
Submitted:
03 July 2023
Posted:
04 July 2023
You are already at the latest version
Abstract
The syntheses of four novel syn-type tricyclic laddersiloxanes bearing eight or six alkenyl groups are presented. These compounds possess reactive alkenyl groups on both the bridged and side silicon atoms and their structures were determined by characterization using multinuclear NMR spectroscopy, mass spectrometry, and elemental analysis techniques. To investigate their reactivity, compounds were subjected to hydrosilylation using two different silanes, and the resulting fully hydrosilylated compounds were analyzed thoroughly. Remarkably, all the synthesized laddersiloxanes displayed high thermal stability, suggesting their potential as promising precursors for the development of new hybrid materials. Additionally, preliminary findings indicate the possibility of exploiting the reactivity difference between the alkenyl groups attached to the D- and T-unit silicon atoms for the synthesis of Janus molecules. These findings highlight the potential of the reported compounds as valuable building blocks in the construction of innovative materials.
Keywords:
tricyclic laddersiloxanes
; Janus molecules
; hydrosilylation
; syn-type conformation
; well-defined structures
1. Introduction
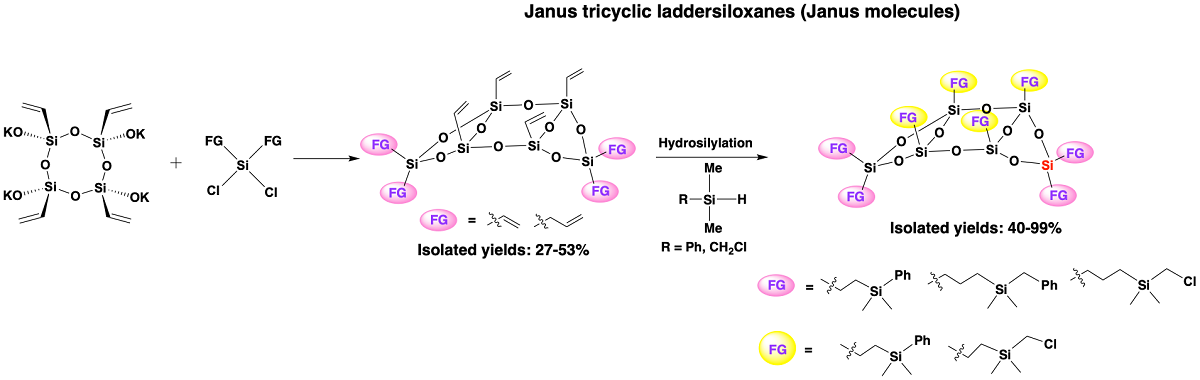
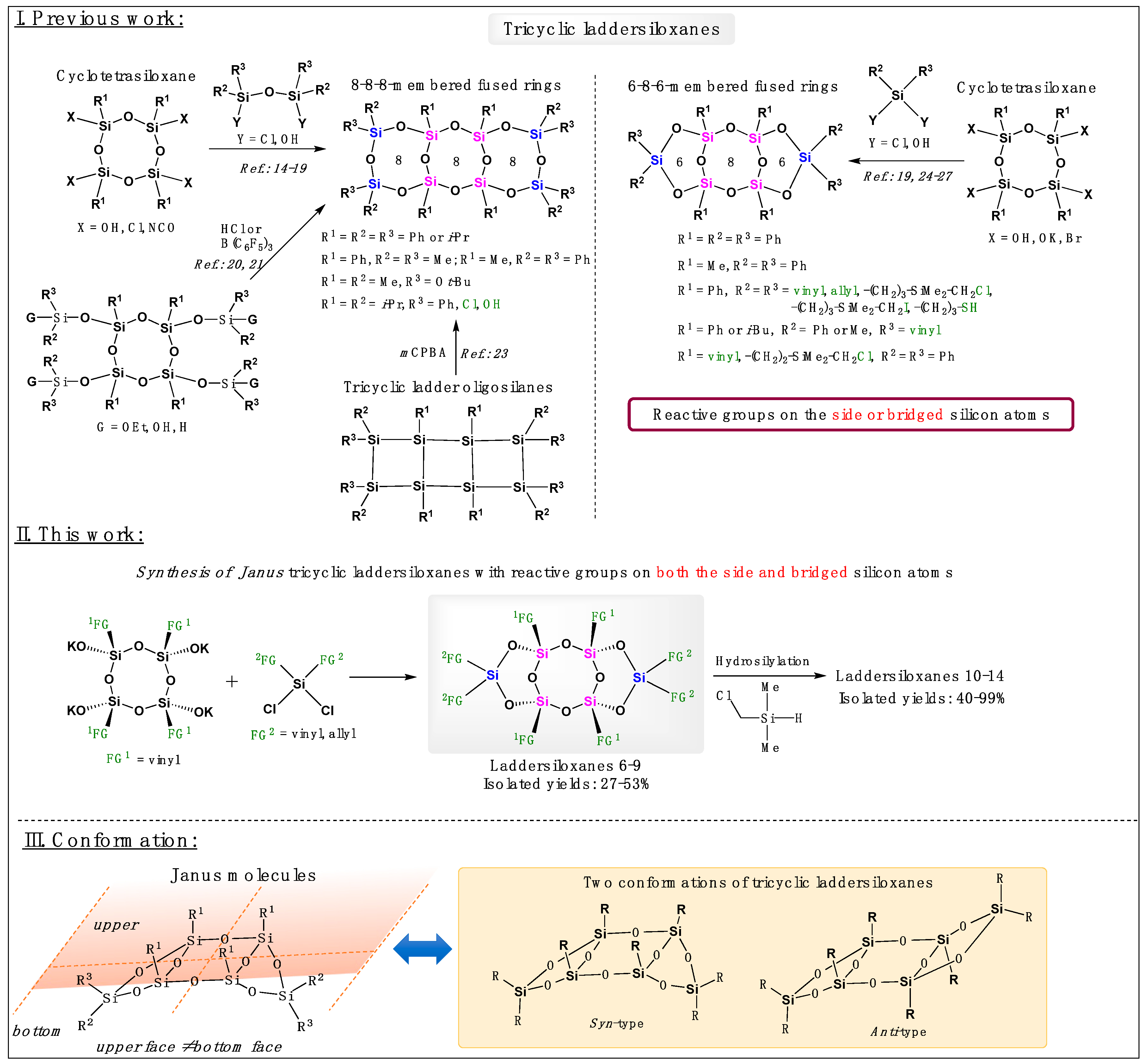
Silsesquioxanes are organosilicon compounds that consist primarily of silicon atoms bonded to three oxygen atoms and one organic substituent. Well-defined silsesquioxanes feature rigid, thermally stable, and chemically inert inorganic siloxane cores, with reactive and easily modifiable organic fragments attached to them [1,2]. These compounds exhibit unique hybrid properties, blending organic and inorganic characteristics, which make them promising building blocks for the development of hybrid materials for various applications [3,4,5,6,7,8,9]. Among them, laddersiloxanes, which are well-defined ladder-type silsesquioxanes, exhibit highly ordered double-chain structures and have the potential to form polymers [10,11]. Additionally, they possess the highest refractive indices among linear siloxanes, cyclic siloxanes, and cage silsesquioxanes [12]. Owing to these exceptional properties, laddersiloxanes have attracted increasing attention over the past decade. The exploration of laddersiloxanes dates back to the 1960s when Brown first proposed an example of this structure [13]. Over the years, improved synthetic routes and characterization methods have been established, leading to the discovery of laddersiloxanes with up to 9 fused rings [10]. Among them, the syn or anti-type tricyclic laddersiloxanes with 6-8-6 or 8-8-8-membered fused rings have been the most intensively investigated (Scheme 1, I.). The synthesis of 8-8-8 laddersiloxanes typically involves cyclotetrasiloxanes and dichloro- or dihydrosiloxanes [14,15,16,17,18,19]. More recently, these compounds have also been directly generated from silylated cyclotetrasiloxanes in the presence of HCl or borane catalyst [20,21]. Notably, this method enabled the synthesis of a unique 12-8-12 tricyclic laddersiloxane, starting from an extended cyclotetrasiloxane [22] Another approach to prepare 8-8-8 tricyclic laddersiloxanes is oxidation of the corresponding tricyclic ladder oligosilanes [23] For the synthesis of 6-8-6 tricyclic laddersiloxanes, cyclotetrasiloxanes and dichloro- or dihydroxysilanes can be employed [19,24,25,26,27]. Of particular interest are syn-type tricyclic laddersiloxanes, which exhibit a structure similar to that of Janus molecules (Scheme 1, III.). Like the Roman god Janus, these molecules possess two distinct faces and have gained significant attention in scientific research and various fields due to their asymmetric nature [28,29,30,31].
In 2019, the first synthesis and characterization of well-defined syn-type 6-8-6 tricyclic laddersiloxanes bearing four peripheral reactive substituents (vinyl or allyl groups) were reported [25]. The introduction of four chloro and four thiol substituents was successfully achieved through organic reactions like hydrosilylation and thiol-ene reactions [25,32]. Later, the synthesis of less symmetrical laddersiloxanes with only one vinyl group on each side silicon atom was described and these compounds were found to be suitable as monomers for polymer preparation [26,33]. The resulting polymers exhibited unconventional conjugation behavior and demonstrated potential for use in semiconducting materials. Furthermore, functional groups were introduced onto the bridged silicon atoms, expanding the range of possibilities [27]. However, despite these advancements, the exploration of syn-type tricyclic laddersiloxanes as Janus molecules remains relatively unexplored. Although previous studies have reported syn-type tricyclic laddersiloxanes with reactive groups on either side or the bridged silicon atoms, compounds featuring reactive groups on both the side and bridged silicon atoms have yet to be reported (Scheme1, I.).
