
Submitted:
12 June 2023
Posted:
13 June 2023
You are already at the latest version
Abstract
Abstract: Cherubism is a rare genetic disorder characterized by painless bilateral expansion of the mandible and maxilla. We present an unusual case of a 50-year-old female patient with Ramon syndrome, a rare variant of cherubism, accompanied by gingival fibromatosis. The patient exhibited symptoms of short stature, mental retardation, rheumatoid arthritis, epilepsy, hirsutism, and gingival enlargement. Clinical examination revealed generalized gingival inflammation with plaque and attachment loss. Radiographically, bilateral defined multilocular radiolucencies were observed in the mandibular body and ramus. The patient underwent a non-surgical phase, including oral hygiene motivation and periodontal debridement, followed by a surgical phase involving gingivoplasty and internal bevel gingivectomy with open flap debridement. Histopathological analysis confirmed the presence of fibroblasts, multinucleated giant cells, and fibrous connective tissue. The patient's son also presented with bilateral facial swelling and similar radiographic findings, indicating familial inheritance of cherubism. The case demonstrates the clinical, radiographic, and histopathological features of cherubism associated with gingival fibromatosis and highlights the challenges in diagnosis and management. Regular follow-up and maintenance are essential for long-term stability and prevention of recurrence. This report contributes to the limited literature on cherubism in older patients and emphasizes the importance of early identification and intervention in affected individuals.
Keywords:
Ramon syndrome
; Cherubism
; Gingival fibromatosis
; Genetic disorder
; Multilocular radiolucencies
; Surgical intervention
; Oral hygiene
; Recurrence prevention
1. Introduction
Cherubism is a rare genetic disorder categorized as a non-neoplastic pathology, which was initially identified by Frangenheim in 1914 [1]. Subsequently, Dr. William Jones documented a case in 1933, describing the condition in three siblings from a Jewish Russian family [1]. The term "cherubism" is derived from the resemblance of the affected individuals’ cherubic or chubby-cheeked facial features, as depicted in Renaissance paintings [2]. It is characterized as a fibro-osseous disorder, primarily involving painless bilateral expansion of the mandible, maxilla, or both, with a higher incidence observed among younger individuals. Furthermore, cherubism is commonly inherited [3].
Radiographic examinations reveal cystic multilocular radiolucencies in the mandibular body, which may extend to the mandibular ramus and angle, with concurrent presence of lesions in the maxilla [4].
Cherubism can also be associated with other medical conditions such as gingival fibromatosis, short stature, mental retardation, epilepsy, rheumatoid arthritis, and hirsutism, collectively referred to as Ramon syndrome, although it is an exceedingly rare occurrence.
This article presents an extraordinary case of Ramon syndrome in an adult female, encompassing the non-surgical phase of treatment and subsequent mucogingival management, with a comprehensive clinical follow-up spanning one year.
2. Case report
In december 2013, a 50-year-old patient presented to the Periodontology Department at the Rabat Dental Consultation and Treatment Center with the chief complaint of generalized gingival enlargement that had persisted for several years, causing functional discomfort.
Upon medical examination, the patient was observed to have mental retardation and short stature. The patient’s medical history revealed a long-standing diagnosis of rheumatoid arthritis and epilepsy since early childhood. Currently, the patient is not taking any medication, and there is no known history of medication associated with gingival overgrowth.
The extraoral examination revealed facial swelling without pain, extending bilaterally between the lower border of the mandible and below the inferior orbital margins. The patient also exhibited prominent maxillae, a bulbous soft nose, and mild hirsutism on the upper lip, cheeks, arms, and legs. No palpable lymph nodes were detected, and there were no signs of skin pigmentation or other visible pathologies (Figure 1).
The intraoral examination revealed generalized marginal gingival inflammation, plaque and calculus accumulation, and attachment loss ranging from 3 to 10 mm. Additionally, the patient exhibited a firm, pale-pink, shiny, and smooth hyperplastic type of generalized gingival growth that covered almost all the teeth without causing pain (Figure 2).
Radiographic examination showed horizontal bone lysis affecting approximately two-thirds of the roots of the teeth, consistent with a moderate chronic generalized periodontitis (AAP 1999) or periodontitis Stage III grade B (AAP 2017). The lamina dura of the teeth appeared normal. Furthermore, bilateral well-defined multilocular radiolucencies were identified in the body and ramus of the mandible (Figure 3).
Peripheral blood test results were within normal limits, and levels of serum alkaline phosphatase, calcium, and phosphorous were also found to be within the normal range.
The treatment plan involved a non-surgical phase, including oral hygiene motivation and prescription of appropriate brushing equipment, as well as periodontal debridement. After three months, a periodontal reassessment revealed significant improvement in individual plaque control, plaque and bleeding indices, and a reduction in periodontal pockets.
