
Submitted:
08 August 2023
Posted:
09 August 2023
You are already at the latest version
Abstract
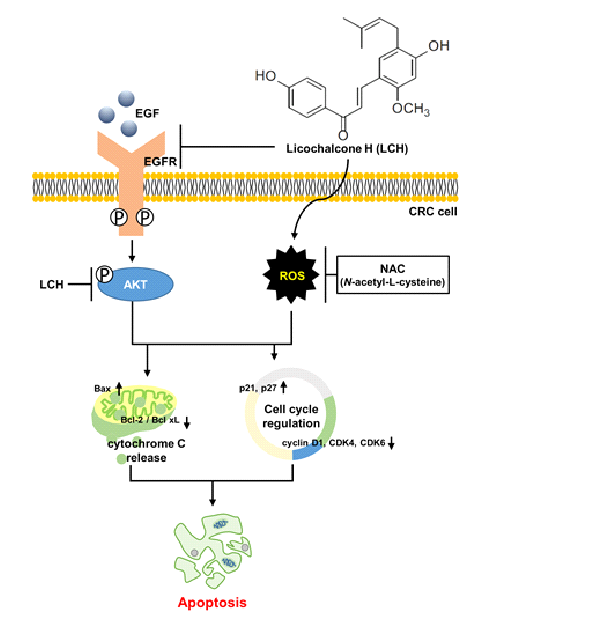
Licochalcone H (LCH), a regioisomer of licochalcone C derived from the root of Glycyrrhiza inflata. CRC treatment has always been challenged by the development of resistance. We investigated the antiproliferative activity of LCH in oxaliplatin (Ox)-sensitive and -resistant CRC cells. LCH significantly inhibited cell viability and colony growth in both Ox-sensitive and Ox-resistant CRC cells. We found that LCH decreased epidermal growth factor receptor (EGFR) and AKT kinase activities and related activating signaling proteins including phospho (p)-EGFR and pAKT. A computational docking model indicated that LCH may interact with EGFR, AKT1, and AKT2 at the ATP-binding sites. LCH induced ROS generation, as verified by N-acetyl-L-cysteine (NAC) treatment, and increased the expression of the ER stress markers. LCH treatment of CRC cells induced depolarization of MMP and increased. Multi-caspase activity was induced by LCH treatment and confirmed by Z-VAD-FMK treatment. LCH increased the number of subG1 cells and arrested the cell cycle at the G1 phase. LCH inhibits the growth of Ox-sensitive and Ox-resistant CRC cells by targeting EGFR and AKT, and inducing ROS generation and ER stress-mediated apoptosis. Therefore, LCH could be a potential therapeutic agent for improving not only Ox-sensitive but also Ox-resistant CRC treatment.
Keywords:
licochalcone H (LCH)
; oxaliplatin
; colorectal cancer
; EGFR
; AKT
; apoptosis
1. Introduction
Licorice, Glycyrrhiza inflata, is a well-known traditional medicine with anti-inflammatory, antioxidant, antiviral, and antimicrobial activities [1,2,3]. So far, the licochalcone components from licorice root that have been reported include licochalcone A, B, C, D, and E. They have been reported to exhibit inhibitory effects against breast, lung, colon, liver, and esophageal cancer [4,5,6,7,8,9]. LCH is a synthetic derivative of licochalcone C that induces apoptosis in skin cancer and oral squamous cell carcinoma by targeting the JAK2/STAT3 or matrin 3 signaling pathways, thereby inhibiting cancer cell growth [10,11,12]. However, further evaluation of its molecular mechanisms of action in other cancer types is required.
Colorectal cancer (CRC) is one of the most dangerous types of cancer worldwide; it is frequently diagnosed and results in high mortality [13,14]. The latest trends in CRC show early onset, with remarkably high incidence rates in patients under 50 years of age. In the USA, it nearly doubled from 11% in 1995 to 20% in 2019 and is predicted to account for 7% of the 52,550 CRC deaths in 2023 [14]. Its major risk factors are obesity, diabetes, smoking, and inflammatory bowel disease [15]. Chemotherapy is a general strategy for the treatment of CRC before and after surgery; 5-fluorouracil, oxaliplatin (Ox), and irinotecan are mainly used alone or in combination with FOLFOX, FOLFIRI, or FOLFRINOX [16]. Ox was the first platinum-based chemotherapeutic agent that improves the moderate response to 5-fluorouracil and limits cisplatin-mediated side effects [17]. However, the responses were lost due to acquired resistance after several months of treatment, thus challenging new strategies.
EGFR is a transmembrane tyrosine kinase receptor that is abnormally expressed and activated by mutations and phosphorylation in cancers, including those of the lung and colon [18,19]. Activation of EGFR affects downstream signaling pathways such as AKT and ERK, thereby promoting cell proliferation and survival. The EGFR/AKT signaling pathway enhances Ox-mediated resistance [20]. Therefore, downregulating the EGFR/AKT signaling pathway might be a good approach to treat Ox resistance.
In this study, we found that LCH inhibited the kinase activity of EGFR and AKT using in vitro kinase assay and its related signaling pathways using western blotting. Thus, LCH treatment of Ox-sensitive HCT116 and Ox-resistant HCT116-OxR cells reduced cell proliferation as shown by the 1-(4,5-dimethylthiazol-2-yl)-3,5-diphenylformazan (MTT) assay, and induced apoptosis as shown by flow cytometry analysis.
2. Materials and Methods
2.1. Materials
LCH (Figure 1A) was synthesized and purified as described previously. RPMI-1640, Dulbecco’s modified Eagle’s medium (DMEM), minimum essential medium (MEM), sodium pyruvate, MEM vitamin solution, and fetal bovine serum (FBS) were purchased from GIBCO (Invitrogen GmbH, Karlsruhe, Germany). The MEM non-essential amino acid solution was purchased from Corning (Corning, NY, USA). Phosphate-buffered saline (PBS), RIPA buffer, and Tris-glycine-sodium dodecyl sulfate (SDS) buffer were purchased from BioSolution (Seoul, Korea). Trypsin and penicillin/streptomycin (p/s) were purchased from HyClone (Logan, UT, USA). Basal Medium Eagle, MTT, dimethyl sulfoxide (DMSO), N-acetyl-L-cysteine (NAC), and N-benzyloxycarbonyl-Val-Ala-Asp(O-Me) fluoromethyl ketone (Z-VAD-FMK; pan-caspase inhibitor) were obtained from Sigma-Aldrich (St. Louis, USA). The antibodies against actin (cat. sc-47778), 78-KDa glucose-regulated protein (GRP78) (cat. sc-1050), C/EBP homologous protein (CHOP) (cat. sc-7351), death receptor (DR)4 (cat. #sc-7863), DR5 (cat. #sc-166624), p21 (cat. #sc-6246), p27 (cat. sc-56338), cyclin D1 (cat. sc-718), CDK4 (cat. sc-70831), CDK6 (cat. #sc-56282), Mcl-1 (cat. sc-819), Bid (cat. sc-56025), Bax (cat. sc-20067), Bcl-xL (cat. sc-8392) Bcl-2 (cat. sc-7382) cytochrome c (cyto c) (cat. #sc-13156), -tubulin (cat. sc-8035) cytochrome c oxidase subunit 4 (COX4) (Cat. sc-69359), apoptotic protease activating factor-1 (Apaf-1) (cat. sc-33870) cleaved poly (ADP-ribose) polymerase (PARP) (cat. sc-7150), and caspase 3 (cat. #sc-7148) were provided by Santa Cruz Biotechnology (Dallas, TX, USA). Anti-Bim antibody (cat. #2933) was purchased from Cell Signaling Technology (Danvers, MA, USA).
2.2. Cell culture
Human CRC cell (HCT116), human keratinocyte (HaCaT), and mouse epidermal cell (JB6) lines were purchased from the American Type Culture Collection (Manassas, VA, USA). HCT116 and HaCaT cells were cultured in complete RPMI-1640 medium and DMEM, respectively, supplemented with 10% FBS and 1% penicillin/streptomycin. JB6 cells were cultured in MEM supplemented with 5% FBS and 1% penicillin/streptomycin. The human oxaliplatin-resistant CRC cell line (HCT116-OxR) was obtained from The University of Texas MD Anderson Cancer Center . HCT116-OxR cells were maintained in MEM medium supplemented with 10% FBS, 1% penicillin/streptomycin, 1% sodium pyruvate, 1% MEM non-essential amino acids solution, 1% MEM vitamin solution, and 2 μM of oxaliplatin. These cells were incubated with 5% CO2 and 95% air in a humidified atmosphere at 37 °C.
2.3. MTT assay
The viability of CRC cells was assessed by MTT assay. HCT116 (5 × 103 cells/well), HCT116-OxR (4 × 103 cells/well), HaCaT (8 × 103 cells/well), and JB6 (a density of 5.5 × 103 cells/well) cells were seeded in 96-well culture plates. After incubation for 24 h, cells were treated with LCH at 0, 4, 8, and 12 μM for 24 h or 48 h. After 30 µL of MTT solution (5 mg/mL) was added to each well, cells were incubated at 37 °C for 1 h. Medium was removed after the incubation and formazan crystals were dissolved in 100 µL of DMSO. The absorbance of each well was measured at 570 nm by using a Microplate Spectrophotometer (Thermo Fisher Scientific). To investigate the role of Reactive oxygen species (ROS) in LCH-induced apoptosis, CRC cells were pre-incubated with 4 mM NAC (ROS scavenging agent) for 3 h before they were treated with LCH.
2.4. Anchorage-independent soft agar assay
For the soft agar assay, 0.6% agar (bottom agar) mixed with culture medium containing Basal Medium Eagle, 10% FBS, 2 mM L-glutamine, and 5 g/mL gentamicin were treated with the indicated concentrations of LCH, Ox (positive control), or DMSO (control) and solidified in six-well plates. Cells (8,000) were suspended in 0.3% agar mixed with the culture medium and treated with LCH (4, 8, and 12 μM), Ox (2 μM), or DMSO. A 1 mL cell suspension (upper agar) was then used to overlay the bottom agar. After culturing for 1–2 weeks, colonies were photographed using a light microscope (Leica Microsystems, Wetzlar, Germany) to analyze their size and number. Colonies with diameters ≥ 50 μm were counted under the light microscope.
