
Submitted:
20 June 2023
Posted:
21 June 2023
You are already at the latest version
Abstract
Matrix vesicles (MVs) are nano-sized extracellular vesicles that are anchored in the extracellular matrix (ECM). In addition to playing a role in biomineralization, osteoblast-derived MVs were recently suggested to have regulatory duties. The aims of this study were to: establish the characteristics of osteoblast-derived MVs in the context of extracellular vesicles like exosomes, assess their role in modulating osteoblast differentiation, and examine their mechanism of uptake. MVs were isolated from the ECM of MG-63 human osteoblast-like cell cultures and characterized via enzyme activity, transmission electron microscopy, nanoparticle tracking analysis, western blot, and small RNA sequencing. Osteoblasts were treated with MVs from two different culture conditions (growth media [GM]; osteogenic media [OM]) to evaluate their effects on differentiation and production of inflammatory markers and on macrophage polarization. MV endocytosis was assessed using a lipophilic, fluorescent dye and confocal microscopy with the role of caveolae determined using methyl-β-cyclodextrin. MVs exhibited a four-fold enrichment in alkaline phosphatase specific activity compared to plasma membranes, were 50-150nm in diameter, possessed exosomal markers CD63, CD81, and CD9 and endosomal markers ALIX, TSG101, and HSP70, and were selectively enriched in microRNA linked to an anti-osteogenic effect and to M2 macrophage polarization. Treatment with GM or OM MVs decreased osteoblast differentiation. Osteoblasts endocytosed MVs by a mechanism that involves caveolae. These results support the hypothesis that osteoblasts produce MVs that participate in the regulation of osteogenesis.
Keywords:
Matrix vesicles
; extracellular vesicles
; osteoblasts
; osteogenesis
; endocytosis
; differentiation
; microRNA
; caveolin
1. Introduction
Populations of various heterogenous, nano-sized, membrane-bound extracellular vesicles are increasingly becoming the focus of regenerative medicine as they contain bioactive lipids, proteins, and nucleic acids [1]. While some populations of these vesicles are found in biological fluids, similar vesicles, termed matrix vesicles (MVs), are also present in the extracellular matrix (ECM) of mineralizing tissues like growth plate cartilage, bone, and dentine. MVs are released by chondrocytes and osteoblasts to become anchored in the ECM via integrin binding to collagen fibrils [2,3,4,5].
Historically, MVs were identified as sites of initial hydroxyapatite formation during endochondral ossification, varying in phospholipid composition and matrix processing enzymes as a function of chondrocyte maturation in the growth plate [3,6,7]. Their composition is regulated genomically, and they exhibit functions independent of calcium phosphate crystal nucleation, suggesting that they are involved in growth plate regulation in ways other than mineral deposition [8,9]. For example, matrix metalloproteinases present in growth plate chondrocyte MVs can activate latent transforming growth factor-β (TGF-β) found in the ECM in a 1,25-dihydroxy vitamin D3-dependent manner [10].
Recent studies found the presence of specific microRNA (miRNA) in MVs produced by growth plate chondrocytes, indicating selective packaging of miRNA cargo into MVs by parent cells. miRNA are short, non-coding RNA approximately 19-25 nucleotides in length that regulate target messenger RNA (mRNA) [11,12]. Sequence complementarity allows one miRNA to target up to hundreds of mRNA, leading to their involvement in the pathogenesis of many disease states, as well as normal physiology [11,12]. MVs from different maturation levels of the growth plate exhibited differences in miRNA enrichment and those differentially upregulated miRNA were found to regulate cell proliferation and maintain an extracellular environment that supported those cellular phenotypes.[4,13,14].
Less is known about MVs that are present in bone. Studies examining MVs produced by osteoblast-lineage cells in culture show they are not identical to MVs produced by chondrocytes, especially with regard to enzyme-enrichment and responsiveness to hormones [3,6,15]. The role of MVs as initial sites of mineralization in the growth plate has been well described, as has initial crystal formation in MVs present in primary bone and osteoid. It is less clear whether MVs play a similar role in mature bone, where nucleation sites exist within the collagen fibrils [2,16,17,18]. Moreover, the heterogeneity in isolated MVs with respect to size and composition suggests that there are different species of MVs with distinct functions in the tissue.
Osteoblast-lineage cells, including perivascular stem cells, bone marrow stromal cells (MSCs), osteoblast-like cell lines, and human osteoblasts, also produce classical exosomes, another class of extracellular vesicles that are not anchored to the ECM. Treatment of MSC cultures with these exosomes upregulated osteogenic markers and enhanced matrix mineralization [19,20,21], supporting the hypothesis that extracellular vesicles from osteoblast-lineage cells can regulate cell-to-cell communication and can affect osteogenic differentiation in target cells [22,23]. These observations suggest that MVs, which are similar to exosomes in many respects, may also play a regulatory role in bone.
The goal of the current study was to characterize MVs produced by osteoblasts and evaluate their role and mechanism of action in bone. The proposed study adds to the growing body of literature regarding extracellular vesicles including specific MV characteristics when derived from an osteoblast-like cell, their impact on osteoblast differentiation and inflammation, and their mechanism of uptake by target cells. We initially hypothesized that MVs would mediate osteogenesis by increasing markers of osteoblastic differentiation similar to exosomes. While MVs produced by MG-63 cells were found to exhibit characteristic size and morphology, their unique miRNA cargo contributed to a role in which they significantly decrease osteoblastic differentiation markers, regardless of their culture conditions, via caveolae-mediated endocytosis, while at the same time promoting the polarization of human macrophages to a pro-regenerative M2 phenotype.
2. Results
2.1. Matrix vesicles display exosomal characteristics.
MVs were isolated at 24 hours post-confluence from the trypsinized ECM of human osteoblast-like MG-63 cells that were cultured in growth media. After pelleting the cells, cells were lysed and plasma membranes (PM) isolated via centrifugation with a sucrose gradient. Alkaline phosphatase specific activity of the MV fraction was 5-fold higher than the PM fraction, validating the purity of the MV isolation (Figure 1A). Transmission electron microscopy confirmed a heterogenous population of vesicles in the MV isolate with a circular morphology and bilaminar membrane (Figure 1B,C). Size distribution was further confirmed by nanoparticle tracking analysis, demonstrating a median size of 105 nm and a mean size of 115 nm with a distribution between 30 and 200 nm (standard deviation ±85 nm; Figure 2D). Western blots of the cell pellet (CP), PM, and MV fractions showed that the tetraspanins CD63, CD9, and CD81, which are classical markers of exosomes, were found only in the MV fraction. Endosomal pathway markers ALIX, HSP70, and TSG101 were present in the CP, PM, and MV fractions, but relative intensities differed depending on the fraction. ALIX and TSG101 were most enriched in the MV fraction with HSP70 displaying the greatest intensity in the CP fraction. Na+-K+ ATPase and annexin V, two proteins responsible for maintaining the calcium gradient within MVs along with other cellular roles, were found in all three fractions as well (Figure 1E). Whereas Na+-K+ ATPase was enriched in the PM and MV fractions, annexin V was enriched in the CP fraction.
2.2. MVs are endocytosed by MG-63 cells.
Confocal microscopy of PKH26-labeled MVs added to counter-stained MG-63 cells (Figure 1F,G) demonstrated visible uptake of vesicles by cells compared to cells incubated with the 0.9% NaCl vehicle control (Figure 1H). When quantified by uptake percentage, 44% of MV-treated cells had at least one fluorescent spot compared to 15% of NaCl-treated cells, constituting a significant difference (Figure 1I).
2.3. microRNAs are selectively packaged into MG-63 MVs.
MV and CP fractions underwent small RNA isolation and sequencing followed by differential expression analysis. Principal component analysis, a measure of variability of the data set with reduced dimensionality, demonstrated distinct grouping of the CP and MV fractions (Figure 2A). The further apart the samples are from one another, the more significance that can be expected in the expression of miRNA. The results indicate that the CP and MV fractions have distinct populations of miRNA with slightly greater variability between samples in the MV group as the samples are more spread apart on the plot. The enriched miRNA, that is those with an absolute Log2 fold change greater than 2 (>2), in the MVs and CPs were then evaluated for their unique expression based on raw read counts. Ninety-six of the differentially expressed miRNA were found exclusively in MVs while 168 were found exclusively in the CP fraction. 229 had increased Log2 fold change between the two fractions but still had some level of raw read counts in the other fraction (Figure 2B). While this diagram does not differentiate between those differentially expressed microRNA that are up- or downregulated, it does demonstrate the specific enrichment in MVs by parent cells as there are miRNA found exclusively in the MV fraction.
2.4. Differentially expressed MV miRNA indicate regulatory roles in osteoblasts and a regenerative role in macrophages.
The differentially expressed miRNA were further characterized, demonstrating the unique populations between MVs and CPs, shown in the heatmap exhibiting clusters based on the similar expression patterns of miRNA (Figure 2C). A volcano plot of those miRNAs with a Log2 fold change >2 and an adjusted p-value less than 0.05 (<0.05) in the MV samples, demonstrated that hsa-miR-122-5p, hsa-miR-451a, and hsa-miR-144-3p had the greatest fold change compared to the CP fraction (Figure 2D). Fourteen miRNAs that were considered the most differentially expressed were further explored in the literature. Nine were linked to anti-osteogenic pathways, found to decrease differentiation and mineralization in vitro, or decrease bone formation in vivo. In contrast, three of the miRNAs were pro-osteogenic, increasing differentiation and mineralization in vitro or increasing bone formation in vivo. Two of the miRNAs, miR-122-5p and miR-451a, were deemed inconclusive as there was published evidence for both pro-osteogenic and anti-osteogenic potential (Table 1, Figure 3A). Additionally, fourteen miRNAs were explored in the literature for any effect on macrophage polarization. Four of the miRNAs were found to promote polarization to an M1 phenotype, associated with the acute inflammatory response to injury or infection. Ten of the miRNAs were found to promote polarization to an M2 phenotype, which is considered the regenerative type of macrophage that promotes an inflammatory environment conducive to cellular turnover and growth (Table 2, Figure 3B).
2.5. Treatment of MG-63 cells with matrix vesicles decreases markers of osteoblast differentiation.
Treatment of MG-63 cells with MVs did not alter DNA content of the cultures (Figure 4A). However, addition of MVs to confluent cultures for 24 or 48 hours caused a decrease in alkaline phosphatase specific activity, an early marker of osteoblast differentiation, and a decrease in osteocalcin production at 48 hours (Figure 4B,C). Osteopontin production was not affected by treatment with MVs (Figure 4D), but there was a significant decrease in production of vascular endothelial growth factor (VEGF), a stimulator of blood vessel formation (Figure 4E). Finally, interleukin-6 (IL-6), a cytokine promoting chronic inflammation, was significantly decreased at 24 and 48 hours (Figure 4F), whereas the regenerative inflammatory cytokine interleukin-10 (IL-10) was not affected (Figure 4G).
2.6. MV composition is sensitive to culture conditions.
MVs were isolated from MG-63 cells cultured for 10 days in osteogenic media (OM) containing ascorbic acid, β-glycerophosphate, and dexamethasone. Treatment of naive cultures with these MVs resulted in a slight decrease in DNA content at 48 hours (Figure 5A). Alkaline phosphatase specific activity was reduced at 48 hours and osteocalcin production was reduced at both 24 and 48 hours of treatment (Figure 5B,C). MVs from the cultures grown in OM had no effect on production of either osteopontin (Figure 5D) or VEGF (Figure 5E). IL-6 was reduced at 24 and 48 hours (Figure 5F), whereas IL-10 was significantly increased at 48 hours (Figure 5G).
2.7. Inhibition of caveolae-mediated endocytosis prevents uptake and effect of MVs on osteoblast-like cells.
MG-63 osteoblast-like cells were treated with methyl-β-cyclodextrin (β-CD), a chemical destroyer of cholesterol in the plasma membrane to inhibit caveolae-mediated endocytosis, followed by treatment with MVs isolated from the trypsinized cell layer of MG-63 cells cultured in growth media. MV uptake was visualized using confocal microscopy. PKH26-labeled MVs were added to counter-stained MG-63 cells pre-treated with vehicle or 10 mM β-CD; significantly more MV fluorescence was observed in the control cells (Figure 6A) than in the β-CD treated cells (Figure 6B). Morphometric analysis confirmed these results, demonstrated MV uptake in 57% of the control cells compared to 1.5% of the β-CD-treated cells (Figure 6C).
Pre-treatment of the cultures with increasing concentrations of β-CD did not alter DNA content of the cultures when the cells were treated with vehicle alone. In contrast, at the highest dose of β-CD (10 mM), MV treatment caused a small decrease in DNA (Figure 6D). MVs caused a reduction in alkaline phosphatase specific activity in the vehicle-treated group, but this effect was lost when the cultures were pre-treated with β-CD (Figure 6E). Together, the results suggest that disruption of the caveolae using β-CD inhibits the uptake of MVs and their downstream effects on osteoblast differentiation.