Subsequently, a surgical phase was scheduled. Under local anesthesia, quadrant-by-quadrant procedures were performed, including gingivoplasty, internal bevel gingivectomy, open flap debridement, and underlying alveolar curettage. Surgeries were spaced approximately three weeks apart. Excised gingival tissue and fragments of the alveolar bone were preserved in formalin and sent to the anatomo-pathology laboratory for analysis.
Post-operative instructions were given to the patient, and a prescription was provided, including a 1g analgesic to be taken twice daily for three days, and a 0.12% chlorhexidine gluconate-based antiseptic to be used twice daily for two weeks following the surgery.
Histopathological examination of the excised gingival tissue revealed numerous fibroblasts and mild chronic inflammatory cells. The overlying epithelium appeared squamous, hyperplastic, and parakeratinized, while the connective tissue exhibited fibrous characteristics and was avascular.
Histopathological analysis of the bone fragments showed a fibrous stroma with numerous multinucleated giant cells.
The clinical, radiologic, and histopathology findings demonstrated the presence of cherubism accompanied by gingival fibromatosis. The collective occurrence of cherubism, gingival fibromatosis, rheumatoid arthritis, mental retardation, epilepsy, and hirsutism is commonly referred to as Ramon syndrome, though it is described as a potential association.
The hereditary aspect of the disease played a contributory role. The patient’s 33-year-old son exhibited bilateral facial swelling similar to that of his mother, which had been diminishing in size since early childhood (Figure 4).
A panoramic radiograph was performed, revealing comparable findings to those of the mother. Both the mother and her son received a diagnosis of cherubism (Figure 5).
The patient has been undergoing regular follow-ups at monthly intervals to monitor plaque control. A surgical reassessment conducted one year postoperatively demonstrated favorable stability of the long-term clinical outcomes, with no recurrence observed and an improvement in the patient’s quality of life (Figure 6).
3. Discussion
Cherubism is a rare genetic disorder recognized by the World Health Organization that primarily affects the jaws of children at an early age. The term "cherubism" derives from the resemblance of affected individuals’ facial appearance to cherubs depicted in Renaissance paintings, conveying a sense of heavenly gaze. This condition is characterized by painless and often symmetrical enlargement of the jaws, as observed in our specific case [3].
The genetic basis of cherubism revolves around candidate genes involved in bone physiology [5]. The majority of research points to the cherubism gene being located on chromosome 4, specifically in band 4p16.3.
The FGFR3 (fibroblast growth factor receptor 3) gene has been eliminated in the study of Stiller [6]. Petschler also demonstrated the absence FGFR3 gene’s mutation in the case of 2 families of cherubism studied over more than three generations [7].
Autosomal dominant inheritance is the prevailing pattern in most reported cases [5]. In our study, both the patient and her son exhibited the same condition, indicating disease inheritance.
However, sporadic cases documented in the literature suggest the possibility of an alternative candidate gene or autosomal recessive transmission. The latest research has shown that the disease is mainly caused by mutations in the SH3domain-binding protein 2 (SH3BP2, MIM#602104) in the SH3BP2 gene [8]. Seven mutations in exon 9 of the gene coding for the SH3BP2 binding protein have been identified by Ueki and al. [9]. The abnormal protein SH3BP2 could be incriminated in the course of the activity of osteoblasts and osteoclasts cells [10].
Cherubism not only affects the bones but also presents dental manifestations, such as agenesis [11]. The MSX1 (Hox7) gene could potentially be implicated in this clinical manifestation. Mutations in this gene have been associated with agenesis of the second premolars and third molars [11]. Although genetic analysis was not conducted in our clinical case due to patient non-approval, the presence of second premolar agenesis in the panoramic X-ray of the patient’s son suggests a potential involvement of the MSX1 gene in the development of this pathology.
The prevalence of cherubism remains poorly specified [10], and it does not exhibit any racial or ethnic preference [12]. It is fully penetrant in males and has a penetrance of 50-70% in females [12]. Due to its rarity, conducting comprehensive analyses is challenging. Published studies have reported fewer than 300 cases, predominantly affecting children and primarily males [12]. Notably, none of the cases reported in the literature have described cherubism as a newly discovered condition in older patients. Therefore, this report contributes value to recent research efforts.
Radiographically, cherubism is characterized by distinct features, including bilaterally symmetrical, well-defined, multilocular radiolucency in the mandible that extends from the molar region towards the midline [4]. Involvement of the maxilla is less common and may regress earlier than mandibular lesions. In severe cases, infiltration of the orbital bone can also occur. Literature describes young patients with bilateral, large multiloculated soap bubble-like cavities at the angle and ramus of the mandible [4]. It is worth noting that some affected areas may remain radiolucent even after the patient has fully recovered from the disease [13].
In this particular case, the mandibular radiolucency appeared to be of medium and small sizes, potentially indicating the absorption of lesions over the years, considering that the patient is an adult.