2.5. Cell apoptosis analysis by flow cytometry
HCT116 (5ⅹ103 cells/well) and HCT116-OxR (4ⅹ103 cells/well) cells were seeded in six-well plates. After 24 h of incubation, cells were treated with LCH at 0, 4, 8, and 12 μM for 48 h. Cells were then harvested and stained with Muse® Annexin V & Dead Cell Reagent (MCH100105, Luminex, Austin, TX, USA). The stained cells were then analyzed on a Muse® Cell Analyzer system (Merck Millipore, Darmstadt, Germany).
2.6. Cell cycle analysis
Cells treated with LCH (0, 4, 8, and 12 μM) were fixed with 70% ethanol at -20 °C overnight. The cells were washed with PBS and the supernatant was removed. DNA content of CRC cells in each stage of the cell cycle was examined using a Muse® cell cycle reagent (MCH100106, Luminex) and detected with a Muse® Cell Analyzer system (Merck Millipore).
2.7. Intracellular ROS detection
Detection of intracellular ROS levels was carried out using a Muse® Oxidative Stress Kit (MCH100111, Luminex) following the manufacturer’s protocol. Briefly, the cells were collected and washed once with 1X PBS. Cells were stained with Muse® Oxidative Stress Reagent working solution for 30 min at 37 °C in the dark. ROS levels in CRC cells were then determined using a Muse® Cell Analyzer system (Merck Millipore).
2.8. Mitochondrial membrane potential (MMP, Δψm) measurement
Mitochondrial membrane depolarization was detected with a Muse® MitoPotential kit (MCH100110, Luminex). Briefly, HCT116 (5ⅹ103 cells/well) and HCT116-OxR (4ⅹ103 cells/well) cells were seeded in six-well plates and incubated at 37 °C for 24 h. Cells then were treated with different concentrations (0, 4, 8, and 12 μM) of LCH for 48 h. After incubation, cells were washed with PBS and resuspended with Muse® MitoPotential working solution, then incubated at 37 °C for 20 min in the dark. After 4 μL Muse® MitoPotential 7-Aminoactinomycin D (7-AAD) was added to cells, plates were incubated at room temperature (RT) for 5 min. MMP measurements were performed by flow cytometry using a Muse® Cell Analyzer system (Merck Millipore).
2.9. Multi-caspase assay
To analyze multi-caspase activity, including that of caspase-1, -3, -4, -5, -6, -7, -8, and -9, HCT116 and HCT116-OxR cells were seeded into 6-well plates and treated with LCH for 48 h prior to analysis. Multi-caspase activities were measured using a Muse® Multi-caspase kit (MCH100109, Luminex) according to the manufacturer's instructions. In brief, 10 μL of the Muse® Multi-caspase reagent working solution were added to 50 μL of cells and incubated at 37 °C for 30 min. Thereafter, cells were resuspended in 125 μL Muse® Caspase 7-AAD working solution and incubated at RT for 5 min. The multi-caspase activity of each sample was determined using a Muse® Cell Analyzer system (Merck Millipore).
2.10. Western blot
Cells were lysed using RIPA buffer (iNtRON Biotechnology, Seongnam, Gyeonggido, South Korea). Proteins (20–40 µg/lane) were separated on 8–15% SDS-polyacrylamide gels (PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). Membranes were blocked with 5% of non-fat milk at RT for 2 h and incubated with specific primary antibodies (diluted 1:1,000) at RT for 2 h at 4 ºC overnight. After washing with PBS containing 1% Tween-20 (PBST) for 5 min, six times, the membranes were then incubated with appropriate Horseradish peroxidase (HRP)-conjugated anti-rabbit (1:10,000), anti-goat (1:5,000), or anti-mouse (1:10,000) immunoglobulin G (IgG) secondary antibody at RT for 2 h. Bound antibodies were treated with Western blotting luminol reagent (Santa Cruz, CA, USA) and visualized with an ImageQuantTM LAS 500 (GE Healthcare, Uppsala, Sweden). Protein levels were quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
2.11. Kinase assay
EGFR and AKT kinase activities were determined using an EGFR (Cat. No. V3831), AKT1 (Cat. No. V1911), and AKT2 (Cat. No. V3861) active kinase enzyme system (Promega, Madison, WI, USA) and the ADP-Glo kinase assay kit (Promega). The EGFR (4.0 ng/μl), AKT1 (3.0 ng/μl), and AKT2 (3.0 ng/μl) active kinases were responded in a 384-well plate with LCH (2, 4, 8, and 12 μM),1 μM of gefitinib (GEF), or 65 nM of MK-2206, in addition to 0.2 μg/μl of substrates, 5 μM of ATP and kinase reaction buffer including 0.1 mg/ml BSA, 50 μM DTT, 20 mM MgCl2, 2 mM MnCl2, 100 μM sodium vanadate and 40 mM Tris (PH 7.5) at RT for 1 h. Then, 5 μl of ADP-Glo reagent (ADP-Glo kinase assay kit; Promega) were added to each well and incubated at RT for 40 min to deplete the remaining ATP and complete the kinase reaction. Ten microliters of kinase detection reagent were added to each reaction. The luminescence was detected using a Centro LB 960 microplate luminometer (Berthold Technologies, Germany) for 0.5 s.
2.12. Molecular modeling
We performed a molecular docking simulation to predict the binding mode between LCH and the kinases EGFR, AKT1, and AKT2. The PDB files 1M17, 6CCY, and 3D0E were downloaded from the protein data bank to obtain the structures of EGFR, AKT1, and AKT2, respectively. The search grid was set to 60 ° in every dimension to allow an unbiased search, and AutoDock Vina was used as previously described . The best modes reported by AutoDock Vina were used for structural depiction.
2.13. Statistical analysis
All data are presented as mean ± standard deviation (SD). Each experiment was performed at least thrice. Multiple comparisons were performed with one-way or two-way analysis of variance (ANOVA) using GraphPad Prism software (version 5.0; San Diego, CA, USA). P-values less than 0.05, 0.01, and 0.001 were used to indicate statistical significance and are marked with asterisks.
3. Results
3.1. LCH reduces cell viability and anchorage-independent colony growth.
To determine the inhibitory effects of LCH on cell viability and colony growth, we performed MTT and soft agar assays on HCT116 and HCT116-OxR cells. Both cell lines were treated with LCH (4, 8, and 12 μM) or oxaliplatin (2 μM) for 24 or 48 h. MTT assay results showed that LCH at 0, 4, 8, and 12 μM decreased the viability of HCT116 cells after 24 h to 93.65, 85.82, and 65.03% and after 48 h to 88.31, 74.49, and 44.67% and that of HCT116-OxR after 24 h to 97.10, 92.90, and 74.89% and after 48 h to 89.34, 72.31, and 40.74%, respectively (Figure 1A). The IC50 values were 10.93 μM for HCT116 and 10.24 μM for HCT116-OxR cells. Meanwhile, oxaliplatin significantly reduced HCT116 cell viability but failed to do so in HCT116-OxR cells (Figure 1A). In addition, LCH did not cause cytotoxicity in both HaCaT and JB6 cells to 12 μM, while oxaliplatin at 2 μM exhibited 75.00% viability after 24 h and 43.08% viability after 48 h in HCT116 cells and 99.13% viability after 24 h and 95.75% viability after 48 h in HCT116-OxR cells. For further validation, we performed anchorage-independent colony growth assays with soft agar after treatment with LCH (Figure 1B-D). The results showed that LCH (0, 4, 8, and 12 μM) significantly decreased the number and size of colonies in both HCT116 and HCT116-OxR cells compared with DMSO-treated controls (Figure 1B-D). Oxaliplatin inhibited colony number and size in HCT116 cells but not in the oxaliplatin-resistant HCT116-OxR cells. In addition, the induction of apoptosis after LCH treatment (4, 8, and 12 μM) in CRC cells was evaluated using annexin V/7-aminoactinomycin D (7-AAD) staining (Figure 1E-F). LCH increased percents of the total apoptotic cell population to 12.05 ± 0.52%, 25.92 ± 2.54% and 33.80 ± 4.24% in HCT116 cells, 11.14 ± 0.74%, 17.66 ± 0.38% and 32.87 ± 0.34% in HCT116-OxR cells (Figure 1E-F). This indicates that LCH decreased the proliferation and caused apoptosis in both HCT116 and HCT116-OxR cells.
3.2. LCH triggers sub-G1 and G1 phase arrest in the cell cycle of CRC cells
Next, we analyzed the effects of LCH on cell cycle progression in HCT116 and HCT116-OxR cells using flow cytometry (Figure 2A-D). LCH treatment resulted in an increase in the number of CRC cells in the sub-G1 and G1 phases (Figure 2A-D). LCH (4, 8, and 12 μM) increased the percentages of the sub-G1 phase in HCT116 cells to 25.33 ± 0.67%, 25.53 ± 1.55%, and 37.67 ± 0.65%, compared to the control (6.7 ± 0.6%). Furthermore, in HCT116-OxR cells, LCH at 4, 8, and 12 μM increased the sub-G1 phase population by 17.3 ± 1.04%, 27.07 ± 0.93%, 42.7 ± 2.43%, respectively, compared to the control (6.97 ± 0.06%), (Figure 2A-B). Moreover, cell cycle arrest at G1 phase was increased after LCH treatment at 4, 8, and 12 μM to 64.53 ± 1.33%, 69.03 ± 0.76%, and 75.20 ± 1.65%, respectively, compared to control (65.60 ± 0.53%) in HCT116 cells and 63.13 ± 0.74%, 67.30 ± 0.75%, and 73.77 ± 1.29%, respectively, compared to control (60.60 ± 0.26%) in HCT116-OxR cells (Figure 2C-D). To verify the changes in the levels of proteins involved in the cell cycle, we performed western blot analysis (Figure 2E). LCH induced p21 and p27 expression in a dose-dependent manner while reducing cyclin D1, CDK6, and CDK4 expression in CRC cells (Figure 2E). These results revealed that LCH contributed to cell cycle arrest at the G1 phase, eventually triggering apoptosis in both Ox-sensitive and Ox-resistant CRC cells.
