Submitted:
30 May 2023
Posted:
01 June 2023
You are already at the latest version
Abstract
Pervasive purifying selection on non-synonymous substitutions is a hallmark of papillomavirus genome history. Whereas the role of selection on, and drift of, non-coding DNA elements on HPV diversification is poorly understood. More than a thousand complete genomes representing Alphapapillomavirus types, lineages and SNP variants were examined phylogenetically and interrogated for the number and position of non-coding DNA sequence motifs using Principal Components Analyses, Ancestral State Reconstructions and Phylogenetic Independent Contrasts. For anciently diverged Alphapapillomavirus types, composition of the 4 nucleotides (A,C,G,T), codon usage, trimer usage and 13 established non-coding DNA sequence motifs revealed phylogenetic clusters consistent with genetic drift. Ancestral state reconstruction and Phylogenetic Independent Contrasts revealed ancient genome alterations, particularly for CpG and APOBEC3 motifs. Each evolutionary analytical method we performed supports the unanticipated conclusion that genetic drift and different evolutionary drivers have structured Alphapapillomavirus genomes in distinct ways during successive epochs, even extending to differences in more recently formed variant lineages
Keywords:
molecular evolution
; genetic drift
; human papillomavirus (HPV)
1. Introduction
Hundreds of different papillomaviruses have been described [1] encompassing the full range of vertebrate hosts from fish [2] and amphibians [3] to birds [4] and especially mammalian host groups [1]. Several dozen species and more than 200 types have been curated from humans [5-8]. Alphapapillomavirus genomes have been the most scrutinized in light of their role in cervical and other cancers [9,10,11]. Alphapapillomavirus 9 species group of human papillomavirus (HPV) types, and HPV16 in particular, stand out as most strongly associated with carcinogenesis [8,12,13,14,15,16]. Thus, understanding how their evolution has resulted in these devastating human pathogens [17,18] is important.
Considerable effort has been devoted to assessing whether there are amino acid differences that would readily explain the carcinogenic properties of HPV16, the species Alphapapillomavirus 9 and the more encompassing high-risk (HR) clade of Alphapapillomavirus [19,20,21,22]. Evolutionary selection is typically measured on the basis of rates of nonsynonymous to synonymous substitutions in codons [23,24,25]. Molecular evolutionary biologists have few other tools to convincingly discover and describe evidence of evolutionary pressures acting at more fundamental genome structural or basic nucleotide composition levels. Thus, current evidence for selection or its counterpart genetic drift are hampered by few analytical methods. Whole genome analyses of various HPV types do not support the notion that pathogenicity is more strongly connected to nonsynonymous substitutions [26] than perhaps to a lack thereof [27,28,29] or to other kinds of substitution in papillomavirus genomes [29,30]. Indeed, elevated oncogenic risk from large epidemiological studies is also associated with variation in non-coding regions and with silent substitutions that are non-randomly distributed across the ~8000 bp genome [30,31,32,33]. Comparative phylogenetic perspectives on papillomavirus genome evolution may yet reveal how disease phenotypes correlate with higher-level clade membership, with species membership, with viral type and even with lineage and sublineage distinctions [31,34,35,36,37].
Chen and colleagues [38] generated UPGMA trees based on k-mer spectra encompassing all papillomaviruses that corresponded surprisingly well with classical alignment-based phylogenetic trees. Those results indicated that changes in amino acid sequences are not the only consequence of evolutionary pressure. Beyond codons, the depletion of CpG sites appeared to be phylogenetically structured [38], which is relevant given the association of CpG content with nucleosome formation and for methylation and deamination [31,39,40,41,42,43,44,45,46,47]. Similarly, host APOBEC3 anti-viral activity should select against TCA and TCT sites [27,28,48,49,50,51]. However, the early open reading frames E6 and E7 upregulate APOBEC3 and host methyltranserferase activity [49,52,53,54,55], but have strikingly different patterns of mutability. Oncogenic HPV16 non-synonymous SNP variants are hypovariable in E7 relative to other open reading frames [28]. In contrast, the E6 locus appears to be able to vary more freely than other early- or late-expressed gene sets [28,56].
In this report, we use phylogenic approaches to investigate non-coding genomic motifs and the evolution of PV genomes. Non-recombining asexual organisms, such as papillomaviruses, under continual mutational burden display some aspects of selective pressure to avoid host primordial defenses, such as cytosine deamination [27,43,57,58]. Here we analyzed intrinsic nucleotide composition (i.e., A, C, G and T) already speculated to be of lineage specific significance [59-62], the number and distribution of CpG sites, sites available to APOBEC3 attack and strand disparities in APOBEC3 sites. We also studied- guanine quadruplexes [63], other guanine-rich motifs (e.g., duplexes) [64], toll-like-receptor 9 (TLR9) stimulatory and suppressing sequence motifs implicated in viral pathogenicity [65,66,67,68,69,70], high-affinity and non-canonical E2-binding sites [71,72], inverted repeats, perfect palindromes, duplicated regions and reverse complementary regions [73,74]. The evidence regarding the evolution of DNA motifs in HPV genomes is most consistent with genetic drift.
2. Materials and Methods
2.1. Virus Genome Data
For higher level analyses at the genus level, all reference genomes for each of 83 Alphapapillomavirus reference types were obtained in Genbank format from the Papillomavirus Episteme database (PAVE, https://pave.niaid.nih.gov ) [5]. For the purposes of phylogenetic and UPGMA analyses, two datasets were established: one unaligned and without modification and a second aligned data set in which each genome was informatically processed to remove both non-coding and overlapping coding regions on the basis of annotations embedded in the Genbank format. For phylogenetic and ancestral state reconstructions in Alphapapillomavirus 9, reference genomes were similarly obtained from PAVE for each variant in each type in Alphapapillomavirus 9 as well as for each variant of HPV18 and HPV45 as outgroup taxa. In order to maximize assessment of variability for Principal Components Analysis within Alphapapillomavirus 9 variants, whole genomes were obtained from NCBI genomes that were complete, annotated for all open reading frames, and in which no more than 25 nucleotides were missing or ambiguous in the URR. The latter comprised a total of 747 for HPV16, 284 for HPV35, 41 for HPV31, 28 for HPV33, 138 for HPV58, and 35 for HPV67. Many deposited complete genomes for HPV52 were found to be incompletely annotated. Relaxing the annotation requirement permitted expansion of the lineage representation for HPV52 to 191 complete genomes.
2.2. Phylogenetic and Cluster Analysis
Phylogenetic trees at the Alphapapillomavirus species and type level were obtained with maximum likelihood (GTR+G) using PAUP 4.0a169 [119] analyzing the concatenated nucleotide sequences from non-overlapping open reading frames for E6, E7, E1, E2, L2 and L1 separately aligned according to the aligned inferred amino acid sequences using the TranslatorX server (translator.co.uk) [120]. Because codon models of selection must faithfully distinguish between synonymous and nonsynonymous nucleotide substitutions in-frame [121], we excluded all overlapping reading frames from those analyses (for example, but not limited to, where E4 and E2 overlap). In addition to phylogenetic trees, separate UPGMA trees were constructed based on each of codon usage, trimer composition and nucleotide composition. For each of the foregoing, Euclidean distances were constructed using reference types from the Papillomavirus Episteme database (PAVE, https://pave.niaid.nih.gov) [5]. Distance matrixes were evaluated with UPGMA in the fitch package of Phylip version 3.695 [122]. Trees were examined with FigTree version 1.4.3 [123].
2.3. Enumeration of Sequence Motifs
Code was written in Python 3 leveraging the Biopython module to determine the number and position of nucleotide sequence motifs across each whole and unaligned genome analyzed, both in total and in a sliding window scan of the genomes. The level of resolution (sliding window size of 150 nucleotides) was chosen to be small enough (i.e., half the size) to resolve the two smallest open reading frames (E6 and E4), while also being large enough to capture degrees of freedom on the occurrence of DNA motifs. For example, to the extent that CpG motifs are suppressed in the Alphapapillomavirus 9 lineages, even if those motifs were randomly distributed, one would expect to find a CpG motif only once every 75 dimers. Each iteration involved a 9-nucleotide widow shift to have at least 30 shifts in E6 and E4. Parameters recovered with regular expression (regex) pattern matching (see Supplementary Document S1) in unaligned genomes as well as base composition on the coding strand (i.e., A,C,T,G), trimer composition on the coding strand, CpG motifs, high affinity E2 binding site motifs on the coding and opposite strand, APOBEC3 binding site motifs on the both strands, toll-like receptor 9 (TLR9) stimulatory and suppressing motifs on both strands, guanine quadruplexes on both strands, palindromes, near-palindromic inverted-repeats allowing from 3 to 50 spacing nucleotides, larger and more distant duplicated or duplicated and reverse-complemented regions being at least 14 nucleotides long and separated by at least 14 nucleotides. Trimer composition was assessed by way of a normalized Euclidean distance in a sliding window relative to that of the whole genome.
2.4. Statistical Analyses
Determinations of confidence limits and statistical significance of local trimer composition, GC content, as well as the relative number of CpG and APOBEC3 sites in each 150 nt sliding window was assessed by comparison of observed values to those obtained from a same-sized window pseudo-sampled from 1000 randomizations of the entire genome. Assessment of differences in relative proportions of covarying residues, and relative proportions of APOBEC sites on opposite strands were accomplished through standard Z score calculations with a Bonferroni correction for multiple comparisons.
Principal Components Analysis (PCA) was accomplished in Python 3 with the SciKit-Learn library, using a standard scaler for preprocessing and Seaborn for visualization.
In order to obtain unbiased branch lengths for each DNA sequence motif and for changes in base composition, ancestral state reconstructions of these continuous variables were fit to the ML topology using a Brownian motion model in Mesquite version 3.70 [124]. Phylogenetic Independent Contrasts (PIC) of reconstructed ancestral states were calculated in Python 3 with the Denropy library. Pearson product-moment correlation p-value among continuous variable PIC were converted to false discovery (Q) values through the ratio of their relative rank to the total number of comparisons.
Non-overlapping reading frame alignments were examined using codon models with HyPhy [85] for overall rates of nonsynonymous to synonymous substitution (with BUSTED), site by site episodic selection (with MEME), and or changes in the strength of selection (RELAX) each on the Datamonkey server [125].
3. Results
3.1. Topological Comparisons
Classical molecular evolution methods are based upon the establishment of homology (i.e., alignment) and tree building. Nonetheless, it can be informative to examine how alignment-free analyses compare to an alignment-based phylogeny [38]. In Figure 1, the Alphapapillomavirus alignment-based tree (Panel A) is largely recapitulated by the alignment-free trees based on trimer composition (Panel B), codon usage (Panel C), and base compositions (Panel D) where the low-risk LR2 viruses (green) did not cluster with the low-risk LR1 (blue) and high-risk (HR) virus types (Figure 1). However, all three alignment-free analyses placed the non-human primate viruses (Alphapapillomavirus 12, grey) within the LR2 clade instead of with the HR group as found in the alignment-based tree. The discordance between alignment-based and alignment-free trees (Figure 1) could indicate driving features of additional information (e.g., non-coding) in the genomes that extends beyond aligned ORF analyses.