Histologically, the presence of various cell lines, including fibroblasts, was observed. Multinuclear giant cells exhibiting osteoclastic activity were identified, surrounded by soft fiber connective tissue. The bone tissue showed replacement by fibrous connective tissue, while the blood vessels exhibited an increase in endothelial cells [14].
In this case, the identification of a fibrous stroma with numerous multinucleated giant cells is a characteristic feature typically associated with cherubism [15].
Cherubism does not typically cause significant abnormalities in peripheral blood results, such as serum calcium, phosphorus, and alkaline phosphatase levels, which was consistent with the normal findings in our clinical case [14].
Based on the clinical presentation, histological examination, molecular analysis, radiographic findings, and considering the family history in our case, a diagnosis of cherubism can be established.
The differential diagnosis should include conditions such as hyperparathyroidism, florid osseous dysplasia of the jaws, giant cell tumor, central and peripheral giant-cell granuloma, Garre’s osteomyelitis, histiocytosis X, ameloblastoma, myxoma, and jaw cysts. Clinical features, radiological imaging, and histologic findings are crucial in distinguishing between these different entities [16].
According to the classification by Ramon and Engelberg, cherubism can be categorized into four grades based on the radiological extent of involvement: grade 1 indicates lesions affecting both mandibular ascending rami, grade 2 involves both maxillary tuberosities, grade 3 signifies extensive involvement of the entire maxilla and mandible except the condylar processes, and grade 4 involves the orbits, leading to orbital compression [17]. The present case would fall under grade 1, as observed in the panoramic X-ray.
Treatment approaches for cherubism remain controversial. In most cases, regression occurs after puberty, but in some instances, the disease may persist into adulthood [18,19]. In this particular case, no treatment was initiated, and the clinical and radiological features continue to persist.
The association between cherubism and other clinical features is rarely documented in the existing literature. Our present case involves an adult female patient who presented with cherubism, along with additional conditions such as short stature, epilepsy, mental retardation, rheumatoid arthritis, hirsutism, bulbous soft nose, and gingival enlargement. The association of these features was first described by Ramon in 1967 [20], and we report a case of Ramon syndrome in this adult female patient.
The recent genetic study of Ramon’s syndrome showed homozygous mutations in ELMO2 [8]. ELMO2 forms a ternary complex with RhoGand DOCK180 to induce the activation of the GTPase RAC1, regulator of cytoskeleton dynamics and activator of a wide range of effector proteins [21].
DOCK4 is one of the DOCK proteins that interact with ELMO2. It has been showed that it is incriminated in the regulation of neurite differentiation [22]. Studies in the litterature [23,24] showed the relation between DOCK4 and human disorders associated with intellectual disabilities and high prevalence of epilepsy. This may be outlined in our case, since the patient suffered from epilepsy in early age.
Gingival fibromatosis is a distinctive characteristic of Ramon syndrome [20]. It represents a heterogeneous group of disorders characterized by progressive and painless enlargement of the gingiva, resulting from an increase in submucosal connective tissue elements [25]. Gingival fibromatosis can manifest as an isolated finding or as part of a genetic syndrome, caused by mutations in the Son of sevenless-1 (SOS1, MIM#182530) gene [8], which may be is the case in our report [26].
The exact etiology of gingival enlargement is not well understood. Some studies suggest that it may occur as a side effect of certain drug therapies [27]. However, in our case, there was no history of medication known to cause gingival overgrowth. Gingival enlargement can contribute to plaque accumulation, thereby promoting periodontitis and bone resorption.
In this study, the diagnosis of gingival fibromatosis was confirmed through histopathologic examination of the excised tissues. Surgical intervention is necessary, and Coletta and Graner have recommended that treatment be performed when the patient is cooperative and demonstrates good oral hygiene practices [28], which was adhered to in our case.
Various techniques can be employed for the excision of enlarged gingiva, including external or internal bevel gingivectomy in combination with gingivoplasty and open flap procedures [29]. In our case, we opted for Camargo and Carranza’s technique [30], which involves an internal bevel gingivectomy with open-flap debridement, as the gingival enlargement was associated with deep pockets and underlying alveolar bone loss. Chaturvedi also reported a case of gingival fibromatosis with generalized aggressive periodontitis using the same protocol [31].
Maintaining good oral hygiene is crucial for achieving the desired stabilization during treatment. However, recurrence of gingival fibromatosis is influenced not only by local factors but also by genetic predisposition [26]. Therefore, predicting the long-term outcomes of gingival fibromatosis treatment, even when combined with ideal oral hygiene practices, is challenging.
In our case, no recurrence was observed after one year. Good oral hygiene was achieved through a combination of monthly examinations with debridement and oral hygiene instructions. The patient was provided with oral hygiene instructions for home maintenance and advised to follow up regularly.
