee (protocol ID: 202112030110), and verbal consent was obtained from farm owners before sampling animals.
Acknowledgments
The authors acknowledge the support from Mr. Ladslav Moonga, Mr. Evans Mulenga, Mr. Chembesonfu Mwelwa, and Mr. Kapila Penjaninge from the School of Veterinary Medicine, University of Zambia, and the veterinary staff from the Ministry of Fisheries and Livestock (MFL) for the help they rendered during sample collection. The authors also acknowledge the support from Mr. Andrew Mukubesa and the support of farmers in making this study a reality.
Conflicts of Interest
The authors declare no conflict of interest
References
- Palombo, E.A. Genetic Analysis of Group A Rotaviruses: Evidence for Interspecies Transmission of Rotavirus Genes. Virus Genes 2002, 24, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Zeller, M.; Patton, J.T.; Heylen, E.; De Coster, S.; Ciarlet, M.; Van Ranst, M.; Matthijnssens, J. Genetic Analyses Reveal Differences in the VP7 and VP4 Antigenic Epitopes between Human Rotaviruses Circulating in Belgium and Rotaviruses in Rotarix and RotaTeq. J. Clin. Microbiol. 2012, 50, 966–976. [Google Scholar] [CrossRef] [PubMed]
- Steele, A.D.; Victor, J.C.; Carey, M.E.; Tate, J.E.; Atherly, D.E.; Pecenka, C.; Diaz, Z.; Parashar, U.D.; Kirkwood, C.D. Experiences with Rotavirus Vaccines: Can We Improve Rotavirus Vaccine Impact in Developing Countries? Hum. Vaccines Immunother. 2019, 15, 1215–1227. [Google Scholar] [CrossRef]
- Heylen, E.; Likele, B.B.; Zeller, M.; Stevens, S.; De Coster, S.; Conceição-Neto, N.; Van Geet, C.; Jacobs, J.; Ngbonda, D.; Van Ranst, M.; et al. Rotavirus Surveillance in Kisangani, the Democratic Republic of the Congo, Reveals a High Number of Unusual Genotypes and Gene Segments of Animal Origin in Non-Vaccinated Symptomatic Children. PLoS One 2014, 9. [Google Scholar] [CrossRef]
- Shachakanza, J.; Zulu, J.M.; Maimbolwa, M. Incidence of Rotavirus Infection among Under-Five Children Attending Health Centres in Selected Communities of Ndola, Copperbelt Province, Zambia. Health (Irvine. Calif). 2019, 11, 298–307. [Google Scholar] [CrossRef]
- Boene, S.S.; João, E.D.; Strydom, A.; Munlela, B.; Chissaque, A.; Bauhofer, A.F.L.; Nabetse, E.; Latifo, D.; Cala, A.; Mapaco, L.; et al. Prevalence and Genome Characterization of Porcine Rotavirus A in Southern Mozambique. Infect. Genet. Evol. 2021, 87, 104637. [Google Scholar] [CrossRef] [PubMed]
- Amimo, J.O.; Junga, J.O.; Ogara, W.O.; Vlasova, A.N.; Njahira, M.N.; Maina, S.; Okoth, E.A.; Bishop, R.P.; Saif, L.J.; Djikeng, A. Detection and Genetic Characterization of Porcine Group A Rotaviruses in Asymptomatic Pigs in Smallholder Farms in East Africa : Predominance of P [ 8 ] Genotype Resembling Human Strains. Vet. Microbiol. 2015, 175, 195–210. [Google Scholar] [CrossRef] [PubMed]
- Malakalinga, J.J.; Misinzo, G.; Msalya, G.M.; Shayo, M.J.; Malakalinga, J.R. Prevalence and Genetic Diversity of Rotavirus Group a in Piglets in Southern Highlands and Eastern Tanzania. SSRN Electron. J. 2022, 8, e11750. [Google Scholar] [CrossRef]
- Stubbs, S.C.B.; Quaye, O.; Acquah, M.E.; Adadey, S.M.; Kean, I.R.L.; Gupta, S.; Blacklaws, B.A. Full Genomic Characterization of a Porcine Rotavirus Strain Detected in an Asymptomatic Piglet in Accra, Ghana. BMC Vet. Res. 2020, 16, 1–18. [Google Scholar] [CrossRef]
