
Submitted:
25 May 2023
Posted:
26 May 2023
You are already at the latest version
Abstract
Toxic advanced glycation end-products (TAGE), formed by glyceraldehyde (GA) as an intermediate in the non-enzymatic reaction with intracellular proteins, are highly cytotoxic and have been implicated in the pathogenesis of various diseases. However, the mechanisms underlying the degradation and removal of TAGE remain largely unknown. In the present study, we identified the checkpoint kinase-1 (CHK1) mutant, d270KD, which was rapidly degraded intracellularly by GA, and showed that its degradation was mainly mediated by the ubiquitin-proteasome pathway. The high-molecular-weight complexes formed by the GA stimulation of d270KD were abundant in the RIPA-insoluble fraction, which also contained high levels of TAGE. The knockdown of p62/SQSTM1 reduced the amount of high-molecular-weight complexes in the RIPA-insoluble fraction, indicating its involvement in the formation of TAGE aggregates. The present results suggest that the ubiquitin-proteasome pathway and p62 play a role in the degradation and aggregation of intracellular TAGE formed by GA. This study provides new insights into the mechanisms underlying TAGE metabolism and may lead to the development of novel therapeutic strategies for diseases associated with TAGE accumulation.
Keywords:
advanced glycation end-products (AGEs)
; glyceraldehyde (GA)
; glyceraldehyde-derived AGEs
; toxic-AGEs (TAGE)
; p62/SQSTM1
1. Introduction
Sugars are a vital source of energy and essential nutrients for living organisms. However, modifications to the lysine and arginine residues of proteins in vivo, which result in the formation of crosslinks, may significantly change the steric structures of proteins, thereby affecting their activities and physical properties. This process is known as glycation or the Maillard reaction and is divided into two stages: an early-stage reaction that produces Amadori transfer products and a late-stage reaction that results in advanced glycation end-products (AGEs) through oxidation, dehydration, and condensation [1]. AGEs have been implicated in the pathogenesis of various diseases, such as diabetes [2], liver disease [3,4], atherosclerosis [5], Alzheimer’s disease [6], and aging [7].
AGEs are generated in vivo not only from glucose, but also from, for example, metabolic intermediates of glucose, degradation products, and Maillard reaction intermediates [8,9,10]. Among AGEs, those derived from glyceraldehyde (GA), a trisaccharide intermediate in fructose and glucose metabolism (toxic AGEs: TAGE), are highly cytotoxic [11] and have been associated with diabetic complications [12], insulin resistance [13], heart disease [14], Alzheimer’s disease [6], hypertension [15], nonalcoholic steatohepatitis [3,16], and cancer [17,18]. The late-stage Maillard reaction is irreversible and once TAGE are formed, there are no known enzymes that specifically remove the sites modified by glycation. The mechanisms by which intracellularly formed TAGE impair cells have not yet been elucidated in detail. Glycation modifications have been suggested to compromise cellular homeostasis by inducing the loss of function of various biomolecules or by forming toxic aggregates, which inactivate other normal essential proteins [19,20,21,22].
The ubiquitin-proteasome pathway and autophagy pathway are intracellular systems that remove defective proteins and aggregates. These two pathways are not independent of each other, they function cooperatively [3,23,24,25,26]. Recent studies suggested that AGEs are closely associated with autophagy [27]. Their effects on autophagy are complex, with some studies indicating that they inhibit autophagy [28,29] and others suggesting that they induce autophagy [30,31,32,33]. Therefore, the coordinated role of the two intracellular degradation mechanisms in the AGE clearance process remains unclear. Although AGEs are formed and accumulate in proteins with a long turnover due to a decrease in the activities of intracellular protein quality control mechanisms with aging, the mechanisms underlying intracellular TAGE degradation and removal have yet to be clarified. A more detailed understanding of these mechanisms is critical for the development of effective therapeutic strategies that mitigate the detrimental effects of TAGE accumulation.
In the present study, we identified d270KD, a mutant lacking the regulatory region of constitutively inactive checkpoint kinase-1 (CHK1), which is rapidly degraded upon a GA stimulation with the formation of high-molecular-weight complexes (typical features indicative of TAGE conversion). d270KD, which is rapidly degraded with TAGE formation, may serve as a useful model of TAGE formation in future research on the molecular mechanisms that act on the clearance of TAGE formed in cells.
2. Materials and Methods
2.1. Antibodies
The primary antibodies used were as follows: anti-mouse IgG2a-magnetic beads (M076-11), anti-DYKDDDDK (Flag)-tag-magnetic beads (M185-11), anti-V5-tag-magnetic beads (M16711), anti-normal rabbit IgG (PM035) for immunoprecipitation, and anti-HA-tag (M132-3) and anti-α-tubulin-HRP for Western blotting from Medical & Biological Laboratories. (MBL, Nagoya, Japan); anti-phospho-SQSTM1/p62 (Ser403) (#39786), anti-SQSTM1/p62 (#5114), and anti-Huwe1 (#5695) from Cell Signaling Technologies (CST, Beverley, MA); anti-V5-HRP (R961-25) from Thermo Fisher Scientific (Waltham, MA, USA); anti-Flag-HRP (015-22391) from Wako Pure Chemicals, (Osaka, Japan); anti-ubiquitin (#AUB01) from Cytoskeleton (Denver, CO). An anti-TAGE antibody was prepared as previously reported [34].
2.2. Cell culture
COS-7 cells were obtained from the American Type Culture Collection and cultured in Dulbecco’s modified Eagle medium (DMEM+GlutaMax medium; Gibco BRL, Gland Island, NY) supplemented with 10% fetal bovine serum (FBS; JRH Biosciences, Lenexa, KS), 100 U/ml penicillin, and 100 µg/ml streptomycin (both from Gibco BRL) at 37 °C in a humidified atmosphere of 5% CO2. HeLa cells (a gift from Dr. Sumiyo Akazawa, Kanazawa Medical University, Japan) were cultured in minimum essential medium (MEM alpha+GlutaMax medium; Gibco BRL) supplemented with 10% FBS, 100 U/ml penicillin, and 100 µg/ml streptomycin at 37 °C in a humidified atmosphere of 5% CO2. Cells were subcultured when they reached 70–80% confluence. In experiments where GA (Nacalai Tesque, Tokyo, Japan) was applied to cells, GA or phosphate buffer was added at the concentrations indicated. In the aminoguanidine (AG)-induced suppression experiment, AG at the indicated concentrations was added to cells 2 h prior to the treatment with GA. Regarding the proteasome inhibitor treatment, cells were treated with 10 µM MG-132 (Sigma-Aldrich, St Louis, MO) or DMSO (Nacalai Tesque) for 6 h.
2.3. Generation of expression plasmid constructs
Flag-tagged CHK1 expression constructs, including the wild-type (WT) form and its kinase-dead mutant form (KDmut), were previously described [35]. Briefly, cDNA encoding full-length rat Chk1 was amplified from rat cardiac myocyte cDNA using a forward primer containing the Flag-tag sequence at the 5' end and then subcloned into the pcDNA4/HisMax vector. Xpress- and 6×His tag sequences at the N terminus were removed from the vector by PCR using the KOD-plus-mutagenesis kit (Toyobo, Osaka, Japan) according to the manufacturer’s instructions. Deletion at the C terminus and point mutations in Flag-Chk1 expression vectors were created by PCR using the KOD-plus-mutagenesis kit. To generate the d270KD-EGFP-V5 expression construct, full-length EGFP cDNA was amplified by PCR using the pEGFP-C1 vector (BD Biosciences, San Jose, CA) and subcloned into the pcDNA3.1 vector (Thermo Fisher Scientific). The open-reading frame of EGFP with the V5-tag and 6×His-tag sequences at the C terminus was then amplified using the vector as a template by PCR using PrimeSTAR Max DNA polymerase, subcloned into the pENTR/D-TOPO vector (Thermo Fisher Scientific) using the TOPO® cloning procedure, and then transformed into the mammalian expression vector pDEST30 (Thermo Fisher Scientific) by LR clonase II enzyme. Fragments containing the N-terminal region of Chk1 (amino acids 1-270) in pFlag-Chk1Wt and its KDmut vectors were cloned in the vector in-frame with the gene that encodes the EGFP-V5/6×His-tag at the 3' end using the In-Fusion® HD PCR cloning kit (Takara, Shiga, Japan). HA-Ubiquitin was a gift from Edward Yeh (Addgene plasmid #18712). All constructs were sequenced to ensure proper ligation in the frame and Taq polymerase fidelity using the ABI PRISM TM 310 genetic analyzer (Applied Biosystems, Foster city, USA).