3.2. Sliding Window Analysis
To look for common patterns in unaligned genomes, we evaluated all Alphapapillomavirus types (n=83) with sliding window analyses, finding the local number of CpG and APOBEC3 motifs in each 150 bp window while also measuring changes in GC content and local trimer composition; these analyses are mapped to nucleotide positions in the HPV16 reference genome (Figure 2A). For viral types infecting humans (Figure 2B), 70% of the windows with significantly high concentrations of CpG sites corresponded to E4 (Figure 2B) which also had the highest GC content (green line in Figure 2A) and atypical trimer usage (blue line Figure 2A). A second region of high CpG concentration was found near the late polyadenylation region within the URR (Figure 2B). Concentrations of APOBEC3 recognition sites accumulate linearly above expectations through the early ORFs with exceptions for regions in E7 and E1 ORFs. The Alphapapillomavirus 12 viruses infecting non-human primates exhibit a pattern that is different from those infecting humans (grey plots Figure 2B).
3.3. Principal Component Analysis
To further examine the alignment-free clustering noted in Figure 1, we investigated Alphapapillomavirus genome content with Principal Components Analysis (PCA). Drift, as predicted by Brownian motion random-walk evolutionary models, results in non-recombining genomes within lineages to diffuse into mutual overlap if given enough time [75-77] (Figure 3). Thus, under drift, the ability to discriminate lineages diminishes with time (Figure 3), as indicated by recently diverged lineages being well-discriminated (see t=1 in Figure 3) and broad overlap for more anciently diverged lineages/clades (see t=3 in Figure 3). In contrast, a common selection pressure should cause narrow convergence on a distinct optimum in the parameter space [78], convincing evidence of which would be the convergence of lineages that are not each-others’ closest relatives (see light blue and light green lineages at t=3 in Figure 3B). Using unaligned Alphapapillomavirus reference genomes [5], PCA of the 13 DNA sequence motifs (Supplementary Document S1) and simple base composition (A, C, G, T) showed a number of similarities (Figure 4A,B). PCA incompletely segregated human high-risk from human LR1 virus types and incompletely segregated LR2 types from NHP-Alpha12 types (NHP) as previously illustrated by alignment-free clustering (Figure 1) and consistent with expectations for anciently diverged clades (see t =3 in Figure 3A).
Tens of millions of years since the divergence of the LR1, LR2 and HR clades, there still remains clustering that likely reflects their common ancestors. However, diffusion of the phylogenetic pattern is evident (Figure 4). Indeed, this is what is predicted with a Brownian motion drift process in which the displacement from an ancestral condition is the product of the parameter variance and time [79,80] (Figure 3). To evaluate patterns of recent type divergence, we leveraged 1,278 complete Alphapapillomavirus 9 HPV genomes (from GenBank, August 2022) including types, lineages, sublineages and SNP variants. PCA of the 13 DNA sequence motifs and of nucleotide base compositions tended to isolate recently diverged types surprisingly well (Figure 5), as also reflected by individual base compositions (Supplementary Document S9). Based on the sequence motifs PCA, the strongest distinctions were for HPV16 (red) and HPV35 (grey) both from each other and from other types, whereas neither HPV52 and HPV67, nor HPV58 and HPV33 were fully discriminated (Figure 5A). Separation of types was more complete based on base composition where only HPV52 and HPV67 failed to discriminate (Figure 5B), and for which the first principal component largely reflected GC content (Supplementary Document S9). In addition to separating types, PCA of base compositions (Figure 5B) also discriminated among some sublineage variants of HPV16, HPV58, HPV31 and HPV67. It should be noted that recently diverged lineages appear to have differentiated in ways that are type-specific (as in Figure 3A), without evidence of common adaptation (as expected in Figure 3B) to a similar ecological niche. In fact, HPV16, the most oncogenic HPV type, best reflects the ancestral nucleotide composition of Alphapapillomavirus 9 (Supplementary Document S9).
The co-location of HPV52 and HPV67 in PCA space (Figure 5A,B) is not explained by recent common ancestry – they do not form a monophyletic group (see tree in. Figure 5). An alternative explanation is that both contain features of their most recent common ancestor (denoted MRCA-x on Figure 5), whereas HPV58 and HPV33 have diverged (Figure 5B). A similar case can be made for the genomes of HPV58 and HPV33. That is, though their base compositions have diverged (Figure 5B), they still overlap in the PCA space based on shared higher-order sequence motifs (Figure 5A) consistent with their recent common ancestor (denoted MRCA-y on Figure 5). The PCA results are consistent with Brownian motion [81] acting upon ancestral states of HPV isolates resulting in drift [76,80,82].
3.4. Ancestral State Reconstruction
To understand how the DNA motifs in HPV genomes have changed over time, we utilized Brownian motion ancestral state reconstruction (see Figure 6, and Supplementary Document S4) [80,83]. Of the 13 DNA sequence motifs, 4 exhibited more ancestral branch change than expected (Figure 6): the number of APOBEC3 recognition sites (11 branches), the number of palindromic regions (8 branches) as well as the number of CpG sites and TLR9 stimulating motifs on the same branches given that the latter are CpG-rich. HPV16 has the least CpG sites and the most palindromic motifs represented by multiple instances of punctuated change since the Alphapapillomavirus MRCA. However, there does not appear to be Alphapapillomavirus-wide coordination of significant changes on ancestral lineages as would be expected from a selection pressure that is common to all of Alphapapillomavirus. In a manner similar to the PCA of recently diverged HPV genomes (Figure 5), ancestral state reconstruction (Figure 6) suggests no obvious common evolutionary trajectory since the Alphapapillomavirus MRCA. For example, some lineages show marked decrement in CpG sites whereas others increase. Similarly, both increases and decreases in TLR9 sites are apparent in the ancestral lineages of LR1 and LR2. Thus, each clade seems to have its own specific history with respect to the evolution of these motifs.
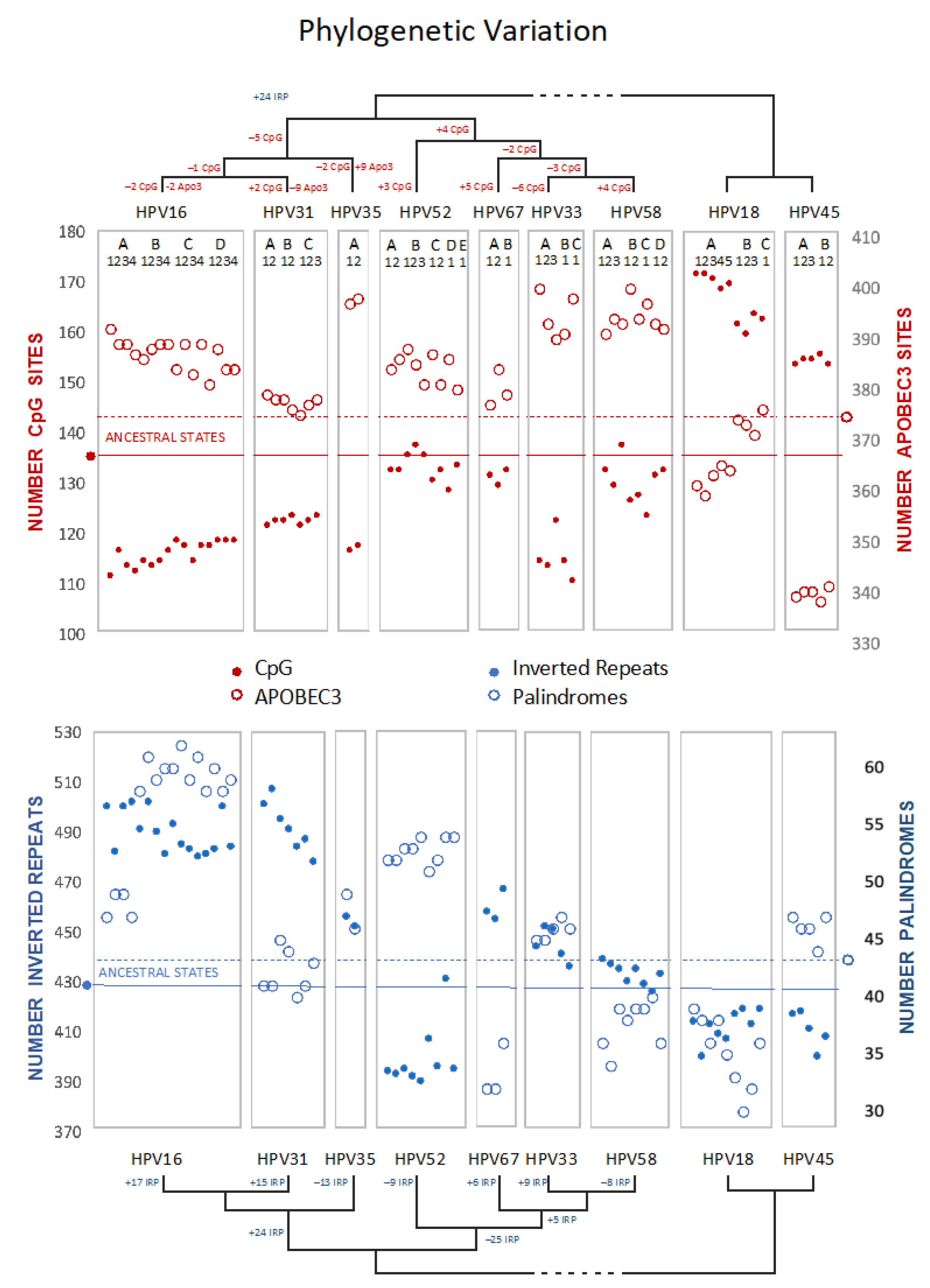
As with PCA (above), to explore recent HPV oncogenic type variant divergences, we compared ancestral states for all Alphapapillomavirus 9 variant reference genomes from PAVE including HPV18 and HPV45 as an outgroup to root the Alphapapillomavirus 9 tree (Figure 7, and Supplementary Document S5). Brownian motion reconstruction revealed that gradual changes for recently diverged variants have resulted in markedly different trajectories. For example, the number of inverted repeats with potential for secondary structure increases in HPV16/31, but was reduced among HPV52 variants, while the number of perfect palindromes increased in both HPV16 and HPV52 variant lineages (and see Supplemental Documents S6, S7, S8). HPV31 and HPV33 have numbers of palindromes that reflect the ancestral condition, implying convergent reduction in HPV67 and HPV58. The number of CpG sites is reduced in variants of HPV16, whereas CpG sites are more numerous in HPV18 and HPV45 than in any Alphapapillomavirus 9 type. HPV31 variants in particular, are closest to the predicted number of ancestral APOBEC3 sites, with increases in these sites for HPV35 and HPV33/58. In the outgroup, HPV45 has markedly fewer APOBEC3 sites in comparison to all other types examined.