3. Discussion
To date, three subtypes of extracellular vesicles have been classified based on their biogenesis pathways: exosomes, microvesicles, and apoptotic bodies. Exosomes are the most extensively studied with biogenesis in the endosomal pathway prior to release into biological fluids [1]. Our results support the hypothesis that osteoblast-derived MVs are a class of exosomes, similar to other matrix associated matrisomes. Electron microscopy demonstrated a circular morphology with a bilaminar membrane and size distribution typical of exosomes: 50-150 nm and 30-150 nm, respectively [1,73]. MVs and exosomes share enrichment of sphingomyelin and annexin A2 in their lipid and protein composition [73]. Western blots of the osteoblast-derived MVs demonstrated classical exosome markers tetraspanins CD63, CD9, and CD81, confirming isolation of an extracellular vesicle population from the trypsinized ECM. Markers of endosomal biogenesis, HSP70, ALIX, and TSG101, were also found in the MV fraction, indicating that their production involved a similar mechanism to that used to produce exosomes. This finding is supported by earlier studies using somatic cell hybrids to demonstrate the specific genesis of MVs as distinct organelles and not due to non-specific budding from the PM [74].
However, it is clear that like MVs isolated from growth plate cartilage, they are distinct from exosomes present in biological fluids. MVs exhibit marked enrichment in alkaline phosphatase specific activity compared to the PMs isolated from their parent osteoblasts. Consistent with previously described MVs, they consist of a heterogenous population between 50-150 nm in size [14,22,75,76]. Earlier studies comparing membrane bound vesicles in the media of chondrocyte cultures to the MVs isolated from the ECM indicate that the two populations are distinctly different, with different lipid composition of their bilayers and different enzyme content. Importantly, the media vesicles did not exhibit an enrichment in alkaline phosphatase specific activity relative to the PM. MVs are tethered to the ECM of mineralizing tissues via integrins, and integrin-tethered exosomes have been reported in other tissues, primarily cancer, suggesting that osteoblast-derived MVs may share similarities with this class of matrisomes [77].
Similar to chondrocyte-derived MVs, those derived from the MG-63 osteoblast-like cell cultures contained small, non-coding miRNA. MVs derived from MC3T3-E1 cell cultures also contain miRNAs and microarray analysis showed that the MC3T3-E1 MVs differed from miRNA identified in chondrocyte- and MSC -derived MVs [22,78]. Our use of small RNA sequencing has higher specificity and sensitivity than microarray, allowing detection of a higher percentage of differentially expressed genes [79]. We found that the MG-63 MVs contained uniquely differentially expressed miRNA from their parent osteoblast-like cells and this miRNA was selectively packaged into MVs, as demonstrated by the heatmap and volcano plot, which both show a distinct pattern of miRNA expression. While the difference in species and degree of osteoblastic differentiation makes a direct comparison difficult, our dataset and that of Minamizaki, et al., have at least 11 similarly expressed miRNA of the 52 we found to be significantly upregulated in osteoblast-derived MVs [78]. It should also be noted that our MG-63 cells and their MC3T3-E1 cells represent different levels of osteoblastic differentiation; therefore, similar to chondrocyte-derived MVs, there may be different expression levels of miRNA depending on parent cell maturity, explaining the 41 possibly unique miRNA [13,78].
When several miRNAs that were significantly upregulated in MG-63 cell MVs were further explored in the literature, the majority of them were found to be anti-osteogenic; that is, they are linked to known anti-osteogenic pathways, found to decrease osteoblastic differentiation and mineralization in vitro, or decrease bone formation in vivo. While three miRNA were pro-osteogenic (promoted differentiation and/or mineralization) and two had conflicting evidence in the literature, the differential expression of miRNA in MVs appears to point toward a role in inhibition of osteoblastic differentiation and slowing of osteogenesis. This was corroborated by the in vitro treatment data with MVs grown in growth medium, as two markers of osteoblastic differentiation, alkaline phosphatase and osteocalcin, were decreased with MV treatment. Mizukami, et al., found similar results when they treated ST2 osteoblast-like cells and primary osteoblasts in vitro with collagenase-released extracellular vesicles from the cell layer of mineralizing osteoblasts and observed decreased messenger RNA expression of osterix and osteocalcin [80]. When MVs were produced from Mg-63 cells cultured in osteogenic medium for a longer period of time, they also decreased osteoblastic differentiation markers, which suggests the anti-osteogenic role is inherent to the MVs themselves.
When compared to the role of exosomes, which are extracellular vesicles released into culture medium, matrix vesicles appear to serve a different role in bone. Exosomes from mineralizing osteoblast-like cells, human osteoblasts, and perivascular stem cells upregulated alkaline phosphatase, osteocalcin, and RUNX2 in vitro and enhanced matrix mineralization within MSC cultures as well as improving bone regeneration and healing speed of critical sized calvarial defects in vivo [19,20,21,81]. Our characterization data, particularly, the presence of microsomal markers evident in the western blots, suggest that MVs and exosomes have a similar biogenesis pathway, size, and morphology. However, it appears that their location within mature bone, anchored to the ECM versus free-floating, does dictate their role. Exosomes may communicate with more distant cells to coordinate osteogenic efforts, while MVs remain close to parent cells to modulate the rate of osteogenesis. Further exploration of MVs compared to exosomes is necessary to resolve this dichotomy.
The role of MVs becomes even more interesting when exploring the significantly upregulated miRNA with respect to macrophage polarization. A significant number of the MV miRNA were linked to M2 polarization and this is corroborated by the in vitro data that demonstrated significant decreases in IL-6 production by osteoblasts treated with MVs and an additional significant increase in IL-10 production caused by MVs grown in osteogenic medium. IL-6 and IL-10 are two cytokines associated with M1 and M2 macrophages and these two pieces of data suggest that osteoblast-derived MVs promote M2 polarization, or the regenerative phenotype of macrophages, over M1 polarization [82]. MVs, while they may not directly promote the osteogenic differentiation of osteoblasts or the production of factors needed for bone formation, still do promote a regenerative environment within bone and may ultimately contribute to the formation of new bone in a more nuanced and complicated manner. This hypothesis is supported by Mizukami, et al., who found increased bone healing and increased chondrogenesis of a femoral defect treated with collagenase-released extracellular vesicles from the cell layer of mineralizing osteoblasts in vivo [80].
Studies using PKH26, a lipophilic, fluorescent dye that stains the membrane of the vesicles, demonstrated that MVs were endocytosed by the MG-63 cells. Confocal microscopy of cells pretreated with methyl-β-cyclodextrin, which depletes cholesterol to inhibit caveolae-mediated endocytosis, had a visibly significant decrease in fluorescence compared to cells treated with vehicle. Furthermore, β-CD-treated cells did not exhibit the decrease in alkaline phosphatase seen with MV treatment. This suggests that MVs are organelles that are endocytosed in a caveolae-dependent manner. Other mechanisms for MV uptake may play a role, particularly if more than one species of MV is present in the heterogeneous population isolated from the ECM. Consensus regarding cellular uptake of extracellular vesicles remains to be reached as some research has identified the involvement of caveolin [83,84], while others support a role of caveolin-independent endocytosis [85,86], such as clathrin-dependent endocytosis [87]. However, the loss of the inhibitory effect on cellular alkaline phosphatase elicited by MVs in the β-CD treated cultures indicates that caveolae are involved.
This study took advantage of a well-characterized cell line, which has been used extensively to study the regulation of osteoblast differentiation by orthopaedic and dental implant topography [88,89]. MG-63 human osteoblast-like osteosarcoma cells were originally isolated from a male patient [90] and have been shown to have characteristics typical of immature osteoblasts [91]. As a result, conclusions as to the role of MVs in regulating mature osteoblasts are limited. We selected these cells because they enabled us to look at early differentiation events and to assess the potential interaction of cells in the osteoblast lineage with immunomodulatory cells that would be in the same microenvironment. Future studies using normal human osteoblasts will provide additional insights into the mechanism of action of MVs in bone.
In summary, osteoblast-derived MVs share many characteristics with MVs from other mineralizing tissues like growth plate cartilage, as well as with extracellular vesicles like cancer-derived exosomes, but they also exhibit properties distinct from exosomes (Figure 7). Our small RNA sequencing demonstrated the enrichment of miRNA within the osteoblast-derived MVs, specifically those that inhibit osteoblast differentiation and promote a regenerative inflammatory state. The results suggest that osteoblast-derived MVs may modulate the rate of osteogenesis as well as contribute in a much more nuanced manner involving M2 polarization of macrophages to a regenerative phenotype. They appear to exert this effect via caveolae-mediated endocytosis. Further studies will continue to explore the impact of MVs on cells within mature bone, their uptake mechanisms to exert these effects, and possible therapeutic application of MVs.
4. Materials and Methods
4.1. Osteoblast Cultures
MG-63 cells were plated in T-175 flasks at a density of 10,000 cells/cm2 in Dulbecco’s modification of Eagle’s medium (DMEM) containing 1.0 g/L glucose (Corning), 10% fetal bovine serum (FBS; Gemini), and 100 U/mL penicillin and streptomycin (pen-strep). Cells were cultured at 37°C in 5% CO2 with media changes 24 hours after plating and every 48 hours thereafter. Cells reached confluence after approximately four days. For matrix vesicle isolation, MG-63 cells were cultured in DMEM full media and were harvested as described below 24 hours after reaching confluence (growth media). Alternatively, MG-63 cells were cultured in DMEM full media supplemented with 10mM β-glycerophosphate, 50 µg/mL ascorbic acid, and 10-8M dexamethasone for 10 days (osteogenic media).
4.2. Cell Harvest and Isolation of Matrix Vesicle and Plasma Membrane Fractions
At harvest, the culture media from each flask of cells were aspirated and discarded. The cell layer, including the extracellular matrix and cells, was washed twice with 10mL of sterile 1X DPBS. The cell layer was then trypsinized with 10mL of sterile 0.25% trypsin and placed in 37°C incubator for four minutes. After four minutes, the cell layer was scraped with a cell scraper and placed back in the incubator for a total time of 10 minutes. The trypsin in each flask was quenched with 10mL of sterile 1x DPBS containing 10% exosome-free FBS. To obtain exosome-free FBS, complete FBS was ultracentrifuged at 180,000xg for 4 hours at 4°C (44,500 RPM in 50.2 Ti rotor using a Beckman Coulter Class R ultracentrifuge). Following ultracentrifugation, the top ½ of the FBS in each tube was selected, leaving behind the pellet and cloudier supernatant, and sterile filtered. Exosome-free FBS was stored at -20°C until the day of harvest.
Following quenching of the trypsin, cells were separated from the digest by centrifugation at 500xg for 10 minutes at 4°C. Supernatant (trypsin-digest supernatant) was separated from the cell pellet and cells were resuspended with sterile filtered 0.9% NaCl and counted.
For the whole cell lysate, 1mL aliquot of cell suspension was taken and re-spun at 500xg for 10 minutes at 4°C. Supernatant was aspirated and discarded and cell pellet was resuspended with 200 µl of NP-40 lysis buffer with 1% proteinase inhibitor cocktail (whole cell pellet lysate).
The remaining cell suspension was used for plasma membrane preparation following the method of Fitzpatrick et al. [92] Briefly, cells were separated from saline by centrifugation at 500xg for 10 minutes at 4°C. Saline supernatant was aspirated and discarded and the cell pellet was resuspended with 5mL of 0.25M sucrose (pH 7.4). Pellet was vortexed and homogenized in a glass, tabletop homogenizer then centrifuged at 1,480xg for 20 minutes at 4°C (3.5K RPM in 50.2 Ti Rotor using Beckman Coulter Class R Ultracentrifuge). Supernatant was discarded and pellet was resuspended with 5mL of 2M sucrose and vortexed, then centrifuged at 20,000xg for 20 minutes at 4°C (12.9K RPM in 50.2 Ti rotor). The supernatant was transferred to a new tube and 20 mL of ice-cold ultrapure H2O was added to each tube. Tubes were vortexed, then centrifuged at 40,000xg for 30 minutes at 4°C (18.2K RPM in 50.2 Ti rotor). The resulting supernatant was discarded and final plasma membrane pellet was re-suspended in sterile-filtered 0.9% NaCl.
MV isolation using differential centrifugation has been previously described [93]. The trypsin-digest supernatant was centrifuged at 25,000xg for 20 minutes at 4°C (16.4K RPM in 50.2 Ti rotor) to pellet any remaining cell debris. The supernatant was transferred to a new tube and matrix vesicles were pelleted via ultracentrifugation at 100,000xg for 70 minutes at 4°C (33K RPM in 50.2 Ti rotor). This supernatant was carefully discarded, and the pellet was resuspended with 20 mL of sterile-filtered 0.9% NaCl for washing and ultracentrifuged again at 100,000xg for 70 minutes at 4°C as above. The final supernatant was carefully discarded, and pellet of matrix vesicles was resuspended in 100 µL sterile-filtered 0.9% NaCl for assays. MVs were measured for their protein content using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA).
