
Submitted:
07 August 2023
Posted:
08 August 2023
You are already at the latest version
Abstract
Herein, we report on a versatile system composed of two Ni(II)-based coordination compounds: a 3D metal-organic framework of {[Ni(μ-3isoani)2]·DMF}n (1) formula and a 0D monomeric [Ni(3isoani)2(H2O)4] (2) complex (where 3isoani = 3-aminoisonicotinato and DMF = dimethylformamide) which show mutual solvent-induced reversible transformations into each other. These 1 ↔ 2 transformations are triggered by the exposure of one compound to the solvent present in the other, that is, the MOF (1) in water and the monomer (2) in DMF. The process is easily followed by the naked eye because it involves a colour change from green (1)-to-brownish green (2), making the system potentially interesting to act as a sensor of those solvents. Moreover, these compounds present very different magnetic properties since 1 shows field-induced single-molecule magnet (SMM) behaviour, which corresponds to one of the few Ni(II)-based compounds showing this property, whereas 2 behaves as a regular paramagnet, all of which is explained according to a careful study of the magnetic properties by means of experimental direct-current (dc) and alternating-current (ac) measurements as well as complete active space self-consistent field (CASSCF) calculations.
Keywords:
Ni(II)-based SIM
; slow magnetic relaxation
; metal-organic framework (MOF)
; Solvent-triggered reversible transformations
; Colorimetric sensing
; CASSCF/NEVPT2
1. Introduction
Metal-organic frameworks (MOFs) represent an appealing class of materials derived from their high porosity in the form of well-defined pores of micrometric size that can be tuned and functionalized not only for their applications in gas adsorption and separation properties, but also in many other fields such as catalysis or healthcare, among others [1,2,3,4,5,6]. In fact, these materials have received large attention of synthetic chemists devoted to develop novel materials because their architectures may be often predesigned to enclose not only record-breaking specific surface areas but also decorate the pore walls with specific chemical functions present in the ligands employed [7,8,9,10]. Taking into account that the great amount of different metal ions available to construct MOFs, and the almost endless options of organic molecules to coordinate, it may be said that the structural diversity of these materials is practically inexhaustible [11,12,13]. During the last decade, attention has been also paid to a particular set of fascinating porous structures consisting of multiple chemically identical framework copies, known as interpenetrated structures, which are quite exclusive for metal-organic and organic compounds [14,15,16,17]. This subclass of MOFs are particularly characterized by a high structural flexibility that may lead to interesting dynamical properties. The origin of this behaviour can be found in the optimization of the framework interactions with the neighbouring counterparts and in their structural response directed by physico-chemical changes in the external medium [18,19]. In this regard, interpenetrated MOFs are prone to guest-induced transformations that may be the basis for good adsorption capacity in case that any of the properties is drastically changed during the process [20].
Among other interesting properties present in MOFs, the occurrence of slow relaxation of the magnetization (SRM) has been particularly studied during the last years [21,22,23], since numerous researchers suggest the possibility that this class of extended metal-organic compounds could behave as single molecule magnets (SMMs) [24,25]. As postulated by Chang and Long [26,27], SRM is manifested for those compounds possessing ground states with high total spin (S) and significant magnetic anisotropy, which is defined by the axial (D) and transverse (E) parameters of the zero-field splitting (zfs) phenomenom. This is a consequence of the increase of the energy barrier for the reversal of magnetization between the two lowest MS = ±S states that governs the SRM according to the expression Ueff = S2D [28]. Therefore, the possibility of possessing an ordered array of paramagnetic metal ions instigated the preparation of MOFs with transition metal ions and lanthanides. These families of coordination polymers are capable of exhibiting such a magnetic anisotropy by their combination with organic linkers that are able to isolate the metal ions in the network avoiding other magnetic exchange interactions [29,30,31]. The quest for MOFs showing SMM behavior using 3d ions has been mainly focused on Mn(III), Fe(I), Fe(II), Fe(III), Co(II) and Ni(I) complexes with variable coordination numbers ranging from hepta- to dicoordinated environments [32]. However, although Ni(II)-based compounds with hexacoordinate environment represent a textbook example of zfs, very few cases have been still found to present SRM [29,33]. Their ground state in the octahedral symmetry, 3A2g, is orbitally non-degenerate, whereas the following two excited states, 3T2g and 3T1g, are orbital triplets, which can be furtherly split into three new states each in the presence of a strong geometric or electronic distortion in the coordination shell. Under this situation, zfs (mainly the D parameter) emerges from the interaction of the ms functions between the ground and excited states. In the case of compounds showing a simple axial distortion, the D parameter sign will be determined by the relative stability of the split excited states (3A1(3T2) and 3E(3T2)) in the case of a simple axial distortion. Moreover, provided that the environment is tetragonally distorted, 3E(3T2) is further split and zfs tensor presents rhombicity (E/D ≠ 0).
Having all this ideas in mind, in a continuation with our previous studies on polymeric metal-organic materials behaving as SMMs [34,35,36,37,38], in the present work we describe two interchangeable Ni(II)-based compounds which are involved in an astonishing structural transformation: from a 3D open framework to a 0D complex. The fact that the transformation is reversible and that it is quite limited to some particular solvents makes of it a system worth studying. The color change occurring during the transformation gives the opportunity of using the system as a colorimetric sensor and the different coordination shell shown by each of the compounds lead to very different magnetic properties in the compounds.
2. Materials and Methods
The chemicals were of reagent grade and employed as obtained from supplier without any further purification.
2.1. Synthesis of {[Ni(μ-3isoani)2]·DMF}n (1)
Ni(NO3)2·6H2O (0.1 mmol, 0.0291 g) and H3isoani (0.1 mmol, 0.0138 g) were introduced in a closed vessel and dissolved in a mixture of DMF:H2O (2:1). Then, the vessel was sonicated for 2 minutes and placed in the oven for 48 h at 130 °C, after which it was slowly cooled down to room temperature. Once the reagent mixture was warm, we obtained a green single crystals corresponding to 1 after one week. Yield: 39-45% (based on metal). Anal. Calcd for C15H17N5NiO5 (%): C, 44.37; H, 4.22; N, 17.25. Found: C, 44.48; H, 4.09; N, 17.18.
2.2. Synthesis of [Ni(3isoani)2(H2O)4] (2)
A polycrystalline sample of 2 was obtained rapidly after the addition of Ni(NO3)2·6H2O (0.0291 g, 0.1mmol) dissolved in water (2 mL) over an aqueous-methanolic solution (2:1, 15 mL) of the H3isoani ligand (0.0276 g, 0.2 mmol) and some drops of Et3N to deprotonate the ligand and ensure its complete dissolution. The light green precipitate was filtered and washed with water and methanol. Yield: 55-60% (based on metal). Anal. Calcd (%) for C12H18N4NiO8 (%): C, 35.59; H, 4.48; N, 13.83. Found: C, 35.45; H, 4.39; N, 13.93.
2.3. Physical measurements
Elemental analyses (C, H, N) were performed using a Euro EA Elemental Analyser. For the collection of the IR spectrum, a FTIR 8400S Shimadzu spectrometer was employed using KBr pellets of the compounds in the 4000-400 cm-1 range. Thermal analysis (TG/DTA) were performed using a TA Instruments SDT 2960 thermal analyser placing the polycrystalline samples under a synthetic atmosphere (79% N2, 21% O2) with a contiuous heating rate of 5 °C/min. Magnetic susceptibility measurements were performed using a Quantum Design SQUID MPMS-7T susceptometer under an external applied magnetic field of 1000 G. The susceptibility data were corrected from the temperature independent paramagnetism, the magnetization of the sample holder and the corrections for the diamagnetism were estimated from Pascal’s Tables [39].
2.4. X-ray Diffraction Data Collection
Crystal structure of 1 was determined with single crystal X-ray diffraction (SCXRD) on a suitable single crystal, and the most relevant data from the refinement can be found in Table 1. SCXRD data collection was performed on a Bruker VENTURE area detector equipped with graphite monochromated Mo Kα radiation (λ = 0.71073 Å) by applying the ω-scan method at a temperature of 100(2) K. Data reduction was done using the APEX2 [40] software and the absorption corrections were made using SADABS [41]. Crystal structures were solved in SHELXT program [42] using direct methods and refined using SHELXL-2018/3 program [43] by full-matrix least-squares on F2 including all reflections and employing the Olex2 (1.5 version) crystallographic package [44]. All hydrogen atoms were located in the difference Fourier map and included as fixed contributions using riding models with isotropic thermal displacement parameters 1.2 times those of their parent atoms for the 3isoani ligand. CCDC 2283006 contains the supplementary crystallographic data of compound 1 for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: deposit@ccdc.cam.ac.uk). On the other hand, powder X-ray diffraction patterns were collected using a Philips X´PERT powder diffractometer in the 5 < 2θ < 50° range with a step size of 0.026° and an acquisition time of 2.5 s per step at 25 °C using Cu-Kα radiation (λ = 1.5418 Å). Indexation of the diffraction patterns was performed by pattern matching analysis using the FULLPROF crystallographic package [45], taking as reference the cell parameters and space group of the single crystal X-ray coordinates of isostructural Co-based MOFs.