These analyses did not suggest a common evolutionary mechanism that would explain how HPV16, HPV18 and HPV58 are among the most prevalent or most oncogenic of the HR-HPV viruses. Nevertheless, visual inspection of CpG and APOBEC3 content across Alphapapillomavirus 9 variants and the outgroup types suggests an inverse relationship (Figure 7). Indeed, with a false-discovery rate of 5%, the extant values for CpG and APOBEC3 motifs on the coding strand of HPV16 and HPV18 variants proved to be inversely correlated (R = –0.655, p = 0.006) (R = –0.955, p = 0.00006), respectively. However, this kind of pairwise correlation fails to account for phylogenetic relatedness and the non-independence of closely related viruses (see also [29]).
3.5. Phylogenetic Independent Contrasts
The inverse pairwise correlation between CpG and APOBEC3 motifs (e.g., HPV18 variants in Figure 7) could result from the close phylogenetic relatedness of variants within lineages compared to the distance between lineages. Phylogenetic Independent Contrasts (PIC) provides a strategy to tease out these possibilities [76,80]. We looked for phylogenetic correlations among the 13 DNA motifs and base composition (A, C, G, T) in viral genomes at 3 hierarchical levels: all 83 Alphapapillomavirus reference types, 25 types in the human HR clade (Alpha5/6/7/9/11), and separately in the variants within Alphapapillomavirus 7, Alphapapillomavirus 9 as well as the clade combining Alphapapillomavirus 5 and Alphapapillomavirus 6 in order to have similar degrees of freedom for variants.
Using the coding strand, base compositions showed correlations with PIC that are expected from Chargaff’s Second Parity Rule [84] (i.e., significant positive correlations for A with T, C with G, and negative for all other combinations) except that in Alphapapillomavirus 9, A and T did not exhibit the expected positive correlation (Supplemental Document S.10). Additional significant correlations (FDR Q < 0.01) using PIC on DNA sequence motifs are shown in Supplementary Document S10 at each hierarchical level. Notably, the inverse correlation of CpG and APOBEC3 sites remains significant within the HR and Alphapapillomavirus 7 clades (Supplementary Document S10). The number of palindromic regions (inverted repeats and perfect palindromes) was inversely related to CpG content across the Alphapapillomavirus and HR clades yet was not significant within any HR species subset. The only commonality among recently diverged HR species groups was that TLR9 and APOBEC3 motifs were significantly correlated in the opposite direction in Alphapapillomavirus 7 and Alphapapillomavirus 9 (Supplementary Document S10). Taken together, results of PIC suggest that coordinated changes among the 13 motifs are not uniform through time as would be expected if alphapapillomaviruses were adapting to the same niche or evolving toward a limited number of selective optimal genome features. This observation is also consistent with drift.
3.6. Codon-based Selection
Lastly, we also evaluated the role of Darwinian (i.e., positive) selection using nonsynonymous to synonymous substitution rates (dN/dS). Examining the aligned non-overlapping ORFs resulted in 8676 aligned nucleotides (of which 6165 were variable), representing 2892 aligned amino acid positions (of which 2106 were variable). Using HyPhy [85] with a GTR model, the phylogeny- and genome-wide dN/dS was estimated to be 0.1743, indicating strong purifying selection. Clade-specific differences in the strength of selection was examined with RELAX [86]. The hypotheses with the highest likelihood ratios (LR) included a significant relaxed selection strength (K) in the LR2 clade (K=0.63; LR=50.72) and a significant increase of selection in the Alphapapillomavirus 12 clade relative to all Alphapapillomavirus (K=1.12; LR=43.26). In terms of selection on aligned sites instead of branches, when considering Alphapapillomavirus as a group, MEME [25] found purifying selection at 277 amino acid sites (P < 0.05) but no sites under diversifying selection, after curation with G-blocks [87] (Supplemental Document S2).
4. Discussion
In this report, we focus on the role of non-coding DNA elements and the evolution of alphapapillomaviruses. Rather than recovering patterns of nucleotide evolution that would suggest a common selective framework for alphapapillomaviruses within the cervicovaginal niche, we document non-coding changes occurring in a highly lineage-specific manner. These results are most consistent with a non-random mutation burden associated with genetic drift [88,89]. Given the rarity of positive Darwinian selection [26] and recombination, new HPV variants emerge containing different combinations of non-coding elements as we describe from our analysis of 13 higher-order nucleotide motifs. Surprisingly, even nucleotide composition alone discriminated closely related types, e.g., alpha-9 types (compare Figure 3A/B and Figure 5). The evolution of HPV genomes represents independent yet successful genetic drift away from ancestral niche-adapted genotypes [90] escaping the accumulation of unfavorable mutations and the speed of Muller’s Ratchet [91].
In addition to alterations to the number of CpG, APOBEC3 and TLR9 sites, there have been significant changes to the number of palindromic regions, each of which can result from simple changes in overall nucleotide (A,C,G,T) composition [92,93]. A progressive loss of CpG sites was identified within the more prevalent HR-HPV types. This included a 30% reduction from the MRCA of all alphapapillomaviruses to the MRCA of HR-HPV, and thereafter an additional 12% decrease from the MRCA of HR-HPV to the MRCA of Alphapapillomavirus 9. Globally, HPV16 is the most prevalent type, and it notably possesses less than half the CpG sites inferred to have been present in the MRCA of all Alphapapillomavirus types. Undoubtedly the viable lower limit of CpG sites is constrained in part by the overlapping reading frames of E2 and E4, either of which may not be able to vary synonymously without altering the other reading frame non-synonymously (Figure 2). Intriguingly, a second region of increased CpG sites across Alphapapillomavirus corresponds to the 5’ URR continaing a nuclear matrix association region –[94–96] where viral genome-wide association studies (VWASs) consistently find SNPs associated with increased carcinogenicity [28,30,32,33].
The diverse family of apolipoprotein B editing and catalytic (APOBEC) enzymes play critical roles in host defense against a wide range of viruses through cytosine deamination of exposed ssDNA. APOBEC3 enzymes are also implicated in precancerous genome instability [51,97-101]. Like methylation, APOBEC3 activity is upregulated in HPV infections [55] and their signature mutation type [102] is associated with viral clearance [27] and the mutational footprint left on neoplastic host cells [48]. Whereas APOBEC3 sites are nonrandomly distributed across many human DNA viruses [27,50], we found more on the transcribed strand (antisense) than on the sense strand of Alphapapillomavirus genomes. This could represent selection to minimize sites on the coding strand, given the tendency of APOBEC3 enzymes to also edit mRNA when they are over-expressed [103]. Alternatively, the coding strand single-stranded state during transcription, and during lagging strand replication of Early ORFs, would imply stronger APOBEC3 mutation pressure on that strand. Interestingly, APOBEC3 sites also are open to attack during repair of deaminated CpG sites [104] which corresponds well to the inverse correlation of these motifs in Phylogenetic Independent Contrasts (Supplemental Document S.10).
It was surprising that something as primordial as base composition recapitulated Alphapapillomavirus phylogeny and also strongly discriminated Alphapapillomavirus 9 types by PCA. Clearly there is more encoded in the genomes of alphapapillomaviruses beyond codon and amino acid usage, alternative splicing of mRNA transcripts [105] and the efficiency afforded by overlapping reading frames [106]. Even simple nucleotide motifs like tandem repeats of guanine are prone to simultaneous oxidation by glutathione [64], the lack of which has been proposed as a risk factor for the development of cervical cancer [107]. In addition to a tight cytosine balance, nucleotide composition could be under selection on silent substitutions [108], for example, through effects on the speed, efficiency and accuracy of transcription and translation [29,109]. Discovering marked differences in the number of palindromic regions (perfect palindromes and inverted repeats) is interesting, given that base composition biases lead to higher palindromic content when the bias favors complementary nucleotides (e.g., A and T) [110]. An increase in the number and diversity of locally arranged inverted repeats could be a source of variation in hairpins, cruciforms, and pseudoknots that are open to evolutionary tinkering given their functional significance for transcription, translation and the influence of miRNA on HPV gene expression [111,112].
The predominance of purifying selection on amino acids is consistent with observations of other dsDNA viruses [113]. We did not find substantial evidence of diversifying selection operating on individual amino acid sites.
Our findings imply something quite different from the notion that Alphapapillomavirus diversity resulted from the onset of diversifying selection with ancestral occupation of a new ecological niche [114]. Such a release from stabilizing selection would be marked by evidence of positive selection [115] which is simply not observed. For example, consider the convergent adoption of nonsynonymous mutations (e.g., K417T, E484K and N501Y) in the spike protein of multiple lineages of SARS-CoV-2 [116] which are under obvious positive natural selection. The emergence and persistence of new variants based to a large extend on drift could result from genomic “robustness” to the mutation pressures imposed by innate intracellular host restriction factors, especially if accompanied by incomplete purifying selection [117]. Were this the case, one would expect the least-mutationally-compromised virus to be the most prevalent; and this seems to be the case when comparing HP16 to other Alphapapillomavirus types (Supplementary Data S9). In conclusion, the evolutionary signature recovered in Alphapapillomavirus HPV genomes is one of marked purifying selection on amino acid changes (which bodes-well for the effectiveness of HPV vaccines), and lack of convergence towards any single common adaptive peak or strategy. Cytosine deamination patterns and strand-asymmetry are consistent with a directional mutation model of neutral molecular evolution [88,118].
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, S1: Regular-Expression Patterns for DNA Sequence Motifs.doc; S2: MEME sites all Alpha.csv; S3: Selection strength RELAX; S4: HPV Reference Types Ancestral Reconstructions.pdf; S5: HPV Alphapapillomavirus 9 Ancestral Reconstructions.pdf; S6: HPV Alpha RefType InvertRepeat.csv; S7: HPV Alpha RefType Duplicated.csv; S8: HPV Alpha RefType Palindromes.csv; S9: HPVAlpha nt changes.pdf; S10: Correlated Phylogenetic Independent Contrasts of DNA Sequence Motifs;.
Author Contributions
Conceptualization, R.B. & R.D.; Methodology, R.D. & R.B.; Software, R.D.; Validation, R.D. and R.B.; Formal Analysis, R.D.; Investigation, R.D., L.M. and R.B.; Resources, R.B.; Data Curation, R.D. & L.M.; Writing—Original Draft Preparation, R.B.; Writing—Review & Editing, R.B., L.M. & R.D.; Supervision, R.B.; Project Administration, R.B.; Funding Acquisition, R.B. & L.M.
Funding
The Intramural Research Program of the United States National Institutes of Health and the NCI (grant number: 5U01CA078527; to Dr. Burk) supported in part the research described in this study, the Albert Einstein Cancer Research Center (P30CA013330, PI Ed Chu, and the CFAR at the Albert Einstein College of Medicine.
