
Submitted:
01 June 2023
Posted:
02 June 2023
You are already at the latest version
Abstract
Treatment of neurological disease is hampered by the lack of validated specific and sensitive biomarkers, resulting in delayed diagnosis and nonspecific disease modifying therapies. For example, in ALS the lack of a sensitive and specific biomarker impedes the ability to administer a treatment prior to or at the onset of motor neuron dysfunction. Although viral or other infectious etiologies have been proposed as a contributing factor to neurodegeneration, it can be argued that these are casual and not causal associations. In the case of ALS, evidence for direct causality would require in vivo validation of specific nucleotide sequences of microbial origin which are known to be neuroinvasive with tropism for motor neurons, such as polio and non-polio. Several viral enteropathogens recapitulate the pathological events in sporadic ALS, including the ability to cleave TDP-43 resulting in accumulation of misfolded proteins in the cytoplasm. This review provides supporting evidence that motor neuron disease is causally related to specific enteroviruses and argues that prospective validation is imperative for effective prevention and treatment.
Keywords:
ALS
; TDP-43
; misfolded proteins
; enterovirus
; polio
; motor neurons
; retrotransposons
; prions
; Alzheimer’s
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterized by the loss of upper and lower motor neurons in the motor cortex, brain stem and the anterior horn of the spinal cord. The etiology ALS is currently unknown, however the incidence in the number of cases across the world is projected to increase by almost 70% in the next two decades [1], suggesting a confluence of genetic and sporadic factors. Among several sporadic factors, a viral etiology has previously been proposed based upon the similarities between the molecular phenotype of ALS and enteroviruses [2].
Several related enteropathogens including polio, Coxsackie and other enteroviruses such as EV-71 have been associated with motor neuron dysfunction, but these are normally acute bases which do not progress in a step wise progression as does ALS. Further, the findings of viral genetic material in the CSF of ALS patients as previously reported can be argued as being incidental and not causal [3]. Causality based biomarkers in ALS require the fulfillment of the revised Koch criterion where a specific nucleic acid sequence corresponding to a putative pathogen is present in most cases of affected patients, but not present in unaffected patients [4]. An additional criterion to fulfill the revised Koch postulate is whether the pathological mechanisms of a particular infection are consistent with protein misfolding of RNA binding proteins which are characteristic hallmarks of ALS. In the following paragraphs we will argue that enteroviruses exert alternative splicing of host RNA resulting in post translational modifications of proteins which recapitulate the hallmark features of neurodegeneration including the accumulation of intrinsically disordered proteins which self-aggregate and cross seed unaffected cells in a prion like manner.
TDP-43 hyperphosphorylation in ALS
Among genes linked to ALS, mutations in the gene encoding for Transactive response DNA binding Protein of 43 kDa (TDP-43) have been linked to familial and sporadic cases [5]. The involvement of TDP-43 in neurodegeneration was first reported when hyperphosphorylated TDP-43 was observed in the majority of patients with ALS and frontal lobar degeneration [6]. As an RNA binding protein, TDP-43 binds to more than 6,000 mRNA targets, representing nearly 30% of the entire transcriptome [7,8,9].
TDP-43 hyperphosphorylation leads to protein aggregation and misfolded RNA binding proteins as the hallmark of ALS and other neurodegenerative diseases, such as Niemann-Pick type C (NPC) [10,11].
As early as 1995, it was noted that TDP-43 acts as a transcriptional repressor of HIV-1 infection by inhibiting viral assembly [12]. More recently, TDP-43 has been shown to affect viral fusion and infection capacities in an epigenetic manner by altering HDAC6 levels and tubulin-deacetylase levels involved in a microtubule function [13]. Subsequent studies confirmed that hyperphosphorylated TDP-43 impairs small interfering RNA silencing, which is the major post-transcriptional mechanism of retrotransposable elements or genomic parasites as a novel mechanism of neurodegeneration in ALS [14]. TDP-43 has been shown to contain a prion-like domain that can form a self-replicating amyloid conformation [15] TDP-43 possesses intrinsically disordered domains whereby the rate α-helix-to-β-sheet transition is dependent upon post translational modifications of phosphorylation-dephosphorylation pathways [16]. Furthermore, prion-like domains may directly originate during viral genomic RNA processing involving nucleocapsid (NC) proteins responsible for virus capsid assembly and structure [17]. To date, approximately 70 human RNA-binding proteins have been reported to possess a prion-like domain and render intracellular proteins more prone to misfolding and aggregation [18].
For example, prion-like RNA binding domains have also been recognized in fused sarcoma (FUS), the most aggressive, early-onset forms of ALS [19]. Mutations in FUS are also associated with misfolded RNA bindings proteins in an identical matter as TDP-43 [20]. In a similar manner, FUS mutations are more sensitive to retroviruses and exacerbate cytoplasmic mislocalization and protein misfolding [21]
The misfolding of proteins in ALS gives rise to hydrophobic PrPSc aggregates which are insoluble filaments comprised of amyloid and tau like plaque. Prior studies have demonstrated that β-amyloid aggregates exhibit many properties indistinguishable from those of prions, including the ability of amyloid fibrils to self-propagate [22].
These observations suggest that common mutations in ALS disinhibit host mediated viral repression by targeting prion like domains, resulting in protein aggregation and motor neuron degeneration in a prion like manner.
Part I: Enteroviruses in the etiology of motor neuron disease
Enteroviruses are now regarded as the most important neurotropic enterovirus in the Asia-Pacific region in the post-polio eradication era [22,23].
Collectively, Enteroviruses are single-stranded positive-sense neurotrophic RNA viruses including polio, coxsackievirus A and B types, and numerous other enteroviruses, including EV-68 and EV-71 [24]. EVs are non-enveloped viruses comprised of a viral capsid that contains a single open reading frame (ORF) which encodes four structural proteins, VP1–4 and seven non-structural proteins (2A–C and 3A–D) including RNA-dependent RNA polymerase (3DPol) [25].