2.5. Computational details
The computational strategy adopted herein for the estimation of the magnetic coupling constant (Jcalc) values has been described and validated elsewhere [46,47,48]. Gaussian 16 package [49] was employed for optimizing two suitable models: i) a dimeric excerpt of compound 1 (the model contains three complete 3isoani ligands surrounding each metal centre in addition to the bridging ligand, see Figure S11a) and ii) a monomeric complex with all surrounding ligands grown (Figure S11b), both of which were taken from the X-ray structure coordinates of 1. For the calculations of compound 2, we took the monomeric complex model from the isostructural Co-based counterpart. The Gaussian 16 package [49] was employed for the determination of the high-spin state and the low-spin broken symmetry state. The correctness of the latter state was ensured by means of its spin density distribution. For these calculations, density functional theory (DFT) methods were employed using the hybrid B3LYP functional and Gaussian-implemented 6-311G++(d,p) basis for non-metallic atoms [50] and LANL2DZ pseudopotentials for the Ni(II) atoms [51]. GaussView 6 [52] software was employed for spin-density surfaces plots. Alternatively, the standard broken symmetry (BS) DFT procedure using the FlipSpin feature of ORCA 5.0.3 [53,54] was followed to estimate the coupling constant value for all main superexchange pathways on dimeric models taken from X-ray coordinates. Ab initio calculations were implemented in ORCA (version 5.0.3) [53,54] to estimate zfs parameters for the monomeric models by means of the B3LYP functional [55,56] and employing the def2-TZVP basis sets for all atoms and def2-QZVPP for the metal atoms, recontracted for zeroth-order regular approximation (ZORA) relativistic approximation [57,58,59,60]. RIJCOSX approximation with appropriate auxiliary basis sets (def2/J) [59] were employed for all calculations. Calculations with state-average complete active space self-consistent field (SA-CASSCF) method were performed incorporating the five d-orbitals and eight electrons. Ten triplets and ten singlets were included according to other previous studies [61]. NEVPT2 calculations were performed on top of SA-CASSCF converged wave functions to include the dynamic correlation [62] according to previous results [61,63].
3. Results and Discussion
3.1. Structural description of compounds 1 and 2
As confirmed by the powder X-ray diffractograms (Figure S2–3), compounds 1 and 2 are isostructural to other previously published compounds with Co(II) and Zn(II) ions and their crystal structures were already reported [64,65,66]. Crystal structure of compound 1 consists of a doubly interpenetrated 3D framework containing voids filled with DMF molecules. The metal center shows a severely distorted octahedral environment established by the N2O4 donor set as confirmed by the continuous shape measures (SOC = 3.157, see Table S4). Nitrogen atoms pertain to two symmetrycally equivalent 3isoani ligands while the oxygen atoms coming from two carboxylate groups of adjacent 3isoani molecules complete the coordination environment, and in the case of the latter forging with the Ni(II) atom a four member chelating ring (Table 2). As a result of the relative arrangement acquired by the four 3isoani ligands around the metal centre, with angles between neighbouring coordination moieties in the 95-107° range, the four-connected unit may be considered as a pseudo-tetrahedral node (Figure 1).
On its part, 3isoani ligand acts as a linear linker between Ni(II) ions by coordinating to them with the μ-κN:κ2O,O', imposing a Ni···Ni distance of ca. 8.8 Å. The pseudo-tetrahedral shape of the metal node, gives rise to a 3D network that can be simplified as a diamond-like architecture (of dia topology and (66) point symbol). The resulting framework structure presents large voids, which are partially occupied by an interpenetrated chemically identical framework. Although this structural interpenetration provides with some flexibility to the overall assembly, the framework shows good stability due to the direct intermolecular hydrogen bonding interactions taking place between the amino and carboxylate groups pertaining to different subnetworks (Table S3). The overall structure is characterized by notable porosity (ca. the 36% of the total cell volume) formed of 1D microchannels running along the [010] crystallographic direction, which are occupied by lattice DMF molecules. These molecules are disordered into two equivalent positions as a consequence of the two-fold axis contained in the c axis of the cell.
Compound 2 exhibits a completely different structure as it consists of a supramolecular architecture based on hydrogen-bonded and π-π stacked isolated complexes with the [Ni(3isoani)2(H2O)4] formula. The centrosymmetric monomeric complex of the latter formula contains the four water molecules coordinated in the equatorial plane while the two 3isoani ligands, linked by their nitrogen atoms, occupy the main axis of the slightly distorted octahedron based on the N2O4 environment (Figure 2). The preference shown by this metal ion for the pyridine nitrogen atoms instead of the carboxylate oxygen atoms seems to obey to the better capacity of the latter to act as hydrogen bonding acceptor, a fact that is key in this structure given the supramolecular nature of the compound. Moreover, that coordination mode also favour that the exocyclic amino group of the ligand is more accessible to establish hydrogen bonding interactions. In detail, carboxylate groups are involved in a large hydrogen bonding network by acting as receptors of both amino groups and water molecules, in such a way that each complex is surrounded by eleven neighbouring complexes. In addition to those bonds, it is worth highlighting that the crystallographically independent 3isoani ligand forms weak π-π interactions with ligands of neighbouring (already hydrogen bonded) complexes, mainly due to the fact that aromatic rings of adjacent ligands do not overlap with each other because of their relative twisted orientation (Figure S5, see ESI).
3.2. Structural dynamics of compounds 1 and 2: solid state transformations
The two previously described compounds show a rapid mutual reversible transformation involving the exchange between the three-dimensional and supramolecular structures, that is, a 1 ↔ 2 transformation. Under ambient conditions in the solid state, sample of compound 1 rapidly shows a colour change from light green to pale brown (Figure 3). The transformation involves the breakage and formation of coordination bonds so that the 3D framework to evolves to the monomeric complexes found in compound 2. This 1 → 2 transformation takes place spontaneously given the high capacity of 1 to capture water from air, in such a way that the entrance of those molecules favours the replacement of most of the coordination shell and forces the recrystallization of compound 2. Interestingly, the reversibility of the reaction was confirmed when sample of 2 was immersed in DMF solvent. It is worth highlighting that the reaction does not require that the previous distillation of the DMF solvent to proceed but it may contain some water. However, the 1 ← 2 transformation does not occur under a saturated atmosphere of DMF (achieved by placing the solid sample inside a closed vessel containing DMF vapours), but it requires soaking the solid into liquid DMF.
To finish up with this section, it is also worth stating that, irrespective of the easy 1 → 2 transformation shown by the MOF, compound 1 may still be somewhat stable when it is kept under dry conditions, which permits the study of its thermal behavior. As detailed in the corresponding section (see Figures S1–2), fresh sample of 1 possesses high thermal stability and the crystal structure is robust enough as to permit the release of DMF molecules and obtain a potentially porous framework. However, it must be remarked that once activated (solvent removed from pores), the transformation dynamics are accelerated and exposing the sample to air triggers its transformation to compound 2.
3.3. Magnetic properties of compounds 1 and 2
The evolution of the magnetic susceptibility over temperature and isothermal magnetization at low temperature were analyzed for polycrystalline samples of both nickel-based compounds. Starting from the susceptibility data, the room temperature χMT product (1.26 and 1.32 cm3 K mol–1, respectively for compounds 1 and 2) shows higher values than that expected for a free Ni(II) ion (1.00 cm3 K mol–1 with g = 2.01) in both compounds. When decreasing the temperature, the χMT value remains constant in both cases but it shows a drastic drop at low temperatures, where it must be distinguished between the more progressive and deeper drop occurring below 100 K in compound 1 (down to 0.57 cm3 K mol–1 at 2 K) and the sudden decrease below 15 K observed in compound 2 (reaching a final value of 0.86 cm3 K mol–1 at 2 K, Figure 4). The χM-1 vs T plot for both compounds describes a linear distribution (although a very small deviation may be appreciated in the plot of compound 1 at below 10 K), from which the fitting gives a set of values (C = 1.28 and 1.42 cm3 K mol–1, θ = -4.62 and -1.29 K for 1 and 2, respectively) that are indicative of weak antiferromagnetic interactions, if any, occurring among Ni(II) ions in the compounds. This is in agreement with the fact that metal ions are quite far apart in both structures (the smallest Ni···Ni distances are of ca. 8.8 Å along 3isoani bridging ligands in 1 and of ca. 6.8 Å through hydrogen bonds in 2). In fact, a DFT calculation by means of the broken symmetry methodology confirms that the exchange coupling in 1 is very weak and antiferromagnetic in nature (J = -0.17 cm-1, Figure S10). Therefore, all these considerations, in addition to the former room temperature values, the magnetic behaviour of both compounds is thus better attributed to the presence of significant magnetic anisotropy of the Ni(II) metal centres. The recorded magnetization curves in the 0-7 T range of applied field at different temperatures in the 2-7 K range do not reach saturation at the maximum 7 T field. Moreover, it must be also highlighted that M(H) curves reach higher values and diverge substantially more with temperature for 1 than for 2, which may be related with a higher magnetic anisotropy for the former compound, as it may be also inferred from the reduced magnetization plots (Figure S8–9). Accordingly, the zfs parameters were estimated by simultaneously fitting the susceptibility and isothermal magnetization data with PHI program [67] to the following Hamiltonian (Equation (1)):
in which S represents the spin of the ground state (S = 1), D and E correspond to the axial and rhombic zfs parameters, respectively and the applied magnetic field is represented as H. The best fitting results (shown in Table 3) are concordant with the magnetostructural correlation performed by Boca et al. [68] and in line with the previous hypothesis, because the magnitude of the D parameter is substantially greater for 1 than 2. It is also worth highlighting the larger uniaxial anisotropy found for 1 compared to 2, in view of the greater E/D ratio observed for both compounds.