4. Conclusions
In conclusion, we presented an unusual case of Ramon syndrome in an adult female patient with cherubism, gingival fibromatosis, short stature, mental retardation, rheumatoid arthritis, hirsutism, and bulbous soft nose. The patient’s son also exhibited signs of cherubism. The clinical, radiographic, and histopathological findings supported the diagnosis of cherubism.
The treatment approach involved a non-surgical phase focusing on oral hygiene motivation and periodontal debridement, followed by surgical intervention using internal bevel gingivectomy with open-flap debridement. The patient showed significant improvement in oral health and quality of life with no recurrence of the condition after one year of follow-up.
This case adds value to the existing literature by highlighting the occurrence of cherubism in older patients and the association with Ramon syndrome. Further research is needed to better understand the genetic mechanisms and long-term outcomes of this rare disorder.
Author Contributions
Conceptualization, S.A.; resources, S.A.; writing—original draft preparation, S.A., A.C., writing—review and editing, S.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding
Institutional Review Board Statement
This study was conducted in accordance with the Declaration of Helsinki. The patient was treated in accordance with his clinical needing. Therefore, no institutional ethics committee approval was required.
Informed Consent Statement
Informed consent was obtained from patients involved in this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Frangenheim. Familiar huperostosen der Kiefer beitrage zurklinishen chirurgie. 1914, 119–139.
- Jones, W.A. Familial multilocular cystic disease of the jaws. Am J Cancer 1933, 17, 946–950. [Google Scholar] [CrossRef]
- Roginsky, V.V.; Ivanov, A.L.; Ovtchinnikov, I.A.; Khonsari, R.H. Familial cherubism: The experience of the Moscow Central Institute for Stomatology and Maxillo-facial Surgery. Int J Oral Maxillofac Sur 2009, 38, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Beaman, F.D.; Bancroft, L.W.; Peterson, J.J.; Kransdorf, M.J.; Murphey, M.D.; Menke, D.M. Imaging characteristics of cherubism. AJR Am J Roentgenol 2004, 182, 1051–1054. [Google Scholar] [CrossRef] [PubMed]
- Mangion, J.; Rahman, N.; Edkins, S.; Barfoot, R.; Nguyen, T.; Sigurdsson, A. The gene for cherubism maps to chromosome 4p16.3. Am J Hum Genet 1999, 65, 151–157. [Google Scholar] [CrossRef]
- Stiller, M.; Urban, M.; Golder, W.; Tiziani, V.; Reichenberger, E.; Frege, J.; Opitz, C.; Peters, H. Craniosynostosis in cherubism. Am J Med Genet 2000, 95, 325–331. [Google Scholar] [CrossRef]
- Petschler, M.; Stiller, M.; Hoffmeister, B.; Witkowski, R.; Opitz, C.; Bill, J.S.; Peters, H. Clinical and molecular genetic observations on families with cherubism over three generations. Mund Kiefer Gesichts Chir 2003, 7, 83–87. [Google Scholar] [CrossRef]
- Mehawej, C.; Hoischen, A.; Farah, R.A.; Marey, I.; David, M.; Stora, S.; Lachlan, K.; Brunner, H.G.; Mégarbané, A. Homozygous mutation in ELMO2 may cause Ramon syndrome. Clin Genet 2018, 93, 703–706. [Google Scholar] [CrossRef]
- Ueki, Y.; Tiziani, V.; Santanna, C.; Fukai, N.; Maulik, C.; Garfinkle, J.; Ninomiya, C.; do Amaral, C.; Peters, H.; Habal, M.; Rhee-Morris, L. Mutations in the gene encoding c-Abl-binding protein SH3BP2 cause cherubism. Nat Genet 2001, 28, 125–126. [Google Scholar] [CrossRef] [PubMed]
- Brix, M.; Peters, H.; Lebeau, J. Le Chérubisme. Rev Stomatol Chir Maxillofac 2009, 110, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Papadaki, M.E.; Lietman, S.A.; Levine, M.A.; Olsen, B.R.; Kaban, L.B.; Reichenberger, E.J. Cherubism: best clinical practice. Orphanet J Rare Dis 2012, 7 (Suppl. 1), S6. [Google Scholar] [CrossRef] [PubMed]
- Peters, W.J. Cherubism: A study of twenty cases from one family. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1979, 47, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Spyridon, T.; Anastasia, I.; Konstantinos, A.; Angelopoulos, C. Cherubism: a case report of a three-generation inheritance and literature review. J Oral Maxillofac Surg 2014, 72, 405.e1–405.e9. [Google Scholar] [CrossRef]