In this work, we describe the synthesis of novel syn-type 6-8-6 tricyclic laddersiloxanes with reactive alkenyl groups on both the bridged and side silicon atoms (Scheme 1, II.). Moreover, the eight or six alkenyl groups on these laddersiloxanes can be fully functionalized through hydrosilylation with silanes containing reactive substituents, thereby demonstrating their potential as precursors for the preparation of new hybrid materials. Furthermore, a selective hydrosilylation trial was conducted on a Janus laddersiloxane bearing four vinyl groups on bridged silicon atoms and four allyl groups on side silicon atoms. The promising results obtained highlight the potential of this compound in the development of new Janus materials.
2. Results and Discussion
2.1. Preparation of Janus tricyclic laddersiloxanes bearing eight or six alkenyl groups
The targeted syn-type tricyclic laddersiloxanes were synthesized from all-cis-tetravinylcyclotetrasiloxanolate [ViSi(OK)O]4 (1) [27]. A mixture of the freshly prepared and pre-dried [ViSi(OK)O]4 (1), distilled triethylamine, and anhydrous THF was prepared and cooled to 0 ˚ C. Next, the corresponding solution of dichlorosilane (specifically, dichlorodivinylsilane (2), diallyldichlorosilane (3), dichloromethylvinylsilane (4), or dichlorophenylvinylsilane (5)) in anhydrous THF was added dropwise at 0 ˚ C under argon. After the addition, the reaction mixture was stirred for one hour at room temperature. Subsequently, the crude product obtained after extraction was purified using gel permeation chromatography (GPC), resulting in the isolation of pure target compounds 6, 7, 8, and 9, in yields ranging from 27 to 53% (Scheme 2). Importantly, the reaction conditions employed preserved the all-cis configuration of the cyclotetrasiloxanolate, and all the synthesized compounds (6-9) exhibited a syn-type conformation.
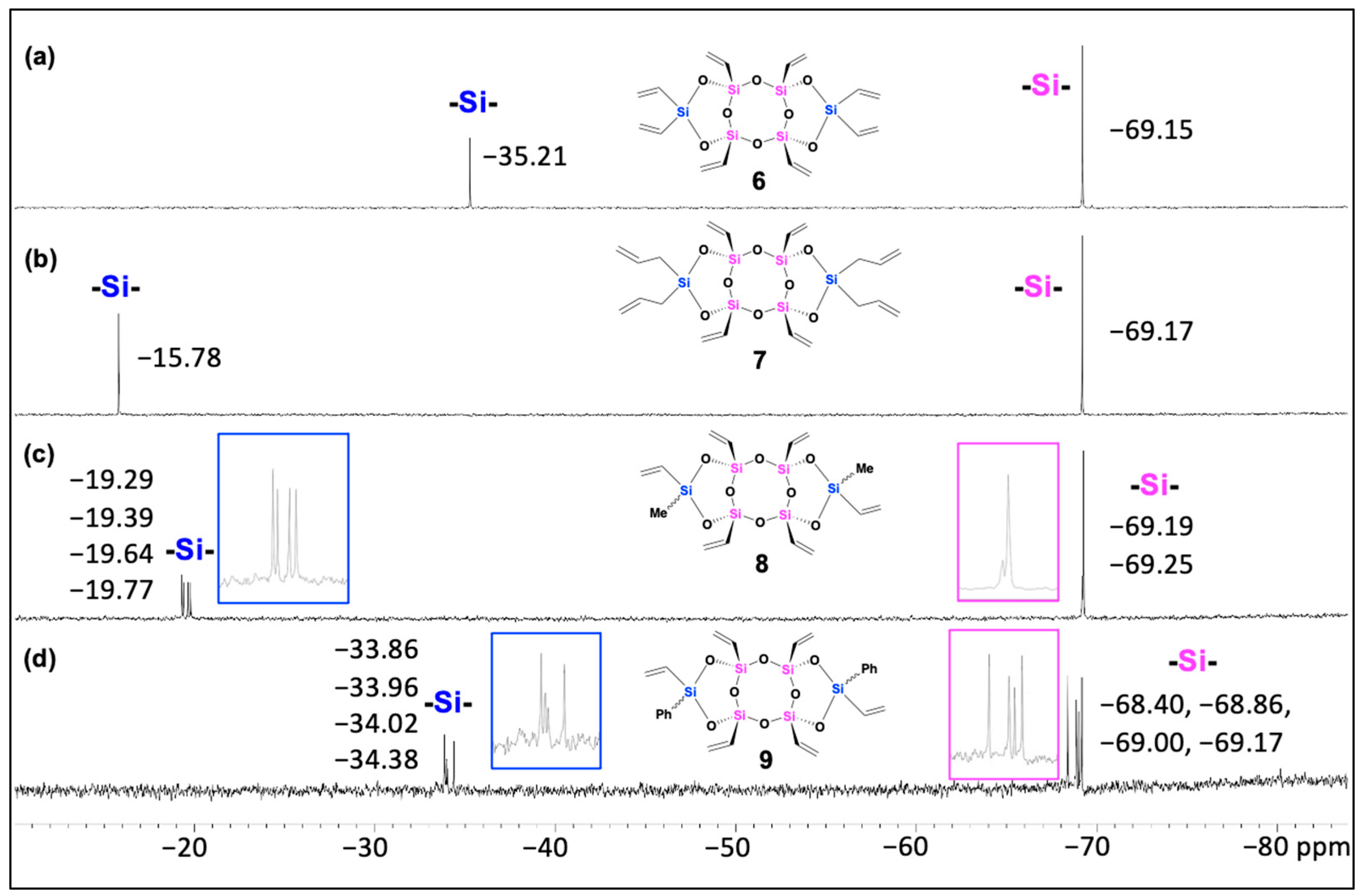
The 29Si NMR spectrum displayed two distinct peaks at −69.15 ppm and −35.21 ppm (Figure 1, a), corresponding to the bridged (T-unit) silicon atoms (Si in pink) and the side (D-unit) silicon atoms (Si in blue), respectively, consistent with previously reported analogous laddersiloxanes [25,27]. Upon analysis of laddersiloxane 6 using 1H NMR (see SI, Figure S1), we observed a range of signals from 5.95 to 6.16 ppm, corresponding to the vinyl groups. The 13C NMR spectrum demonstrated that the two vinyl substituents connected to the same D-unit silicon atom exhibited magnetic inequivalence (see SI, Figure S2), which is in accordance with previous reports on that type of structures [25]. There were four distinct signals for the carbon atoms of the peripheral vinyl groups, and a total of six carbon signals for compound 6.
Moving on to laddersiloxane 7, the 29Si NMR spectrum showed two single peaks at −69.17 (T-unit silicon) and −15.78 (D-unit silicon) ppm (Figure 1, b), matching well with the values reported for analogous laddersiloxanes [25,27]. The 1H NMR spectrum atoms (see SI, Figure S4) clearly distinguished signals assigned to the vinyl and allyl groups. Specifically, the multiplets at 4.93-5.04 ppm and 5.75-5.84 ppm represented the allyl groups connected to the D-unit silicon atoms, while the three clear doublet-doublet (dd) patterns at 5.95, 6.07, 6.14 ppm corresponded to the four chemically equivalent vinyl groups on the T-unit silicon atoms. Additionally, two distinctive doublet signals at 1.74 ppm and 1.80 ppm indicated the presence of two types of methylene groups adjacent to the D-unit silicon. The 13C NMR spectrum (see SI, Figure S5) exhibited two signals for the carbon atoms of the methylene groups and six signals for the carbon atoms of the olefinic moieties, confirming the syn-type conformation of tricyclic laddersiloxane 7.
Compounds 8 and 9 possessed two different substituents on the D-unit silicon atoms, resulting in less symmetrical structures. Each compound has three possible stereoisomers [26]. The 29Si NMR spectrum of compound 8 clearly revealed four singlets for the D-unit silicon atoms at −19.29, −19.39, −19.64, and −19.77 ppm, as well as T-unit silicon atoms between −69.19 and −69.25 ppm, indicating the presence of three isomers (Figure 1, c). This was further supported by the 1H and 13C NMR analyses (see SI, Figures S7 & S8), which displayed four signals for the methyl groups. The formation of three isomers of compound 9 was also observed in the corresponding 29Si NMR spectrum, which showed four singlets for both the D-unit (−33.86, −33.96, −34.02, −34.38 ppm) and T-unit (−68.40, −68.86, −69.00, −69.17 ppm) silicon atoms (Figure 2, d).
To verify the structures of these four laddersiloxanes, matrix-assisted laser desorption/ionization coupled time-of-flight (MALDI-TOF) mass spectrometry and elemental analysis were preformed, which yielded experimental results in good accordance with the calculated values (see SI, Figure S31-S34).
2.2. Full functionalization of Janus tricyclic laddersiloxanes bearing eight or six alkenyl groups
Syn-type tricyclic laddersiloxanes (6-9) possess either eight or six reactive double bonds, making them potentially valuable precursors for the creation of new hybrid materials. To assess the reactivity of the surrounding alkenyl substituents, compounds 6, 7, and 8 were hydrosilylated with dimethylphenylsilane (65 ˚C, 18 h) (Scheme 3). 1H NMR analysis of compounds 7 and 8 enabled to confirm that the reaction quantitatively occurred, with the disappearance of signals corresponding to alkenyl groups. In the case of compound 6, an increase of the temperature from 65 to 100 ˚C was required to complete the transformation of the vinyl groups. After purification using GPC, pure functionalized products 10 and 11 were successfully obtained (82 and 40%, respectively), while compound 12 was obtained quantitatively (99%) as a mixture of three stereoisomers without GPC purification.
