Submitted:
01 August 2023
Posted:
02 August 2023
You are already at the latest version
Abstract
In the last decades, studies on the inflammatory signaling pathways in multiple pathological contexts revealed new targets for novel therapies. Among the family of G-protein-coupled Proteases Activated Receptors, PAR2 was identified as a driver of the inflammatory cascade in many pathologies, ranging from autoimmune disease to cancer metastasis. For this reason, many efforts have been focused on the development of potential antagonists of PAR2 activity. This work focuses on a small molecule, 1-Piperidine Propionic Acid (1-PPA), previously described to be active against inflammatory processes, but whose target is still unknown. Stabilization effects observed by cellular thermal shift assay coupled to in-silico investigations, including molecular docking and Molecular Dynamics simulations, suggested that 1-PPA binds PAR2 in an allosteric pocket of the receptor inactive conformation. In addition, this compound was found able to inhibit platelet aggregation, a process mediated by PAR family members, including PAR2. Since the allosteric pocket binding 1-PPA is highly conserved in all the members of the PAR family, the evidence reported here suggests that 1-PPA could represent a promising new small molecule targeting PARs with antagonistic activity.
Keywords:
G protein-coupled receptors
; Protease Activated Receptor2
; Allosteric modulator
; 1-piperidinepropionic acid
; Molecular Dynamics.
1. Introduction
Alterations of the extracellular environment modulate cellular activities, leading to the activation of different responses to adapt to a new condition. G-protein coupled receptors (GPCRs) play a crucial role in the transduction of such extracellular stimuli. GPCRs are endowed with broad tissue specificity and possess the ability to bind a vast range of activating molecules, including hormones, neurotransmitters, ions, photons, and odorants [1]. Despite these remarkable features, GPCRs are structurally conserved and characterized by the well-described 7-helices transmembrane domain (7TMD), connected by loops spanning the extracellular and intracellular environment [2]. The sequences and structures of these loops are crucial for GPCRs activity and specialization, as they are responsible for mediating signal transduction, even though all the five classes of GPCRs (A, B, B1, C, F) share this feature in a highly conserved fashion [3].
In recent years, structural and computational studies have significantly improved the understanding of the mechanisms of action of GPCRs and the development of new pharmaceuticals targeting them [2], especially for what concerns Class A, or Rhodopsin-like Class, to which belongs most of the drugged GPCRs. Due to the intrinsic allosteric nature of GPCRs, and thanks to the advancement of bioinformatic tools, it was possible to further map and understand the changes that these receptors undergo to transduce the signals. These techniques also allowed to decipher the translation from the inactive to the active conformation, revealing new sites for allosteric modulation [4,5]. Furthermore, they revealed that most of Class A GPCRs are modulated into an intermediate conformation by the presence of a sodium ion coordination among the helices, which are stabilized in a pose similar to the inactive conformation [6,7].
Among GPCRs in this Class, Protease-activated receptors (PARs) define a small family, composed of four members, well-studied for their peculiar activation mechanism, which is protease-induced. As a matter of fact, these receptors are activated upon removal of their N-terminal signal peptide by protease cleavage [8,9].
The main role of these receptors has been widely described in processes related to the inflammation of the endothelium and the coagulation cascade, where they have been discovered for the first time [10]. PAR2, a member of this family, was found to play an important role in other stress responses, including cell proliferation and differentiation and its activity has been linked to epithelial-mesenchymal transition (EMT) in gastrointestinal and pancreatic cancers [11,12]. Therefore, in the last few years, many efforts have been devoted to the design of potent PAR2 inhibitors that could be applicable to cardiovascular inflammatory diseases and cancer. To reach this goal, the structure of human PAR2 in complex with inhibitors has already been experimentally determined by X-ray diffraction experiments (PDB ID: 5NDD, 5NDZ, 5NJ6) [13,14].
In this work, the small molecule 1-piperidinepropionic acid (1-PPA) was taken in consideration for its anti-inflammatory and anticancer properties described in different patents [15,16], where it was described as an inhibitor of proteins production, including cytokines and the serin-protease inhibitor SerpinB3, although its cellular target and mechanism of action are yet unclear. A recent work has reviewed the compounds targeting PAR2, highlighting the need of the development of novel PAR2 modulators to treat PAR2-mediated diseases [17]. Our goal was to investigate the capability of 1-PPA to bind PAR2 by exploiting in silico studies complemented with in vitro and cellular assays. Our results demonstrate that PAR2 represents a cellular target for 1-PPA while docked models here described let us recapitulate the modulation that such small molecule can exert at the level of PAR2 activation and signaling.
2. Materials and Methods
Chemicals
1-Piperidin propionic acid (1-PPA) was purchased at SIGMA and solubilized in Sterile PBS pH 7.4 for CETSA assays.
Cell culture and transfection
The Episomal Expression System for Recombinant Protein Production in Chines Hamster Ovary Cells (ExpiCHO) was used. CHO cells were grown in suspension (160 rpm) at 37°C and 5% CO2, in ProCHO5 medium (Lonza) supplemented with 4 mM Glutamine (Thermofisher Scientific) until they reached a concentration of 5x106 cells/ml. Cells were then harvested by centrifugation and resuspended in fresh media at a final concentration of 2.5x106 cells/ml. The day after, cells were transfected with 15 µg/ml Polyethylenimine hydrochloride 40000 (FisherScientific) and 3µg/ml of pcDNA3.0 plasmid, containing PAR2 FLAG and HA tagged at the N- and C- terminus, respectively (cat. #53228, AddGene). Protein expression was performed by keeping the culture in constant shaking for 48h at 30°C in a humidified atmosphere of 5% of CO2 in air. Finally, cells were harvested by centrifugation (500g) for 5 minutes at room temperature.
Cellular Thermal Shift Assay
Thermal Shift Assay (CETSA) protocol was adapted from [18]. ExpiCHO pellet was washed once with PBS pH 7.4 (137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, and 2 mM KH2PO4) and then centrifuged at 500g for 5 minutes. Pellet was resuspended in PBS pH 7.4, containing protease inhibitor cocktail (539132-1SET, Sigma-Aldrich), to a final concentration of 1.5x108 cells/ml. Samples were heated for 5 minutes in a thermocycler with a temperature gradient ranging from 37 to 95 °C, chilled at room temperature for 1 minute, and diluted with ice-cold 1% NP-40 in PBS pH 7.4 till a final concentration of 0.2% (v/v) detergent. Samples were then exposed to 10 freeze-thaw cycles in liquid nitrogen. Finally, soluble fraction was isolated by centrifuging samples at 17,000g for 30 minutes at 4°C. Protein concentration was quantified by Bradford assay on the untreated control (37°C). A total amount of 20 µg of protein, for each sample, was treated with PNGase F (NEB, cat. n.#P0711S) for 1 hour at 50°C to remove N-glycosylation. Samples were subjected to SDS-PAGE followed by Western Blotting.