- Steyer, A.; Poljšak-Prijatelj, M.; Barlič-Maganja, D.; Marin, J. Human, Porcine and Bovine Rotaviruses in Slovenia: Evidence of Interspecies Transmission and Genome Reassortment. J. Gen. Virol. 2008, 89, 1690–1698. [Google Scholar] [CrossRef] [PubMed]
- Amimo, J.O.; Vlasova, A.N.; Saifa, L.J. Detection and Genetic Diversity of Porcine Group a Rotaviruses in Historic (2004) and Recent (2011 and 2012) Swine Fecal Samples in Ohio: Predominance of the G9P[13] Genotype in Nursing Piglets. J. Clin. Microbiol. 2013, 51, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Weldegebriel, G.; Mwenda, J.M.; Chakauya, J.; Daniel, F.; Masresha, B.; Parashar, U.D.; Tate, J.E. Impact of Rotavirus Vaccine on Rotavirus Diarrhoea in Countries of East and Southern Africa. Vaccine 2018, 36, 7124–7130. [Google Scholar] [CrossRef] [PubMed]
- Malakalinga, J.J.; Misinzo, G.; Msalya, G.M.; Kazwala, R.R. Rotavirus Burden, Genetic Diversity and Impact of Vaccine in Children under Five in Tanzania. Pathogens 2019, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mokoena, F.; Esona, M.D.; Seheri, L.M.; Nyaga, M.M.; Magagula, N.B.; Mukaratirwa, A.; Mulindwa, A.; Abebe, A.; Boula, A.; Tsolenyanu, E.; et al. Whole Genome Analysis of African G12P[6] and G12P[8] Rotaviruses Provides Evidence of Porcine-Human Reassortment at NSP2, NSP3, and NSP4. Front. Microbiol. 2021, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Holmes, J.L.; Kirkwood, C.D.; Gerna, G.; Clemens, J.D.; Rao, M.R.; Naficy, A.B. Characterization of Unusual G8 Rotavirus Strains Isolated from Egyptian Children. 1999, 144, 1381–1396. [Google Scholar] [CrossRef] [PubMed]
- Esona, M.D.; Armah, G.E.; Geyer, A.; Steele, A.D. Detection of an Unusual Human Rotavirus Strain with G5P [ 8 ] Specificity in a Cameroonian Child with Diarrhea. 2004, 42, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Wandera, E.A.; Hatazawa, R.; Tsutsui, N.; Kurokawa, N.; Kathiiko, C.; Mumo, M.; Waithira, E.; Wachira, M.; Mwaura, B.; Nyangao, J.; et al. Genomic Characterization of an African G4P[6] Human Rotavirus Strain Identified in a Diarrheic Child in Kenya: Evidence for Porcine-to-Human Interspecies Transmission and Reassortment. Infect. Genet. Evol. 2021, 96, 105133. [Google Scholar] [CrossRef] [PubMed]
- Tacharoenmuang, R.; Guntapong, R.; Upachai, S.; Singchai, P.; Fukuda, S.; Ide, T.; Hatazawa, R.; Sutthiwarakom, K.; Kongjorn, S.; Onvimala, N.; et al. Full Genome-Based Characterization of G4P[6] Rotavirus Strains from Diarrheic Patients in Thailand: Evidence for Independent Porcine-to-Human Interspecies Transmission Events. Virus Genes 2021, 57, 338–357. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, Y.H.; Ghosh, S.; Tang, W.F.; Pang, B.B.; Liu, M.Q.; Peng, J.S.; Zhou, D.J.; Kobayashi, N. Genomic Characterization of G3P[6], G4P[6] and G4P[8] Human Rotaviruses from Wuhan, China: Evidence for Interspecies Transmission and Reassortment Events. Infect. Genet. Evol. 2015, 33, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Degiuseppe, J.I.; Beltramino, J.C.; Millán, A.; Stupka, J.A.; Parra, G.I. Complete Genome Analyses of G4P[6] Rotavirus Detected in Argentinean Children with Diarrhoea Provides Evidence of Interspecies Transmission from Swine. Clin. Microbiol. Infect. 2013, 19. [Google Scholar] [CrossRef]
- Platts-Mills, J.A.; Steele, A.D. Rotavirus Vaccine Impact in Africa: Greater than the Sum of Its Parts? Lancet Glob. Heal. 2018, 6, e948–e949. [Google Scholar] [CrossRef] [PubMed]
- Saif, L.J.; Vlasova, A.N.; Morrow, W.E.M. Rotaviral Diarrhea in Pigs. 1980, 1–6.
