Submitted:
27 May 2023
Posted:
30 May 2023
You are already at the latest version
Abstract
Next Generation Sequencing (NGS), now widely used in the clinical setting, offers an efficient and comprehensive molecular approach for patients with Familial hypercholesterolemia (FH). Although the dominant form of disease is mostly due to low-density lipoprotein receptor (LDLR) small-scale pathogenic variants, approximately 10% of molecularly defined FH cases are due to Copy Number Variations (CNVs). Here, we report a novel large deletion of the LDLR gene involving exons 4–18, identified by bioinformatic analysis of NGS data in an Italian family. A long PCR strategy was employed for breakpoint region analysis where an insertion of 6 nucleotides (TTCACT) was found. Two Alu sequences, identified within intron 3 and exon 18, could underlie the identified rearrangement by a Nonallelic Homologous Recombination (NAHR) mechanism. NGS proves to be a powerful tool used to precisely identify CNVs, in addition to small-scale variants in FH-related genes. At this purpose, the use and implementation of this cost-effective, efficient molecular approach meets the clinical need for personalized diagnosis in FH cases.
Keywords:
Familial hypercholesterolemia
; Next Generation Sequencing
; LDL-cholesterol
; LDLR gene
; Copy number variations (CNVs)
; Alu sequences
1. Introduction
Familial hypercholesterolemia (FH) is a dominant hereditary metabolic disorder mostly caused by disease-causing variants in LDLR (low-density lipoprotein receptor), APOB (apolipoprotein B) or PCSK9 (proprotein convertase subtilisin/kexin type 9) genes. The respective prevalence of heterozygous FH (HeFH) and homozygous FH (HoFH) was initially assumed to be 1:500 and 1:1,000,000. Nowadays, it has been estimated that HeFH affects 1:313 individuals worldwide, while HoFH is still being an ultrarare condition (prevalence of 1:160–400,000) [1,2].
The well-known clinical hallmarks of FH are high levels of total and LDL cholesterol. Early detection of such cases and the prompt beginning of lipid-lowering treatment is crucial for ASCVD (Atherosclerotic Cardiovascular Disease) prevention, with the final aim of reduction of LDL cholesterol level below 55 mg/dL (1.42 mmol/L) or 70 mg/dL (1.81 mmol/L), depending on the specific risk (3).The most widely adopted diagnostic algorithms are Dutch Lipid Clinical Network Criteria (DLCN), Simon Broome (SB), and Making Early Diagnosis Prevents Early Death (MEDPED). However, regardless of the specific criteria, genetic testing is unequivocal and a key part of any clinical diagnosis [4]. Actually, after a critical assessment of the usefulness of FH molecular evaluation, the Familial Hypercholesterolemia Foundation recommended the use of genetic testing as standard of disease management in subjects with a definitive or suggestive FH diagnosis, and for their at-risk relatives [5,6].
The FH phenotype is mainly caused by loss-of-function variants in the LDLR (60–80% of patients) and APOB (5–10%) genes or by gain-of-function variants in PCSK9 gene (<1%). Rarely, patients harbor pathogenic variants in the APOE gene or in other genes (LDLRAP1, LIPA, SCAP) [7]. The prevalent types of mutation in FH-related genes, are missense, indels, and splicing but approximately 10% of mutated FH cases are due to Copy Number Variations (CNVs). CNVs are genomic structural variants that include deletions and duplications larger than 50 bp in size and their possible role in dyslipidemias is still being investigated. In the 2018, Iacocca et al. reported about 56 unique deletions and 27 unique duplications in the LDLR gene [8]. This finding is probably related to the presence of 98 Alu repeats within the LDLR gene: 95 are intronic and three are within the 3’ untranslated region (UTR) (9). LDLR is hence especially susceptible to CNV rearrangements with breakpoints mostly located within the introns, leading to whole-exon deletion events [9]. The gold standard for CNVs detection are MLPA (Multiplex Ligation-dependent Probe Amplification) or aCGH (array-based Comparative Genome Hybridization) techniques. However, also the use of bioinformatics prediction of CNVs events from Next Generation Sequencing (NGS) data is now a common laboratory practice.
In this study, we report an Italian patient, with a personal and familial history of hypercholesterolemia, carrier of a novel large-scale variant (exons 4_18 loss) in the LDLR gene.
2. Materials and Methods
2.1. Case presentation and family history
The proband is a 34-year-old Italian woman referred to our Center for Endocrine and Metabolic Diseases in November 2022. Her medical history began when she was 6-year-old (1994), on routine blood tests that revealed a hypercholesterolemic status indicated by elevated plasma lipid values corresponding to Total Cholesterol (TC) 374 mg/dl, High Density Lipoprotein (HDL) 93 mg/dl, triglyceride (TGs) 281 mg/dl, Low Density Lipoprotein (LDL-C) 225 mg/dl as shown in Table 1.