Data Availability Statement
Alignments of nucleotide sequences, amino acid sequences and matrices of continuous variables with corresponding trees can be found with accession number XXXXXX at Treebase (www.trabase.org).
Acknowledgments
In this section, you can acknowledge any support given which is not covered by the author contribution or funding sections. This may include administrative and technical support, or donations in kind (e.g., materials used for experiments).
Conflicts of Interest
The authors declare no conflict of interest.The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Bernard, H.-U.; Burk, R.D.; Chen, Z.; Van Doorslaer, K.; zur Hausen, H.; de Villiers, E.-M. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 2010, 401, 70–79, . [CrossRef]
- Kraberger, S.; Austin, C.; Farkas, K.; Desvignes, T.; Postlethwait, J.H.; Fontenele, R.S.; Schmidlin, K.; Bradley, R.W.; Warzybok, P.; Van Doorslaer, K.; et al. Discovery of novel fish papillomaviruses: From the Antarctic to the commercial fish market. Virology 2022, 565, 65–72, . [CrossRef]
- Russo, A.G.; Harding, E.F.; Yan, G.J.H.; Selechnik, D.; Ducatez, S.; DeVore, J.L.; Zhou, J.; Sarma, R.R.; Lee, Y.P.; Richardson, M.F.; et al. Discovery of Novel Viruses Associated With the Invasive Cane Toad (Rhinella marina) in Its Native and Introduced Ranges. Front. Microbiol. 2021, 12, 733631. [CrossRef]
- Truchado, D.A.; Williams, R.A.; Benítez, L. Natural history of avian papillomaviruses. Virus Res. 2018, 252, 58–67, . [CrossRef]
- Van Doorslaer, K.; Li, Z.; Xirasagar, S.; Maes, P.; Kaminsky, D.; Liou, D.; Sun, Q.; Kaur, R.; Huyen, Y.; McBride, A.A. The Papillomavirus Episteme: a major update to the papillomavirus sequence database. Nucleic Acids Res. 2017, 45, D499–D506, . [CrossRef]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Dempsey, D.M.; Dutilh, B.E.; Harrach, B.; Harrison, R.L.; Hendrickson, R.C.; et al. Changes to virus taxonomy and the Statutes ratified by the International Committee on Taxonomy of Viruses (2020). Arch. Virol. 2020, 165, 2737–2748, . [CrossRef]
- Mühr, L.S.A.; Eklund, C.; Dillner, J. Towards quality and order in human papillomavirus research. Virology 2018, 519, 74–76, . [CrossRef]
- Bruni L, A.G., Serrano B, Mena M, Collado JJ, Gómez D, Muñoz J, Bosch FX, de Sanjosé S., ICO/IARC Information Centre on HPV and Cancer (HPV Information Centre). Human Papillomavirus and Related Diseases in the World. 2021.
- zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350, . [CrossRef]
- Kidd, L.C.; Chaing, S.; Chipollini, J.; Giuliano, A.R.; Spiess, P.E.; Sharma, P. Relationship between human papillomavirus and penile cancer—implications for prevention and treatment. Transl. Androl. Urol. 2017, 6, 791–802, . [CrossRef]
- Sabatini, M.E.; Chiocca, S. Human papillomavirus as a driver of head and neck cancers. Br. J. Cancer 2020, 122, 306–314, . [CrossRef]
- Münger, K.; Baldwin, A.; Edwards, K.M.; Hayakawa, H.; Nguyen, C.L.; Owens, M.; Grace, M.; Huh, K. Mechanisms of Human Papillomavirus-Induced Oncogenesis. J. Virol. 2004, 78, 11451–60, . [CrossRef]
- Schiffman, M.; Rodriguez, A.C.; Chen, Z.; Wacholder, S.; Herrero, R.; Hildesheim, A.; Desalle, R.; Befano, B.; Yu, K.; Safaeian, M.; et al. A Population-Based Prospective Study of Carcinogenic Human Papillomavirus Variant Lineages, Viral Persistence, and Cervical Neoplasia. Cancer Res 2010, 70, 3159–3169, . [CrossRef]
- Byun, J.M.; Jeong, D.H.; Kim, Y.N.; Jung, E.J.; Lee, K.B.; Sung, M.S.; Kim, K.T. Persistent HPV-16 infection leads to recurrence of high-grade cervical intraepithelial neoplasia. Medcine 2018, 97, e13606, doi:10.1097/md.0000000000013606.
- Wei, F., et al., Age-specific prevalence of anal and cervical HPV infection and high-grade lesions in 11 177 women by HIV status: a collaborative pooled analysis of 26 studies. J Infect Dis, 2022.
- Arbyn, M.; Weiderpass, E.; Bruni, L.; de Sanjosé, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of incidence and mortality of cervical cancer in 2018: a worldwide analysis [published correction appears in Lancet Glob Health. 2022 Jan;10,e41]. Lancet Glob. Health 2020, 8, e191–e203, doi:10.1016/s2214-109x(19)30482-6.
- Schiffman, M.; Herrero, R.; DeSalle, R.; Hildesheim, A.; Wacholder, S.; Rodriguez, A.C.; Bratti, M.C.; Sherman, M.E.; Morales, J.; Guillen, D.; et al. The carcinogenicity of human papillomavirus types reflects viral evolution. Virology 2005, 337, 76–84, . [CrossRef]
- Burk, R.D.; Chen, Z.; Van Doorslaer, K. Human Papillomaviruses: Genetic Basis of Carcinogenicity. Public Heal. Genom. 2009, 12, 281–290, . [CrossRef]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part B: biological agents. Lancet Oncol. 2009, 10, 321–322, doi:10.1016/s1470-2045(09)70096-8.
- Chen, Z.; Schiffman, M.; Herrero, R.; DeSalle, R.; Anastos, K.; Segondy, M.; Sahasrabuddhe, V.V.; Gravitt, P.E.; Hsing, A.W.; Burk, R.D. Evolution and Taxonomic Classification of Human Papillomavirus 16 (HPV16)-Related Variant Genomes: HPV31, HPV33, HPV35, HPV52, HPV58 and HPV67. PLOS ONE 2011, 6, e20183, . [CrossRef]
- Clifford, G.M.; Howell-Jones, R.; Franceschi, S. Judging the carcinogenicity of human papillomavirus types by single/multiple infection ratio in cervical cancer. Int. J. Cancer 2010, 129, 1792–1794, . [CrossRef]
- Chen, Z.; Schiffman, M.; Herrero, R.; DeSalle, R.; Anastos, K.; Segondy, M.; Sahasrabuddhe, V.V.; Gravitt, P.E.; Hsing, A.W.; Chan, P.K.; et al. Classification and evolution of human papillomavirus genome variants: Alpha-5 (HPV26, 51, 69, 82), Alpha-6 (HPV30, 53, 56, 66), Alpha-11 (HPV34, 73), Alpha-13 (HPV54) and Alpha-3 (HPV61). Virology 2018, 516, 86–101, . [CrossRef]
- Fitch, W.M. Rate of change of concomitantly variable codons. J. Mol. Evol. 1971, 1, 84–96, . [CrossRef]
- Suzuki, Y. New Methods for Detecting Positive Selection at Single Amino Acid Sites. J. Mol. Evol. 2004, 59, 11–19, . [CrossRef]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Pond, S.L.K. Detecting Individual Sites Subject to Episodic Diversifying Selection. PLOS Genet. 2012, 8, e1002764, . [CrossRef]
- King, K.M.; Rajadhyaksha, E.V.; Tobey, I.G.; Van Doorslaer, K. Synonymous nucleotide changes drive papillomavirus evolution. Tumour Virus Res. 2022, 14, 200248, . [CrossRef]
- Zhu, B.; Xiao, Y.; Yeager, M.; Clifford, G.; Wentzensen, N.; Cullen, M.; Boland, J.F.; Bass, S.; Steinberg, M.K.; Raine-Bennett, T.; et al. Mutations in the HPV16 genome induced by APOBEC3 are associated with viral clearance. Nat. Commun. 2020, 11, 886, . [CrossRef]
- Mirabello, L.; Yeager, M.; Yu, K.; Clifford, G.M.; Xiao, Y.; Zhu, B.; Cullen, M.; Boland, J.F.; Wentzensen, N.; Nelson, C.W.; et al. HPV16 E7 Genetic Conservation Is Critical to Carcinogenesis. Cell 2017, 170, 1164–1174.e6, . [CrossRef]
- Shackelton, L.A.; Parrish, C.R.; Holmes, E.C. Evolutionary Basis of Codon Usage and Nucleotide Composition Bias in Vertebrate DNA Viruses. J. Mol. Evol. 2006, 62, 551–563, . [CrossRef]
- Pinheiro, M.; Harari, A.; Schiffman, M.; Clifford, G.M.; Chen, Z.; Yeager, M.; Cullen, M.; Boland, J.F.; Raine-Bennett, T.; Steinberg, M.; et al. Phylogenomic Analysis of Human Papillomavirus Type 31 and Cervical Carcinogenesis: A Study of 2093 Viral Genomes. Viruses 2021, 13, 1948, . [CrossRef]
- Mirabello, L.; Yeager, M.; Cullen, M.; Boland, J.F.; Chen, Z.; Wentzensen, N.; Zhang, X.; Yu, K.; Yang, Q.; Mitchell, J.; et al. HPV16 Sublineage Associations With Histology-Specific Cancer Risk Using HPV Whole-Genome Sequences in 3200 Women. JNCI J. Natl. Cancer Inst. 2016, 108, . [CrossRef]
- Pinheiro, M.; Gage, J.C.; Clifford, G.M.; Demarco, M.; Cheung, L.C.; Chen, Z.; Yeager, M.; Cullen, M.; Boland, J.F.; Chen, X.; et al. Association of HPV35 with cervical carcinogenesis among women of African ancestry: Evidence of viral-host interaction with implications for disease intervention. Int. J. Cancer 2020, 147, 2677–2686, . [CrossRef]
- Bee, K.J.; Gradissimo, A.; Chen, Z.; Harari, A.; Schiffman, M.; Raine-Bennett, T.; Castle, P.E.; Clarke, M.; Wentzensen, N.; Burk, R.D. Genetic and Epigenetic Variations of HPV52 in Cervical Precancer. Int. J. Mol. Sci. 2021, 22, 6463, . [CrossRef]
- Kovacic, M.B.; Castle, P.E.; Herrero, R.; Schiffman, M.; Sherman, M.E.; Wacholder, S.; Rodriguez, A.C.; Hutchinson, M.L.; Bratti, M.C.; Hildesheim, A.; et al. Relationships of Human Papillomavirus Type, Qualitative Viral Load, and Age with Cytologic Abnormality. Cancer Res 2006, 66, 10112–10119, . [CrossRef]
- Berumen, J.; Ordoñez, R.M.; Lazcano, E.; Salmeron, J.; Galvan, S.C.; Estrada, R.A.; Yunes, E.; Garcia-Carranca, A.; Gonzalez-Lira, G.; la Campa, A.M.-D. Asian-American Variants of Human Papillomavirus 16 and Risk for Cervical Cancer: a Case-Control Study. JNCI J. Natl. Cancer Inst. 2001, 93, 1325–1330, . [CrossRef]
- Hildesheim, A.; Herrero, R.; E Castle, P.; Wacholder, S.; Bratti, M.C.; E Sherman, M.; Lorincz, A.T.; Burk, R.D.; Morales, J.; Rodriguez, A.C.; et al. HPV co-factors related to the development of cervical cancer: results from a population-based study in Costa Rica. Br. J. Cancer 2001, 84, 1219–1226, . [CrossRef]
- Xi, L.F.; Koutsky, L.A.; Hildesheim, A.; Galloway, D.A.; Wheeler, C.M.; Winer, R.L.; Ho, J.; Kiviat, N.B. Risk for High-Grade Cervical Intraepithelial Neoplasia Associated with Variants of Human Papillomavirus Types 16 and 18. Cancer Epidemiology Biomarkers Prev. 2007, 16, 4–10, . [CrossRef]
- Chen, Z., et al., K-Mer Analyses Reveal Different Evolutionary Histories of Alpha, Beta, and Gamma Papillomaviruses. Int J Mol Sci, 2021. 22(17).