The 29Si NMR spectrum of compound 10 revealed three groups of signals representing the silicon atoms of the T-unit (Si in pink), D-unit (Si in blue), and carbosilane moieties (Si in green) (Figure 2, a). The chemical shifts of T- and D-unit silicon atoms in compound 10 were downfield shifted to −55.06 and −7.94 ppm, respectively, compared to −69.15 and −35.21 ppm in compound 6, owing to the reduction of vinyl groups. The appearance of new signals at −1.12, −1.18, and −1.31 ppm in the 29Si NMR spectrum indicated the presence of silicon atoms in the three types of carbosilane moieties (Figure 2, a). Similarly, in compound 11, a downfield movement of both the chemical shifts of the T- and D-unit silicon atoms (−55.41 and −8.46 ppm) was observed compared to −69.17 and −15.78 ppm in compound 7, indicating the successful reduction of all the olefins (Figure 2, b). The 29Si NMR spectrum of compound 11 showed three singlets corresponding to the silicon atoms of the carbosilane parts: one (Si in green) at −1.16 ppm connected to the T-unit silicon atoms, and two (Si in red) at −3.80 and −3.91 ppm attached to the D-unit silicon atoms (Figure 2, b). In the case of compound 12, the 29Si NMR spectrum exhibited four distinct singlets at −54.95, −54.99, −55.31, and −55.39 ppm, indicating the presence of three isomers, with two patterns of signals around −1.1 and −6.5 ppm assigned to the carbosilane and D-unit silicon atoms, respectively (Figure 2, c). The 1H and 13C NMR spectra of compounds 10, 11, and 12 exhibited signals corresponding to methylene groups resulting from the hydrosilylation of olefinic substituents (see SI, Figures S13, S14, S16, S17, S19, and S20).
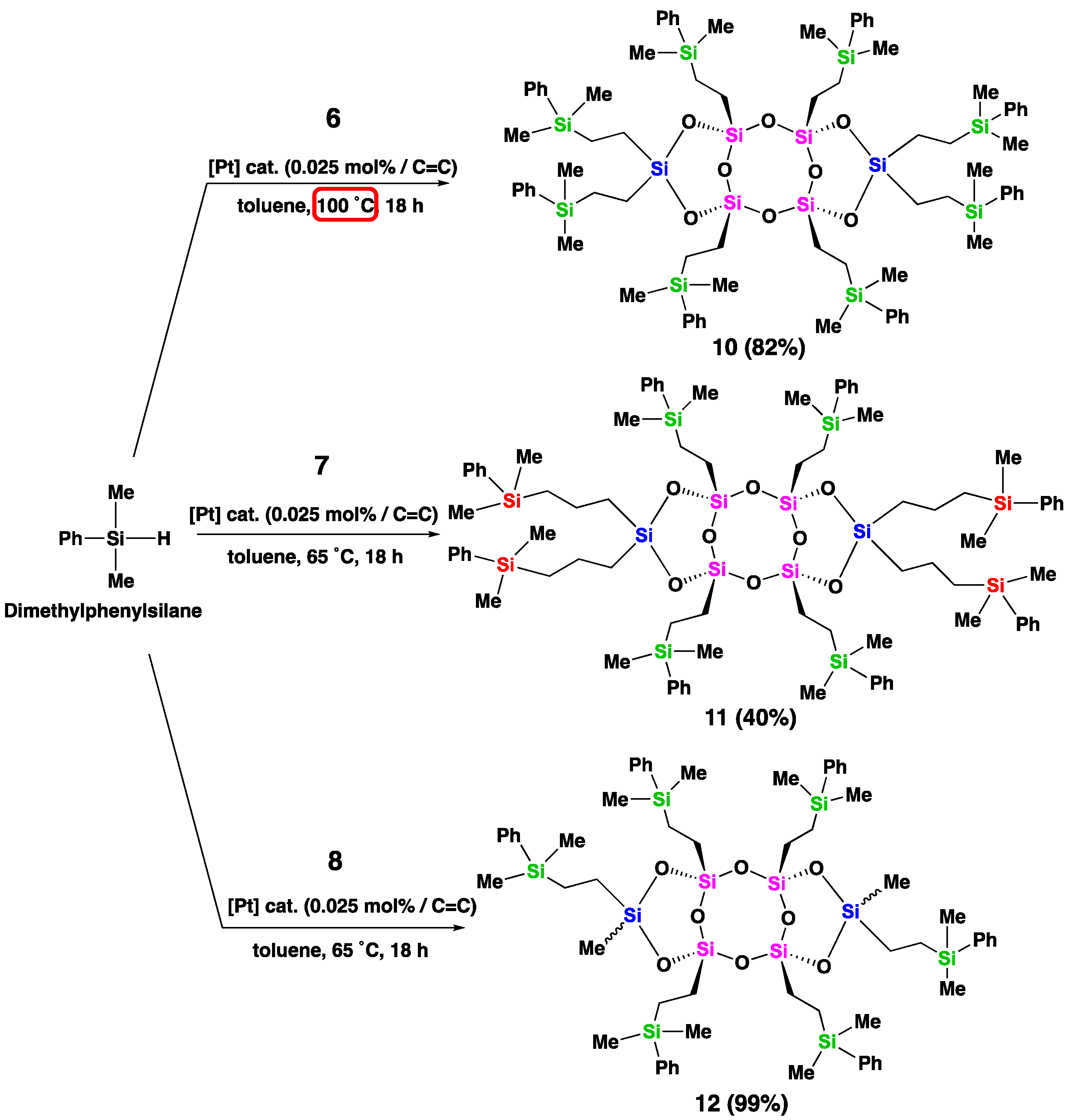
Compounds 7 and 8, which were more easily hydrosilylated, were selected for hydrosilylation with chloromethyl(dimethyl) silane, which contains a reactive chloro group. The reaction conditions were similar to those used for dimethylphenylsilane, but at a lower temperature (40 ˚C) (Scheme 4). After 18 h, the reaction with both compounds 7 and 8 was complete, and all olefinic groups were fully consumed. The 29Si NMR spectrum of compound 13 displayed downfield shifted peaks for the T- and D-unit silicon atoms (−55.58 and −8.58 ppm, respectively) compared to compound 7, as well as three peaks at 5.38 ppm assigned to the silicon atoms of carbosilane on the bridged silicon atoms, and 2.99 and 2.93 ppm assigned to the silicon atoms of carbosilane on the side silicon atoms (Figure 2, d). 1H NMR analysis of compound 13 (see SI, Figure S22) revealed three singlets at 2.79, 2.774, and 2.770 ppm, corresponding to the three types of protons adjacent to the chloro groups. The signals of the methylene groups between two silicon atoms, resulting from the reduction of olefins, appeared in the range of 0.63-0.75 ppm and 1.44-1.56 ppm. The hydrosilylated product (14) from compound 8 was formed as a mixture of three stereoisomers, which was supported by the multinuclear NMR results. For instance, in the 29Si NMR spectrum of 14, three distinct singlets were observed at −55.12, −55.22, and −55.58 ppm for the T-unit silicon atoms, and at −6.17, −6.57, and −6.92 ppm for the D-unit silicon atoms (Figure 2, e). It should be noted that theoretically, four singlets were expected, but two of them appeared to overlap in the experimental spectrum. The functionalized laddersiloxanes (10-14) obtained through hydrosilylation were also characterized using MALDI-TOF mass spectrometry and elemental analysis. The experimental results were consistent with the theoretically calculated results (see SI, Figures S35-S39).
2.3. Thermal properties of laddersiloxanes 6-14
To investigate the thermal properties of the synthesized laddersiloxanes (6-14), thermogravimetric analysis (TGA) was performed under nitrogen flow (250 mL min-1) at a rate of 10 ˚C min-1. Figure 3 and Figure 4 illustrate the TGA results for each laddersiloxane.
Laddersiloxanes (6-9) containing either eight or six alkenyl groups exhibited a two-step weight loss profile (Figure 3). The first step corresponds to the cleavage of the C-Si bond, whereas the second step could be attributed to the cleavage of the side Si-O bond. Compounds 7 and 9 demonstrated similar initial decomposition temperatures, both higher than that of compound 6 which in turn was higher than that of compound 8. Additionally, the decomposition temperatures at 5% weight loss of the initial mass (Td5) of compounds 7 and 9 were higher (196 ˚C and 198 ˚C, respectively) than that of compound 6 (172 ˚C), and compound 8 exhibited the lowest Td5 value (148 ˚C). Of particular interest is the residue weight of compound 6 at 1000˚C, which amounts to 80% of the initial weight, surpassing the weight percentage of the siloxane core (Si+O: 58%) by approximately 20%. This significant value suggests that compound 6 may undergo self-polymerization at high temperatures because of the presence of two adjacent vinyl groups on each side of the silicon atom. As a result, highly thermally stable polymers were formed.
The thermal stability of the hydrosilylated laddersiloxanes (10-14) was also explored using TGA. These compounds showed a one-step weight loss profile (Figure 4). The order of the initial decomposition temperatures was as follows: compound 10 ≈ compound 11 > compound 12 > compound 13 > compound 14. Compound 11 exhibited the highest Td5 value (422 ˚C), among all synthesized laddersiloxanes. Compounds 10 and 12 displayed comparable Td5 values at 340 ˚C and 348 ˚C, respectively. In the absence of phenyl groups, compounds 13 and 14 exhibited slightly lower thermal stabilities, with Td5 values of 333 and 323 ˚C, respectively. The similar effect of phenyl groups on thermal stability has already been observed [26].
2.4. Trials of selective functionalization of Janus tricyclic laddersiloxane 7
All the synthesized syn-type tricyclic laddersiloxanes exhibited distinct up and down faces when considering the plan of the central cyclotetrasiloxane core (Scheme 1). Of particular interest is compound 7, which possesses two types of reactive substituents: vinyl groups on the upper face and allyl groups on the bottom face. Although vinyl and allyl substituents share the same functional group, namely a double bond, their reactivity towards organic transformations may vary.