She hasn’t been treated until age of 18-year-old when she started a treatment with a daily 10 mg dose of Ezetimibe. At 20-year-old, a low dose of statin was added to Ezetimibe by her endocrinologist. The treatment was soon interrupted due to the onset of intolerable muscle pain. Since then, she has been treated with Ezetimibe in monotherapy. In 2019, her lipid profile remained unchanged, with very high levels of TC and LDL-C, as showed in Table 1.
In 2021, she started treatment with the anti-PCSK-9 monoclonal antibody Alirocumab 75 mg every two weeks, then increased at 150 mg every two weeks that is still ongoing.
As reported in Table 1, despite the treatment with Alirocumab, her lipid profile is not yet at target levels, according to the ESC/EAS guidelines of 2016 and 2019 [3], therefore we decided to try again low-dose statin therapy adding Rosuvastatin 5 mg daily to Alirocumab and Ezetimibe.
On physical examination, she showed no tendon xanthoma or corneal arcus. She had a body mass index of 30 kg/m2 and no previous history of coronary heart disease; she was normotensive, non-diabetic and did not smoke. Since her lipid profile was strongly suggestive for FH and, based on DCLN diagnostic criteria for FH [10], the score reached by our patient was 12, a definite diagnosis of FH was made. For this reason and according to EAS/European Society of Cardiology (ESC) guidelines, we performed an ad hoc genetic evaluation as explained later in the text. As a matter of fact, the above-mentioned guidelines recommend genetic testing in presence of the following criteria: TC >_8 mmol/L (>310 mg/dL) without treatment in an adult or adult family member (or >95th percentile by age and gender for country); premature CHD in the patient or a family member; tendon xanthomas in the patient or a family member; or sudden premature cardiac death in a family member [3].
The family pedigree is reported in Figure 1. The proband’s paternal great-grandmother was the first family member known to suffer from hypercholesterolemia. She had five children, four sons and one daughter. Three of them died for acute myocardial infarction between the age of 37 and 50 years, among these, the proband’s grandfather who died at the age of 46 years.
The proband inherited the hypercholesterolemia from her father currently treated with Rosuvastatin 10 mg and Ezetimibe 10 mg daily. He has two siblings: one brother dead at the age of 46 years for myocardial infarction and one sister, still alive, treated with statin. Written informed consent was obtained before genetic test.
2.2. NGS and Large Genomic Rearrangement (LGR) Detection
Genomic DNA was isolated from peripheral blood using MagCore® Genomic DNA whole blood kit (RBC Bioscience, New Taipei City, Taiwan) on the automated MagCore® HF16Plus instrument (Diatech Lab Line, Jesi, Italy) following the manufacturer’s instructions. The quantitation of the extracted DNA was performed using the Qubit dsDNA BR fluorimetric assays (Life Technologies, Gaithersburg, MD, USA).
The 6-genes panel named Devyser FH NGS kit v2 (Devyser, Hagersten, Sweden) was used for the targeted NGS (tNGS) approach. The multi-gene panel covers the coding regions and splicing junctions of the following genes: LDLR (NM_000527.4), APOB (NM_000384.2), PCSK9 (NM_174936.3), APOE (NM_000041.3), LDLRAP1 (NM_015627.2), and STAP1 (NM_012108.3) and the sequence determination of the following polygenic single Single Nucleotide Polymorphisms (SNPs) related to FH and/or statin treatment response: rs629301, rs1564348, rs1800562, rs2479409, rs3757354, rs4299376, rs6511720, rs8017377, rs11220462, rs1367117, rs429358, rs7412, rs646776, rs4149056, rs3798220, rs10455872. NGS was performed in paired-ends reads mode (2X151) on the Illumina MiSeq® NGS platform (Illumina, San Diego, USA). FASTQ files were analysed by the CE-IVD Amplicon Suite Software (SmartSeq, Novara, Italy), as previuolsy reported [11].
The MLPA assay was performed as a confirmatory method of the new rearrangement. The SALSA MLPA kit for LDLR (P062; MRC Holland, Amsterdam, The Netherlands) was used according to the manufacturer’s instructions. Amplicons were run on an ABI 3500 Genetic Analyzer (Thermo Fisher Scientific, Foster City, CA, USA), and the collected data were analyzed using Coffalyser.NET Software (MRC Holland). Three healthy males and three healthy females were included in the analysis as wild-type controls.