- Meta-analysis, C.A.; Mwila-kazimbaya, K.; Bosomprah, S.; Simuyandi, M.; Chisenga, C.C.; Munsaka, S. IMedPub Journals Efficacy and Effectiveness of Rotavirus Vaccine on Incidence of Diarrhoea Among. 2018, 3, 1–11. [Google Scholar] [CrossRef]
- Todd, S.; Page, N.A.; Steele, A.D.; Peenze, I.; Cunliffe, N.A. Rotavirus Strain Types Circulating in Africa: Review of Studies Published during 1997-2006. J. Infect. Dis. 2010, 202. [Google Scholar] [CrossRef] [PubMed]
- Nakagomi, O.; Nakagomi, T. Genetic Diversity and Similarity among Mammalian Rotaviruses in Relation to Interspecies Transmission of Rotavirus. Arch. Virol. 1991, 120, 43–55. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Heylen, E.; Zeller, M.; Rahman, M.; Lemey, P.; Van Ranst, M. Phylodynamic Analyses of Rotavirus Genotypes G9 and G12 Underscore Their Potential for Swift Global Spread. Mol. Biol. Evol. 2010, 27, 2431–2436. [Google Scholar] [CrossRef]
- Theuns, S.; Desmarets, L.M.B.; Heylen, E.; Zeller, M.; Dedeurwaerder, A.; Roukaerts, I.D.M.; Van Ranst, M.; Matthijnssens, J.; Nauwynck, H.J. Porcine Group a Rotaviruses with Heterogeneous VP7 and VP4 Genotype Combinations Can Be Found Together with Enteric Bacteria on Belgian Swine Farms. Vet. Microbiol. 2014, 172, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Matthijnssens, J.; Ciarlet, M.; McDonald, S.M.; Attoui, H.; Bányai, K.; Brister, J.R.; Buesa, J.; Esona, M.D.; Estes, M.K.; Gentsch, J.R.; et al. Uniformity of Rotavirus Strain Nomenclature Proposed by the Rotavirus Classification Working Group (RCWG). Arch. Virol. 2011, 156, 1397–1413. [Google Scholar] [CrossRef]
- Maes, P.; Matthijnssens, J.; Rahman, M.; Van Ranst, M. RotaC: A Web-Based Tool for the Complete Genome Classification of Group A Rotaviruses. BMC Microbiol. 2009, 9, 2–5. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Van Ranst, M. Genotype Constellation and Evolution of Group A Rotaviruses Infecting Humans. Curr. Opin. Virol. 2012, 2, 426–433. [Google Scholar] [CrossRef]
- Mukherjee, A.; Ghosh, S.; Bagchi, P.; Dutta, D.; Chattopadhyay, S.; Kobayashi, N.; Chawla-Sarkar, M. Full Genomic Analyses of Human Rotavirus G4P[4], G4P[6], G9P[19] and G10P[6] Strains from North-Eastern India: Evidence for Interspecies Transmission and Complex Reassortment Events. Clin. Microbiol. Infect. 2011, 17, 1343–1346. [Google Scholar] [CrossRef]
- Zaraket, H.; Charide, R.; Kreidieh, K.; Dbaibo, G.; Melhem, N.M. Update on the Epidemiology of Rotavirus in the Middle East and North Africa. Vaccine 2017, 35, 6047–6058. [Google Scholar] [CrossRef] [PubMed]
- Vlasova, A.N.; Amimo, J.O.; Saif, L.J. Porcine Rotaviruses: Epidemiology, Immune Responses and Control Strategies. Viruses 2017, 9, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Alaoui Amine, S.; Melloul, M.; El Alaoui, M.A.; Boulahyaoui, H.; Loutfi, C.; Touil, N.; El Fahime, E. Evidence for Zoonotic Transmission of Species A Rotavirus from Goat and Cattle in Nomadic Herds in Morocco, 2012–2014. Virus Genes 2020, 56, 582–593. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Qian, Y.; Huang, T.; Zhu, R.; Zhao, L.; Zhang, Y.; Li, R. Infection, Genetics and Evolution Identification of Circulating Porcine – Human Reassortant G4P [ 6 ] Rotavirus from Children with Acute Diarrhea in China by Whole Genome Analyses. Infect. Genet. Evol. 2013, 20, 155–162. [Google Scholar] [CrossRef]