Enteroviruses are primarily transmitted through the fecal–oral route where infections can be limited to febrile illnesses but in more severe cases can result in meningitis, brainstem encephalitis, and acute flaccid myelitis through retrograde transmission from the gut to the brain [26]. In the case of Polio, the enterovirus specifically targets motor neurons, resulting in cases that are clinically indistinguishable from ALS. Coxsackie and enterovirus 71 also target motor neurons and have been associated with outbreaks of acute flaccid paralysis in several regions of the globe, including in the US [27].
The establishment of a causal relationship between enteroviruses and motor neuron disease has resulted from epidemiological studies where the isolation of viral particles was discovered in spinal cord samples of affected patients with motor neuron disease [28]. In non-polio enteroviruses, amplification of a 414 base RNA target sequence in the conserved enterovirus 5' untranslated region was found in eight of 11 cases of sporadic motor neuron disease, suggesting that the class of enteroviruses display neurotrophic features similar to polio [29]. Subsequently, in later studies, qRT-PCR analysis of cerebrospinal fluid of EV-71, a newly emergent enterovirus detection was detected in 14.5% of 242 ALS patients and 7.6% of 354 controls [3].
It is believed that EV-71 emerged in the mid-20th century, as indicated by an analysis of viral capsid sequences similar to those found in other enterovirus genotypes belonging to common ancestral genogroups. The emergence of EV-71 was likely due to a recombination event between a portion of the genome of CV-A16 and the 5′-NTR of poliovirus. [30,31].
These shared nucleotide sequences between phenotypically different viral pathogens are likely based upon evolutionarily events between host and pathogen. Darwinian like pressures most likely have driven enteroviruses to “best fit strategies” in the context of population bottlenecks imposed by competitive forces in host- infection dynamics and mitochondrial energy barriers. As enteroviruses have high mutation rates, “best fit” sub-strains of enteroviruses categorically lead to the formation of quorums, where structural genes within the viral capsid maintain a degree of molecular fidelity across different viral species, which are noted as quasi-species [32].
In neurotrophic viruses including all enteroviruses, redundant RNA sequences within the viral capsid act as an “ancestral” hub for successful retroviral replication by encoding Activity-regulated cytoskeleton-associated protein (Arc-Gag complexes). The Arc-Gag complex facilitates the assemble of viral RNA genomes into a spherical virion [33] Interestingly, evolutionary analysis indicates that Arc protein complexes are derived from retrotransposons, which are also ancestral to retroviruses with highly conserved homology to human protein isoforms, repurposed during evolution to mediate intra and intercellular communication at synaptic junctions via specific RNA sequences [34].
Part IV: Unresolved questions
One of the most perplexing questions regarding the potential pathogenesis of ALS as a chronic infectious disease is why only certain individuals develop motor neuron disease and not others as these pathogens are ubiquitous in the population? According to the Koch hypothesis, only individuals who have evidence of a microbial infection should manifest a particular disease whereas those who do not have symptoms will not as otherwise the infection is not causal. Enteroviruses are common but only a small number of infected individuals develop signs and symptoms. Thus, viral infections are necessary, but not sufficient to trigger ALS. The likely explanation regarding only a subset of patients with enterovirus relates to the inherent capacity to develop ALS secondary to immunosenescent states imposed upon by viral mediated editing of adenosine to inosine, resulting in Adenosine deaminase deficiency and aberrant activation of antiviral innate immune For example, in ALS neuronal astrocytic progenitor cells with C9orf72 or sporadic mutation have demonstrated defects in the conversion of adenosine to inosine secondary to adenosine deaminase deficiency [67].
In conclusion, viral induced neurodegeneration involves a pathological cascade beginning with the Arc-Gag protein complexes released by retrotransposons and retroviruses, which encodes specific viral helicases shared by HIV, and enteroviruses such as polio and others, such as EV-71. Release of these helicases results in abnormal RNA editing of RNA based proteins such as TDP-43 and adenosine deaminases which disrupt host defense mechanisms, resulting in viral derepression to enhance viral replication. The fact that viral enteropathogens recapitulate motor neuron disease through similar mechanisms that involves PolyP resulting in the formation of insoluble protein aggregates in an amyloid like manner, supports our hypothesis that ALS is virally-mediated.
Part V: Concluding remarks: A call to Arms:
The prospective validation of the viral hypothesis of ALS can be enabled by viral specific nucleic acids may be quantified using qPCR methods or by newer digital PCR methods. These emerging technologies can detect spliced versus un-spliced RNA in affected individuals at risk or displaying evidence of neurodegeneration. However, most current viral sequencing technologies provide no coverage of noncoding regions, for example in the untranslated region of polioviruses [68]. These new sequencing methods will ultimately fulfill the Koch hypothesis by proving causality, and in the event, offering the promise of targeted therapies for neurodegeneration. Prospective studies of the CSF of ALS patients involving elevations of inorganic polyphosphates should be considered to replicate previous findings by Arredondo, et al [62]. Various FDA-approved phosphatases, in this regard, act to reduce PolyP by promoting CD28-response elements by enhancing calcineurin, which in turn can target TDP43 as a disease modifying therapy. As ALS is an invariably fatal disease without a cure, the primary goal of this paper is to implore clinicians and researchers to look beyond our current theories of ALS pathogenesis and consider a broader explanation regarding the cause of ALS being infectious mediated.