2.3. Analysis of Breakpoint Region
To characterize the breakpoint region, deletion-specific PCR primers (DF 5′–ACCGCTGCATTCCTCAGTTCTG–3′ and DR 5′–AACCTGAAGTCCCGTCAAAC–3′) were designed. PCR reactions were performed using a long-range PCR kit (Expand Long Template PCR System, Roche Applied Science, Monza, Italy). The PCR product was sequenced using a BigDye Terminator Cycle Sequencing Kit v3.1 (Thermo Fisher Scientific) and an ABI 3500 Genetic Analyzer (Thermo Fisher Scientific). Internal primer sequence is available on request. Results were analyzed with the SeqScape v2.5 software package (Thermo Fisher Scientific) using NG_009060.1 as reference. The Repeat Masker program was employed to identify Alu sequences at breakpoint junctions [12].
3. Results
3.1. NGS Analysis and LRG Detection
No small-scale pathogenic variants were detected in the 6 FH-related genes investigated by the Devyser FH NGS kit. However, NGS CNV prediction analysis identified a large LDLR deletion, involving exons 4–18 (Figure 2a). This result was confirmed by performing the MLPA assay on a fresh DNA sample (Figure 2b). Successively, the proband’s father, screened by LDLR MLPA analysis, resulted carrier of the deletion.
3.2. Analysis of Breakpoint Region
A PCR fragment of 891 bp, containing the breakpoint region, showed a wild-type sequence until the nucleotide g.19163T (NG_009060.1) of LDLR intron 3. The following sequence showed an upstream insertion of 6 nucleotides (TTCACT) to the sequence corresponding to the LDLR exon 18 region starting from the g.48821A (NG_009060.1) nucleotide (Figure3a). According to HGVS nomenclature, we report the rearrangement as NG_009060.1:g.19164_48820delinsTTCACT. The Repeat Masker program identified two Alu sequences around breakpoint junctions: an AluSx within inton 3 and an AluJb in exon 18 of LDLR gene. Figure 3b shows the sequence homology near the breakpoint site of LDLR 4_18del.
4. Discussion
In this study, the molecular workflow allowing the diagnosis of a patient with a personal and family history of FH was reported. A tNGS-based approach with appropriate bioinformatics analysis detected a previously unreported large-scale deletion in the LDLR gene. The structural rearrangement identified involves a 29.6-kb region, causing the loss of exon 4 to exon 18 gene regions. In addition, by a long-PCR strategy, we also established the exact genomic coordinates of the deletion characterized by a 6-nucleotide insertion (TTCACT) at breakpoint site: NG_009060.1:g.19164_48820delinsTTCACT (Figure 3a). Since most of the functional domains of the protein are lost, this rearrangement is considered pathogenic, supporting the molecular diagnosis of HeFH in our patient.
The LDLR locus is localized on human chromosome 19 (19p13.1-3), that is the one presenting the highest content in Alu repeats (13,14). Moreover, Alu repeats represent 65% of LDLR intronic sequences, reaching 85% of genomic sequence outside exon-intron junctions [9]. Soon after the cloning of the LDLR gene, the first Southern blotting experiments revealed that Alu repeats would give rise to unequal meiosic cross-over and to large gene rearrangements causing FH [15]. Basically, these sequences offer many opportunities for homologous recombinations, and Nonallelic homologous recombination (NAHR) represents the most common mechanism underlying disease associated to genome rearrangements [9,16]. We identified two Alu sequences located around the breakpoint junctions, an AluSx within intron 3 and an AluJb in exon 18 of LDLR gene, supporting the hypothesis that a homologous recombination event could underlie the identified rearrangement. However, as reported in Figure 3b, a low homology was found close to the breakpoint site. In addition, an insertion of six nucleotide (TTCACT) characterizes the novel rearrangement (Figure 3).
This case report emphasizes how advances in genetic sequencing technology have resulted in remarkable improvements in speed, throughput, and identification of all causing disease defects in FH related genes [17]. To date, it is known that about 10% of the disease-causing variants consist of LDLR CNVs, so the ability to detect the full spectrum of LDLR variants is critical to achieving a definitive molecular diagnosis of FH. Although MLPA assay remains the reference method for assessment of CNVs, Iacocca et al. demonstrated that NGS data from FH patient have a 100% of concordance rate for large-scale LDLR CNV calling using MLPA as the "gold standard" reference method [8].
We emphasize that the accurate identification of CNVs from NGS data is a relevant topic, considering that not all the sequencing facilities and also the clinical laboratories have the resources, time, or interest to set up an ad hoc parallel MLPA workflow to detect them. In particular, using NGS data to detect CNVs would delete the cost for LDLR MLPA analysis, approximately $80 USD per sample. For this reason, it is important that all laboratories that already use NGS technology, or that are currently in the process of doing so, implement their workflow also to the NGS-based CNV evaluation. First of all, an internal validation of the CNV calling should be performed on previously genotyped samples, so that the sensitivity and specificity of the method can be calculated [18,19].