2.4. Transfection
Regarding transient plasmid DNA transfection, cells were seeded on 35-, 60-, 100-mm, or 6-well tissue culture plates, cultured in complete growth medium, and then transfected using FuGENE® 4K (Promega, Madison, WI) according to the manufacturer’s protocol. The transfection of siRNA was performed using RNAiMax Transfection reagent (Thermo Fisher Scientific) according to the manufacturer’s recommendations. The final siRNA concentration was 5 nM. Silencer Select siRNA (Ambion, Valencia, California) targeting Huwe1 (siRNA ID: s19596 and s19597), SQSTM1 (siRNA ID: s16962), or scrambled siRNA (Silencer Select Negative Control #1, catalogue #4390843) was used.
2.5. Western blot
Transfected COS-7 and HeLa cells were lysed in lysis buffer (CelLytic™M cell lysis reagent; Sigma-Aldrich) containing proteinase inhibitors and phosphatase inhibitors (both from Nacalai Tesque). After cellular debris was removed by centrifugation, protein concentrations in the supernatants were measured using the Qubit protein assay kit (Thermo Fisher Scientific). To detect TAGE or high-molecular-weight complexes, cells were lysed in RIPA buffer (50 mM Tris-HCl pH7.5, 150 mM NaCl, 1% NP-40, 0.5% Na deoxycholate, and 0.1% SDS), centrifuged, and separated into their supernatant (soluble fraction) and pellet (insoluble fraction). After the addition of SDS sample buffer to each fraction, the RIPA-insoluble fraction was sonicated. All samples were boiled at 95 °C for 5 min, separated on 5-12 or 12.5% SDS-PAGE gels, and analyzed by Western blotting. Target proteins were visualized by Chemi-Lumi One L, Super (Nacalai Tesque) or Immobilon Forte Western HRP Substrate (Millipore, Bedford, MA).
2.6. In vitro TAGE modification assay
The in vitro TAGE modification of the d270KD protein was performed with some modifications to previous methods. Briefly, HeLa cells transfected with Flag-tag fused d270KD (Flag-d270KD) were lysed in CelLytic™M cell lysis reagent containing proteinase inhibitors and phosphatase inhibitors. After cellular debris was removed by centrifugation, total cellular proteins were incubated with anti-Flag antibody-immobilized magnetic beads at 4 °C overnight, and the resulting immunoprecipitates were washed three times with lysis reagent. After the elution of Flag-d270KD with 40 µl of Flag peptide (2 mg/ml), 200 µl of PBS was added. Eluted samples, including Flag-d270KD were then ultrafiltrated and concentrated with Amicon Ultra 0.5 (10K) (Millipore) to remove the FLAG peptide. This purified Flag-d270KD recombinant protein (2.5 µg) was incubated in 50 µl of PBS for 20 h with or without the addition of 4 mM GA. After the addition of SDS sample buffer to these reaction mixtures, they were boiled at 95 °C for 5 minutes.
2.7. In vivo ubiquitination assay
In in vivo ubiquitylation assays, the d270KD-EGFP expression vector d270KD-EGFP was transfected with COS-7 cells (100-mm plates) for 48 h. Cells were treated with 10 µM MG132 and then harvested after 8 h. Cell pellets were resuspended in denaturing buffer (1.5% SDS, 50 mM Tris-HCl pH7.5, and 5 mM DTT) followed by boiling at 10 min, and were then diluted 10-fold with CelLytic™M cell lysis reagent containing proteinase inhibitors and phosphatase inhibitors. After cellular debris was removed by centrifugation, extracted proteins were immunoprecipitated with anti-V5 antibody-immobilized magnetic beads and subjected to the assay described above. Immunoprecipitates were analyzed by Western blotting with anti-ubiquitin antibodies or anti-V5 antibodies.
2.8. Luciferase assay
The gene encoding luciferase was amplified by PCR using the pGL4.54 vector (Promega) as a template with the forward primer (5'-ACAAACACACTTAACATGGAAGATGCCAAAAAC-3') and reverse primer (5'-GTAACAGGCCTTCTACACGGGCGATCTTGCCGCCC-3'). The pFlag-d270KD plasmid vector was amplified by PCR using the forward primer (5'-TA GAAGGGCCTGTACCTAGGATCCAGT-3') and reverse primer (5'-GTTAAGTGGTTTGTTATACCATCTA-3') for amplification by PCR to form a linear strand. The luciferase gene was then inserted downstream of the d270KD gene using the InFusion HD cloning kit (Clontech, Palo Alto, CA). The accuracy of the gene insertion site was confirmed by a sequence analysis. Twenty-four hours after the transfection of the d270KD fusion luciferase expression vector (d270KD-Luc) into cells cultured in 6-well plates, the Firefly luciferase reporter assay was performed using the Luciferase Reporter Assay System (Promega). Three independent assays were performed under each condition.
2.9. Fluorescence imaging analysis
COS-7 cells were grown in six-well plates containing collagen-coated glass coverslips (diameter of 12 mm, Iwaki) and transfected with the d270KD-EGFP expression vector at 60% confluence using FuGENE® 4K as described above. After an incubation for 24 h in complete medium, cells were treated with or without GA (2 mM) for the indicated time and fixed with ice-cold 4% paraformaldehyde in PBS for 10 min. Fixed cells were washed three times for 5 min each with PBS and then mounted with Prolong gold antifade reagent/DAPI (Thermo Fisher Scientific). All fluorescence images were obtained using a digital high-definition microscope system (BZ-9000, Keyence, Osaka, Japan) with the following filter sets: OP-66834, Ex360/40 Em460/50; OP-66836, Ex470/40 Em535/50; OP-66838, Ex560/40 Em630/60.
2.10. Statistical analysis
All numerical results are reported as the mean ± SEM of at least three measurements. Statistical analyses were performed using a one-way ANOVA, followed by Dunnett’s post hoc test with GraphPad Prism software (Version 5.0, GraphPad Prism Software, San Diego, CA). Differences were considered to be significant at p <0.01 and were denoted by an asterisk in the graphs.
3. Results
3.1. Identification of a constitutive KDmut of CHK1 that is rapidly modified with TAGE and degraded in cells after a GA stimulation
We previously reported that the prolonged stimulation of cells with GA, an intermediate metabolite of TAGE, induced DNA damage stress and necrosis [36]. CHK1 is a serine-threonine kinase that plays a central role in cell cycle checkpoints and DNA stress responses [37]. However, the effects of the GA stimulation on CHK1 (TAGE modifications) in cells remain unclear. Furthermore, the mechanisms by which conformational changes in the kinase affect modifications by glycation have not yet been elucidated. Therefore, we introduced CHK1 expression vectors with different molecular sizes and enzyme activities, such as WT CHK1 (CHK1WT), KDmut, or its C-terminal deletion mutant (d270KD), into HeLa cells, expressed these recombinant proteins in cells, stimulated cells with GA, and the TAGE of these proteins were then analyzed by Western blotting. The results obtained showed that the GA treatment of CHK1WT and KDmut resulted in a strong band of high-molecular-weight complexes, which is characteristic of TAGE-modified proteins (Figure 1a). On the other hand, high-molecular-weight bands were not detected in d270KD, and the monomer band almost disappeared after 20 h of the GA treatment.