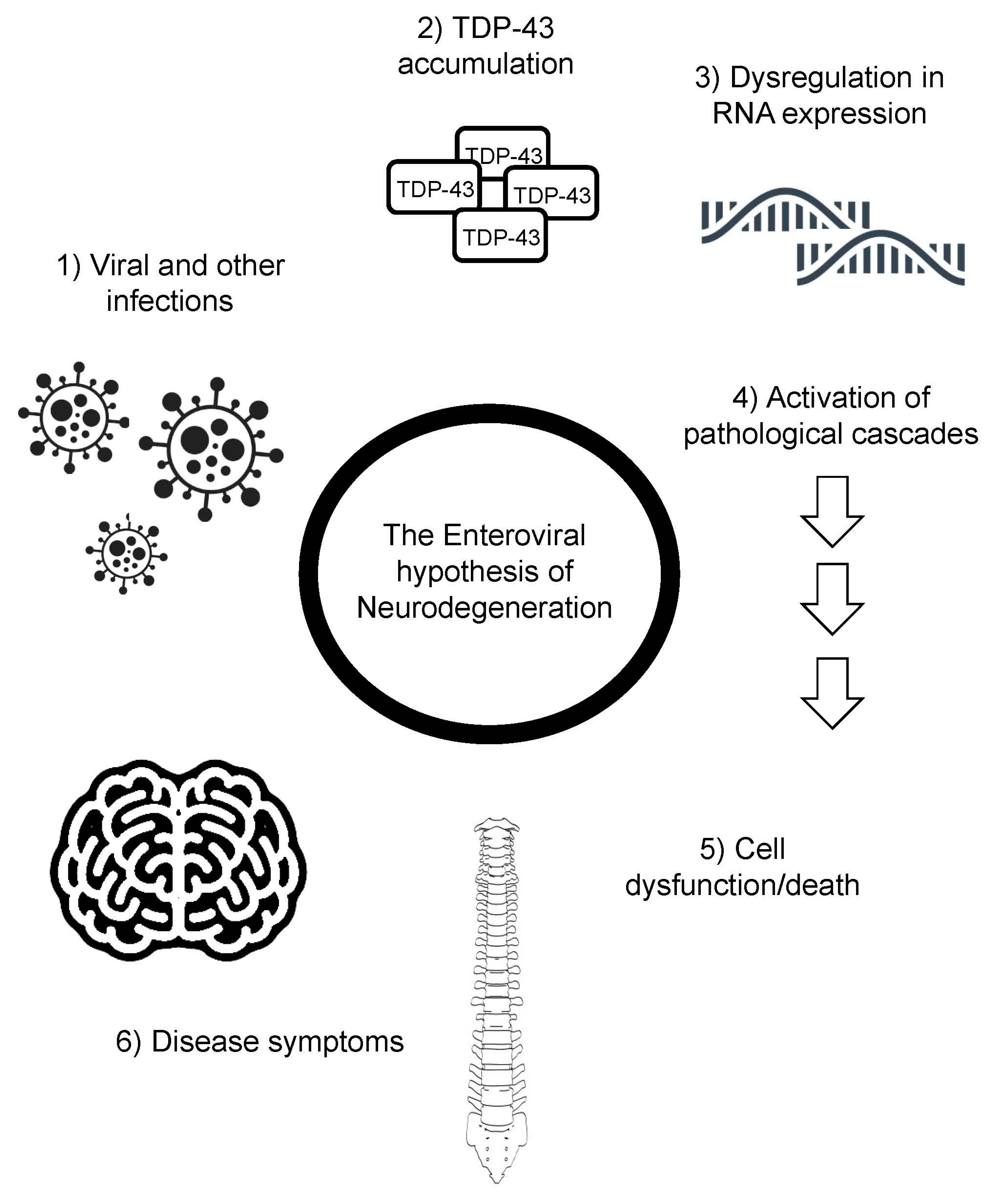
Figure 3.
The enteroviral hypothesis of neurodegeneration. Viral infections lead to TDP-43 phosphorylation and aggregation, altering RNA expression, activating pathological cascades, leading to cell dysfunction/death, and the characteristic symptoms of ALS.
Figure 3.
The enteroviral hypothesis of neurodegeneration. Viral infections lead to TDP-43 phosphorylation and aggregation, altering RNA expression, activating pathological cascades, leading to cell dysfunction/death, and the characteristic symptoms of ALS.
Author Contributions
J.L conceptualized and wrote the manuscript. A.D.K. wrote the article and made the figures. Both authors edited the text.
Funding
A.D.K. is funded by Fondecyt grant 1230317.
Acknowledgment
The authors thanks Martha Beach Oleksandra Shchebet, for helpful discussions and all the wonderful researchers and clinicians at the Center for Neuroimmunology at Nova University in FL for their commitment to cure ALS.
Conflicts of Interest
The authors declare that there are no competing interests.
References
- Arthur, K.C.; Calvo, A.; Price, T.R.; Geiger, J.T.; Chiò, A.; Traynor, B.J. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat. Commun. 2016, 7, 12408. [Google Scholar] [CrossRef] [PubMed]
- Xue YC, Feuer R, Cashman N, Luo H. Enteroviral Infection: The Forgotten Link to Amyotrophic Lateral Sclerosis? Front Mol Neurosci [Internet]. Frontiers Media SA; 2018 [cited 2023 Apr 25];11. Available from: /pmc/articles/PMC5857577/.
- Vandenberghe, N.; Leveque, N.; Corcia, P.; Brunaud-Danel, V.; Salort-Campana, E.; Besson, G.; Tranchant, C.; Clavelou, P.; Beaulieux, F.; Ecochard, R.; et al. Cerebrospinal fluid detection of enterovirus genome in ALS: A study of 242 patients and 354 controls. Amyotroph. Lateral Scler. 2010, 2010 11, 277–282. [Google Scholar] [CrossRef]
- Fredericks DN, Relman DA. Sequence-based identification of microbial pathogens: a reconsideration of Koch’s postulates. Clin Microbiol Rev [Internet]. Clin Microbiol Rev; 1996 [cited 2023 Apr 17];9:18–33. Available from: https://pubmed.ncbi.nlm.nih.gov/8665474/.
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Sanelli, T.; Dib, S.; Sheps, D.; Findlater, J.; Bilbao, J.; Keith, J.; Zinman, L.; Rogaeva, E.; Robertson, J. RNA targets of TDP-43 identified by UV-CLIP are deregulated in ALS. Mol. Cell. Neurosci. 2011, 47, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci [Internet]. Nat Neurosci; 2011 [cited 2023 Apr 17];14:452–8. Available from: https://pubmed.ncbi.nlm.nih.gov/21358640/.
- Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci [Internet]. Nat Neurosci; 2011 [cited 2023 Apr 17];14:459–68. Available from: https://pubmed.ncbi.nlm.nih.gov/21358643/.