In order to confirm the reliability of the experimental fitting, the magnetic properties of both compounds were additionally computed by CASSCF/NEVPT2 calculations, from which the results in Table 3 were obtained. As observed, the calculations reproduce fairly well the experimental results by giving zfs parameters of the same sign and magnitude. In agreement with the largely distorted octahedral environment of 1, the ground state is dominated by the (dx2-y2)2(dz2)2(dyz)2(dxy)1(dxz)1 configuration that shares more characteristics with a tetrahedral than an octahedral environment, although the dyz orbital is quite far from the almost degenerated dxy and dxz orbitals (Figure 5a). Such an orbital splitting is in agreement with the largely distorted environment found in the compound and also with the fact that the environment imposes a pseudo-tetrahedral connectivity around Ni(II) ion. In fact, four of the positions of the coordination shell (those of the carboxylate oxygen atoms) correspond to chelating moieties that, somehow, represent a ligand field featuring a distorted tetrahedron with two extra weakly bonded oxygen atoms. On the contrary, the less distorted octahedral environment observed in compound 2 imposes the (dxy)2(dxz)2(dyz)2(dz2)1(dx2-y2)1 ground state configuration, where evident splittings of the ideal t2g and eg groups are found (Figure 5b). In addition to the latter, another remarkable difference between the energy levels in both compounds is that the energy gap between HOMO and LUMO orbitals is greater in 2 than in 1, which a larger separation from the ground to the excited states that somehow disables the transitions to the excited states (see Tables S5–8).
In view of the large magnetic anisotropy present in both compounds, magnetic susceptibility was also measured upon a small oscillating magnetic field (3.5 Oe) derived from an alternating current (ac) to explore the potential SMM behavior. When there is no external magnetic field applied, none of the compounds revealed maxima even at a very high frequency of the ac field (10000 Hz), which confirmed the absence of slow magnetic relaxation possibly due to the quantum tunneling of the magnetization (QTM) effect. On contrary, in the presence of a dc field of 1000 Oe, a nice set of frequency-dependent maxima were observed for compound 1 (Figure 6) although the data of compound 2 showed no change compared to that collected at Hdc = 0 Oe (Figure S12). Therefore, further measurements were conducted on the polycrystalline sample of 1 to study its SMM behavior. As observed in Figure 6b, the low temperature tiles below the maxima (peaking at 2.8–3.8 K) still indicate that QTM could be operative at this Hdc, for which we first inspected the behavior of the slow magnetic relaxation with respect to the dc field by measuring the magnetic susceptibility at 2.6 K for all the frequencies. As observed in Figure S13, the relaxation time rapidly increases from 250 Oe on and reaches a maximum at 1000 Oe, in such a way that we may consider that the data shown in Figure 6 corresponds to the optimal dc field.
Firstly, taking a look to the χM’’ vs frequency plot, it can be observed a unique maximum that is progressively shifted toward higher frequencies with the increasing temperature, a behavior associated with SMMs. Cole-Cole plots for 1 show semicircular-shaped curves which were fitted according to the generalized Debye model in the 2.3-3.5 K temperature range. Estimated α values are in the 0.48-0.20 range. Despite the fact that these quite high values suggest that the SRM could imply several relaxation pathways, the plot of the relaxation time according to the temperature follows the Arrhenius law (Equation (2)) by showing an almost rectilinear distribution covering the whole inspected temperature range.
τ = τ0 exp(Ueff/κBT)
The best fitting results give an energy barrier value (Ueff) of 26.1 K (18.1 cm-1) with a τ0 of 1.20 x 10-7 s for compound 1. The value of the energy barrier fits very well with the calculated axial zfs parameter (D = 17.3 cm-1), which is concordant with the fact that, by definition, the energy barrier for the reversal of magnetization for a Ni(II)-based compound equals to the value of D parameter (Ueff = S2D). Therefore, we can assume that the SRM mainly follows the Orbach mechanism, although the possibility that QTM could be present is not be totally discarded.
In any case, irrespective of the origin of the SMM behaviour in 1, the most interesting feature of the Ni3isoani system is the drastic overall change occurring in the magnetic properties of compounds 1 and 2, given the presence or absence of field-induced SRM. Moreover, taking into account that the change of the magnet behaviour is also linked to a green-to-pale green colour change triggered by the system as a consequence of the 1 ↔ 2 transformation, it must be highlighted that Ni/3isoani system is a material that could have potential in the field of sensors to detect changes in the mentioned solvents (DMF and H2O).
4. Conclusions
Two coordination compounds based on Ni(II) and 3isoani ligand are reported, one of which consists of a 3D metal-organic framework (compound 1) and the other one is a monomer-based supramolecular architecture (compound 2). The MOF is formed by a doubly interpenetrated network that shows 1D microchannels filled with DMF lattice molecules whereas the monomeric complex is formed by coordination water molecules which sustain the supramolecular hydrogen bonding network of the compounds. Interestingly, these compounds take part in an equilibrium in which their crystal structures are transformed into each other when the dry powdered samples are exposed to the appropriate solvents (1 → 2 when 1 is exposed to water, even simply at an ambient moisture at room temperature; and 1 ← 2 by soaking the sample in liquid DMF). The mentioned transformation is accompanied by a noticeable colour change between green (1) ↔ brownish green (2). In spite of the easy 1 → 2 transformation occurring for the MOF, the structure is robust enough and possesses good thermal stability under dry conditions (up to ca. 400 °C) that allow its activation (release of DMF molecules).