Since the absence of high-molecular-weight bands in d270KD indicated that the structure was not susceptible to modifications by GA, we performed an in vitro TAGE experiment in the absence of a degradation system. d270KD fused with a Flag tag was expressed in cells, and the recombinant protein was purified by immunoprecipitation with a Flag antibody and then reacted with GA in a tube for 20 h. The results obtained showed that the GA stimulation resulted in the appearance of high-molecular-weight complexes, which were detected by anti-TAGE antibodies (Figure 1b). Therefore, the d270KD mutant was considered to be TAGE-modified by GA as well as CHK1WT; presumably, high-molecular-weight complexes were not detected in these cells because GA-stimulated degradation was accelerated. The time course of the GA-stimulated degradation of d270KD was analyzed by Western blotting. The results obtained showed that most of the bands disappeared after the GA stimulation at 4 mM for approximately 6 h (Figure 2a). Additionally, the GA stimulation for 6 h resulted in significant degradation from a concentration of 1.5 mM (Figure 2b). These results indicate that d270KD was rapidly degraded upon intracellular TAGE modifications by a short-term GA stimulation.
3.2. The intracellular destabilizing property of d270KD upon a GA stimulation may be added by fusion with other proteins
We examined whether the rapid degradation of d270KD induced by the GA stimulation is a property retained by fusion to other proteins. d270KD-EGFP, a fusion of EGFP to the C terminus of d270KD, was expressed in COS-7 cells and then stimulated with 4 mM GA. A Western blot analysis showed that d270KD-EGFP was markedly degraded after the GA stimulation for approximately 6 h (Figure 3a).
Slightly different from the degradation pattern of Flag-d270KD, high-molecular-weight complex bands were clearly observed in the EGFP fusion 2 h after the GA stimulation. A fluorescence imaging analysis was performed using cells expressing d270KD-EGFP. The results obtained showed that when cells were not stimulated, their fluorescence was extensively observed in the cytoplasm, whereas after the GA stimulation for 6 h, the fluorescent area decreased and only small granule-like signals were noted (Figure 3b). Furthermore, when d270KD-Luc, a luciferase fused to the C terminus of d270KD, was expressed in HeLa cells, rapid proteolysis was observed with the appearance of high-molecular-weight bands upon the GA stimulation, similar to Flag-d270KD (Figure 4a). Consistent with the proteolysis of d270KD-Luc, its luciferase activity also decreased in concentration- and time-dependent manners upon the GA stimulation (Figure 4b). We also investigated the effects of AG, a known inhibitor of AGE formation, on the GA-stimulated rapid degradation of 270KD-Luc. We found that the AG pretreatment inhibited the decrease stimulated by GA in luciferase activity in a concentration-dependent manner (Figure 4c). The Western blot analysis confirmed that this effect was due to the inhibition of the GA-induced proteolysis of d270KD-Luc (Figure 4d).
3.3. The pathway by which d270KD undergoes ubiquitination and degradation, stimulated by GA, does not appear to involve Mule/HUWE1 E3 ubiquitin ligase
The autophagy pathway is known to be inhibited in cells stimulated with GA, along with an increase in intracellular TAGE levels. However, the effects of a GA stimulation on the ubiquitin-proteasome pathway have remained unclear. Flag-d270KD began to degrade within 2 h of the GA stimulation (Figure 2a), and presumably, intracellular TAGE levels were low at that time, suggesting that the degradation system by the ubiquitin-proteasome pathway and autophagy pathway was functioning. The pre-administration of MG132, a proteasome inhibitor, completely blocked the degradation of d270KD by GA (Figure 5a). Additionally, a stimulation with MG132 and GA promoted the formation of the high-molecular-weight d270KD-EGFP complex of d270KD-EGFP and increased its level of ubiquitination more than the stimulation with MG132 alone (Figure 5b). Collectively, these results suggest that the GA stimulation induced TAGE modifications with the rapid ubiquitination of d270KD, which was mainly degraded through the proteasome pathway.
The E3 ubiquitin ligase that acts in the ubiquitination of d270KD has not yet been identified. Mule/HUWE1, a Homologous to E6-AP Carboxyl Terminus (HECT) E3 ubiquitin ligase, was previously shown to be involved in regulating the basal level of the CHK1 protein [38]. In the experimental system of HeLa cells used in the present study, when Mule/HUWE1 was knocked down with siRNA, an increase in endogenous CHK1 levels and a decrease in turnover were observed in the chase experiment with cycloheximide (Figure 6a). Similar results were obtained with the transiently expressed Flag-fused CHK1 protein (Figure 6b). We then investigated whether Mule/HUWE1 played a role in the GA-stimulated degradation of d270KD. The knockdown of Mule/HUWE1 did not suppress the degradation of d270KD (Figure 6c). It also did not affect the formation of high-molecular-weight complexes. Therefore, another ubiquitin ligase besides Mule/HUWE1 may be involved in the GA-stimulated d270KD degradation pathway. Alternatively, the GA-stimulated degradation of d270KD may involve a different pathway from that regulated at the basal level of CHK1WT because GA-stimulated d270KD undergoes TAGE modifications as well as ubiquitination.
3.4. GA-stimulated high-molecular-weight protein complexes and TAGE-modified protein complexes become resistant to detergents
The d270KD protein expressed in HeLa cells appeared exclusively as high-molecular-weight complexes in the insoluble fraction of RIPA buffer over time after the GA stimulation (Figure 7a). Based on this result, it was assumed that not only the d270KD-derived TAGE-modified protein, but also many other TAGE are abundant in the RIPA-insoluble fraction. Therefore, a Western blot analysis of RIPA-insoluble fractions of cells treated or not with 2 mM GA for 8 h revealed that GA-treated samples were enriched with high-molecular-weight complexes that were strongly detected by TAGE antibodies (Figure 7b). Therefore, when intracellular proteins are modified with TAGE by GA, they appear to accumulate intracellularly as aggregates that are resistant to common detergents, such as SDS.
3.5. p62 is involved in the formation of high-molecular-weight d270KD complexes of d270KD by GA
We hypothesized that the bulk degradation mechanism by the autophagy pathway, rather than the proteasome pathway, may function in the degradation of high-molecular-weight aggregates. Therefore, we investigated changes in the GA-stimulated behavior of p62, which plays an important role in selective autophagy by binding to ubiquitinated proteins and transporting them to autophagosomes. A single band of p62 was detected in soluble and insoluble fractions of RIPA; however, no significant changes were observed in its expression level upon the GA stimulation (Figure 7a). On the other hand, the phosphorylated form of serine 403, which increases binding affinity to ubiquitinated proteins [39,40], appeared to increase in the RIPA-insoluble fraction over time upon the GA stimulation. This result suggested that the GA stimulation induced the binding of p62 to TAGE-modified high-molecular-weight complexes that were abundant in the RIPA-insoluble fraction.