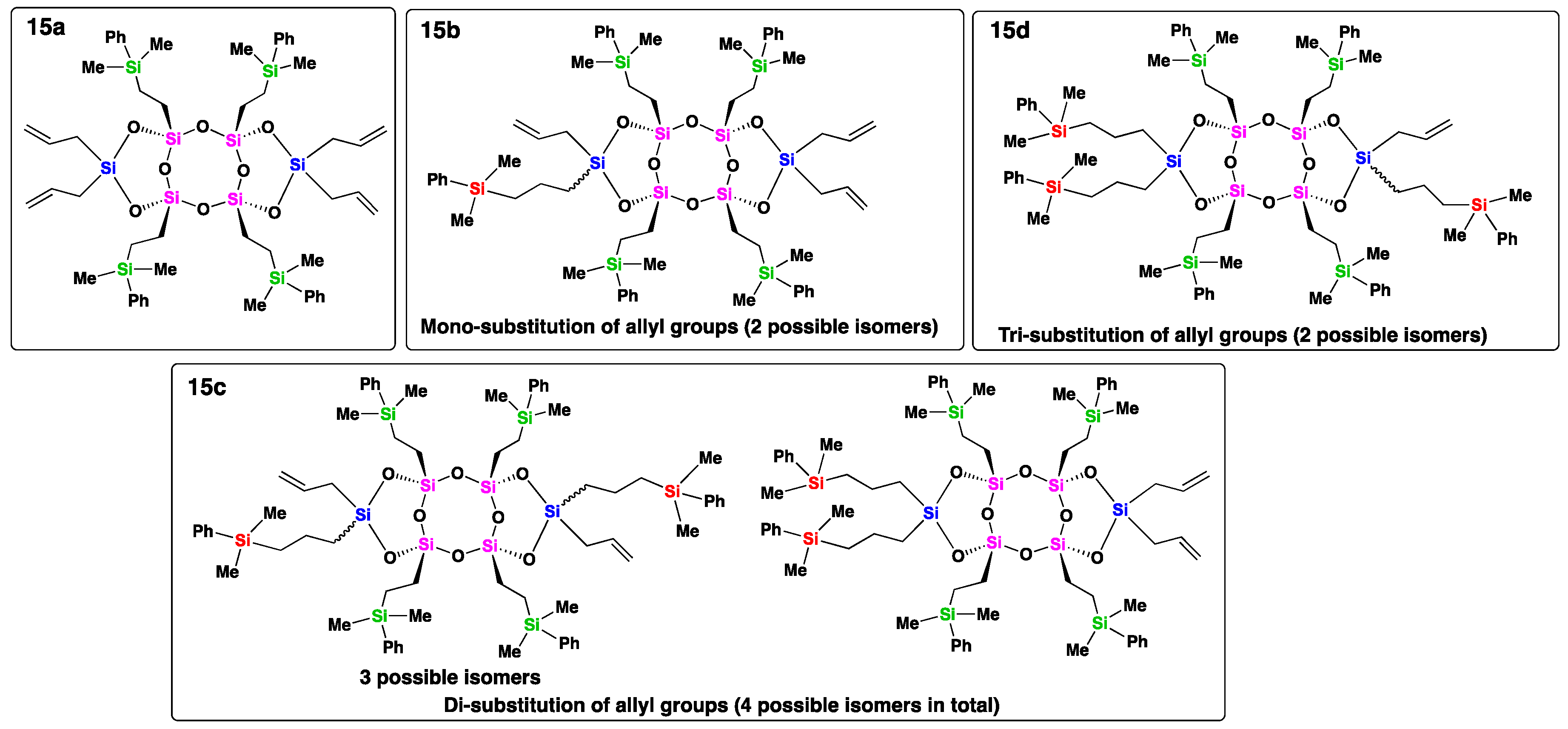
To demonstrate this concept, we conducted a hydrosilylation reaction using compound 7 and dimethylphenylsilane (insufficient amount) under reaction conditions similar to those described above. The 1H NMR analysis of the resulting crude product (15) revealed a complete disappearance of signals corresponding to the vinyl groups in the range of 5.95-6.14 ppm, whereas the peaks corresponding to the allyl groups remained partially observable (Figure 5). In the 29Si NMR spectrum of crude product 15 (see SI, Figure S30), the peak at −69.17 ppm assigned to the T-unit silicon atoms connected to the vinyl groups of compound 7 vanished, while new peaks appeared around −55.2 ppm and −1.1 ppm, corresponding to the T-unit and carbosilane silicon atoms resulting from the hydrosilylation of the vinyl groups. Additionally, the signals of D-unit silicon atoms (−15.78 ppm) connected to the allyl groups of compound 7 shifted to approximately −8.4 ppm, −12.9 ppm and −17.2 ppm, and new signals were observe around −3.8 ppm. These changes in the 29Si NMR chemical shifts indicate that after hydrosilylation with four equivalents of dimethylphenylsilane, all four vinyl groups on the bridged silicon atoms were completely substituted, while the four allyl groups on the side silicon atoms were only partially substituted, resulting in different substituted products (Figure 6). This result suggests that the reactivity of the vinyl groups on the bridged silicon atoms in the hydrosilylation reaction is higher than that of the allyl groups on the side silicon atoms. Furthermore, MALDI-TOF mass spectrometry was performed on crude product 15 (see SI, Figure S40), which supported the NMR results. Mass spectrometry analysis revealed that the main products were compounds 15b and 15c, corresponding to mono- and di-substituted allyl groups, respectively. Theoretically, compound 15b has two stereoisomers, whereas 15c has four stereoisomers. The presence of multiple signals for each type of silicon atom suggested the formation of stereoisomers for each compound. Additionally, minor products, such as compounds 15a and 15d, which contained four and three allyl groups, respectively, were detectable as well. Although the isolation of different compounds from crude product 15 has not been tested, these results indicate that the functionalization of compound 7 is tunable and that this compound holds promise as a precursor for the preparation of new Janus materials.
3. Materials and Methods
3.1. General considerations
All reactions were performed under an argon atmosphere using the standard Schlenk technique unless otherwise noted. THF and toluene were dried using an mBRAUN purification system. Triethylamine was distilled from potassium hydroxide and stored on potassium hydroxide under argon atmosphere with protection from light. Triethoxyvinylsilane was purchased from Asahikasei Wacker Silicon Co., Ltd.; dichlorodivinylsilane was purchased from Shin-Etsu Chemical Co., Ltd.; diallyldichlorosilane and dichlorophenylvinylsilane were purchased from Gelest, Inc.; dichloromethylvinylsilane, dimethylphenylsilane, and chloromethyl(dimethyl)silane were purchased from TCI Co., Ltd.; Karstedt’s catalyst (in xylene, 2% Pt) was purchased from Sigma-Aldrich. All the reagents were used as received without further purification.
Fourier transform nuclear magnetic resonance (NMR) spectra were obtained using a JEOL JNM-ECA 600 (1H at 600.17 MHz, 13C at 150.91 MHz, 29Si at 119.24 MHz) NMR instrument. For 1H NMR, chemical shifts were reported as δ units (ppm) relative to SiMe4 (TMS), and the residual solvent peaks were used as standards. For 13C NMR and 29Si NMR, chemical shifts were reported as δ units (ppm) relative to SiMe4 (TMS). The residual solvent peaks were used as standards and the spectra were obtained by complete proton decoupling. Matrix-assisted laser desorption/ionization coupled time-of-flight (MALDI-TOF) mass analyses were performed with a Shimadzu AXIMA Performance instrument using 2,5-dihydroxybenzoic acid (dithranol) as the matrix and AgNO3 as the ion source. All the reagents used were of analytical grade. Elemental analyses were performed at the Center for Material Research by Instrumental Analysis (CIA), Gunma University, Japan. Infrared (IR) spectra were measured using a Shimadzu IRSpirit FTIR spectrometer. TGA was performed using a Rigaku thermogravimetric analyzer (Thermoplus TG-8120). The investigations were carried out under nitrogen flow (250 mL min-1) or air flow (300 mL min-1) at a heating rate of 10 ˚C min-1. All samples were measured at temperatures ranging from 50 to 1000 ˚C, where they remained for 5 min. The weight loss and heating rate were continuously recorded during the experiment. Gel permeation chromatography (GPC) was performed using a Japan Analytical Industry LaboACE LC-5060.
3.2. Synthetic procedures of compounds 6-15
- Synthesis of 6-8-6 tricyclic laddersiloxane (6)
An argon-purged three necked flask equipped with a magnetic stirring bar and an addition funnel was charged with all-cis-[ViSi(OK)O]4 (0.5 g, 1.0 mmol), anhydrous THF (20 mL), and distilled Et3N (0.42 mL, 3.0 mmol). A solution of dichlorodivinylsilane (0.42 mL, 3.0 mmol) in anhydrous THF (45 mL) was introduced into the addition funnel under an argon atmosphere. Then, a solution of dichlorodivinylsilane was added dropwise to the flask at 0 ℃. After the addition, the reaction mixture was stirred at 25 ˚C for 1 h. Then, a saturated NH4Cl aqueous solution was added to the reaction medium to neutralize Et3N, and chloroform was added to extract the product three times. The gathered organic layer was then washed with brine three times, dried over anhydrous Na2SO4, and concentrated on a rotary evaporator to afford the crude product as a slightly yellow viscous oil, which was purified by GPC (CHCl3) to give pure product 6 as a colorless solid (0.15 g, 0.30 mmol, 30%).