- Badal, S.; Badal, V.; E Calleja-Macias, I.; Kalantari, M.; Chuang, L.S.; Li, B.F.; Bernard, H.-U. The human papillomavirus-18 genome is efficiently targeted by cellular DNA methylation. Virology 2004, 324, 483–492, . [CrossRef]
- Mirabello, L.; Sun, C.; Ghosh, A.; Rodriguez, A.C.; Schiffman, M.; Wentzensen, N.; Hildesheim, A.; Herrero, R.; Wacholder, S.; Lorincz, A.; et al. Methylation of Human Papillomavirus Type 16 Genome and Risk of Cervical Precancer in a Costa Rican Population. JNCI J. Natl. Cancer Inst. 2012, 104, 556–565, . [CrossRef]
- Ekanayake Weeramange, C.E.; Tang, K.D.; Vasani, S.; Langton-Lockton, J.; Kenny, L.; Punyadeera, C. DNA Methylation Changes in Human Papillomavirus-Driven Head and Neck Cancers. Cells 2020, 9, doi:10.3390/cells9061359.
- Wentzensen, N.; Sun, C.; Ghosh, A.; Kinney, W.; Mirabello, L.; Wacholder, S.; Shaber, R.; LaMere, B.; Clarke, M.; Lorincz, A.T.; et al. Methylation of HPV18, HPV31, and HPV45 Genomes and Cervical Intraepithelial Neoplasia Grade 3. JNCI J. Natl. Cancer Inst. 2012, 104, 1738–1749, . [CrossRef]
- Upadhyay, M.; Samal, J.; Kandpal, M.; Vasaikar, S.; Biswas, B.; Gomes, J.; Vivekanandan, P. CpG Dinucleotide Frequencies Reveal the Role of Host Methylation Capabilities in Parvovirus Evolution. J. Virol. 2013, 87, 13816–13824, . [CrossRef]
- Xi, L.F.; Jiang, M.; Shen, Z.; Hulbert, A.; Zhou, X.-H.; Lin, Y.-Y.; Kiviat, N.B.; Koutsky, L.A. Inverse Association between Methylation of Human Papillomavirus Type 16 DNA and Risk of Cervical Intraepithelial Neoplasia Grades 2 or 3. PLOS ONE 2011, 6, e23897, . [CrossRef]
- Piyathilake, C.J.; Macaluso, M.; Alvarez, R.D.; Chen, M.; Badiga, S.; Edberg, J.C.; Partridge, E.E.; Johanning, G.L. A higher degree of methylation of the HPV 16 E6 gene is associated with a lower likelihood of being diagnosed with cervical intraepithelial neoplasia. Cancer 2010, 117, 957–963, . [CrossRef]
- Clarke, M.A.; Wentzensen, N.; Mirabello, L.; Ghosh, A.; Wacholder, S.; Harari, A.; Lorincz, A.; Schiffman, M.; Burk, R.D. Human Papillomavirus DNA Methylation as a Potential Biomarker for Cervical Cancer. Cancer Epidemiology Biomarkers Prev. 2012, 21, 2125–2137, . [CrossRef]
- Feng, C.; Dong, J.; Chang, W.; Cui, M.; Xu, T. The Progress of Methylation Regulation in Gene Expression of Cervical Cancer. Int. J. Genom. 2018, 2018, 8260652, . [CrossRef]
- Vartanian, J.-P.; Guétard, D.; Henry, M.; Wain-Hobson, S. Evidence for Editing of Human Papillomavirus DNA by APOBEC3 in Benign and Precancerous Lesions. Science 2008, 320, 230–233, . [CrossRef]
- Warren, C.J.; Westrich, J.A.; Van Doorslaer, K.; Pyeon, D. Roles of APOBEC3A and APOBEC3B in Human Papillomavirus Infection and Disease Progression. Viruses 2017, 9, 233, . [CrossRef]
- Poulain, F.; Lejeune, N.; Willemart, K.; Gillet, N.A. Footprint of the host restriction factors APOBEC3 on the genome of human viruses. PLOS Pathog. 2020, 16, e1008718, . [CrossRef]
- Xu, W.K.; Byun, H.; Dudley, J.P. The Role of APOBECs in Viral Replication. Microorganisms 2020, 8, 1899, . [CrossRef]
- A Burgers, W.; Blanchon, L.; Pradhan, S.; de Launoit, Y.; Kouzarides, T.; Fuks, F. Viral oncoproteins target the DNA methyltransferases. Oncogene 2007, 26, 1650–1655, . [CrossRef]
- Au Yeung, C.L.; Tsang, W.P.; Tsang, T.Y.; Co, N.N.; Yau, P.L.; Kwok, T.T. HPV-16 E6 upregulation of DNMT1 through repression of tumor suppressor p53.. Oncol. Rep. 2010, 24, 1599–1604, . [CrossRef]
- Sen, P.; Ganguly, P.; Ganguly, N. Modulation of DNA methylation by human papillomavirus E6 and E7 oncoproteins in cervical cancer (Review). Oncol. Lett. 2018, 15, 11–22, . [CrossRef]
- Wallace, N.A.; Münger, K. The curious case of APOBEC3 activation by cancer-associated human papillomaviruses. PLOS Pathog. 2018, 14, e1006717, . [CrossRef]
- Zhe, X.; Xin, H.; Pan, Z.; Jin, F.; Zheng, W.; Li, H.; Li, D.; Cao, D.; Li, Y.; Zhang, C.; et al. Genetic variations in E6, E7 and the long control region of human papillomavirus type 16 among patients with cervical lesions in Xinjiang, China. Cancer Cell Int. 2019, 19, 65, . [CrossRef]
- Woo, P.C.; Wong, B.H.; Huang, Y.; Lau, S.K.; Yuen, K.-Y. Cytosine deamination and selection of CpG suppressed clones are the two major independent biological forces that shape codon usage bias in coronaviruses. Virology 2007, 369, 431–442, . [CrossRef]
- Van Doorslaer, K. Evolution of the Papillomaviridae. Virology 2013, 445, 11–20, . [CrossRef]
- Liu, P.; Qiu, Y.; Xing, C.; Zhou, J.-H.; Yang, W.-H.; Wang, Q.; Li, J.-Y.; Han, X.; Zhang, Y.-Z.; Ge, X.-Y. Detection and genome characterization of two novel papillomaviruses and a novel polyomavirus in tree shrew (Tupaia belangeri chinensis) in China. Virol. J. 2019, 16, 35, . [CrossRef]
- Latsuzbaia, A.; Wienecke-Baldacchino, A.; Tapp, J.; Arbyn, M.; Karabegović, I.; Chen, Z.; Fischer, M.; Mühlschlegel, F.; Weyers, S.; Pesch, P.; et al. Characterization and Diversity of 243 Complete Human Papillomavirus Genomes in Cervical Swabs Using Next Generation Sequencing. Viruses 2020, 12, 1437, . [CrossRef]
- Lou, H.; Boland, J.F.; Torres-Gonzalez, E.; Albanez, A.; Zhou, W.; Steinberg, M.K.; Diaw, L.; Mitchell, J.; Roberson, D.; Cullen, M.; et al. The D2 and D3 Sublineages of Human Papilloma Virus 16–Positive Cervical Cancer in Guatemala Differ in Integration Rate and Age of Diagnosis. Cancer Res 2020, 80, 3803–3809, . [CrossRef]
- Liu, W.; Li, J.; Du, H.; Ou, Z. Mutation Profiles, Glycosylation Site Distribution and Codon Usage Bias of Human Papillomavirus Type 16. Viruses 2021, 13, 1281, . [CrossRef]
- Tlučková, K.; Marušič, M.; Tóthová, P.; Bauer, L.; Šket, P.; Plavec, J.; Viglasky, V. Human Papillomavirus G-Quadruplexes. Biochemistry 2013, 52, 7207–7216, . [CrossRef]
- Cadet, J.; Douki, T.; Ravanat, J.-L. One-electron oxidation of DNA and inflammation processes. Nat. Chem. Biol. 2006, 2, 348–349, . [CrossRef]
- Krieg, A.M.; Wu, T.; Weeratna, R.; Efler, S.M.; Love-Homan, L.; Yang, L.; Yi, A.-K.; Short, D.; Davis, H.L. Sequence motifs in adenoviral DNA block immune activation by stimulatory CpG motifs. Proc. Natl. Acad. Sci. 1998, 95, 12631–12636, . [CrossRef]
- Miller, L.S. Toll-Like Receptors in Skin. Adv. Dermatol. 2008, 24, 71–87, . [CrossRef]
- Hasan, U.A.; Zannetti, C.; Parroche, P.; Goutagny, N.; Malfroy, M.; Roblot, G.; Carreira, C.; Hussain, I.; Müller, M.; Taylor-Papadimitriou, J.; et al. The Human papillomavirus type 16 E7 oncoprotein induces a transcriptional repressor complex on the Toll-like receptor 9 promoter. J. Exp. Med. 2013, 210, 1369–1387, . [CrossRef]
- Pohar, J.; Yamamoto, C.; Fukui, R.; Cajnko, M.-M.; Miyake, K.; Jerala, R.; Benčina, M. Selectivity of Human TLR9 for Double CpG Motifs and Implications for the Recognition of Genomic DNA. J. Immunol. 2017, 198, 2093–2104, . [CrossRef]
- Cannella, F.; Pierangeli, A.; Scagnolari, C.; Cacciotti, G.; Tranquilli, G.; Stentella, P.; Recine, N.; Antonelli, G. TLR9 is expressed in human papillomavirus-positive cervical cells and is overexpressed in persistent infections. Immunobiology 2015, 220, 363–368, . [CrossRef]
- King, K.; Larsen, B.B.; Gryseels, S.; Richet, C.; Kraberger, S.; Jackson, R.; Worobey, M.; Harrison, J.S.; Varsani, A.; Van Doorslaer, K. Coevolutionary Analysis Implicates Toll-Like Receptor 9 in Papillomavirus Restriction. mBio 2022, 13, e0005422, . [CrossRef]
- Rogers, A.; Waltke, M.; Angeletti, P.C. Evolutionary variation of papillomavirus E2 protein and E2 binding sites. Virol. J. 2011, 8, 379–379, . [CrossRef]
- Newhouse, C.D.; Silverstein, S.J. Orientation of a Novel DNA Binding Site Affects Human Papillomavirus-Mediated Transcription and Replication. J. Virol. 2001, 75, 1722–1735, . [CrossRef]
- Gao, R.; Kim, C.; Sei, E.; Foukakis, T.; Crosetto, N.; Chan, L.-K.; Srinivasan, M.; Zhang, H.; Meric-Bernstam, F.; Navin, N. Nanogrid single-nucleus RNA sequencing reveals phenotypic diversity in breast cancer. Nat. Commun. 2017, 8, 228, . [CrossRef]
- Ganapathiraju, M.K.; Subramanian, S.; Chaparala, S.; Karunakaran, K.B. A reference catalog of DNA palindromes in the human genome and their variations in 1000 Genomes. Hum. Genome Var. 2020, 7, 40, . [CrossRef]
Figure 1.