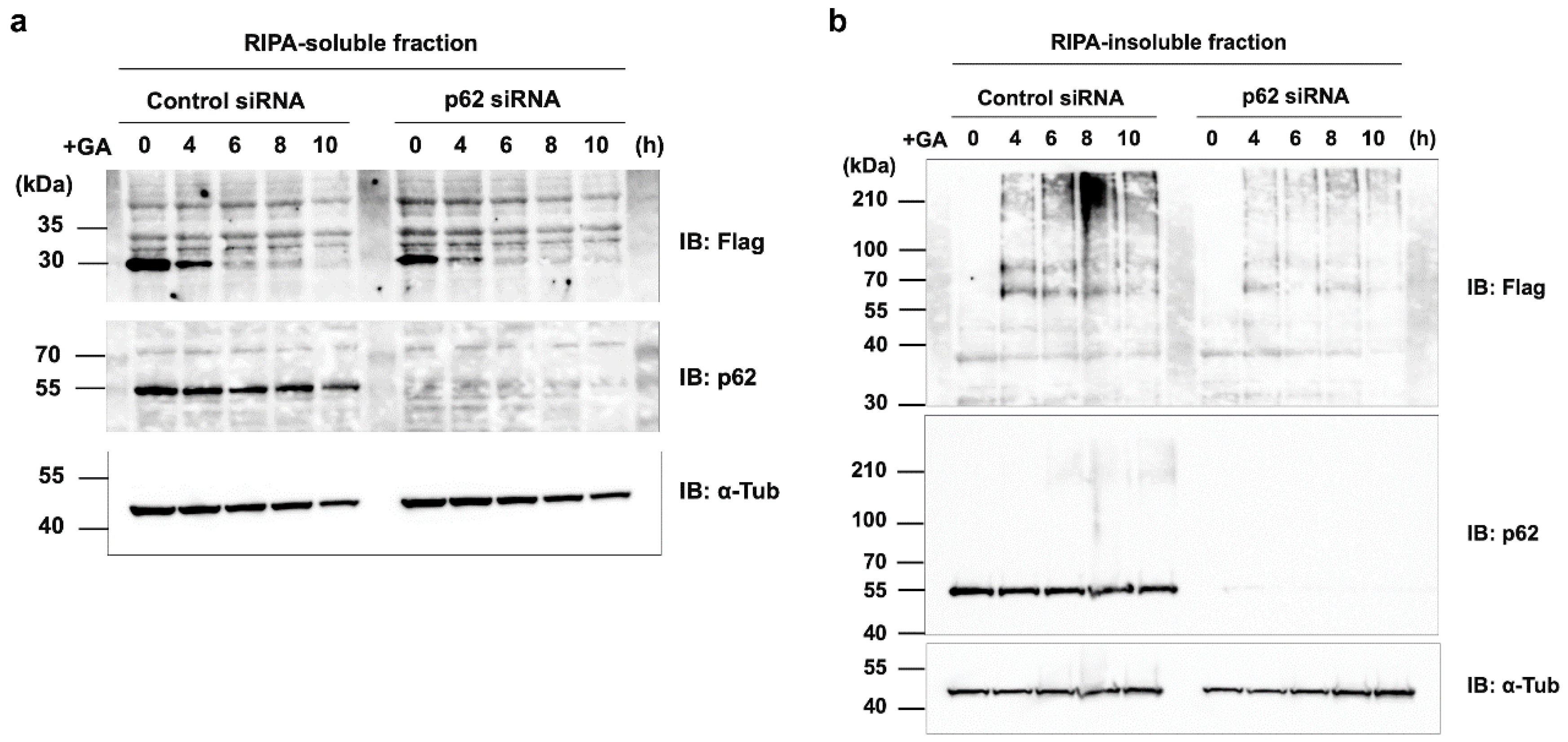
We also examined the effects of the p62 siRNA knockdown on the GA-induced rapid degradation of d270KD. Contrary to our expectations, the p62 knockdown did not affect the degradation of d270KD in the RIPA-soluble fraction (Figure 8a), while a similar study on the RIPA-insoluble fraction showed a slight decrease in the high-molecular-weight complex of d270KD in cells in which p62 was knocked down (Figure 8b). These results suggest that p62 was partially responsible for the GA-stimulated formation of high-molecular-weight complexes.
4. Discussion
Under hyperglycemic conditions, glucose, fructose, and its metabolic intermediates bind non-enzymatically to intracellular proteins to produce AGEs. Among these AGEs, GA-derived AGEs, known as TAGE, are highly cytotoxic and have been implicated in various pathological conditions, such as diabetic complications, insulin resistance, heart disease, Alzheimer’s disease, hypertension, nonalcoholic steatohepatitis, obesity, and cancer [7,41,42,43]. TAGE predominantly form intracellularly as a result of glycation reactions between GA and intracellular proteins, resulting in a wide range of TAGE molecules with different sizes and properties. The mechanisms by which TAGE exert their cytotoxic effects have not yet been elucidated in detail; however, previous studies suggested that TAGE toxicity was attributed to oxidative stress damage [36], the loss of protein functions due to glycation modifications [44], and the aggregation and accumulation of TAGE [20].
To the best of our knowledge, this is the first study to show the rapid degradation (less than 2 h) of a protein in an experimental model in which intracellular AGEs were formed by the administration of glycating agents, such as GA and methylglyoxal (MGO) to cells. Even when d270KD was fused with relatively stable proteins, such as EGFP or luciferase, the GA-responsive rapid degradation property was retained (Figure 3 and Figure 4), indicating that d270KD functions as a degradation element induced by glycated substances. Furthermore, a reporter assay using luciferase activity in d270KD-Luc-transfected cells allowed the degradation process to be monitored without a Western blot analysis, and GA-induced reductions in luciferase activity were attenuated by inhibitors of AGE formation in a concentration-dependent manner (Figure 4c). Therefore, this reporter assay using d270KD-Luc-transfected cells may be a useful tool for the rapid screening of natural compounds that inhibit the formation of AGE.
The etiology of TAGE toxicity is attributed to the functional impairment of the proteins themselves as a result of TAGE modifications. Previous studies demonstrated that chaperone molecules, such as Hsc70 [22], and a mediator of apoptosis, caspase-3 formed high-molecular-weight complexes that led to a loss of activity due to TAGE modifications [16]. In the present study, we observed the similar formation and accumulation of high-molecular-weight complexes in CHK1 after the GA stimulation, as shown in Figure 1. CHK1 is a well-known key player in the cellular stress response to DNA damage and the normal progression of the cell cycle under non-stress conditions [37]. Therefore, CHK1 may be a target of TAGE modifications, and its functional impairment may be responsible for the induction of TAGE-mediated cell death. A previous study reported that Mule/HUWE1, a HECT-type ubiquitin E3 ligase, ubiquitinated CHK1 and played a critical role in regulating the basal level of CHK1 [38]. In our HeLa cell model, we noted a decrease in endogenously and transiently expressed CHK1 turnover after the elimination of Mule, as shown in Figure 6a and 6b. However, the knockdown of Mule did not affect the degradation of d270KD in response to the GA stimulation or suppress the formation of high-molecular-weight complexes, as shown in Figure 6c. These results suggest that the degradation mechanism of d270KD, which is dependent on glycation modifications, involves a distinct pathway from the steady-state degradation mechanism of CHK1.
An important result from our Western blot analysis is that Mule was also affected by GA-induced glycation, as shown in Figure 6c (Control siRNA). Specifically, the GA stimulation for 6 h resulted in a conspicuous smear of the putative monomer form of Mule. However, it remains unclear whether the decrease in single bands was a consequence of TAGE-modified Mule degradation induced by the GA stimulation or the formation of high-molecular-weight complexes. This uncertainty is attributed to the technical challenges associated with analyzing the behavior of TAGE-modified Mule over time because the transfer efficiency of high-molecular-weight proteins to a PVDF membrane is very low in a conventional Western blot analysis. The loss of function of ubiquitin ligases due to TAGE modifications may lead to the accumulation of proteins that need to be degraded, further exacerbating non-enzymatic glycation reactions and disrupting protein homeostasis. Therefore, a more comprehensive and detailed analysis of the effects of GA-induced glycation on ubiquitin ligases is warranted in the future.