- Synthesis of 6-8-6 tricyclic laddersiloxane (7)
An argon-purged, three necked, round bottom flask equipped with a magnetic stirring bar and an addition funnel was charged with all-cis-[ViSi(OK)O]4 (0.5 g, 1.0 mmol), anhydrous THF (20 mL), and distilled Et3N (0.42 mL, 3.0 mmol). A solution of diallyldichlorosilane (0.51 mL, 3.0 mmol) in anhydrous THF (45 mL) was introduced into the addition funnel under an argon atmosphere. Then, a solution of diallyldichlorosilane was added dropwise to the flask at 0 ℃. The reaction mixture was stirred at 25 ˚C for 1 h. Then, a saturated NH4Cl aqueous solution was added to the reaction medium to neutralize Et3N, and chloroform was added to extract the product three times. The gathered organic layer was then washed with brine three times, dried over anhydrous Na2SO4, and concentrated on a rotary evaporator to afford the crude colorless product as a colorless viscous oil, which was purified by GPC (CHCl3) to give pure product 7 as a colorless liquid (0.30 g, 0.53 mmol, 53%).
- Synthesis of 6-8-6 tricyclic laddersiloxane (8)
An argon-purged three necked flask equipped with a magnetic stirring bar and an addition funnel was charged with all-cis-[ViSi(OK)O]4 (0.5 g, 1.0 mmol), anhydrous THF (25 mL), and distilled Et3N (0.56 mL, 3.75 mmol). A solution of dichloromethylvinylsilane (0.39 mL, 3.0 mmol) in anhydrous THF (45 mL) was introduced into the addition funnel under an argon atmosphere. Then, a solution of dichloromethylvinylsilane was added dropwise to the flask at 0 ℃. The reaction mixture was stirred at 25 ˚C for 1 h. Then, a saturated NH4Cl aqueous solution was added into the reaction medium to neutralize Et3N, and chloroform was added to extract the product three times. The gathered organic layer was then washed with brine three times, dried over anhydrous Na2SO4, and concentrated on a rotary evaporator to afford the crude as a slightly yellow viscous oil, which was purified by GPC (CHCl3) to give product 8 as a colorless solid (0.20 g, 0.4 mmol, 41%).
- Synthesis of 6-8-6 tricyclic laddersiloxane (9)
An argon-purged three necked flask equipped with a magnetic stirring bar and an addition funnel was charged with all-cis-[ViSi(OK)O]4 (0.5 g, 1.0 mmol), anhydrous THF (20 mL), and distilled Et3N (0.51 mL, 3.0 mmol). A solution of dichlorophenylvinylsilane (0.39 mL, 3.0 mmol) in anhydrous THF (45 mL) was introduced into the addition funnel under an argon atmosphere. Then, a solution of dichlorophenylvinylsilane was added dropwise to the flask at 0 ℃. The reaction mixture was stirred at 25 ˚C for 1 h. Then, a saturated NH4Cl aqueous solution was added into the reaction medium to neutralize Et3N, and chloroform was added to extract the product three times. The gathered organic layer was then washed with brine three times, dried over anhydrous Na2SO4, and concentrated on a rotary evaporator to afford the crude product as white wax, which was purified by GPC (CHCl3) to give pure product 9 as a colorless solid (0.166 g, 27%).
- Synthesis of 6-8-6 tricyclic laddersiloxane (10):
An argon-purged Schlenk equipped with a stir bar was charged with 6 (0.026 g, 0.05 mmol), anhydrous toluene (0.3 mL), and dimethylphenylsilane (92 µL, 0.6 mmol). Karstedt’s catalyst (2% Pt, commercial bottle, diluted 100 times in anhydrous toluene under argon, 115 μL, 0.1 μmol Pt) was added to the mixture under an argon atmosphere at 25 °C. After addition, the mixture was heated to 100 °C and stirred at 100 ˚C for 20 h. After the reaction, the mixture was cooled to room temperature and passed through a silica plug, which was then washed with toluene and dichloromethane. The solvents were removed using a rotary evaporator to afford the crude product as a viscous colorless oil (87 mg). The crude product was purified using GPC (eluent: CHCl3), and pure product 10 was obtained as a colorless liquid (66 mg, 82%).
- Synthesis of 6-8-6 tricyclic laddersiloxane (11):
An argon-purged Schlenk equipped with a stir bar was charged with 7 (0.028 g, 0.050 mmol), anhydrous toluene (0.30 mL), and dimethylphenylsilane (92 µL, 0.60 mmol). Karstedt’s catalyst (2% Pt, commercial bottle, diluted 100 times in anhydrous toluene under argon, 115 μL, 0.1 μmol Pt) was added to the mixture under an argon atmosphere at 25 °C. After addition, the mixture was heated to 65 °C and stirred at 65 °C for 18 h. After the reaction, the mixture was cooled to room temperature and passed through a silica plug, which was then washed with toluene and dichloromethane. The solvents were removed using a rotary evaporator to obtain viscous brown oil (90 mg). The crude product was purified using GPC (CHCl3), and pure product 11 was obtained as a colorless liquid (32 mg, 40%).
- Synthesis of 6-8-6 tricyclic laddersiloxane (12):
An argon-purged Schlenk equipped with a stir bar was charged with 8 (0.024 g, 0.05 mmol), anhydrous toluene (0.3 mL), and dimethylphenylsilane (61 µL, 0.45 mmol). Karstedt’s catalyst (2% Pt, commercial bottle, diluted 100 times in anhydrous toluene under argon, 57 μL, 0.05 μmol Pt) was added to the mixture under an argon atmosphere at 25 °C. After addition, the mixture was heated to 65 °C and stirred at 65 ˚C for 18 h. After the reaction, the mixture was cooled to room temperature and passed through a silica plug, which was then washed with toluene and dichloromethane. The solvents were removed using a rotary evaporator to afford pure product 12 as viscous yellow oil (64 mg, 99%).
- Synthesis of 6-8-6 tricyclic laddersiloxane (13):
An argon-purged Schlenk equipped with a stir bar was charged with 7 (0.028 g, 0.050 mmol), anhydrous toluene (0.30 mL), and chloromethyl(dimethyl)silane (73 µL, 0.60 mmol). Karstedt’s catalyst (2% Pt, commercial bottle, diluted 100 times in anhydrous toluene under argon, 115 μL, 0.1 μmol Pt) was added to the mixture under an argon atmosphere at 25 °C. After addition, the mixture was heated to 40 °C and stirred at 40 °C for 18 h. After the reaction, the mixture was cooled to room temperature and passed through a silica plug, which was then washed with toluene and dichloromethane. The solvents were removed using a rotary evaporator to give 13 as a colorless solid (42 mg, 60%).
- Synthesis of 6-8-6 tricyclic laddersiloxane (14):
An argon-purged Schlenk equipped with a stir bar was charged with 8 (0.024 g, 0.05 mmol), anhydrous toluene (0.3 mL), and chloromethyl(dimethyl)silane (55 µL, 0.45 mmol). Karstedt’s catalyst (2% Pt, commercial bottle, diluted 100 times in anhydrous toluene under argon, 57 μL, 0.05 μmol Pt) was added to the mixture under an argon atmosphere at 25 °C. After addition, the mixture was heated to 40 °C and stirred at 40 ˚C for 18 h. After the reaction, the mixture was cooled to room temperature and passed through a silica plug, which was then washed with toluene and dichloromethane. The solvents were removed using a rotary evaporator to afford pure product 14 as viscous colorless oil (57 mg, 99%).
- Synthesis of 6-8-6 tricyclic laddersiloxane (15):
An argon-purged Schlenk equipped with a stir bar was charged with 7 (0.056 g, 0.1 mmol), anhydrous toluene (0.3 mL), and dimethylphenylsilane (92 µL, 0.6 mmol). Karstedt’s catalyst (2% Pt, commercial bottle, diluted 100 times in anhydrous toluene under argon, 57 μL, 0.05 μmol Pt) was added to the mixture under an argon atmosphere at 25 °C. After addition, the mixture was heated to 65 °C and stirred at 65 ˚C for 18 h. After the reaction, the mixture was cooled to room temperature and passed through a silica plug, which was then washed with toluene and dichloromethane. The solvents were removed using a rotary evaporator to afford crude product 15 as viscous yellow oil (135 mg).
4. Conclusions
In conclusion, this study presents the successful synthesis and comprehensive characterization of four novel syn-type Janus tricyclic laddersiloxanes (6-9) bearing either eight or six alkenyl groups. The structures of these compounds were determined using multinuclear NMR spectroscopy, mass spectrometry, and elemental analysis techniques. Furthermore, the hydrosilylation reactions of compounds 6-8 with two different silanes were successfully carried out, resulting in the formation of fully hydrosilylated compounds (10-14). Notably, all the synthesized laddersiloxanes exhibited high thermal stability, indicating their potential as promising precursors for the development of new hybrid materials. Moreover, preliminary results indicate that the possibility of exploiting the reactivity difference between the alkenyl groups attached to the D- or T-unit silicon atoms for the development of Janus materials. This opens opportunities for further exploration and utilization of compound 7 in the design and fabrication of innovative materials. Overall, this is the first time that syn-type tricyclic laddersiloxanes with reactive groups on both bridged and side silicon atoms have been synthesized. The findings presented in this study contribute to expanding knowledge in the field of laddersiloxanes and pave the way for future research and application opportunities in materials science and related disciplines.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. characterization data for pure synthetic compounds 6-14, 1H, 13C, 29Si NMR and MALDI-TOF mass spectra of compounds 6-15 (Figure S1-S40); thermogravimetry/differential thermal analysis (TG/DTA) spectra for compounds 6-14 (Figure S41-S49); Table S1: Thermal properties for compounds 6-14 under N2.
Author Contributions
Conceptualization: A. O., Y. L., M. U.; Methodology: Y. L., A. O.; Formal analysis and investigation: Y. L., M. T., N. T., A. O.; Writing – original draft preparation: Y. L.; Writing – review and editing: A. O., M. U.; Funding acquisition: A. O., M. U.