- Peñarrocha, M.; Bonet, J.; Mínguez, J.M.; Bagán, J.V.; Vera, F.; Mínguez, I. Cherubism: A clinical, radiographic and histopathologic comparison of 7 cases. J Oral Maxillofac Surg 2006, 64, 924–930. [Google Scholar] [CrossRef]
- Meng, X.M.; Yu, S.F.; Yu, G.Y. Clinicopathologic study of 24 cases of cherubism. Int J Oral Maxillofac Surg 2005, 34, 350–356. [Google Scholar] [CrossRef]
- Timosca, G.; Galesanu, R.; Cotutiu, C.; Grigoras, M. Aggressive form of cherubism: Report of a case. J Oral Maxillofac Surg 2000, 58, 336–344. [Google Scholar] [CrossRef]
- Ramon, Y.; Engelberg, I.S. An unusually extensive case of cherubism. J Oral Maxillofac Surg 1986, 44, 325–328. [Google Scholar] [CrossRef]
- Carvalbo-Silva, E.; Carvalbo-Silva, G.C.; Vieira, T. Cherubism: Clinicoradiographic features, treatment and long-term follow up of 8 cases. J Oral Maxillofac Surg 2007, 65, 517–522. [Google Scholar] [CrossRef]
- Kozakiewicz, M.; Perczynska-Partyka, W.; Kobos, J. Cherubism--Clinical picture and treatment. Oral Dis 2001, 7, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Ramon, Y.; Berman, W.; Bubus, J.J. Gingival fibromatosis combined with cherubism. Oral Surg Oral Med Oral Pathol 1967, 24, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Katoh, H.; Negishi, M. RhoG activates Rac1 by direct interaction with the Dock180-binding protein Elmo. Nature 2003, 424, 461–464. [Google Scholar] [CrossRef]
- Xiao, Y.; Peng, Y.; Wan, J.; Tang, G.; Chen, Y.; Tang, J.; Ye, W.C.; Ip, N.Y.; Shi, L. The atypical guanine nucleotideexchange factor Dock4 regulates neurite differentiation throughmodulation of Rac1 GTPase and actin dynamics. J Biol Chem 2013, 288, 20034–20045. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Negishi, M.; Katoh, H. Rac GEF Dock4 interacts with cortactinto regulate dendritic spine formation. Mol Biol Cell 2013, 24, 1602–1613. [Google Scholar] [CrossRef] [PubMed]
- Swann, J.W.; Al-Noori, S.; Jiang, M.; Lee, C.L. Spine loss and other dendritic abnormalities in epilepsy. Hippocampus 2000, 10, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Pappachan, B.; Narayan, J.; Nayak, A. Idiopathic gingival bromatosis: A neglected case. Indian J Radiol Imaging 2002, 12, 335–338. [Google Scholar]
- Coletta, R.D.; Graner, E. Hereditary gingival bromatosis: A systematic review. J Periodontol 2006, 77, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Suhanya, J.; Aggarwal, C.; Mohideen, K.; Jayachandran, S.; Ponniah, I. Cherubism combined with epilepsy, mental retardation and gingival bromatosis (Ramon syndrome): A case report. Head Neck Pathol 2010, 4, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Bozzo, L.; Machado, M.A.; de Almeida, O.P.; Lopes, M.A.; Coletta, R.D. Hereditary gingival fibromatosis: Report of three cases. J Clin Pediatr Dent 2000, 25, 41–46. [Google Scholar] [CrossRef]
- Howe, L.C.; Palmer, R.M. Periodontal and restorative treatment in a patient with familial gingival bromatosis: A case report. Quintessence Int 1991, 22, 871–872. [Google Scholar]
- Camargo, P.M.; Carranza, F.A. Treatment of gingival enlargement. In Clinical Periodontology, 9th ed.; Newman, M.G., Takei, H.H., Carranza, F.A., Eds.; Saunders: Philadelphia, PA, USA, 2002; pp. 754–761. [Google Scholar]
- Brown, R.S.; Trejo, P.M.; Weltman, R.; Pinero, G. Treatment of a patient with hereditary gingival fibromatosis: A case report. Spec Care Dentist 1995, 15, 149153. [Google Scholar] [CrossRef]
Figure 1.
Clinical appearance of puffiness face of bilateral sweeling, prominent maxillae, bulbous soft nose and mild hirsutism on the upper lip and cheeks.
Figure 1.
Clinical appearance of puffiness face of bilateral sweeling, prominent maxillae, bulbous soft nose and mild hirsutism on the upper lip and cheeks.

Figure 2.
Generalized marginal gingival inflammation, accumulation of plaque and calculus, generalized gingival growth, hyperplastic type, firm consistency, pale-pink, shiny and smooth in appearance, painless and pedicled covering almost all the teeth.
Figure 2.
Generalized marginal gingival inflammation, accumulation of plaque and calculus, generalized gingival growth, hyperplastic type, firm consistency, pale-pink, shiny and smooth in appearance, painless and pedicled covering almost all the teeth.