Another possible cause of cellular damage due to TAGE is proteopathy, a broad term that refers to the crosslinking of glycated proteins between molecules over time, resulting in the formation of larger high-molecular-weight complexes that gradually aggregate and accumulate, disrupting normal protein function [19]. The proper folding of intracellular proteins is crucial for their biological functions, while aberrantly folded proteins may accumulate and aggregate in cells, leading to cellular stress. In response to this stress, cells activate mechanisms to monitor protein folding, such as the unfolded protein response in the endoplasmic reticulum [45], which induces autophagy, an intracellular degradation mechanism that recycles damaged proteins and protects cells. Non-enzymatic glycation modifications to intracellular proteins may lead to structural mutations and the formation of abnormal proteins, which may be recognized as aberrant by intracellular protein quality control mechanisms.
Extracellular AGEs have also been shown to activate intracellular autophagy pathways, which often have a cytoprotective role [30,31,32,33]. Intracellularly formed AGEs were previously found to induce autophagy [27]. For example, in a model in which cells were treated with MGO, a glycating substance, to accumulate intracellular AGEs (MG-H1), autophagy was induced through a pathway that involved p62. Importantly, while the loss of p62 promotes the accumulation of AGEs, p62 itself appears to be affected by MGO-induced glycation over time, forming high-molecular-weight complexes and losing its function. In other words, the autophagy-lysosome pathway (and possibly the ubiquitin-proteasome degradation pathway) remains functional in the early stages of intracellular AGE formation, allowing for the proper clearance of AGEs. However, as glycation reactions continue, degradation pathways and other potential protective factors gradually transform into AGEs, resulting in a dysfunction in the AGE clearance pathway. This late stage of AGE formation may eventually induce apoptosis or passive, unprogrammed cell death by necrosis in affected cells [16]. To the best of our knowledge, the induction of autophagy in the early stages of TAGE formation in cells has not yet been demonstrated.
We previously reported that the stimulation of a human pancreatic beta cell line (1.4E7 cells) with GA resulted in prominent cell death, accompanied by decreases in the autophagosome markers, microtubule-associated protein 1 light chain 3 (LC3-I) and LC3-II as well as p62 [28]. These findings were observed after a prolonged stimulation (24 h) with 2 mM GA, at which stage cell death was already induced. Therefore, important factors involved in various vital functions were modified by TAGE, aggregated, and accumulated, suggesting that the TAGE degradation pathway was already disrupted in the late stage. To confirm the existence of an endogenous degradation and removal mechanism for TAGE, a detailed analysis of the behavior of degradation-related factors from the initial stage of a glycation stimulation is needed.
In the present study, we investigated the behavior of p62, a known selective autophagy receptor, over 10 h, starting 2 h after a stimulation with 2 mM GA. The results obtained revealed a slight change in total endogenous p62 levels (Figure 7a). Since autophagy is responsible for the degradation of p62 when degrading its substrates, the accumulation of p62 is often observed when autophagy is impaired [27]. However, total p62 levels remained unchanged after 10 h of the stimulation with 2 mM GA; therefore, the complete failure of autophagy had not yet occurred. p62 plays a key role in substrate degradation by recruiting ubiquitinated cargo to autophagosomes. The phosphorylation of the 403rd serine (Ser403) in the UBA domain of p62 by casein kinase-2 or TANK-binding kinase 1 increased its binding affinity to the ubiquitin chain, thereby facilitating selective autophagy [39,40].
Interestingly, after the GA stimulation for 2 h, a gradual decrease in the phosphorylated form of p62 Ser403 was observed in the RIPA-soluble fraction, in contrast to an increase over time in the RIPA-insoluble fraction (Figure 7a). Most of the d270KD high-molecular-weight complexes formed by the GA stimulation were present in the RIPA-insoluble fraction, and the level of d270KD high-molecular-weight complexes in this fraction peaked at approximately 6 h of the GA stimulation, followed by a gradual decrease (Figure 7a). Furthermore, the RIPA-insoluble fraction of cells that did not express d270KD but were stimulated with GA for 6 h also showed the abundant localization of high-molecular-weight complexes that were positive for TAGE antibodies (Figure 7b). This result suggests that intracellular proteins modified with TAGE by GA form aggregates that have acquired resistance to detergents, such as SDS, and are more likely to become insoluble intracellularly. Furthermore, the binding of phosphorylated p62 to ubiquitinated TAGE may have been increased in the insoluble fraction, suggesting that the degradation system by selective autophagy was still partially functional during the initial stage of the GA stimulation, despite the formation of TAGE-modified aggregates.
Overall, the present results highlight the importance of analyzing the behavior of degradation-related factors, such as p62, from the initial stage of a glycation stimulation to elucidate endogenous TAGE degradation and removal mechanisms. Further research is needed to clarify the complex interplay between TAGE formation, autophagy, and cellular responses to proteopathy, which will provide a more detailed understanding of the pathogenesis of TAGE-related cellular damage. Since proteasome inhibitors completely suppressed the GA-stimulated rapid degradation of d270KD (Figure 5a), the ubiquitin-proteasome pathway may also work in conjunction with the autophagy pathway during the early stages of TAGE formation to maintain intracellular protein homeostasis, which is disrupted by glycation modifications. The knockdown of p62 did not affect the degradation of d270KD by GA, but modestly reduced the formation of high-molecular-weight complexes derived from d270KD in the insoluble fraction. Therefore, p62 may play a partial role in the formation of TAGE aggregates in the insoluble fraction. One hypothesis is that p62 may be involved in sequestering randomly formed TAGE throughout the cell by aggregating them, thereby preventing interference with the function of normal proteins and potentially protecting the cell. A more detailed analysis is needed on the subcellular localization of ubiquitinated and insoluble TAGE aggregates.
In conclusion, the present study provides novel insights into the intracellular degradation mechanism of TAGE and proposes potential therapeutic strategies to prevent or mitigate TAGE-related diseases. Further research in this area will contribute to the development of effective interventions for diseases associated with protein aggregation and the accumulation of AGEs, including diabetes, neurodegenerative diseases, and cardiovascular diseases.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: title.
Author Contributions
Conceptualization, K.T. and M.T.; methodology, K.T.; investigation, K.T.; writing—original draft preparation, K.T.; writing—review and editing, A.S.-S, M.T., and K.T.; project administration, A.S.-S., K.K., and M.T.; funding acquisition, K.T., A.S.-S., and M.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by JSPS KAKENHI grant numbers 20K08410 (K.T.) and 21H04865 (M.T.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data for this study may be found in the main article or in the supplementary material.
Acknowledgments
The authors would like to thank Sumiyo Akazawa for generously providing us with HeLa cells.
Conflicts of Interest
The authors declare that they have no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced glycation end-products: a review. Diabetologia 2001, 44, 129-146. https://doi.org/10.1007/s001250051591. [CrossRef]
- Meerwaldt, R.; Links, T.; Zeebregts, C.; Tio, R.; Hillebrands, J.L.; Smit, A. The clinical relevance of assessing advanced glycation endproducts accumulation in diabetes. Cardiovasc Diabetol 2008, 7, 29. https://doi.org/10.1186/1475-2840-7-29. [CrossRef]
- Hyogo, H.; Yamagishi, S.; Iwamoto, K.; Arihiro, K.; Takeuchi, M.; Sato, T.; Ochi, H.; Nonaka, M.; Nabeshima, Y.; Inoue, M.; et al. Elevated levels of serum advanced glycation end products in patients with non-alcoholic steatohepatitis. J Gastroenterol Hepatol 2007, 22, 1112-1119. https://doi.org/10.1111/j.1440-1746.2007.04943.x. [CrossRef]
- Lyu, C.; Kong, W.; Liu, Z.; Wang, S.; Zhao, P.; Liang, K.; Niu, Y.; Yang, W.; Xiang, C.; Hu, X.; et al. Advanced glycation end-products as mediators of the aberrant crosslinking of extracellular matrix in scarred liver tissue. Nat Biomed Eng 2023. https://doi.org/10.1038/s41551-023-01019-z. [CrossRef]
- Mao, L.; Yin, R.; Yang, L.; Zhao, D. Role of advanced glycation end products on vascular smooth muscle cells under diabetic atherosclerosis. Front Endocrinol (Lausanne) 2022, 13, 983723. https://doi.org/10.3389/fendo.2022.983723. [CrossRef]
- Choei, H.; Sasaki, N.; Takeuchi, M.; Yoshida, T.; Ukai, W.; Yamagishi, S.; Kikuchi, S.; Saito, T. Glyceraldehyde-derived advanced glycation end products in Alzheimer's disease. Acta Neuropathol 2004, 108, 189-193. https://doi.org/10.1007/s00401-004-0871-x. [CrossRef]
- Grillo, M.A.; Colombatto, S. Advanced glycation end-products (AGEs): involvement in aging and in neurodegenerative diseases. Amino Acids 2008, 35, 29-36. https://doi.org/10.1007/s00726-007-0606-0. [CrossRef]
- Bucala, R.; Cerami, A. Advanced glycosylation: chemistry, biology, and implications for diabetes and aging. Adv Pharmacol 1992, 23, 1-34. https://doi.org/10.1016/s1054-3589(08)60961-8. [CrossRef]
- Takeuchi, M.; Makita, Z. Alternative routes for the formation of immunochemically distinct advanced glycation end-products in vivo. Curr Mol Med 2001, 1, 305-315. https://doi.org/10.2174/1566524013363735. [CrossRef]
- Vlassara, H.; Bucala, R.; Striker, L. Pathogenic effects of advanced glycosylation: biochemical, biologic, and clinical implications for diabetes and aging. Lab Invest 1994, 70, 138-151.