- Wandera, E.A.; Hatazawa, R.; Tsutsui, N.; Kurokawa, N.; Kathiiko, C.; Mumo, M.; Waithira, E.; Wachira, M.; Mwaura, B.; Nyangao, J.; et al. Genomic Characterization of an African G4P[6] Human Rotavirus Strain Identified in a Diarrheic Child in Kenya: Evidence for Porcine-to-Human Interspecies Transmission and Reassortment. Infect. Genet. Evol. 2021, 96, 105133. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Choi, S.; Kim, J.S.; Lee, E.J.; Hyun, J.; Kim, H.S. Whole-Genome Analysis of Rotavirus G4P[6] Strains Isolated from Korean Neonates: Association of Korean Neonates and Rotavirus P[6] Genotypes. Gut Pathog. 2019, 11. [Google Scholar] [CrossRef]
- Simwaka, J.C.; Mpabalwani, E.M.; Seheri, M.; Peenze, I.; Monze, M.; Matapo, B.; Parashar, U.D.; Mufunda, J.; Mphahlele, J.M.; Tate, J.E.; et al. Diversity of Rotavirus Strains Circulating in Children under Five Years of Age Who Presented with Acute Gastroenteritis before and after Rotavirus Vaccine Introduction, University Teaching Hospital, Lusaka, Zambia, 2008 – 2015. Vaccine 2018, 3–7, in press. [Google Scholar] [CrossRef]
- Sasaki, M.; Kajihara, M.; Changula, K.; Mori-Kajihara, A.; Ogawa, H.; Hang’ombe, B.M.; Mweene, A.S.; Simuunza, M.; Yoshida, R.; Carr, M.; et al. Identification of Group A Rotaviruses from Zambian Fruit Bats Provides Evidence for Long-Distance Dispersal Events in Africa. Infect. Genet. Evol. 2018, 63, 104–109. [Google Scholar] [CrossRef]
- Kishimoto, M.; Kajihara, M.; Tabata, K.; Itakura, Y.; Toba, S.; Ozono, S.; Sato, Y.; Suzuki, T.; Ito, N.; Changula, K.; et al. Isolation and Characterization of Distinct Rotavirus A in Bat and Rodent Hosts. J. Virol. 2023, 97. [Google Scholar] [CrossRef]
- Elschner, M.; Prudlo, J.; Hotzel, H.; Otto, P.; Sachse, K. Nested Reverse Transcriptase-Polymerase Chain Reaction for the Detection of Group A Rotaviruses. J. Vet. Med. Ser. B 2002, 49, 77–81. [Google Scholar] [CrossRef]
- Engeset, R.V.; Udnæs, H.C.; Guneriussen, T.; Koren, H.; Malnes, E.; Solberg, R.; Alfnes, E. Improving Runoff Simulations Using Satellite-Observed Time-Series of Snow Covered Area. Nord. Hydrol. 2003, 34, 281–294. [Google Scholar] [CrossRef]
- Ngomane, T.; Seheri, M. Genetic Diversity of Group a Rotavirus Strains Circulating in Porcine from Five Provinces in South Africa during 2007, 2008 and 2015. 2015, 2015. [Google Scholar]
- Hasan, M.A.; Mouw, C.; Jutla, A.; Akanda, A.S. Quantification of Rotavirus Diarrheal Risk Due to Hydroclimatic Extremes Over South Asia: Prospects of Satellite-Based Observations in Detecting Outbreaks. GeoHealth 2018, 2, 70–86. [Google Scholar] [CrossRef]
- Amimo, J.O.; El Zowalaty, M.E.; Githae, D.; Wamalwa, M.; Djikeng, A.; Nasrallah, G.K. Metagenomic Analysis Demonstrates the Diversity of the Fecal Virome in Asymptomatic Pigs in East Africa. Arch. Virol. 2016, 161, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Mpabalwani, E.M.; Simwaka, J.C.; Mwenda, J.M.; Matapo, B.; Parashar, U.D.; Tate, J.E. Sustained Impact of Rotavirus Vaccine on Rotavirus Hospitalisations in Lusaka, Zambia, 2009–2016. Vaccine 2018, 36, 7165–7169. [Google Scholar] [CrossRef] [PubMed]
- Atii, D.J.I.; Ojeh, C.K. Subgroup Determination of Group A Rotaviruses Recovered from Piglets in Nigeria. 1995, 8, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Yahiro, T.; Takaki, M.; Chandrasena, T.G.A.N.; Rajindrajith, S.; Iha, H.; Ahmed, K. Human-Porcine Reassortant Rotavirus Generated by Multiple Reassortment Events in a Sri Lankan Child with Diarrhea. Infect. Genet. Evol. 2018, 65, 170–186. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Map of Zambia showing Lusaka Province and districts of sample collection.