- Liu, E.A.; Mori, E.; Hamasaki, F.; Lieberman, A.P. TDP-43 proteinopathy occurs independently of autophagic substrate accumulation and underlies nuclear defects in Niemann-Pick C disease. Neuropathol. Appl. Neurobiol. 2021, 47, 1019–1032. [Google Scholar] [CrossRef]
- Dardis, A.; Zampieri, S.; Canterini, S.; Newell, K.L.; Stuani, C.; Murrell, J.R.; Ghetti, B.; Fiorenza, M.T.; Bembi, B.; Buratti, E. Altered localization and functionality of TAR DNA Binding Protein 43 (TDP-43) in niemann- pick disease type C. Acta Neuropathol. Commun. 2016, 4, 1–13. [Google Scholar] [CrossRef]
- Ou, S.H.; Wu, F.; Harrich, D.; García-Martínez, L.F.; Gaynor, R.B. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J. Virol. 1995, 69, 3584–3596. [Google Scholar] [CrossRef]
- Cabrera-Rodríguez, R.; Pérez-Yanes, S.; Montelongo, R.; Lorenzo-Salazar, J.M.; Estévez-Herrera, J.; García-Luis, J.; Íñigo-Campos, A.; Rubio-Rodríguez, L.A.; Muñoz-Barrera, A.; Trujillo-González, R.; et al. Transactive Response DNA-Binding Protein (TARDBP/TDP-43) Regulates Cell Permissivity to HIV-1 Infection by Acting on HDAC6. Int. J. Mol. Sci. 2022, 23, 6180. [Google Scholar] [CrossRef]
- Krug, L.; Chatterjee, N.; Borges-Monroy, R.; Hearn, S.; Liao, W.-W.; Morrill, K.; Prazak, L.; Rozhkov, N.; Theodorou, D.; Hammell, M.; et al. Retrotransposon activation contributes to neurodegeneration in a Drosophila TDP-43 model of ALS. PLOS Genet. 2017, 13, e1006635. [Google Scholar] [CrossRef]
- Smethurst, P.; Sidle, K.C.L.; Hardy, J. Review: Prion-like mechanisms of transactive response DNA binding protein of 43 kDa (TDP-43) in amyotrophic lateral sclerosis (ALS). Neuropathol. Appl. Neurobiol. 2015, 41, 578–597. [Google Scholar] [CrossRef] [PubMed]
- Chiang WC, Lee MH, Chen TC, Huang JR. Interactions between the Intrinsically Disordered Regions of hnRNP-A2 and TDP-43 Accelerate TDP-43′s Conformational Transition. Int J Mol Sci [Internet]. Multidisciplinary Digital Publishing Institute (MDPI); 2020 [cited 2023 Apr 22];21:1–11. Available from: /pmc/articles/PMC7460674/.
- Monette, A.; Mouland, A.J. Zinc and Copper Ions Differentially Regulate Prion-Like Phase Separation Dynamics of Pan-Virus Nucleocapsid Biomolecular Condensates. Viruses 2020, 12, 1179. [Google Scholar] [CrossRef]
- Harrison, A.F.; Shorter, J. RNA-binding proteins with prion-like domains in health and disease. Biochem. J. 2017, 474, 1417–1438. [Google Scholar] [CrossRef]
- Gitler, A.D.; Shorter, J. RNA-binding proteins with prion-like domains in ALS and FTLD-U. Prion 2011, 5, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Chen C, Ding X, Akram N, Xue S, Luo SZ. Fused in Sarcoma: Properties, Self-Assembly and Correlation with Neurodegenerative Diseases. Molecules 2019, Vol 24, Page 1622 [Internet]. Multidisciplinary Digital Publishing Institute; 2019 [cited 2023 Apr 17];24:1622. Available from: https://www.mdpi.com/1420-3049/24/8/1622/htm.
- Bellmann J, Monette A, Tripathy V, Sójka A, Abo-Rady M, Janosh A, et al. Viral Infections Exacerbate FUS-ALS Phenotypes in iPSC-Derived Spinal Neurons in a Virus Species-Specific Manner. Front Cell Neurosci. Frontiers Media S.A.; 2019;13:480.
- Watts JC, Prusiner SB. β-Amyloid Prions and the Pathobiology of Alzheimer’s Disease. Cold Spring Harb Perspect Med [Internet]. Cold Spring Harbor Laboratory Press; 2018 [cited 2023 Apr 25];8. Available from: /pmc/articles/PMC5554751/.
- Bible, J.M.; Pantelidis, P.; Chan, P.K.S.; Tong, C.Y.W. Genetic evolution of enterovirus 71: epidemiological and pathological implications. Rev. Med Virol. 2007, 17, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Simmonds P, Gorbalenya AE, Harvala H, Hovi T, Knowles NJ, Lindberg AM, et al. Recommendations for the nomenclature of enteroviruses and rhinoviruses. Arch Virol [Internet]. Arch Virol; 2020 [cited 2023 Apr 17];165:793–7. Available from: https://pubmed.ncbi.nlm.nih.gov/31980941/.
- Kitamura, N.; Semler, B.L.; Rothberg, P.G.; Larsen, G.R.; Adler, C.J.; Dorner, A.J.; Emini, E.A.; Hanecak, R.; Lee, J.J.; van der Werf, S.; et al. Primary structure, gene organization and polypeptide expression of poliovirus RNA. Nature 1981, 291, 547–553. [Google Scholar] [CrossRef]
- Wells AI, Coyne CB. Enteroviruses: A Gut-Wrenching Game of Entry, Detection, and Evasion. Viruses [Internet]. Multidisciplinary Digital Publishing Institute (MDPI); 2019 [cited 2023 Apr 19];11. Available from: /pmc/articles/PMC6563291/.
- Sejvar JJ, Lopez AS, Cortese MM, Leshem E, Pastula DM, Miller L, et al. Acute Flaccid Myelitis in the United States, August-December 2014: Results of Nationwide Surveillance. Clin Infect Dis [Internet]. Clin Infect Dis; 2016 [cited 2023 Apr 19];63:737–45. Available from: https://pubmed.ncbi.nlm.nih.gov/27318332/. 20 December.