Furthermore, CNV analysis can be extended to all FH-associated genes as APOB, PCSK9, LDLRAP1, and APOE at no extra cost. Although causative CNVs in these genes are expected to be rare, they have long remained uninvestigated because MLPA methods are either not available or not applied for genes outside the LDLR gene. Extending CNV analysis to all these FH-associated genes will increase the ability to identify all genetic aberrations that can explain FH cases [20,21].
Finally, we believe that the progressive, and hopefully inexorable, consolidation of NGS as widespread method of choice in multiple diagnostics context, will have a strong impact on the knowledge of FH disease, enabling the development of public health approaches aimed primarily at early clinical diagnosis and treatment of FH. This will benefit all populations, even those little studied populations for which the genetics of FH is still unexplored and poorly understood.
5. Conclusions
This study showed that the integrated care model routinely adopted in our laboratory for FH molecular diagnosis led to a discovery of a novel LDLR 29.6-kilobase deletion in an Italian family, contributing to broaden the mutational LDLR landscape in our population.
NGS-based approach has the potential to identify large-scale variants in LDLR and can be further applied to extend CNV screening to other FH-related genes. Nevertheless, the outcomes from the bioinformatic approach still need to be confirmed by the MLPA method, and this remains the reference method for assessing CNV.
Author Contributions
Conceptualization, P.C., E.D.P., A.M; methodology, M.E.O; C.R.T.; M.D.B., M.R; software, P.C, C.R.; validation, A.M., C.S., S.C; investigation, L.S., C.R.; data curation, S.M. A.G., S.C.; writing—original draft preparation, P.C., E.D.P., A.M.; writing—review and editing, P.C, E.D.P., C.R.; supervision, A.G, A.U. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding
Institutional Review Board Statement
all procedures were in accordance with the ethical standards of the Ethics Committee of “Fondazione Policlinico Universitario A. Gemelli IRCCS” of Rome.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study. Written informed consent has been obtained from the patient(s) to publish this paper.
Data Availability Statement
Data are available from the Authors upon request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Beheshti, S.O. , Madsen C.M., Varbo A., Nordestgaard B.G.. Worldwide Prevalence of Familial Hypercholesterolemia: Meta-Analyses of 11 Million Subjects. J. Am. Coll. Cardiol. 2020;75:2553–2566. [CrossRef]
- Vallejo-Vaz, A.J. , Stevens C.A.T., Lyons A.R.M., Dharmayat K.I., Freiberger T., Hovingh G.K., Mata P., Raal F.J., Santos R.D., Soran H., et al. Global Perspective of Familial Hypercholesterolaemia: A Cross-Sectional Study from the EAS Familial Hypercholesterolaemia Studies Collaboration (FHSC) Lancet. 2021;398:1713–1725.
- Mach, F. , Baigent C., Catapano A.L., Koskinas K.C., Casula M., Badimon L., Chapman M.J., de Backer G.G., Delgado V., Ference B.A., et al. 2019 ESC/EAS Guidelines for the Management of Dyslipidaemias: Lipid Modification to Reduce Cardiovascular Risk. Eur. Heart J. 2020;41:111–188. [CrossRef]
- Haralambos, K. , Ashfield-Watt P., McDowell I.F. Diagnostic scoring for familial hypercholesterolaemia in practice. Curr Opin Lipidol. 2016 Aug;27(4):367-74. [CrossRef]
- Sturm A.C., Knowles J.W., Gidding S.S. et al Convened by the Familial Hypercholesterolemia Foundation. Clinical Genetic Testing for Familial Hypercholesterolemia: JACC Scientific Expert Panel. J Am Coll Cardiol. 2018 Aug 7;72(6):662-680. [CrossRef]
- Vallejo-Vaz A.J., De Marco M., Stevens C.A.T. et al Overview of the current status of familial hypercholesterolaemia care in over 60 countries - The EAS Familial Hypercholesterolaemia Studies Collaboration (FHSC). Atherosclerosis. 2018 Oct;277:234-255. [CrossRef]
- Moffa, S. , Onori M.E., De Paolis E., Ricciardi Tenore C., Perrucci A., Pontecorvi A., Giaccari A., Urbani A., Minucci A. A novel low-density lipoprotein receptor variant in a Ukrainian patient: a case report and overview of the disease-causing low-density lipoprotein receptor variants associated to familial hypercholesterolemia. Mol Biol Rep. 2022 Feb;49(2):1623-1630. [CrossRef]
- Iacocca, M.A. , Hegele R.A. Role of DNA Copy Number Variation in Dyslipidemias. Curr. Opin. Lipidol. 2018;29:125–132. [CrossRef]
- Amsellem, S. , Briffaut D., Carrié A., Rabés J.P., Girardet J.P., Fredenrich A., Moulin P., Krempf M., Reznik Y., Vialettes B., et al. Intronic Mutations Outside of Alu-Repeat-Rich Domains of the LDL Receptor Gene Are a Cause of Familial Hypercholesterolemia. Hum. Genet. 2002;111:501–510. [CrossRef]
- Defesche, J.C. , Lansberg P.J., Umans-Eckenhausen M.A.W. Advanced method for the identification of patients with inherited hypercholesterolemia. Semin Vasc Med. 2004 Feb;4(1):59-65. [CrossRef]
- Moffa, S. , Mazzuccato G., De Bonis M., De Paolis E., Onori M.E., Pontecorvi A., Urbani A., Giaccari A., Capoluongo E., Minucci A. Identification of two novel LDLR variants by Next Generation Sequencing. Ann Ist Super Sanita. 2020 Jan-Mar;56(1):122-127. [CrossRef]
- https://www. repeatmasker.org. Accessed 11 May 2023.