Relatedness of each Alphapapillomavirus reference type inferred from maximum likelihood (ML) phylogenetic analysis of aligned nucleot ides in codons that are free to vary excluding those in overlapping reading frames (A), as well as UPGMA analysis of unaligned genomes for trimer composition (B), codon usage (C) and base composition (D). Branch lengths are proportional to amount of change. The internal branch uniting the high risk (HR) clade is denoted with an asterisk (*). HR HPV types in the Alpha5/6/7/9/11 clade are colored pink, brown, orange, red, and gold, respectively; low-risk (LR) HPV type groups 1 and 2 are shown in blue and green, respectively; non-human primate HPV types in Alpha12 are shown in grey.
Figure 1.
Relatedness of each Alphapapillomavirus reference type inferred from maximum likelihood (ML) phylogenetic analysis of aligned nucleot ides in codons that are free to vary excluding those in overlapping reading frames (A), as well as UPGMA analysis of unaligned genomes for trimer composition (B), codon usage (C) and base composition (D). Branch lengths are proportional to amount of change. The internal branch uniting the high risk (HR) clade is denoted with an asterisk (*). HR HPV types in the Alpha5/6/7/9/11 clade are colored pink, brown, orange, red, and gold, respectively; low-risk (LR) HPV type groups 1 and 2 are shown in blue and green, respectively; non-human primate HPV types in Alpha12 are shown in grey.
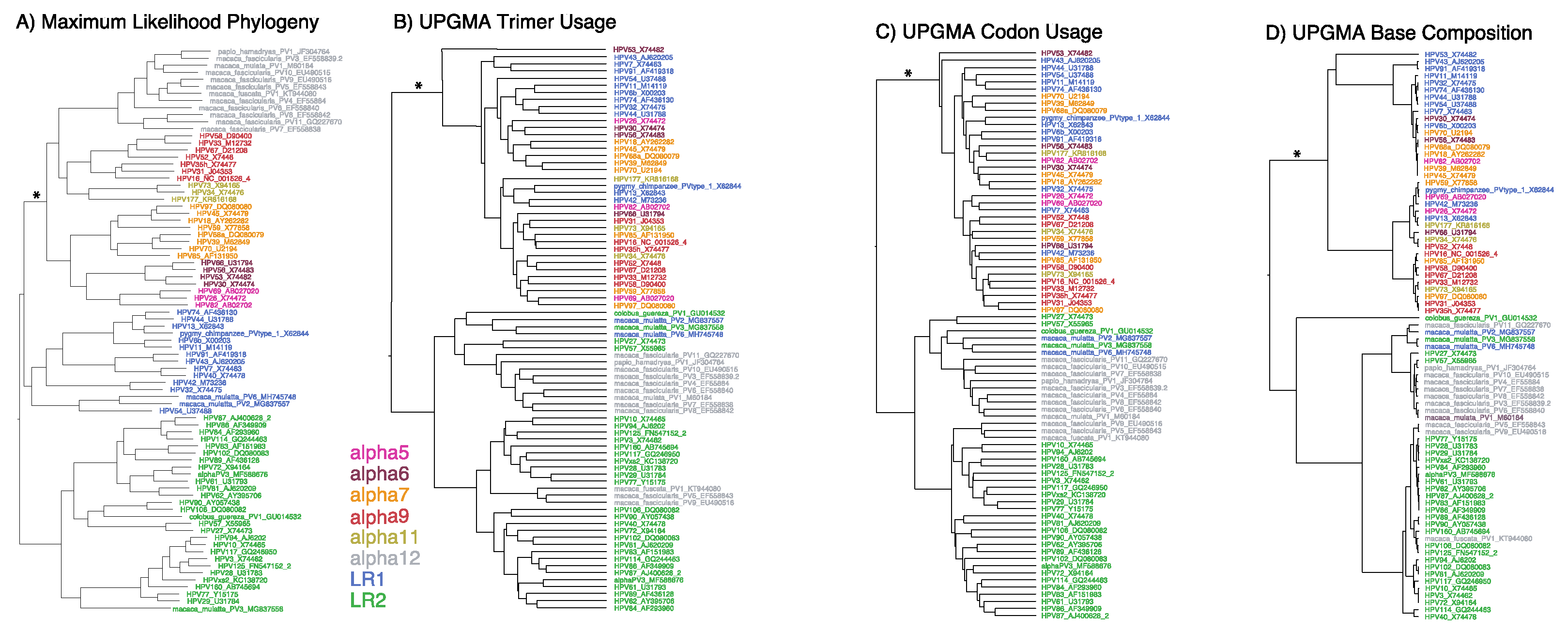
Figure 2.
Sliding window analyses: (A) HPV16, deviation from expectation (dashed line) for each of genome trimer composition (3mer, purple), GC composition (GC, green), CpG number (CpG, red) and local number of APOBEC3 motifs (Apo3, grey). The 95% confidence limits (dotted lines) are depicted for trimers composition and GC content relative to genome-wide expectations (dashed line). Positions with significant clustering of CpG or APOBEC3 sites are marked with asterisks. Promoter and polyadenylation sites are marked with arrows. (B) Pooled across all 83 Alphapapillomavirus reference types, regions exhibiting significant clustering of CpG (solid circles) and APOBEC3 (open circles) sites. Genome position was standardized against the first codon of E6 in HPV16. Data from HPV Alphapapillomavirus types are in red. Data from non-human-primate-specific Alphapapillomavirus types are in grey. Red lines indicate expected cumulative distributions should sites be randomly distributed lacking clustering (solid, on the abscissa) and should sites exhibit random clustering (dashed).
Figure 2.
Sliding window analyses: (A) HPV16, deviation from expectation (dashed line) for each of genome trimer composition (3mer, purple), GC composition (GC, green), CpG number (CpG, red) and local number of APOBEC3 motifs (Apo3, grey). The 95% confidence limits (dotted lines) are depicted for trimers composition and GC content relative to genome-wide expectations (dashed line). Positions with significant clustering of CpG or APOBEC3 sites are marked with asterisks. Promoter and polyadenylation sites are marked with arrows. (B) Pooled across all 83 Alphapapillomavirus reference types, regions exhibiting significant clustering of CpG (solid circles) and APOBEC3 (open circles) sites. Genome position was standardized against the first codon of E6 in HPV16. Data from HPV Alphapapillomavirus types are in red. Data from non-human-primate-specific Alphapapillomavirus types are in grey. Red lines indicate expected cumulative distributions should sites be randomly distributed lacking clustering (solid, on the abscissa) and should sites exhibit random clustering (dashed).
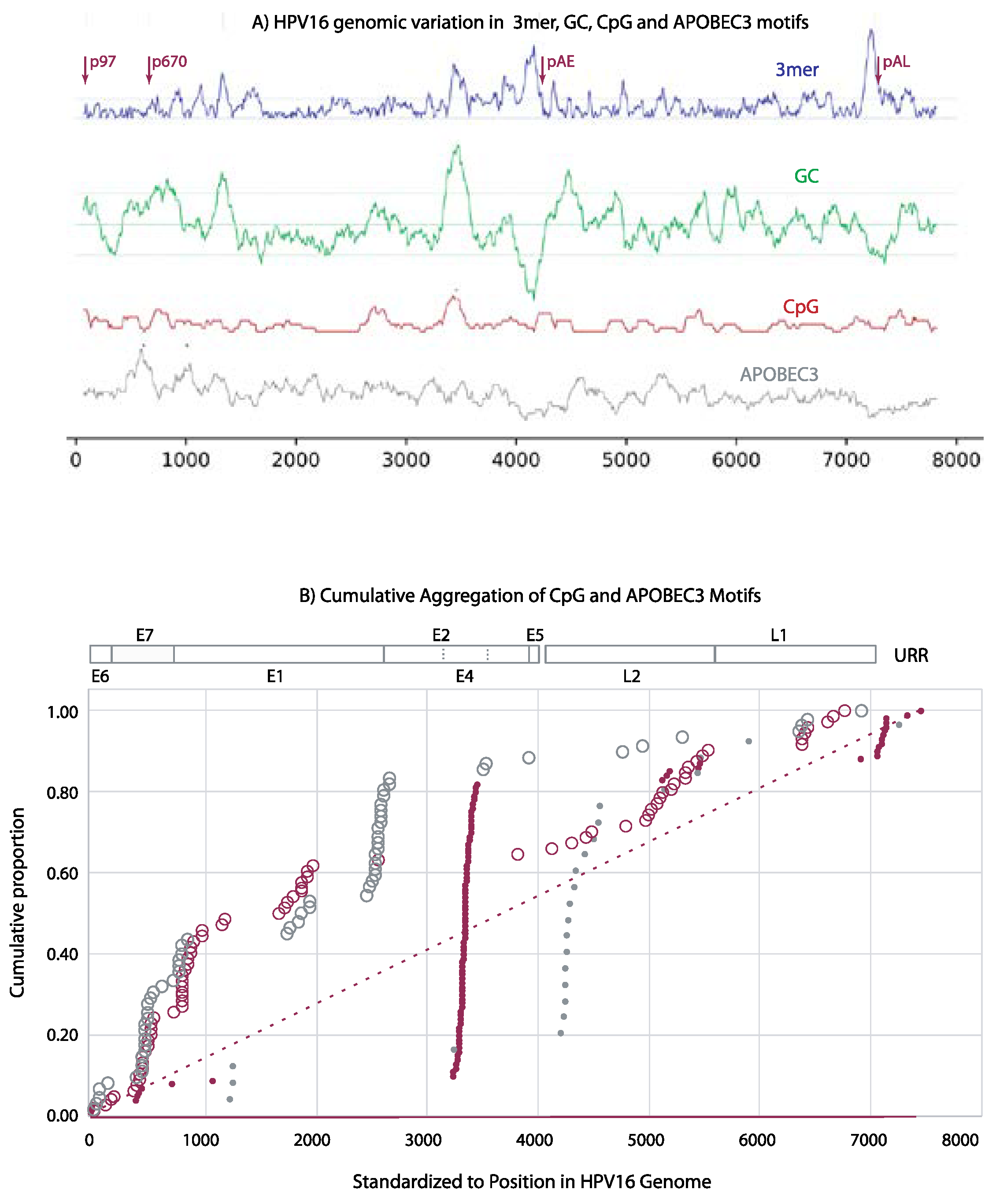
Figure 3.