4.3. Alkaline Phosphatase Specific Activity Assay
Following whole cell lysate, plasma membrane, and MV isolation, each fraction was assayed for alkaline phosphatase specific activity via the formation of para-nitrophenol from para-nitrophenylphosphate at pH 10.2. Protein content for normalization was quantified using a Pierce BCA Protein Assay Kit as above.
4.4. Transmission Electron Microscopy
Isolated matrix vesicles were fixed in Karnovsky fixative (Electron Microscopy Sciences, Hatfield, PA, USA). A single drop of fixed MVs was placed on parafilm and a formvar-carbon-coated copper grid (Electron Microscopy Sciences, Hatfield, PA, USA) was floated in the suspension, carbon coated-side in the suspension, for 20 minutes. The grid was then washed in droplets of ultrapure water for 2 minutes per droplet for a total of 8 washes. After washing, the grid was transferred to a droplet of uranyl-oxalate solution for 5 minutes. Uranyl-oxalate was prepared by combining equal parts 2% Uranyl acetate (Electron Microscopy Sciences, Hatfield, PA, USA) with 0.075M oxalic acid and adjusting the pH to 7 with 25% NH4OH. Finally, the grid was embedded in a droplet of 0.2% uranyl acetate and 1.8% methylcellulose for 10 minutes on ice. The excess liquid was removed and the grid was dried at room temperature. The grid was viewed at 50,000x and 100,000x magnification and 200kV using an electron microscope (JEM-F200 Cold FEG Electron Microscope, Jeol, Peabody, MA, USA).
4.5. Nanoparticle Tracking Analysis
The size distribution of the matrix vesicles was determined via nanoparticle tracking analysis. 10 µl of matrix vesicle suspension was mixed with 10 µL of 0.02% paraformaldehyde in 1x phosphate buffered saline (PBS). MVs were then diluted 10x in paraformaldehyde-PBS solution before completing 3 repeats of nanoparticle tracking analysis using a Zetaview (Analytik, Cambridge, United Kingdom) per the manufacturer’s instructions.
4.6. Western Blot
From each cell pellet lysate, plasma membrane, and MV fraction, a total of 3 µg of protein were subjected to 4-20% mini-Protean TGX gels (Bio-Rad, Hercules, CA, USA) and transferred to a PVFD membrane (Bio-Rad). After 1 hour of blocking in 5% nonfat milk in PBS containing 0.1% Tween-20 (PBS-T) at room temperature, membranes were incubated overnight with antibodies. Membranes were washed in PBS-T and incubated with species-specific horseradish peroxidase-labeled secondary antibody for 1 hour at room temperature. After a second washing with PBS-T, an ECL chemiluminescence system (Pierce) was used to develop membranes to detect bound antibodies.
4.7. PKH26 Labeling and Confocal Microscopy
Purified MVs were labeled with PKH26 red fluorescent cell membrane label (Sigma-Aldrich, St. Louis, MO, USA). Briefly, the PKH26 dye in manufacturer’s diluent was mixed with MV suspension or sterile 0.9% sodium chloride (negative control) and pipetted to mix once per minute for five minutes. After 5 minutes, 1 mL of sterile 1% bovine serum albumin was added to each tube to quench unbound dye. A total of 8 mL of basal medium was added to each tube, tubes were balanced, and ultracentrifuged at 100,000xg for 70 minutes at 4°C (33K RPM in 50.2 Ti r). The supernatant was carefully removed, and the pellet was resuspended in 100 mL of sterile DPBS. A total of 8x103 cells were plated in each well of a CCS-8 well culture slide. MVs (0.85µg protein) were added to the antibiotic-free culture media when cells reached 70% confluence. After 24 hours of incubation, culture media were removed, cells were washed twice with sterile DPBS and then fixed with 10% neutral buffered formalin. The nuclei (DRAQ5, Abcam, Cambridge, United Kingdom) and plasma membrane (CellBrite Green, Biotium, Fremont, CA) were stained according to manufacturer’s instructions. Twelve random fields of view from each group were imaged using a confocal microscope (LSM 980, Zeiss, Oberkochen, Germany). Two independent observers reviewed all 24 images for total cell number and total number of cells with at least one distinct spot of PKH26 fluorescence.
4.8. RNA Extraction and Detection
microRNAs from MG-63 MVs and whole cells were extracted using QIAzol (Qiagen, Hilden, Germany) and the miRNeasy micro extraction kit (Qiagen, Hilden, Germany) per manufacturer’s instructions. Briefly, QIAzol lysis reagent homogenized each sample. Chloroform addition followed by centrifugation separated the homogenate into aqueous and organic phases. The aqueous phase with the RNA was isolated from the DNA interphase and protein organic phase. Isopropanol precipitated the RNA from the aqueous phase with miRNeasy spin column collection. Using the spin column, the RNA was washed with ethanol. Finally, RNA was eluted in RNase-free water and RNA quantity and quality were assessed via bioanalyzer analysis with an RNA 6000 Pico kit (Agilent Technologies, Santa Clara, CA, USA) and an Agilent 2100 Bioanalyzer per manufacturer’s instructions (Agilent Technologies). Eighteen independent MV samples were pooled into three samples for sequencing with at least 750 ng of RNA per sample. Corresponding cell samples were pooled in the same manner for a total volume of 60 µL.
4.9. microRNA Sequencing (RNA-Seq)
Following verification of RNA quality and quantity, samples were sent to GENEWIZ for next-generation sequencing (Azenta Life Sciences, Burlington, MA, USA). Library preparation was completed using size fractionation with adapter ligation to the 5’ phosphate of the microRNA. Libraries were pooled and sequenced 2x150 base pair (bp) on the Illumina HiSeq 2500. Raw RNA-seq reads (FASTQ) were aligned to the respective H. sapiens genome using miARma-seq. The read count data were then normalized, and statistical tests were performed to determine differentially expressed genes (adjusted P value using Benjamin-Hochberg method, <0.05 and fold change ≥2) using DESeq2.
4.10. Pathway Analysis of microRNA Associations
microRNAs were selected from the differential expression list using the Log2 fold change (>2) and the adjusted p-value (<0.05). Those with the greatest Log2 fold change were searched within PubMed for their miRNA along with the terms: “osteoblast”, “bone”, or “osteogenesis” to begin the literature review. Peer-reviewed publications were selected based on title and abstract, then further reviewed for association between selected miRNA, osteogenic potential, and macrophage polarization. The resulting classifications were based on at least one publication or deemed inconclusive if two or more publications were found with conflicting results.
4.11. MV Treatment and Osteogenic Response
MG-63 osteoblast-like cells were cultured as described. After reaching confluence, culture media were replaced with fresh DMEM with 10% FBS without pen-strep and cells were treated with 5 µg/mL (based on protein content) of MVs (either GM or OM as described above) for 24 or 48 hours. At the time of harvest, conditioned media were collected, cells were washed with 1 mL of phosphate-buffered saline (PBS), and lysed with 0.05% Triton X-100 in 1x PBS. After a single freeze-thaw, cells were sonicated. From the cell lysate, DNA quantification and alkaline phosphatase specific activity were assessed using Quantifluor dsDNA system (Promega, Madison, WI, USA) and via the formation of para-nitrophenol from para-nitrophenylphosphate at pH 10.2, respectively. Osteocalcin (R&D, Minneapolis, MN, USA), osteopontin (R&D Systems, Minneapolis, MN, USA), vascular endothelial growth factor (R&D Systems), interleukin-6 (Peprotech, Cranbury, NJ, USA), and interleukin-10 (Peprotech) levels were measured from the conditioned media using enzyme-linked immunoassay (ELISA) following manufacturer’s instructions.
4.12. Methyl-β-cyclodextrin Endocytosis Inhibition Treatment
MG-63 osteoblast-like cells were cultured as described above in 24-well plates. After reaching confluence, culture media were replaced with fresh basal DMEM supplemented with methyl-β-cyclodextrin (β-CD; Thermo Fisher Scientific, Waltham, MA, USA). Briefly, stock 100 mM β-CD was prepared in sterile ultrapure H2O. The stock solution was then serially diluted into 10 mM, 1 mM, and 0.1 mM β-CD in basal DMEM and added to cells for 30 minutes, based on previous studies demonstrating that this incubation time destroyed caveolae but was not cytotoxic [94]. A vehicle of ultrapure H2O at 10 mM was also prepared as a control. After 30 minutes, cells were washed twice with basal DMEM followed by treatment with 5 µg protein/mL MVs or an equivalent amount of sterile 0.9% NaCl in DMEM with 10% FBS and without pen-strep for 48 hours. At the end of MV treatment, cells were harvested for conditioned media and cell lysate in 0.05% Triton X-100 in 1x PBS as described above. DNA quantification and alkaline phosphatase specific activity were assessed using Quantifluor dsDNA system (Promega, Madison, WI, USA) and via the formation of para-nitrophenol from para-nitrophenylphosphate at pH 10.2, respectively.
4.13. Statistical Analysis
The results of the culture study are presented as mean ± standard error for six independent cultures per variable. Between-group differences with only two groups were calculated by unpaired t-test, while between-group differences with three or more groups were calculated by one-way ANOVA with post-hoc Tukey HSD. A p-value less than 0.05 was considered significant. All statistically significant t-tests are denoted by * and all statistically significant ANOVA analyses are denoted by groups not sharing a letter. Experimental observations were validated with at least one experimental repeat. Statistical analysis and presentation for all experiments were carried out using GraphPad Prism 9.3.1 and JMP Pro 16.
Author Contributions
Conceptualization, Anne Skelton, D. Cohen and Zvi Schwartz; Formal analysis, Anne Skelton, D. Cohen, Barbara Boyan and Zvi Schwartz; Funding acquisition, Barbara Boyan; Investigation, Anne Skelton and D. Cohen; Project administration, Zvi Schwartz; Supervision, Barbara Boyan; Writing – original draft, Anne Skelton; Writing – review & editing, D. Cohen, Barbara Boyan and Zvi Schwartz.
Funding
This work is supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institute of Health under Award Number R01AR072500.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
All data are available in the manuscript. Additional information will be provided following receipt of reasonable written request.
Acknowledgments
Support was provided by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institute of Health under Award Number R01AR072500. The content is solely the authors’ responsibility and does not necessarily represent the official views of the National Institutes of Health. We acknowledge the use of the facilities within the Nanomaterials Characterization Core at Virginia Commonwealth University (NCC). We would like to acknowledge assistance from Dr. Massimo Bertino and Dr. Carl Mayer from the NCC for assistance with transmission electron microscopy. Services in support of the research project were generated by the VCU Massey Cancer Center Microscopy Shared Resource, supported, in part, with funding from NIH-NCI Cancer Center Support Grant P30 CA016059. High performance computing resources provided by the High-Performance Research Computing (HPRC) core facility at Virginia Commonwealth University (https://hprc.vcu.edu) were used for conducting the research reported in this work. The study is in partial fulfillment of the Ph.D. degree for Anne M. Skelton.
Conflicts of Interest
The authors declare no conflict of interest related to this work.
