
Submitted:
18 July 2023
Posted:
19 July 2023
You are already at the latest version
Abstract
The interest on amorphous carbon thin films with low secondary electron yield (SEY) has been increasing in the last years, to mitigate electron multipacting in particle accelerators and in RF devices. Previous works found that the SEY increases with the hydrogen amount and correlates with the Tauc gap. In this work, we analyse films produced by magnetron sputtering with different contents of hydrogen and deuterium, incorporated via target poisoning and sputtering of CxDy molecules. XPS was implemented to estimate the phase composition of the films. The maximal SEY was found to decrease linearly with the fraction of the graphitic phase in the films. These results are supported by Raman scattering and UPS. The graphitic phase decreases almost linearly for hydrogen and deuterium concentrations between 12% and 46% (at.), but abruptly decreases when the concentration reaches 53%. This vanishing of the graphitic phase is accompanied by a strong increase of SEY and of the Tauc gap. These results suggest that the SEY is not dictated directly by the concentration of H/D but by the fraction of graphitic phase. The results are supported by a model used to calculate SEY of films consisting of a mixture of graphitic and polymeric phases.
Keywords:
amorphous carbon
; particle accelerators
; SEY
; XPS
; Raman spectroscopy
1. Introduction
The emission of secondary electrons from surfaces plays a key role on the generation of electron multipacting in high power radio frequency devices and particle accelerators with positively charged beams. In particle accelerators, it can result in the formation of clouds of electrons, causing beam instabilities, deterioration of the vacuum, or heat loads to cryogenics parts of the system, limiting the overall efficiency of the accelerator (i.e. the beam luminosity) [1,2,3,4,5,6]. A possible cure to this problem is to reduce the electron emission by coating the internal surfaces of the vacuum chambers with a thin film of low Secondary Electron Yield (SEY) material. Amorphous carbon (a-C) coatings have been successfully used in the Super Proton Synchrotron at CERN (European Organisation for Nuclear Research) to mitigate the electron multipacting [7], and it is now in the baseline for the High Luminosity upgrade of the Large Hadron Collider, (CERN, Switzerland), and the Electron Ion Collider, (Brookhaven National Laboratory, USA) [8,9].
Secondary electron emission can be conveniently described as follows [10]. Primary electrons that penetrate a material mainly lose their energy by different types of electron excitations (via plasmon excitations or by a direct energy transfer to valence and core electrons). This results in the generation of secondary electrons inside the material, with energy above the vacuum level. On their way out, these internal secondary electrons can efficiently lose their energy only by excitation of valence electrons. Finally, once they reach the surface, only the internal secondary electrons which still have energy above the vacuum level have a chance to be emitted. The whole process is strongly affected by the electronic structure of the material, which determines the energy loss processes of both primary and internal secondary electrons. Particularly important is the latter, which can be reduced by opening the energy gap. This is the origin of the high SEY of dielectric materials [11].
The efficiency of a-C coatings for suppressing electron multipacting depends on structural properties: only coatings with pronounced electrical conductivity can have a low SEY. It has been well established that the presence of hydrogen impurities is particularly harmful in that respect [12,13]. The effects of hydrogen and nitrogen impurities on the secondary electron emission properties of a-C coatings were a subject of two recent studies [14,15]. It was clearly demonstrated that adding hydrogen or deuterium in the discharge gas increases the SEY of carbon films, while the presence of nitrogen has an opposite effect. By combining SEY measurements with Ion Beam Analysis (IBA), a correlation between the deuterium content and the maximum SEY (SEYmax) was established. SEYmax increases linearly, from 1 to 1.4, with the overall hydrogen and deuterium (H + D) relative content up to 47%. Further increase of the H + D amount up to 54% is accompanied by a steep SEYmax growth to 2.2. The samples were also investigated by Optical Absorption Spectroscopy (OAS), enabling estimation of the optical energy gap using the procedure established by Tauc (so-called Tauc gap). These results reveal a strong correlation between the SEYmax and the Tauc gap, apparently offering a straightforward explanation for the SEYmax increase. However, a-C films are often non-uniform, i.e., they may consist of various regions with different composition and electronic structure (e.g., graphitic, diamond-like and hydrocarbon domains, the latter being a result of highly common hydrogen contamination) [16]. Such materials have different local energy gaps, limiting the interpretation of Tauc plots [17].
Robertson summarized measurements of the Tauc gap performed on different types of a-C (pure and hydrogenated) in his seminal review paper [18]. He concluded that the Tauc gap of a film is not determined by the hydrogen content, but by the relative amount and properties of the sp2 carbon phase. Local energy gaps of diamond-like and various hydrocarbon phases are too large to be related with a Tauc gap below ≈2 eV. The gap is therefore related to the configuration of π states on the sp2 sites. In a planar cluster model, the band gap of a given cluster is inversely proportional to the square root of the number of the hexagonal rings in the cluster [19]. Different types of defects (e.g., formation of pentagons or heptagons) will also open the gap. From this perspective, deviation from the linear dependence at low photon energies in Tauc plots should not be attributed to the so-called tail states (i.e., Urbach tail) but to the small quantity of graphitic clusters with small local energy gaps.
The abrupt increase of SEYmax, when the H+D relative concentration changes from 47% to 54%, suggests that the hydrogen content may not be directly responsible for the change of the secondary electron emission (SEE) properties of a-C. This doubt is further supported by the high correlation between the SEYmax and the Tauc gap [14,15], knowing that the Tauc gap can be independent of the hydrogen concentration [18]. This aspect motivates our study: since the Tauc gap in a-C is known to be affected by the concentration and size of graphitic domains, could the latter also influence the SEE?
In our recent work we performed deposition of a-C coatings using magnetron sputtering in Ar discharges with several fractions of D2, identified the mechanism behind the incorporation of deuterium in the films during the production phase and its impact on the SEY [15]. In this work we report detailed analysis of the same samples by means of different electron and vibrational spectroscopic techniques. X-ray and UV Photoelectron Spectroscopies (XPS and UPS, respectively) were used to determine the surface composition, to identify different phases in the a-C films, and obtain information on their electronic structure. Further insights are provided by Raman scattering, Fourier Transform Infrared Spectroscopy (FTIR) and High Resolution Electron Energy Loss Spectroscopy (HREELS). Raman scattering, enabling the detection of the presence of graphitic carbon, was particularly valuable in supporting the XPS results. FTIR and HREELS were used to identify the character of C-D bonds in the most contaminated samples. These results are then compared with the corresponding SEY, OAS and IBA measurements, enabling us to reveal the actual mechanism behind the SEY increase by the hydrogen contamination.
2. Materials and Methods
All details of the thin film deposition procedure are explained in [15]. Briefly, the films were deposited by magnetron sputtering from a 50 mm diameter graphite target. The total operating pressure was set to 2 Pa, consisting of Ar and small quantities of D2, to study deuterium incorporation into the films. The substrates were mounted 93 mm away from the graphite target and the discharge power fixed at 30 W. Five sets of samples were deposited on Si single crystal and quartz substrates for different characterisations: the reference (without D2 added to the discharge gas), 0.2D (0.2 vol% of added D2), 0.5D (0.5 vol% of D2), 1D (1.3 vol% of D2) and 10D (10.9 vol% of D2). D2 was added to distinguish deliberately introduced contaminants from hydrogen contamination from the residual gas. The system was baked prior to each run for 24 h at 230 °C to minimize the natural contamination, keeping the residual gas pressure in the low 10-6 Pa range or better (N2 equivalent). A new graphite target was used for each run. The deposition rate was 10-15 nm/h, generally growing with the D2 partial pressure, pD2. The thin film thicknesses are in the range from 486–719 nm (measured by SEM). The samples were transferred to different laboratories in stainless steel vacuum chambers, pre-evacuated by a turbomolecular pump and filled with N2 gas.
UPS and XPS measurement s of ‘as-received’ samples were performed on AXIS SUPRA setup (Kratos Analytical), containing both UPS and monochromatic XPS sources. XPS measurements were performed using the monochromatic Al Kα line (photon energy of 1486.7 eV), and the spectrometer pass energy of 80 eV (survey spectra) and 5 eV (high resolution spectra). UPS measurements were performed by means of the He I α line (photon energy of 21.22 eV) and the spectrometer pass energy of 5 eV. The binding energy scale for UPS and XPS measurements were both calibrated using a sputter-cleaned Ag sample, based on the position of the Ag 3d5/2 line and of the top of the valence band. Fitting of graphitic contributions was based on the XPS measurements of freshly cleaved Highly Oriented Pyrolytic Graphite (HOPG). A correction of the raw UPS spectra was made by removing the contributions of the He I β line (energy shift of 1.87 eV, intensity of 1.2% with respect to the He I α line). All samples were conductive, apart from the sample 10D, which was measured with charge compensation using an electron flood gun.
Raman spectroscopy measurements were carried out at room temperature in a backscattering geometry on an alpha 300 R confocal Raman microscope (WITec) using a 532 nm Nd:YAG laser (2nd harmonic) as well as a 633 nm He-Ne laser for excitation. The laser beam with a power of 0.7 mW was focused on the sample by a ×50 lens (Zeiss), providing a spot with a diameter of about 1 μm. The spectra were collected with a 600 groove/mm grating using 5 acquisitions with a 2 s acquisition time. The same setup was used to perform photoluminescence measurements with two excitation wavelengths, 532 nm and 633 nm. For each sample, the spectra were acquired on several spots to check their lateral uniformity.
FTIR measurements were performed in vacuum in the wave number range 500-5000 cm-1, using a Vertex 80v system (Bruker) in conventional reflection and transmission mode, as well as in the attenuated total reflection (ATR) configuration using a Ge crystal.
HREELS measurements were performed in a UHV system (operating pressure in the 10-7 Pa range) using a LK Technologies 2000R spectrometer. Further details on this experimental setup are provided in the Appendix A, together with the experimental results.
3. Results
3.1. Composition, Electron Emission and Optical Properties of the Coatings
In this section, we summarise the main results of the thin films obtained by different techniques and reported in [15]. Thin film composition analysis performed by IBA revealed the presence of C, D, H and O uniformly distributed along the film depth, as confirmed by Secondary Ion Mass Spectrometry measurements. Results of the quantitative composition, the mass densities of the films, estimated from the IBA measurements and the film thicknesses (obtained by Scanning Electron Microscopy), as well as the results of SEY and Tauc gap (ET) measurements are summarised in Table 1. The amount of incorporated deuterium steadily increases with D2 partial pressure in the discharge gas (pD2), getting relatively close to the theoretical maximum of 65 at.%, corresponding to saturated hydrocarbons. The dependence of the maximum secondary electron yield (SEYmax) on pD2 in the films has a distinctively different trend: an increase of the H/D amount from 10 to 46 at.% changes SEYmax from 0.99 to 1.38, while additional 7 at.% of H/D increases the SEYmax abruptly to 2.2. SEYmax and the Tauc gap are strongly correlated. It appears that small changes of the D/H content in samples 1D and 10D are accompanied by the very different electronic structures, which govern both the secondary electron emission and optical properties. The latter is also clear from the fact that the sample 1D was conductive in contrast to the sample 10D. Namely the latter motivated usage of different approaches for the SEY measurements of the two samples [15].