- Lander, E.S. , Linton L.M., Birren B., Nusbaum C., Zody M.C., et al. International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001 Feb 15;409(6822):860-921. [CrossRef]
- Venter, J.C. , Adams M.D., Myers E.W., Li P.W., Mural R.J., Sutton G.G., et al. The sequence of the human genome. Science. 2001 Feb 16;291(5507):1304-51. [CrossRef]
- Hobbs, H.H. , Russell D.W., Brown M.S., Goldstein J.L. The LDL receptor locus in familial hypercholesterolemia: mutational analysis of a membrane protein. Annu Rev Genet. 1990;24:133-70. [CrossRef]
- Goldmann, R. , Tichý L., Freiberger T., Zapletalová P., Letocha O., Soska V., Fajkus J., Fajkusová L. Genomic characterization of large rearrangements of the LDLR gene in Czech patients with familial hypercholesterolemia. BMC Med Genet. 2010 Jul 27;11:115. [CrossRef]
- Dron, J.S. , Wang J., McIntyre A.D., Iacocca M.A., Robinson J.F., Ban M.R., Cao H., Hegele R.A. Six years' experience with LipidSeq: clinical and research learnings from a hybrid, targeted sequencing panel for dyslipidemias. BMC Med Genomics. 2020 Feb 10;13(1):23. [CrossRef]
- Iacocca, M. A, Wang J., Dron J.S., Robinson J.F., McIntyre A.D., Cao H., Hegele R.A. Use of next-generation sequencing to detect LDLR gene copy number variation in familial hypercholesterolemia. J Lipid Res. 2017 Nov;58(11):2202-2209.
- Concolino, P. , Rizza R., Mignone F., Costella A., Guarino D., Carboni I., Capoluongo E., Santonocito C., Urbani A., Minucci A. A comprehensive BRCA1/2 NGS pipeline for an immediate Copy Number Variation (CNV) detection in breast and ovarian cancer molecular diagnosis. Clin Chim Acta. 2018 May;480:173-179. [CrossRef]
- Lazarte, J. , Berberich A.J, Wang J., Hegele R.A. A cautionary tale: Is this APOB whole-gene duplication actually pathogenic? J Clin Lipidol. 2020 Sep-Oct;14(5):631-63. [CrossRef]
- Iacocca, M.A. , Wang J., Sarkar S., Dron J.S., Lagace T., McIntyre A.D., Lau P., Robinson J.F., Yang P., et al. Whole-Gene Duplication of PCSK9 as a Novel Genetic Mechanism for Severe Familial Hypercholesterolemia. Can J Cardiol. 2018 Oct;34(10):1316-1324. [CrossRef]
Figure 1.
The figure illustrates the proband’s family tree. Molecular genetic diagnosis was performed only in the proband (indicated by the arrow) and her father. Several family members prematurely died for CVD (Cardio Vascular Disease).
Figure 1.
The figure illustrates the proband’s family tree. Molecular genetic diagnosis was performed only in the proband (indicated by the arrow) and her father. Several family members prematurely died for CVD (Cardio Vascular Disease).