3.3. LCH directly inhibits EGFR and AKT kinases activity and EGFR-mediated signaling axis.
To identify whether LCH (Figure 3A) inhibited EGFR, AKT1, and AKT2 kinase activity, we performed an in vitro kinase assay using active EGFR, AKT1, and AKT2 kinases (Figure 3B-C). The results showed that LCH significantly decreased EGFR kinase activity to 102.4, 63.5, 54.7, and 24.0% with increasing concentrations of 2, 4, 8, and 12 μM, respectively, and that gefitinib, a positive-control, reduced EGFR kinase activity to 2.8% at 1 μM (Figure 3B). Moreover, AKT1 and AKT2 kinase activities were reduced to 2.2% and 18% with LCH (12 μM) and 1.2% and 13% with MK-2206 (AKT inhibitor; 65 nM), respectively (Figure 3C). To examine the interaction between LCH and EGFR, AKT1, or AKT2, we performed computational docking analysis (Figure 3D-F). Molecular modeling using Autodock Vina indicated that LCH might bind to the ATP-binding sites of both the kinases EGFR and AKT1. In EGFR, the backbone amino group from Met769 formed a hydrogen bond with the four hydroxyl groups in LCH. In addition, the aliphatic amino acids Leu694, Val702, Alla719, Leu764, and Leu768 were close to LCH for possible hydrophobic interactions. For interaction with AKT1, the side chains of Lys179 and Asp292 were close to the four hydroxyl groups of LCH, and Phe161, Leu181, and Ile186 were in close contact with LCH for hydrophobic interactions. Similarly, Lys288 and Glu279 were predicted to form hydrogen bonds with the four hydroxyl groups of LCH, and Phe163, Val166, Val187, and Val188 were close enough to LCH for hydrophobic interactions (Figure 3D-F). Then, we measured the EGFR-mediated signaling proteins using western blotting after HCT116 and HCT116-OxR cells treated with 4, 8, and 12 μM of LCH for 48 h. The results revealed that LCH suppressed the expression of pEGFR and pAKT without altering the total forms of EGFR and AKT in both cell lines (Figure 4A-C). This indicates that LCH inhibited EGFR, AKT1, and AKT2 kinase activities, thus decreasing EGFR/AKT signaling.
3.4. LCH increases ROS production in both HCT116 and HCT116-OxR cells.To identify
Next, we examined ROS generation by treatment with LCH using a cell analyzer system (Figure 5A). After treatment of LCH (4, 8 and 12 μM), the ROS levels were increased to 20.00 ± 1.12%, 34.45 ± 0.80%, and 70.18 ± 0.87% compared to control (6.60 ± 0.52%) in HCT116 cells, and 6.68 ± 0.66%, 19.89 ± 0.77%, and 44.77 ± 1.10% compared to control (6.50 ± 0.11%) in HCT116-OxR cells, respectively (Figure 5A). To verify ROS production-mediated cell death, we treated cells with NAC, a ROS scavenger, and measured cell viability, apoptosis, and multi-caspase activity (Figure 5B-D). LCH (12 μM) significantly inhibited cell viability and induced annexin V/7-ADD staining and caspase activity in both HCT116 and HCT116-OxR cells; however, these effects were lost when the cells were treated with NAC (4 mM) (Figure 5B-D). This indicated that LCH increased ROS generation and induced apoptosis in CRC cells.
3.5. LCH induces mitochondrial dysfunction and ER stress in CRC cells
To determine whether LCH regulates mitochondrial function, we analyzed MMP depolarization using flow cytometry after treatment of HCT116 and HCT116-OxR cells with LCH. As shown in Figure 6A-B, LCH increased the number of depolarized/live cells (lower left) and depolarized/dead cells (upper left) compared with the controls (Figure 6A-B). LCH treatment (4, 8, and 12 μM) increased the total depolarized cells to 4.80 ± 0.47%, 23.21 ± 1.08%, and 43.15 ± 0.95% in HCT116 cells, and 6.22 ± 0.60%, 35.94 ± 0.52%, and 53.48 ± 0.89% in HCT116-OxR cells. To identify the underlying mechanism, we examined the expression of ER stress and mitochondria-mediated apoptosis markers, including GRP78, CHOP, DR4, DR5, Bim, Mcl-1, Bid, Bax, Bcl-xL, and Bcl-2, after LCH treatment of HCT116 and HCT116-OxR cells (Figure 6C). The expression of ER stress markers GRP78, CHOP, DR4 and DR5, and apoptosis inducing markers Bim and Bax were increased, while the levels of the anti-apoptotic markers Mcl-1, Bid, Bcl-xL, and Bcl-2 were decreased by LCH treatment in a dose-dependent manner (Figure 6C). Moreover, LCH treatment increased the cytosolic cytochrome c levels and decreased the mitochondrial fraction in a dose-dependent doses (Figure 6C). In addition, apoptosis signaling proteins, Apaf-1 and cleaved (c)-PARP, were upregulated, and the pro-apoptosis protein caspase3 was downregulated by LCH treatment in CRC cells (Figure 6C). These results indicated that LCH induces apoptosis by mediating ER stress and mitochondrial dysfunction through the regulation of related proteins in CRC cells.
3.6. LCH induces apoptosis by increasing the caspases activities in CRC cells
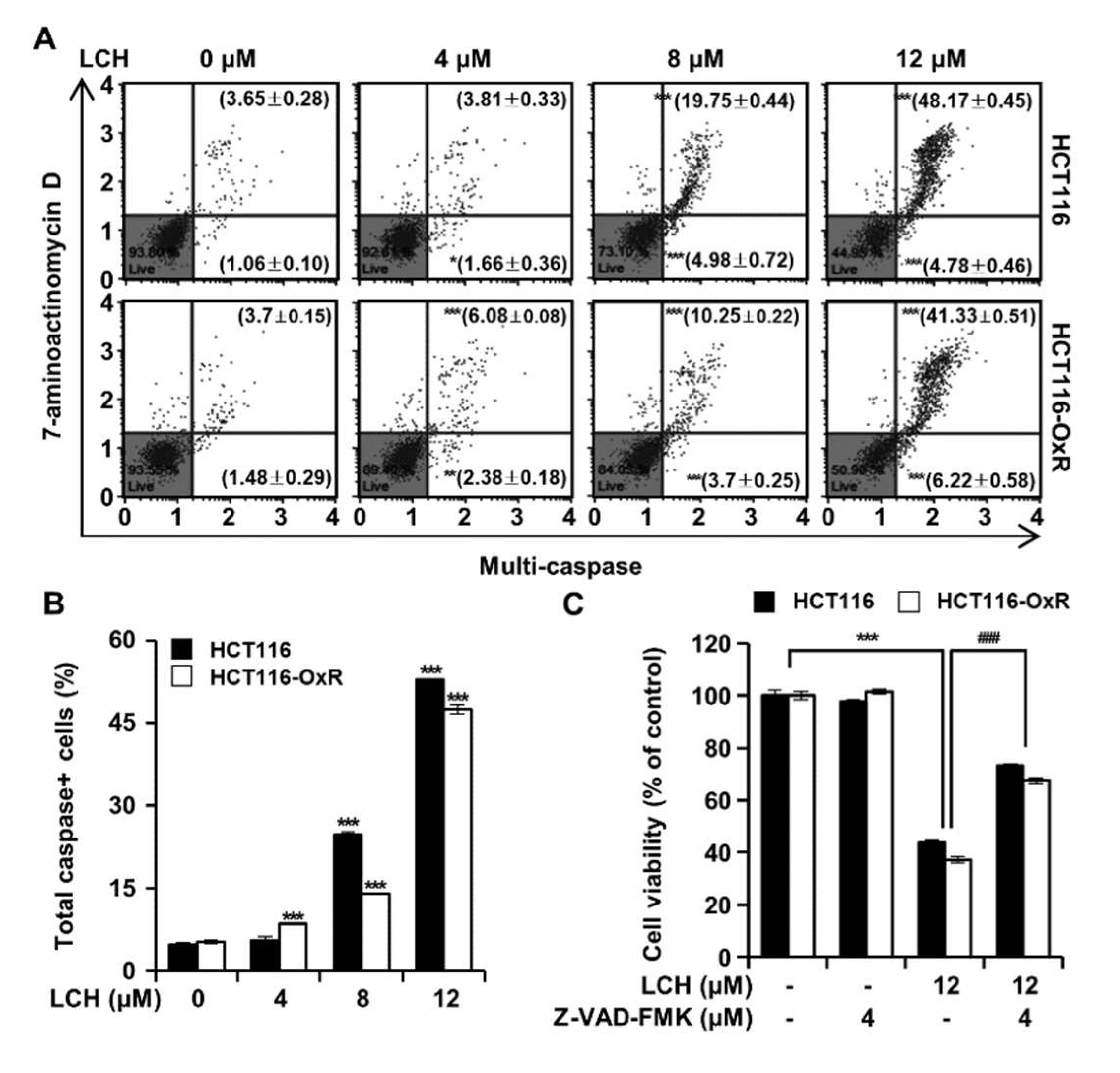
To further verify the induction of apoptosis following LCH treatment in HCT116 and HCT116-OxR cells, we measured caspase activity (Figure 7). Flow cytometry analysis results showed that LCH (4, 8, and 12 μM) increase the percentage of total caspase positive cells to 5.47 ± 0.69%, 24.73 ± 0.58% and 52.95 ± 0.18% in HCT116 cells, 8.47 ± 0.16%, 13.95 ± 0.15% and 47.55 ± 0.82% in HCT116-OxR cells, respectively (Figure 7A-B). Moreover, these results were confirmed by treatment with the pan-caspase inhibitor Z-VAD-FMK (Figure 7C). The treatment of HCT116 and HCT116-OxR cells with LCH (12 μM) only, LCH (12 μM) plus Z-VAD-FMK (4 μM), or Z-VAD-FMK (4 μM) only exhibited that LCH only treatment significantly inhibited the cell viability, and that the effect was reversed by Z-VAD-FMK treatment (Figure 7C). These results indicate that LCH induces apoptosis in CRC cells by increasing caspase activity.
4. Discussion
CRC is the third most common cancer worldwide [13,24]. Although the overall incidence has decreased with early detection by endoscopy, the number of younger-onset patients is increasing due to risk factors such as lifestyle and inflammatory disease [15,25]. For CRC therapy, 5-fluorouracil, Ox, irinotecan, or a combination of these three drugs is currently used. However, such regimens are still not safe due to their side effects and development of resistance. Ox is a third-generation DNA adduct-forming agent that is used as a first-line chemotherapy for CRC [17,26,27]. It contains a platinum moiety and binds to DNA bases, such as guanine or adenine, resulting in cell apoptosis due to DNA replication deficiency. However, treatment with Ox for several months is challenged by the development of resistance [28]. Therefore, we investigated whether LCH, a chalcone derivative in licorice, induces apoptosis in Ox-sensitive and Ox-resistant CRC cells.