From the mass densities and the Tauc gaps of the films we can already make a first estimation of their structure using general guidelines summarized in [20] and by considering that deuterated carbon films are generally somewhat denser than the equivalent hydrogenated ones. It appears that the reference, 0.2D and 0.5D samples, with their Tauc gaps clearly lower than 1 eV, are most probably dominated by graphitic and partially deuterated graphite-like (a-C:D) regions. The sample 1D with ET of 1.38 eV, high density, and large H+D content, might be closest to diamond-like a-C:D. Finally, the sample 10D, characterised by a low density and ET > 2 eV, could be a typical example of polymer-like a-C:D.
3.2. XPS Measurements
Apart from carbon, the survey XPS spectra of all samples show the presence of oxygen at the surface, estimated to be about 8 at.% in all cases. The samples were not charging during the measurements apart from the sample 10D, which was measured utilising an electron flood gun for charge compensation. The O 1s line in the first four samples was situated at 532.6 eV. Assuming that the O 1s line position is the same in the sample 10D, the latter was used as a binding energy reference for that sample.
Hydrogenated/deuterated a-C is expected to be a mixture of graphitic (pure sp2 hybridised) carbon, diamond-like carbon and different hydrocarbons in which carbon could be both in sp2 and sp3 hybridisation. Resolving these distributions in the C 1s photoelectron line is a complex problem due to their quite small chemical peak shifts, as extensively discussed in the literature [21,22,23,24]. The graphitic contribution is located at 284.3-284.5 eV, while a pure sp3 C contribution is usually encountered at about 285.2 eV [22]. The latter feature overlaps with different hydrocarbon contributions, around 285.0 eV, while at higher binding energies, further contributions related to various carbon bonds with O can be expected [24,25]. All peaks should be symmetric apart the one from the pure graphitic contribution, which is highly asymmetric [23,24]. The peak model for the graphitic contribution could be established by measuring the C 1s photoelectron line of the freshly cleaved Highly Oriented Pyrolytic Graphite (HOPG). Unfortunately, the asymmetry of the graphitic contribution is becoming less pronounced with the reduction of the cluster size and with the increase of the amount of defects in this phase. All this makes bond identification from the C 1s line fitting a challenging task without a straightforward solution. However, the elemental O concentration can be used as an additional constraint, which the carbon-oxygen bond contributions in the C 1s line need to follow. This approach, applied for all samples, except for the 10D (see below), strengthens the reliability of the interpretation of the spectra taken from the a-C coatings.
High resolution spectra of the C 1s photoelectron line of a freshly cleaved HOPG and the five samples considered in this study, together with the corresponding fittings, are presented in Figure 1. In all cases the background was calculated using the Shirley algorithm. As in [14], we again observe that the C 1s line becomes more symmetric with the increase of the D content in the films. One possible explanation of this trend is that the relative amount of graphitic C is being reduced and/or that the clusters are getting smaller with the increase of the D content.
The C 1s spectrum of HOPG (Figure 1a) was fitted using three contributions. The main peak (graphite), at 284.45 eV has asymmetric profile with a nomenclature LA(1.2, 2.5, 5) in CasaXPS software [26] and full width at half maximum (FWHM) of 0.34 eV. It should be stressed that the same peak profile was used by M. Biesinger, failing to secure perfect agreement with the measured spectrum [24]. Since a similar problem appeared in our spectra, we introduced the second peak (graphite sat), with the same profile and a FWHM of 1.3 eV. This peak is shifted with respect to the first towards higher binding energies by 1.47 eV, while their intensity ratio is 0.106. Finally, there is a well-known wide symmetric peak at about 291.2 eV attributed to the π-π* satellite always present in spectra of pure graphite [23,24].
It might be tempting to interpret the peak graphite sat as evidence of defects, such as sp3 carbon or C-H bonds. However, one should have in mind that such contributions would be symmetric. The asymmetry of this contribution would then imply non-negligible presence of other contaminants (typically O). Knowing that XPS is distinctively more sensitive to such contaminants than to C [27], the corresponding photoelectron lines would be clearly observable. Since this is not the case, we assume that both contributions originate from perfect graphite. They are introduced to better describe the shape of the C 1s line of HOPG, without attributing any physical interpretation to this separation. In our fittings of the graphitic contribution, we fixed the relative positions of the first two peaks and kept their intensity ratio approximately constant (see below). The intensity of the third peak was considered to be a free parameter (the latter is affected by the electronic structure and size of the graphitic clusters). The profile of all symmetric peaks (attributed to less conductive phases in a-C films) was of pseudo-Voigt type GL(30). They are attributed to sp3 carbon and C-H(D) bonds with both sp2 and sp3 carbon (284.8-285.2 eV), C-O (≈286.6 eV), C=O (287.8-288.2 eV) and O-C=O (≈289.0 eV) bonds [22,25]. All symmetric peaks within one spectrum have FWHM fixed to the same value.
The essential element of the fitting procedure was to maintain the carbon-oxygen bonds of C 1s in accordance with the O:C ratio obtained from the quantitative analysis, being 8±0.8% for all samples. In this approach we consider that the C:O stoichiometric ratio is 1:1 for the C-O and C=O contributions and 2:1 for the COO contribution. Consistency between the composition and the C 1s fitting could only be achieved by successively decreasing the asymmetry of the graphitic line as the H+D amount increases. The degree of asymmetry is characterised here by the ratio of the parameters α and β of the line shape LA(α,β,m), which determine the spread of the tail on the high and low binding energy side, respectively (α/β being below 1 for asymmetric line shapes). In addition, final asymmetry tuning of the graphitic contribution was performed by slight modification of the intensity ratio between the main peak and the graphite sat in the range 0.078 – 0.106. This peak fitting model did not provide consistent results only for the sample 10D, as can be seen in Table 2 where the XPS fitting results are summarised. For this sample, the oxygen content is overestimated, most probably due to the charging problems that affected the shape of the C 1s line.
Fitting results of the C 1s line taken from the 5 samples and the relative content of hydrogen and deuterium taken from Table 1 are summarized in Table 2. The following general trends with the increase of the H+D relative content appear:
- The relative contribution of the graphitic component decreases and even disappears in the sample 10D;
- The relative contribution attributed to the sp3 carbon and the hydrocarbons increases;
- The asymmetry of the graphitic component becomes less pronounced.
Therefore, increasing the amount of incorporated H/D in the films is accompanied by the higher content of hydrocarbons and reduction of the graphitic content. Besides, the graphitic regions are becoming smaller and/or with higher defect concentration.
At this point, total disappearance of the graphitic content in the sample 10D is not certain. We can only claim that there is no need to introduce the asymmetric graphitic component to obtain a good fit of the spectrum presented in Figure 1f. Since the sample 10D was charging, one could expect a deformation of the line shape. Moreover, the latter certainly took place since it was not possible to perform a good fit that would justify the correct content of the oxygen amount. While one cannot reliably exclude the presence of some graphitic regions in that sample, their relative amount most probably does not exceed ≈10%.
3.3. UPS Measurements
In Figure 2 we present UPS spectra close to the Fermi level (binding energy of 0 eV) taken from the reference 0.2D, 0.5D and 1D samples, as well as of the freshly cleaved HOPG. The spectra are normalised with respect to their intensities at 2 eV. The same structure cannot be reliably taken from the sample 10D due to the charging problems.
The very top of the valence band of HOPG (binding energy range 0-1 eV) can be attributed to π electrons from the K point of the Brillouin zone [28]. Since the diamond-like and hydrocarbon regions have energy gaps well above 2 eV [18], the presence of the signal in other samples is additional evidence of the graphitic regions in the a-C samples. The intensity of this structure gradually decreases with the H+D content, just as previously concluded from the XPS spectra. Closer inspection of the spectra reveals that the top of the valence band is shifting towards higher binding energy with the H+D content, verifying opening of the local energy gap attributed to the graphitic regions. The gap opening implies size reduction of the graphitic regions [19,29].
The difference between the top of the valence band and the Fermi level (the latter corresponds to the 0 eV binding energy) in the spectra presented in Figure 2 is in the range 0.0-0.2 eV. Assuming that the Fermi level is around mid-gap position, the energy gap of the graphitic regions can be roughly estimated to be ≈0.2 eV for the reference and 0.2D samples, and ≈0.4 eV for the samples 0.5D and 1D. In a first approximation, the UPS value for the reference sample corresponds to its Tauc gap (Table 1), while the mismatches between the UPS and Tauc values for the other samples (particularly for 1D) are much higher. The latter is due to the character of the Tauc gap, which corresponds to the electronic properties of dominant components in the film, whereas UPS detects the highest occupied states. Therefore, reasonable agreement between the Tauc gap measurements and the UPS results of the reference and 0.2D samples is additional evidence of their graphitic character. On the other hand, the mismatch in the case of the other two samples confirms a lower graphitic content as already revealed by XPS.
3.4. Raman Spectroscopy
Further insight into the structure of the thin films was obtained from Raman scattering spectroscopy. Results of the Raman spectroscopy performed on the samples deposited on silicon using the 532 nm laser beam are summarised in Figure 3. The representative spectra of each sample (apart from the sample 10D) are presented in Figure 3a. The steady increase of the Raman signal with the D2 content in the discharge gas is most probably due to the drop of the optical absorption [15], which is also manifested in the increase of the Tauc gap (cf. Table 1), combined with the monotonic increase of the film thickness with the H+D content.
A feature in the range 1100-1700 cm-1 corresponds to the overlapping G and D bands, being typical for various carbon phases [30] (including deuterated a-C films [31]). The G band is attributed to sp2 carbon atoms (it is not restricted to sixfold carbon rings in graphite nor to pure carbon phases). The D band is a fingerprint of isolated sixfold carbon aromatic rings, being forbidden in a perfect graphite [19]. Co-existence of both bands therefore implies presence of graphitic regions separated by disordered phases.
Apart from the G and D bands, an intense wide structure at about 2800 cm-1 can be also observed in Figure 3a. Its intensity relative to the G and D bands increases with the amount of deuterium incorporated in the samples. In the sample 10D, the intensity of this structure is about 10 times greater than in the other samples measured under the same conditions, thus hindering eventual identification of the graphitic carbon (more details on this matter are provided in the Supplementary Information S1). Raman spectra of carbon-based materials may have some bands in this range. For instance, graphene is characterised by the 2D mode at around 2700 cm-1 and a second order D+D’ band at around 2900 cm-1 [32]; Raman spectra taken from polymeric amorphous carbon have in this range a C-H stretching band [30,33]. To clarify the origin of this band, Raman measurements were also performed with a He-Ne laser (wavelength of about 632.8 nm) resulting in the lack of this structure (cf. Supplementary information S1). The absolute energy position of this feature is ≈1.97 eV, corresponding well to the position of the broad emission band (1.9-2.5 eV) observed in a photoluminescence of hydrogenated a-C [34]. Therefore, we conclude that this feature is not a Raman signal but evidence of photoluminescence i.e. it is related to the excitation of electronic states. The latter also explains why this feature increases with the amount of incorporated deuterium in the sample.