- Messacar, K.; Asturias, E.J.; Hixon, A.M.; Van Leer-Buter, C.; Niesters, H.G.M.; Tyler, K.L.; Abzug, M.J.; Dominguez, S.R. Enterovirus D68 and acute flaccid myelitis—Evaluating the evidence for causality. Lancet Infect. Dis. 2018, 18, e239–e247. [Google Scholar] [CrossRef] [PubMed]
- Woodall, C.J.; Riding, M.H.; I Graham, D.; Clements, G.B. Sequences specific for enterovirus detected in spinal cord from patients with motor neurone disease. BMJ 1994, 308, 1541–1543. [Google Scholar] [CrossRef]
- Tee, K.K.; Lam, T.T.-Y.; Chan, Y.F.; Bible, J.M.; Kamarulzaman, A.; Tong, C.Y.W.; Takebe, Y.; Pybus, O.G. Evolutionary Genetics of Human Enterovirus 71: Origin, Population Dynamics, Natural Selection, and Seasonal Periodicity of the VP1 Gene. J. Virol. 2010, 84, 3339–3350. [Google Scholar] [CrossRef]
- McMinn PC. An overview of the evolution of enterovirus 71 and its clinical and public health significance. FEMS Microbiol Rev. No longer published by Elsevier; 2002;26:91–107.
- Domingo E. Molecular basis of genetic variation of viruses: error-prone replication. Virus as Populations [Internet]. Elsevier; 2020 [cited 2023 Apr 22];35. Available from: /pmc/articles/PMC7153327/.
- Bharat, T.A.; Noda, T.; Riches, J.D.; Kraehling, V.; Kolesnikova, L.; Becker, S.; Kawaoka, Y.; Briggs, J.A. Structural dissection of Ebola virus and its assembly determinants using cryo-electron tomography. Proc. Natl. Acad. Sci. USA 2012, 109, 4275–4280. [Google Scholar] [CrossRef]
- Pastuzyn, E.D.; Day, C.E.; Kearns, R.B.; Kyrke-Smith, M.; Taibi, A.V.; McCormick, J.; Yoder, N.; Belnap, D.M.; Erlendsson, S.; Morado, D.R.; et al. The Neuronal Gene Arc Encodes a Repurposed Retrotransposon Gag Protein that Mediates Intercellular RNA Transfer. Cell 2018, 172, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Hantak, M.P.; Einstein, J.; Kearns, R.B.; Shepherd, J.D. Intercellular Communication in the Nervous System Goes Viral. Trends Neurosci. 2021, 44, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Wang, P.; Qin, C.-F.; Hu, Y.; Zhou, X.; Wang, G.-C.; Yang, J.; Sun, X.; Wu, W.; Qiu, Y.; et al. Human Enterovirus Nonstructural Protein 2CATPase Functions as Both an RNA Helicase and ATP-Independent RNA Chaperone. PLoS Pathog. 2015, 11, e1005067. [Google Scholar] [CrossRef]
- Leung HW, Foo G, VanDongen A. Arc Regulates Transcription of Genes for Plasticity, Excitability and Alzheimer’s Disease. Biomedicines [Internet]. Multidisciplinary Digital Publishing Institute (MDPI); 2022 [cited 2023 Apr 17];10. Available from: /pmc/articles/PMC9405677/.
- Wu, J.; Petralia, R.S.; Kurushima, H.; Patel, H.; Jung, M.-Y.; Volk, L.; Chowdhury, S.; Shepherd, J.D.; Dehoff, M.; Li, Y.; et al. Arc/Arg3.1 Regulates an Endosomal Pathway Essential for Activity-Dependent β-Amyloid Generation. Cell 2011, 147, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chuang, Y.-A.; Na, Y.; Ye, Z.; Yang, L.; Lin, R.; Zhou, J.; Wu, J.; Qiu, J.; Savonenko, A.; et al. Arc Oligomerization Is Regulated by CaMKII Phosphorylation of the GAG Domain: An Essential Mechanism for Plasticity and Memory Formation. Mol. Cell 2019, 75, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Haolong, C.; Du, N.; Hongchao, T.; Yang, Y.; Wei, Z.; Hua, Z.; Wenliang, Z.; Lei, S.; Po, T. Enterovirus 71 VP1 Activates Calmodulin-Dependent Protein Kinase II and Results in the Rearrangement of Vimentin in Human Astrocyte Cells. PLoS ONE 2013, 8, e73900. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lovell, S.; Tiew, K.-C.; Mandadapu, S.R.; Alliston, K.R.; Battaile, K.P.; Groutas, W.C.; Chang, K.-O. Broad-Spectrum Antivirals against 3C or 3C-Like Proteases of Picornaviruses, Noroviruses, and Coronaviruses. J. Virol. 2012, 86, 11754–11762. [Google Scholar] [CrossRef]
- Rahic Z, Buratti E, Cappelli S. Reviewing the Potential Links between Viral Infections and TDP-43 Proteinopathies. Int J Mol Sci [Internet]. Multidisciplinary Digital Publishing Institute (MDPI); 2023 [cited 2023 Apr 17];24. Available from: /pmc/articles/PMC9867397/.
- Fung, G.; Shi, J.; Deng, H.; Hou, J.; Wang, C.; Hong, A.; Zhang, J.; Jia, W.; Luo, H. Cytoplasmic translocation, aggregation, and cleavage of TDP-43 by enteroviral proteases modulate viral pathogenesis. Cell Death Differ. 2015, 22, 2087–2097. [Google Scholar] [CrossRef]
- Pathinayake PS, Hsu ACY, Wark PAB. Innate Immunity and Immune Evasion by Enterovirus 71. Viruses [Internet]. Multidisciplinary Digital Publishing Institute (MDPI); 2015 [cited 2023 Apr 22];7:6613. Available from: /pmc/articles/PMC4690884/.