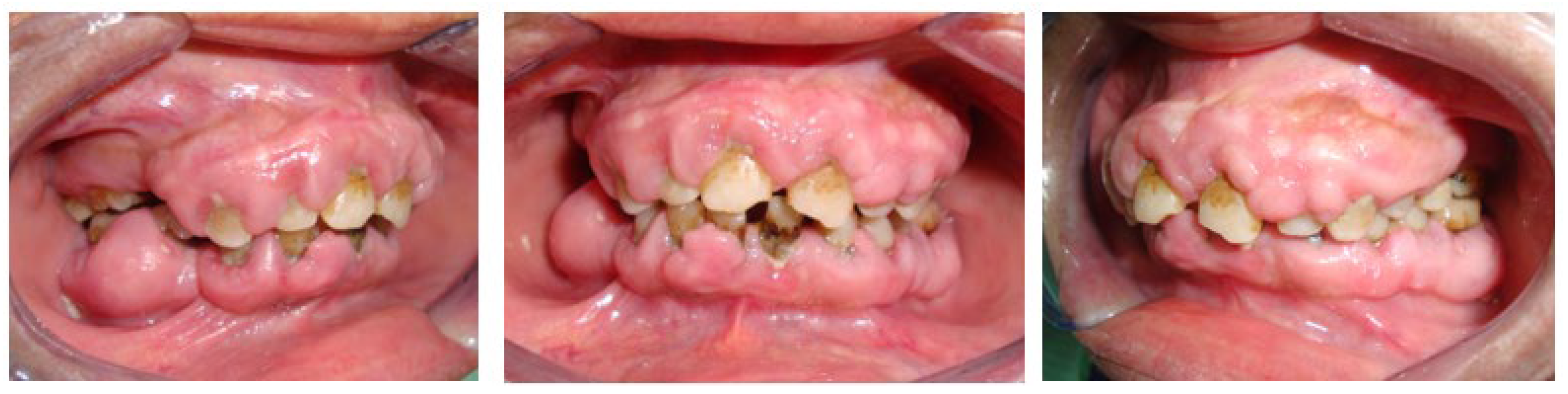
Figure 3.
Horizontal bone lysis reaching the average of 2/3 of the teeth’s roots. Normal teeth’s lamina dura. Bilateral well-defined multilocular radiolucenies in the body and ramus of the mandible.
Figure 3.
Horizontal bone lysis reaching the average of 2/3 of the teeth’s roots. Normal teeth’s lamina dura. Bilateral well-defined multilocular radiolucenies in the body and ramus of the mandible.
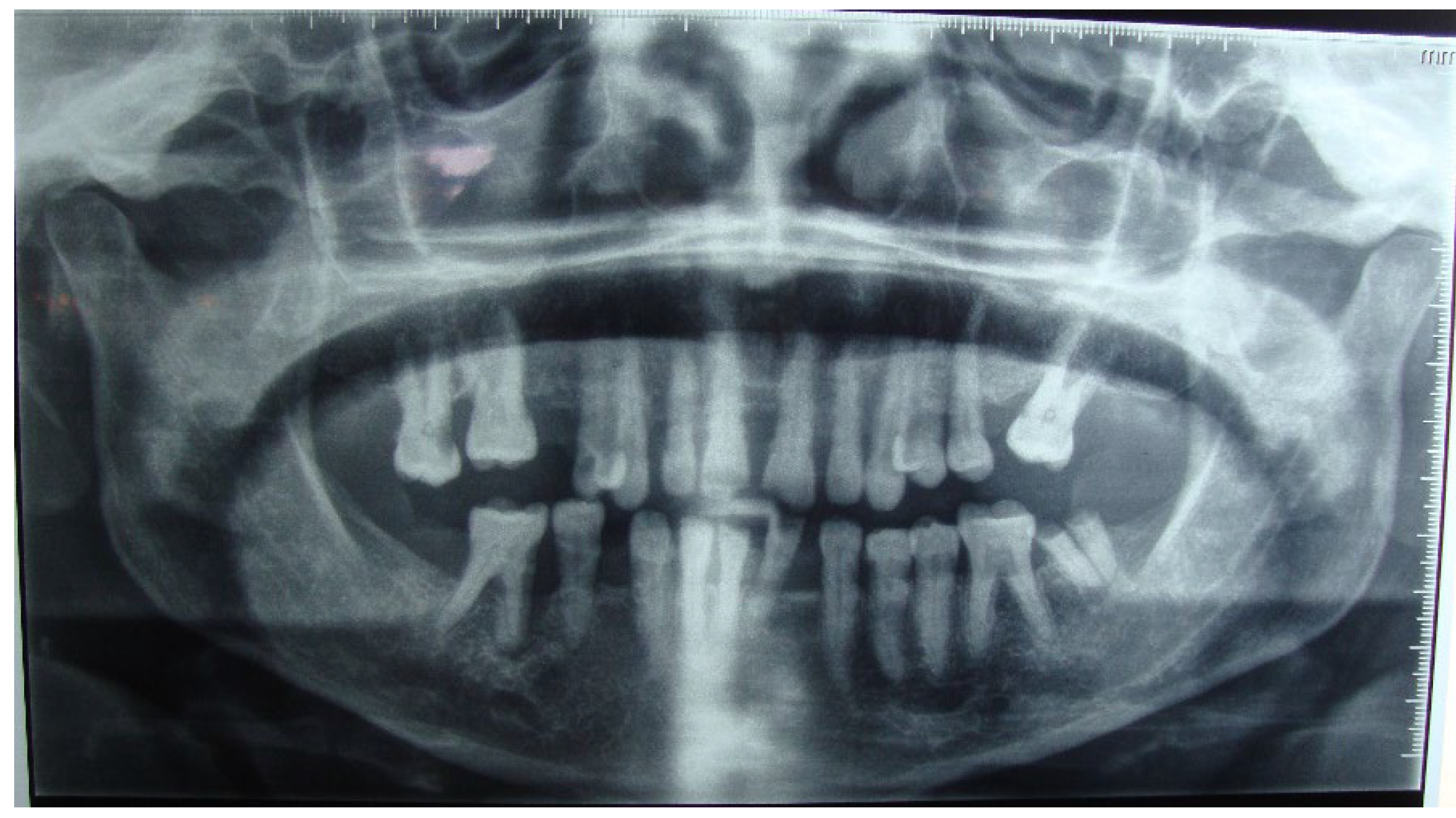
Figure 4.