Fitting of the Raman spectra is demonstrated in Figure 3b on the example of the sample 1D, while other fitting details can be found in the Supplementary Information S1. The spectra were fitted using 4 Lorentzian peaks after removing a linear background. Although this approach did not provide a perfect fit out of the range of interest (the latter being 1100-3000 cm-1), it allows extraction of the most valuable information characterising the a-C samples. Two of the Lorentzians are readily attributed to the G and D bands at about 1550 cm-1 and 1360 cm-1, respectively. The peak widths and the position of the D band do not change significantly among the studied samples. There is, however, a substantial difference between the relative peak intensities from different samples as well as a clearly pronounced shift of the G band. The wide hump related to the photoluminescence (PL), was fitted by two contributions, at about 2800 and 2150 cm-1. The latter one could also be, at least partially, attributed to the CDx stretching band [30] (PL2/C-D). The measurements performed with 632.8 nm support this interpretation (cf. Supplementary Information S1).
Most of the findings concerning different a-C systems extracted from Raman spectra are related with the G and D bands. These bands have a large width (≈200 cm-1) in all samples, being a characteristic of disordered systems [35]. The ratio of the intensities of the D and G bands, ID/IG, and the position of the G band are plotted as a function of the relative H+D content (Figure 3c). The increase of the H+D content in the films reduces the ID/IG intensity ratio from 0.75 to 0.45 (a similar trend has been reported in [36] due to the increase of the hydrogen content), while the G band position σD is shifted from 1563 to 1540 cm-1. While the observed trends are ambiguous when considered separately, a unique interpretation of this joint information is that the increase of the deuterium content corresponds to stage two of the well-known amorphisation trajectory of graphite as proposed by Ferrari and Robertson [19]: transformation of nano-graphite into amorphous carbon. In this amorphisation stage, the ratio ID/IG is directly proportional to the average number of carbon atoms in the graphitic clusters i.e. to the square root of their diameter.
The fitting approach shown in Figure 3b was not applicable to the sample 10D due to the intense curvilinear background, mostly caused by the intense photoluminescence signal, which makes the extraction of signal in the range of the D and G bands unreliable. Nevertheless, such an attempt has been made by removing the background of a polynomial form, which resulted in an asymmetric structure that could be mainly interpreted as the G band, while the D band is apparently lacking. Although of a limited reliability, this result implies that the graphitic phase in the sample 10D is extremely small or even absent (see above). Details concerning the analysis of the Raman spectra taken from the sample 10D can be found in the Supplementary information S1.
In addition, an attempt was made to identify the character of C-D bonds using other vibrational spectroscopies, namely FTIR and HREELS. These results are presented in the supplementary material S2 and the appendix A, respectively. In the case of FTIR, the signal attributed to C-D bonds was identified only in the case of the sample 10D, revealing that about 60% of deuterium atoms is bonded to sp2 carbon. A similar conclusion was obtained from the measurement of the sample 1D by HREELS, confirming that the character of C-D bonds, being practically the same, is not responsible for the large difference between the electronic structures of the two samples.
4. Discussion
The XPS results and the mismatch between the Tauc plot measurements and UPS spectra evidence that the samples are generally non-uniform. Hence, the films should be considered as mixtures of different carbon phases (being a frequent situation [19]), each characterised with their own local energy gaps. The steady increase of the films’ density with the amount of incorporated deuterium up to the sample 1D can be understood as the increase of the sp3 phases (cf. Table 1). At the same time the sample with the lowest density is 10D, despite the similar relative composition with that of 1D, clearly indicating formation of a polymeric phase [20]. These estimations are fully supported by the results of the XPS, UPS and Raman analyses, revealing that increase of the deuterium content is accompanied by a decrease of the overall relative amount of the graphitic regions and their size reduction (i.e., transition from nanocrystalline graphitic phase into amorphous carbon). This is particularly evident when comparing the samples 1D and 10D, characterized with the similar relative content of deuterium (Table 1) and practically the same character of the C-D bonds. The abrupt increase of the SEY and the Tauc gap from sample 1D to 10D can be only explained by the dramatic diminishing of the graphitic component (Table 1).
A similar conclusion has been obtained by Robertson [18], demonstrating a nearly linear dependence of the Tauc gap with the sp2 fraction csp2 for its relative contents below 80%, practically independent from the hydrogen relative amount. Since the data presented in [18] are compiled from the works of different authors and measured on various a-C and a-C:H samples, the points are rather scattered. Nevertheless, the line ET = 3 – 2.5∙csp2 represents well the observed trend with the Tauc gap uncertainty bar of about ΔET = ±0.25 eV [37]. The dependence of the Tauc gap and the SEYmax vs. the graphitic content, obtained from the XPS measurements, is shown in Figure 4. It can be observed that both ET and SEYmax also decrease with the relative amount of graphitic regions. Moreover, the trend of the Tauc plot fits well with that of Robertson, presented by a dashed blue line, for the graphitic contents below 70%. The systematic shift between the two trends, which is within the uncertainty ΔET, can be explained by different means of measuring the graphitic content in [37] (using electron energy loss spectroscopy and nuclear magnetic resonance). Besides, at csp2 > 70% we observe faster drop of ET, as expected having the non-existent band gap of graphite. These agreements imply that the methodology used to extract the graphitic fraction from the XPS spectra is quite reliable and can be also used to correlate the SEY with the graphitic content.
The decrease of the SEY with the increase of the graphitic content (observed in Figure 4) is in accordance with the expectations: higher the fraction of the sp2 phase, lower the SEY. A simple model, based on the same assumptions as those used to derive the well-known semi-empirical equation for SEY [38], was developed to support the observed experimental dependence. This equation, originally derived for uniform samples, was recently modified to encompass multilayer systems [39]. The latter approach was used as a starting point to model non-uniform a-C films as mixtures of two different phases: a graphitic one, and a polymeric one. Therefore, the polymeric phase is considered as a representative of all non-graphitic phases in the films.
In the frame of the semi-empirical theory, the number of internal secondary electrons generated in a depth range (z, z+dz) is equal to S(z)∙dz/ε(z), where S and ε represent the stopping power of the primary electrons and the effective energy required to create one internal secondary electron, respectively. The secondary electrons created at depth z will be then emitted with a probability equal to 0.5∙exp(-z/λ) [40], where λ is the mean escape depth of secondary electrons, taken from the literature [11]. Since S, λ and ε are material dependent, they will be changing from point to point of the sample interior.
The first step is to determine the parameters that characterize the electron emission properties of the graphitic and polymeric regions. For that purpose, the SEY dependencies on the primary electron energy of the reference sample and the sample 10D [15] were used as representatives of pure graphitic and pure polymeric material, respectively. Then, for a defined relative graphitic content, it is possible to estimate the range of primary electrons R. The samples were then modelled as a multilayer system with predefined graphitic content. However, the depth distribution of different phases within each sample is unknown. Another difficulty in establishing this distribution is in the lateral non-uniformity of the samples. To overcome this problem, a set of M depth distributions were generated (here designated as configurations), each containing the same amount of graphitic carbon, using a Monte Carlo approach. Finally, for each of the configuration, the SEY(E) was calculated by generating S(E,z)∙dz/ε(z) electrons at all depths z in the range [0, R], and calculating their escape probabilities affected by the multilayer structure in the range [0, z]. The whole procedure is detailed in the Appendix B.
The result of the SEY model, obtained using M = 1000 configurations and for the primary electron energies in the range 50-1000 eV is presented in Fig. 4. Having in mind the simplifications introduced in the frame of the model, and that the calculated SEY curves were obtained without any adjustments to the existing experimental data, the agreement with the measured values is very good. There is also a built-in systematic error in the model due to the consideration that the reference sample would be purely graphitic, while its actual graphitic content is only 90%. This approximation is responsible for an overestimation of the SEY for the highest graphitic contents. Although the effective energy ε is indeed smaller in the polymeric (40.8 eV) than in the graphitic (60.4 eV) regions, the major difference between the characteristics of the two regions is the mean escape depth λ, being more than two times smaller in graphite (4.9 nm) than in polymers (11 nm). That is probably the reason why the theoretical SEYmax(sp2 content) dependence can be described by an exponential decay function.
In a sample with sufficiently high content of the graphitic phase, the regions with large energy gap (e.g., diamond-like or hydrocarbon regions) will not contribute to the Tauc gap: photons that may penetrate large energy gap regions will be most likely absorbed once they enter a sufficiently large graphitic region. The reduction of the amount and size of the graphitic phase increases the optical transparency of the films and, consequently, the Tauc gap. A similar effect takes place during the transport of secondary electrons through the samples. The escape depth of internal secondary electrons, which is directly related with their energy loss mechanisms, is strongly affected by the electronic structure of a material. The efficient energy loss is secured by sufficiently long trajectories of the secondary electrons through graphitic regions characterised by very narrow energy gap. As in the case of light transmission, the graphitic phase serves as energy absorber. The increase of the graphitic content directly reduces the escape depth of internal secondary electrons and, therefore, the secondary electron yield.
Although the presence of hydrogen and deuterium in the thin films is not directly responsible for the SEY increase, it plays a key role in reducing the graphitic amount. Indeed, the H/D incorporation will naturally reduce the relative contents of both graphitic and diamond-like phases in a-C:H(D). It was revealed in the previous work that the deuterium incorporation is a consequence of target poisoning, yielding in the sputtering of CD and CD2 molecules in parallel with C atoms [15]. At the same time, pure carbon phases can only be deposited by magnetron sputtering of C atoms. The relative amount of pure carbon phases is directly related with the flux ratio of C and CDx particles leaving the target and travelling towards the substrate. In the case of the sample 10D, this ratio was very low due to the strong target poisoning, resulting in a typical polymeric film.
5. Conclusions
Amorphous carbon coatings, deliberately modified by deuterium incorporation during the magnetron sputtering deposition, were thoroughly analysed using different electron and vibrational spectroscopies. This data was combined with previous composition and optical measurements [15], to reveal the role of H/D incorporation on the change of the secondary electron emission characteristics.
Both XPS and UPS (in combination with the Tauc gap) measurements demonstrate non-uniform character of the samples, being common for a-C materials [16]. The increase of the deuterium amount in the films is accompanied with the decrease of the fraction of the graphitic component and the cluster size. We conclude that a high content of graphitic phases in the films is responsible for a low secondary electron emission, just as it is for the low Tauc gap [18]. This phase has a local energy gap approaching to 0 eV, which reduces the escape depth of electrons travelling through it and, consequently, the secondary electron yield.
The formation of pure carbon phases (graphitic or diamond like) and hydrocarbon regions are competing processes. In our experiments, the latter is the result of target contamination by D atoms formed in the plasma, followed by the physical sputtering of CDx molecules representing building blocks of hydrocarbon phases [15]. Therefore, the flux ratio of CDx and C particles approaching the substrate is detrimental in this case for the relative graphitic content in the films.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
“Conceptualization, N.B. and P.C.P.; methodology, N.B and P.C.P; software, N.B.; validation, A.F., M.H. and M.V.; formal analysis, N.B., J.D., M.C., A.R.; investigation, C.A., J.D., M.C., A.R., N.P.B, Y.D., H.N., M.R.; writing—original draft preparation, N.B. and P.C.P.; writing—review and editing, M.H., O.T., C.A., A.R.; visualization, C.A.; supervision, M.V., I.F., E.A. All authors have read and agreed to the published version of the manuscript.”
Funding
This research was funded by the Portuguese Foundation for Science and Technology (FCT) in the frame of the project CERN/FIS-TEC/0039/2019. The support of FCT via the grants UIDB/00068/2020, UIDP/00068/2020 UIDB/04565/2020, UIDP/04565/2020 and LA/P/0140/2020 is also gratefully acknowledged. A.M. F. wishes to thank Instituto Superior Técnico for the Scientific Employment contract IST-ID/131/2018.