- Takeuchi, M.; Yamagishi, S. TAGE (toxic AGEs) hypothesis in various chronic diseases. Med Hypotheses 2004, 63, 449-452. https://doi.org/10.1016/j.mehy.2004.02.042. [CrossRef]
- Nakamura, K.; Yamagishi, S.; Adachi, H.; Matsui, T.; Kurita-Nakamura, Y.; Takeuchi, M.; Inoue, H.; Imaizumi, T. Serum levels of soluble form of receptor for advanced glycation end products (sRAGE) are positively associated with circulating AGEs and soluble form of VCAM-1 in patients with type 2 diabetes. Microvasc Res 2008, 76, 52-56. https://doi.org/10.1016/j.mvr.2007.09.004. [CrossRef]
- Tahara, N.; Yamagishi, S.; Matsui, T.; Takeuchi, M.; Nitta, Y.; Kodama, N.; Mizoguchi, M.; Imaizumi, T. Serum levels of advanced glycation end products (AGEs) are independent correlates of insulin resistance in nondiabetic subjects. Cardiovasc Ther 2012, 30, 42-48. https://doi.org/10.1111/j.1755-5922.2010.00177.x. [CrossRef]
- Yasuda, Y.; Aoki, H.; Fujita, W.; Fujibayashi, K.; Wakasa, M.; Kawai, Y.; Nakanishi, H.; Saito, K.; Takeuchi, M.; Kajinami, K. Glyceraldehyde-derived advanced glycation end-products are associated with left ventricular ejection fraction and brain natriuretic peptide in patients with diabetic adverse cardiac remodeling. Scand Cardiovasc J 2022, 56, 208-216. https://doi.org/10.1080/14017431.2022.2095013. [CrossRef]
- Tahara, N.; Yamagishi, S.; Takeuchi, M.; Honda, A.; Tahara, A.; Nitta, Y.; Kodama, N.; Mizoguchi, M.; Kaida, H.; Ishibashi, M.; et al. Positive association between serum level of glyceraldehyde-derived advanced glycation end products and vascular inflammation evaluated by [(18)F]fluorodeoxyglucose positron emission tomography. Diabetes Care 2012, 35, 2618-2625. https://doi.org/10.2337/dc12-0087. [CrossRef]
- Sakasai-Sakai, A.; Takata, T.; Takino, J.I.; Takeuchi, M. Impact of intracellular glyceraldehyde-derived advanced glycation end-products on human hepatocyte cell death. Sci Rep 2017, 7, 14282. https://doi.org/10.1038/s41598-017-14711-3. [CrossRef]
- Kan, H.; Yamagishi, S.; Ojima, A.; Fukami, K.; Ueda, S.; Takeuchi, M.; Hyogo, H.; Aikata, H.; Chayama, K. Elevation of serum levels of advanced glycation end products in patients with non-B or non-C hepatocellular carcinoma. J Clin Lab Anal 2015, 29, 480-484. https://doi.org/10.1002/jcla.21797. [CrossRef]
- Mao, Z.; Baker, J.R.; Takeuchi, M.; Hyogo, H.; Tjønneland, A.; Eriksen, A.K.; Severi, G.; Rothwell, J.; Laouali, N.; Katzke, V.; et al. Prediagnostic serum glyceraldehyde-derived advanced glycation end products and mortality among colorectal cancer patients. Int J Cancer 2023, 152, 2257-2268. https://doi.org/10.1002/ijc.34449. [CrossRef]
- Nasu, R.; Furukawa, A.; Suzuki, K.; Takeuchi, M.; Koriyama, Y. The effect of glyceraldehyde-derived advanced glycation end products on β-tubulin-inhibited neurite outgrowth in SH-SY5Y human neuroblastoma cells. Nutrients 2020, 12. https://doi.org/10.3390/nu12102958. [CrossRef]
- Ooi, H.; Nasu, R.; Furukawa, A.; Takeuchi, M.; Koriyama, Y. Pyridoxamine and aminoguanidine attenuate the abnormal aggregation of β-tubulin and suppression of neurite outgrowth by glyceraldehyde-derived toxic advanced glycation end-products. Front Pharmacol 2022, 13, 921611. https://doi.org/10.3389/fphar.2022.921611. [CrossRef]
- Sakasai-Sakai, A.; Takata, T.; Takeuchi, M. The association between accumulation of toxic advanced glycation end-products and cytotoxic effect in MC3T3-E1 cells. Nutrients 2022, 14. https://doi.org/10.3390/nu14050990. [CrossRef]
- Takino, J.; Kobayashi, Y.; Takeuchi, M. The formation of intracellular glyceraldehyde-derived advanced glycation end-products and cytotoxicity. J Gastroenterol 2010, 45, 646-655. https://doi.org/10.1007/s00535-009-0193-9. [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885-889. https://doi.org/10.1038/nature04724. [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324-332. https://doi.org/10.1038/nature10317. [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880-884. https://doi.org/10.1038/nature04723. [CrossRef]
- Uchiki, T.; Weikel, K.A.; Jiao, W.; Shang, F.; Caceres, A.; Pawlak, D.; Handa, J.T.; Brownlee, M.; Nagaraj, R.; Taylor, A. Glycation-altered proteolysis as a pathobiologic mechanism that links dietary glycemic index, aging, and age-related disease (in nondiabetics). Aging Cell 2012, 11, 1-13. https://doi.org/10.1111/j.1474-9726.2011.00752.x. [CrossRef]
- Aragonès, G.; Dasuri, K.; Olukorede, O.; Francisco, S.G.; Renneburg, C.; Kumsta, C.; Hansen, M.; Kageyama, S.; Komatsu, M.; Rowan, S.; et al. Autophagic receptor p62 protects against glycation-derived toxicity and enhances viability. Aging Cell 2020, 19, e13257. https://doi.org/10.1111/acel.13257. [CrossRef]
- Takata, T.; Sakasai-Sakai, A.; Takeuchi, M. Intracellular toxic advanced glycation end-products in 1.4E7 cell line induce death with reduction of microtubule-associated protein 1 light chain 3 and p62. Nutrients 2022, 14. https://doi.org/10.3390/nu14020332. [CrossRef]
- Zhao, B.; Bower, M.J.; McDevitt, P.J.; Zhao, H.; Davis, S.T.; Johanson, K.O.; Green, S.M.; Concha, N.O.; Zhou, B.B. Structural basis for Chk1 inhibition by UCN-01. J Biol Chem 2002, 277, 46609-46615. https://doi.org/10.1074/jbc.M201233200. [CrossRef]
- Liang, B.; Zhou, Z.; Yang, Z.; Liu, J.; Zhang, L.; He, J.; Li, H.; Huang, Y.; Yang, Q.; Xian, S.; et al. AGEs-RAGE axis mediates myocardial fibrosis via activation of cardiac fibroblasts induced by autophagy in heart failure. Exp Physiol 2022, 107, 879-891. https://doi.org/10.1113/EP090042. [CrossRef]
- Liu, Z.; Huang, S.; Hu, P.; Zhou, H. The role of autophagy in advanced glycation end product-induced proliferation and migration in rat vascular smooth muscle cells. Iran J Basic Med Sci 2018, 21, 634-638. https://doi.org/10.22038/IJBMS.2018.20266.5305. [CrossRef]
- Mei, Y.M.; Li, L.; Wang, X.Q.; Zhang, M.; Zhu, L.F.; Fu, Y.W.; Xu, Y. AGEs induces apoptosis and autophagy via reactive oxygen species in human periodontal ligament cells. J Cell Biochem 2020, 121, 3764-3779. https://doi.org/10.1002/jcb.29499. [CrossRef]
- Meng, H.Z.; Zhang, W.L.; Liu, F.; Yang, M.W. Advanced glycation end products affect osteoblast proliferation and function by modulating autophagy via the receptor of advanced glycation end products/Raf protein/mitogen-activated protein kinase/extracellular signal-regulated kinase kinase/extracellular signal-regulated kinase (RAGE/Raf/MEK/ERK) pathway. J Biol Chem 2015, 290, 28189-28199. https://doi.org/10.1074/jbc.M115.669499. [CrossRef]
- Takeuchi, M.; Makita, Z.; Bucala, R.; Suzuki, T.; Koike, T.; Kameda, Y. Immunological evidence that non-carboxymethyllysine advanced glycation end-products are produced from short chain sugars and dicarbonyl compounds in vivo. Mol Med 2000, 6, 114-125.