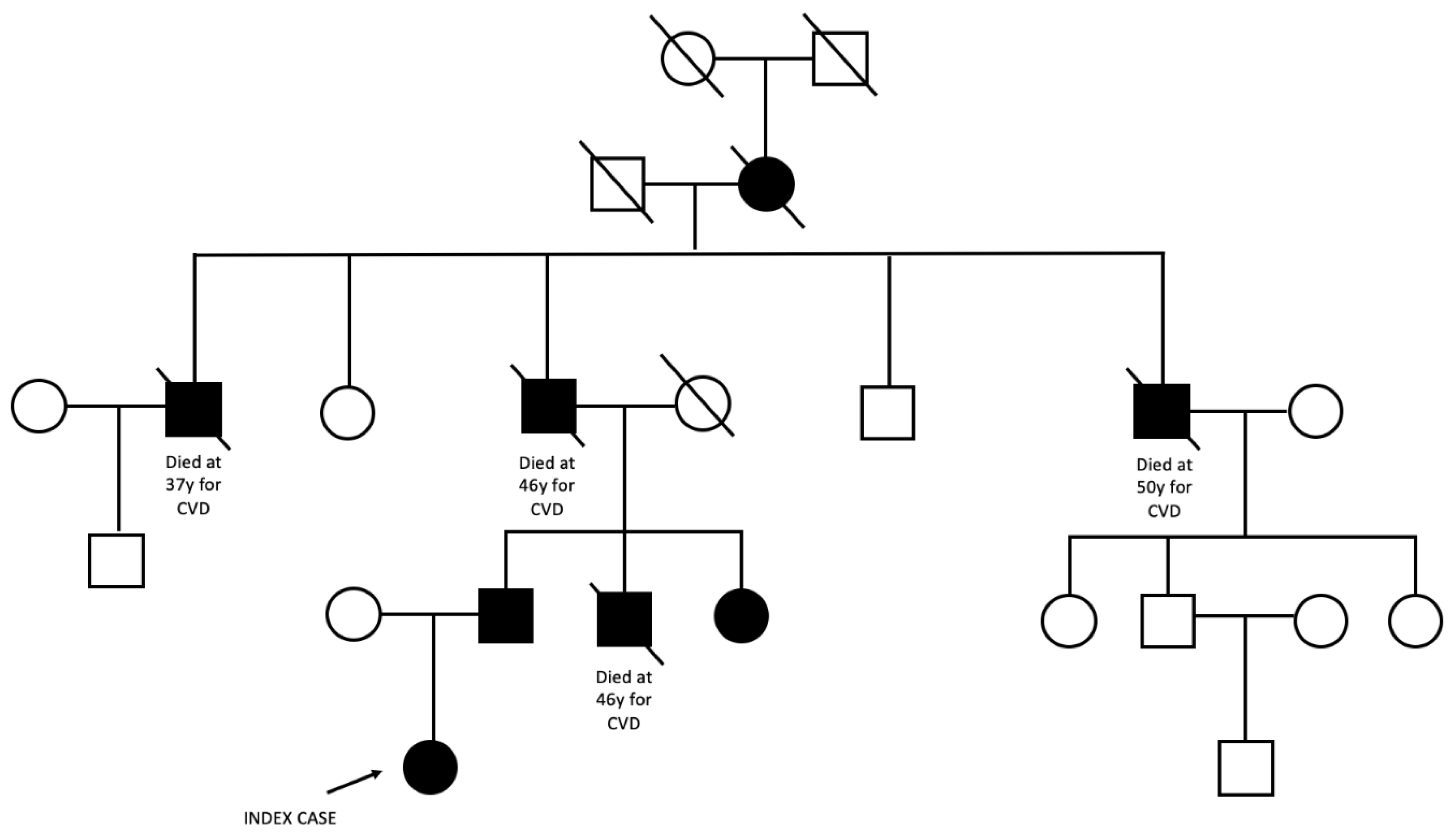
Figure 2.
Panel A: Bioinformatics CNVs prediction of LDLR gene performed by Amplicon Suite® tool. Copy Number plot of LDLR gene with indication of heterozygous deletion of exons 4-18. Panel B: MLPA results from comparative analysis experiment obtained by Coffalyser.NET Software (MRC Holland, Amsterdam, Netherlands). The final ratio of LDLR exons 4-18 probes was about 0.50 (normal range 0.80<FR<1.20), indicating a heterozygous deletion.
Figure 2.
Panel A: Bioinformatics CNVs prediction of LDLR gene performed by Amplicon Suite® tool. Copy Number plot of LDLR gene with indication of heterozygous deletion of exons 4-18. Panel B: MLPA results from comparative analysis experiment obtained by Coffalyser.NET Software (MRC Holland, Amsterdam, Netherlands). The final ratio of LDLR exons 4-18 probes was about 0.50 (normal range 0.80<FR<1.20), indicating a heterozygous deletion.