Mechanistic study
PAR2 Transfected ExpiCHO cells were resuspended as described in the previous section, equally distributed in clean PCR tubes and subjected to thermal treatment at 53°C for 3 minutes. Then, 1-PPA was added to each sample at increasing concentrations (0, 2.5, 5, 12.5, 25, 50, 75, 125 µM) and samples were incubated for 2 minutes. Samples were then chilled at room temperature for 1 minute and diluted with ice cold 1% (v/v) NP-40 in PBS pH7.4 to final concentration of 0.2% (v/v). Total protein concentration was quantified based on untreated control (0 µM 1-PPA) and 20 µg of total protein were treated with PNGase F. Proteins were spotted onto nitrocellulose membrane and immunoblotted as described in the next section.
Gel electrophoresis and Western Blot
Protein samples were loaded on linear gradient polyacrylamide gel (SurePAGE 4-12%, GeneScript) and were run in MES SDS Running Buffer (M00677, GeneScript) for 45 minutes at 180 V. Protein bands were then transferred to Nitrocellulose Membrane (Amersham Protran 0.2 NC, Cytiva) in Tris-Glycine Transfer Buffer (25 mM Tris, 192 mM Glycine pH 8.3, 15% (v/v) Methanol) applying 20 V for 1.5 hours at room temperature.
Immunoblotting
Nitrocellulose membranes, either after SDS-PAGE transfer procedure or direct sample spotting, were blocked for 1 hour in Tris Saline Buffer with Tween-20 0.05% (TTBS) and milk. Primary antibody incubations were performed at 4°C overnight. Membranes were washed three times with T-TBS and incubated with secondary antibody, HRP conjugated, for 2 hours at RT (Table 1). Bands detection was performed by the addition of anti-HA (Invitrogen, 2-2.2.14) diluted 1:5000, while 1:5000 dilution of HL1964 (GeneTex, GTX637857) was employed for Vinculin detection. Bands were revealed with chemiluminescent substrate (SuperSignal™ West Pico PLUS Chemiluminescent Substrate, Thermofisher Scientific) with VWR Imager CHEMI Premium (VWR). Bands intensity was quantified based on chemiluminescence count per square millimeter (I = counts per mm2) using ImageJ-win 64 (Fiji) software [19] and normalized to loading control. For CETSA studies, data were also normalized to the lower temperature or maximum compound concentration. Data obtained were fitted with Boltzmann sigmoidal and analyzed with “[Inhibitor] vs. response” equation in GraphPad Prism 9.5.0 (GraphPad Software, Boston, Massachusetts USA, www.graphpad.com).
Protein modelling and docking
A model for apo PAR2 was generated by editing the crystal structure of human PAR2 in complex with AZ8838 (PDB ID: 5NDD) [13]. Firstly, experimentally introduced mutations were reverted using PyMOL mutagenesis wizard [20]. Then, ligands and solubilization moieties were removed, so that only the region in between Val61 and Arg362 was maintained. Finally, gap in the crystal structure were modelled with MODELLER interface in UCSF Chimera, using the DOPE-HR algorithm [21]. CASTp 3.0 webserver was used to detect solvent accessible pockets using a probe of 1.4 Å diameter [22].
Molecular Dynamics
Molecular dynamics (MD) was performed with Gromacs 2022.3 [25,26] using the Charmm36-jul2021 force field [27,28]. Forcefield parameters for AZ8838, PPA, and RO54464 were derived using cgenff and manually added to complexes' topology [29,30]. Simulations of 1 µs were performed as previously described [31]. Briefly, models were solvated with the TIP3P water model in a rectangular box with a minimum distance of 1 nm between the protein complex and the border. 0.15 M of NaCl was added to simulate physiological ionic strength. System energy was minimized by 5000 steps of steepest descent energy minimization, with a tolerance of 1000 kJ mol-1 nm-1. Subsequently, a 200 ps NVT MD simulation was used to heat the system from 0 to 310 K and equilibrated to 1 atm during a 1 ns NPT simulation. Energy minimization and equilibration simulations were performed with 1000 kJ mol-1 nM-2 on all atoms; however, they were removed for the production run. The V-rescale thermostat was used to equilibrate the temperature, whereas the C-rescale barostat was used to control the pressure [32,33]. Newton’s equation of motion was integrated using a leapfrog algorithm with a 2 fs time step. The particle mesh Ewald (PME) method was used to compute the long-range electrostatic force [34,35]. Rotational and translational motions of the system were removed, and all bonds were constrained with the LINCS algorithm. Protein conformation variations were analyzed by estimating global values of Root Mean Square Deviation (RMSD) and Root Mean Square Fluctuation (RMSF) from the original structure with Gromacs built-in rms and rmsf tools, respectively. Covariance matrices were obtained from Gromacs anaeig tool and converted into cross-correlation matrices. Finally, similarity between cross-correlation matrices was assessed with Spearman rho test.
TRAPtest
After informed consent 5mL of venous blood was collected from four healthy donors after overnight fasting, into syringes pre-filled with 0.5 mL of sodium citrate 109 mM with 21-gauge needles, without applying venostasis. Platelet aggregation testing was performed on the Multiplate® function analyzer (Roche Diagnostics GmbH, Mannheim, Germany). The instrument continuously measures the changes of the electrical resistance (called “impedance”) between two copper wires. The greater the area the more platelets aggregate. Briefly, 300 μL of whole citrated blood were added to an equal amount of saline solution, preheated at 37°C, and platelet aggregation was tested after specific activation with a thrombin analogue (called TRAPtest) and with or without 1-PPA (10-1000ng/ml) as inhibitor. Platelet aggregation was electronically measured for 6 minutes and expressed as units of area under the curve (AUC) plotted over time in arbitrary units (U) (1 U = 10 AU*min) [36]. Normal reference range for TRAPtest is 86–159 (U).
3. Results and Discussion
3.1. CETSA assay suggests that 1-PPA has a stabilizing effect on PAR2
To explore the hypothesis that 1-PPA can interact with PAR2 receptor, we first tested 1-Piperidine Propionic Acid effect on PAR2 stability by the CETSA experiment, previously used to identify AZ8838 compound as a PAR2 receptor inhibitor [18].
CETSA is appropriate for membrane and difficult-to-purify proteins. This method relies on the possibility to determine the protein target aggregation temperature (Tagg) by exposing cell extracts or even whole cells, overexpressing the protein of interest, to a temperature gradient. The readout obtained corresponds to the fraction of protein kept in solution at any step of the gradient. The presence of the cellular context further ensures to observe stabilization and eventual interactions in conditions resembling the physiological ones.