Traits evolving under a Brownian motion drift model that readily distinguish recently diverged (asexual, nonrecombining) viral types (t=1) are expected to broaden their distributions through random walk (t=2) resulting in overlapping distributions for more anciently diverged clades (t=3). In contrast, the imposition of a common selective force will draw at least some unrelated lineages (light blue and light green lineages in B) to a more narrow, distinct and coincident region of the parameter space.
Figure 3.
Traits evolving under a Brownian motion drift model that readily distinguish recently diverged (asexual, nonrecombining) viral types (t=1) are expected to broaden their distributions through random walk (t=2) resulting in overlapping distributions for more anciently diverged clades (t=3). In contrast, the imposition of a common selective force will draw at least some unrelated lineages (light blue and light green lineages in B) to a more narrow, distinct and coincident region of the parameter space.
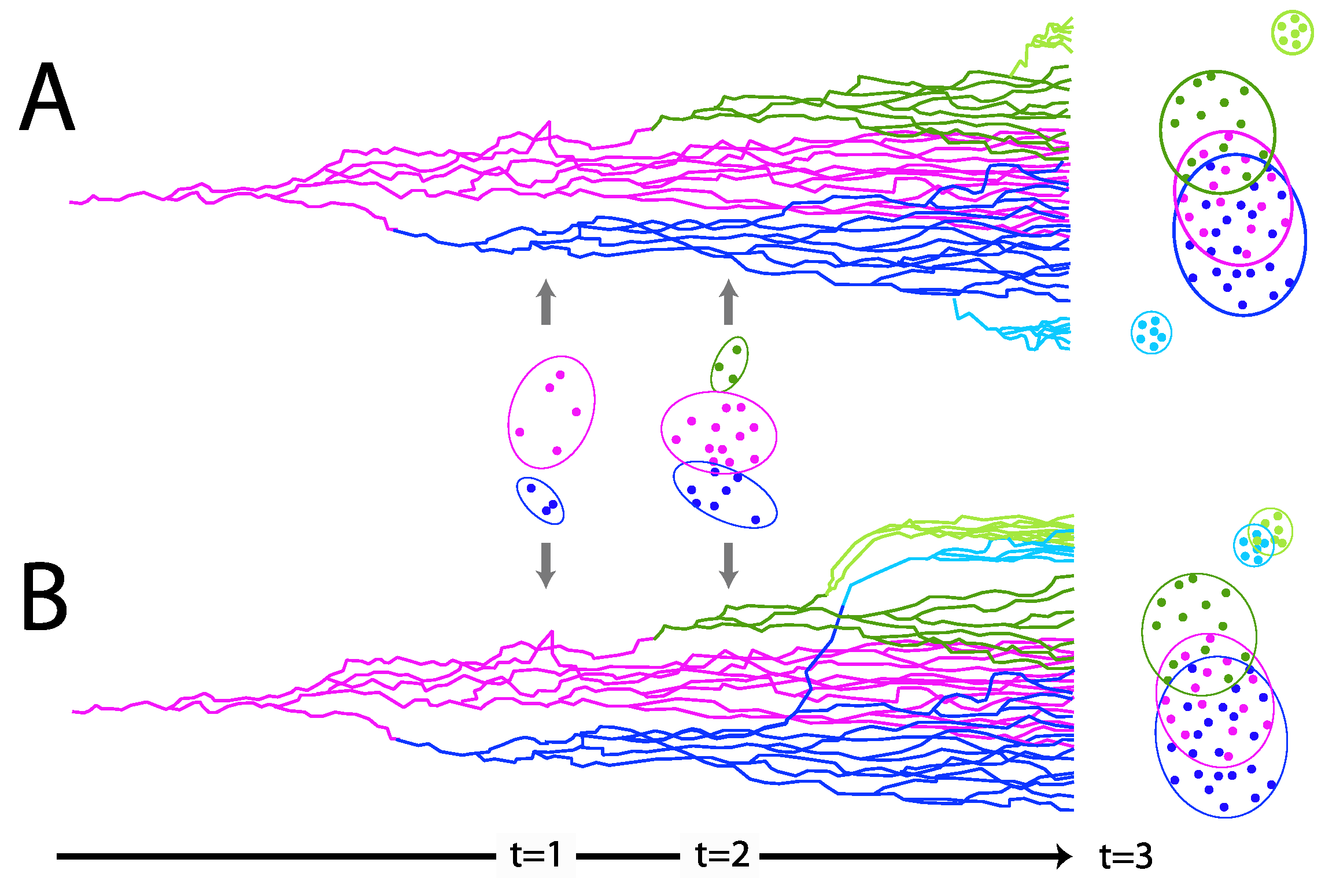
Figure 4.
Plot of second versus first principal components for 83 Alphapapillomavirus reference type genomes summarizing variation across 13 DNA sequence motifs (A), and summarizing variation for base compositions alone (B). Each point represents an Alphapapillomavirus type genome. Colors correspond to Alphapapillomavirus species as in Figure 1. Ellipses circumscribe each of the two Low Risk (LR1 and LR2), High Risk (HR) and Non-Human-Primate-infecting (NHP) groups. Percentages of variation explained by each principal component are in parentheses.
Figure 4.
Plot of second versus first principal components for 83 Alphapapillomavirus reference type genomes summarizing variation across 13 DNA sequence motifs (A), and summarizing variation for base compositions alone (B). Each point represents an Alphapapillomavirus type genome. Colors correspond to Alphapapillomavirus species as in Figure 1. Ellipses circumscribe each of the two Low Risk (LR1 and LR2), High Risk (HR) and Non-Human-Primate-infecting (NHP) groups. Percentages of variation explained by each principal component are in parentheses.
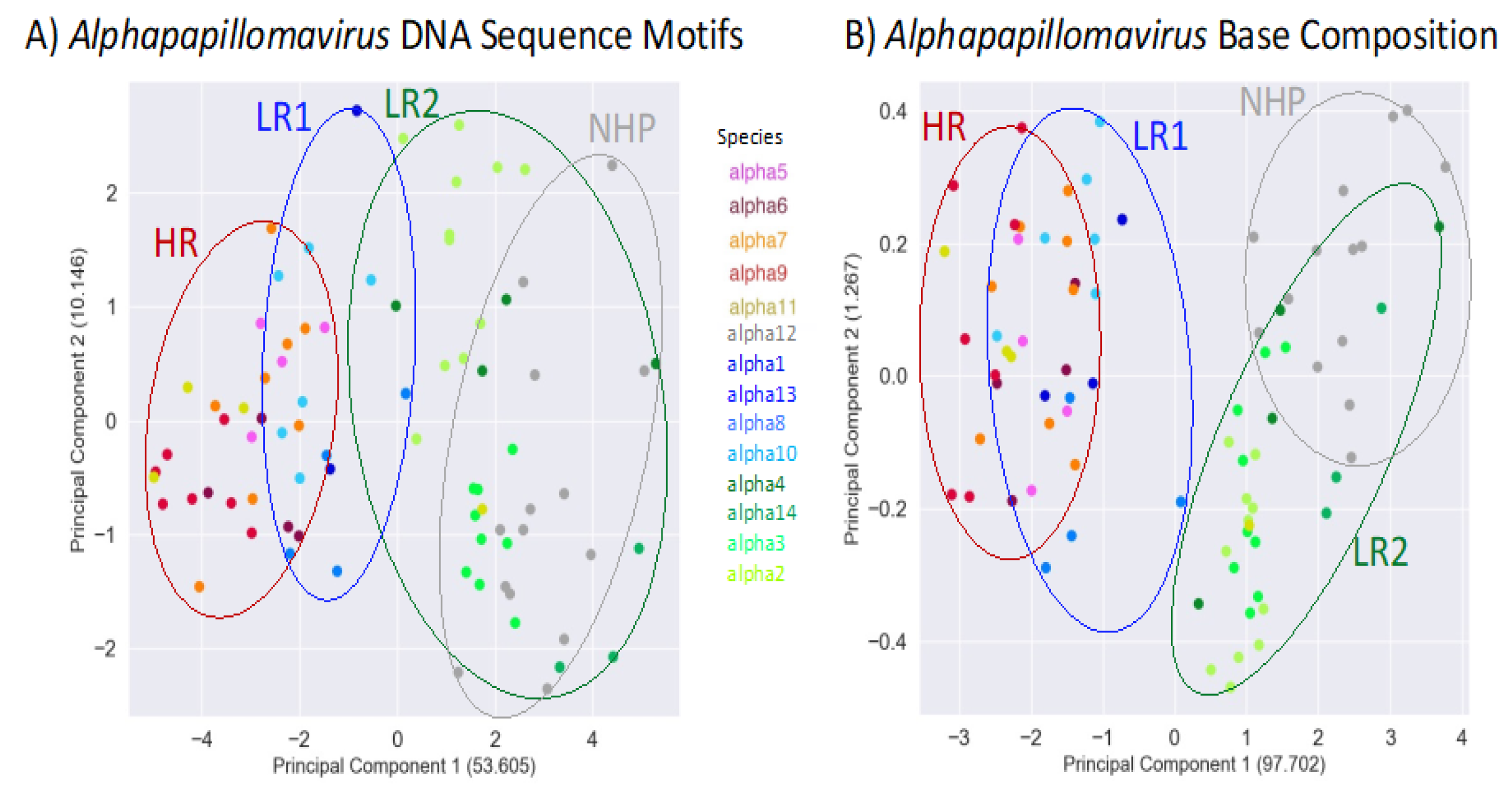
Figure 5.
Plot of second versus first principal components for lineage variants covering 7 viral types and their variants in the Alphapapillomavirus 9 species summarizing variation across 13 DNA sequence motifs (A), and summarizing variation for base compositions alone (B). Each point represents one of 1278 genomes. Colors correspond to HPV type and subtype as indicated on the phylogenetic tree (at right). Percentages of variation explained by each principal component are in parentheses.