Data Availability Statement
Not applicable.
Acknowledgments
We acknowledge Mauro Taborelli, from CERN, for discussions and suggestions during the work on the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A. HREELS Measurements
A.1 Experimental Setup
HREELS measurements were performed in a UHV system (operating pressure in the 10-7 Pa range) using a LK Technologies 2000R spectrometer. Incidence and analysis angles, measured relative to the surface normal, were 60° and 30º, respectively. Such geometry secures very high surface sensitivity. The analysis of the sample 1D was performed, as the one with potentially the highest deuterium content that can be measured (it appeared that the sample 10D was charging, as explained below). The nominal primary electron energy was 3 eV (with the elastic peak Full Width at Half Maximum (FWHM) of 12 meV). The cut-off of the spectra was measured at about 3.8 eV, corresponding to the true primary electron energy. The spectra were acquired with an energy step of 0.916 meV (7.38 cm-1).
A.2 Results
In a HREELS experiment, a monoenergetic electron beam interacts with the surface region in different ways exciting vibrational modes [41] and electronic states [42]. For non-specular conditions (different incident and analysis angles) the dominant excitation mechanism is via impact (short range) interactions, occurring at distances below 1 nm. In this regime, the technique is mainly sensitive to the molecular groups in the first atomic layer, providing information on both vibrational [43] and electronic [44] excitations.
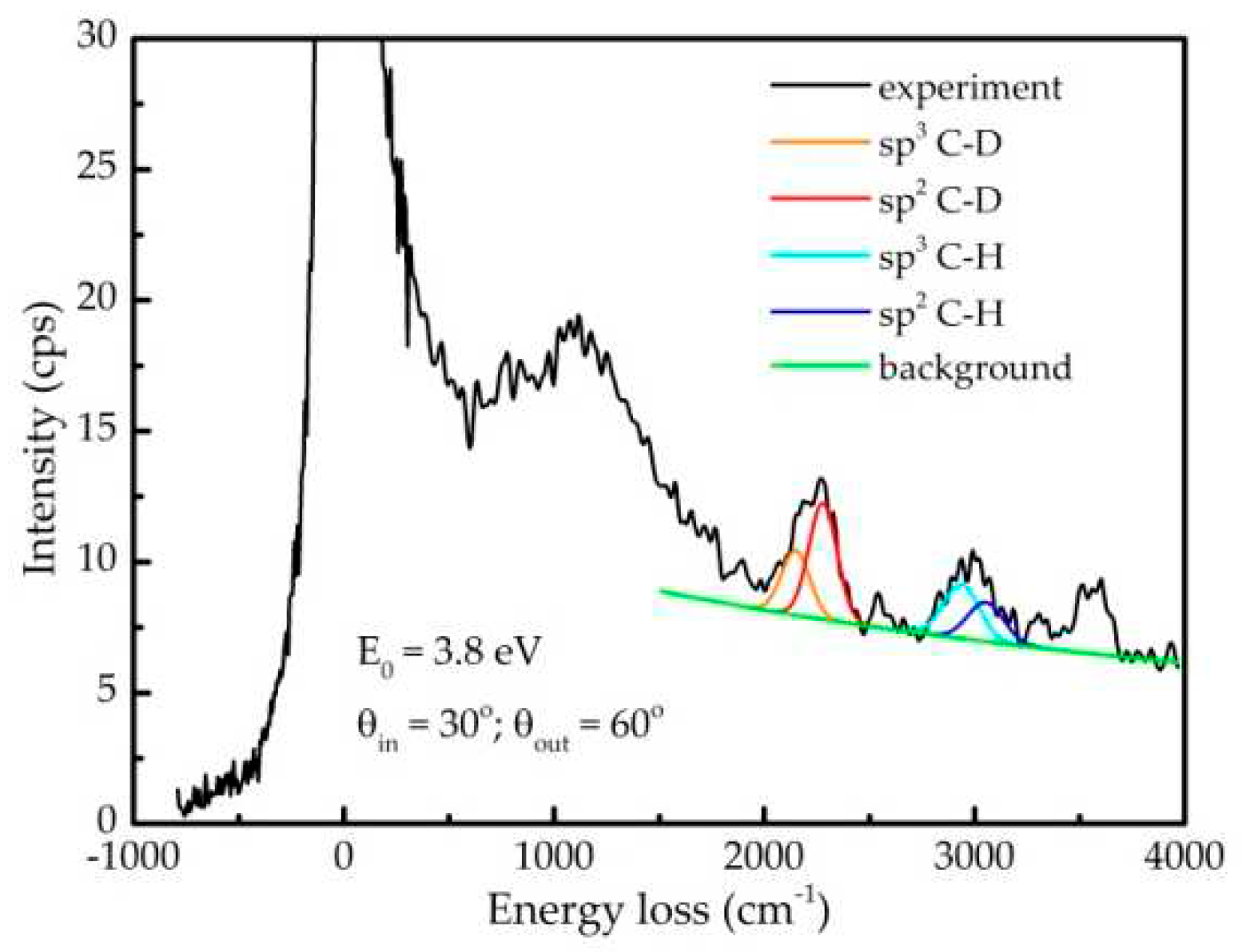
Figure A1 displays the spectrum taken from the sample 1D obtained in the vibrational energy loss range. The elastic peak is asymmetric due to the excitation of low energy phonons. From 600 to ~2000 cm-1 a complex, not resolvable structure exists due to IR active modes corresponding to deformation vibrations of C-D and C-H bonds as well as to the Raman modes which can also be excited in a non-specular geometry [45]. In the stretching modes region, C-D (2000 to 2450 cm-1 range) and C-H (2750 to 3200 cm-1 range) bonds are detected [46]. Even with some uncertainty of the background, each of those two structures can be fitted to two components. The first ones at 2279 and 2150 cm-1 can be assigned to the stretching vibrations of D bonded to sp2 and sp3 carbon, respectively. This result is well correlated with the similar structures observed in FTIR measurements of the sample 10D (Suplementary Information S4). Indeed, perdeuterated benzene exhibits an IR peak at 2286 cm-1 [47]. In addition, in our previous work dealing with selectively deuterated polystyrenes, peaks at those frequencies were also found [48]. The equivalent C-H structure can be fitted to two components at 3054 and 2938 cm-1, which can be assigned to hydrogen bonds with sp2 and sp3 hybridised carbon, respectively. At higher energy loss (3558 cm-1) another peak is detected, that we assigned to the O-H stretching in an alcohol group. The equivalent deuterated group, which should be present between the C-D and C-H structures, was not detected. One possible reason is the corresponding energy loss cross section is lower for the O-D, similarly to the C-D bonds as compared to the C-H bonds (see below). In addition, lack of this bond may imply that OH groups mainly originate from the surface contamination due to air exposure that took place after the deposition. This interpretation is supported by the facts that the vacuum system was baked prior to the deposition and had very low residual gas pressure (below 1·10-8 Pa), and that the oxygen amount measured by XPS was higher than that obtained by IBA [15].
Figure A1.
Vibrational HREELS spectrum taken from the sample 1D. The partial fittings of C-H and C-D stretching modes are also included.
Figure A1.
Vibrational HREELS spectrum taken from the sample 1D. The partial fittings of C-H and C-D stretching modes are also included.
The ratio of surface concentrations of D (nD) and H (nH) atoms can be estimated from the ratios of the respective peak areas AD and AH, and the inelastic loss cross sections attributed to the C-D (σD) and C-H (σH) bonds:
The corresponding cross-section ratios σH/σD in IR spectroscopy are around 1.81 (aromatic bonds) and 2.17 (aliphatic bonds), while the equivalent magnitudes are somewhat lower in HREELS [43]. Having in mind experimental difficulties in calibrating HREELS in that work, we prefer to use the cross-section ratios from the IR spectroscopy in the attempt to determine the concentration ratios nD/nH. Since the cross-section ratios do not depend strongly on the bonding character, their average (1.99) was used, yielding nD:nH = 3.0. The same magnitude in the sample 1D is about 2 times greater when measured by IBA, most probably due to very different information depths.
In further analysis, we consider that the cross sections for H/D bonded with sp2 and sp3 carbon are approximately the same. If so, the peak intensity ratios obtained from the fittings directly correspond to the fractions of D and H bonded with sp2 and sp3 hybridized carbon atoms. By implementing this approach, it appears that about 64 % of D atoms are bonded with sp2 carbon, while this percentage is only 42 % for H atoms. One should, however, have in mind that this ratio does not correspond directly to the relative ratios of hydrocarbon species with sp2 and sp3 carbon, since the relative amount of H/D in saturated aliphatic hydrocarbons is certainly higher than in compounds containing sp2 hybridised carbon atoms.
When comparing the D:H ratio restricted to sp2 or sp3 carbon (using the actual intensity ratios of the corresponding cross-sections), we get the values of 4.23 and 2.03, respectively. While the ratio for the bonds with sp2 carbon approaches the corresponding values obtained by IBA [15], the ratio for the sp3 carbon is clearly lower. Again, as for the O-H, the results obtained can be explained by the surface contamination that took place after the deposition, which mainly consists of aliphatic saturated hydrocarbons (airborne). Namely this is the reason for the reduced nD:nH ratio at the surface as compared to the values measured by IBA, but also for the different ratios in the case of the sp2 and sp3 carbon hybridisations.
Appendix B. Modelling SEY from Non-Uniform a-Carbon Samples
B.1 Semi-Empirical Theory of Secondary Electron Emission and Its Application to Graphitic and Polymeric Samples
The semi-empirical theory of secondary electron emission are based on the following assumptions [49]:
- all primary electrons have the same range R in a material, which can be described by the power law R = b∙En, with E being the energy of incident electrons, while b is the material constant;
- the number of internal secondary electrons generated in the depth interval (z, z + Δz) is directly proportional to the stopping power of primary electrons S(z) = -dE/dz, ΔN(z) = S(z)Δz/ε, where ε is the effective energy required to create one internal secondary electron;
- electron stopping power is assumed to be constant, and frequently its average value along the trajectory E/R(E) is used;
- the probability that an internal secondary electron will be emitted is 0.5∙exp(-z/λ), where λ is an electron escape depth (a material dependent parameter).