Ac susceptibility measurements show that only compound 1 presents SRM when an optimal external field of 1000 Oe is applied, whereas compound 2 shows no sign of that behaviour. This fact seems to be derived from the larger magnetic anisotropy present in compound 1 compared to compound 2, as suggested by the fitting of the magnetic susceptibility and isothermal magnetization measurements. In this sense, 1 presents high uniaxial anisotropy (D = -17.3 cm-1 and E/D = 0.05), probably as a consequence of the severely distorted octahedral N2O4 environment of 1, which contains two chelating carboxylate moieties. In contrast, that of 2 is substantially lower (D = -4.8 cm-1 and E/D = 0.15) in agreement with the quite regular octahedron shown by the N2Ow4 present in the compound. CASSCF/NEVPT2 calculations confirm those properties and reveal quite different orbital splitting scenarios in the compounds concordant with the mentioned distortion of the coordination environment: pseudo-tetrahedral in 1 vs perfectly octahedral in 2. Fitting of the variable temperature relaxation times obtained for compound 1 suggest that the SRM obeys the Arrhenius law and, although the presence of QTM cannot be discarded, it proceeds through an Orbach relaxation with an energy barrier of 26.1 K, thus in agreement with the theoretical approach of Ueff = S2D.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Elemental analysis and TG/DTA curves of compound 1; Table S2: Elemental analysis and TG/DTA curves of compound 2; Figure S1: IR spectra of H3isoani and compounds 1 and 2; Figure S2: Simulated and experimental PXRD of 1 with the full profile pattern matching analysis; Figure S3: Comparison of the experimental PXRD of 2 with the simulated diffractogram of the Ni-based isostructural counterpart reported in reference [1]. Full profile pattern matching analysis of 2 is also shown as an inset; Figure S4: View of the assembly of the building unit into the interpenetrated diamond-like framework of 1. Note that its independent subnetwork is shown in a different colour; Figure S5: View of the supramolecular network of 2 showing the hydrogen bonding network and π-π stacking interactions; Table S3: Structural parameters (Å, º) of hydrogen bonds (Å, º) in compound 1; Table S4: CShMs for the coordination environment of the optimized fragments of compounds 1 and 2. The lowest SHAPE values for each ion are shown highlighted indicating best fits. Note that this calculation for 2 has been performed over the optimized structure starting from X-ray coordinates of the isostructural Co(II) compound; Figure S6: χM-1 vs T of 1 showing fitting of Curie-Weiss law in the 300-50 K range; Figure S7: χM-1 vs T of 2 showing fitting of Curie-Weiss law; Figure S8: Reduced magnetization curves in the 2-7 K and 1-7 T region for compound 1; Figure S9: Reduced magnetization curves in the 2-7 K and 1-7 T region for compound 2; Figure S10: Calculated spin density distributions for suitable dimeric models of high (S = 2) and low (S = 0) spin states of compound 1; Table S5: Calculated CAS(8,5)/NEVPT2 transition energies and SOC splitting of the ground state for the model complex of compound 1; Table S6: Calculated CAS(8,5)/NEVPT2 transition energies and SOC splitting of the ground state for the model complex of compound 2; Table S7: Individual contributions to D and E of the excited triplet and singlet states computed at the CAS(8,5)/NEVPT2 level for the model complex of compound 1; Table S8: Individual contributions to D and E of the excited triplet and singlet states computed at the CAS(8,5)/NEVPT2 level for the model complex of compound 2; Figure S11: Monomeric models of compounds 1 and 2 used for the computational calculations of the magnetic properties. Note that the model of compound 2 was optimized starting from the isostructural Co-based counterpart [2]; Figure S12: χM’ (circles) and χM’’ (squares) vs T plots under Hdc = 1000 Oe for compound 2 at 10000 Hz of frequency for the oscillating field; Figure S13: Optimization of the dc field for compound 1 in the form of τ vs H; Figure S14: Cole-Cole plot for compound 1 showing the best fit for the selected temperature region; Figure S15: Variable-temperature frequency dependence of the χM´´ signal of 1.
Author Contributions
Conceptualization, J.C. and J.M.S.; synthesis of the samples, O.P.C.; structural analysis, A.R.D. and I.V.Y.; validation, J.C. and J.M.S.; formal analysis, resources, J.C.; data curation, O.P.C., J.C. and I.V.Y.; writing—original draft preparation, O.P.C and J.C.; writing—review and editing, J.C.; supervision, J.C. and J.M.S.; project administration, J.C. and A.R.D.; funding acquisition, J.C. and A.R.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Gobierno Vasco/Eusko Jaurlaritza (IT1755-22) and Junta de Andalucía (B-FQM-734-UGR20, ProyExcel_00386 and FQM-394).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data supporting this study’s findings are available from the corresponding author upon reasonable request.
Acknowledgments
The authors are thankful for the technical and human support provided by SGIker of UPV/EHU and European funding (ERDF and ESF), and wish to acknowledge the terrific help of all reviewers of the present manuscript whose comments helped to improve the quality of the work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jiao, L.; Seow, J.Y.R.; Skinner, W.S.; Wang, Z.U.; Jiang, H.-L. Metal–organic frameworks: Structures and functional applications. Mater. Today 2019, 27, 43–68. [CrossRef]
- Li, J.R.; Sculley, J.; Zhou, H.C. Metal-organic frameworks for separations. Chem. Rev. 2012, 112, 869–932. [CrossRef]
- Li, H.; Wang, K.; Sun, Y.; Lollar, C.T.; Li, J.; Zhou, H.-C. Recent advances in gas storage and separation using metal–organic frameworks. Mater. Today 2018, 21, 108–121. [CrossRef]
- Liu, J.; Chen, L.; Cui, H.; Zhang, J.; Zhang, L.; Su, C.Y. Applications of metal-organic frameworks in heterogeneous supramolecular catalysis. Chem. Soc. Rev. 2014, 43, 6011–6061. [CrossRef]
- Cepeda, J.; Pérez-Mendoza, M.; Calahorro, A.J.; Casati, N.; Seco, J.M.; Aragones-Anglada, M.; Moghadam, P.Z.; Fairen-Jimenez, D.; Rodr\’\iguez-Diéguez, A. Modulation of pore shape and adsorption selectivity by ligand functionalization in a series of {\textquotedblleft}rob{\textquotedblright}-like flexible metal{\textendash}organic frameworks. J. Mater. Chem. A 2018. [CrossRef]
- Yang, D.; Gates, B.C. Catalysis by Metal Organic Frameworks: Perspective and Suggestions for Future Research. ACS Catal. 2019, 9, 1779–1798. [CrossRef]
- Yaghi, O.M.; O’Keeffe, M.; Ockwig, N.W.; Chae, H.K.; Eddaoudi, M.; Kim, J. Reticular synthesis and the design of new materials. Nature 2003, 423, 705–714. [CrossRef]
- Farha, O.K.; Eryazici, I.; Jeong, N.C.; Hauser, B.G.; Wilmer, C.E.; Sarjeant, A.A.; Snurr, R.Q.; Nguyen, S.T.; Yazaydın, A.Ö.; Hupp, J.T. Metal–Organic Framework Materials with Ultrahigh Surface Areas: Is the Sky the Limit? J. Am. Chem. Soc. 2012, 134, 15016–15021. [CrossRef]
- Feng, L.; Wang, K.-Y.; Lv, X.-L.; Yan, T.-H.; Zhou, H.-C. Hierarchically porous metal–organic frameworks: Synthetic strategies and applications. Natl. Sci. Rev. 2019, 7, 1743–1758. [CrossRef]
- Liu, X.; Zhang, L.; Wang, J. Design strategies for MOF-derived porous functional materials: Preserving surfaces and nurturing pores. J. Mater. 2021, 7, 440–459. [CrossRef]
- Guillerm, V.; Kim, D.; Eubank, J.F.; Luebke, R.; Liu, X.; Adil, K.; Lah, M.S.; Eddaoudi, M. A supermolecular building approach for the design and construction of metal-organic frameworks. Chem. Soc. Rev. 2014, 43, 6141–6172. [CrossRef]
- Eddaoudi, M.; Sava, D.F.; Eubank, J.F.; Adil, K.; Guillerm, V. Zeolite-like metal–organic frameworks (ZMOFs): Design{,} synthesis{,} and properties. Chem. Soc. Rev. 2015, 44, 228–249. [CrossRef]