References
- Azoidis, I.; Cox, S.C.; Davies, O.G. The role of extracellular vesicles in biomineralisation: Current perspective and application in regenerative medicine. J. Tissue Eng. 2018, 9, 2041731418810130. [Google Scholar] [CrossRef] [PubMed]
- Anderson, H.C.; Garimella, R.; Tague, S.E. The role of matrix vesicles in growth plate development and biomineralization. Front. Biosci. 2005, 10, 822–837. [Google Scholar] [CrossRef]
- Dean, D.D.; Schwartz, Z.; Bonewald, L.; Muniz, O.E.; Morales, S.; Gomez, R.; Brooks, B.P.; Qiao, M.; Howell, D.S.; Boyan, B.D. Matrix Vesicles produced by osteoblast-like cells in culture become significantly enriched in proteoglycan-degrading metalloproteinases after addition of β-glycerophosphate and ascorbic acid. Calcif. Tissue Int. 1994, 54, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Rodriguez, N.E.; Zhao, J.; Ramey, A.N.; Hyzy, S.L.; Boyan, B.D.; Schwartz, Z. Selective enrichment of microRNAs in extracellular matrix vesicles produced by growth plate chondrocytes. Bone 2016, 88, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Nahar, N.N.; Missana, L.R.; Garimella, R.; Tague, S.E.; Anderson, H.C. Matrix vesicles are carriers of bone morphogenetic proteins (BMPs), Vascular endothelial growth factor (VEGF), and noncollagenous matrix proteins. J. Bone Miner. Metab. 2008, 26, 514–519. [Google Scholar] [CrossRef]
- Boyan, B.D.; Schwartz, Z.; Swain, L.D.; Khare, A. Role of lipids in calcification of cartilage. Anat. Rec. 1989, 224, 211–219. [Google Scholar] [CrossRef]
- Dean, D.D.; Boyan, B.D.; Muniz, O.E.; Howell, D.S.; Schwartz, Z. Vitamin D metabolites regulate matrix vesicle metalloproteinase content in a cell maturation-dependent manner. Calcif. Tissue Int. 1996, 59, 109–116. [Google Scholar] [CrossRef]
- Sylvia, V.L.; Schwartz, Z.; Holmes, S.C.; Dean, D.D.; Boyan, B.D. 24,25-(OH)2D3 regulation of matrix vesicle protein kinase C occurs both during biosynthesis and in the extracellular matrix. Calcif. Tissue Int. 1997, 61, 313–321. [Google Scholar] [CrossRef]
- Schwartz, Z.; Sylvia, V.L.; Larsson, D.; Nemere, I.; Casasola, D.; Dean, D.D.; Boyan, B.D. 1α,25(OH)2D3 regulates chondrocyte matrix vesicle protein kinase C (PKC) directly via G-protein-dependent mechanisms and indirectly via incorporation of PKC during matrix vesicle biogenesis. J. Biol. Chem. 2002, 277, 11828–11837. [Google Scholar] [CrossRef]
- Boyan, B.D.; Schwartz, Z.; Park-Snyder, S.; Dean, D.D.; Yang, F.; Twardzik, D.; Bonewald, L.F. Latent transforming growth factor-β is produced by chondrocytes and activated by extracellular matrix vesicles upon exposure to 1,25-(OH)2D3. J. Biol. Chem. 1994, 269, 28374–28381. [Google Scholar] [CrossRef]
- Mohr, A.M.; Mott, J.L. Overview of microRNA biology. Semin. Liver Dis. 2015, 35, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.X.; Rothenberg, M.E. MicroRNA. J. Allergy Clin. Immunol. 2018, 141, 1202–1207. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; McClure, M.J.; Zhao, J.; Ramey, A.N.; Asmussen, N.; Hyzy, S.L.; Schwartz, Z.; Boyan, B.D. MicroRNA contents in matrix vesicles produced by growth plate chondrocytes are cell maturation dependent. Sci. Rep. 2018, 8, 3609. [Google Scholar] [CrossRef] [PubMed]
- Asmussen, N.C.; Cohen, D.J.; Lin, Z.; McClure, M.J.; Boyan, B.D.; Schwartz, Z. Specific microRNAs found in extracellular matrix vesicles regulate proliferation and differentiation in growth plate chondrocytes. Calcif. Tissue Int. 2021. [Google Scholar] [CrossRef] [PubMed]
- Bonewald, L.F.; Schwartz, Z.; Swain, L.D.; Boyan, B.D. Stimulation of matrix vesicle enzyme activity in osteoblast-like cells by 1,25(OH)2D3 and transforming growth factor beta (TGF Beta). Bone Miner. 1992, 17, 139–144. [Google Scholar] [CrossRef]
- Hasegawa, T. Ultrastructure and biological function of matrix vesicles in bone mineralization. Histochem. Cell Biol. 2018, 149, 289–304. [Google Scholar] [CrossRef]
- Boyan, B.D.; Schwartz, Z.; Swain, L.D. Matrix vesicles as a marker of endochondral ossification. Connect. Tissue Res. 1990, 24, 67–75. [Google Scholar] [CrossRef]
- Bonucci, E. Bone mineralization. Front. Biosci. 2012, 17, 100. [Google Scholar] [CrossRef]
- Cui, Y.; Luan, J.; Li, H.; Zhou, X.; Han, J. Exosomes derived from mineralizing osteoblasts promote ST2 cell osteogenic differentiation by alteration of microRNA expression. FEBS Lett. 2016, 590, 185–192. [Google Scholar] [CrossRef]
- Qin, Y.; Wang, L.; Gao, Z.; Chen, G.; Zhang, C. Bone marrow stromal/stem cell-derived extracellular vesicles regulate osteoblast activity and differentiation in vitro and promote bone regeneration in vivo. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Xu, J.; Wang, Y.; Hsu, C.Y.; Gao, Y.; Meyers, C.A.; Chang, L.; Zhang, L.; Broderick, K.; Ding, C.; Peault, B.; et al. Human perivascular stem cell-derived extracellular vesicles mediate bone repair. Elife 2019, 8, 1–23. [Google Scholar] [CrossRef]
- Yoshiko, Y.; Minamizaki, T. Emerging roles of microRNAs as extracellular vesicle cargo secreted from osteoblasts. J. Oral Biosci. 2020, 62, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Bjørge, I.M.; Kim, S.Y.; Mano, J.F.; Kalionis, B.; Chrzanowski, W. Extracellular vesicles, exosomes and shedding vesicles in regenerative medicine-a new paradigm for tissue repair. Biomater. Sci. 2018, 6, 60–78. [Google Scholar] [CrossRef]
- Gu, H.; Shi, S.; Xiao, F.; Huang, Z.; Xu, J.; Chen, G.; Zhou, K.; Lu, L.; Yin, X. miR-1-3p regulates the differentiation of mesenchymal stem cells to prevent osteoporosis by targeting secreted frizzled-related protein 1. Bone 2020, 137, 115444. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.G.; Konicke, K.; Kristianto, J.; Gustavson, A.; Garbo, R.; Wang, X.; Yuan, B.; Blank, R.D. Endothelin signaling regulates mineralization and posttranscriptionally regulates SOST in TMOb cells via miR 126-3p. Physiol. Rep. 2017, 5, e13088. [Google Scholar] [CrossRef] [PubMed]
- Bedene, A.; Mencej Bedrač, S.; Ješe, L.; Marc, J.; Vrtačnik, P.; Preželj, J.; Kocjan, T.; Kranjc, T.; Ostanek, B. miR-148a the epigenetic regulator of bone homeostasis is increased in plasma of osteoporotic postmenopausal women. Wien. Klin. Wochenschr. 2016, 128, 519–526. [Google Scholar] [CrossRef]
- Ji, Y.; Fang, Q.Y.; Wang, S.N.; Zhang, Z.W.; Hou, Z.J.; Li, J.N.; Fu, S.Q. Lnc-RNA BLACAT1 regulates differentiation of bone marrow stromal stem cells by targeting miR-142-5pin osteoarthritis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 2893–2901. [Google Scholar] [CrossRef]
- Tu, M.; Tang, J.; He, H.; Cheng, P.; Chen, C. miR-142-5p promotes bone repair by maintaining osteoblast activity. J. Bone Miner. Metab. 2017, 35, 255–264. [Google Scholar] [CrossRef]
- Zhao, R.; Zhu, Y.; Sun, B. Exploration of the effect of mmu-miR-142-5p on osteoblast and the mechanism. Cell Biochem. Biophys. 2015, 71, 255–260. [Google Scholar] [CrossRef]
- Wang, Q.; Li, Y.; Zhang, Y.; Ma, L.; Lin, L.; Meng, J.; Jiang, L.; Wang, L.; Zhou, P.; Zhang, Y. LncRNA MEG3 inhibited osteogenic differentiation of bone marrow mesenchymal stem cells from postmenopausal osteoporosis by targeting miR-133a-3p. Biomed. Pharmacother. 2017, 89, 1178–1186. [Google Scholar] [CrossRef]
- Li, M.; Shen, Y.-J.; Chai, S.; Bai, Y.-L.; Li, Z.-H. miR-133a-3p inhibits the osteogenic differentiation of bone marrow mesenchymal stem cells by regulating ankyrin repeat domain 44. Gen. Physiol. Biophys. 2021, 40, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Bourgery, M.; Ekholm, E.; Hiltunen, A.; Heino, T.J.; Pursiheimo, J.P.; Bendre, A.; Yatkin, E.; Laitala, T.; Määttä, J.; Säämänen, A.M. signature of circulating small non-coding RNAs during early fracture healing in mice. Bone Reports 2022, 17, 101627. [Google Scholar] [CrossRef]
- Peng, J.; Zhan, Y.; Zong, Y. METTL3-Mediated LINC00657 promotes osteogenic differentiation of mesenchymal stem cells via miR-144-3p/BMPR1B axis. Cell Tissue Res. 2022, 388, 301–312. [Google Scholar] [CrossRef]
- Li, N.; Liu, L.; Liu, Y.; Luo, S.; Song, Y.; Fang, B. miR-144-3p suppresses osteogenic differentiation of BMSCs from patients with aplastic anemia through repression of TET2. Mol. Ther. - Nucleic Acids 2020, 19, 619–626. [Google Scholar] [CrossRef]
- Sun, Z.; Wu, F.; Yang, Y.; Liu, F.; Mo, F.; Chen, J.; Wang, G.; Zhang, B. miR-144-3p inhibits bmsc proliferation and osteogenic differentiation via targeting FZD4 in steroid-associated osteonecrosis. Curr. Pharm. Des. 2019, 25, 4806–4812. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.X.; Zhang, J.F.; Xu, J.; Xu, L.L.; Wu, T.Y.; Wang, B.; Pan, X.H.; Li, G. MicroRNA-144-3p inhibits bone formation in distraction osteogenesis through targeting Connexin 43. Oncotarget 2017, 8, 89913–89922. [Google Scholar] [CrossRef]
- Liu, N.; Sun, Y. MicroRNA-148a-3p-targeting P300 protects against osteoblast differentiation and osteoporotic bone reconstruction. Regen. Med. 2021, 16, 435–449. [Google Scholar] [CrossRef]
- Tian, L.; Zheng, F.; Li, Z.; Wang, H.; Yuan, H.; Zhang, X.; Ma, Z.; Li, X.; Gao, X.; Wang, B. miR-148a-3p regulates adipocyte and osteoblast differentiation by targeting lysine-specific demethylase 6b. Gene 2017, 627, 32–39. [Google Scholar] [CrossRef]
- Almeida, M.I.; Silva, A.M.; Vasconcelos, D.M.; Almeida, C.R.; Caires, H.; Pinto, M.T.; Calin, G.A.; Santos, S.G.; Barbosa, M.A. miR-195 in human primary mesenchymal stromal/stem cells regulates proliferation, osteogenesis and paracrine effect on angiogenesis. Oncotarget 2016, 7, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.Y.; Deng, R.; Qiu, L.; Wen, Q.; Zeng, Y.; Gao, L.; Zhang, C.; Kong, P.; Zhong, J.; Zeng, N.; et al. miR-203a-3p.1 is involved in the regulation of osteogenic differentiation by directly targeting Smad9 in MM-MSCs. Oncol. Lett. 2019, 18, 6339–6346. [Google Scholar] [CrossRef]