To set the experiment, we first determined the aggregation temperature (Tagg) of the receptor in phosphate buffer saline (PBS) without any additional compound, which resulted to be 49° C (Figure 1a). Afterward, we performed the experiments keeping the temperature at 53° C to ensure that more than 50% the receptor is denatured and let us observing the effects of 1-PPA compound on the receptor, if any. We evaluated the effect at different compound concentrations, ranging from 2.5 to 125 µM. As shown in Figure 1b, PAR2 is more soluble at higher concentrations of 1-PPA compared to the control one in PBS only, where no protein could be detected in Dot Blot. The curve suggests that 1-PPA interacts with the protein and stabilizes its structure, maintaining it soluble at temperatures higher than that of Tagg.
3.2. Molecular dynamics simulations suggest that 1-PPA occupies an allosteric pocket buried within the PAR2 receptor
To get insights into the interaction between PAR2 and 1-PPA, we applied molecular docking combined to MD simulations. The crystal structure of human PAR2 transmembrane domain has been previously obtained in complex with the inhibitor compound AZ8838 (PDB ID: 5NDD; [1]). Since the construct used for structural studies includes extra fusion domains and few stabilizing mutations to be crystallized, we modelled wtPAR2 (Val61 to – Arg362) by reconstructing and refining it in silico, as described in the methods section, and used the resulting model in all the subsequent computational analyses.
The putative binding poses of 1-PPA were first predicted by docking simulation. Most reliable poses were further filtered by an accessible volume analysis, performed by CASTp 3.0. Indeed, only 3 out of the 42 different solvent-accessible pockets present in the structure resulted accessible to at least 1-PPA, based on the compound volume and calculated cavities (Suppl. Figure S1a, S1b). Two final poses fulfilled the criteria, one at the very peripheral entrance (close to ECL2 loop) and a second one, corresponding to the pocket occupied by AZ8838 (Suppl. Figure S1b, S1c). As a control, this latter compound was docked to PAR2, following the same unbiased docking procedure. Protocol applied here was able to predict, within the most reliable ones, the same position experimentally determined in the crystal structure (5NDD), thus supporting the robustness of the adopted procedure.
To further validate our results, microsecond-scale MD simulations were applied both to PAR2 receptor model and 1-PPA or AZ8838 complexes. A third, unrelated molecule (Ro5-4864) was forcefully placed in the orthosteric pocket occupied by 1-PPA and AZ8838 to be used as control. This compound was chosen in that its size, overall structure and charge state resemble those of AZ8838, but it does not belong to PAR2 known ligands [18].
Interestingly, the simulation allowed us to observe a significative shift in the 1-PPA binding mode. Indeed, 1-PPA reached an allosteric pocket, more engulfed within PAR2 transmembrane helices, roughly 9 Å apart from the initially occupied orthosteric one (Figure 2(a-b) and Figure 3(b)). Neither AZ8838 nor the negative control molecule experienced the same repositioning: the lead molecule AZ8838 maintained its original place, while the negative control Ro5-4864 resulted unstable, hence it was pushed out of the orthosteric pocket toward the extracellular space (Suppl. Figure S2).
The novel pocket occupied by 1-PPA is hidden by the transmembrane helices, and the propionic moiety is placed 5 Å above the sodium cation, highly conserved among GPCRs [6,37]. 1-PPA binding is stabilized both by hydrogen bonds and polar contacts in the pocket. The propionic moiety forms hydrogen bonds between its oxygen atoms and the side chains of Ser1242.53 and Arg1212.50 (Ballesteros and Weinstein nomenclature is indicated in superscript hereafter to allow the comparison with other GPCRs structures and models [38]). It is noteworthy to consider that Arg1212.50 side chain forms a hydrogen bond with one of the propionic acid oxygens, while the other coordinates Na+. Relevant non-bonded contacts are formed with the carbons of the piperidine ring and the side chains of residues Met1593.36, Phe3006.48 and Thr3347.43 (Figure 2c).
Binding of AZ8838 to PAR2 has been previously reported to result in allosteric rearrangement of ECL2 loop (residues 212-235) limiting the solvent accessibility of His227ECL2 [13]. While no broad rearrangements of the ECL2 loop have been observed in the time scale of our simulations, suggesting that rearrangement may happen starting from the mid-microsecond scale, the root mean square fluctuation (RMSF) indicates a higher mobility for the ECL2 region in the 1-PPA and AZ8838 bound complexes (Figure 3d). In general, the observed larger mobility of the apo PAR2 compared to the holo protein states could be related to a stabilizing effect induced by the presence of the ligands, able to trap the molecule in an inactive conformation [37]. On the other side, the negative control does not show any mobility in the ECL2 region, which is replaced by a higher mobility of 196-207 residue, within transmembrane helix 4.
Furthermore, Figure 3c shows that both 1-PPA and AZ8838 compounds induce similar conformational adaptations in cross-correlated motions that let describe how local protein motions in different regions of a protein are correlated. On the contrary, the pattern produced by simulation of the Ro5-4864 complex is completely different from both the apo and holo forms, further supporting the robustness of binding simulations about 1-PPA.
When considering the stability of each compound throughout the simulations, we found that while the AZ8838 compound remained tightly bound to the original pocket for all simulations, 1-PPA moved to a more buried one within the first 50 ns of simulation, where it is engaged by the previously described interactions (Figure 3b,f). Since 1-PPA detached from the initial docked pose and then remained constant for all the time of the simulation, we can deduce that such pose is the most favored one for 1-PPA binding. Of note, 1-PPA is driven by a slight reshaping of the internal cavities of PAR2 into a more continuous funnel, as an effect of slight motions of helices TM1 and TM7. Such motion is observed in all MD trajectory independently on the presence of a docked compound, suggesting it is an intrinsic property of PAR2 rather than a 1-PPA specific effect. (Figure 3a).
3.3. 1-PPA inhibits platelet aggregation
Given the binding and stabilizing effect of the compound described in our simulations, we decided to investigate the antagonistic properties suggested by MD using a cellular assay. We have chosen the platelet aggregation assay as cellular model to unvail the properties exerted by 1-PPA on the receptor since platelets are the cell fragments with the highest level of PAR receptors on their membrane [9]. Cooperating with endothelial cells of the blood vessels, they start the coagulation cascade upon activation of PAR receptors by proteolytic cleavage of their N-terminal region [40]. In particular, PAR2 is activated by trypsin, tryptase, kallikreins, coagulation factors, but also by cathepsin-S or elastase.
Given the potential of 1-PPA as allosteric inhibitor of PAR2 and of the other family members, we have analyzed the degree of conservation in the four PARs isoforms, focusing our attention on the residues defining the pocket occupied by 1-PPA, discovered in our simulations (Figure 4).
The main residues forming H-bonds and establishing contacts with the ligand (Arg1212.50, Met1593.36, Phe3006.48, Thr3347.43) are all conserved or replaced by homologous amino acids except for Ser1242.53, which is substituted by hydrophobic residues in the other family members. Such high similarity between PARs is conserved also in the hortosteric pocket occupied by AZ8838, suggesting 1-PPA might display a broad-spectrum of activity toward this class of receptors and make platelets a good model to test its activity.