- Yang J, Li Y, Wang S, Li H, Zhang L, Zhang H, et al. The SARS-CoV-2 main protease induces neurotoxic TDP-43 cleavage and aggregates. Signal Transduct Target Ther [Internet]. Nature Publishing Group; 2023 [cited 2023 Apr 22];8. Available from: /pmc/articles/PMC9998009/.
- Gumna, J.; Purzycka, K.J.; Ahn, H.W.; Garfinkel, D.J.; Pachulska-Wieczorek, K. Retroviral-like determinants and functions required for dimerization of Ty1 retrotransposon RNA. RNA Biol. 2019, 16, 1749–1763. [Google Scholar] [CrossRef]
- Waheed, A.A.; Freed, E.O.; Li, G.; Theys, K.; Verheyen, J.; Pineda-Peña, A.-C.; Khouri, R.; Piampongsant, S.; Eusébio, M.; Ramon, J.; et al. HIV Type 1 Gag as a Target for Antiviral Therapy. AIDS Res. Hum. Retroviruses 2012, 28, 54–75. [Google Scholar] [CrossRef]
- Campbell, S.; Rein, A. In Vitro Assembly Properties of Human Immunodeficiency Virus Type 1 Gag Protein Lacking the p6 Domain. J. Virol. 1999, 73, 2270–2279. [Google Scholar] [CrossRef] [PubMed]
- Ao, Z.; Jayappa, K.D.; Wang, B.; Zheng, Y.; Wang, X.; Peng, J.; Yao, X. Contribution of Host Nucleoporin 62 in HIV-1 Integrase Chromatin Association and Viral DNA Integration. J. Biol. Chem. 2012, 287, 10544–10555. [Google Scholar] [CrossRef] [PubMed]
- Lingemann, M.; McCarty, T.; Liu, X.; Buchholz, U.J.; Surman, S.; Martin, S.E.; Collins, P.L.; Munir, S. The alpha-1 subunit of the Na+,K+-ATPase (ATP1A1) is required for macropinocytic entry of respiratory syncytial virus (RSV) in human respiratory epithelial cells. PLOS Pathog. 2019, 15, e1007963. [Google Scholar] [CrossRef] [PubMed]
- Krieger, C.; Wang, S.J.H.; Yoo, S.H.; Harden, N. Adducin at the Neuromuscular Junction in Amyotrophic Lateral Sclerosis: Hanging on for Dear Life. Front. Cell. Neurosci. 2016, 10, 11. [Google Scholar] [CrossRef]
- Gallardo G, Barowski J, Ravits J, Siddique T, Lingrel JB, Robertson J, et al. An α2-Na/K ATPase/α-adducin complex in astrocytes triggers non–cell autonomous neurodegeneration. Nature Neuroscience 2014 17:12 [Internet]. Nature Publishing Group; 2014 [cited 2023 Apr 17];17:1710–9. Available from: https://www.nature.com/articles/nn.3853.
- Mann, C.N.; Devi, S.S.; Kersting, C.T.; Bleem, A.V.; Karch, C.M.; Holtzman, D.M.; Gallardo, G. Astrocytic α2-Na + /K + ATPase inhibition suppresses astrocyte reactivity and reduces neurodegeneration in a tauopathy mouse model. Sci. Transl. Med. 2022, 14, eabm4107–eabm4107. [Google Scholar] [CrossRef]
- Graham, J.F.; Kurian, D.; Agarwal, S.; Toovey, L.; Hunt, L.; Kirby, L.; Pinheiro, T.J.T.; Banner, S.J.; Gill, A.C. Na+/K+-ATPase Is Present in Scrapie-Associated Fibrils, Modulates PrP Misfolding In Vitro and Links PrP Function and Dysfunction. PLOS ONE 2011, 6, e26813. [Google Scholar] [CrossRef]
- Gallardo G, Barowski J, Ravits J, Siddique T, Lingrel JB, Robertson J, et al. An α2-Na/K ATPase/α-adducin complex in astrocytes triggers non–cell autonomous neurodegeneration. Nature Neuroscience 2014 17:12 [Internet]. Nature Publishing Group; 2014 [cited 2023 Apr 25];17:1710–9. Available from: https://www.nature.com/articles/nn.3853.
- Li, L.; Rao, N.N.; Kornberg, A. Inorganic polyphosphate essential for lytic growth of phages P1 and fd. Proc. Natl. Acad. Sci. 2007, 104, 1794–1799. [Google Scholar] [CrossRef]
- Lempart J, Jakob U. Role of Polyphosphate in Amyloidogenic Processes. Cold Spring Harb Perspect Biol [Internet]. Cold Spring Harbor Laboratory Press; 2019 [cited 2023 Apr 19];11. Available from: /pmc/articles/PMC6496346/.
- Johnson, R.S.; Strausbauch, M.; McCloud, C. An NTP-driven mechanism for the nucleotide addition cycle of Escherichia coli RNA polymerase during transcription. PLOS ONE 2022, 17, e0273746. [Google Scholar] [CrossRef]
- Maiolino, M.; O'Neill, N.; Lariccia, V.; Amoroso, S.; Sylantyev, S.; Angelova, P.R.; Abramov, A.Y. Inorganic Polyphosphate Regulates AMPA and NMDA Receptors and Protects Against Glutamate Excitotoxicity via Activation of P2Y Receptors. J. Neurosci. 2019, 39, 6038–6048. [Google Scholar] [CrossRef]
- Chrysanthopoulou A, Kambas K, Stakos D, Mitroulis I, Mitsios A, Vidali V, et al. Interferon lambda1/IL-29 and inorganic polyphosphate are novel regulators of neutrophil-driven thromboinflammation. J Pathol [Internet]. J Pathol; 2017 [cited 2023 Apr 25];243:111–22. Available from: https://pubmed.ncbi.nlm.nih.gov/28678391/.
- Odessky L, Rosenblatt P, Sands IJ, Schiff I, Dubin FM, Spielsinger D, et al. Cerebrospinal fluid inorganic phosphorus in acute poliomyelitis; study of one hundred four patients. AMA Arch Neurol Psychiatry [Internet]. AMA Arch Neurol Psychiatry; 1955 [cited 2023 Apr 17];73:255–66. Available from: https://pubmed.ncbi.nlm.nih.gov/14349427/.