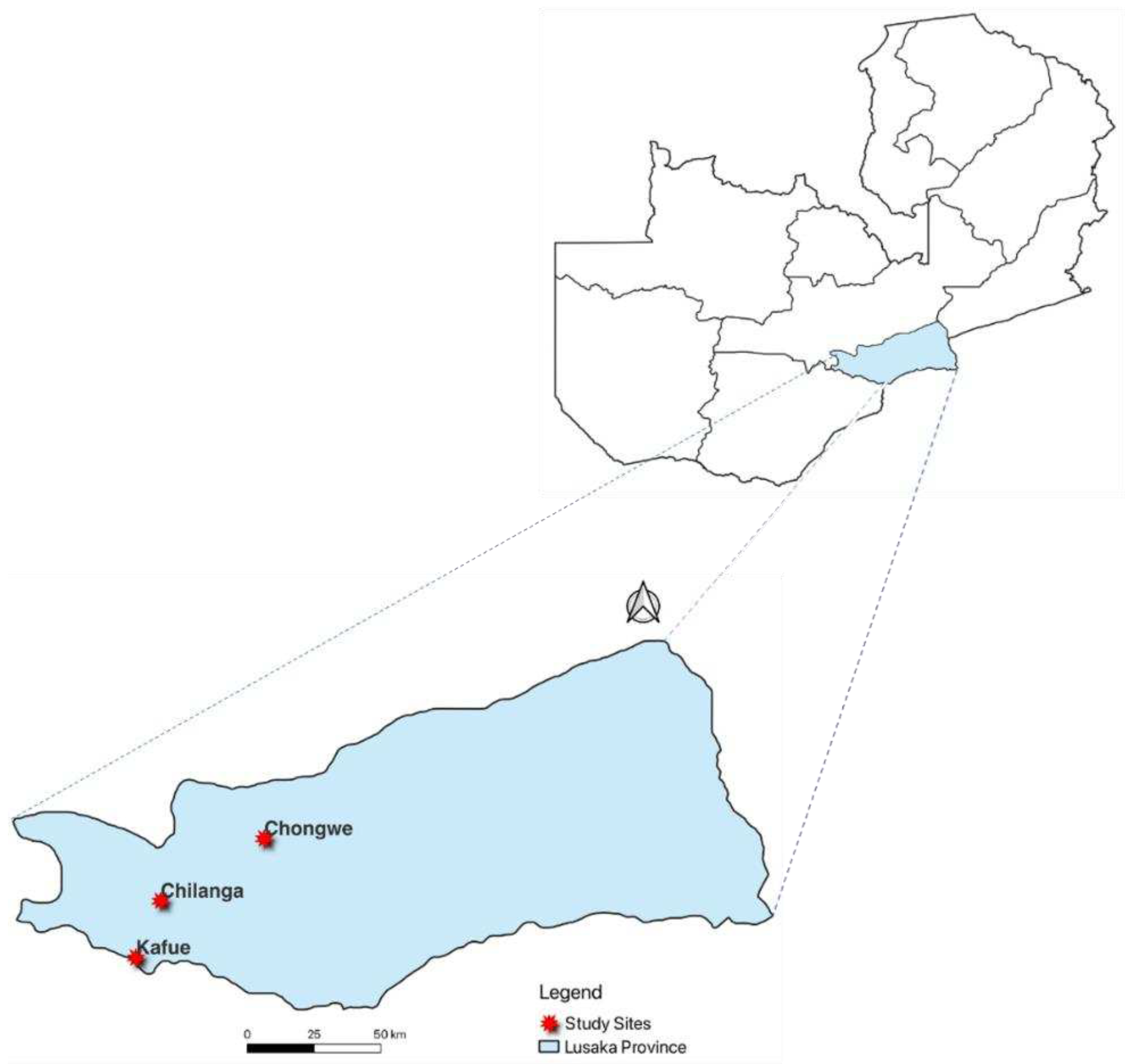
Figure 2.
Phylogenetic tree of VP7 genes. The analysis was based on 784 nucleotides. Bootstrap values on branch nodes ≥ 50% are shown. The GenBank accession number/ Rotavirus group/Species origin/country of origin / G/P-types represent the reference sequences included in the tree. The viruses characterized in this study with their accession number are shown in red text. The vertical lines represent Genotypes.
Figure 2.
Phylogenetic tree of VP7 genes. The analysis was based on 784 nucleotides. Bootstrap values on branch nodes ≥ 50% are shown. The GenBank accession number/ Rotavirus group/Species origin/country of origin / G/P-types represent the reference sequences included in the tree. The viruses characterized in this study with their accession number are shown in red text. The vertical lines represent Genotypes.