For this study platelets from plasma samples of healthy donors were stimulated with a thrombin substitute in presence or absence of different concentrations of 1-PPA. The presence of 1-PPA determined a progressive reduction of the level of aggregation compared to the untreated samples (Figure 5). The addition of 1-PPA reduced up to 50% platelet aggregation, suggesting that this small molecule might impact on the PAR receptors by blocking the cascade of events activated by such receptor family.
5. Conclusions
Targeting GPCR to regulate pathological conditions has been demonstrated to be one of the most valuable strategies in the pharmaceutical field in the last decades. Recent studies have shown the important role of Protease Activated Receptors, in particular of PAR2, which is involved in many inflammatory and autoimmune disease as well as in cancer. In this work we have demonstrated that it is possible to target this receptor with the small molecule 1-piperidinepropionic acid, previously described for its anti-inflammatory and anticancer properties.
Through direct interaction measurements, such as CETSA, we were able to describe a stabilizing effect induced by the presence of the compound, even at high temperature. Computational analysis allowed us to suggest an allosteric mechanism of action, that blocks the receptor in an inactive conformation. Cell based experiments, and comparison with the other members of the PAR family allowed the disclosure of a pan-inhibitor activity across all the members, due to the high conservation of the allosteric site.
Our work opens the route to further investigations on the impact of 1-PPA activity at the cellular level. Its potential role as an allosteric inhibitor of PAR2 encourages future studies to develop potential drugs for the treatment of inflammatory conditions and cancer progression.
6. Patents
Italian Patent Application N. 102022000014593 filed by the University of Padova on July 12, 2022; PTC/IB2023/057138 filed on July 12, 2023.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Suppl. Figure S1: Binding pockets and Docking of 1-PPA and AZ8838; Suppl. Figure S2: Movements of compounds during MD simulations.
Author Contributions
Conceptualization, L.C. and P.P.; methodology, M.C., L.C., L.S. and P.P..; software and formal analysis, M.C. and M.G.; investigation, M.C., M.G., S.Q., M.R., A.B., L.S.; resources, L.C. and P.P.; data curation, M.C., M.G., S.Q. and M.R.; writing—original draft preparation, M.C., M.G., L.C. and P.P.; writing—review and editing, M.C., M.G., S.Q., M.R., A.B. L.S., F.F., L.C. and P.P.; supervision L.C. and P.P.; funding acquisition, P.P. All authors have read and agreed to the published version of the manuscript.
Funding
Please add: This research was funded in part by the National Ministry of Health, grant number RF-2019-12369984 (P.P.) and by CaRiPaRo foundation (PhD fellowship to M.G.).
Informed Consent Statement
Informed consent was obtained from all healthy volunteers involved in the study.
Data Availability Statement
The data presented in this study are available upon request from the corresponding author.
Acknowledgments
We thank Angela Pavan and Filippo Vascon, respectively for their precious contribution in cell culture maintenance and critical review and suggestion on experiments and data graphical representation.
Conflicts of Interest
Pontisso P, Biasiolo A, Cendron L, Chinellato M, Quarta, S, Ruvoletto M. are inventors of the Patent Application of the University of Padova N. 102022000014593. No conflict of interest exists for the other authors.
References
- Hilger, D.; Masureel, M.; Kobilka, B.K. Structure and Dynamics of GPCR Signaling Complexes. Nature Structural & Molecular Biology 2017 25:1 2018, 25, 4–12. [Google Scholar] [CrossRef]
- Tesmer, J.J.G. Hitchhiking on the Heptahelical Highway: Structure and Function of 7TM Receptor Complexes. Nature Reviews Molecular Cell Biology 2016 17:7 2016, 17, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Song, G.; de Graaf, C.; Stevens, R.C. Structure and Function of Peptide-Binding G Protein-Coupled Receptors. J. Mol. Biol. 2017, 429, 2726–2745. [Google Scholar] [CrossRef]
- Hedderich, J.B.; Persechino, M.; Becker, K.; Heydenreich, F.M.; Gutermuth, T.; Bouvier, M.; Bünemann, M.; Kolb, P. The Pocketome of G-Protein-Coupled Receptors Reveals Previously Untargeted Allosteric Sites. Nat Commun 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Latorraca, N.R.; Venkatakrishnan, A.J.; Dror, R.O. GPCR Dynamics: Structures in Motion. Chem. Rev. 2017, 117, 139–155. [Google Scholar] [CrossRef]
- Miller-Gallacher, J.L.; ¤a, R.N.; Warne, T.; Edwards, P.C.; Schertler, G.F.X.; Leslie, A.G.W.; Tate, C.G.; Van Veen, H.W. The 2.1 Å Resolution Structure of Cyanopindolol-Bound Β1-Adrenoceptor Identifies an Intramembrane Na+ Ion That Stabilises the Ligand-Free Receptor. PLoS One 2014, 9, e92727. [Google Scholar] [CrossRef]
- Manglik, A.; Kobilka, B. The Role of Protein Dynamics in GPCR Function: Insights from the Β2AR and Rhodopsin. Curr. Opin. Cell Biol. 2014, 27, 136–143. [Google Scholar] [CrossRef]
- Chandrabalan, A.; Ramachandran, R.; Ramachandran, C.R. Molecular Mechanisms Regulating Proteinase-Activated Receptors (PARs). [CrossRef]
- Heuberger, D.M.; Schuepbach, R.A. Protease-Activated Receptors (PARs): Mechanisms of Action and Potential Therapeutic Modulators in PAR-Driven Inflammatory Diseases. Thromb J 2019, 17, 1–24. [Google Scholar] [CrossRef]
- Rasmussen, U.B.; Vouret-Craviari, V.; Jallat, S.; Schlesinger, Y.; Pages, G.; Pavirani, A.; Lecocq, J.-P.; Pouyssegur, J.; Van Obberghen-Schilling, E. DNA Cloning and Expression of a Hamster A-Thrombin Receptor Coupled to &la2 + Mobilization. 1991, 288. [CrossRef]
- Kanke, T.; Takizawa, T.; Kabeya, M.; Kawabata, A. Physiology and Pathophysiology of Proteinase-Activated Receptors (PARs): PAR-2 as a Potential Therapeutic Target. J Pharmacol Sci 2005, 97, 38–42. [Google Scholar] [CrossRef]
- Suhaj, P.; Olejar, T.; Matej, R.; Suhaj, P. PAR2: The Cornerstone of Pancreatic Diseases. [CrossRef]
- Cheng, R.K.; Fiez-Vandal, C.; Schlenker, O.; Edman, K.; Aggeler, B.; Brown, D.G.; Brown, G.A.; Cooke, R.; Dumelin, C.E.; Doré, A.S.; et al. Structural Insight into Allosteric Modulation of Protease-Activated Receptor 2. 2017. [CrossRef]
- Kennedy, A.J.; Sundström, L.; Geschwindner, S.; Poon, E.K.Y.; Jiang, Y.; Chen, R.; Cooke, R.; Johnstone, S.; Madin, A.; Lim, J.; et al. Protease-Activated Receptor-2 Ligands Reveal Orthosteric and Allosteric Mechanisms of Receptor Inhibition. [CrossRef]
- Pontisso, P.; Biasiolo, A.; Cappon, A.; Martini, A.; Quarta, S.; Ruvoletto, M.; Turato, C.; Villano, G. Università degli Studi di Padova. Italian Patent N.102017000026858 issued July 3, 2019, US Patent N.11,628,163 issued April 18, 2023. [Google Scholar]
- Pontisso, P.; Biasiolo, A.; Martini, A.; Quarta, S.; Ruvoletto, M.; Turato, C.; Villano, G. Università degli Studi di. Padova. Patent IT102019000012930 issued July 14, 2021, PTC pending.