We examined whether LCH could inhibit Ox-sensitive (HCT116) and Ox-resistant (HCT116-OxR) CRC cell growth. As shown in Figure 1A, LCH treatment significantly suppressed CRC cell viability. In addition, the colony growth of both cell types was decreased by LCH treatment in a dose-dependent manner, although Ox failed to inhibit HCT116-OxR cell growth (Figure 1B-D). Moreover, LCH induced apoptosis in both CRC cell lines (Figure 1E-F). In this study, we investigated the in vitro anticancer effects of LCH on Ox-sensitive and Ox-resistant CRC cell proliferation.
The cell cycle is composed of G1, S, and G2/M phases, and each phase progresses with cyclin/cyclin-dependent kinase (CDKs) complexes, such as cyclin D/CDK4 or 6, cyclinB/CDK1, and cyclin A or E/CDK2, and is finely regulated by CDK inhibitors, including p16, p21, and p27 [29]. LCH induced the G1 phase (Figure 2C-D) and increased the expression of the late G1 regulators p21 and p27 (Figure 2E). Thus, LCH contributes to cell death by regulating the cell cycle.
Ox-resistant cells have been reported to overexpress or activate EGFR [28,30] and its downstream kinase protein phosphatidylinositol 3-kinase (PI3K)/AKT [31,32]. In the signaling pathway, EGFR binds to its ligand EGF and induces autophosphorylation, resulting in its activation via conformational changes [33]. Activated EGFR transfers signals to downstream cascades, such as the PI3K/AKT pathway. We analyzed kinase activity with different concentrations of LCH and the positive control, confirmed that LCH inhibited EGFR and AKT activities, and predicted LCH and EGFR or AKT interactions by computational simulation (Figure 3B-F). We found that LCH directly inhibited the expression of the active forms, pEGFR and pAKT, compared to the intact forms, EGFR and AKT (Figure 4).
Recently, oxidative stress has been shown to promote cancer progression [34]. ROS levels are increased in cancer tissues, such as in colon, lung, and esophageal cancers [35,36,37]. Additionally, ROS and EGFR are closely associated with cancer growth and anticancer drug resistance. ROS play a role in sustaining EGFR-mediated signaling pathways by increasing the half-life of EGFR [38,39,40]. In contrast, enhanced oxidative stress induces apoptosis in cancer cells as a therapeutic strategy [41,42]. Here, we revealed that LCH increased ROS levels and the expression of ER stress proteins, including GRP78, CHOP, DR4, and DR5, in a dose-dependent manner (Figure 5-6). In addition, high ROS levels inhibit calcium transport into the mitochondria, resulting in mitochondrial membrane potential dysfunction. Cytochrome c is then mechanically released into the cytoplasm and forms an apoptosome complex with Apaf-1, activating downstream caspases, and eventually inducing apoptosis [41]. LCH increased the total number of depolarized cells in both Ox-sensitive and Ox-resistant CRC cells (Figure 6A-B). These markers were regulated by LCH treatment in a dose-dependent manner. Bim, Bax, cytosolic cytochrome c, Apaf-1, and cPARP increased, while Mcl-1, Bid, Bcl-2, mitochondrial cytochrome c, and intact caspase3 decreased (Figure 6C).
5. Conclusions
In conclusion, our results indicate that LCH inhibits EGFR and AKT kinase activity and related signal expression, including pEGFR and pAKT, thereby retarding cell proliferation and inducing apoptotic pathways in Ox-sensitive (HCT116) and-resistant (HCT116-OxR) CRC cells. Specifically, LCH increases ER stress, MMP dysfunction, and multi-caspase activation, and arrests the G1 phase of the cell cycle together with each biomarker, including GRP78, CHOP, DR4, DR5, Bim, Mcl-1, Bid, Bax, Bcl-xL, Bcl-2, cyto c, Apaf-1, cPARP, caspase3, p21, p27, cyclin D1, and CDK4/6. Our results suggest that LCH could be a chemotherapeutic agent for both Ox-sensitive and Ox-resistant CRC treatment. This provides a new perspective for drug-resistant CRC therapy.
Author Contributions
Seung-On Lee: conceptualization, data curation, formal analysis, methodology, validation, visualization, investigation, writing–original draft, writing–review, and editing. Mee-Hyun Lee: conceptualization, data curation, formal analysis, methodology, validation, visualization, investigation, writing–original draft, writing–review, and editing. Ah-Won Kwak: conceptualization, data curation, formal analysis, methodology, validation, investigation, software, resources. Jin-Young Lee: conceptualization, data curation, formal analysis, methodology, validation, investigation, software, resources. Goo Yoon: conceptualization, data curation, formal analysis, methodology, validation, investigation, software, resources. Sang Hoon Joo: conceptualization, data curation, formal analysis, methodology, validation, investigation, software, resources. Yung Hyun Choi: conceptualization, data curation, formal analysis, methodology, validation, investigation, software, resources. Jin Woo Park: project administration, resources, supervision, and funding acquisition. Jung-Hyun Shim: Project administration, resources, supervision, funding acquisition. All the data were generated in-house. All authors agree to be accountable for all aspects of the work and to ensure their integrity and accuracy.
Funding
This study was funded by the Basic Science Research Program of the National Research Foundation of Korea (NRF) (No. 2019R1A2C1005899) and an NRF grant from the Korean Government (MSIT) (No. 2022R1A5A8033794).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the article.
Acknowledgments
We greatly appreciated it using the Convergence Research Laboratory (established by the MNU Innovation Support Project in 2019) to conduct this research.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chiu, Y.J.; Lee, C.M.; Lin, T.H.; Lin, H.Y.; Lee, S.Y.; Mesri, M.; Chang, K.H.; Lin, J.Y.; Lee-Chen, G.J.; Chen, C.M. Chinese Herbal Medicine Glycyrrhiza inflataReduces Abeta Aggregation and Exerts Neuroprotection through Anti-Oxidation and Anti-Inflammation. Am J Chin Med 2018, 1–25. [Google Scholar] [CrossRef]
- van Dinteren, S.; Meijerink, J.; Witkamp, R.; van Ieperen, B.; Vincken, J.P.; Araya-Cloutier, C. Valorisation of liquorice (Glycyrrhiza) roots: antimicrobial activity and cytotoxicity of prenylated (iso)flavonoids and chalcones from liquorice spent (G. glabra, G. inflata, and G. uralensis). Food Funct 2022, 13, 12105–12120. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xu, G.; Li, Z.; Xiao, X.; Tang, J.; Bai, Z. NLRP3 Inflammasome Pharmacological Inhibitors in Glycyrrhiza for NLRP3-Driven Diseases Treatment: Extinguishing the Fire of Inflammation. J Inflamm Res 2022, 15, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.H.; Seo, J.H.; Oh, H.; Yoon, G.; Chae, J.I.; Shim, J.H. Licochalcone A Suppresses Specificity Protein 1 as a Novel Target in Human Breast Cancer Cells. J Cell Biochem 2017, 118, 4652–4663. [Google Scholar] [CrossRef]
- Kwak, A.W.; Choi, J.S.; Liu, K.; Lee, M.H.; Jeon, Y.J.; Cho, S.S.; Yoon, G.; Oh, H.N.; Chae, J.I.; Shim, J.H. Licochalcone C induces cell cycle G1 arrest and apoptosis in human esophageal squamous carcinoma cells by activation of the ROS/MAPK signaling pathway. J Chemother 2020, 32, 132–143. [Google Scholar] [CrossRef]
- Liu, X.; Xing, Y.; Li, M.; Zhang, Z.; Wang, J.; Ri, M.; Jin, C.; Xu, G.; Piao, L.; Jin, H.; et al. Licochalcone A inhibits proliferation and promotes apoptosis of colon cancer cell by targeting programmed cell death-ligand 1 via the NF-kappaB and Ras/Raf/MEK pathways. J Ethnopharmacol 2021, 273, 113989. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.N.; Lee, M.H.; Kim, E.; Kwak, A.W.; Yoon, G.; Cho, S.S.; Liu, K.; Chae, J.I.; Shim, J.H. Licochalcone D Induces ROS-Dependent Apoptosis in Gefitinib-Sensitive or Resistant Lung Cancer Cells by Targeting EGFR and MET. Biomolecules 2020, 10. [Google Scholar] [CrossRef]
- Oh, H.N.; Lee, M.H.; Kim, E.; Yoon, G.; Chae, J.I.; Shim, J.H. Licochalcone B inhibits growth and induces apoptosis of human non-small-cell lung cancer cells by dual targeting of EGFR and MET. Phytomedicine 2019, 63, 153014. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Feng, P.P.; Wang, H.F.; Zhang, H.; Liang, T.; Hao, X.S.; Wang, F.Z.; Fei, H.R. Licochalcone B induces DNA damage, cell cycle arrest, apoptosis, and enhances TRAIL sensitivity in hepatocellular carcinoma cells. Chem Biol Interact 2022, 365, 110076. [Google Scholar] [CrossRef]
- Nho, S.H.; Yoon, G.; Seo, J.H.; Oh, H.N.; Cho, S.S.; Kim, H.; Choi, H.W.; Shim, J.H.; Chae, J.I. Licochalcone H induces the apoptosis of human oral squamous cell carcinoma cells via regulation of matrin 3. Oncol Rep 2019, 41, 333–340. [Google Scholar] [CrossRef]