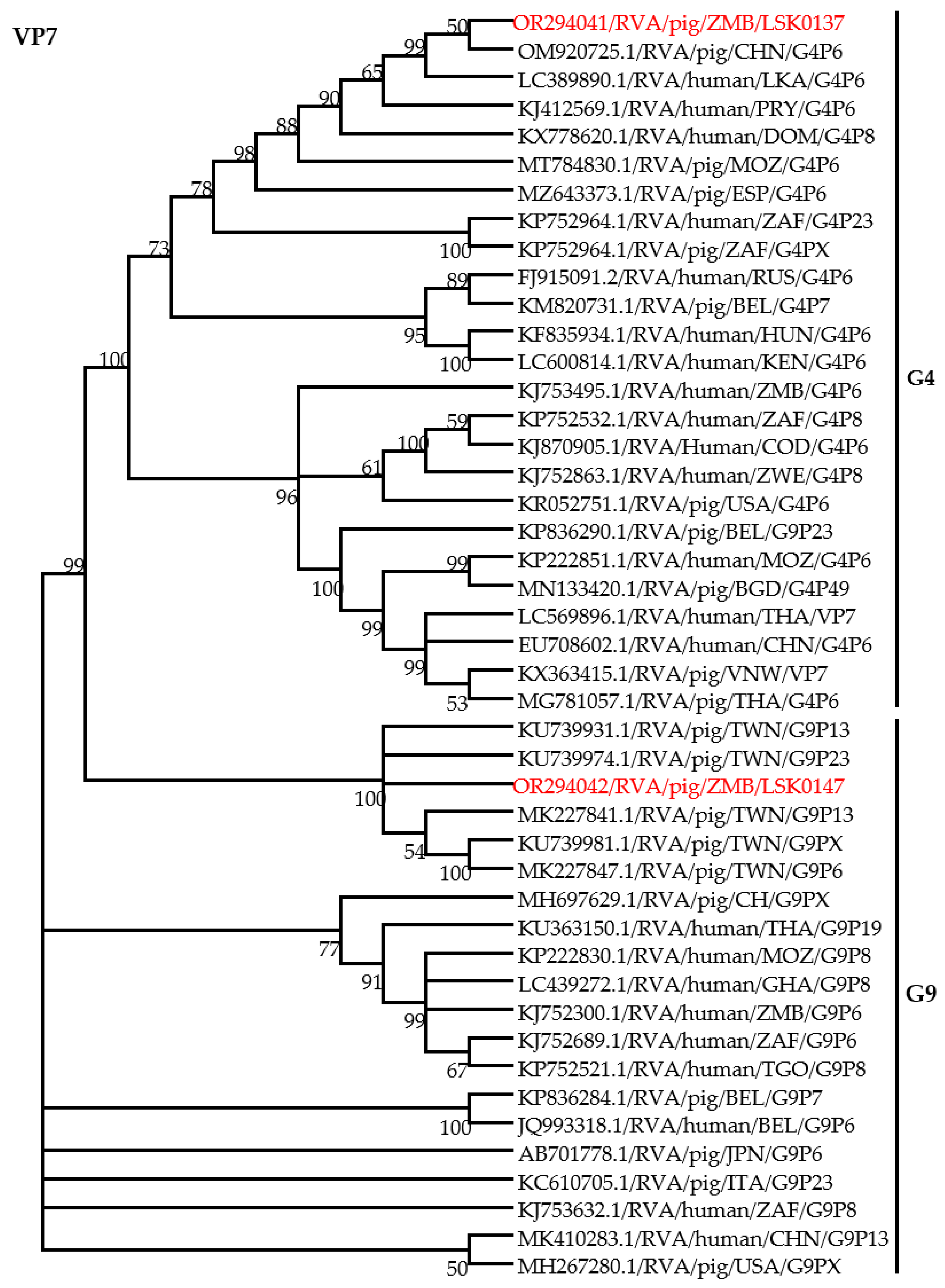
Figure 3.
Phylogenetic tree of VP6 genes. The analysis was based on 1,222 nucleotides. Bootstrap values on branch nodes ≥ 50% are shown. The GenBank accession number/ Rotavirus group/Species origin/country of origin / G/P-types represent the reference sequences included in the tree. The viruses characterized in this study with their Bioproject accession number are shown in red text. The vertical lines represent Genotypes.
Figure 3.
Phylogenetic tree of VP6 genes. The analysis was based on 1,222 nucleotides. Bootstrap values on branch nodes ≥ 50% are shown. The GenBank accession number/ Rotavirus group/Species origin/country of origin / G/P-types represent the reference sequences included in the tree. The viruses characterized in this study with their Bioproject accession number are shown in red text. The vertical lines represent Genotypes.

Figure 4.
Phylogenetic tree of VP4 genes that belong to genotype P6. The analysis was based on 2,075 nucleotides. Bootstrap values on branch nodes ≥ 50% are shown. The GenBank accession number/ Rotavirus group/Species origin/country of origin / G/P-types represent the reference sequences included in the tree. The virus characterized in this study with its Bioproject accession number is shown in red text.