- Zhang, M.; Bosch, M.; Gentle, T.; Zhou, H.C. Rational design of metal-organic frameworks with anticipated porosities and functionalities. CrystEngComm 2014, 16, 4069–4083. [CrossRef]
- Jiang, H.-L.; Makal, T.A.; Zhou, H.-C. Interpenetration control in metal–organic frameworks for functional applications. Coord. Chem. Rev. 2013, 257, 2232–2249. [CrossRef]
- Gong, Y.-N.; Zhong, D.-C.; Lu, T.-B. Interpenetrating metal–organic frameworks. CrystEngComm 2016, 18, 2596–2606. [CrossRef]
- Fernandez, B.; Beobide, G.; Sanchez, I.; Carrasco-Marin, F.; Seco, J.M.; Calahorro, A.J.; Cepeda, J.; Rodriguez-Dieguez, A. Controlling interpenetration for tuning porosity and luminescence properties of flexible MOFs based on biphenyl-4,4 ’-dicarboxylic acid. Crystengcomm 2016, 18, 1282–1294. [CrossRef]
- Kang, X.; Wu, X.; Han, X.; Yuan, C.; Liu, Y.; Cui, Y. Rational synthesis of interpenetrated 3D covalent organic frameworks for asymmetric photocatalysis. Chem. Sci. 2020, 11, 1494–1502. [CrossRef]
- Nasrollahi, M.; Nabipour, H.; Valizadeh, N.; Mozafari, M. Chapter 5 - The role of flexibility in MOFs. In Metal-Organic Frameworks for Biomedical Applications; Mozafari, M., Ed.; Woodhead Publishing, 2020; pp. 93–110 ISBN 978-0-12-816984-1. [CrossRef]
- Zhao, P.; Tsang, S.C.E.; Fairen-Jimenez, D. Structural heterogeneity and dynamics in flexible metal-organic frameworks. Cell Reports Phys. Sci. 2021, 2, 100544. [CrossRef]
- Garcı́a-Valdivia, A.A.; Pérez-Mendoza, M.; Choquesillo-Lazarte, D.; Cepeda, J.; Fernández, B.; Souto, M.; González-Tejero, M.; Garcı́a, J.A.; Espallargas, G.M.; Rodrı́guez-Diéguez, A. Interpenetrated Luminescent Metal–Organic Frameworks based on 1H-Indazole-5-carboxylic Acid. Cryst. Growth Des. 2020, 20, 4550–4560. [CrossRef]
- Echenique-Errandonea, E.; Rojas, S.; Cepeda, J.; Choquesillo-Lazarte, D.; Rodríguez-Diéguez, A.; Seco, J.M. Slow Magnetic Relaxation and Modulated Photoluminescent Emission of Coordination Polymer Based on 3-Amino-4-hydroxybenzoate Zn and Co Metal Ions. Molecules 2023, 28, 1846. [CrossRef]
- Castells-Gil, J.; Baldoví, J.J.; Martí-Gastaldo, C.; Mínguez Espallargas, G. Implementation of slow magnetic relaxation in a SIM-MOF through a structural rearrangement. Dalt. Trans. 2018, 47, 14734–14740. [CrossRef]
- Huang, G.; Fernandez-Garcia, G.; Badiane, I.; Camarra, M.; Freslon, S.; Guillou, O.; Daiguebonne, C.; Totti, F.; Cador, O.; Guizouarn, T.; et al. Magnetic Slow Relaxation in a Metal–Organic Framework Made of Chains of Ferromagnetically Coupled Single-Molecule Magnets. Chem. – A Eur. J. 2018, 24, 6983–6991. [CrossRef]
- Maheswaran, S.; Chastanet, G.; Teat, S.J.; Mallah, T.; Sessoli, R.; Wernsdorfer, W.; Winpenny, R.E.P. Phosphonate Ligands Stabilize Mixed-Valent {MnIII20−xMnIIx} Clusters with Large Spin and Coercivity. Angew. Chem. Int. Ed. 2005, 44, 5044–5048. [CrossRef]
- Aromí, G.; Aguilà, D.; Gamez, P.; Luis, F.; Roubeau, O. Design of magnetic coordination complexes for quantum computing. Chem. Soc. Rev. 2012, 41, 537–546. [CrossRef]
- Boča, R. Zero-field splitting in metal complexes. Coord. Chem. Rev. 2004, 248, 757–815. [CrossRef]
- Bar, A.K.; Pichon, C.; Sutter, J.P. Magnetic anisotropy in two- to eight-coordinated transition–metal complexes: Recent developments in molecular magnetism. Coord. Chem. Rev. 2016, 308, 346–380. [CrossRef]
- Gatteschi, D.; Sessoli, R. Quantum Tunneling of Magnetization and Related Phenomena in Molecular Materials. Angew. Chemie Int. Ed. 2003, 42, 268–297. [CrossRef]
- Fortea-Pérez, F.R.; Vallejo, J.; Mastropietro, T.F.; De Munno, G.; Rabelo, R.; Cano, J.; Julve, M. Field-Induced Single-Ion Magnet Behavior in Nickel(II) Complexes with Functionalized 2,2′:6′-2″-Terpyridine Derivatives: Preparation and Magneto-Structural Study. Molecules 2023, 28, 4423. [CrossRef]
- Meng, Y.-S.; Jiang, S.-D.; Wang, B.-W.; Gao, S. Understanding the Magnetic Anisotropy toward Single-Ion Magnets. Acc. Chem. Res. 2016, 49, 2381–2389. [CrossRef]
- Baldoví, J.J.; Coronado, E.; Gaita-Ariño, A.; Gamer, C.; Giménez-Marqués, M.; Mínguez Espallargas, G. A SIM-MOF: Three-Dimensional Organisation of Single-Ion Magnets with Anion-Exchange Capabilities. Chem. – A Eur. J. 2014, 20, 10695–10702. [CrossRef]
- Frost, J.M.; Harriman, K.L.M.; Murugesu, M. The rise of 3-d single-ion magnets in molecular magnetism: Towards materials from molecules? Chem. Sci. 2016, 7, 2470–2491. [CrossRef]
- Miklovič, J.; Valigura, D.; Boča, R.; Titiš, J. A mononuclear Ni(II) complex: A field induced single-molecule magnet showing two slow relaxation processes. Dalt. Trans. 2015, 44, 12484–12487. [CrossRef]
- Pajuelo-Corral, O.; García, J.A.; Castillo, O.; Luque, A.; Rodríguez-Diéguez, A.; Cepeda, J. Single-ion magnet and photoluminescence properties of lanthanide(Iii) coordination polymers based on pyrimidine-4,6-dicarboxylate. Magnetochemistry 2021, 7, 8. [CrossRef]
- Razquin-Bobillo, L.; Pajuelo-Corral, O.; Artetxe, B.; Zabala-Lekuona, A.; Choquesillo-Lazarte, D.; Rodríguez-Diéguez, A.; Sebastian, E.S.; Cepeda, J. Combined experimental and theoretical investigation on the magnetic properties derived from the coordination of 6-methyl-2-oxonicotinate to 3d-metal ions. Dalt. Trans. 2022, 51, 9780–9792. [CrossRef]
- Rodriguez-Dieguez, A.; Perez-Yanez, S.; Ruiz-Rubio, L.; Seco, J.M.; Cepeda, J. From isolated to 2D Coordination Polymers based on 6-aminonicotinate and 3d-Metal Ions: Towards Field-Induced Single-Ion-Magnets. CrystEngComm 2017, 19, 2229–2242. [CrossRef]
- García-Valdivia, A.A.; Seco, J.M.; Cepeda, J.; Rodríguez-Diéguez, A. Designing Single-Ion Magnets and Phosphorescent Materials with 1-Methylimidazole-5-carboxylate and Transition-Metal Ions. Inorg. Chem. 2017, 56, 13897–13912. [CrossRef]
- Cepeda, J.; Navas, A.; Jannus, F.; Fernández, B.; Cepeda, J.; O’Donnell, M.M.; Díaz-Ruiz, L.; Sánchez-González, C.; Llopis, J.; Seco, J.M.; et al. Designing Single-Molecule Magnets as Drugs with Dual Anti-Inflammatory and Anti-Diabetic Effects. Int. J. Mol. Sci. 2020. [CrossRef]
- Bain, G.A.; Berry, J.F. Diamagnetic Corrections and Pascal’s Constants. J. Chem. Educ. 2008, 85, 532–536. [CrossRef]
- Bruker APEX2; Inc., B.A., Ed.; Madison, Wisconsin, USA, 2012.
- Sheldrick, G.M. SADABS empirical absorption program; 1996.
- Sheldrick, G.M. {\it SHELXT} {--} Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with {\it SHELXL}. Acta Crystallogr. Sect. C 2015, 71, 3–8. [CrossRef]
- Dolomanov, O. V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.K.; Puschmann, H. {\it OLEX2}: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [CrossRef]
- Rodríguez-Carvajal, J. Recent advances in magnetic structure determination by neutron powder diffraction. Phys. B Condens. Matter 1993, 192, 55–69. [CrossRef]
- Rudberg, E.; Sałek, P.; Rinkevicius, Z.; Ågren, H. Heisenberg Exchange in Dinuclear Manganese Complexes: A Density Functional Theory Study. J. Chem. Theory Comput. 2006, 2, 981–989. [CrossRef]
- Ruiz, E.; Cano, J.; Alvarez, S.; Alemany, P. Broken symmetry approach to calculation of exchange coupling constants for homobinuclear and heterobinuclear transition metal complexes. J. Comput. Chem. 1999, 20, 1391–1400. [CrossRef]
- Ruiz, E.; Rodríguez-Fortea, A.; Cano, J.; Alvarez, S.; Alemany, P. About the calculation of exchange coupling constants in polynuclear transition metal complexes. J. Comput. Chem. 2003, 24, 982–989. [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian16 {R}evision {C}.01 2016.
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. J. Chem. Phys. 1980, 72, 5639–5648. [CrossRef]
- Yang, Y.; Weaver, M.N.; Merz, K.M. Assessment of the “6-31+G** + LANL2DZ” Mixed Basis Set Coupled with Density Functional Theory Methods and the Effective Core Potential: Prediction of Heats of Formation and Ionization Potentials for First-Row-Transition-Metal Complexes. J. Phys. Chem. A 2009, 113, 9843–9851. [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView {V}ersion {6} 2019.