- Yau, M.K.; Lim, J.; Liu, L.; Fairlie, D.P. Protease Activated Receptor 2 (PAR2) Modulators: A Patent Review (2010–2015). 2016, 26, 471–483. [Google Scholar] [CrossRef]
- Kawatkar, A.; Schefter, M.; Hermansson, N.-O.; Snijder, A.; Dekker, N.; Brown, D.G.; Lundbä, T.; Zhang, A.X.; Castaldi, M.P. CETSA beyond Soluble Targets: A Broad Application to Multipass Transmembrane Proteins. 2019. [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Schrödinger, LLC The {PyMOL} Molecular Graphics System, Version~1.8. 2015.
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J Mol Biol 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed Atlas of Surface Topography of Proteins. Nucleic Acids Res 2018, 46, W363–W367. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. Fast Docking Using the CHARMM Force Field with EADock DSS. J Comput Chem 2011, 32, 2149–2159. [Google Scholar] [CrossRef]
- Lien Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a Protein-Small Molecule Docking Web Service Based on EADock DSS. [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A Message-Passing Parallel Molecular Dynamics Implementation. Comput Phys Commun 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J Comput Chem 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Soteras Gutiérrez, I.; Lin, F.-Y.; Vanommeslaeghe, K.; Lemkul, J.A.; Armacost, K.A.; Brooks, C.L.; MacKerell, A.D. Parametrization of Halogen Bonds in the CHARMM General Force Field: Improved Treatment of Ligand–Protein Interactions. Bioorg Med Chem 2016, 24, 4812–4825. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; MacKerell, A.D. Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing. J Chem Inf Model 2012, 52, 3144–3154. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell, A.D. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of Bonded Parameters and Partial Atomic Charges. J Chem Inf Model 2012, 52, 3155–3168. [Google Scholar] [CrossRef]
- Zorzan, M.; Castellan, M.; Gasparotto, M.; de Melo, G.D.; Zecchin, B.; Leopardi, S.; Chen, A.; Rosato, A.; Angelini, A.; Bourhy, H.; et al. Antiviral Mechanisms of Two Broad-Spectrum Monoclonal Antibodies for Rabies Prophylaxis and Therapy. Front Immunol 2023, 14, 1186063. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J Chem Phys 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Bernetti, M.; Bussi, G. Pressure Control Using Stochastic Cell Rescaling. J Chem Phys 2020, 153, 114107. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N·log(N) Method for Ewald Sums in Large Systems. J Chem Phys 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J Chem Phys 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Braun, S.; Jawansky, S.; Vogt, W.; Mehilli, J.; Schömig, A.; Kastrati, A.; vonBeckerath, N.; DirkSibbing, D. Assessment of ADP-Induced Plateletaggregationwithlight Transmission Aggregometryand Multiple Electrodeplatelet Aggregometrybeforeand Afterclopidogrel Treatment Platelets AndBlood Cells. ThrombHaemost 2008, 99, 121–126. [Google Scholar] [CrossRef]
- Thal, D.M.; Glukhova, A.; Sexton, P.M.; Christopoulos, A. Structural Insights into G-Protein-Coupled Receptor Allostery. Nature 2018. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Weinstein, H. Integrated Methods for the Construction of Three-Dimensional Models and Computational Probing of Structure-Function Relations in G Protein-Coupled Receptors. In Methods in Neurosciences; Academic Press, 1995; Vol. 25, pp. 366–428.
- Schwartz, R.; Ruthstein, S.; Major, D.T. Molecular Dynamics Simulations of the Apo and Holo States of the Copper Binding Protein CueR Reveal Principal Bending and Twisting Motions. Journal of Physical Chemistry B 2021, 125, 9417–9425. [Google Scholar] [CrossRef]
- Camerer, E.; Qazi, A.A.; Duong, D.N.; Cornelissen, I.; Advincula, R.; Coughlin, S.R. Platelets, Protease-Activated Receptors, and Fibrinogen in Hematogenous Metastasis. Blood 2004, 104, 397–401. [Google Scholar] [CrossRef]
Figure 1.
CETSA assay on PAR2 in absence and presence of 1-PPA. (a) PAR2 thermal denaturation performed on live cells shows a decrease in soluble protein as the temperature increase. Data of protein content were initially normalized on Vinculin content and then on the untreated sample (38°C); (b) 1-PPA at increasing concentration sorts a solubilizing effect on PAR2 even at temperature higher of the determined Tagg. No effects are sorted on the Vinculin, which remains overall constant for each 1-PPA concentration. Therefore, data were normalized on Vinculin levels and then normalized on the highest concentration of 1-PPA (125 µM).
Figure 1.
CETSA assay on PAR2 in absence and presence of 1-PPA. (a) PAR2 thermal denaturation performed on live cells shows a decrease in soluble protein as the temperature increase. Data of protein content were initially normalized on Vinculin content and then on the untreated sample (38°C); (b) 1-PPA at increasing concentration sorts a solubilizing effect on PAR2 even at temperature higher of the determined Tagg. No effects are sorted on the Vinculin, which remains overall constant for each 1-PPA concentration. Therefore, data were normalized on Vinculin levels and then normalized on the highest concentration of 1-PPA (125 µM).