- Zhang, B.; Huo, S.; Cen, X.; Pan, X.; Huang, X.; Zhao, Z. CircAKT3 positively regulates osteogenic differentiation of human dental pulp stromal cells via miR-206/CX43 axis. Stem Cell Res. Ther. 2020, 11, 531. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chang, B.; Zheng, G.; Du, S.; Li, X. quercetin stimulates osteogenic differentiation of bone marrow stromal cells through miRNA-206/Connexin 43 pathway. Am. J. Transl. Res. 2020, 12, 2062–2070. [Google Scholar] [PubMed]
- Chen, Y.; Yang, Y.R.; Fan, X.L.; Lin, P.; Yang, H.; Chen, X.Z.; Xu, X.D. miR-206 inhibits osteogenic differentiation of bone marrow mesenchymal stem cells by targetting glutaminase. Biosci. Rep. 2019, 39, BSR20181108. [Google Scholar] [CrossRef] [PubMed]
- Inose, H.; Ochi, H.; Kimura, A.; Fujita, K.; Xu, R.; Sato, S.; Iwasaki, M.; Sunamura, S.; Takeuchi, Y.; Fukumoto, S.; et al. A microRNA regulatory mechanism of osteoblast differentiation. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 20794–20799. [Google Scholar] [CrossRef]
- Li, W.; Li, L.; Cui, R.; Chen, X.; Hu, H.; Qiu, Y. Bone marrow mesenchymal stem cells derived exosomal Lnc TUG1 promotes bone fracture recovery via miR-22-5p/Anxa8 axis. Hum. Cell 2023, 36, 1041–1053. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, Y.; Li, Y.; Wen, T. MALAT1 enhanced the proliferation of human osteoblasts treated with ultra-high molecular weight polyethylene by targeting VEGF via miR-22-5p. Int. J. Mol. Med. 2018, 41, 1536–1546. [Google Scholar] [CrossRef]
- Wei, J.; Shi, Y.; Zheng, L.; Zhou, B.; Inose, H.; Wang, J.; Guo, X.E.; Grosschedl, R.; Karsenty, G. miR-34s inhibit osteoblast proliferation and differentiation in the mouse by targeting SATB2. J. Cell Biol. 2012, 197, 509–521. [Google Scholar] [CrossRef]
- Garcia, J.; Smith, S.S.; Karki, S.; Drissi, H.; Hrdlicka, H.H.; Youngstrom, D.W.; Delany, A.M. miR-433-3p suppresses bone formation and mRNAs critical for osteoblast function in mice. J. Bone Miner. Res. 2021, 36, 1808–1822. [Google Scholar] [CrossRef]
- Kim, E.J.; Kang, I.H.; Lee, J.W.; Jang, W.G.; Koh, J.T. miR-433 mediates ERRγ-suppressed osteoblast differentiation via direct targeting to Runx2 mRNA in C3H10T1/2 cells. Life Sci. 2013, 92, 562–568. [Google Scholar] [CrossRef]
- Liao, W.; Ning, Y.; Xu, H.J.; Zou, W.Z.; Hu, J.; Liu, X.Z.; Yang, Y.; Li, Z.H. BMSC-derived exosomes carrying microRNA-122-5p promote proliferation of osteoblasts in osteonecrosis of the femoral head. Clin. Sci. 2019, 133, 1955–1975. [Google Scholar] [CrossRef]
- Lu, X.D.; Han, W.X.; Liu, Y.X. Suppression of miR-451a accelerates osteogenic differentiation and inhibits bone loss via Bmp6 signaling during osteoporosis. Biomed. Pharmacother. 2019, 120, 109378. [Google Scholar] [CrossRef] [PubMed]
- Karvande, A.; Kushwaha, P.; Ahmad, N.; Adhikary, S.; Kothari, P.; Tripathi, A.K.; Khedgikar, V.; Trivedi, R. Glucose dependent miR-451a expression contributes to parathyroid hormone mediated osteoblast differentiation. Bone 2018, 117, 98–115. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Jin, Y.; Zhao, S.; Yuan, H.; Shi, J.; Zhao, H. Hypoxic ADSC-derived exosomes enhance wound healing in diabetic mice via delivery of circ-Snhg11 and induction of M2-like macrophage polarization. Biomed. Pharmacother. 2022, 153, 113463. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.W.; Hu, Y.R.; Zhao, J.Y.; Li, S.F.; Ma, X.; Wu, S.G.; Lu, J.B.; Qiu, Y.R.; Sha, Y.H.; Wang, Y.C.; et al. An agomir of miR-144-3p accelerates plaque formation through impairing reverse cholesterol transport and promoting pro-inflammatory cytokine production. PLoS One 2014, 9, e94997. [Google Scholar] [CrossRef]
- Bai, X.; Li, J.; Li, L.; Liu, M.; Liu, Y.; Cao, M.; Tao, K.; Xie, S.; Hu, D. Extracellular vesicles from adipose tissue-derived stem cells affect Notch-miR148a-3p axis to regulate polarization of macrophages and alleviate sepsis in mice. Front. Immunol. 2020, 11, 1391. [Google Scholar] [CrossRef]
- Huang, F.; Zhao, J.L.; Wang, L.; Gao, C.C.; Liang, S.Q.; An, D.J.; Bai, J.; Chen, Y.; Han, H.; Qin, H.Y. miR-148a-3p mediates Notch signaling to promote the differentiation and M1 activation of macrophages. Front. Immunol. 2017, 8, 1327. [Google Scholar] [CrossRef]
- Wei, Y.; Nazari-Jahantigh, M.; Chan, L.; Zhu, M.; Heyll, K.; Corbalán-Campos, J.; Hartmann, P.; Thiemann, A.; Weber, C.; Schober, A. The microRNA-342-5p fosters inflammatory macrophage activation through an Akt1- and microRNA-155-dependent pathway during atherosclerosis. Circulation 2013, 127, 1609–1619. [Google Scholar] [CrossRef]
- Liu, N.; Wang, X.; Steer, C.J.; Song, G. MicroRNA-206 promotes the recruitment of CD8+ T cells by driving M1 polarisation of kupffer cells. Gut 2022, 71, 1642–1655. [Google Scholar] [CrossRef]
- Wu, J.; Liao, Y.; Li, D.; Zhu, Z.; Zhang, L.; Wu, Z.; He, P.; Wang, L. Extracellular vesicles derived from Trichinella Spiralis larvae promote the polarization of macrophages to M2b type and inhibit the activation of fibroblasts. Front. Immunol. 2022, 13, 974332. [Google Scholar] [CrossRef]
- Li, K.; Yan, G.; Huang, H.; Zheng, M.; Ma, K.; Cui, X.; Lu, D.; Zheng, L.; Zhu, B.; Cheng, J.; et al. Anti-inflammatory and immunomodulatory effects of the extracellular vesicles derived from human umbilical cord mesenchymal stem cells on osteoarthritis via M2 macrophages. J. Nanobiotechnology 2022, 20, 38. [Google Scholar] [CrossRef]
- Coulson, D.J.; Bakhashab, S.; Latief, J.S.; Weaver, J.U. miR-126, IL-7, CXCR1/2 receptors, inflammation and circulating endothelial progenitor cells: The study on targets for treatment pathways in a model of subclinical cardiovascular disease (Type 1 diabetes mellitus). J. Transl. Med. 2021, 19, 140. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ming, J.; Li, Y.; Li, B.; Deng, M.; Ma, Y.; Chen, Z.; Zhang, Y.; Li, J.; Liu, S. Exosomes derived from miR-126-3p-overexpressing synovial fibroblasts suppress chondrocyte inflammation and cartilage degradation in a rat model of osteoarthritis. Cell Death Discov. 2021, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Njock, M.S.; O’Grady, T.; Nivelles, O.; Lion, M.; Jacques, S.; Cambier, M.; Herkenne, S.; Muller, F.; Christian, A.; Remacle, C.; et al. Endothelial extracellular vesicles promote tumour growth by tumour-associated macrophage reprogramming. J. Extracell. Vesicles 2022, 11, e12228. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Chen, M.; Ouyang, L.; Wang, Q.; Guo, Y.; Huang, L.; Jiang, S. miR-142-5p and miR-130a-3p regulate pulmonary macrophage polarization and asthma airway remodeling. Immunol. Cell Biol. 2020, 98, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Tong, L.; Liu, F.; Liu, A.; Zeng, S.; Xiong, Q.; Yang, Z.; He, X.; Sun, Y.; Xu, C. Tumor-infiltrating M2 macrophages driven by specific genomic alterations are associated with prognosis in bladder cancer. Oncol. Rep. 2019, 42, 581–594. [Google Scholar] [CrossRef]
- Mao, M.; Xu, Y.; Zhang, X.Y.; Yang, L.; An, X. Bin; Qu, Y.; Chai, Y.N.; Wang, Y.R.; Li, T.T.; Ai, J. MicroRNA-195 prevents hippocampal microglial/macrophage polarization towards the M1 phenotype induced by chronic brain hypoperfusion through regulating CX3CL1/CX3CR1 signaling. J. Neuroinflammation 2020, 17, 244. [Google Scholar] [CrossRef]
- Bras, J.P.; Silva, A.M.; Calin, G.A.; Barbosa, M.A.; Santos, S.G.; Almeida, M.I. miR-195 inhibits macrophages pro-inflammatory profile and impacts the crosstalk with smooth muscle cells. PLoS One 2017, 12, e0188530. [Google Scholar] [CrossRef]
- Youn, G.S.; Park, J.K.; Lee, C.Y.; Jang, J.H.; Yun, S.H.; Kwon, H.Y.; Choi, S.Y.; Park, J. MicroRNA-22 negatively regulates LPS-induced inflammatory responses by targeting HDAC6 in macrophages. BMB Rep. 2020, 53, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhong, Y.; Zhou, W.; Song, Y.; Li, W.; Jin, Q.; Gao, T.; Zhang, L.; Xie, M. Low-intensity pulsed ultrasound (LIPUS) enhances the anti-inflammatory effects of bone marrow mesenchymal stem cells (BMSCs)-derived extracellular vesicles. Cell. Mol. Biol. Lett. 2023, 28, 9. [Google Scholar] [CrossRef]
- Cheng, J.; Hao, J.; Jiang, X.; Ji, J.; Wu, T.; Chen, X.; Zhang, F. Ameliorative effects of miR-423-5p against polarization of microglial cells of the M1 phenotype by targeting a NLRP3 inflammasome signaling pathway. Int. Immunopharmacol. 2021, 99, 108006. [Google Scholar] [CrossRef]
- Li, R.; Li, D.; Wang, H.; Chen, K.; Wang, S.; Xu, J.; Ji, P. Exosomes from adipose-derived stem cells regulate M1/M2 macrophage phenotypic polarization to promote bone healing via miR-451a/MIF. Stem Cell Res. Ther. 2022, 13, 149. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, D.; Wang, H.; Ren, Q.; Li, B.; Wang, L.; Zheng, G. MiR-451a enhances the phagocytosis and affects both M1 and M2 polarization in macrophages. Cell. Immunol. 2021, 365, 104377. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, I.M.; Landis, W.J.; Risbud, M. V. Matrix vesicles: Are they anchored exosomes? Bone 2015, 79, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Leach, R.J.; Schwartz, Z.; Johnson-Pais, T.L.; Dean, D.D.; Luna, M.; Boyan, B.D. Osteosarcoma hybrids can preferentially target alkaline phosphatase activity to matrix vesicles: Evidence for independent membrane biogenesis. J. Bone Miner. Res. 1995, 10, 1614–1624. [Google Scholar] [CrossRef] [PubMed]