Funding
This research was funded by New Energy and Industrial Technology Development Organization (NEDO, project No. JPNP06046) for funding. CNRS is greatly thank for supporting the collaboration (International Research Project between Institute Charles Gerhardt Montpellier and Gunma University).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data and material described in this work are available in this article or in a Supplementary Materials.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Not available.
References
- Baney, R.H.; Itoh, M.; Sakakibara, T. Silsesquioxanes. Chem. Rev. 1995, 95, 1409-1430.
- Kickelbick, G. Silsesquioxanes in Functional Molecular Silicon Compounds I; Structure and Bonding; Scheschkewitz, D., Ed.; Springer: Berlin/Heidelberg, Germany, 2014; 155, pp. 1-28.
- Cordes, D.B.; Lickiss, P.D.; Rataboul, F. Recent Developments in the Chemistry of Cubic Polyhedral Oligosilsesquioxanes. Chem. Rev. 2010, 110, 2081-2173. [CrossRef]
- Laine, R.M.; Roll, M.F. Polyhedral Phenylsilsesquioxanes. Macromolecules 2011, 44, 1073-1109. [CrossRef]
- Zhou, H.; Ye, Q.; Xu, J. Polyhedral oligomeric silsesquioxane-based hybrid materials and their applications. Mater. Chem. Front. 2017, 1, 212-230. [CrossRef]
- Chen, F.; Lin, F.; Zhang, Q.; Cai, R.; Wu, Y.; Ma, X. Polyhedral Oligomeric Silsesquioxane Hybrid Polymers: Well-Defined Architectural Design and Potential Functional Applications. Macromol. Rapid Commun. 2019, 40, 1900101. [CrossRef]
- Du, Y.; Liu, H. Cage-like silsesquioxanes-based hybrid materials. Dalton Trans. 2020, 49, 5396-5405. [CrossRef]
- Calabrese, C.; Aprile, C.; Gruttadauria, M.; Giacalone, F. POSS nanostructures in catalysis. Catal. Sci. Technol. 2020, 10, 7415-7447. [CrossRef]
- Dudziec, B.; Marciniec, B. Double-decker Silsesquioxanes: Current Chemistry and Applications. Curr. Org. Chem. 2017, 21, 2794-2813. [CrossRef]
- Unno, M.; Suto, A.; Matsumoto, T. Laddersiloxanes−silsesquioxanes with defined ladder structure. Russ. Chem. Rev. 2013, 82, 289-302. [CrossRef]
- Kim, M. J.; Heo, Y. M.; Cho, J. H. Ladder-type silsesquioxane copolymer gate dielectrics for gating solution-processed IGZO field-effect transistors. Org. Electron. 2017, 43, 41-46. [CrossRef]
- Endo, H.; Takeda, N.; Takanashi, M.; Imai, T.; Unno, M. Refractive Indices of Silsesquioxanes with Various Structures. Silicon 2015, 7, 127-132. [CrossRef]
- Brown Jr., J.F.; Vogt Jr., L.H.; Katchman, A.; Eustance, J.W.; Kiser, K.M.; Krantz, K.W. Double Chain Polymers of Phenylsilsesquioxane. J. Am. Chem. Soc. 1960, 82, 6194-6195.
- Unno, M.; Suto, A.; Takada, K.; Matsumoto, H. Synthesis of Ladder and Cage Silsesquioxanes from 1,2,3,4-Tetrahydroxycyclotetrasiloxane. Bull. Chem. Soc. Jpn. 2000, 73, 215-220. [CrossRef]
- Unno, M.; Suto, A.; Matsumoto, H. Pentacyclic Laddersiloxane. J. Am. Chem. Soc. 2002, 124, 1574-1575. [CrossRef]
- Suyama, K.; Gunji, T.; Arimitsu, K.; Abe, Y. Synthesis and Structure of Ladder Oligosilsesquioxanes: Tricyclic Ladder Oligomethylsilsesquioxanes. Organometallics 2006, 25, 5587-5593. [CrossRef]
- Unno, M.; Matsumoto, T.; Matsumoto, H. Synthesis of laddersiloxanes by novel stereocontrolled approach. J. Organomet. Chem. 2007, 692, 307-312. [CrossRef]
- Seki, H.; Abe, Y.; Gunji, T. Stereochemistry of the reaction of cis, trans, cis-2,4,6,8-tetraisocyanato-2,4,6,8-tetramethylcyclotetrasiloxane with triphenylsilanol and 1,1,3,3-tetraphenyldisiloxane-1,3-diol. J. Organomet. Chem. 2011, 696, 846-851. [CrossRef]
- Endo, H.; Takeda, N.; Unno, M. Synthesis and Properties of Phenylsilsesquioxanes with Ladder and Double-Decker Structures. Organometallics 2014, 33, 4148-4151. [CrossRef]
- Sugiyama, T.; Shiba, H.; Yoshikawa, M.; Wada, H.; Shimojima, A.; Kuroda, K. Synthesis of Polycyclic and Cage Siloxanes by Hydrolysis and Intramolecular Condensation of Alkoxysilylated Cyclosiloxanes. Chem. Eur. J. 2019, 25, 2764-2772. [CrossRef]
- Chaiprasert, T.; Liu, Y.; Takeda, N.; Unno, M. Janus ring siloxane: versatile precursor of extended Janus ring and tricyclic laddersiloxanes. Dalton Trans. 2020, 49, 13533-13537. [CrossRef]
- Chaiprasert, T.; Liu, Y.; Intaraprecha, P.; Kunthom, R.; Takeda, N.; Unno, M. Synthesis of Tricyclic Laddersiloxane with Various Ring Sizes (Bat Siloxane). Macromol. Rapid Commun. 2021, 42, 2000608.
- Unno, M.; Tanaka, R.; Tanaka, S.; Takeuchi, T.; Kyushin, S.; Matsumoto, H. Oligocyclic Ladder Polysiloxanes: Alternative Synthesis by Oxidation. Organometallics 2005, 24, 765-768. [CrossRef]
- Unno, M.; Chang, S.; Matsumoto, H. cis-trans-cis-Tetrabromotetramethylcyclotetrasiloxane: a Versatile Precursor of Ladder Silsesquioxanes. Bull. Chem. Soc. Jpn. 2005, 78, 1105-1109. [CrossRef]
- Liu, Y.; Onodera, K.; Takeda, N.; Ouali, A.; Unno, M. Synthesis and Characterizatioin of Functionalizable Silsesquioxanes with Ladder-type Structures. Organometallics 2019, 38, 4373-4376.
- Liu, Y.; Endo, A.; Zhang, P.; Takizawa, A.; Takeda, N.; Ouali, A.; Unno, M. Synthesis, Characterization, and Reaction of Divinyl-substituted Laddersiloxanes. Silicon 2022, 14, 2723-2730.
- Liu, Y.; Katano, M.; Yingsukkamol, P.; Takeda, N.; Unno, M.; Ouali, A. Tricyclic 6-8-6 laddersiloxanes derived from all-cis-tetravinylcyclotetrasiloxanolate: Synthesis, characterization and reactivity. J. Organomet. Chem. 2022, 959, 122213.
- Pang, X.; Wan, C.; Wang, M.; Lin, Z. Strictly Biphasic Soft and Hard Janus Structures: Synthesis, Properties, and Applications. Angew. Chem. Int. Ed. 2014, 53, 5524-5538. [CrossRef]
- Yi, Y.; Sanchez, L.; Gao, Y.; Yu, Y. Janus particles for biological imaging and sensing. Analyst 2016, 141, 3526-3539. [CrossRef]
- Ng, S.-W.; Noor, N.; Zheng, Z. Graphene-based two-dimensional Janus materials. NPG Asia Materials 2018, 10, 217-237. [CrossRef]
- Peng, Z.; Huang, J.; Guo, Z. Anisotropic Janus materials: from micro-/nanostructures to applications. Nanoscale 2021, 13, 18839-18864.
- Liu, Y.; Kigure, M.; Okawa, R.; Takeda, N.; Unno, M.; Ouali, A. Synthesis and characterization of tetrathiol-substituted double-decker or ladder silsesquioxane nano-cores. Dalton Trans. 2021, 50, 3473-3478. [CrossRef]
- Guan, J.; Sun, Z.; Ansari, R.; Liu, Y.; Endo, A.; Unno, M.; Ouali, A.; Mahbub, S.; Furgal, J.C.; Yodsin, N.; Jungsuttiwong, S.; Hashemi, D.; Kieffer, J.; Laine, R.M. Conjugated Copolymers That Shouldn’t Be. Angew. Chem. Int. Ed. 2021, 60, 11115-11119. [CrossRef]
Scheme 1.
Synthesis of tricyclic laddersiloxanes and their conformations.
Scheme 2.
Synthesis of syn-type tricyclic laddersiloxanes (6-9) using all-cis-tetravinylcyclotetrasiloxanolate [ViSi(OK)O]4 (1) and dichlorosilanes (2-5).
Scheme 2.
Synthesis of syn-type tricyclic laddersiloxanes (6-9) using all-cis-tetravinylcyclotetrasiloxanolate [ViSi(OK)O]4 (1) and dichlorosilanes (2-5).