Starting from these assumptions, one can easily derive the following equation for the energy dependence of SEY:
After introducing the relative energy Er = E/Em, where Em is the energy at which SEY has maximum, and a parameter x = R(Em)/λ, the expression (B1) can be transformed into
Since x is the solution of the transcendent equation [50], the shape of the SEY(E) curve is fully described by the single parameter n, which can be then obtained by fitting the experimental SEY(E) curve to the eq. B2 [51]. By combining the eqs. B1 and B2, the knowledge of n enables to calculate the energy ε as
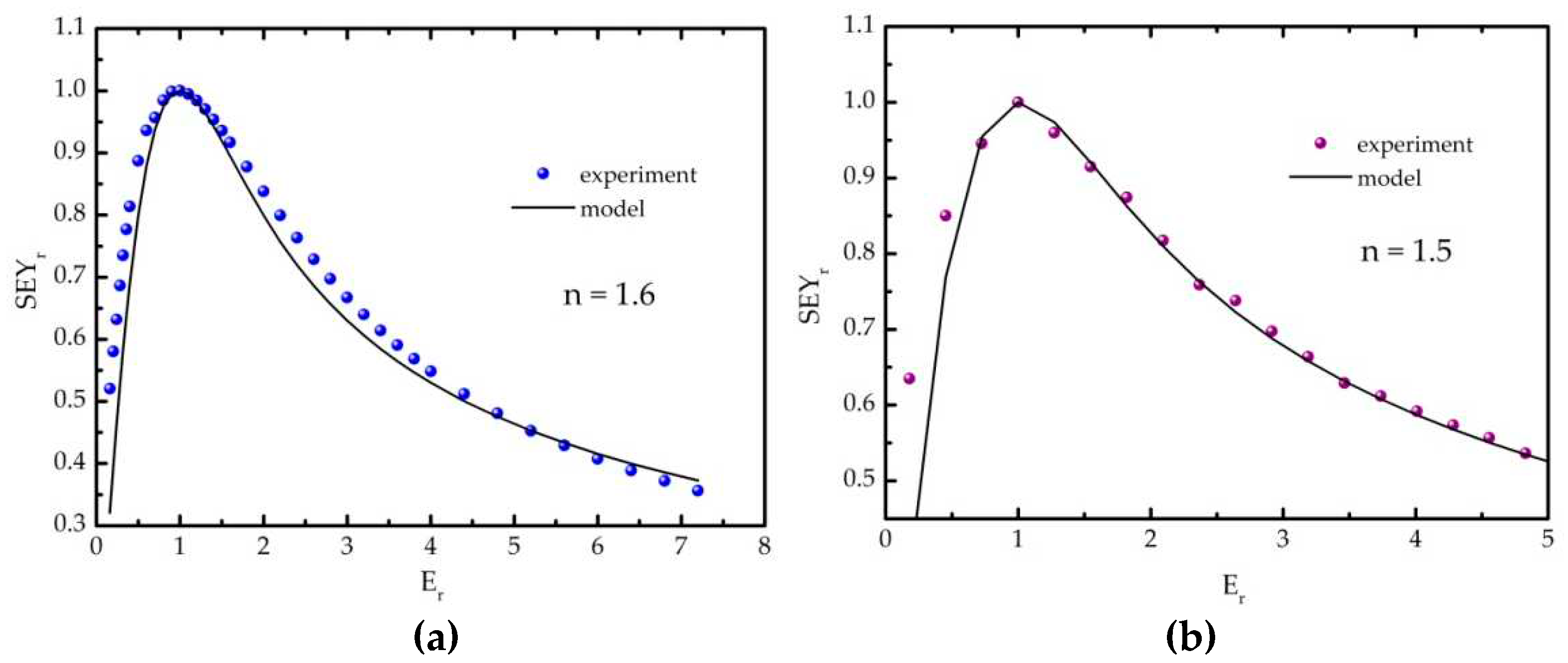
In our approach, we consider the reference and the 10D samples as representatives of dominantly graphitic and polymeric samples, respectively. Their SEYr(Er) curves (obtained from the SEY(E) curves from [15]), presented in Figure B1, can be well described within the semi-empirical theory using the values n = 1.6 (the reference) and n = 1.5 (10D). The corresponding parameters x were then calculated by solving the above mentioned transcendent equation, as well as the effective energies ε using the eq. B3 and the experimental values for Em and SEYmax. The obtained values for the reference and 10D samples are 60.4 and 40.8 eV, respectively. The escape depths used for graphitic and organic carbon samples, taken from [11], are 4.9 and 11 nm, respectively. After calculating the factors b as x∙λ/Emn (from the assumed R(E) dependence and the definition of the parameter x), we were able to calculate the SEY curves in the frame of the semi-empirical theory.
Figure B1.
Determination of the parameter n in the case of the a) reference and b) 10D sample, representing dominantly graphitic and polymeric samples, respectively.
Figure B1.
Determination of the parameter n in the case of the a) reference and b) 10D sample, representing dominantly graphitic and polymeric samples, respectively.
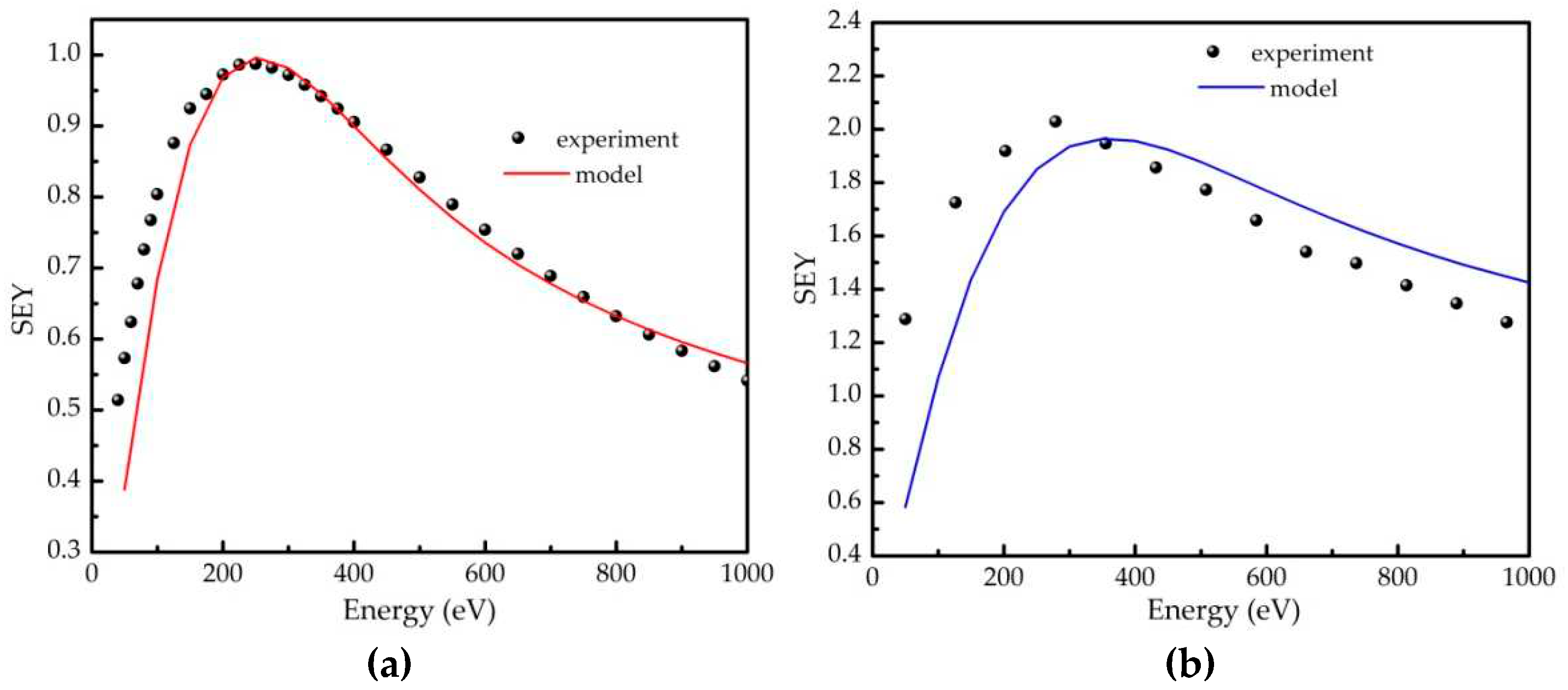
The experimental and modelled SEY curves are compared in Figure B2. Although experimental curves were used to determine some of the parameters, namely n and ε, it should be stressed that there were no adjustments of calculated values to the experimental SEY curves. With this in mind, the semi-empirical theory provides very good description of the experimental SEY curves. This is particularly valid for the reference sample (Figure B2a), for which an excellent agreements for both SEYmax and Em are obtained. That is not the case with the polymeric sample, particularly concerning the maximum position. Possible reason for such discrepancy could be in the different measurement approach for the sample 10D, which was affected by charging (see Section 2 for details). Nevertheless, even for that sample the model curve appears to be a good representative of the experimental dependence.
Figure B2.
Comparison of the experimental and calculated SEY in the case of the a) reference and b) 10D sample.
Figure B2.
Comparison of the experimental and calculated SEY in the case of the a) reference and b) 10D sample.
B.2 Model Description
The semi-empirical theory, including its recent upgrade for multilayer systems [39], is a 1D continuous model. Therefore, a non-uniform material can be only considered as a multilayer system. For a fixed sample composition, there is a huge number of depth distributions of polymeric and graphitic layers (here designated as configurations) with very different SEY. Some of the possible configurations in the case of a sample consisting of 30% of graphitic component are presented in Figure B3. Extreme configurations, consisting of only 2 layers (one for each component), would have very different SEY, depending which phase is on the top. Since a representative configuration cannot be determined, we considered this problem in the frame of the Monte Carlo approach.
Figure B3.
Illustration of different configurations ck (k = 1, 2…), each containing the same relative amounts of graphitic (grey) and polymeric (brown) regions, which will be characterised with very different SEY curves.
Figure B3.
Illustration of different configurations ck (k = 1, 2…), each containing the same relative amounts of graphitic (grey) and polymeric (brown) regions, which will be characterised with very different SEY curves.
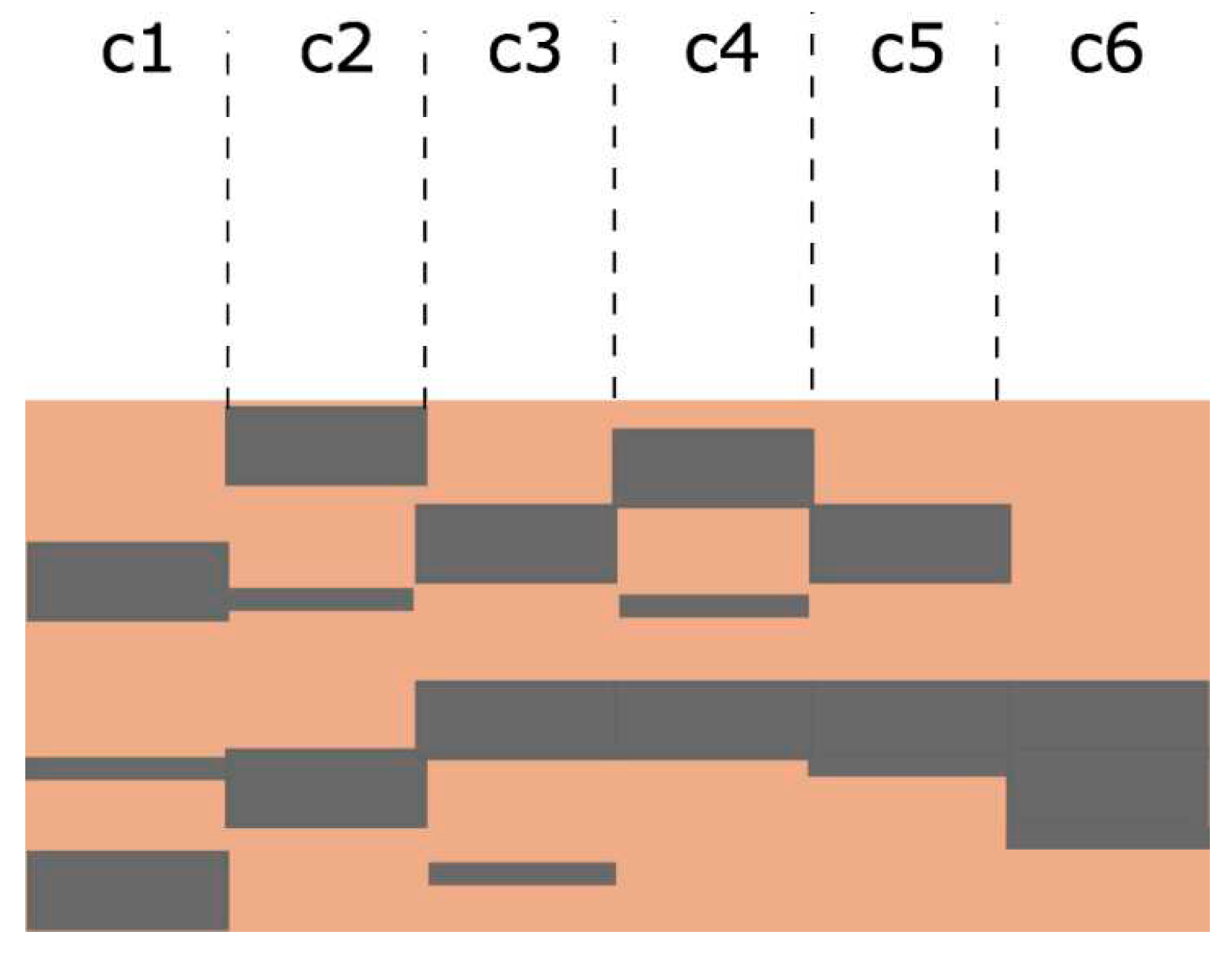
Figure B3. Illustration of different configurations ck (k = 1, 2…), each containing the same relative amounts of graphitic (grey) and polymeric (brown) regions, which will be characterised with very different SEY curves.