- Asmussen, N.; Lin, Z.; McClure, M.J.; Schwartz, Z.; Boyan, B.D. Regulation of extracellular matrix vesicles via rapid responses to steroid hormones during endochondral bone formation. Steroids 2019, 142, 43–47. [Google Scholar] [CrossRef]
- Bottini, M.; Mebarek, S.; Anderson, K.L.; Strzelecka-Kiliszek, A.; Bozycki, L.; Simão, A.M.S.; Bolean, M.; Ciancaglini, P.; Pikula, J.B.; Pikula, S.; et al. Matrix vesicles from chondrocytes and osteoblasts: Their biogenesis, properties, functions and biomimetic models. Biochim. Biophys. acta. Gen. Subj. 2018, 1862, 532–546. [Google Scholar] [CrossRef]
- Soe, Z.Y.; Park, E.J.; Shimaoka, M. Integrin regulation in immunological and cancerous cells and exosomes. Int. J. Mol. Sci. 2021, 22, 1–22. [Google Scholar] [CrossRef]
- Minamizaki, T.; Nakao, Y.; Irie, Y.; Ahmed, F.; Itoh, S.; Sarmin, N.; Yoshioka, H.; Nobukiyo, A.; Fujimoto, C.; Niida, S.; et al. The matrix vesicle cargo miR-125b accumulates in the bone matrix, inhibiting bone resorption in mice. Commun. Biol. 2020, 3, 1–2. [Google Scholar] [CrossRef]
- Rao, M.S.; Van Vleet, T.R.; Ciurlionis, R.; Buck, W.R.; Mittelstadt, S.W.; Blomme, E.A.G.; Liguori, M.J. Comparison of RNA-Seq and microarray gene expression platforms for the toxicogenomic evaluation of liver from short-term rat toxicity studies. Front. Genet. 2018, 9, 636. [Google Scholar] [CrossRef]
- Mizukami, Y.; Kawao, N.; Takafuji, Y.; Ohira, T.; Okada, K.; Jo, J.-I.; Tabata, Y.; Kaji, H. Matrix vesicles promote bone repair after a femoral bone defect in mice. PLoS One 2023, 18, e0284258. [Google Scholar] [CrossRef]
- Li, W.; Liu, Y.; Zhang, P.; Tang, Y.; Zhou, M.; Jiang, W.; Zhang, X.; Wu, G.; Zhou, Y. Tissue-engineered bone immobilized with human adipose stem cells-derived exosomes promotes bone regeneration. ACS Appl. Mater. Interfaces 2018, 10, 5240–5254. [Google Scholar] [CrossRef] [PubMed]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Costa Verdera, H.; Gitz-Francois, J.J.; Schiffelers, R.M.; Vader, P. Cellular uptake of extracellular vesicles Is mediated by clathrin-independent endocytosis and macropinocytosis. J. Control. Release 2017, 266, 100–108. [Google Scholar] [CrossRef]
- Svensson, K.J.; Christianson, H.C.; Wittrup, A.; Bourseau-Guilmain, E.; Lindqvist, E.; Svensson, L.M.; Mörgelin, M.; Belting, M. Exosome uptake depends on ERK1/2-Heat Shock Protein 27 signaling and lipid raft-mediated endocytosis negatively regulated by caveolin-1. J. Biol. Chem. 2013, 288, 17713–17724. [Google Scholar] [CrossRef]
- Tian, T.; Zhu, Y.L.; Zhou, Y.Y.; Liang, G.F.; Wang, Y.Y.; Hu, F.H.; Xiao, Z.D. Exosome uptake through clathrin-mediated endocytosis and macropinocytosis and mediating miR-21 delivery. J. Biol. Chem. 2014, 289, 22258–22267. [Google Scholar] [CrossRef]
- Hazan-Halevy, I.; Rosenblum, D.; Weinstein, S.; Bairey, O.; Raanani, P.; Peer, D. Cell-specific uptake of mantle cell lymphoma-derived exosomes by malignant and non-malignant B-lymphocytes. Cancer Lett. 2015, 364, 59–69. [Google Scholar] [CrossRef]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.Y.; Schwartz, Z.; Hummert, T.W.; Schraub, D.M.; Simpson, J.; Lankford, J.; Dean, D.D.; Cochran, D.L.; Boyan, B.D. Effect of titanium surface roughness on proliferation, differentiation, and protein synthesis of human osteoblast-like cells (MG63). J. Biomed. Mater. Res. 1995, 29, 389–401. [Google Scholar] [CrossRef]
- Schwartz, Z.; Lohmann, C.H.; Oefinger, J.; Bonewald, L.F.; Dean, D.D.; Boyan, B.D. Implant surface characteristics modulate differentiation behavior of cells in the osteoblastic lineage. Adv. Dent. Res. 1999, 13, 38–48. [Google Scholar] [CrossRef]
- Heremans, H.; Billiau, A.; Cassiman, J.J.; Mulier, J.C.; De Somer, P. In vitro cultivation of human tumor tissues II. Morphological and virological characterization of three cell lines. Oncol. 1978, 35, 246–252. [Google Scholar] [CrossRef]
- Czekanska, E.M.; Stoddart, M.J.; Richards, R.G.; Hayes, J.S. In search of an osteoblast cell model for in vitro research. Eur. Cells Mater. 2012, 24, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, D.F.; Davenport, G.R.; Forte, L.; Landon, E.J. Characterization of plasma membrane proteins in mammalian kidney. I. Preparation of a membrane fraction and separation of the protein. J. Biol. Chem. 1969, 244, 3561–3569. [Google Scholar] [CrossRef] [PubMed]
- Boyan, B.D.; Schwartz, Z.; Swain, L.D.; Carnes Jr., D. L.; Zislis, T. Differential expression of phenotype by resting zone and growth region costochondral chondrocytes in vitro. Bone 1988, 9, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Boyan, B.D.; Wong, K.L.; Wang, L.; Yao, H.; Guldberg, R.E.; Drab, M.; Jo, H.; Schwartz, Z. Regulation of growth plate chondrocytes by 1,25-dihydroxyvitamin D 3 requires caveolae and caveolin-1. J. Bone Miner. Res. 2006, 21, 1637–1647. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Matrix vesicles display extracellular vesicle characteristics and demonstrate uptake by osteoblast-like cells. Quantification of alkaline phosphatase specific activity for cell pellet lysate (CP), plasma membrane (PM), and matrix vesicle (MV) isolates (A); Transmission electron microscopy at 50K magnification of MVs demonstrated circular vesicles with a bilaminar membrane (B, scale bar = 100 nm); TEM of green inset in B at 100K magnification (C, scale bar = 50 nm); Size distribution of osteoblast-derived MVs using nanoparticle tracking analysis with median of 105 nm (green line), mean of 115 nm (red line), and standard deviation of ±85 nm (blue dashed lines), results are mean of three independent measurements (D); Western blot analysis of protein expression in CP, PM, and MV isolates (E); Osteoblasts treated with PKH26-stained MVs (red fluorescence), DRAQ5 nuclear counterstain (blue fluorescence) and plasma membrane counterstain (yellow fluorescence) using confocal microscopy (F, scale bar = 50 µm); Region of interest to highlight PKH26-stained MVs surrounding osteoblast nuclei (G, scale bar = 10 µm); NaCl control-treated osteoblasts with DRAQ5 nuclear counterstain and plasma membrane counterstain (H, scale bar = 50 µm); Quantification of cells with a minimum of one spot of PKH26-stained MV fluorescence between MV and NaCl control images (I). Results are expressed as the mean ± SEM of 2 independent reviewers. Groups not sharing a letter are considered statistically different at α = 0.05 by one-way ANOVA with Tukey post-hoc test. Group with * is significantly different from the vehicle at an α = 0.05 by unpaired t-test.
Figure 1.
Matrix vesicles display extracellular vesicle characteristics and demonstrate uptake by osteoblast-like cells. Quantification of alkaline phosphatase specific activity for cell pellet lysate (CP), plasma membrane (PM), and matrix vesicle (MV) isolates (A); Transmission electron microscopy at 50K magnification of MVs demonstrated circular vesicles with a bilaminar membrane (B, scale bar = 100 nm); TEM of green inset in B at 100K magnification (C, scale bar = 50 nm); Size distribution of osteoblast-derived MVs using nanoparticle tracking analysis with median of 105 nm (green line), mean of 115 nm (red line), and standard deviation of ±85 nm (blue dashed lines), results are mean of three independent measurements (D); Western blot analysis of protein expression in CP, PM, and MV isolates (E); Osteoblasts treated with PKH26-stained MVs (red fluorescence), DRAQ5 nuclear counterstain (blue fluorescence) and plasma membrane counterstain (yellow fluorescence) using confocal microscopy (F, scale bar = 50 µm); Region of interest to highlight PKH26-stained MVs surrounding osteoblast nuclei (G, scale bar = 10 µm); NaCl control-treated osteoblasts with DRAQ5 nuclear counterstain and plasma membrane counterstain (H, scale bar = 50 µm); Quantification of cells with a minimum of one spot of PKH26-stained MV fluorescence between MV and NaCl control images (I). Results are expressed as the mean ± SEM of 2 independent reviewers. Groups not sharing a letter are considered statistically different at α = 0.05 by one-way ANOVA with Tukey post-hoc test. Group with * is significantly different from the vehicle at an α = 0.05 by unpaired t-test.
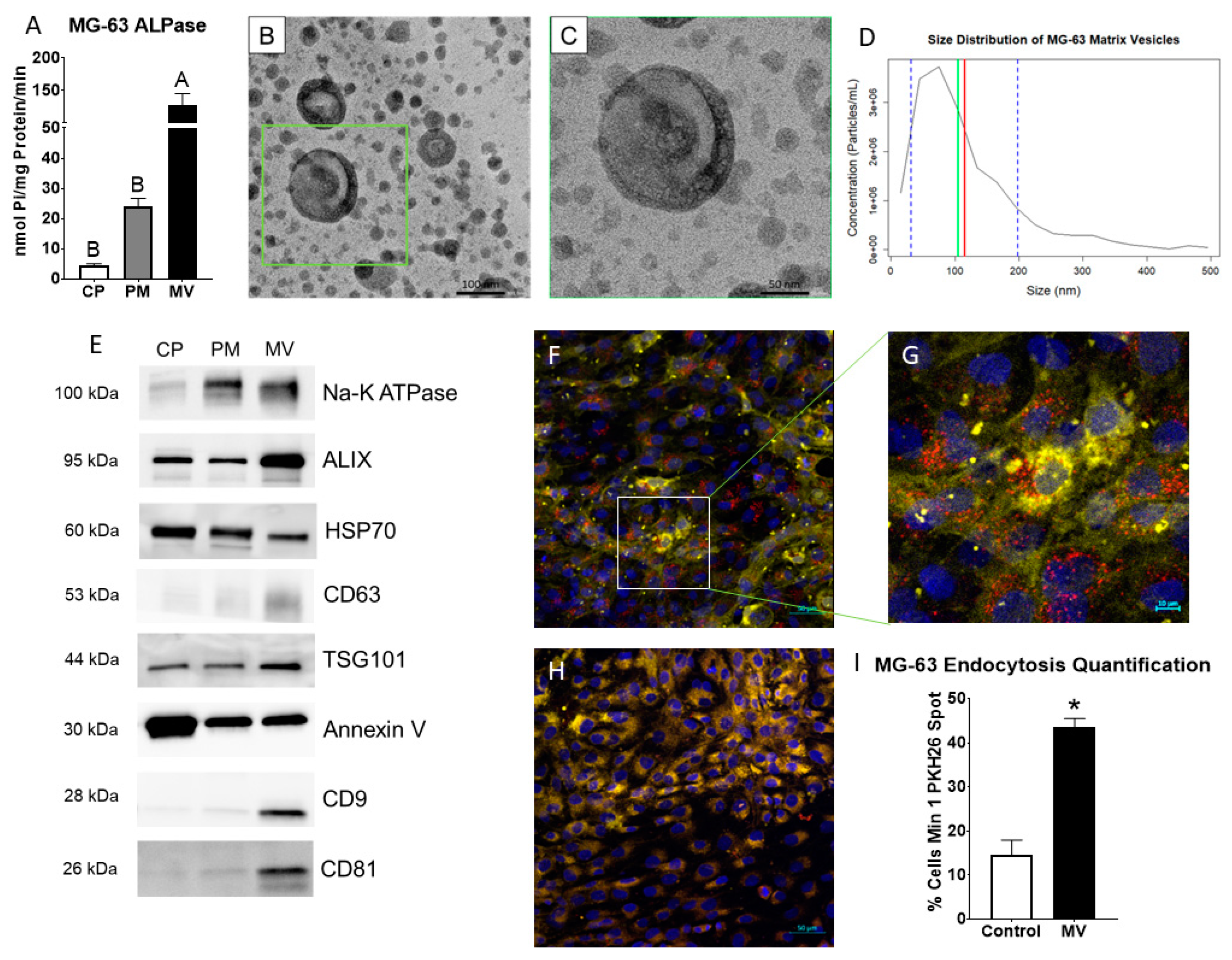
Figure 2.
Small RNA sequencing suggests selective packaging of microRNA into MG-63 matrix vesicles. Principal component analysis plot of components 1 & 2 for all six samples with grouping of cell lysate (CP) samples (pink) and matrix vesicle (MV) samples (teal) (A); Venn diagram displaying overlap of unique microRNA with differential expression between CP and MV samples (B); Heatmap of sample z-scores clustered with Euclidean distance measure (C); Volcano plot of microRNA with Log2 fold change greater than ±2 and adjusted p-value less than 0.05 (D). .
Figure 2.
Small RNA sequencing suggests selective packaging of microRNA into MG-63 matrix vesicles. Principal component analysis plot of components 1 & 2 for all six samples with grouping of cell lysate (CP) samples (pink) and matrix vesicle (MV) samples (teal) (A); Venn diagram displaying overlap of unique microRNA with differential expression between CP and MV samples (B); Heatmap of sample z-scores clustered with Euclidean distance measure (C); Volcano plot of microRNA with Log2 fold change greater than ±2 and adjusted p-value less than 0.05 (D). .
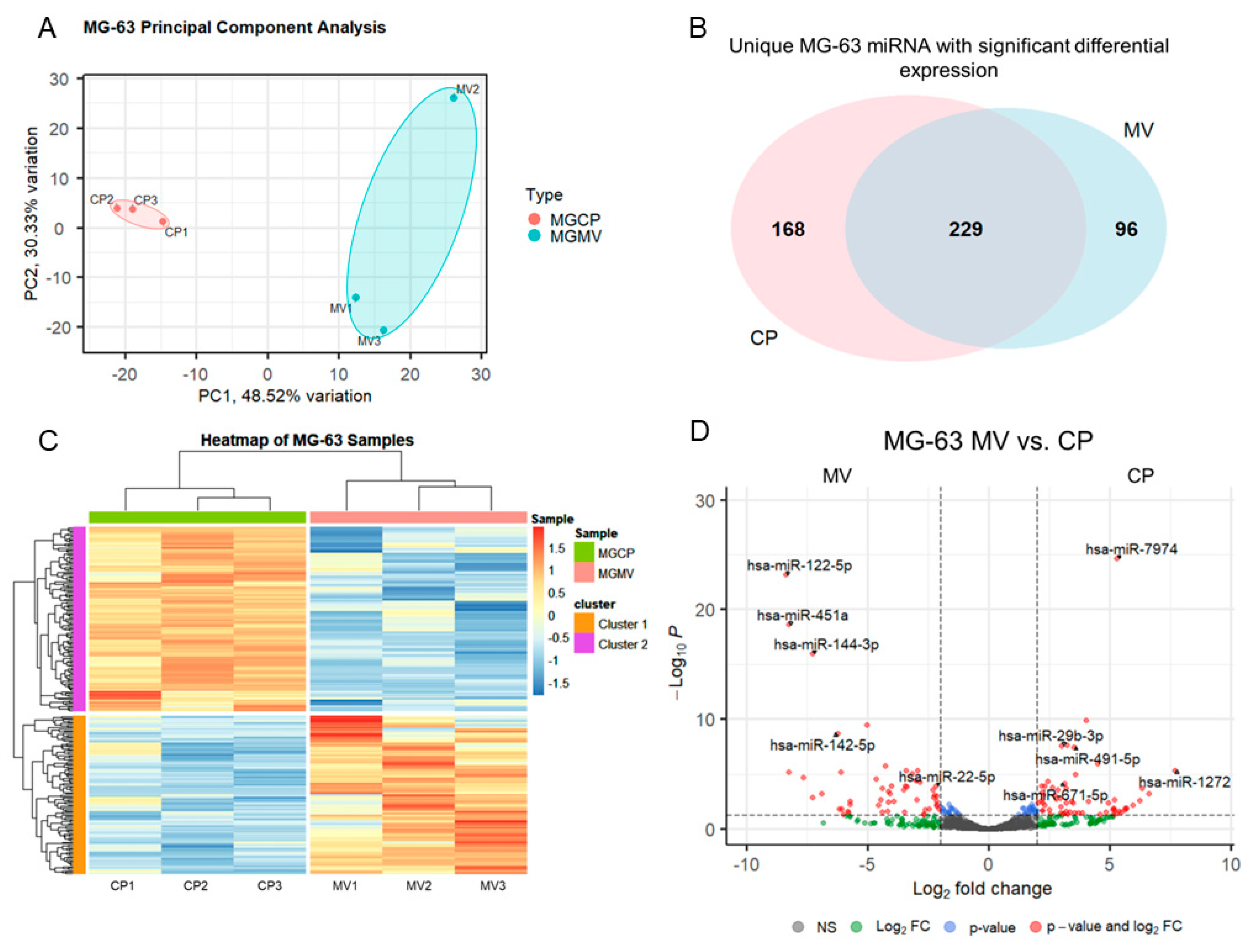
Figure 3.