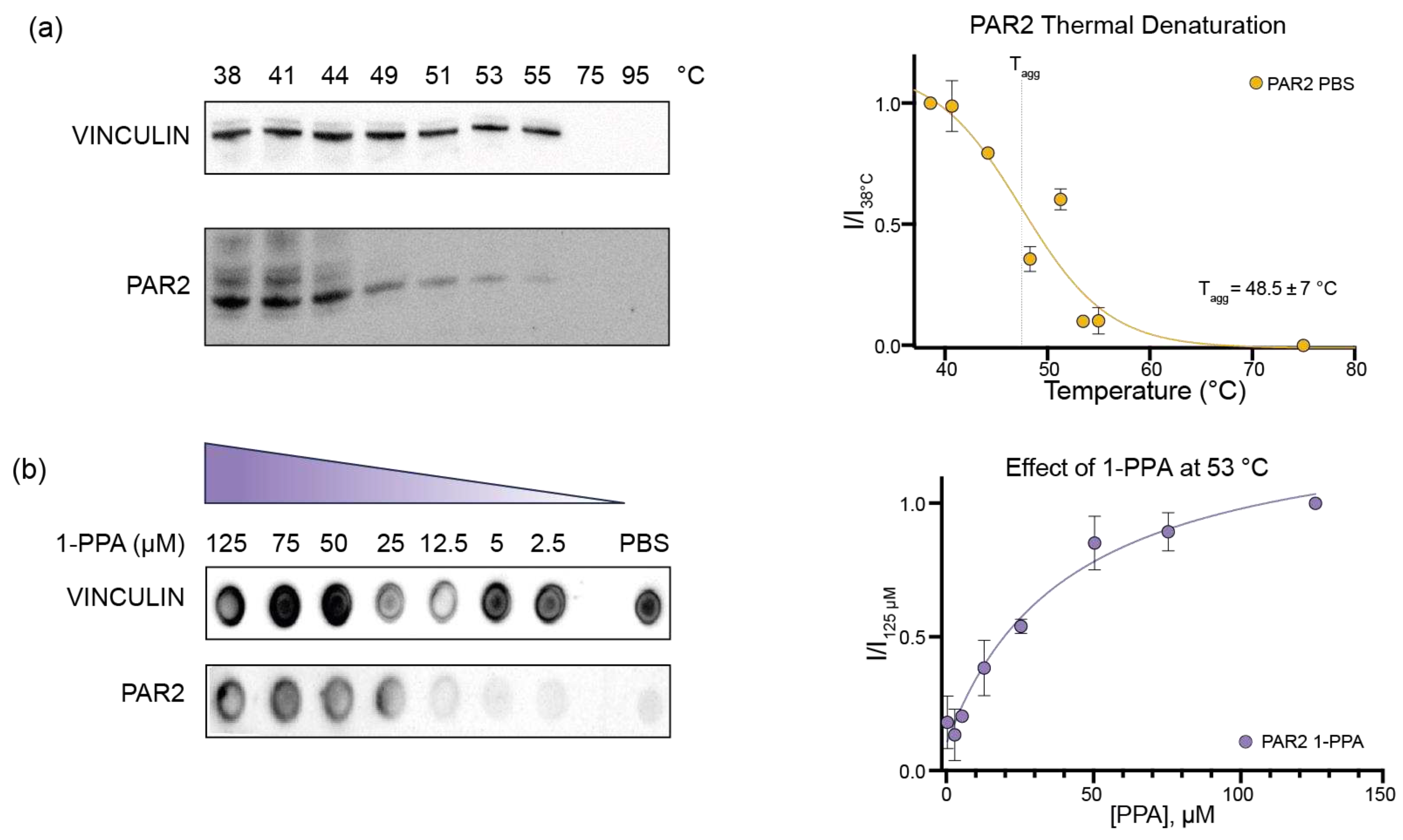
Figure 2.
1-PPA binding site in PAR2 based on Molecular Dynamics. (a) wtPAR2 model of the transmembrane domain (Val61-Arg362) in white. Among the 7TMD, 1-PPA and AZ8838 are represented in purple and light green, respectively. AZ8838 is positioned in the orthostatic pocket as described in 5NDD, whereas 1-PPA is positioned in the allosteric pocket above the Na+ pocket; (b) Cut-through representation of the binding pockets, PAR2 surface is showing in gray while 1-PPA e AZ8838 are indicated by the arrows; (c) Top view of the allosteric pocket containing 1-PPA (in purple) residues interacting with it are shown as white sticks and are indicated with Ballesteros-Weinstein numeration. Hydrogen bonds formed with D121 and S124 are shown as black dashed lines. Na+ ion is shown in green; (b) Schematic representation of 1-piperidinepropionic acid structure and molecular weight (MW).
Figure 2.
1-PPA binding site in PAR2 based on Molecular Dynamics. (a) wtPAR2 model of the transmembrane domain (Val61-Arg362) in white. Among the 7TMD, 1-PPA and AZ8838 are represented in purple and light green, respectively. AZ8838 is positioned in the orthostatic pocket as described in 5NDD, whereas 1-PPA is positioned in the allosteric pocket above the Na+ pocket; (b) Cut-through representation of the binding pockets, PAR2 surface is showing in gray while 1-PPA e AZ8838 are indicated by the arrows; (c) Top view of the allosteric pocket containing 1-PPA (in purple) residues interacting with it are shown as white sticks and are indicated with Ballesteros-Weinstein numeration. Hydrogen bonds formed with D121 and S124 are shown as black dashed lines. Na+ ion is shown in green; (b) Schematic representation of 1-piperidinepropionic acid structure and molecular weight (MW).