Figure 5.
Plot of second versus first principal components for lineage variants covering 7 viral types and their variants in the Alphapapillomavirus 9 species summarizing variation across 13 DNA sequence motifs (A), and summarizing variation for base compositions alone (B). Each point represents one of 1278 genomes. Colors correspond to HPV type and subtype as indicated on the phylogenetic tree (at right). Percentages of variation explained by each principal component are in parentheses.
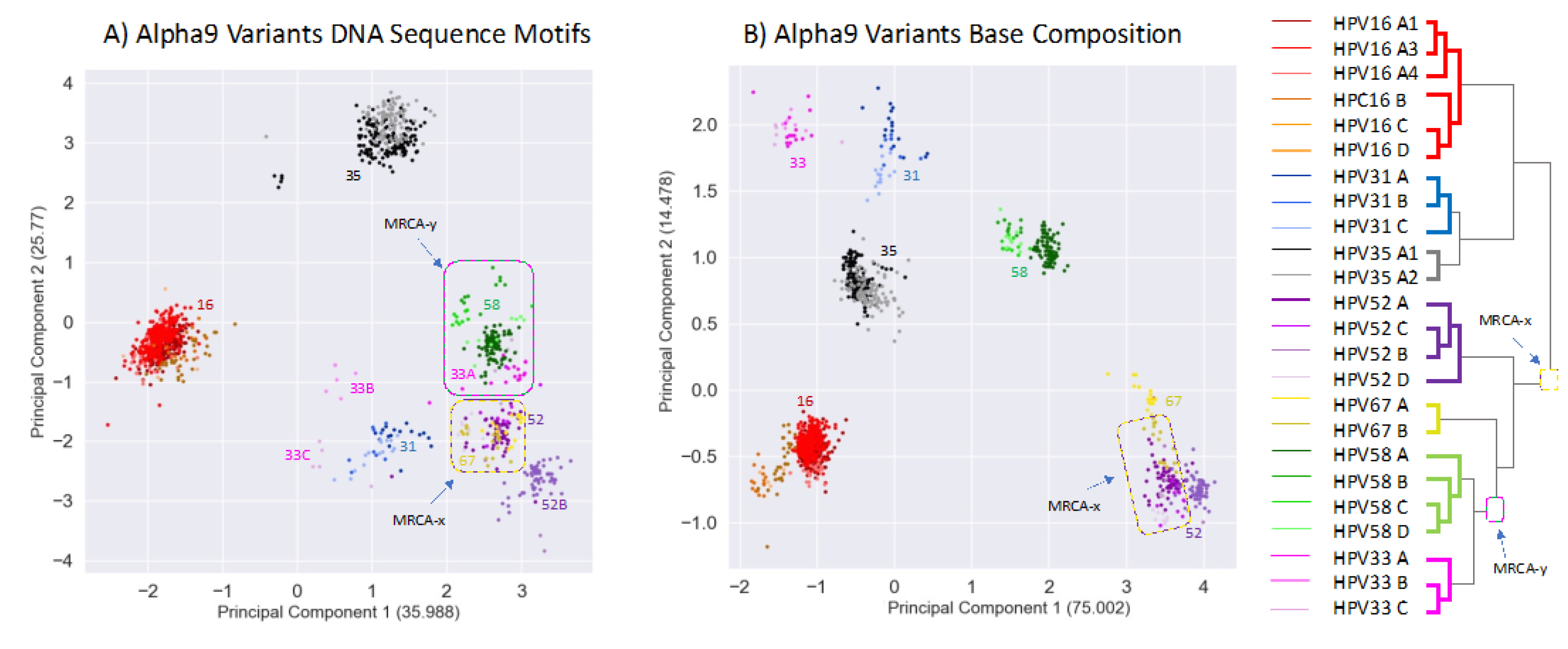
Figure 6.
Significant changes in ancestral states reconstructed on the ML tree for number of CpG sites (CpG), number of APOBEC3 sites (Apo3), number of Toll-like receptor 9 stimulating motifs (TLR) and the combined number of inverted repeats and perfect palindromes (IRP). Colored lines illustrates the ancestral paths from the most recent common ancestor (MRCA) of Alphapapillomavirus to the HPV type with the most extreme number of CpG sites (blue), number of APOBEC3 sites (magenta), number of Toll-like receptor 9 stimulating motifs (brown) and the combined number of inverted repeats and perfect palindromes (olive). Branch lengths are proportional to total genomic nucleotide change.
Figure 6.
Significant changes in ancestral states reconstructed on the ML tree for number of CpG sites (CpG), number of APOBEC3 sites (Apo3), number of Toll-like receptor 9 stimulating motifs (TLR) and the combined number of inverted repeats and perfect palindromes (IRP). Colored lines illustrates the ancestral paths from the most recent common ancestor (MRCA) of Alphapapillomavirus to the HPV type with the most extreme number of CpG sites (blue), number of APOBEC3 sites (magenta), number of Toll-like receptor 9 stimulating motifs (brown) and the combined number of inverted repeats and perfect palindromes (olive). Branch lengths are proportional to total genomic nucleotide change.
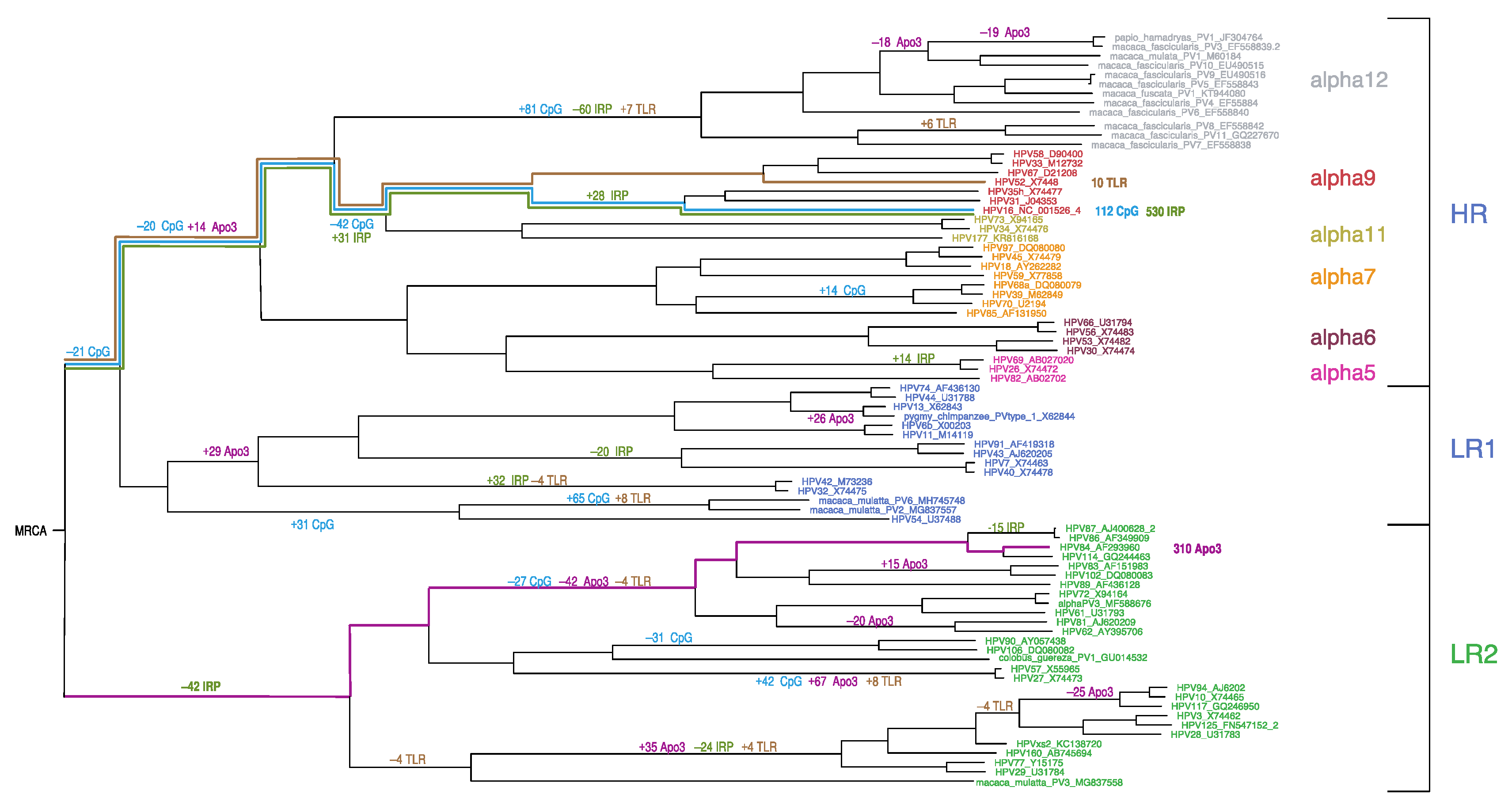
Figure 7.
Ancestral state variation across HPV types and variants in the Alphapapillomavirus 9 species contrasting methylation (CpG) with deamination (APOBEC3) sites (top), and inverted repeats with palindromes (bottom). The ancestral state inferred for each parameter is represented by a horizontal line. Each boxed group represents an HPV type in phylogenetic order with each point inside a box representing an HPV variant in alphabetical order (i.e., HPV16: A1/A2/A3/A4/B1/B2/B3/B4/C1/C2/C3/C4/D1/D2/D3/D4; HPV31: A1/A2/B1/B2/C1/C2/C3; HPV35: A1/A2; HPV52: A1/A2/B1/B2/B3/C1/C2/D1/E1; HPV67: A1/A2/B1; HPV33: A1/A2/A3/B1/C1; HPV58: A1/A2/A3/B1/B2/C1/D1/D2; HPV18: A1/A2/A3/A4/A5/B1/B2/B3/C; HPV45: A1/A2/A3/B1/B2).
Figure 7.
Ancestral state variation across HPV types and variants in the Alphapapillomavirus 9 species contrasting methylation (CpG) with deamination (APOBEC3) sites (top), and inverted repeats with palindromes (bottom). The ancestral state inferred for each parameter is represented by a horizontal line. Each boxed group represents an HPV type in phylogenetic order with each point inside a box representing an HPV variant in alphabetical order (i.e., HPV16: A1/A2/A3/A4/B1/B2/B3/B4/C1/C2/C3/C4/D1/D2/D3/D4; HPV31: A1/A2/B1/B2/C1/C2/C3; HPV35: A1/A2; HPV52: A1/A2/B1/B2/B3/C1/C2/D1/E1; HPV67: A1/A2/B1; HPV33: A1/A2/A3/B1/C1; HPV58: A1/A2/A3/B1/B2/C1/D1/D2; HPV18: A1/A2/A3/A4/A5/B1/B2/B3/C; HPV45: A1/A2/A3/B1/B2).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