Differentially expressed microRNA of matrix vesicles suggest an anti-osteogenic role in osteoblasts and regenerative role in macrophages. Differentially expressed microRNA (adjusted p-value < 0.05 & absolute Log2 fold change > 2) with pro-, anti-, or unclear osteogenic potential based on literature search (A); Differentially expressed microRNA with promotion of M1 or M2 macrophage polarization according to literature search (B).
Figure 3.
Differentially expressed microRNA of matrix vesicles suggest an anti-osteogenic role in osteoblasts and regenerative role in macrophages. Differentially expressed microRNA (adjusted p-value < 0.05 & absolute Log2 fold change > 2) with pro-, anti-, or unclear osteogenic potential based on literature search (A); Differentially expressed microRNA with promotion of M1 or M2 macrophage polarization according to literature search (B).
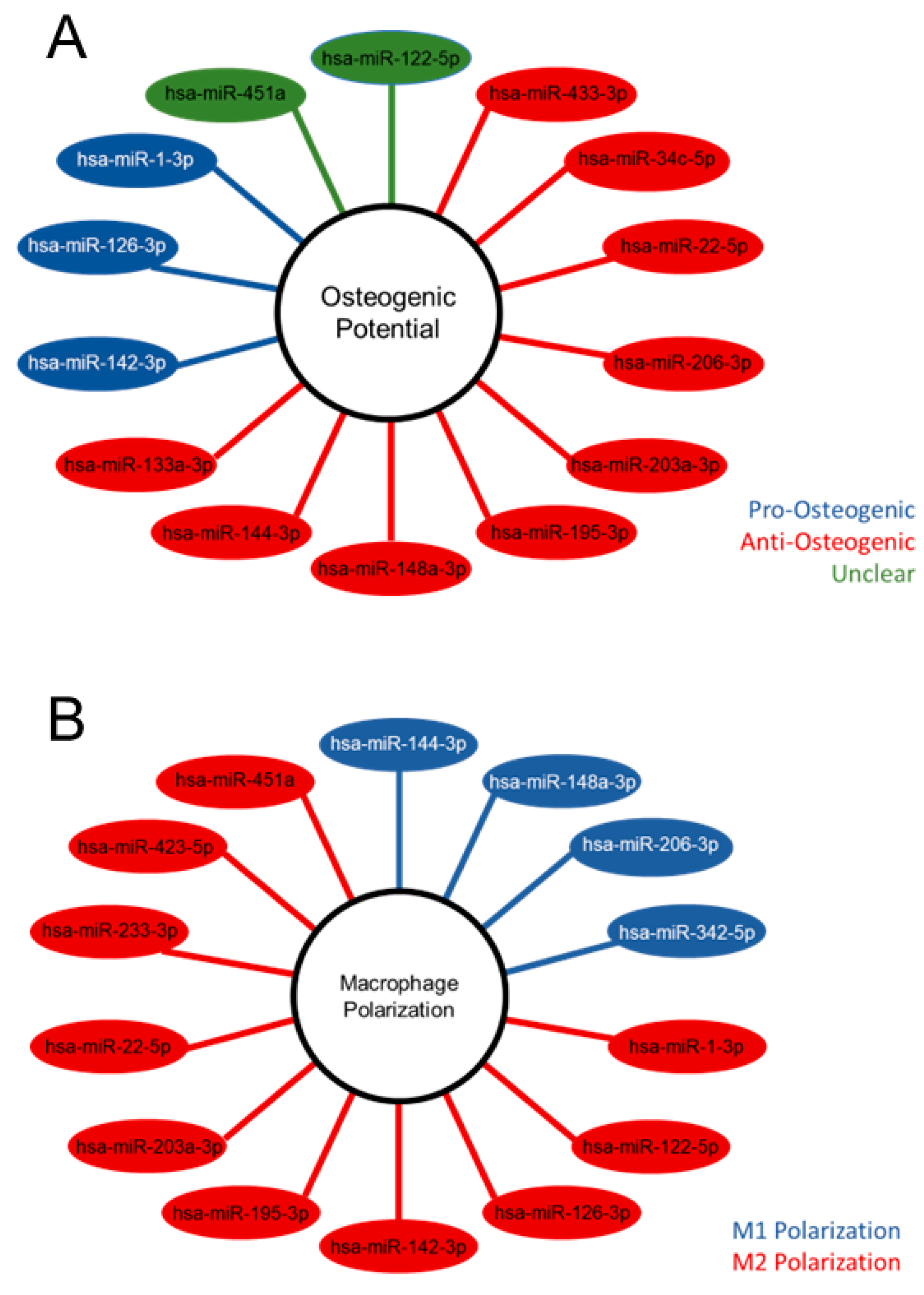
Figure 4.
Treatment of MG-63 cells with matrix vesicles decreases markers of differentiation. Quantification of DNA (A), alkaline phosphatase specific activity (B), osteocalcin (C), osteopontin (D), vascular endothelial growth factor (E), interleukin-6 (F), and interleukin-10 (G) in MG-63 cells treated for 24 or 48 hours with or without 5 µg/mL matrix vesicles (MV) harvested from MG-63s grown in growth media until 24 hours after confluence. Data are from a representative experiment and are shown as mean ± SEM of n=6 per group. Groups not sharing a letter are considered statistically different at α = 0.05 by one-way ANOVA with Tukey post-hoc test. .
Figure 4.
Treatment of MG-63 cells with matrix vesicles decreases markers of differentiation. Quantification of DNA (A), alkaline phosphatase specific activity (B), osteocalcin (C), osteopontin (D), vascular endothelial growth factor (E), interleukin-6 (F), and interleukin-10 (G) in MG-63 cells treated for 24 or 48 hours with or without 5 µg/mL matrix vesicles (MV) harvested from MG-63s grown in growth media until 24 hours after confluence. Data are from a representative experiment and are shown as mean ± SEM of n=6 per group. Groups not sharing a letter are considered statistically different at α = 0.05 by one-way ANOVA with Tukey post-hoc test. .
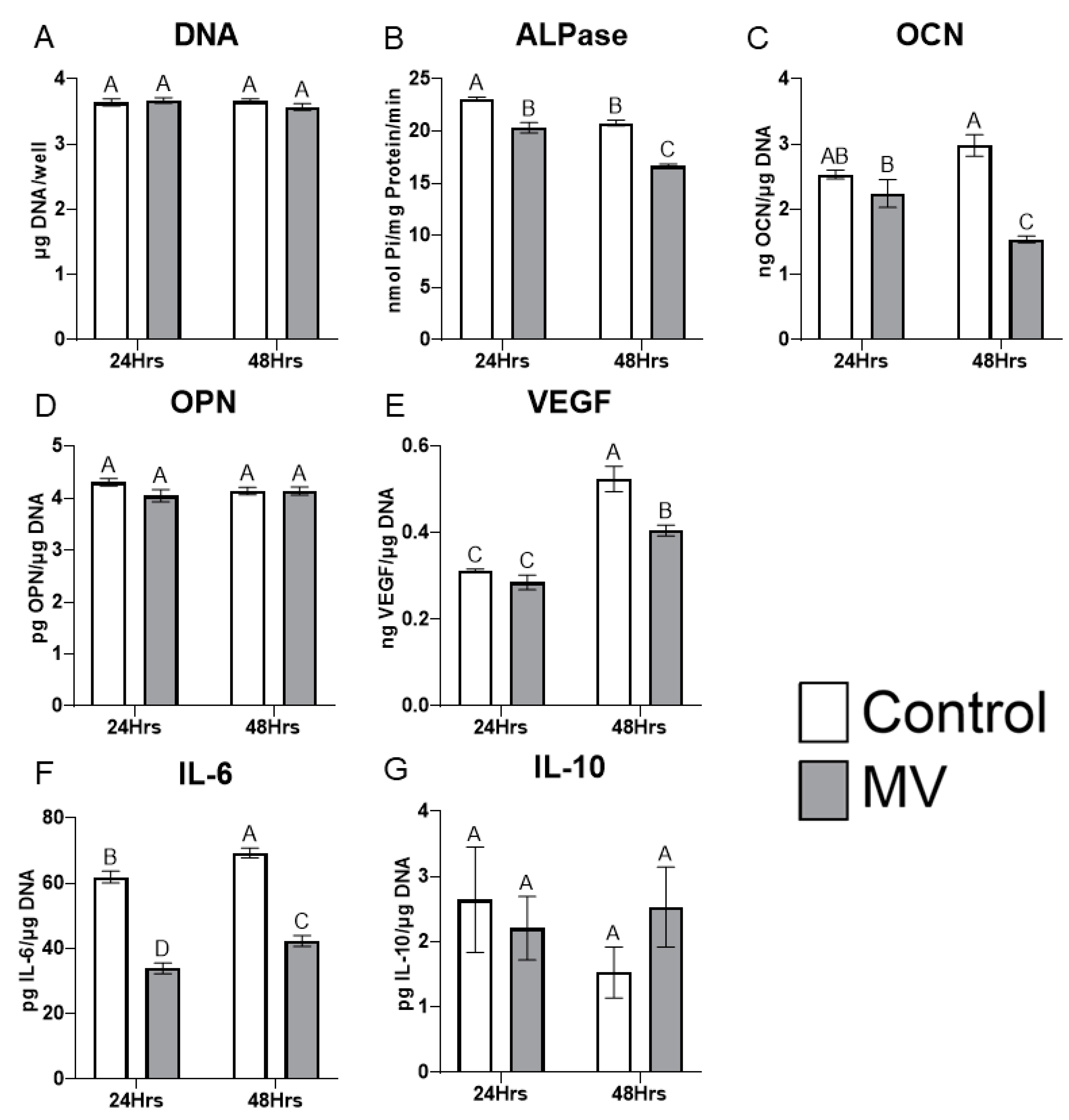
Figure 5.
Treatment of MG-63 cells with matrix vesicles from osteogenic culture conditions reduces osteoblastic differentiation markers and promotes a regenerative inflammation phenotype. Quantification of DNA (A), alkaline phosphatase specific activity (B), osteocalcin (C), osteopontin (D), vascular endothelial growth factor (E), interleukin-6 (F), and interleukin-10 (G) in MG-63 cells treated for 24 or 48 hours with or without 5 µg/mL matrix vesicles (MV) harvested from MG-63s grown in osteogenic media for 10 days. Data are from a representative experiment and are shown as mean ± SEM of n=6 per group. Groups not sharing a letter are considered statistically different at α = 0.05 by one-way ANOVA with Tukey post-hoc test.
Figure 5.
Treatment of MG-63 cells with matrix vesicles from osteogenic culture conditions reduces osteoblastic differentiation markers and promotes a regenerative inflammation phenotype. Quantification of DNA (A), alkaline phosphatase specific activity (B), osteocalcin (C), osteopontin (D), vascular endothelial growth factor (E), interleukin-6 (F), and interleukin-10 (G) in MG-63 cells treated for 24 or 48 hours with or without 5 µg/mL matrix vesicles (MV) harvested from MG-63s grown in osteogenic media for 10 days. Data are from a representative experiment and are shown as mean ± SEM of n=6 per group. Groups not sharing a letter are considered statistically different at α = 0.05 by one-way ANOVA with Tukey post-hoc test.