Clinical appearance of patient’s son. Bilateral painless swelling of the face and bulbous soft nose.
Figure 4.
Clinical appearance of patient’s son. Bilateral painless swelling of the face and bulbous soft nose.
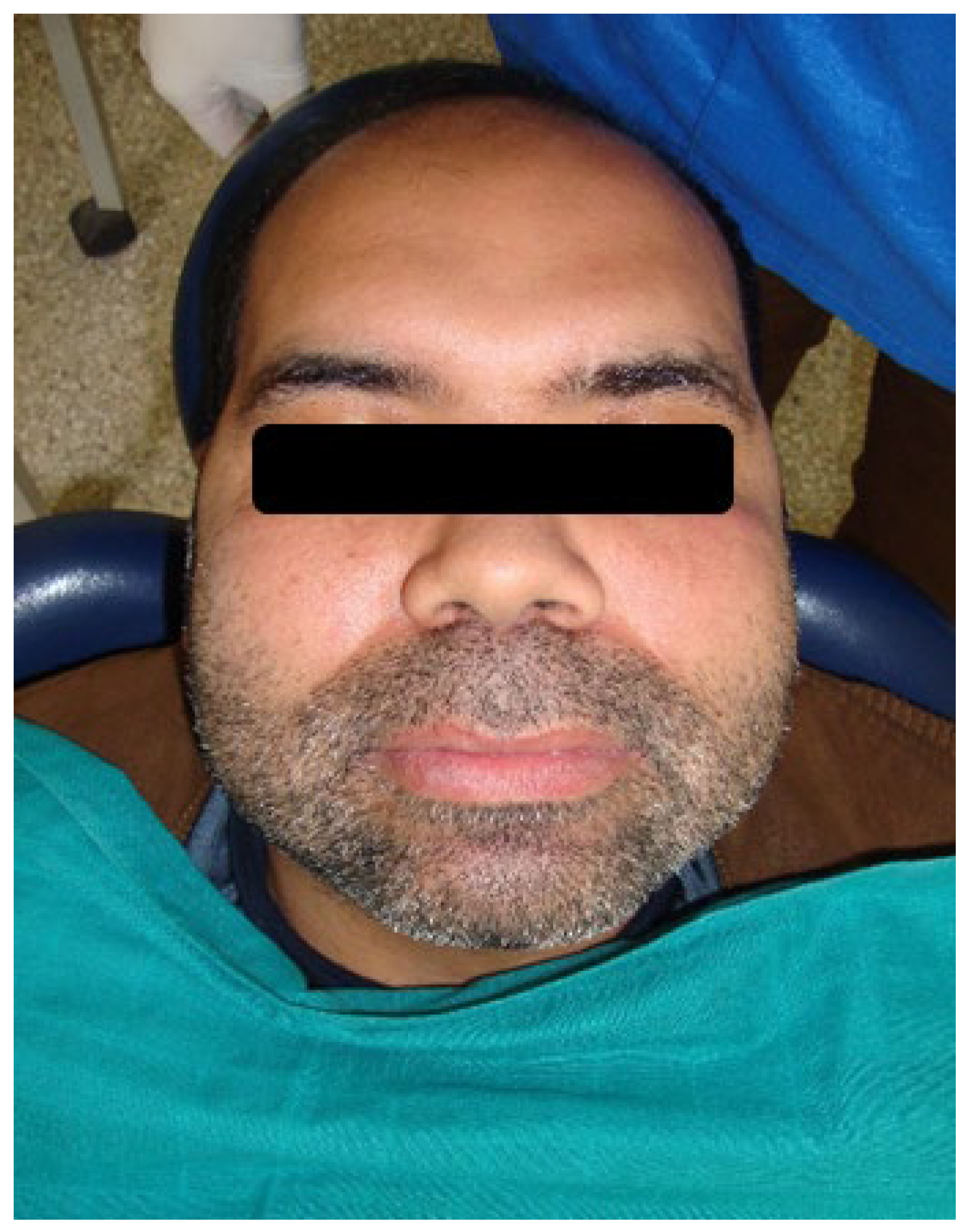
Figure 5.