The first step was to determine range of the primary electrons using the assumptions built into the semi-empirical theory: the stopping power of electrons in each region i is constant and can be determined as Si = E/Ri, where Ri is the range in a uniform sample consisting of the component i. Let Lp and Lg be the total lengths of trajectories of primary electrons passing through the polymeric and graphitic regions, respectively. The range of primary electrons will then be R = Lp + Lg. Since all of the primary electron energy is transferred to the material after the primary electrons pass the trajectory of length R, we can write , i.e. . We assume that the fraction of trajectory passing through the component i equals the relative concentration of that component ni i.e. Li/R = ni. In that case, the range of primary electrons through the two-component sample can be written as
The sample is considered to have thickness equal to the range R obtained for the maximum primary electron energy, calculated from the known composition, using eq. B4. The sample is divided in 100 layers of equal thickness, Δz = R/100. The composition of each layer is determined using the Monte Carlo approach, knowing that the probability that a particular layer is graphitic/polymeric equals ng/(1 – ng). Once a configuration is formed, the SEY is calculated by creating in each layer of index i Si∙Δz/εi internal secondary electrons, which will leave the material with a probability (the indices i and j increase with the layer depth). For a particular configuration, SEY is calculated for different primary electron energies. This procedure is then repeated for M different configurations characterized by the same sample composition, and the final SEY(E) is obtained as an average of all curves obtained for different configurations. The results presented in the paper are obtained for M = 1000.
References
- O. Gröbner, Bunch induced multipactoring, CM-P0064850, CERN Libraries, Geneva, Switzerland, presented at the 10th International Conference on High-Energy Accelerators, 11th-17th of July 1977, Serpukhov, USSR.
- R. Cimino, I.R. Collins, M.A. Furman, M. Pivi, F. Ruggiero, G. Rumolo, F. Zimmermann, Can low-energy electrons affect high-energy physics accelerators?, Phys. Rev. Lett. 2004, 93, 014801. [CrossRef]
- R. A. Kishek, Y.Y. Lau, L.K. Ang, A. Valfells, R.M. Gilgenbach, Multipactor discharge on metals and dielectrics: historical review and recent theories, Phys. Plasmas 1998, 5, 2120–2126. [CrossRef]
- J. Puech, et al., A multipactor threshold in waveguides: theory and experiment, in: J.L. Hirshfield, M.I. Petelin (Eds.), Quasi-Optical Control of Intense Microwave Transmission. NATO Science Series II: Mathematics, Physics, and Chemistry vol. 203, Springer, Dordrecht, 2006.
- J. Hillairet, M. Goniche, N. Fil, M. Belhaj, and J. Puech, Multipactor in High Power Radio-Frequency Systems for Nuclear Fusion, presented at the International Workshop on Multipactor, Corona and Passive Intermodulation, MULCOPIM 2017, 5th-7th of April 2017 Noordwijk, The Netherlands.
- G. Skripka, G. Iadarola, L. Mether, G. Rumolo, Non-monotonic dependence of heat loads induced by electron cloud on bunch population at the LHC, Eur. Phys. J. Plus 2022, 137, 849. [CrossRef]
- C. Yin Vallgren, G. Arduini, J. Bauche, S. Calatroni, P. Chiggiato, K. Cornelis, P. Costa Pinto, B. Henrist, E. Métral, H. Neupert, G. Rumolo, E. Shaposhnikova, M. Taborelli, Amorphous carbon coatings for the mitigation of electron cloud in the CERN super proton synchrotron, Phys. Rev. ST Accel. Beams 2011, 14, 071001. [CrossRef]
- F. Willeke, J. F. Willeke, J. Beebe-Wang, Electron Ion Collider Conceptual Design Report 2021. United States: N. p., 2021. Web. [CrossRef]
- S. Verdú-Andrés et at., A beam screen to prepare the RHIC vacuum chamber for EIC hadron beams: conceptual design and requirements, 12th International Particle Accelerator Conference - IPAC’21, May 24 - 28, 2021 Web. https://www.osti.gov/servlets/purl/1784486.
- R.F. Egerton, Electron-Energy Loss Spectroscopy in the Electron Microscope 3rd edn. Springer, New York, 2011.
- S. Ono, K. Kanaya, The energy dependence of secondary emission based on the range-energy retardation power formula, J. Phys. D: Appl. Phys. 1979, 12, 619.
- P. Costa Pinto, et al., Carbon coating of the SPS dipole chambers. AIP Conf. Proc, C 2013, 12, 141–148.
- A. Santos, N. Bundaleski, B.J. Shaw, A.G. Silva, O.M.N.D. Teodoro, Increase of Secondary Electron Yield of Amorphous Carbon Coatings under High Vacuum Conditions. Vacuum 2013, 98, 37–40. [CrossRef]
- H. M. Fernández, M. Himmerlich, P. Costa Pinto, J. Coroa, D. Sousa, A. Baris, M. Taborelli, The impact of H2 and N2 on the material properties and secondary electron yield of sputtered amorphous carbon films for anti-multipacting applications, Appl. Surf. Sci. 2021, 542, 148552. [CrossRef]
- C. F. Adame, E. Alves, N.P. Barradas, P. Costa Pinto, Y. Delaup, I.M.M. Ferreira, H. Neupert, M. Himmerlich, S. Pfeiffer, M. Rimoldi, M. Taborelli, O.M.N.D. Teodoro, N. Bundaleski, Amorphous carbon thin films: mechanisms of hydrogen incorporation during magnetron sputtering and consequences for the secondary electron emission, J. Vac. Sci. Technol. A 2023, 41, 043412. [CrossRef]
- A. C. Ferrari, J. Robertson, Interpretation of Raman spectra of disordered and amorphous carbon, Phys. Rev. B 2000, 61, 14095. [CrossRef]
- D. Roy, G. F. Samub, M. K. Hossain, C. Janáky, K. Rajeshwar, On the measured optical bandgap values of inorganic oxide semiconductors for solar fuels generation, Catalysis Today 2018, 300, 136–144. [CrossRef]
- J. Robertson, Diamond-like amorphous carbon, Mat. Sci. & Engineering R 2002, 37, 129–281. [CrossRef]
- A. C. Ferrari, J. Robertson, Interpretation of Raman spectra of disordered and amorphous carbon, Phys. Rev. B, 2000, 61, 14095. [CrossRef]
- L. Zhang, X. Wei, F. Wang, A ternary phase diagram for amorphous carbon, Carbon 2015, 94, 202–213. [CrossRef]
- S. Kaciulis, Spectroscopy of carbon: from diamond to nitride films, Surf. Interface Anal. 2012, 44, 1155–1161. [CrossRef]
- P. Mérel, M. Tabbal, M. Chaker, S. Moisa, J. Morgot, Direct evaluation of the sp3 content in diamond-like-carbon films by XPS, Appl. Surf. Sci. 1998, 136, 105–110. [CrossRef]
- D. J. Morgan, Comments on the XPS analysis of carbon materials, C 2021, 7, 51. [CrossRef]
- M. C. Biesinger, Accessing the robustness of adventitious carbon for charge referencing (correction) purposes in XPS analysis: Insights from a multi-user facility data review, Appl. Surf. Sci. 2022, 597, 153681. [CrossRef]
- G. Beamson, D. Briggs, High Resolution XPS of Organic Polymers - The Scienta ESCA300 Database, Wiley Interscience, 1992.
- N. Fairley, A. Carrick, The Casa Cookbook – Part 1: Recipes for XPS data processing, Acolyte Science, Cheshire, 2005.
- J. H. Scofield, Hartree-Slater subshell photoionisation cross-sections at 1254 and 1487 eV, J. Elec. Spec. Rel. Phenom. 1976, 8, 129–137.
- A. Law, M. Johnson, and H. Hughes, Synchrotron-radiation-excited angle-resolved photoemission from single-crystal graphite, Phys. Rev. B 1986, 34, 4289.
- I. Ozfidan, M. Karkusinski, P. Hawrylak, Electronic properties and electron-electron interactions in graphene quantum dots, Phys. Status Solidi RRL 2016, 10, 13–23.
- A. C. Ferrari, J. Robertson, Raman spectroscopy of amorphous, nanostructured, diamond-like carbon, and nanodiamond, Phil. Trans. R. Soc. London A 2004, 362, 2477–2512. [CrossRef]
- M. E.H. Maia da Costa, F.L. Freire Jr., Deuterated amorphous carbon films: Film growth and properties, Surf. Coat. Technol. 2010, 204, 1993–1996. [CrossRef]
- A. C. Ferrari, D.M. Basko, Raman spectroscopy as a versatile tool for studying the properties of graphene, Nature Nanotechnology 2013, 8, 235–246. [CrossRef]
- S. Nakahara, S. Stauss, H. Miyazoe, T. Shizuno, M. Suzuki, H. Kataoka, T. Sasaki, K. Terashima, Pulsed Laser Ablation Synthesis of Diamond Molecules in Supercritical Fluids, App. Phys. Express 2010, 2, 096201. [CrossRef]
- Rusli and, J. Robertson, G. A.J. Amaratunga, Photoluminescence behavior of hydrogenated amorphous carbon, J. Appl. Phys. 1996, 80, 2998–3003. [Google Scholar] [CrossRef]
- A. Merlen, J.G. Buijnsters, C. Pardanaud, A Guide to and Review of the Use of Multiwavelength Raman Spectroscopy for Characterizing Defective Aromatic Carbon Solids: from Graphene to Amorphous Carbons, Coatings 2017, 7, 153.