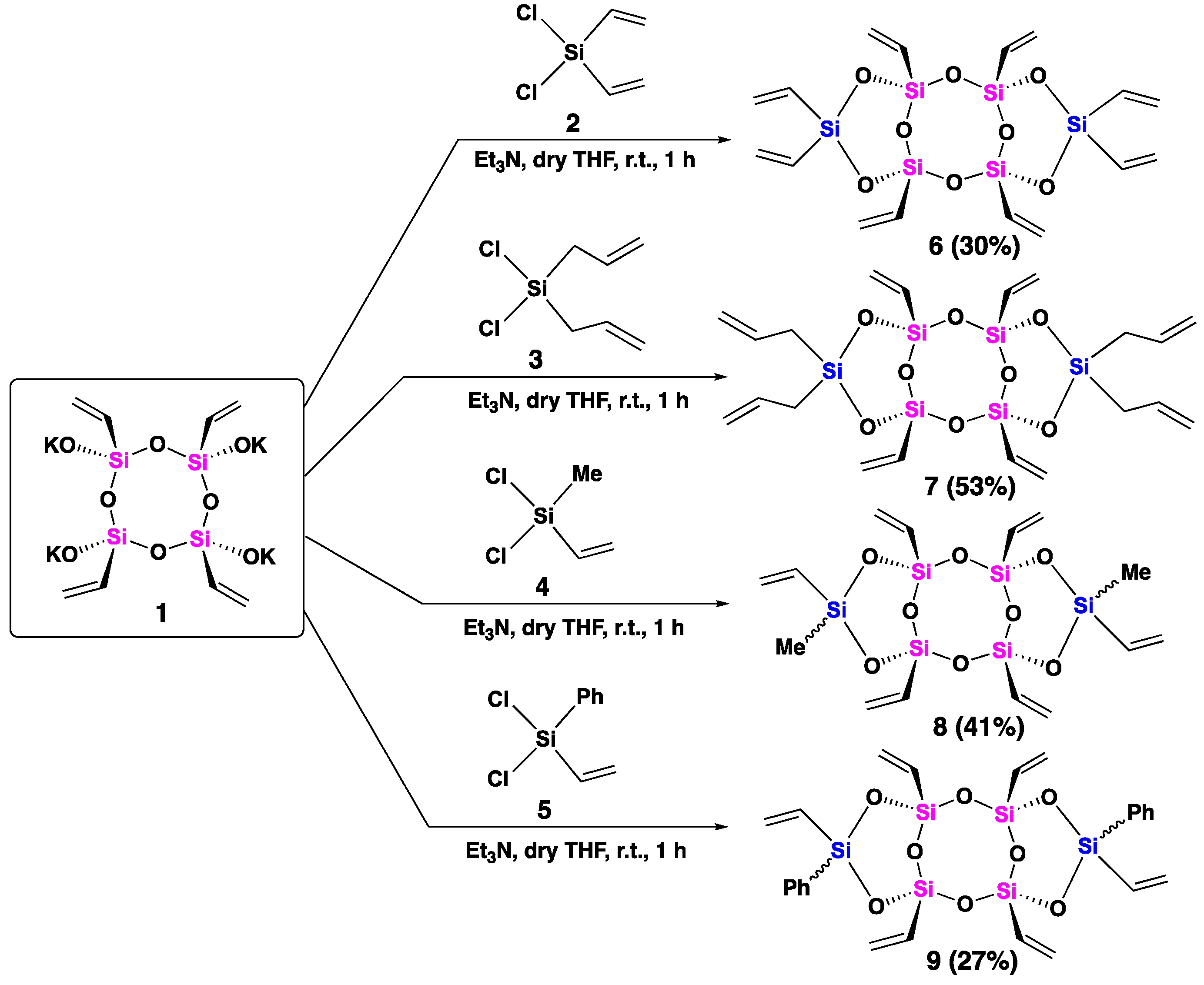
Figure 1.
29Si NMR spectra (CDCl3) of laddersiloxanes 6 (a), 7 (b), 8 (c), and 9 (d).
Scheme 3.
Hydrosilylation of laddersiloxanes 6-8 with dimethylphenylsilane.
Scheme 4.
Hydrosilylation of laddersiloxanes 7 and 8 with chloromethyl(dimethyl)silane.
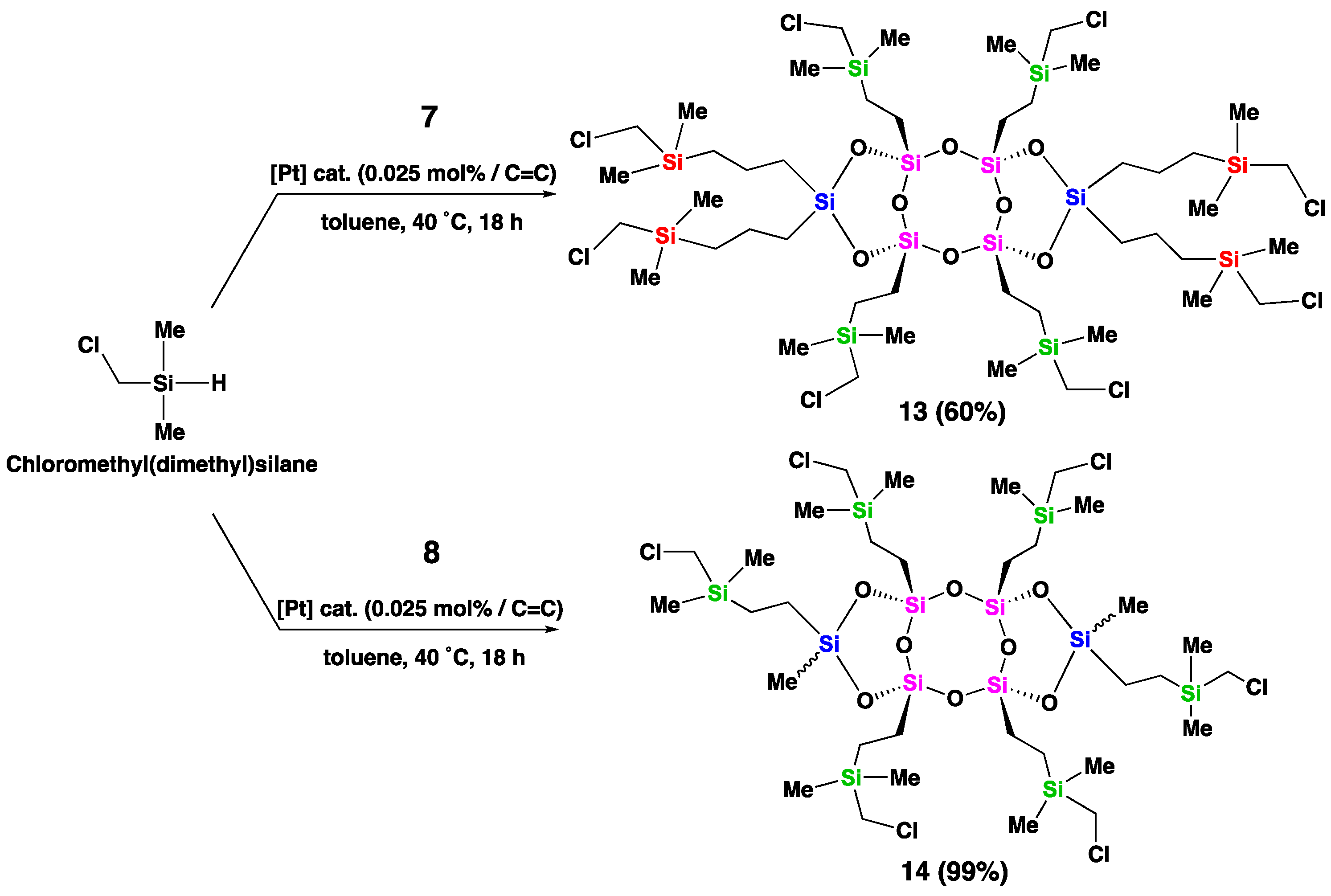
Figure 2.
29Si NMR spectra (CDCl3) of laddersiloxanes 10 (a), 11 (b), 12 (c), 13 (d), and 14 (e).
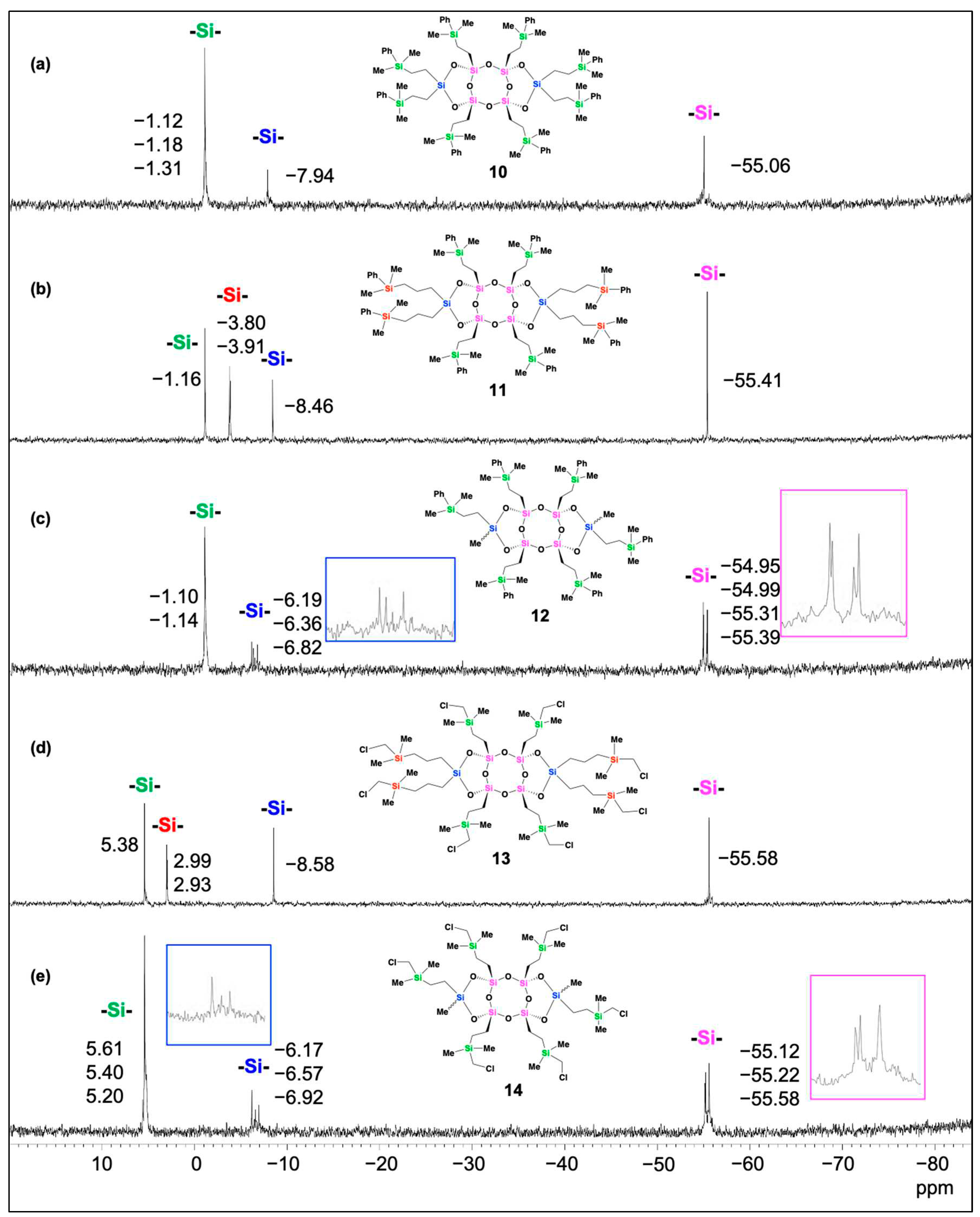
Figure 3.