- Oh, H.N.; Oh, K.B.; Lee, M.H.; Seo, J.H.; Kim, E.; Yoon, G.; Cho, S.S.; Cho, Y.S.; Choi, H.W.; Chae, J.I.; et al. JAK2 regulation by licochalcone H inhibits the cell growth and induces apoptosis in oral squamous cell carcinoma. Phytomedicine 2019, 52, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Park, K.H.; Joo, S.H.; Seo, J.H.; Kim, J.; Yoon, G.; Jeon, Y.J.; Lee, M.H.; Chae, J.I.; Kim, W.K.; Shim, J.H. Licochalcone H Induces Cell Cycle Arrest and Apoptosis in Human Skin Cancer Cells by Modulating JAK2/STAT3 Signaling. Biomol Ther (Seoul) 2022, 30, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.; Arnold, M.; Gini, A.; Lorenzoni, V.; Cabasag, C.J.; Laversanne, M.; Vignat, J.; Ferlay, J.; Murphy, N.; Bray, F. Global burden of colorectal cancer in 2020 and 2040: incidence and mortality estimates from GLOBOCAN. Gut 2023, 72, 338–344. [Google Scholar] [CrossRef]
- Siegel, R.L.; Wagle, N.S.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2023. CA Cancer J Clin 2023. [CrossRef]
- Gausman, V.; Dornblaser, D.; Anand, S.; Hayes, R.B.; O'Connell, K.; Du, M.; Liang, P.S. Risk Factors Associated With Early-Onset Colorectal Cancer. Clin Gastroenterol Hepatol 2020, 18, 2752–2759. [Google Scholar] [CrossRef]
- Tsubaki, M.; Takeda, T.; Matsuda, T.; Kishimoto, K.; Takefuji, H.; Taniwaki, Y.; Ueda, M.; Hoshida, T.; Tanabe, K.; Nishida, S. Statins enhances antitumor effect of oxaliplatin in KRAS-mutated colorectal cancer cells and inhibits oxaliplatin-induced neuropathy. Cancer Cell Int 2023, 23, 73. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.; Mushin, M.; Kirkpatrick, P. Oxaliplatin. Nat Rev Drug Discov 2004, 3, 11–12. [Google Scholar] [CrossRef]
- Brinzan, C.S.; Aschie, M.; Cozaru, G.C.; Deacu, M.; Dumitru, E.; Burlacu, I.; Mitroi, A. KRAS, NRAS, BRAF, PIK3CA, and AKT1 signatures in colorectal cancer patients in south-eastern Romania. Medicine (Baltimore) 2022, 101, e30979. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Gu, T.; Lee, M.H.; Dong, Z. Challenge and countermeasures for EGFR targeted therapy in non-small cell lung cancer. Biochim Biophys Acta Rev Cancer 2022, 1877, 188645. [Google Scholar] [CrossRef]
- Lin, L.; Li, X.; Pan, C.; Lin, W.; Shao, R.; Liu, Y.; Zhang, J.; Luo, Y.; Qian, K.; Shi, M.; et al. ATXN2L upregulated by epidermal growth factor promotes gastric cancer cell invasiveness and oxaliplatin resistance. Cell Death Dis 2019, 10, 173. [Google Scholar] [CrossRef]
- Wang, Z.; Cao, Y.; Paudel, S.; Yoon, G.; Cheon, S.H. Concise synthesis of licochalcone C and its regioisomer, licochalcone H. Arch Pharm Res 2013, 36, 1432–1436. [Google Scholar] [CrossRef] [PubMed]
- Bose, D.; Zimmerman, L.J.; Pierobon, M.; Petricoin, E.; Tozzi, F.; Parikh, A.; Fan, F.; Dallas, N.; Xia, L.; Gaur, P.; et al. Chemoresistant colorectal cancer cells and cancer stem cells mediate growth and survival of bystander cells. Br J Cancer 2011, 105, 1759–1767. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J Clin 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Fernandez-Rozadilla, C.; Timofeeva, M.; Chen, Z.; Law, P.; Thomas, M.; Schmit, S.; Diez-Obrero, V.; Hsu, L.; Fernandez-Tajes, J.; Palles, C.; et al. Deciphering colorectal cancer genetics through multi-omic analysis of 100,204 cases and 154,587 controls of European and east Asian ancestries. Nat Genet 2023, 55, 89–99. [Google Scholar] [CrossRef]
- Meyerhardt, J.A.; Mayer, R.J. Systemic therapy for colorectal cancer. N Engl J Med 2005, 352, 476–487. [Google Scholar] [CrossRef]
- Comella, P.; Casaretti, R.; Sandomenico, C.; Avallone, A.; Franco, L. Role of oxaliplatin in the treatment of colorectal cancer. Ther Clin Risk Manag 2009, 5, 229–238. [Google Scholar] [CrossRef]
- Temraz, S.; Mukherji, D.; Alameddine, R.; Shamseddine, A. Methods of overcoming treatment resistance in colorectal cancer. Crit Rev Oncol Hematol 2014, 89, 217–230. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Ekblad, L.; Johnsson, A. Cetuximab sensitivity associated with oxaliplatin resistance in colorectal cancer. Anticancer Res 2012, 32, 783–786. [Google Scholar]
- Hsu, C.P.; Kao, T.Y.; Chang, W.L.; Nieh, S.; Wang, H.L.; Chung, Y.C. Clinical significance of tumor suppressor PTEN in colorectal carcinoma. Eur J Surg Oncol 2011, 37, 140–147. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Li, A.J.; Han, Y.; Yin, L.; Lin, M.B. Inhibition of Girdin enhances chemosensitivity of colorectal cancer cells to oxaliplatin. World J Gastroenterol 2014, 20, 8229–8236. [Google Scholar] [CrossRef]
- Purba, E.R.; Saita, E.I.; Maruyama, I.N. Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The "Rotation Model". Cells 2017, 6. [Google Scholar] [CrossRef]
- Sreevalsan, S.; Safe, S. Reactive Oxygen Species and Colorectal Cancer. Curr Colorectal Cancer Rep 2013, 9, 350–357. [Google Scholar] [CrossRef]
- Kwak, A.W.; Lee, J.Y.; Lee, S.O.; Seo, J.H.; Park, J.W.; Choi, Y.H.; Cho, S.S.; Yoon, G.; Lee, M.H.; Shim, J.H. Echinatin induces reactive oxygen species-mediated apoptosis via JNK/p38 MAPK signaling pathway in colorectal cancer cells. Phytother Res 2023, 37, 563–577. [Google Scholar] [CrossRef]
- Kwak, A.W.; Lee, M.J.; Lee, M.H.; Yoon, G.; Cho, S.S.; Chae, J.I.; Shim, J.H. The 3-deoxysappanchalcone induces ROS-mediated apoptosis and cell cycle arrest via JNK/p38 MAPKs signaling pathway in human esophageal cancer cells. Phytomedicine 2021, 86, 153564. [Google Scholar] [CrossRef]
- Kim, H.S.; Oh, H.N.; Kwak, A.W.; Kim, E.; Lee, M.H.; Seo, J.H.; Cho, S.S.; Yoon, G.; Chae, J.I.; Shim, J.H. Deoxypodophyllotoxin Inhibits Cell Growth and Induces Apoptosis by Blocking EGFR and MET in Gefitinib-Resistant Non-Small Cell Lung Cancer. J Microbiol Biotechnol 2021, 31, 559–569. [Google Scholar] [CrossRef]
- Weng, M.S.; Chang, J.H.; Hung, W.Y.; Yang, Y.C.; Chien, M.H. The interplay of reactive oxygen species and the epidermal growth factor receptor in tumor progression and drug resistance. J Exp Clin Cancer Res 2018, 37, 61. [Google Scholar] [CrossRef]
- Truong, T.H.; Carroll, K.S. Redox regulation of epidermal growth factor receptor signaling through cysteine oxidation. Biochemistry 2012, 51, 9954–9965. [Google Scholar] [CrossRef]
- Goldkorn, T.; Balaban, N.; Matsukuma, K.; Chea, V.; Gould, R.; Last, J.; Chan, C.; Chavez, C. EGF-Receptor phosphorylation and signaling are targeted by H2O2 redox stress. Am J Respir Cell Mol Biol 1998, 19, 786–798. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin Cell Dev Biol 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Gupta, S.C.; Tyagi, A.K. Reactive oxygen species (ROS) and cancer: Role of antioxidative nutraceuticals. Cancer Lett 2017, 387, 95–105. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
LCH suppresses proliferation and induces apoptosis of HCT116 and HCT116-OxR CRC cells. (A) Cell viability was examined by MTT assay. HCT116, HCT116-OxR, HaCaT and JB6 cells were treated with 0, 4, 8, and 12 μM of LCH or 2 μM of oxaliplatin for 24 and 48 h. (B-D) Anchorage-independent colony growth was determined by soft agar assay in HCT116 and HCT116-OxR CRC cells treated with 0, 4, 8, and 12 μM of LCH or 2 μM of oxaliplatin for 14 days. Colonies were captured and counted under microscope. (B) Representative pictures. (C) Histogram of colonies number. (D) Histogram of over 50 μm diameter size. (E-F) Cells were treated with or without LCH at 4, 8 and 12 µM for 48 h. Flow cytometric analysis was performed using annexin V and 7-aminoactionomycin D (7-AAD) staining in CRC cells. (E) The results of flow cytometric analysis. (F) A histogram of annexin V-FITC/7-AAD stained cell population (%). Total apoptotic cells included Annexin V positive/7-AAD negative cells and Annexin V positive/7-AAD positive cells. All data are expressed as mean ± SD. *p < 0.05, **p < 0.01, and ***p < 0.001.
Figure 1.
LCH suppresses proliferation and induces apoptosis of HCT116 and HCT116-OxR CRC cells. (A) Cell viability was examined by MTT assay. HCT116, HCT116-OxR, HaCaT and JB6 cells were treated with 0, 4, 8, and 12 μM of LCH or 2 μM of oxaliplatin for 24 and 48 h. (B-D) Anchorage-independent colony growth was determined by soft agar assay in HCT116 and HCT116-OxR CRC cells treated with 0, 4, 8, and 12 μM of LCH or 2 μM of oxaliplatin for 14 days. Colonies were captured and counted under microscope. (B) Representative pictures. (C) Histogram of colonies number. (D) Histogram of over 50 μm diameter size. (E-F) Cells were treated with or without LCH at 4, 8 and 12 µM for 48 h. Flow cytometric analysis was performed using annexin V and 7-aminoactionomycin D (7-AAD) staining in CRC cells. (E) The results of flow cytometric analysis. (F) A histogram of annexin V-FITC/7-AAD stained cell population (%). Total apoptotic cells included Annexin V positive/7-AAD negative cells and Annexin V positive/7-AAD positive cells. All data are expressed as mean ± SD. *p < 0.05, **p < 0.01, and ***p < 0.001.