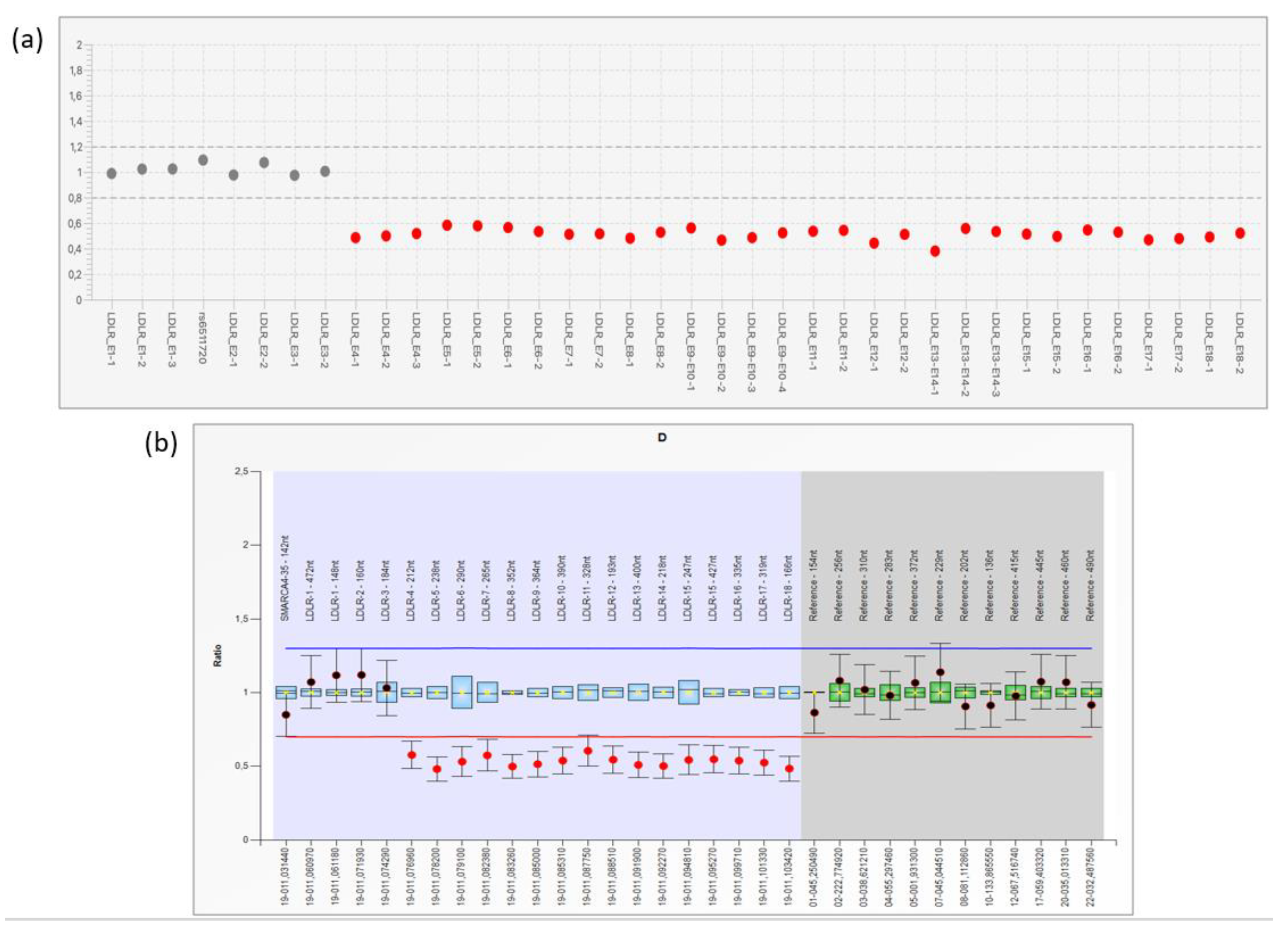
Figure 3.
Panel A: Schematic representation of the deletion of LDLR exon 4_18. The panel shows: the gene region spanning from exon 3 to exon 18, as well as the location of the Alu sequences (arrowed box) involved in the deletion; the gene region resulting from the deletion; the electropherogram of the breakpoint: LDLR NG_009060.1:g.19164_48820delinsTTCACT. The red boxes represent coding sequence, white boxes 3’UTR region and lines introns. Panel B: Sequence alignment of LDLR Intron 3 and LDLR 3’–UTR regions. The figure depicts the sequence homology near the breakpoint site of LDLR 4_18del.
Figure 3.
Panel A: Schematic representation of the deletion of LDLR exon 4_18. The panel shows: the gene region spanning from exon 3 to exon 18, as well as the location of the Alu sequences (arrowed box) involved in the deletion; the gene region resulting from the deletion; the electropherogram of the breakpoint: LDLR NG_009060.1:g.19164_48820delinsTTCACT. The red boxes represent coding sequence, white boxes 3’UTR region and lines introns. Panel B: Sequence alignment of LDLR Intron 3 and LDLR 3’–UTR regions. The figure depicts the sequence homology near the breakpoint site of LDLR 4_18del.