Thermogravimetric analysis (TGA) for compounds 6, 7, 8, and 9.
Figure 4.
Thermogravimetric analysis (TGA) for compounds 10, 11, 12, 13, and 14.
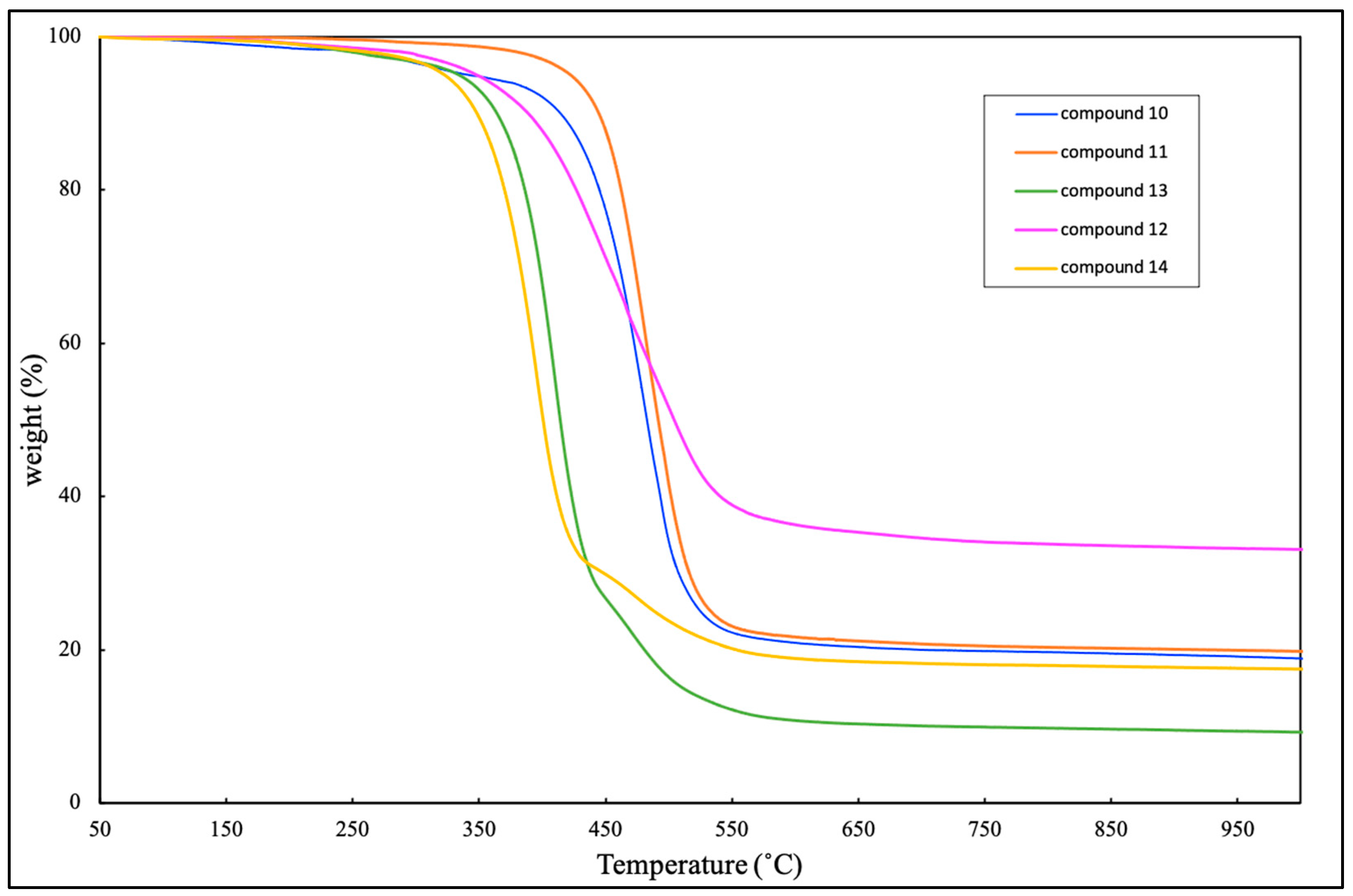
Figure 5.
1H NMR spectra (CDCl3) of laddersiloxanes 7 (up) and the crude product 15 (down).
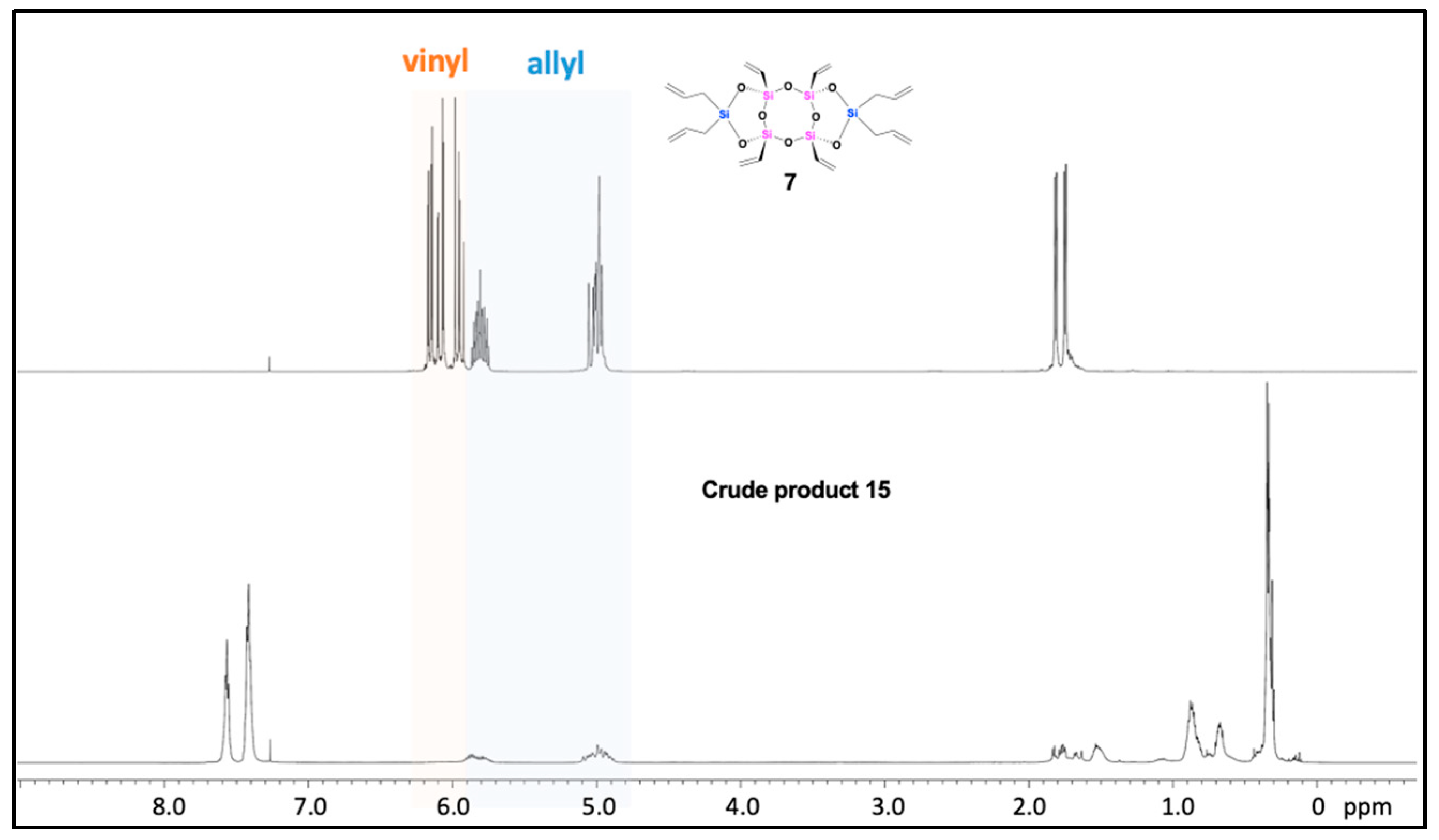
Figure 6.
Structures of the present substituted compounds within the crude product 15.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