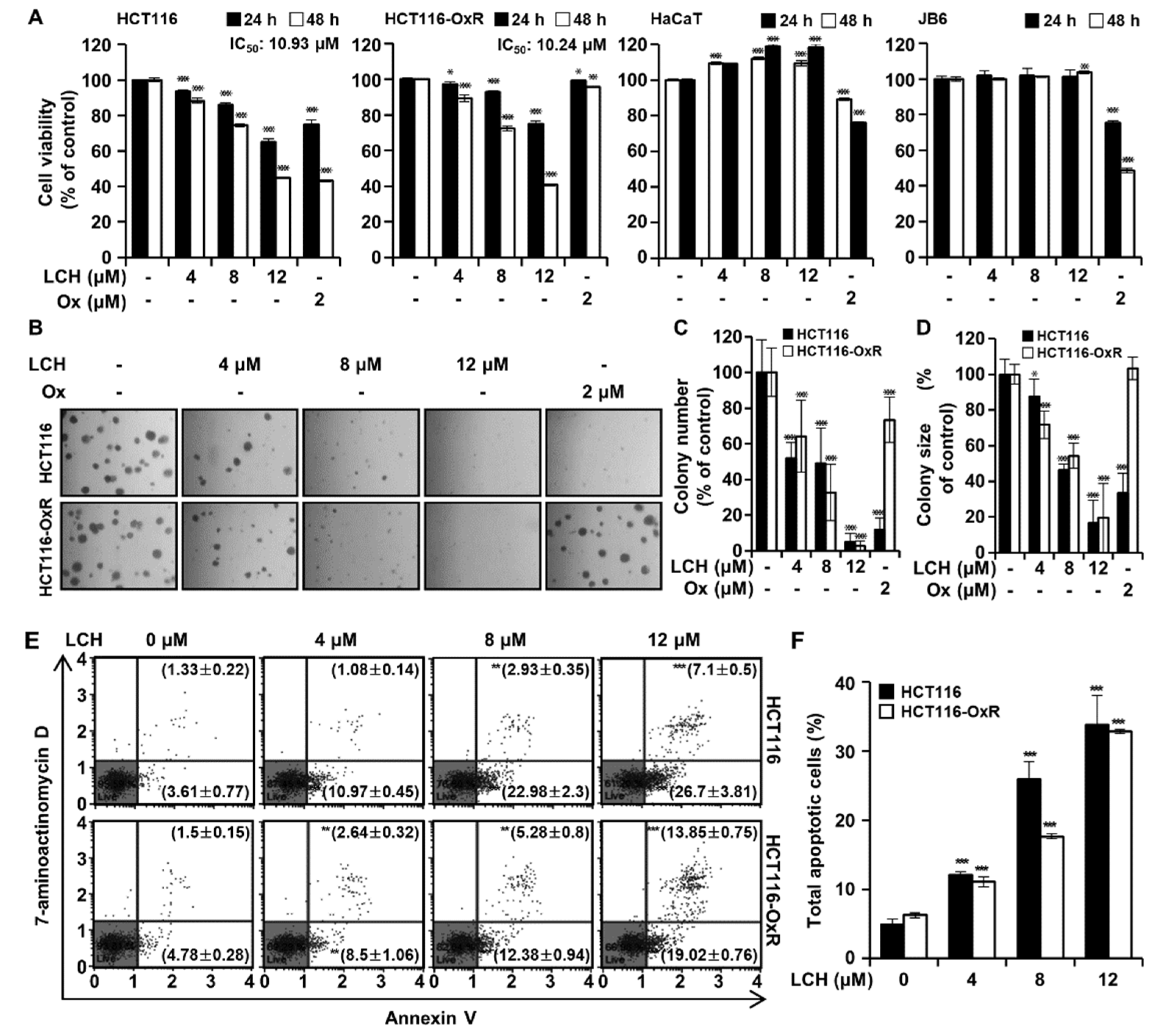
Figure 2.
LCH induces sub-G1 population and arrests G1 phase of cell cycle in HCT116 and HCT116-OxR CRC cells. Cells were treated with indicated concentrations of LCH for 48 h. (A) Cells were analyzed by the Muse® Cell Analyzer to determine cell cycle distributions. (B) Representative histogram of the sub-G1 population. (C-D) Representative histograms of the cell cycle distributions in HCT116 and HCT116-OxR cells. Data are presented as the mean ± SD. *p < 0.05, **p < 0.01, or ***p < 0.001 compared to the controls. (E) Cell lysates were examined for the expression of G1 phase-related proteins by Western blotting against antibodies, p21, p27, cyclin D1, CDK6, and CDK4. Actin was used as the loading control.
Figure 2.
LCH induces sub-G1 population and arrests G1 phase of cell cycle in HCT116 and HCT116-OxR CRC cells. Cells were treated with indicated concentrations of LCH for 48 h. (A) Cells were analyzed by the Muse® Cell Analyzer to determine cell cycle distributions. (B) Representative histogram of the sub-G1 population. (C-D) Representative histograms of the cell cycle distributions in HCT116 and HCT116-OxR cells. Data are presented as the mean ± SD. *p < 0.05, **p < 0.01, or ***p < 0.001 compared to the controls. (E) Cell lysates were examined for the expression of G1 phase-related proteins by Western blotting against antibodies, p21, p27, cyclin D1, CDK6, and CDK4. Actin was used as the loading control.
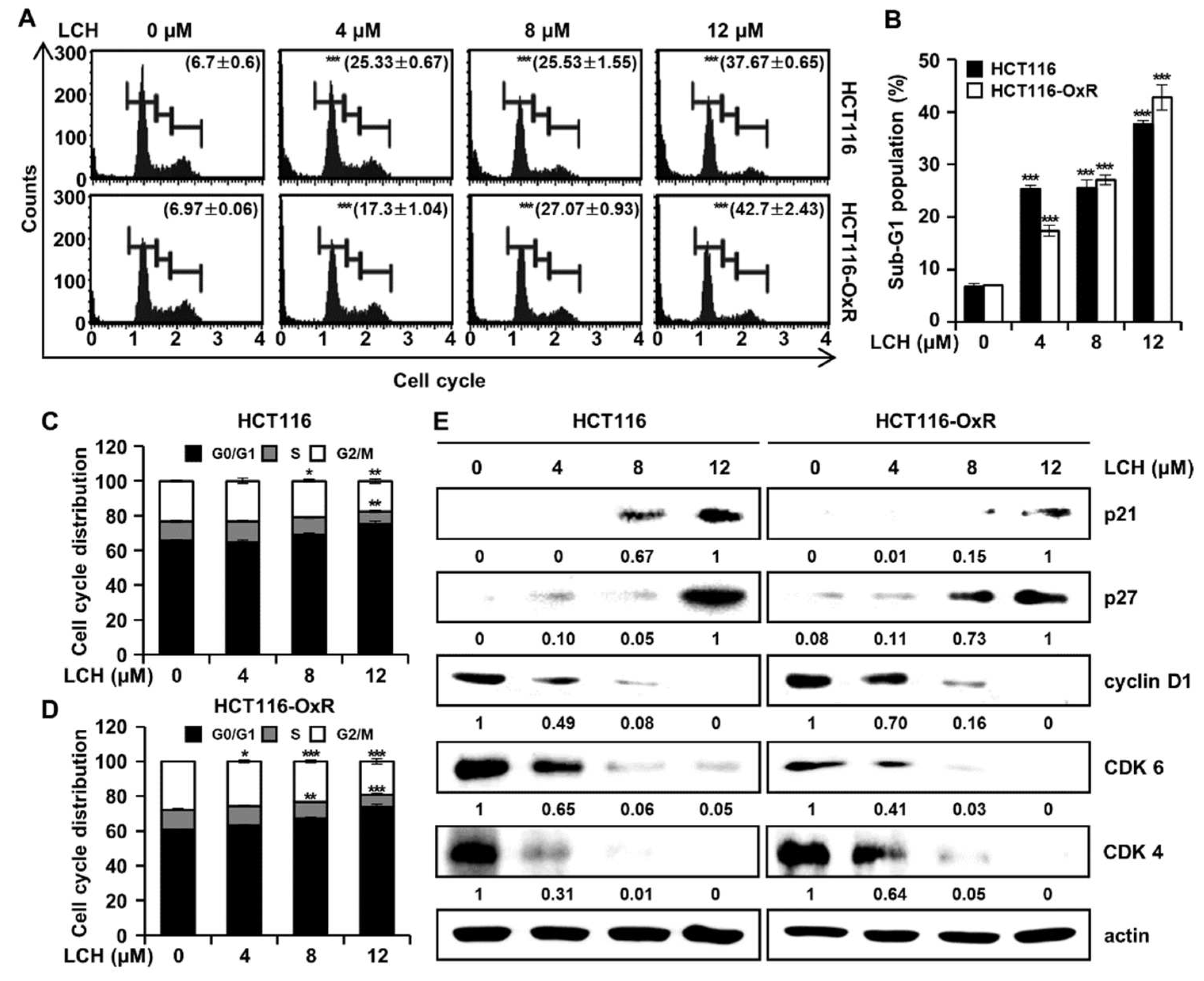
Figure 3.
LCH binds with EGFR and inhibits EGFR and AKT kinase activity. (A) LCH structure. (B) In vitro ADP-Glo kinase assay was performed using EGFR kinase enzyme system. Gefitinib used as a positive control. The values indicated mean ± SD. (C) In vitro ADP-Glo kinase assay was performed using AKT1 and AKT2 kinase enzyme system. MK2206 used as a positive control. The values indicated mean ± SD. (D-F) The predicted binding sites of LCH in EGFR (D), AKT1 (E) and AKT2 (F). The ATP binding pockets of both kinases, shown with surface representation, were occupied tightly by LCH (spheres). The binding pockets were zoomed in. LCH (light pink) and the amino acids (purple) in 4 Å are drawn with sticks. Those amino acids with possible hydrophobic interactions are circled.
Figure 3.
LCH binds with EGFR and inhibits EGFR and AKT kinase activity. (A) LCH structure. (B) In vitro ADP-Glo kinase assay was performed using EGFR kinase enzyme system. Gefitinib used as a positive control. The values indicated mean ± SD. (C) In vitro ADP-Glo kinase assay was performed using AKT1 and AKT2 kinase enzyme system. MK2206 used as a positive control. The values indicated mean ± SD. (D-F) The predicted binding sites of LCH in EGFR (D), AKT1 (E) and AKT2 (F). The ATP binding pockets of both kinases, shown with surface representation, were occupied tightly by LCH (spheres). The binding pockets were zoomed in. LCH (light pink) and the amino acids (purple) in 4 Å are drawn with sticks. Those amino acids with possible hydrophobic interactions are circled.