Figure 4.
Phylogenetic tree of VP4 genes that belong to genotype P6. The analysis was based on 2,075 nucleotides. Bootstrap values on branch nodes ≥ 50% are shown. The GenBank accession number/ Rotavirus group/Species origin/country of origin / G/P-types represent the reference sequences included in the tree. The virus characterized in this study with its Bioproject accession number is shown in red text.

Figure 5.
Phylogenetic tree of NSP3 genes that belong to genotype T1. The analysis was based on 656 nucleotides. Bootstrap values on branch nodes ≥ 50% are shown. The GenBank accession number/ Rotavirus group/Species origin/country of origin / G/P-types represent the reference sequences included in the tree. The viruses characterized in this study with their Bioproject accession number are shown in red text.
Figure 5.
Phylogenetic tree of NSP3 genes that belong to genotype T1. The analysis was based on 656 nucleotides. Bootstrap values on branch nodes ≥ 50% are shown. The GenBank accession number/ Rotavirus group/Species origin/country of origin / G/P-types represent the reference sequences included in the tree. The viruses characterized in this study with their Bioproject accession number are shown in red text.

Figure 6.
Phylogenetic tree of NSP4 genes that belong to genotype E1. The analysis was based on 674 nucleotides. Bootstrap values on branch nodes ≥ 50% are shown. The GenBank /accession number/ Rotavirus group/Species origin/country of origin / G/P-types represent the reference sequences included in the tree. The virus characterized in this study with its Bioproject accession number is shown in red text.
Figure 6.
Phylogenetic tree of NSP4 genes that belong to genotype E1. The analysis was based on 674 nucleotides. Bootstrap values on branch nodes ≥ 50% are shown. The GenBank /accession number/ Rotavirus group/Species origin/country of origin / G/P-types represent the reference sequences included in the tree. The virus characterized in this study with its Bioproject accession number is shown in red text.


Table 1.
Univariate logistic regression showing the selection of variables for multivariate regression.
Table 1.
Univariate logistic regression showing the selection of variables for multivariate regression.
| Variable | Levels | PCR “+ve” |
PCR “-ve” |
Odds ratio | 95% CI | p-value |
|---|---|---|---|---|---|---|
| Season | Dry | 11 | 72 | 1 | ||
| Rainy | 23 | 42 | 3.58 | 1.62 – 8.34 | 0.00209 | |
| Diarrhea | No | 6 | 41 | 1 | ||
| Yes | 28 | 73 | 2.62 | 1.06 – 7.47 | 0.0494 | |
| Age | Grower | 1 | 9 | 1 | ||
| Piglet | 18 | 31 | 5.23 | 0.87 – 100.40 | 0.1310 | |
| Weaner | 15 | 74 | 1.82 | 0.31 – 34.91 | 0.5817 |
Table 2.
Multivariate logistic regression showing the dependence of rotavirus infection on various variables.
Table 2.
Multivariate logistic regression showing the dependence of rotavirus infection on various variables.
| Variable | Levels | Odds Ratio | 95% CI | p-Value |
|---|---|---|---|---|
| Season | Dry | 1 | ||
| Rainy | 3.13 | 1.37 – 7.47 | 0.00781 | |
| Diarrhea | No | 1 | ||
| Yes | 1.61 | 0.59 – 4.88 | 0.36649 | |
| Age | Grower | 1 | ||
| Piglet | 4.18 | 0.62 – 83.71 | 0.20829 | |
| Weaner | 1.68 | 0.25 – 33.39 | 0.64534 |
Table 3.
Most identical genotypes of segment 9 of the VP7 genes of Zambian rotavirus detected in pigs and their percentage identities by BLASTn.
Table 3.
Most identical genotypes of segment 9 of the VP7 genes of Zambian rotavirus detected in pigs and their percentage identities by BLASTn.
| Sample ID | GenBank Accession no. |
Stool | Closest Sequence | |||
|---|---|---|---|---|---|---|
| Country | Host | Genotype VP7 Gene |
% Identity | |||
| 14 | OR294031 | Diarrhea | China | Pig | G9 | 96.17 |
| 18 | OR294032 | Diarrhea | China | Human | G5 | 94.68 |
| 31 | OR294033 | Diarrhea | China | Pig | G9 | 96.38 |
| 32 | OR294034 | Diarrhea | China | Pig | G9 | 96.05 |
| 34 | OR294035 | Diarrhea | Ghana | Pig | G5 | 87.52 |
| 41 | OR294036 | Diarrhea | China | Pig | G9 | 96.42 |
| 42 | OR294037 | Diarrhea | China | Pig | G9 | 95.84 |
| 71 | OR294038 | Diarrhea | China | Human | G5 | 94.48 |
| 72 | OR294039 | Diarrhea | China | Human | G5 | 94.28 |
| 77 | OR294040 | Normal | China | Human | G5 | 94.01 |
| 137 | OR294041 | Diarrhea | China | Human | G4 | 97.1 |
| 147 | OR294042 | Diarrhea | Taiwan | Human | G9 | 92.2 |
Table 4.