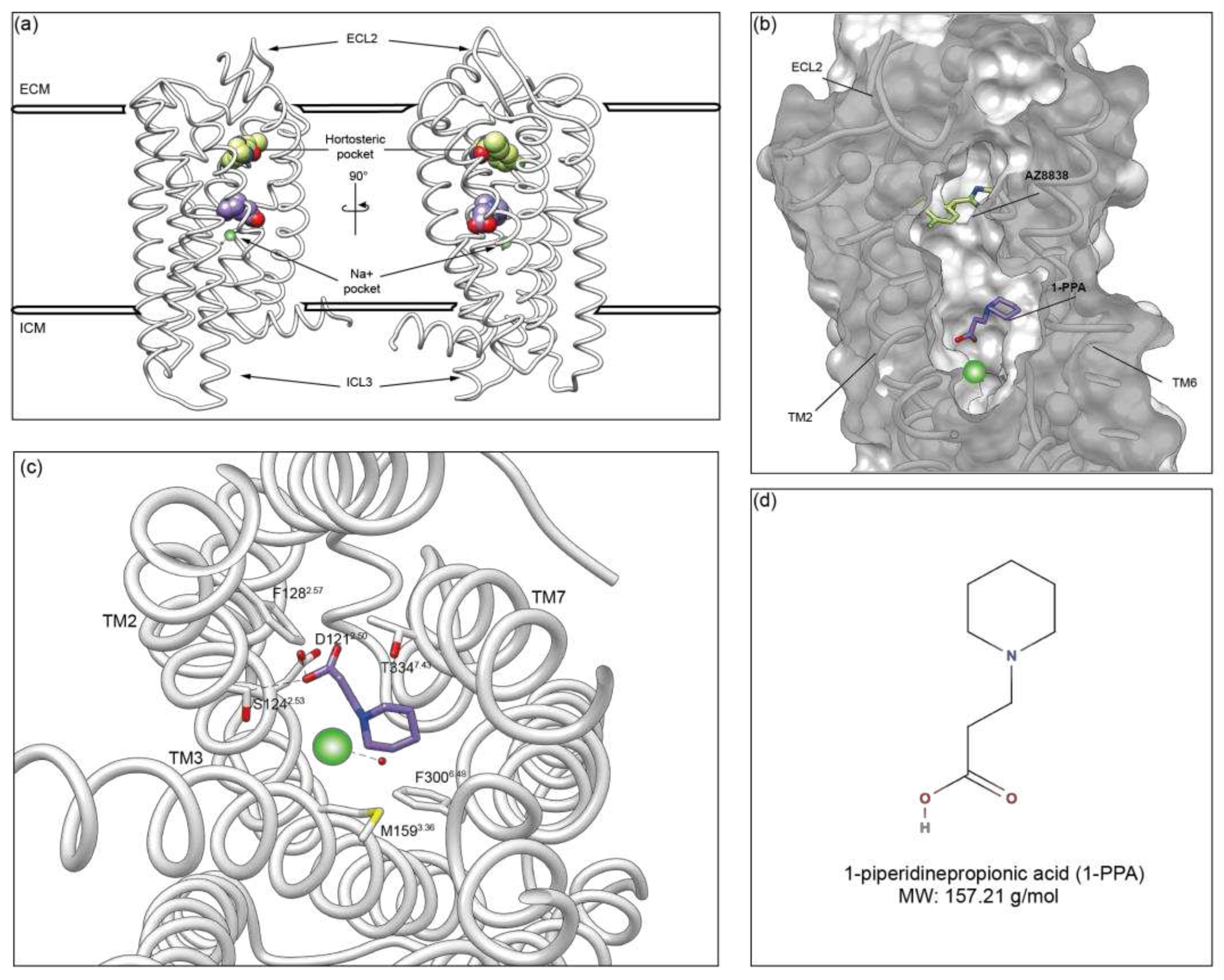
Figure 3.
Molecular Dynamics analysis. (a) Sliced view of wtPAR2 model shows the 3 pockets previously identified in the S0 (Initial State), in yellow, of the molecular dynamics compared to the model that underwent the simulation (S1, Final State) in purple. Pocket 1, or entrance, at the end of the simulation is united with the other two in a single funnel; (b) Sliced of wtPAR2 model, in white, shows the engulfment of 1-PPA (purple) after the simulations, in the allosteric site given by the allosteric pocket 2; (c) Cross-correlation matrix showing that the modification apported to wtPAR2 are more similar between AZ8838 and 1-PPA indicating a similar modulation toward a stabilized structure; (d) RMSF plot representing the residue mobility, dotted lines shows the portion of the ECL2 while continuous lines box the ICL2-3; (e) RMSD variation during simulation time shows no significant variation in each model; (f) Distance variation of each compound respect the initial position shows that AZ8838 does not move from the orthosteric pocket, while negative control Ro5-4864 and 1-PPA move to another position. .
Figure 3.
Molecular Dynamics analysis. (a) Sliced view of wtPAR2 model shows the 3 pockets previously identified in the S0 (Initial State), in yellow, of the molecular dynamics compared to the model that underwent the simulation (S1, Final State) in purple. Pocket 1, or entrance, at the end of the simulation is united with the other two in a single funnel; (b) Sliced of wtPAR2 model, in white, shows the engulfment of 1-PPA (purple) after the simulations, in the allosteric site given by the allosteric pocket 2; (c) Cross-correlation matrix showing that the modification apported to wtPAR2 are more similar between AZ8838 and 1-PPA indicating a similar modulation toward a stabilized structure; (d) RMSF plot representing the residue mobility, dotted lines shows the portion of the ECL2 while continuous lines box the ICL2-3; (e) RMSD variation during simulation time shows no significant variation in each model; (f) Distance variation of each compound respect the initial position shows that AZ8838 does not move from the orthosteric pocket, while negative control Ro5-4864 and 1-PPA move to another position. .
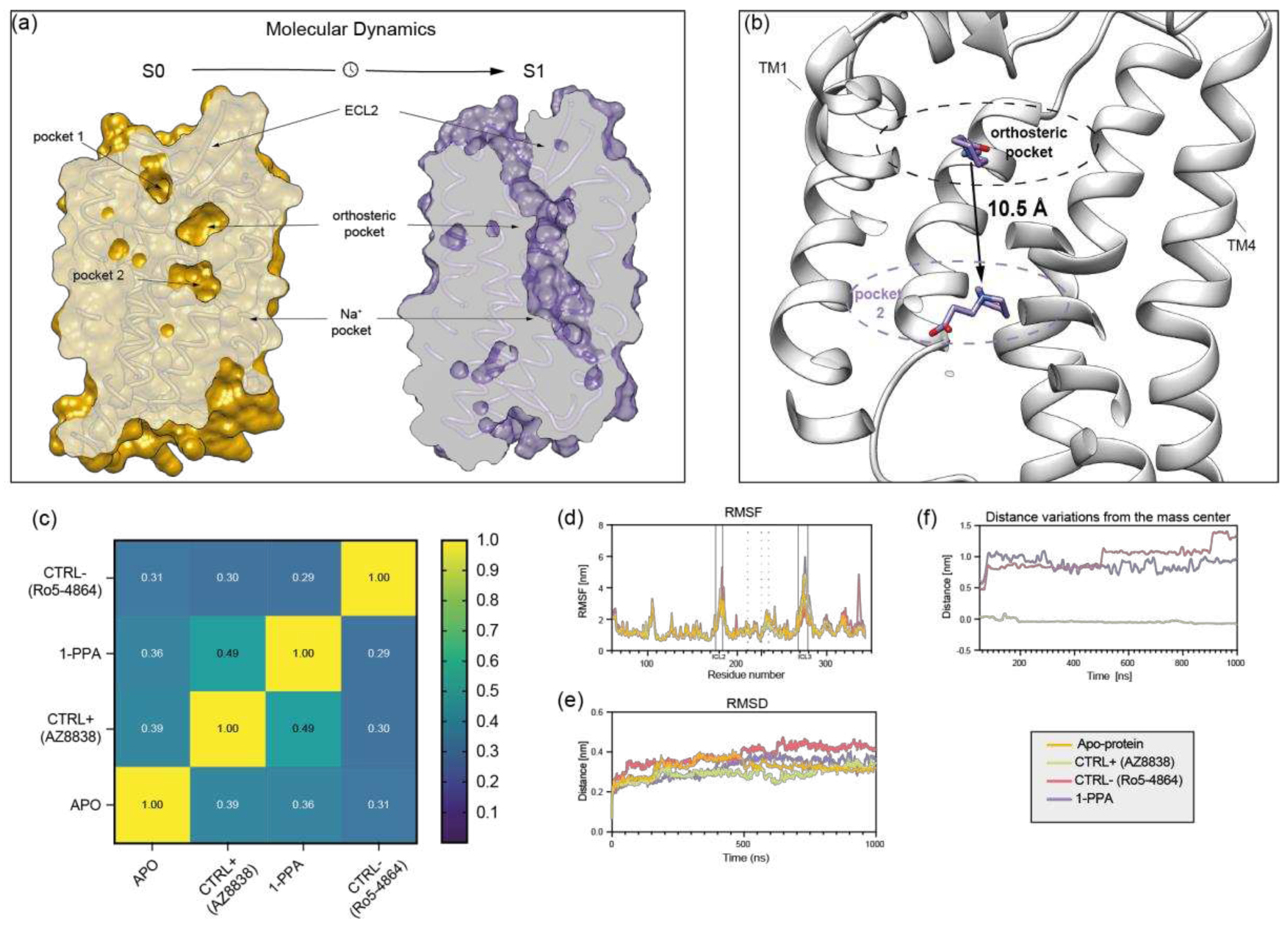
Figure 4.