- Arredondo, C.; Cefaliello, C.; Dyrda, A.; Jury, N.; Martinez, P.; Díaz, I.; Amaro, A.; Tran, H.; Morales, D.; Pertusa, M.; et al. Excessive release of inorganic polyphosphate by ALS/FTD astrocytes causes non-cell-autonomous toxicity to motoneurons. Neuron 2022, 110, 1656–1670. [Google Scholar] [CrossRef]
- Wickramasinghe, S.P.; Lempart, J.; Merens, H.E.; Murphy, J.; Huettemann, P.; Jakob, U.; Rhoades, E. Polyphosphate Initiates Tau Aggregation through Intra- and Intermolecular Scaffolding. Biophys. J. 2019, 117, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.S.; Boutej, H.; Soucy, G.; Bareil, C.; Kumar, S.; Picher-Martel, V.; Dupré, N.; Kriz, J.; Julien, J.-P. Transmission of ALS pathogenesis by the cerebrospinal fluid. Acta Neuropathol. Commun. 2020, 8, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Britson, K.A.; Ling, J.P.; Braunstein, K.E.; Montagne, J.M.; Kastenschmidt, J.M.; Wilson, A.; Ikenaga, C.; Tsao, W.; Pinal-Fernandez, I.; Russell, K.A.; et al. Loss of TDP-43 function and rimmed vacuoles persist after T cell depletion in a xenograft model of sporadic inclusion body myositis. Sci. Transl. Med. 2022, 14, eabi9196–eabi9196. [Google Scholar] [CrossRef] [PubMed]
- Henriques, A.; Kastner, S.; Chatzikonstantinou, E.; Pitzer, C.; Plaas, C.; Kirsch, F.; Wafzig, O.; Krã¼Ger, C.; Spoelgen, R.; De Aguilar, J.-L.G.; et al. Gene expression changes in spinal motoneurons of the SOD1G93A transgenic model for ALS after treatment with G-CSF. Front. Cell. Neurosci. 2015, 8. [Google Scholar] [CrossRef]
- Allen, S.P.; Hall, B.; Castelli, L.M.; Francis, L.; Woof, R.; Siskos, A.P.; Kouloura, E.; Gray, E.; Thompson, A.G.; Talbot, K.; et al. Astrocyte adenosine deaminase loss increases motor neuron toxicity in amyotrophic lateral sclerosis. Brain 2019, 142, 586–605. [Google Scholar] [CrossRef]