- Takeda, K.; Takata, T.; Kawai, Y.; Ishigaki, Y.; Kajinami, K. Chk1-mediated phosphorylation of receptor-associated late transducer at serine 250 increases its stability by stimulating its interaction with 14-3-3. Genes Cells 2013, 18, 369-386. https://doi.org/10.1111/gtc.12043. [CrossRef]
- Sakasai-Sakai, A.; Takata, T.; Takeuchi, M. Intracellular toxic advanced glycation end-products promote the production of reactive oxygen species in HepG2 cells. Int J Mol Sci 2020, 21. https://doi.org/10.3390/ijms21144861. [CrossRef]
- Dai, Y.; Grant, S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin Cancer Res 2010, 16, 376-383, doi:1078-0432.CCR-09-1029 [pii]10.1158/1078-0432.CCR-09-1029. [CrossRef]
- Cassidy, K.B.; Bang, S.; Kurokawa, M.; Gerber, S.A. Direct regulation of Chk1 protein stability by E3 ubiquitin ligase HUWE1. FEBS J 2020, 287, 1985-1999. https://doi.org/10.1111/febs.15132. [CrossRef]
- Matsumoto, G.; Wada, K.; Okuno, M.; Kurosawa, M.; Nukina, N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell 2011, 44, 279-289. https://doi.org/10.1016/j.molcel.2011.07.039. [CrossRef]
- Pilli, M.; Arko-Mensah, J.; Ponpuak, M.; Roberts, E.; Master, S.; Mandell, M.A.; Dupont, N.; Ornatowski, W.; Jiang, S.; Bradfute, S.B.; et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 2012, 37, 223-234. https://doi.org/10.1016/j.immuni.2012.04.015. [CrossRef]
- Sato, T.; Shimogaito, N.; Wu, X.; Kikuchi, S.; Yamagishi, S.; Takeuchi, M. Toxic advanced glycation end products (TAGE) theory in Alzheimer's disease. Am J Alzheimers Dis Other Demen 2006, 21, 197-208. https://doi.org/10.1177/1533317506289277. [CrossRef]
- Takeuchi, M.; Sakasai-Sakai, A.; Takata, T.; Takino, J.I.; Koriyama, Y. Effects of toxic AGEs (TAGE) on human health. Cells 2022, 11. https://doi.org/10.3390/cells11142178. [CrossRef]
- Takeuchi, M.; Sakasai-Sakai, A.; Takata, T.; Takino, J.I.; Koriyama, Y.; Kikuchi, C.; Furukawa, A.; Nagamine, K.; Hori, T.; Matsunaga, T. Intracellular toxic AGEs (TAGE) triggers numerous types of cell damage. Biomolecules 2021, 11. https://doi.org/10.3390/biom11030387. [CrossRef]
- Sakasai-Sakai, A.; Takata, T.; Takino, J.I.; Takeuchi, M. The relevance of toxic AGEs (TAGE) cytotoxicity to NASH pathogenesis: a mini-review. Nutrients 2019, 11. https://doi.org/10.3390/nu11020462. [CrossRef]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat Cell Biol 2015, 17, 829-838. https://doi.org/10.1038/ncb3184. [CrossRef]
Figure 1.
Effects of the GA treatment on TAGE formation in wild-type CHK1 and its constitutively inactive mutants. (a) Various CHK1 expression vectors with different molecular sizes and enzymatic activities, including wild-type CHK1 (Flag-Chk1WT), a constitutive kinase-dead mutant (Flag-KDmut), and its C-terminal deletion mutant (Flag-d270KD), were transfected into HeLa cells. Cells were stimulated with 4 mM GA for 20 h. Cytoplasmic proteins were extracted and TAGE modifications were analyzed by Western blotting. (b) In vitro TAGE modifications to the d270KD recombinant protein. A Flag-d270KD expression vector was transfected into HeLa cells and cell lysates were extracted 24 h later. Lysates were immunoprecipitated with an anti-Flag antibody to purify the Flag-d270KD recombinant protein, which was then reacted with 4 mM GA in vitro for 20 h. TAGE modifications to the d270KD recombinant protein were analyzed by Western blotting.
Figure 1.
Effects of the GA treatment on TAGE formation in wild-type CHK1 and its constitutively inactive mutants. (a) Various CHK1 expression vectors with different molecular sizes and enzymatic activities, including wild-type CHK1 (Flag-Chk1WT), a constitutive kinase-dead mutant (Flag-KDmut), and its C-terminal deletion mutant (Flag-d270KD), were transfected into HeLa cells. Cells were stimulated with 4 mM GA for 20 h. Cytoplasmic proteins were extracted and TAGE modifications were analyzed by Western blotting. (b) In vitro TAGE modifications to the d270KD recombinant protein. A Flag-d270KD expression vector was transfected into HeLa cells and cell lysates were extracted 24 h later. Lysates were immunoprecipitated with an anti-Flag antibody to purify the Flag-d270KD recombinant protein, which was then reacted with 4 mM GA in vitro for 20 h. TAGE modifications to the d270KD recombinant protein were analyzed by Western blotting.