PAR family sequence alignment. ClustalO alignment of sequences from Protease Activated Receptor family members. Purple boxes highlight the main residues involved in the interaction between PAR2 and 1-PPA. Lined boxes refer to residues that are fully conserved in the other proteins in the same position, while pale blue boxes identify residues conserved for their side chain properties. Gray boxes indicate residues, in the binding site, that completely diverge compared to PAR2.
Figure 4.
PAR family sequence alignment. ClustalO alignment of sequences from Protease Activated Receptor family members. Purple boxes highlight the main residues involved in the interaction between PAR2 and 1-PPA. Lined boxes refer to residues that are fully conserved in the other proteins in the same position, while pale blue boxes identify residues conserved for their side chain properties. Gray boxes indicate residues, in the binding site, that completely diverge compared to PAR2.
Figure 5.
Platelet aggregation assay. (a) TRAPtest plot showing the reduction of the platelet aggregation level during time compared to the reference level of untreated platelets; (b) Histograms representing the percentage of aggregation in absence or in presence of different concentrations of 1-PPA.
Figure 5.
Platelet aggregation assay. (a) TRAPtest plot showing the reduction of the platelet aggregation level during time compared to the reference level of untreated platelets; (b) Histograms representing the percentage of aggregation in absence or in presence of different concentrations of 1-PPA.
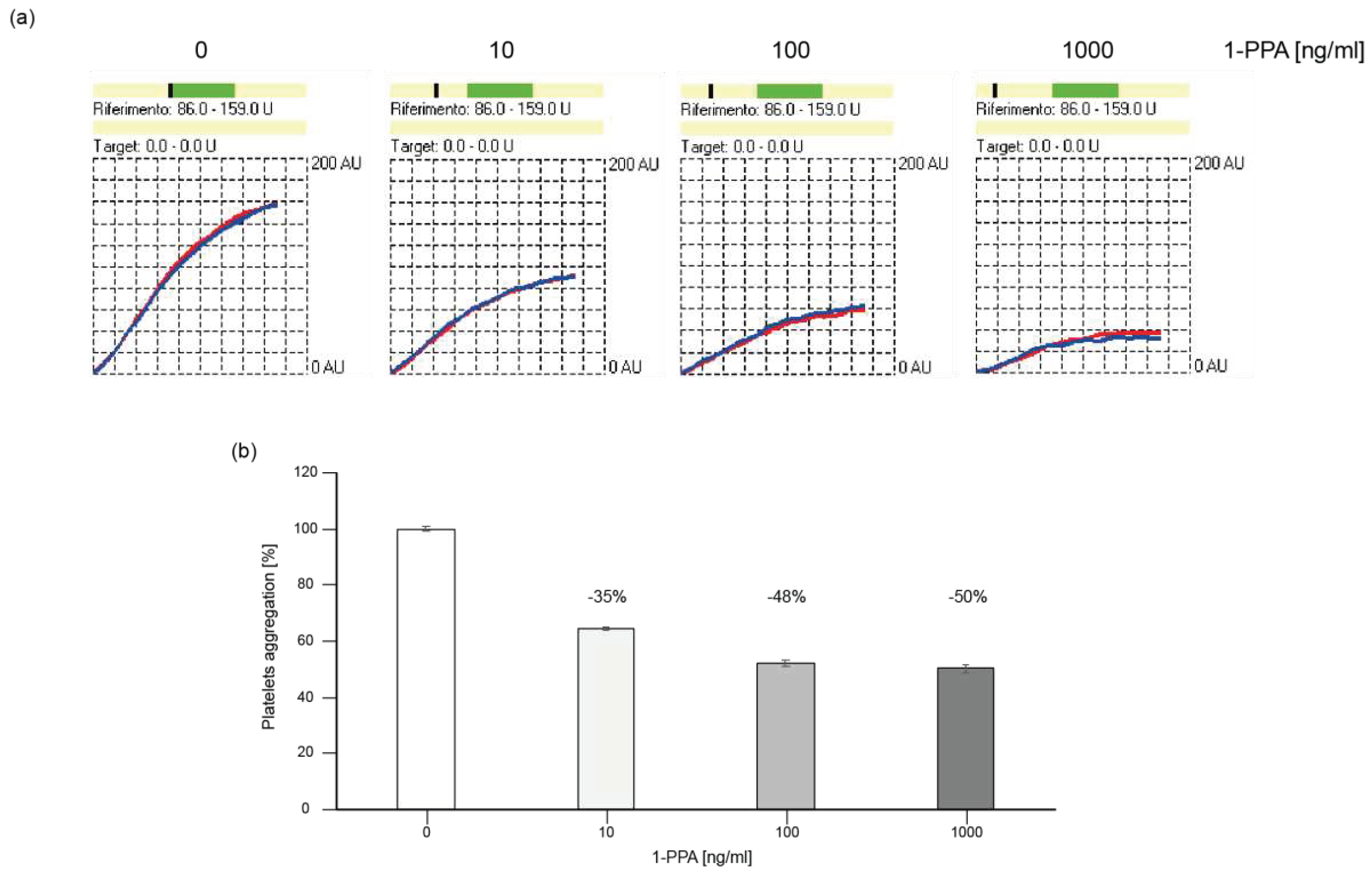

Table 1.
Antibodies applied for Western and Dot Blot analysis.
| Target | Dilution | Code | Producer |
|---|---|---|---|
| hPAR2 (HA tag) | 1:5000 | 2-2.2.14 | Invitrogen |
| Vinculin | 1:5000 | HL1964 | GTX637857 |
| Mouse IgG | 1:10000 | A16066 | Invitrogen |
| Rabbit IgG | 1:10000 | SSA004 | SinoBiological |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