- C. Pardanaud, C. Martin, P. Roubin, G. Giacometti, C. Hopf, T. Schwarz-Selinger, W. Jacob, Raman spectroscopy investigation of the H content of heated hard amorphous layers, Diam. Relat. Mat. 2013, 34, 100–104. [CrossRef]
- J. Robertson, Photoluminescence Mechanism in amorphous hydrogenated carbon, Diamond and Related Materials 1996, 5, 457–460. [CrossRef]
- R. G. Lye, A.J. Dekker, Theory of Secondary Emission, Phys. Rev. 1957, 107, 977–981. [CrossRef]
- Y. He, T. Shen, Q. Wang, G. Miao, C. Bai, B. Yu, J. Yang, G. Feng, T. Hu, X. Wang, W. Cui, Effect of atmospheric exposure on secondary electron yield of inert metal and its potential impact on the threshold of multipactor effect, Appl. Surf. Sci. 2020, 520, 146320. [CrossRef]
- Y. Lin, D.C. Joy, A new examination of secondary electron yield data, Surf. Interface Anal. 2005, 37, 895–900. [CrossRef]
- H. Ibach, D. L. Mills, Electron Energy Loss Spectroscopy and Surface Vibrations (Acad. Press, New York, 1982).
- A. M. Botelho do Rego, M. Rei Vilar, J. Lopes da Silva, Mechanisms of vibrational and electronic excitations of polystyrene films in high resolution electron energy loss spectroscopy, J. Elec. Spectr. Rel. Phenom. 1997, 85, 81–91. [CrossRef]
- M. Rei Vilar, A.M. Botelho do Rego, J. Lopes da Silva, F. Abel, V. Quillet, M. Schott, S. Petitjean, R. Jérôme, Quantitative Analysis of Polymer Surfaces and Films Using Elastic Recoil Detection Analysis (ERDA), Fourier Transform Infrared Spectroscopy (FTIRS), and High-Resolution Electron Energy Loss Spectroscopy (HREELS) Macromolecules 1994, 27, 5900–5906.
- A. M. Botelho do Rego, A.M. Ferraria, M. Rei Vilar, Grafting of Cobaltic Protoporphyrin IX on Semiconductors toward Sensing Devices: Vibrational and Electronic High-Resolution Electron Energy Loss Spectroscopy and X-ray Photoelectron Spectroscopy Study, J. Phys. Chem. C. 2013, 117, 22298–22306. [CrossRef]
- A. M. Botelho do Rego, J.L. da Silva, M. Rei Vilar, R. Voltz, Resonance mechanisms in electron energy loss spectra of stearic acid Langmuir-Blodgett monolayers revealed by isotopic effects, J. Electron. Spectrosc. 1998, 87, 221–230. [CrossRef]
- M. Diem, Modern Vibrational Spectroscopy and Micro-Spectroscopy, 2nd edition (John Wiley&Sons Ltd, The Atrium, 2015).
- NIST Standard Reference Database SRD Number 69. Last access to data: May 2023. [CrossRef]
- M. Rei Vilar, M. Schott, J.J. Pireaux, C. Grégoire, P.A. Thiry, R. Caudano, A. Lapp, A.M. Botelho do Rego, J. Lopes da Silva, Study of Polymer Film Surfaces by EELS Using Selectively Deuterated Polystyrene, Surf. Sci. 1987, 189/190, 927–934.
- S. Clerc, J.R. Dennison, R. Hoffmann, J. Abbott, On the computation of secondary electron emission models, IEEE Trans. Plasma Sci. 2006, 34, 2219–2225. [CrossRef]
- G. F. Dionne, Origin of secondary electron emission yield curve parameters, J. Appl. Phys. 1975, 46, 3347–3351. [CrossRef]
- N. Bundaleski, M. Belhaj, T. Gineste, O.M.N.D. Teodoro, Calculation of the angular dependence of the total electron yield, Vacuum 2015, 122, 255–259. [CrossRef]
Figure 1.
High resolution spectra of the C 1s state of (a) freshly cleaved HOPG, (b) reference a-C sample, (c) sample 0.2D, (d) sample 0.5D, (e) sample 1D and (f) sample 10D, and the corresponding fittings.
Figure 1.
High resolution spectra of the C 1s state of (a) freshly cleaved HOPG, (b) reference a-C sample, (c) sample 0.2D, (d) sample 0.5D, (e) sample 1D and (f) sample 10D, and the corresponding fittings.
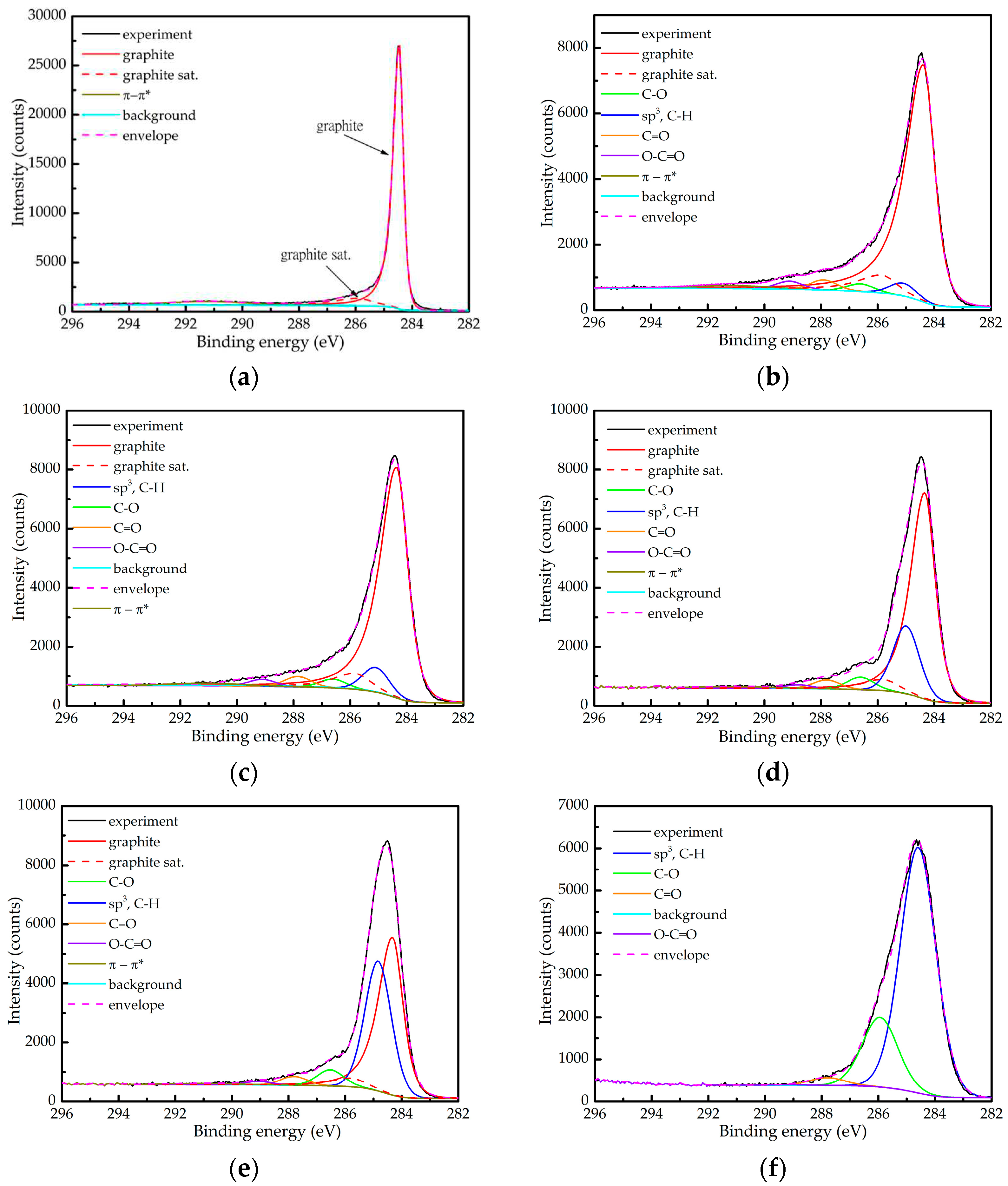
Figure 2.
Valence bands of HOPG and produced a-C coatings normalised to their values at binding energy of 2 eV.
Figure 2.
Valence bands of HOPG and produced a-C coatings normalised to their values at binding energy of 2 eV.
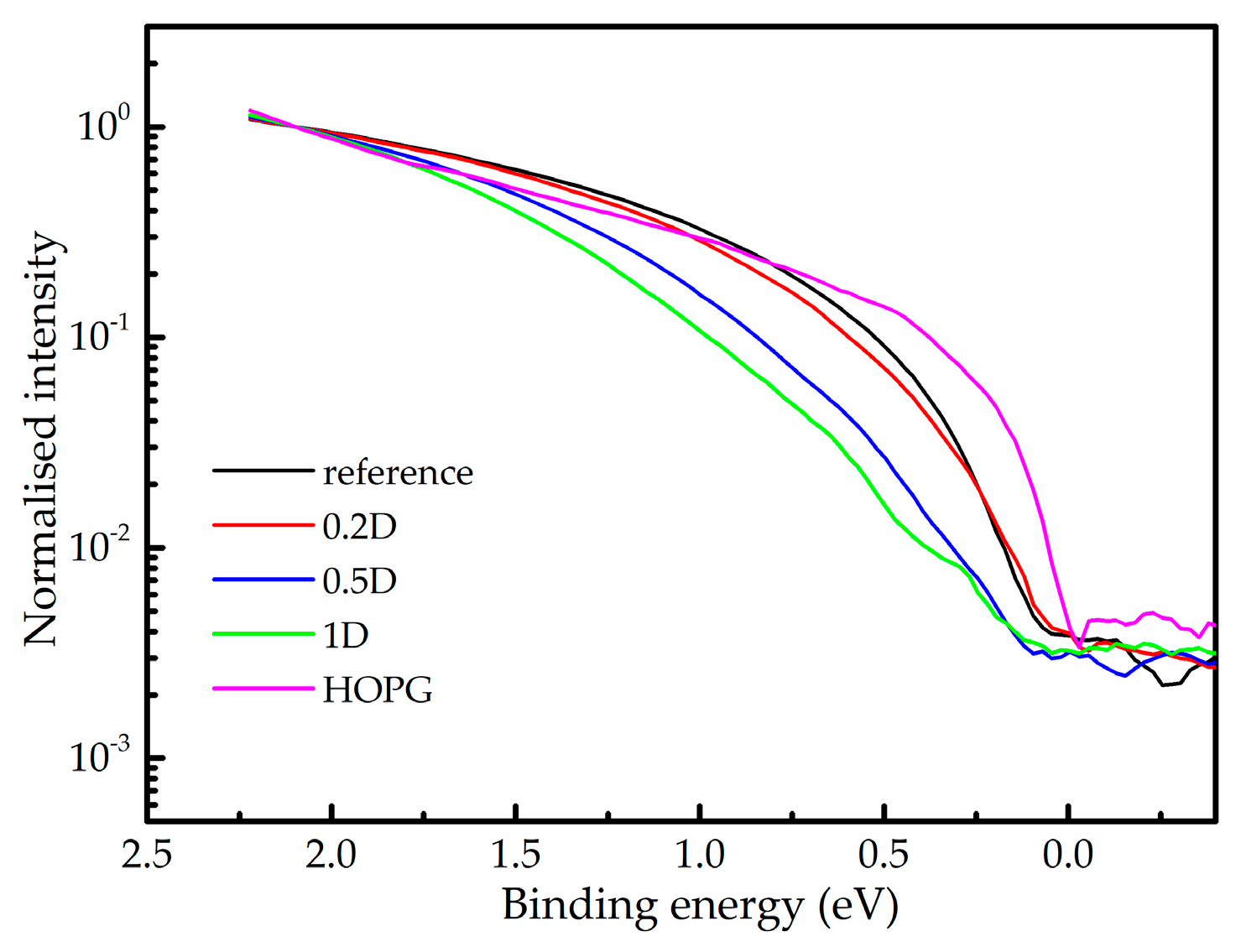
Figure 3.