- Briese, T.; Kapoor, A.; Mishra, N.; Jain, K.; Kumar, A.; Jabado, O.J.; Lipkin, W.I. Virome Capture Sequencing Enables Sensitive Viral Diagnosis and Comprehensive Virome Analysis. mBio 2015, 6, e01491–15. [Google Scholar] [CrossRef]
Figure 1.
Virus-mediated excitotoxicity. The Arc-Gag complex normally interacts with the plasma membrane, weakening excitatory synapsis. CamKIIa and b phosphorylates the Arc-Gag complex of the virions, decreasing its canonical function, leading to the clustering and activation of glutamate receptors and calcium efflux, which mediates excitotoxicity.
Figure 1.
Virus-mediated excitotoxicity. The Arc-Gag complex normally interacts with the plasma membrane, weakening excitatory synapsis. CamKIIa and b phosphorylates the Arc-Gag complex of the virions, decreasing its canonical function, leading to the clustering and activation of glutamate receptors and calcium efflux, which mediates excitotoxicity.
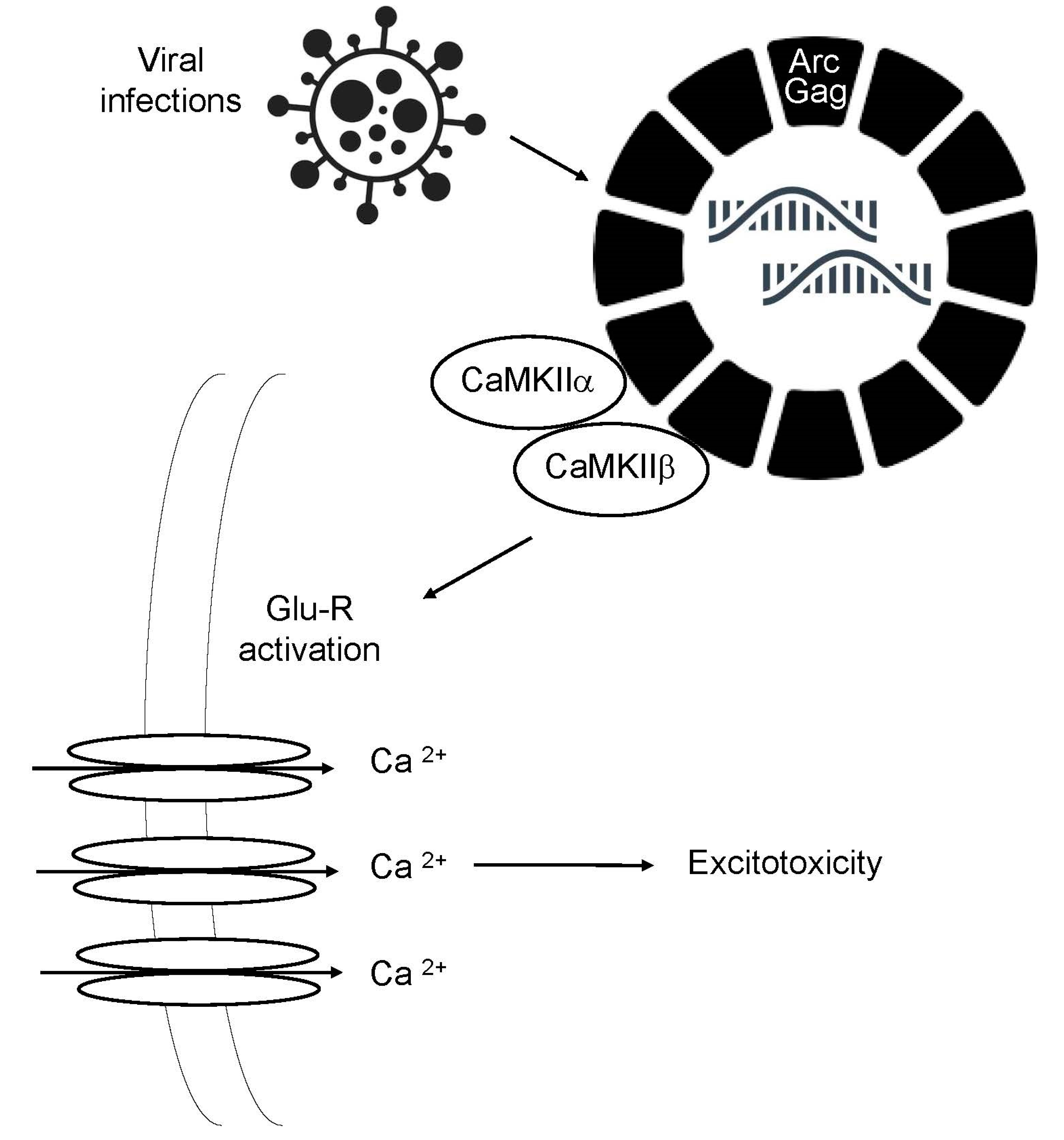
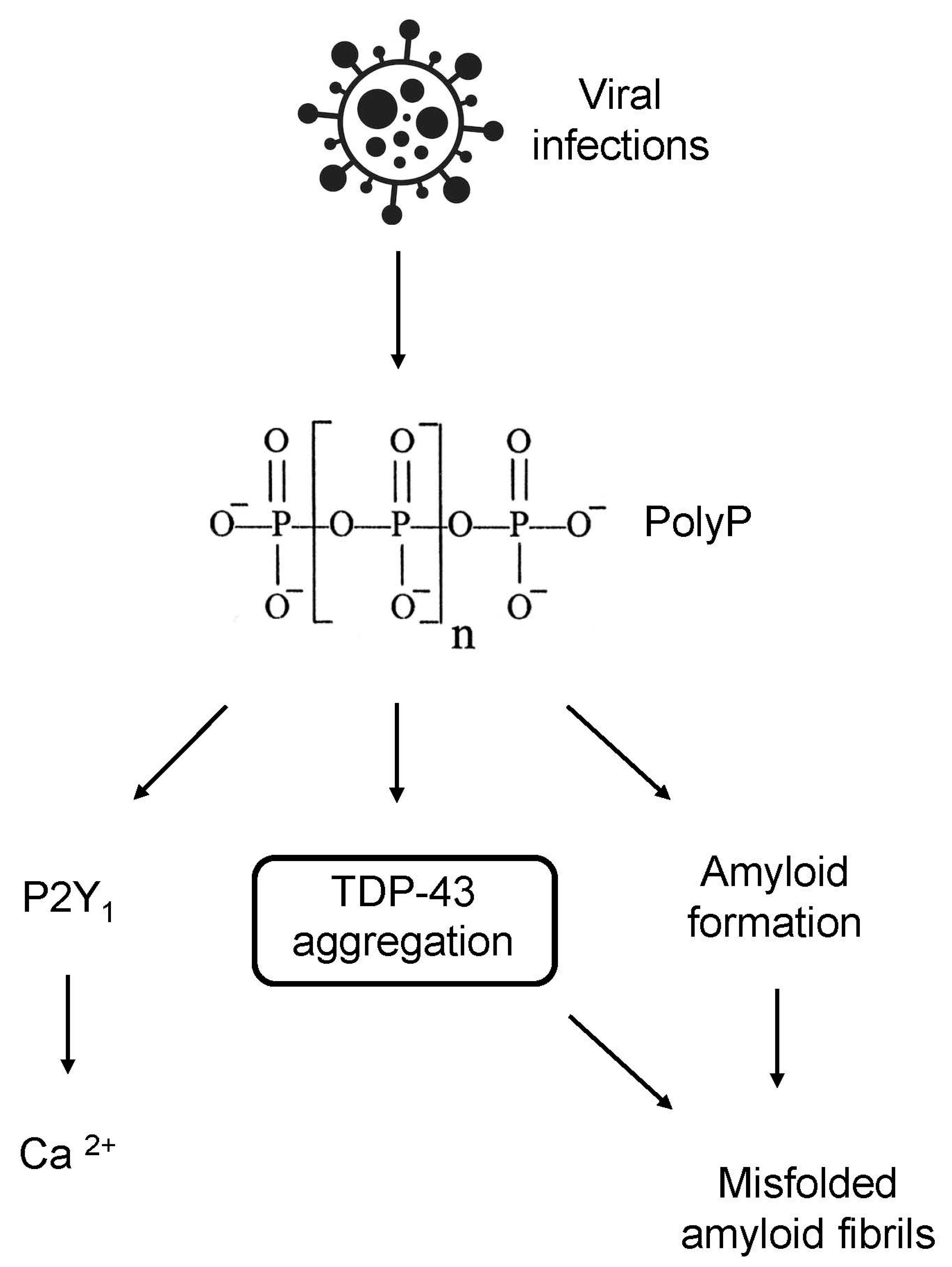
Figure 2.
PolyP in neurodegeneration. Viral infections lead to increased levels of PolyP, triggering the phosphorylation and aggregation of TDP-43, the formation of misfolded amyloid fibrils, and activation of P2Y1 receptors and, subsequently, calcium influx. TDP-43 buildup also contributes to the formation of toxic amyloid fibrils.
Figure 2.
PolyP in neurodegeneration. Viral infections lead to increased levels of PolyP, triggering the phosphorylation and aggregation of TDP-43, the formation of misfolded amyloid fibrils, and activation of P2Y1 receptors and, subsequently, calcium influx. TDP-43 buildup also contributes to the formation of toxic amyloid fibrils.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