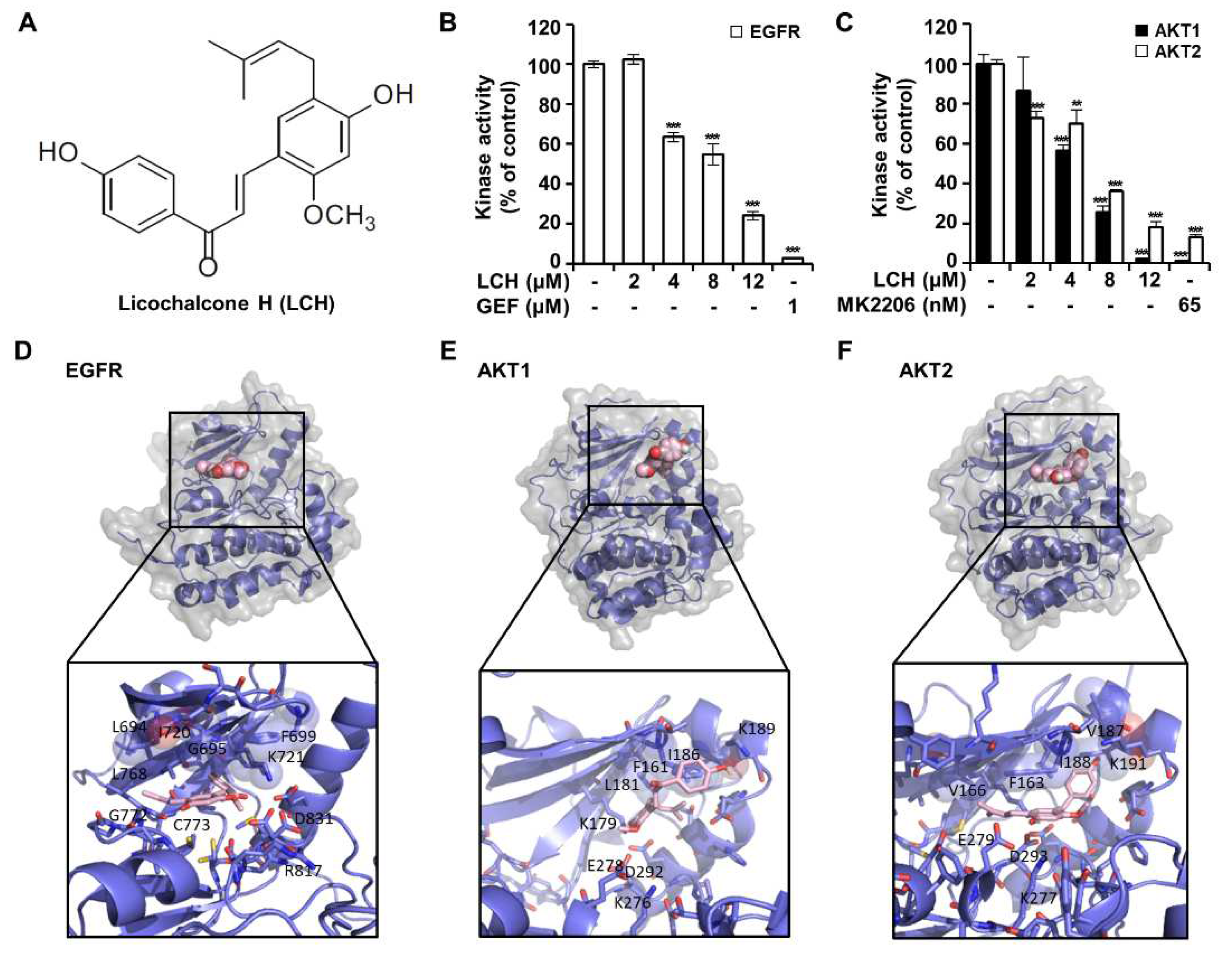
Figure 4.
LCH inhibits the expression of active form p-EGFR and p-AKT kinases. (A) Cells were treated with LCH at the indicated concentration for 48 h, then the protein levels were determined by Western blotting against phospho (p)-EGFR (Tyr1068), EGFR, p-AKT (Ser473), AKT antibodies. Actin was used as an internal control for equal loading. (B) Quantitation of active form p-EGFR and p-AKT compared to each total form. **p<0.01 and ***p<0.001 compared with the control.
Figure 4.
LCH inhibits the expression of active form p-EGFR and p-AKT kinases. (A) Cells were treated with LCH at the indicated concentration for 48 h, then the protein levels were determined by Western blotting against phospho (p)-EGFR (Tyr1068), EGFR, p-AKT (Ser473), AKT antibodies. Actin was used as an internal control for equal loading. (B) Quantitation of active form p-EGFR and p-AKT compared to each total form. **p<0.01 and ***p<0.001 compared with the control.
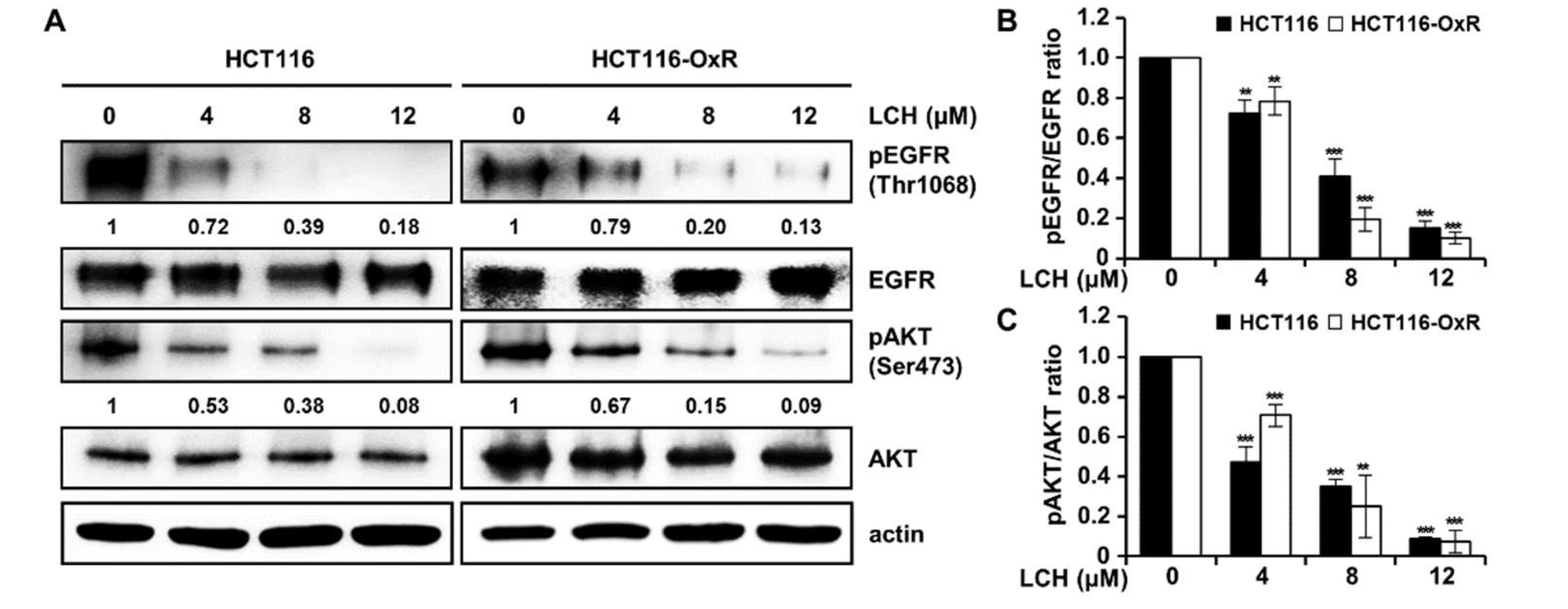
Figure 5.
LCH induces ROS production in HCT116 and HCT116-OxR CRC cells. (A) ROS generation by LCH treatment (0, 4, 8, and 12 μM) was measured using a Muse® Cell Analyzer. M1 and M2 represent ROS-negative and ROS-positive cell populations, respectively. Based on the bar in the graph, the left represents ROS-negative and the right represents ROS-positive. (B-D) Cells were pre-treated with NAC (4 mM) or vehicle for 3 h, then incubated with LCH (12 μM) for 48 h. (B) Cell viability was measured using the MTT assay. (C) Apoptosis was analyzed by flowcytometry. (D) Multi-caspase activity was analyzed using flowcytometry. Data are expressed as mean ± SD (*p < 0.05 and ***p < 0.001). ##p < 0.01 and ###p < 0.001 compared to the cells treated with LCH only.
Figure 5.
LCH induces ROS production in HCT116 and HCT116-OxR CRC cells. (A) ROS generation by LCH treatment (0, 4, 8, and 12 μM) was measured using a Muse® Cell Analyzer. M1 and M2 represent ROS-negative and ROS-positive cell populations, respectively. Based on the bar in the graph, the left represents ROS-negative and the right represents ROS-positive. (B-D) Cells were pre-treated with NAC (4 mM) or vehicle for 3 h, then incubated with LCH (12 μM) for 48 h. (B) Cell viability was measured using the MTT assay. (C) Apoptosis was analyzed by flowcytometry. (D) Multi-caspase activity was analyzed using flowcytometry. Data are expressed as mean ± SD (*p < 0.05 and ***p < 0.001). ##p < 0.01 and ###p < 0.001 compared to the cells treated with LCH only.