Genotypes and similar strains from the Genbank of all the segments of samples LSK0137 and LSK0147 analyzed from Rotavirus A Genotyping tool Version 0.1 and BLASTn.
Table 4.
Genotypes and similar strains from the Genbank of all the segments of samples LSK0137 and LSK0147 analyzed from Rotavirus A Genotyping tool Version 0.1 and BLASTn.
| Sample ID | Bioproject Accession No. |
Gene | Genotype | Cut Off % Value |
BLASTn %Identity |
Similar Strain from GenBank |
|---|---|---|---|---|---|---|
| LSK0137 | PRJNA997783 | VP7 | G4 | 80 | 97.24 | RVA/Human-wt/CHN/2018/G4P[6] |
| LSK0137 | PRJNA997783 | VP4 | P[6] | 80 | 95.58 | RVA/Human-wt/CHN/B24-R2/2019/P6 VP4 |
| LSK0137 | PRJNA997783 | VP6 | I5 | 85 | 96.02 | RVA/Pig-wt/VNM/14150_53/VP6 |
| LSK0137 | PRJNA997783 | VP1 | R1 | 83 | 95.20 | RVA/Human-tc/VNM/NT0042/2007/G4P[6] |
| LSK0137 | PRJNA997783 | VP2 | C1 | 84 | 96.39 | RVA/Human-wt/CHN/R1954/2013/G4P[6] |
| LSK0137 | PRJNA997783 | VP3 | M1 | 81 | 95.74 | RVA/Human-wt/RUS/2015 VP3 |
| LSK0137 | PRJNA997783 | NSP1 | A8 | 79 | 97.35 | RVA/Human-tc/VNM/NT0042/2007/G4P[6] |
| LSK0137 | PRJNA997783 | NSP2 | N1 | 85 | 96.76 | RVA/Human-tc/VNM/NT0042/2007/G4P[6] |
| LSK0137 | PRJNA997783 | NSP3 | T1 | 85 | 95.91 | Human rotavirus A strain GX54 |
| LSK0137 | PRJNA997783 | NSP4 | E1 | 85 | 97.97 | RVA/Pig/China/FJSH01/2021/G26P[23 |
| LSK0137 | PRJNA997783 | NSP5 | H1 | 91 | 98.83 | RVA/Human-wt/BRA/HST327/1999/G4P[6] |
| LSK0147 LSK0147 LSK0147 LSK0147 LSK0147 LSK0147 LSK0147 LSK0147 LSK0147 LSK0147 LSK0147 |
PRJNA997783 PRJNA997783 PRJNA997783 PRJNA997783 PRJNA997783 PRJNA997783 PRJNA997783 PRJNA997783 PRJNA997783 PRJNA997783 PRJNA997783 |
VP7 VP4 VP6 VP1 VP2 VP3 NSP1 NSP2 NSP3 NSP4 NSP5 |
G9 P[x] I5 R1 C1 M1 Ax N1 T1 E1 H1 |
80 80 85 83 84 81 79 85 85 85 91 |
94.22 93.15 94.54 96.97 92.25 98.68 96.31 96.85 97.60 98.21 99.58 |
RVA/Human-wt/TWN/G9P19 RVA/Pig-wt/VNM/14150_54/VP4 Porcine rotavirus strain JN-1 VP6 RVA/Human-wt/THA/PK2015-1-0001 VP1 RVA/Human-wt/LKA/R1207/2009/G4P[6] RVA/Pig-wt/VNM/14225_44/VP3 RVA/Human-wt/CHN/E931/2008/G4P[6] Porcine rotavirus A isolate GDJM1NSP2 Pig-wt/CHN/CN127/2021/G12P[7] NSP3 RVA/Human-tc/VNM/NT0001/2007/G3P[6] RVA/Human-wt/LKA/R1207/2009/G4P[6] |
RVA= Rotavirus A; VP = Viral proteins; NSP = Nonstructural protein; X= not genotyped.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