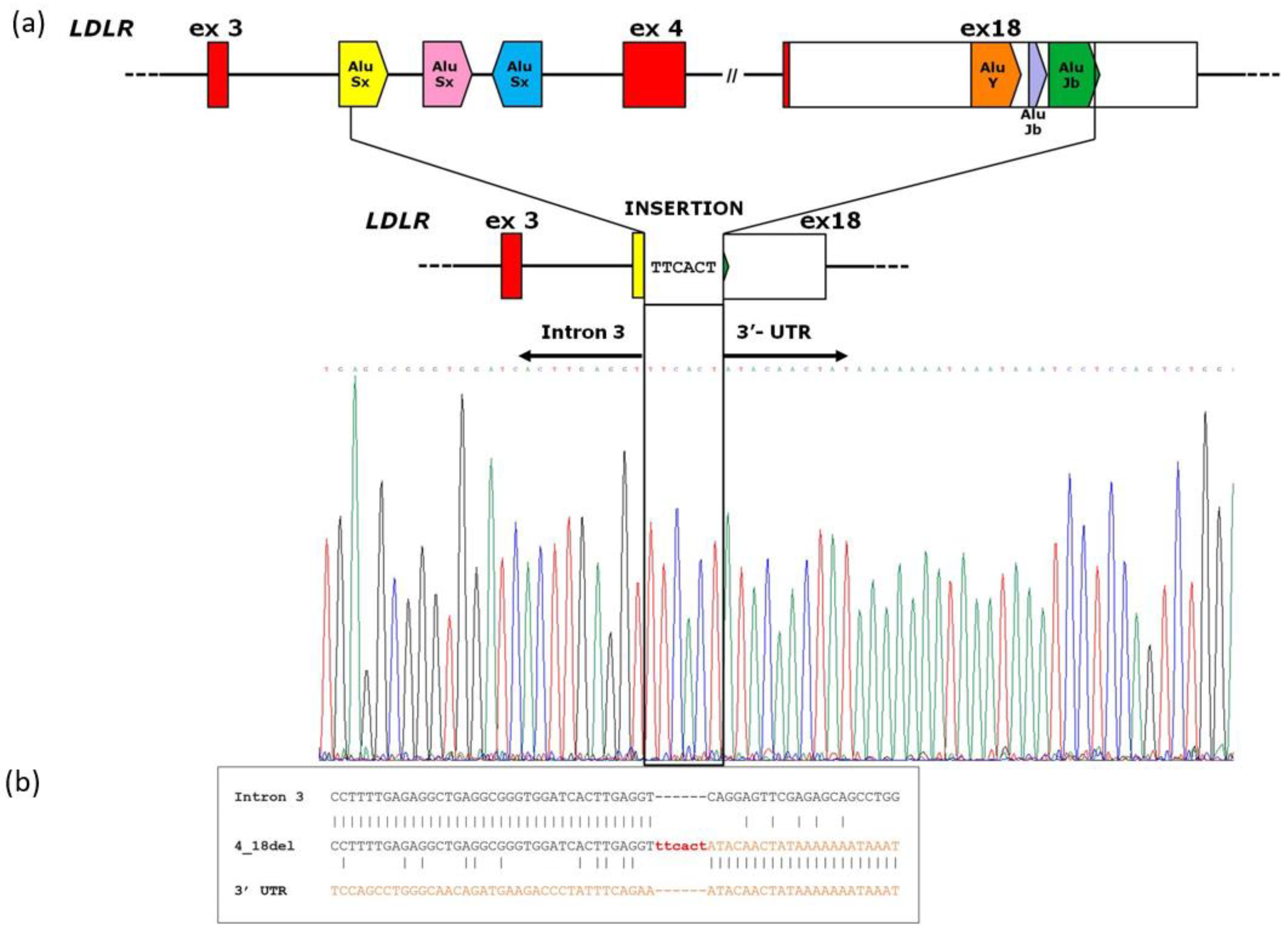

Table 1.
Lipid profiles of the proband. The first column shows the lipid profile off therapy, when she was 6-year old, the second one shows the results with Ezetimibe 10 mg therapy, and the last column indicates the lipid profile after adding Alirocumab 150 mg every two weeks.
Table 1.
Lipid profiles of the proband. The first column shows the lipid profile off therapy, when she was 6-year old, the second one shows the results with Ezetimibe 10 mg therapy, and the last column indicates the lipid profile after adding Alirocumab 150 mg every two weeks.
| Lipid Profile | Years | Normal range | ||
|---|---|---|---|---|
| 1994 | 2019 | 2022 | ||
| Total Cholesterol (mg/dl) | 374 | 357 | 321 | 130-200 mg/dl |
| HDL | 93 | 68 | 66 | female>45; male>40 |
| Triglycerides (mg/dl) | 281 | 124 | 95 | 20-170 mg/dl |
| LDL (mg/dl) | 225 | 264 | 236 | <130 mg/dl |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