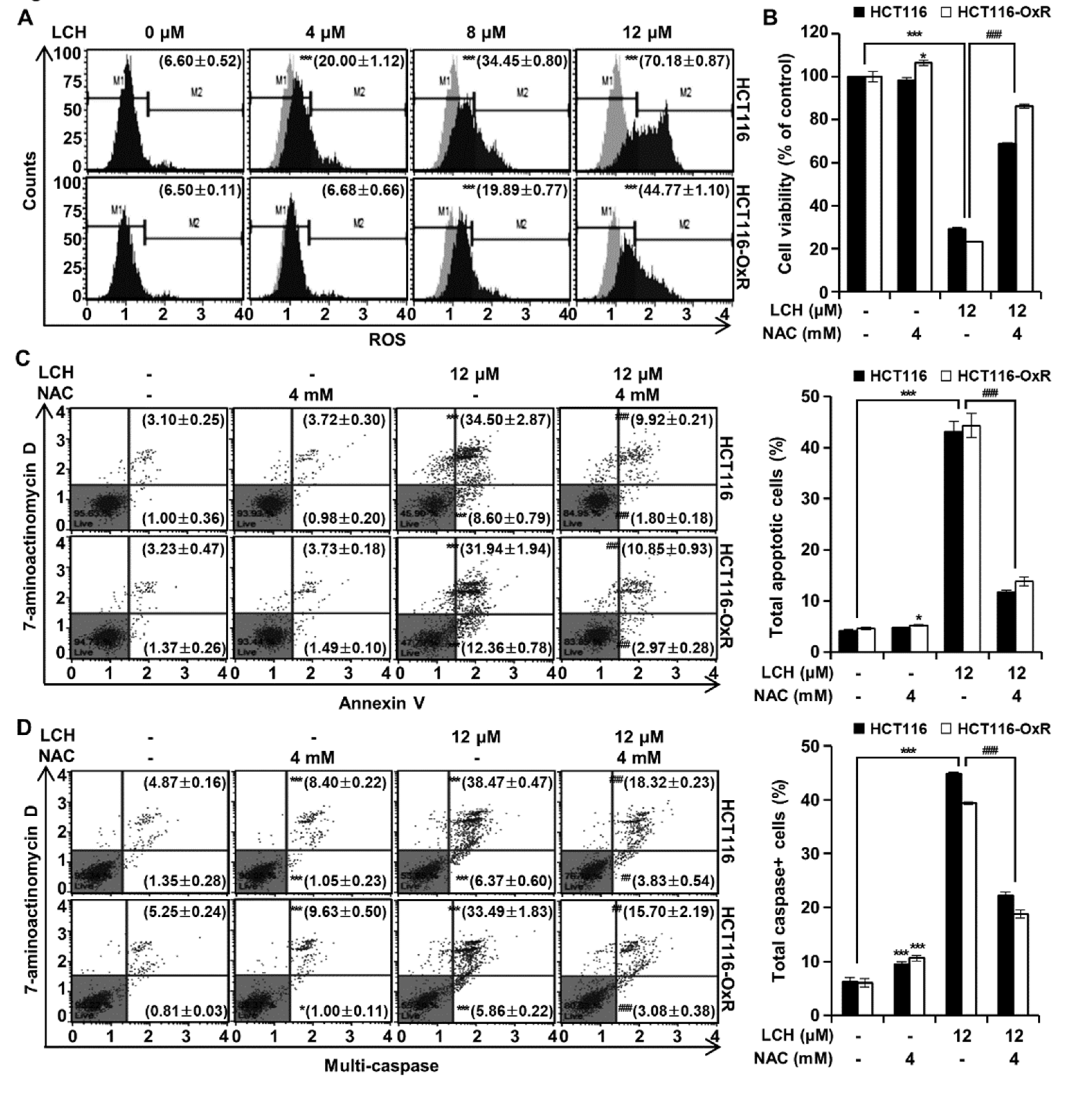
Figure 6.
LCH increases mitochondrial membrane potential (MMP) dysfunction in HCT116 and HCT116-OxR CRC cells. (A) Cells were treated with or without 4, 8 and 12 µM of LCH for 48 h, and then MMP depolarization was determined using flow cytometric analysis after staining with a Muse® MitoPotential kit. (B) The histogram of total MMP depolarized cells (%). Results are presented as mean ± SD (*p < 0.05, **p < 0.01, and ***p < 0.001 compared to the control group). (C) The expression of ER stress (GRP78, CHOP, DR4, and DR5), mitochondria-mediated apoptosis (Bim and Bax) and antiapoptosis (Mcl-1, Bcl-xL, and Bcl-2) proteins was examined by Western blotting. And the expression of apoptosis-associated proteins was examined. For determining the cyto c levels, cytosol and mitochondrial fraction proteins were separated, and -Tubulin and COX4 were used as cytosolic and mitochondrial controls, respectively. Actin was used as an internal control for equal loading.
Figure 6.
LCH increases mitochondrial membrane potential (MMP) dysfunction in HCT116 and HCT116-OxR CRC cells. (A) Cells were treated with or without 4, 8 and 12 µM of LCH for 48 h, and then MMP depolarization was determined using flow cytometric analysis after staining with a Muse® MitoPotential kit. (B) The histogram of total MMP depolarized cells (%). Results are presented as mean ± SD (*p < 0.05, **p < 0.01, and ***p < 0.001 compared to the control group). (C) The expression of ER stress (GRP78, CHOP, DR4, and DR5), mitochondria-mediated apoptosis (Bim and Bax) and antiapoptosis (Mcl-1, Bcl-xL, and Bcl-2) proteins was examined by Western blotting. And the expression of apoptosis-associated proteins was examined. For determining the cyto c levels, cytosol and mitochondrial fraction proteins were separated, and -Tubulin and COX4 were used as cytosolic and mitochondrial controls, respectively. Actin was used as an internal control for equal loading.
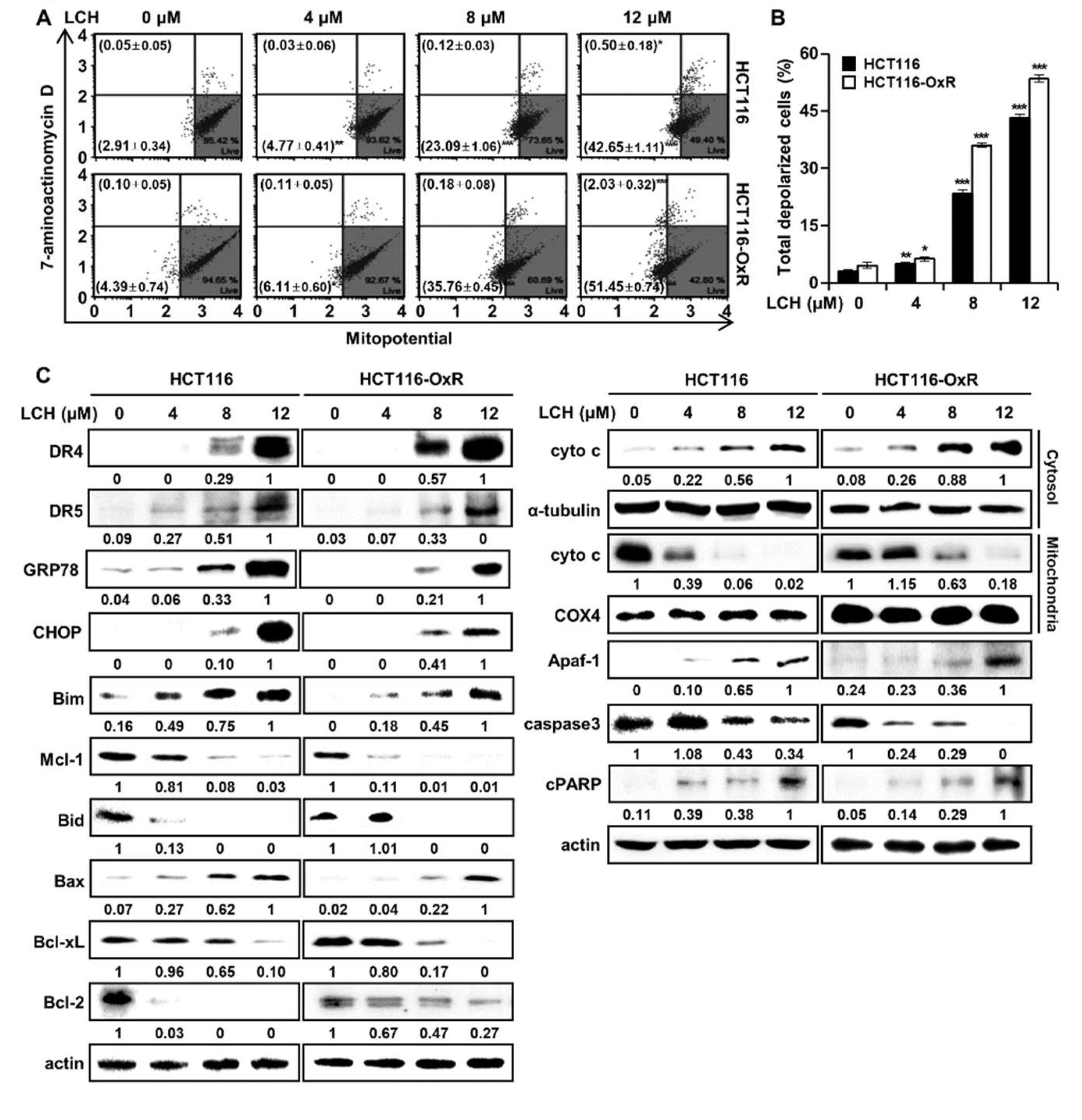
Figure 7.
LCH increases multi-caspases activity HCT116 and HCT116-OxR cells. (A) Cells were treated with LCH (0, 4, 8 and 12 µM) for 48 h and stained with a Muse® MultiCaspase Kit, then the activity were measured using a Muse® Cell Analyzer. (B) The histogram of total caspase+ cells percentage. (C) Cells were pre-treated with pan-caspase inhibitor, Z-VAD-FMK (4 µM) for 3 h and then treated with or without 12 µM of LCH for 48 h. Cell viability was measured by the MTT assay. Results are presented as mean ± SD. *p < 0.05, **p < 0.01, and ***p < 0.001 compared to the control group. ###p < 0.001 compared to the cells treated with LCH only.
Figure 7.
LCH increases multi-caspases activity HCT116 and HCT116-OxR cells. (A) Cells were treated with LCH (0, 4, 8 and 12 µM) for 48 h and stained with a Muse® MultiCaspase Kit, then the activity were measured using a Muse® Cell Analyzer. (B) The histogram of total caspase+ cells percentage. (C) Cells were pre-treated with pan-caspase inhibitor, Z-VAD-FMK (4 µM) for 3 h and then treated with or without 12 µM of LCH for 48 h. Cell viability was measured by the MTT assay. Results are presented as mean ± SD. *p < 0.05, **p < 0.01, and ***p < 0.001 compared to the control group. ###p < 0.001 compared to the cells treated with LCH only.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