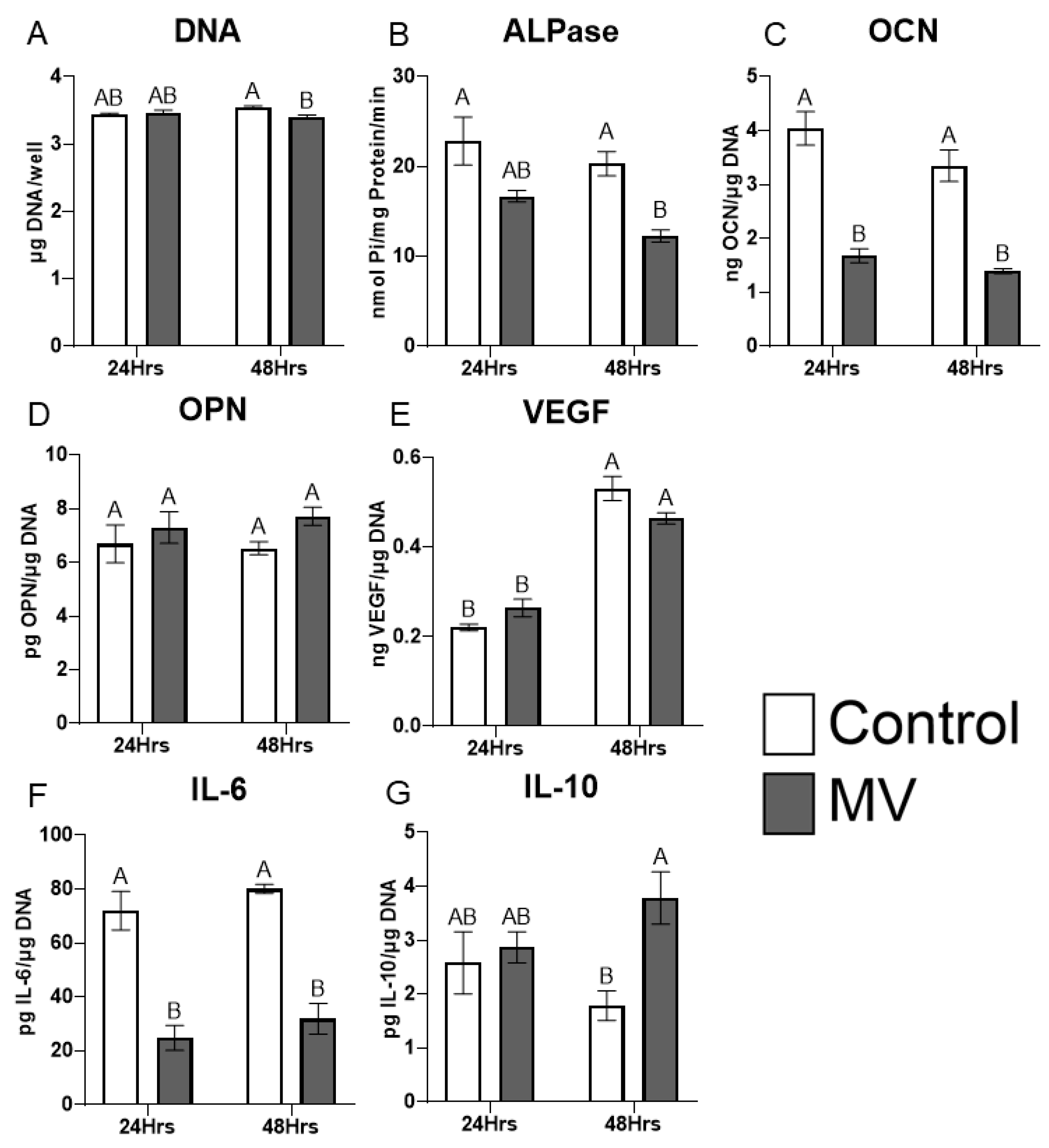
Figure 6.
Inhibition of caveolin-mediated endocytosis prevents uptake and effect of matrix vesicles on osteoblast-like cells. MG-63 cells pre-treated with 10 mM vehicle (A) or methyl-β-cyclodextrin (β-CD; B) followed by treatment with PKH26-stained MVs (red fluorescence), DRAQ5 nuclear counterstain (blue fluorescence) and plasma membrane counterstain (yellow fluorescence) using confocal microscopy (scale bar = 50 µm); Quantification of cells with a minimum of one spot of PKH26-stained MV fluorescence between vehicle and β-CD images (C); Quantification of DNA (D) and alkaline phosphatase specific activity (E) in MG-63 cells pre-treated with increasing concentrations of β-CD followed by treatment for 48 hours with or without 5 µg/mL matrix vesicles (MV) harvested from MG-63s grown in growth media until 24 hours after confluence. Results are expressed as the mean ± SEM of 2 independent reviewers. Group with * is significantly different from the vehicle at an α = 0.05 by unpaired t-test. Groups not sharing a letter are considered statistically different at α = 0.05 by one-way ANOVA with Tukey post-hoc test.
Figure 6.
Inhibition of caveolin-mediated endocytosis prevents uptake and effect of matrix vesicles on osteoblast-like cells. MG-63 cells pre-treated with 10 mM vehicle (A) or methyl-β-cyclodextrin (β-CD; B) followed by treatment with PKH26-stained MVs (red fluorescence), DRAQ5 nuclear counterstain (blue fluorescence) and plasma membrane counterstain (yellow fluorescence) using confocal microscopy (scale bar = 50 µm); Quantification of cells with a minimum of one spot of PKH26-stained MV fluorescence between vehicle and β-CD images (C); Quantification of DNA (D) and alkaline phosphatase specific activity (E) in MG-63 cells pre-treated with increasing concentrations of β-CD followed by treatment for 48 hours with or without 5 µg/mL matrix vesicles (MV) harvested from MG-63s grown in growth media until 24 hours after confluence. Results are expressed as the mean ± SEM of 2 independent reviewers. Group with * is significantly different from the vehicle at an α = 0.05 by unpaired t-test. Groups not sharing a letter are considered statistically different at α = 0.05 by one-way ANOVA with Tukey post-hoc test.
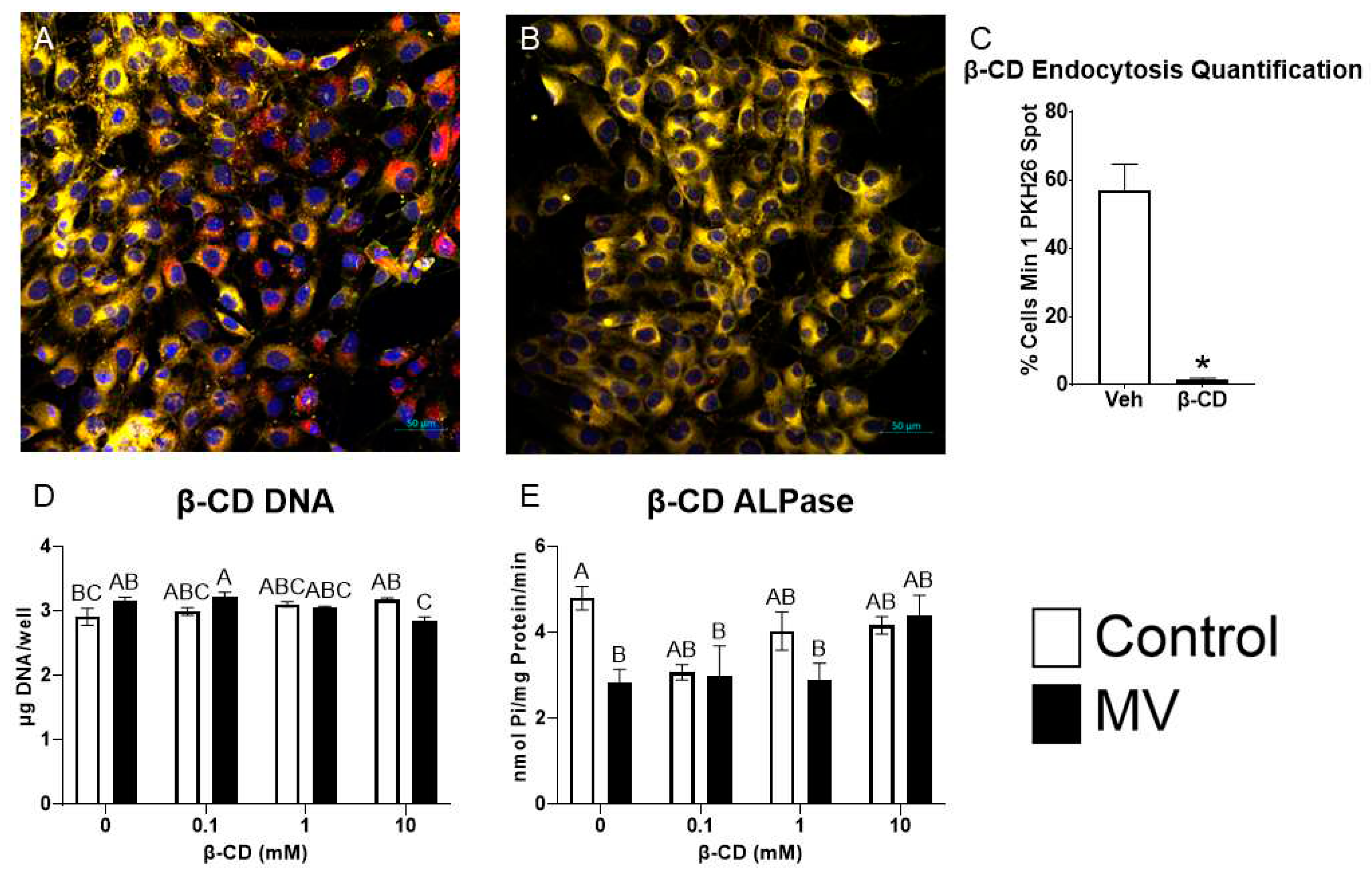
Figure 7.
Schematic Representation of Osteoblast-derived Matrix Vesicles. Matrix vesicles (MVs), with their protein and microRNA (miRNA) enrichment, are produced by osteoblasts and become embedded in the mineralized extracellular matrix. During osteoclast-mediated resorption of bone, MVs are released from the mineralized matrix to act in an autocrine and/or paracrine fashion on nearby osteoblasts. These MVs are taken up by osteoblasts via caveolin-mediated endocytosis, where they are able to release their miRNA cargo intracellularly. miRNA interact with target messenger RNA (mRNA) to active or inhibit translation into proteins, slowing production of osteoblastic differentiation markers. Image created with BioRender.com.
Figure 7.
Schematic Representation of Osteoblast-derived Matrix Vesicles. Matrix vesicles (MVs), with their protein and microRNA (miRNA) enrichment, are produced by osteoblasts and become embedded in the mineralized extracellular matrix. During osteoclast-mediated resorption of bone, MVs are released from the mineralized matrix to act in an autocrine and/or paracrine fashion on nearby osteoblasts. These MVs are taken up by osteoblasts via caveolin-mediated endocytosis, where they are able to release their miRNA cargo intracellularly. miRNA interact with target messenger RNA (mRNA) to active or inhibit translation into proteins, slowing production of osteoblastic differentiation markers. Image created with BioRender.com.

Table 1.
Differentially Expressed microRNA, Osteogenic Potential, and Linked Pathways.
| miRNA | Osteogenic Potential | References |
|---|---|---|
| hsa-miR-1-3p | Pro-Osteogenic | [24] |
| hsa-miR-126-3p | Pro-Osteogenic | [25,26] |
| hsa-miR-142-5p | Pro-Osteogenic | [27,28,29] |
| hsa-miR-133a-3p | Anti-Osteogenic | [30,31,32] |
| hsa-miR-144-3p | Anti-Osteogenic | [33,34,35,36] |
| hsa-miR-148a-3p | Anti-Osteogenic | [26,37,38] |
| hsa-miR-195-3p | Anti-Osteogenic | [39] |
| hsa-miR-203a-3p | Anti-Osteogenic | [40] |
| hsa-miR-206 | Anti-Osteogenic | [41,42,43,44] |
| hsa-miR-22-5p | Anti-Osteogenic | [45,46] |
| hsa-miR-34c-3p | Anti-Osteogenic | [47] |
| hsa-miR-433-3p | Anti-Osteogenic | [48,49] |
| hsa-miR-122-5p | Unclear | [14,50] |
| hsa-miR-451a | Unclear | [51,52] |
Table 2.
Differentially Expressed microRNA and Inflammatory Modulation.
| miRNA | M1/M2 Polarization | References |
|---|---|---|
| hsa-miR-144-3p | M1 | [53,54] |
| hsa-miR-148a-3p | M1 | [55,56] |
| hsa-miR-342-5p | M1 | [57] |
| hsa-miR-206-3p | M1 | [58] |
| hsa-miR-1-3p | M2b | [59] |
| hsa-miR-122-5p | M2 | [60] |
| hsa-miR-126-3p | M2 | [61,62] |
| hsa-miR-142-5p | M2 | [63,64,65] |
| hsa-miR-195-3p | M2 | [66,67] |
| hsa-miR-22-5p | M2 | [68] |
| hsa-miR-223-3p | M2 | [65] |
| hsa-miR-328-5p | M2 | [69] |
| hsa-miR-423-5p | M2 | [70] |
| hsa-miR-451a | M2 | [71,72] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