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [CrossRef]
- van Wüllen, C. Molecular density functional calculations in the regular relativistic approximation: Method, application to coinage metal diatomics, hydrides, fluorides and chlorides, and comparison with first-order relativistic calculations. J. Chem. Phys. 1998, 109, 392–399. [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence{,} triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [CrossRef]
- Hellweg, A.; Hättig, C.; Höfener, S.; Klopper, W. Optimized accurate auxiliary basis sets for RI-MP2 and RI-CC2 calculations for the atoms Rb to Rn. Theor. Chem. Acc. 2007, 117, 587–597. [CrossRef]
- Deng, Y.-F.; Singh, M.K.; Gan, D.; Xiao, T.; Wang, Y.; Liu, S.; Wang, Z.; Ouyang, Z.; Zhang, Y.-Z.; Dunbar, K.R. Probing the Axial Distortion Effect on the Magnetic Anisotropy of Octahedral Co(II) Complexes. Inorg. Chem. 2020, 59, 7622–7630. [CrossRef]
- Angeli, C.; Borini, S.; Cestari, M.; Cimiraglia, R. A quasidegenerate formulation of the second order n-electron valence state perturbation theory approach. J. Chem. Phys. 2004, 121, 4043–4049. [CrossRef]
- Maganas, D.; Sottini, S.; Kyritsis, P.; Groenen, E.J.J.; Neese, F. Theoretical Analysis of the Spin Hamiltonian Parameters in Co(II)S4 Complexes, Using Density Functional Theory and Correlated ab initio Methods. Inorg. Chem. 2011, 50, 8741–8754. [CrossRef]
- Shao, Z.; Yu, C.; Xie, Q.; Wu, Q.; Zhao, Y.; Hou, H. Porous functionalized MOF self-evolution promoting molecule encapsulation and Hg2+ removal. Chem. Commun. 2019, 55, 13382–13385. [CrossRef]
- Gantzler, N.; Kim, M.-B.; Robinson, A.; Terban, M.W.; Ghose, S.; Dinnebier, R.E.; York, A.H.; Tiana, D.; Simon, C.M.; Thallapally, P.K. Computation-informed optimization of Ni(PyC)2 functionalization for noble gas separations. Cell Reports Phys. Sci. 2022, 3, 101025. [CrossRef]
- Pajuelo-Corral, O.; Pérez-Yáñez, S.; Vitorica-Yrezabal, I.J.; Beobide, G.; Zabala-Lekuona, A.; Rodríguez-Diéguez, A.; Seco, J.M.; Cepeda, J. A metal-organic framework based on Co(II) and 3-aminoisonicotinate showing specific and reversible colourimetric response to solvent exchange with variable magnet behaviour. Mater. Today Chem. 2022, 24, 100794. [CrossRef]
- Chilton, N.F.; Anderson, R.P.; Turner, L.D.; Soncini, A.; Murray, K.S. PHI: A powerful new program for the analysis of anisotropic monomeric and exchange-coupled polynuclear d- and f-block complexes. J. Comput. Chem. 2013, 34, 1164–1175. [CrossRef]
- Titiş, J.; Boça, R. Magnetostructural D correlation in Nickel(II) complexes: Reinvestigation of the zero-field splitting. Inorg. Chem. 2010, 49, 3971–3973. [CrossRef]
Figure 1.
Coordination polyhedron, building unit and crystal packing of the structure of 1. Note that lattice DMF molecules have been omitted for clarity in the packing. Nickel atoms are represented in green, nitrogen in blue, carbon in grey, oxygen in red and hydrogens in white.
Figure 1.
Coordination polyhedron, building unit and crystal packing of the structure of 1. Note that lattice DMF molecules have been omitted for clarity in the packing. Nickel atoms are represented in green, nitrogen in blue, carbon in grey, oxygen in red and hydrogens in white.
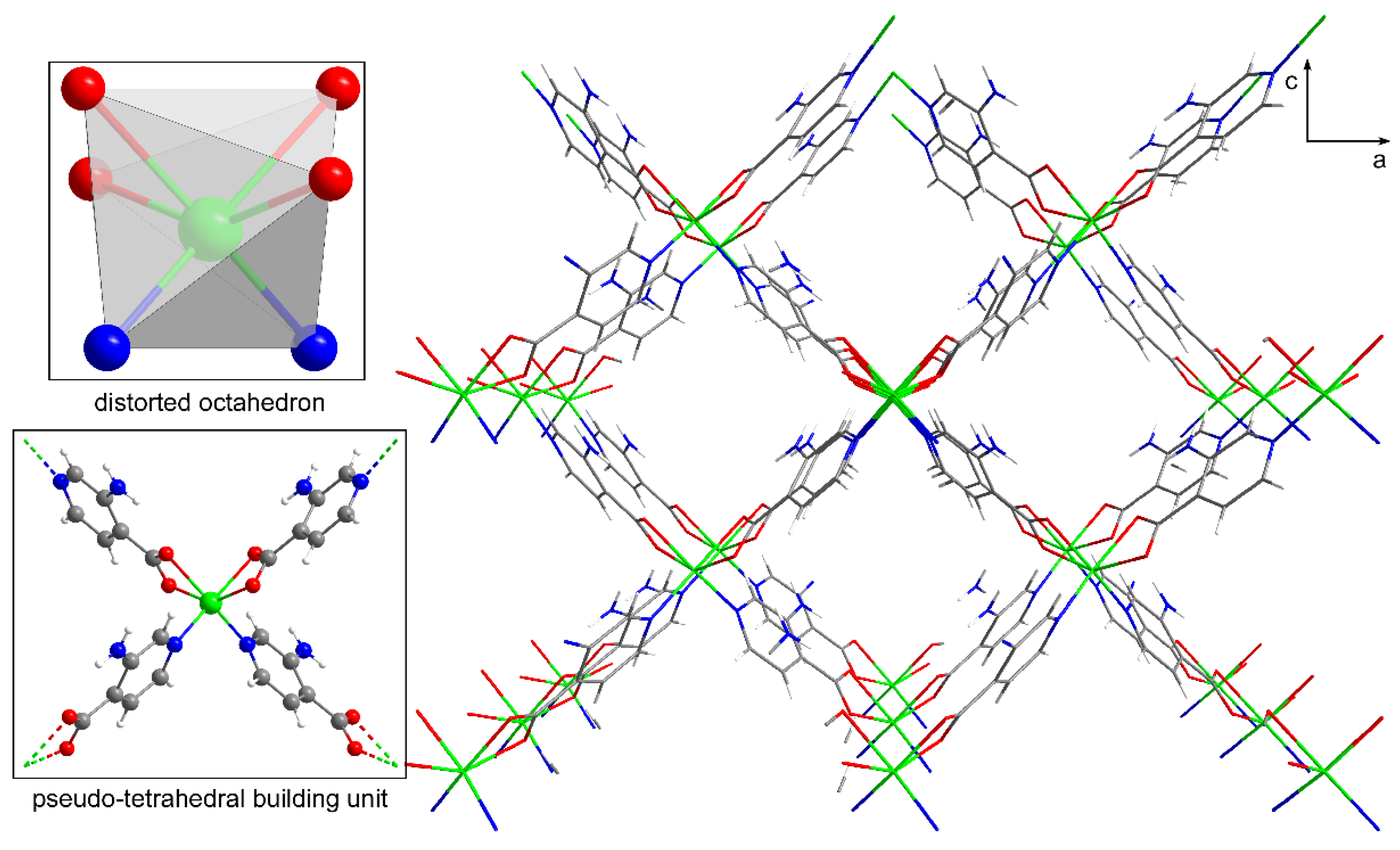
Figure 2.
Monomeric complex and crystal packing of compound 2 based on X-ray coordinates of the Co(II) isostructural counterpart [66]. Colour code as Figure 1.