Bilateral well-defined multilocular radiolucenies in the body and ramus of the mandible. Agenisis of maxillary second premolars.
Figure 5.
Bilateral well-defined multilocular radiolucenies in the body and ramus of the mandible. Agenisis of maxillary second premolars.
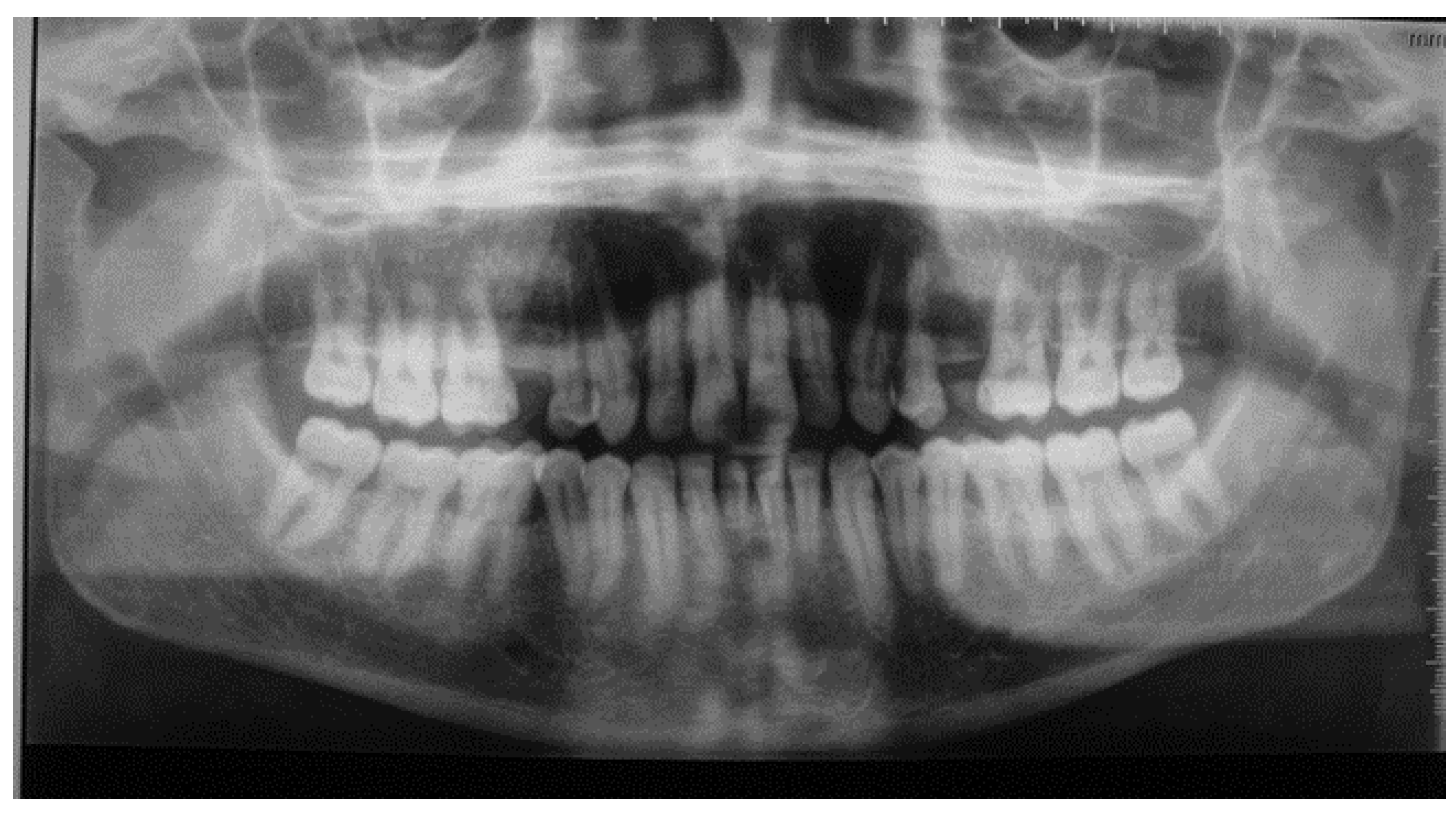
Figure 6.
One year follow-up with stability of clinical results and absence of any recurrence.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