(a) Raman spectra of the reference, 0.2D, 0.5D and 1D samples deposited on Si substrate measured by 0.7 W 532 nm laser beam; (b) Fitting illustration on the example of the sample 1D; (c) intensity ratio of the D and G bands ID/IG and position of the G band maximum σD vs. the relative H+D film content.
Figure 3.
(a) Raman spectra of the reference, 0.2D, 0.5D and 1D samples deposited on Si substrate measured by 0.7 W 532 nm laser beam; (b) Fitting illustration on the example of the sample 1D; (c) intensity ratio of the D and G bands ID/IG and position of the G band maximum σD vs. the relative H+D film content.
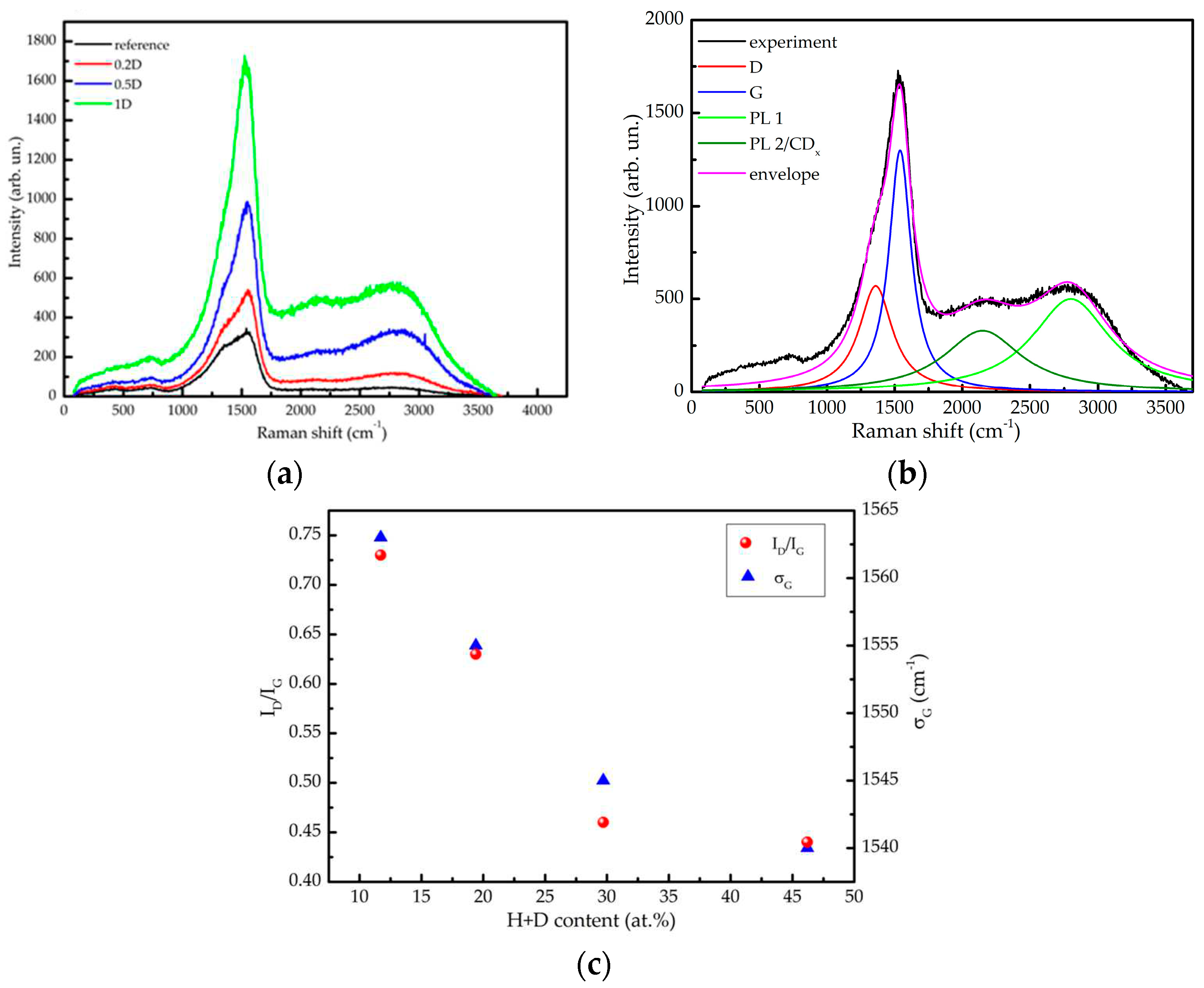
Figure 4.
Dependences of the Tauc gap and the SEYmax vs. the graphitic content. The dashed blue line represents the linear dependence of the Tauc gap estimated by Robertson [37]. The dashed red line is the prediction of the modified semi-empirical model for the SEY developed in this work.
Figure 4.
Dependences of the Tauc gap and the SEYmax vs. the graphitic content. The dashed blue line represents the linear dependence of the Tauc gap estimated by Robertson [37]. The dashed red line is the prediction of the modified semi-empirical model for the SEY developed in this work.
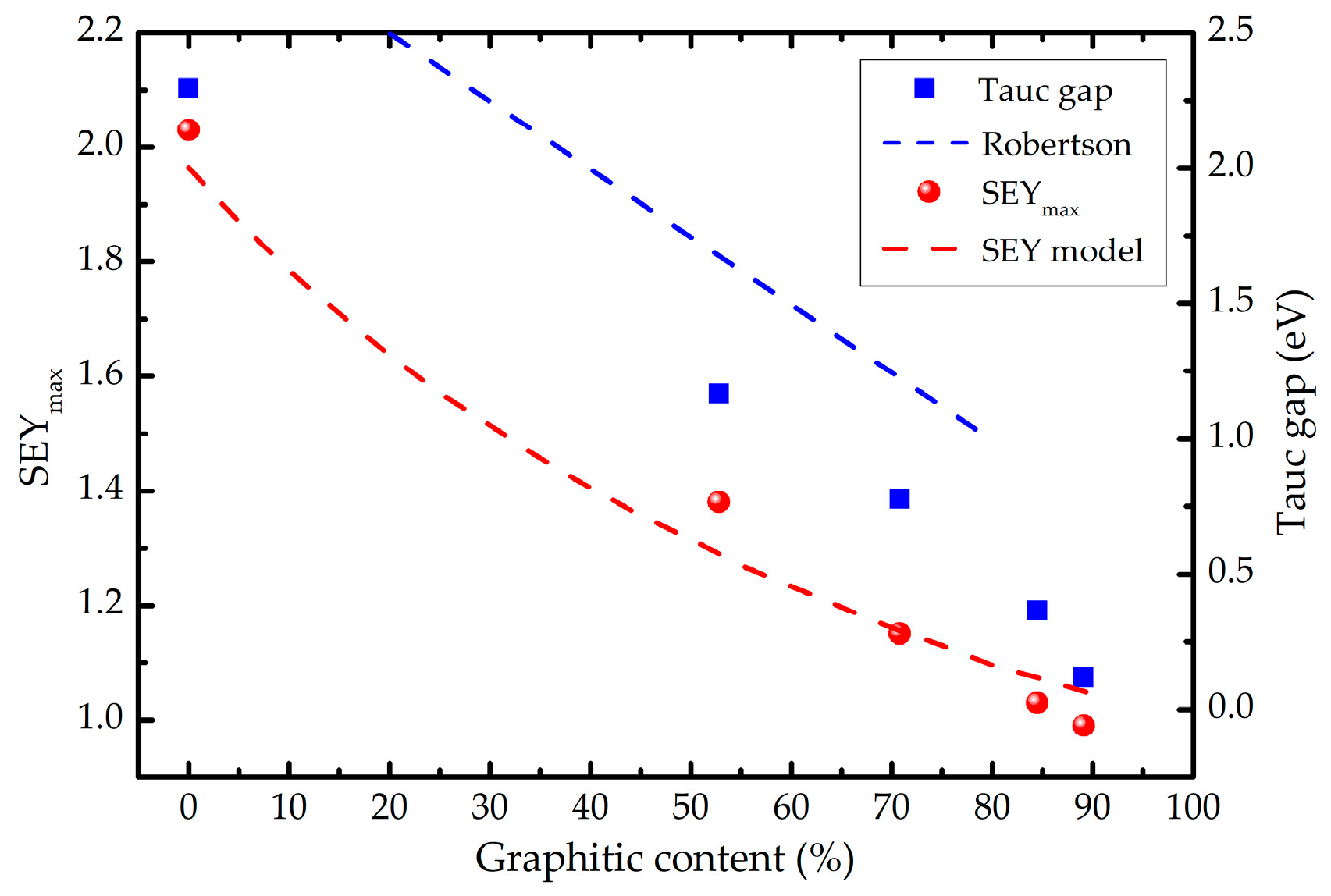

Table 1.
Composition (in at.%), maximum SEY and the Tauc gap of the a-C thin films [15].
Table 1.
Composition (in at.%), maximum SEY and the Tauc gap of the a-C thin films [15].
| Sample | pD2 (Pa) | C (%) | H (%) | D (%) | O (%) | SEYmax | ET (eV) | ρ(g/cm3) |
|---|---|---|---|---|---|---|---|---|
| Reference | 0 | 83.4 | 11.7 | 0 | 4.9 | 0.99 | 0.12 | 1.44 |
| 0.2D | 3.8·10-3 | 77.9 | 9.8 | 9.6 | 2.7 | 1.03 | 0.37 | 1.99 |
| 0.5D | 1.1·10-2 | 67.8 | 6.0 | 23.7 | 2.5 | 1.15 | 0.78 | 1.91 |
| 1D | 2.6·10-2 | 52.2 | 6.3 | 39.9 | 1.6 | 1.38 | 1.17 | 2.34 |
| 10D | 2.2·10-1 | 45.2 | 2.8 | 50.4 | 1.6 | 2.03 | 2.29 | 1.15 |
Table 2.
Summary of the C 1s line fittings and the relative H+D contents.
| Sample | α/β | graphitic (%) | sp3 C-H (%) | C-O (%) | C=O (%) | -(C=O)-O- (%) | H+D content (%) |
|---|---|---|---|---|---|---|---|
| reference | 0.45 | 88.14 | 3.29 | 1.91 | 2.55 | 2.01 | 11.7 |
| 0.2D | 0.50 | 85.17 | 6.43 | 2.41 | 2.57 | 1.73 | 19.4 |
| 0.5D | 0.58 | 71.09 | 20.4 | 3.75 | 2.46 | 1.05 | 29.7 |
| 1D | 0.58 | 52.75 | 38.11 | 4.83 | 2.46 | 0.96 | 46.2 |
| 10D | 1.00 | 0 | 75.99 | 21.53 | 2.35 | 0.13 | 53.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