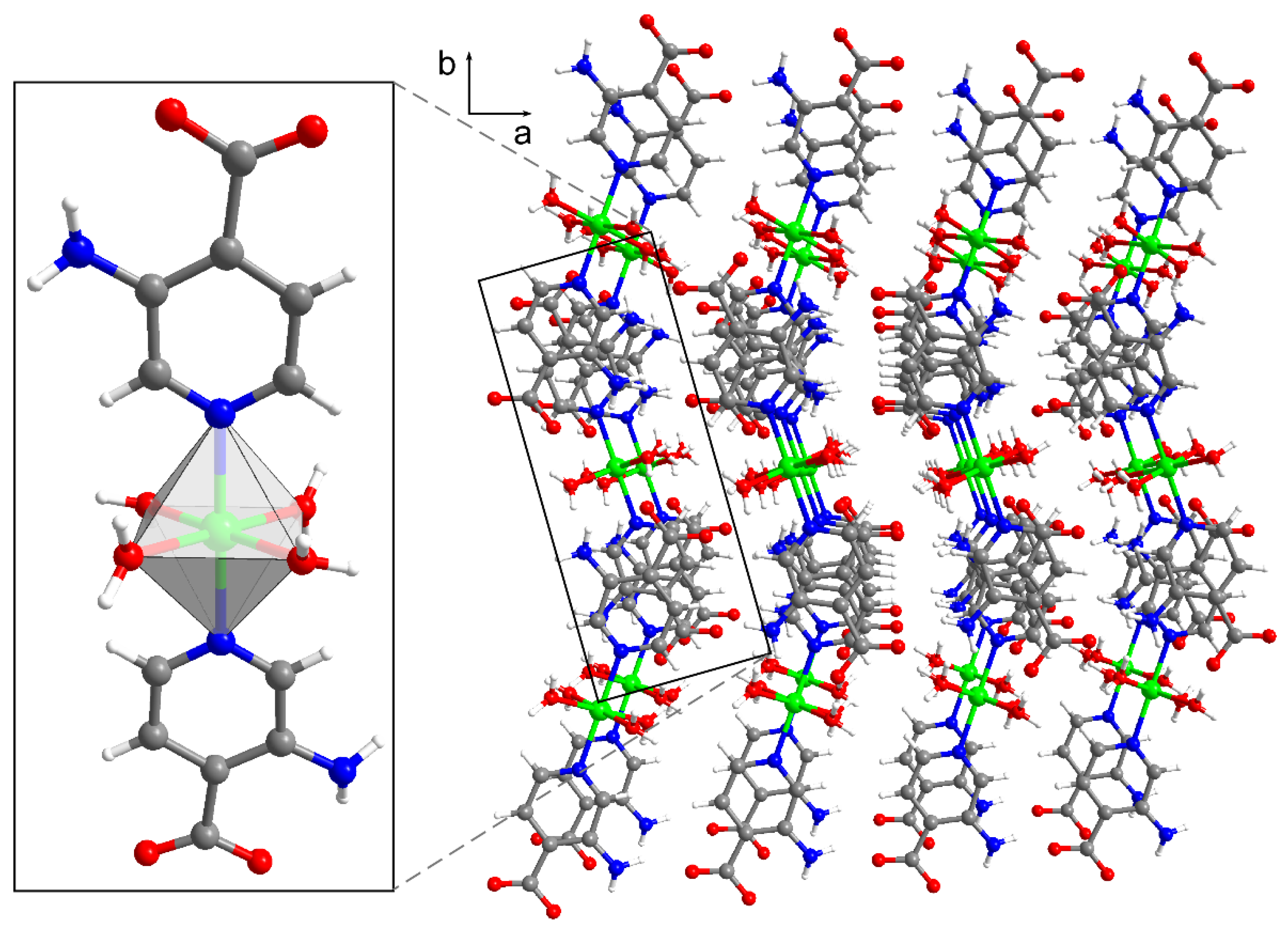
Figure 3.
Photographs of the solid samples of compounds 1 and 2 summarizing the conditions of the transformations taking place.
Figure 3.
Photographs of the solid samples of compounds 1 and 2 summarizing the conditions of the transformations taking place.
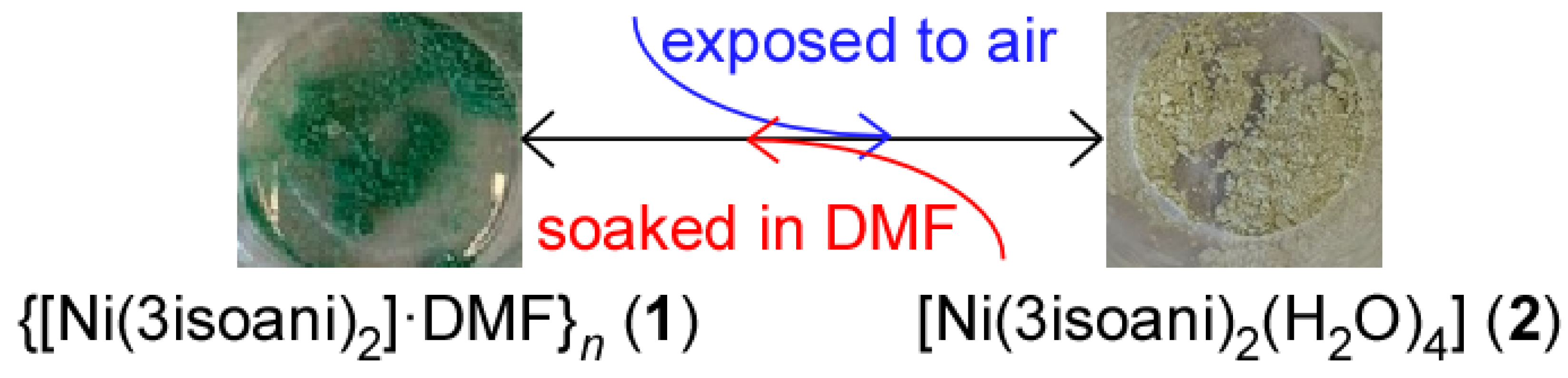
Figure 4.
Plots of the variable-temperature evolution of the χMT product and isothermal magnetization vs field with the simultaneous susceptibility and magnetization fitting for (a) compound 1 and (b) compound 2.
Figure 4.
Plots of the variable-temperature evolution of the χMT product and isothermal magnetization vs field with the simultaneous susceptibility and magnetization fitting for (a) compound 1 and (b) compound 2.
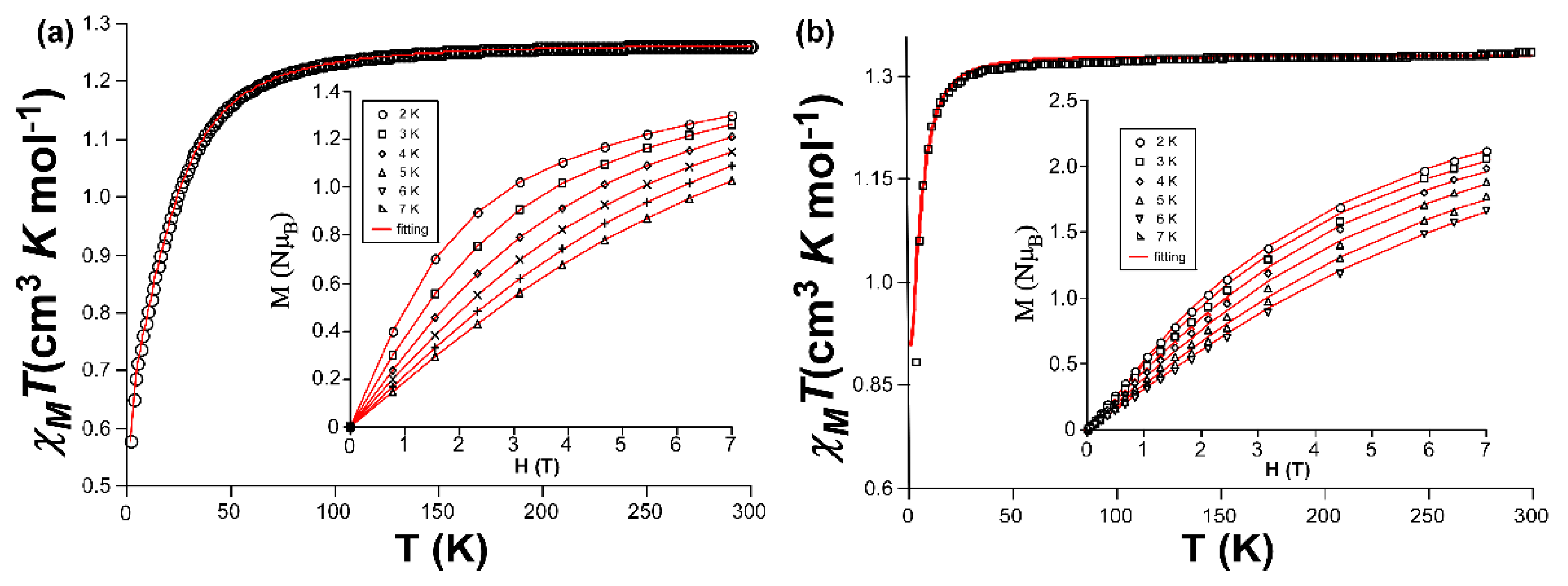
Figure 5.
Energy diagrams of the orbital splitting and configuration of the main contribution of the ground state for compounds (a) 1 and (b) 2.
Figure 5.
Energy diagrams of the orbital splitting and configuration of the main contribution of the ground state for compounds (a) 1 and (b) 2.
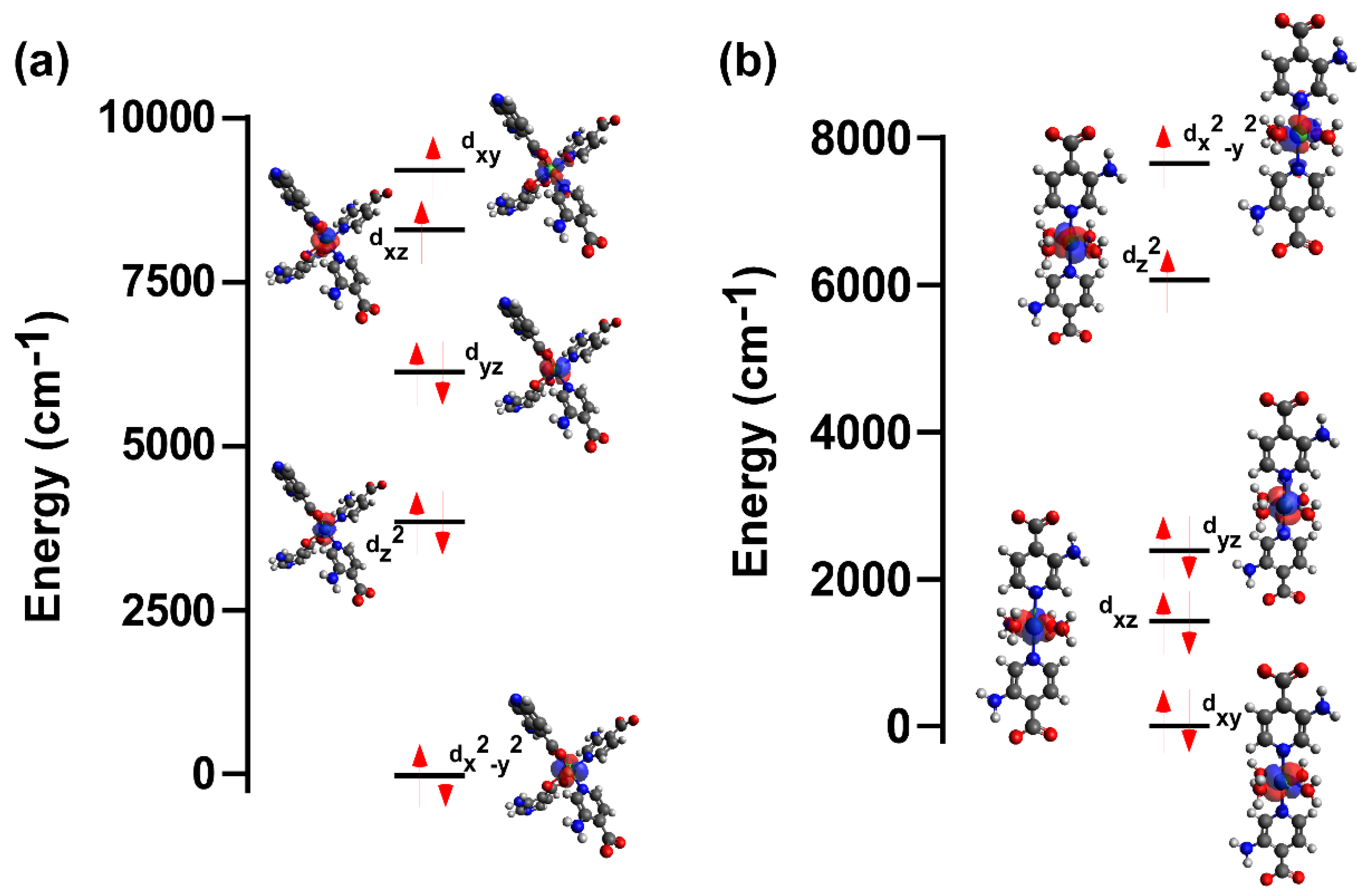
Figure 6.
Variable temperature a) in-phase and b) out-of-phase ac molar susceptibility signals when a dc field of 1000 Oe is applied for compound 1. Inset in plot b) corresponds to the Arrhenius plot with the variable-temperature relaxation times estimated from and best fitting results.
Figure 6.
Variable temperature a) in-phase and b) out-of-phase ac molar susceptibility signals when a dc field of 1000 Oe is applied for compound 1. Inset in plot b) corresponds to the Arrhenius plot with the variable-temperature relaxation times estimated from and best fitting results.
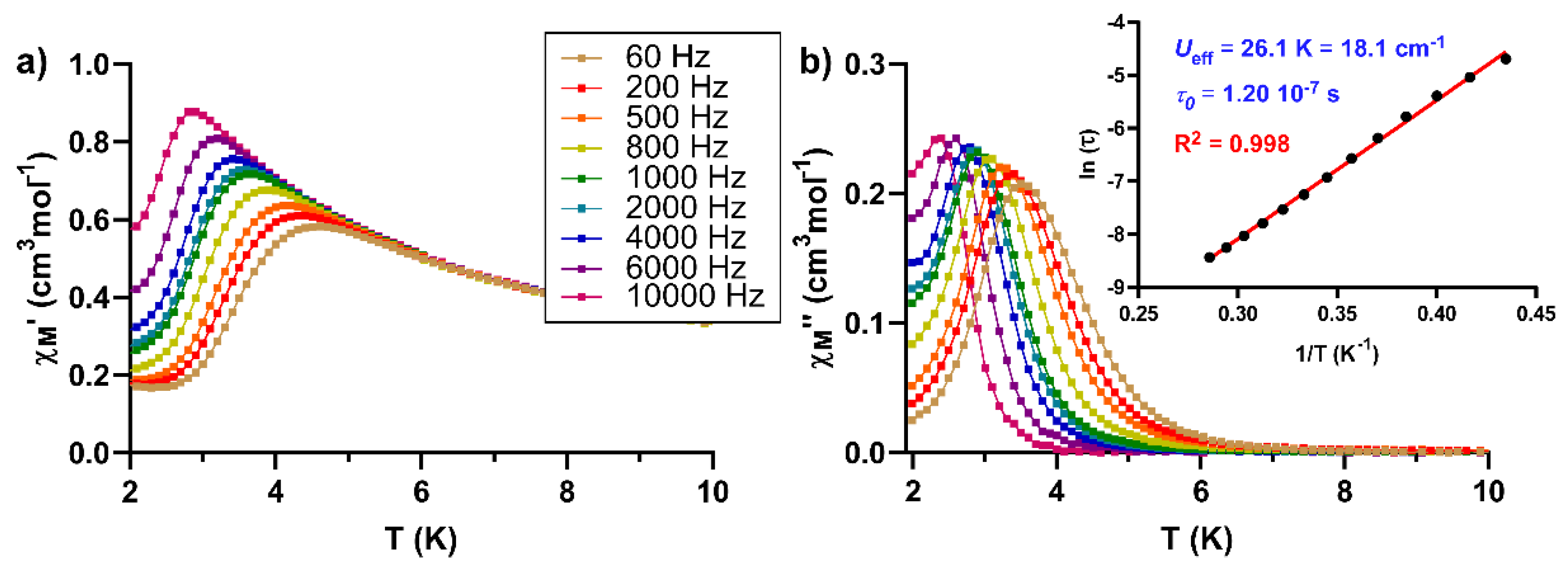

Table 1.
Single crystal X-ray diffraction data and structure refinement details of compound 1.
| Compound | 1 |
|---|---|
| Empirical formula | C15H17N5NiO5 |
| Formula weight (g mol-1) | 406.04 |
| Crystal system | Orthorhombic |
| Space group | Pnn2 |
| a (Å) | 11.944(2) |
| b (Å) | 6.740(1) |
| c (Å) | 10.661(2) |
| V (Å3) | 858.2(3) |
| Reflections collected | 6467 |
| Unique data/parameters | 2814/136 |
| Rint | 0.0521 |
| GoF (S)a | 1.075 |
| R1b/wR2 [I > 2σ(I)]c | 0.0410/0.0869 |
| R1b/wR2 [all]c | 0.0486/0.0941 |
a S = [∑w(F02 – Fc2)2 / (Nobs – Nparam)]1/2. b R1 = ∑||F0|–|Fc|| / ∑|F0|; c wR2 = [∑w(F02 – Fc2)2 / ∑wF02]1/2; w = 1/[σ2(F02) + (aP)2 + bP] where P = (max(F02,0) + 2Fc2)/3 with a = 0.0189 and b = 1.5121.
Table 2.
Selected bond lengths for compound 1 (Å).
| Ni1–O1 | 2.123(2) | Ni1–O2A (i) | 2.081(2) |
| Ni1–O1A (i) | 2.123(2)) | Ni1–N1A | 2.028(3) |
| Ni1–O2A | 2.081(2) | Ni1–N1A (i) | 2.028(3) |
1 Symmetries: (i) –x+1, -y+1, z.
Table 3.
Best fitting results for magnetic data of compounds 1 and 2 using Equation (1).
| Compound | Exp. | Calc. | ||||
|---|---|---|---|---|---|---|
| D | E/D | giso | D | E/D | g2 | |
| 1 | -17.3(1) | 0.05 | 2.25(1) | -21.1 | 0.07 | 2.24, 2.26, 2.38 (2.27) |
| 2 | -4.8(3) | 0.15 | 2.37(5) | -6.7 | 0.12 | 2.34, 2.35, 2.39 (2.38) |
1 The values are given in cm-1. 2 These calculated values are listed as gxx, gyy, gzz and (giso).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