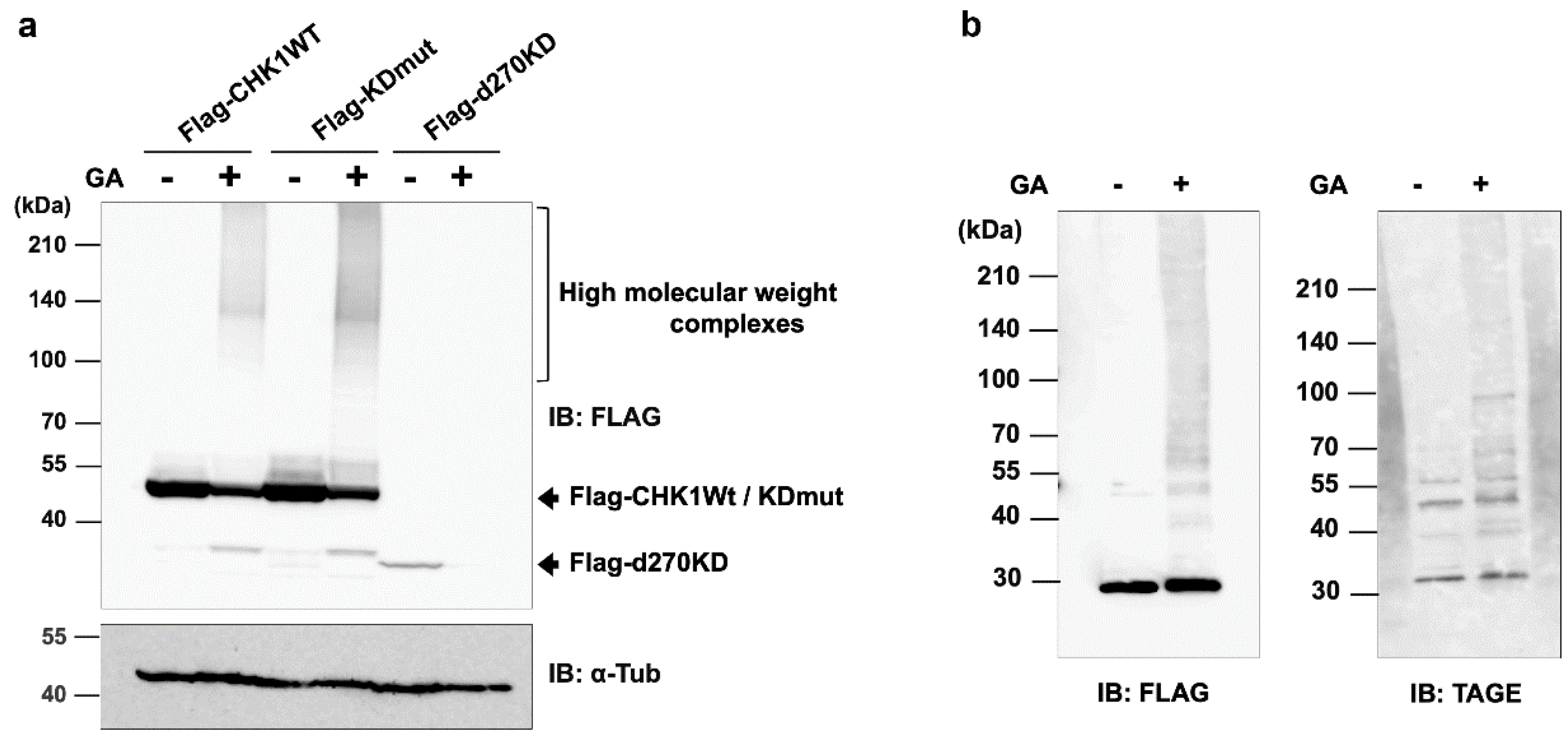
Figure 2.
GA-stimulated rapid degradation of the intracellular d270KD protein. (a) A Flag-d270KD expression vector was transfected into HeLa cells, and cell lysates were collected at the indicated times after 2 mM GA was applied to the cells 24 h later. Lysates were subjected to a Western blot analysis using the indicated antibodies. (b) A Flag-d270KD expression vector was transfected into HeLa cells and cells were stimulated 24 h later with the indicated concentration of GA for 6 h. A Western blot analysis was performed on lysates collected from these cells as described above.
Figure 2.
GA-stimulated rapid degradation of the intracellular d270KD protein. (a) A Flag-d270KD expression vector was transfected into HeLa cells, and cell lysates were collected at the indicated times after 2 mM GA was applied to the cells 24 h later. Lysates were subjected to a Western blot analysis using the indicated antibodies. (b) A Flag-d270KD expression vector was transfected into HeLa cells and cells were stimulated 24 h later with the indicated concentration of GA for 6 h. A Western blot analysis was performed on lysates collected from these cells as described above.
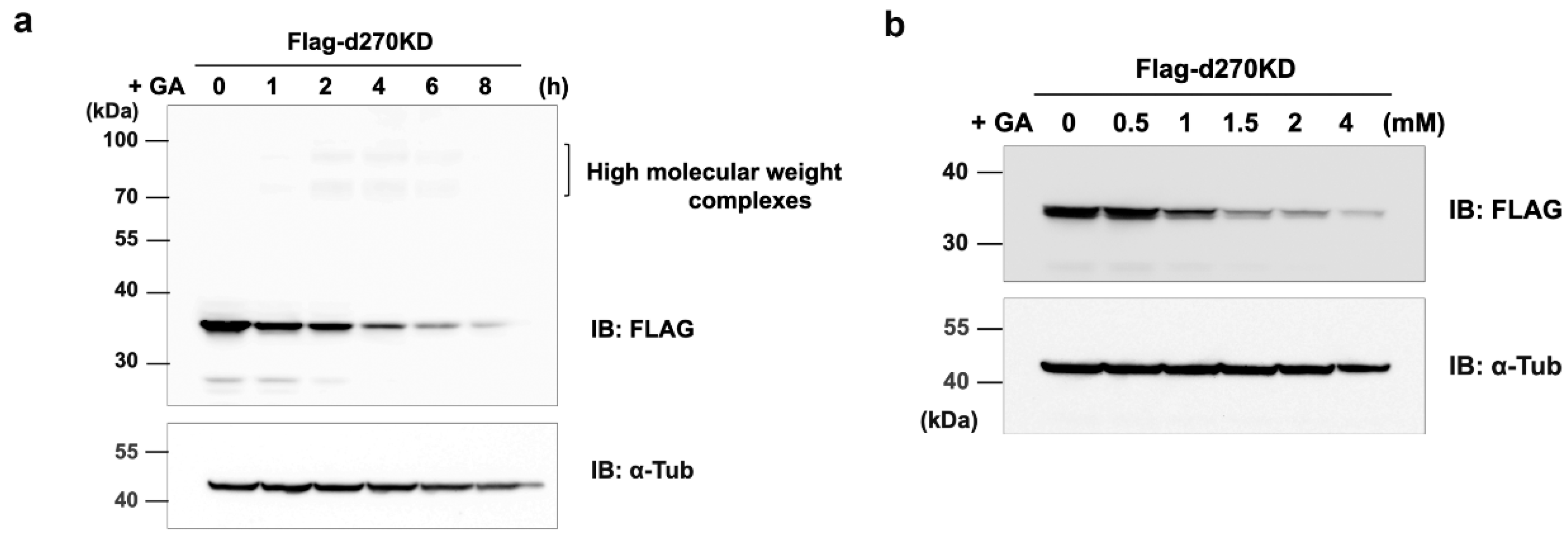
Figure 3.
GA-stimulated rapid intracellular degradation of d270KD fusion proteins to EGFP. (a) The expression vector of d270KD fusion EGFP (d270KD-EGFP) was transfected into COS-7 cells and stimulated with 4 mM GA 48 h later. Cell lysates were collected at the indicated times and subjected to a Western blot analysis. (b) COS-7 cells were transfected with the d270KD-EGFP expression vector for 24 h, stimulated with phosphate buffer (control) or 4 mM GA for 6 h, and then fixed. Nuclei were counterstained with DAPI and analyzed by fluorescence microscopy. Scale bars = 50 μm.
Figure 3.
GA-stimulated rapid intracellular degradation of d270KD fusion proteins to EGFP. (a) The expression vector of d270KD fusion EGFP (d270KD-EGFP) was transfected into COS-7 cells and stimulated with 4 mM GA 48 h later. Cell lysates were collected at the indicated times and subjected to a Western blot analysis. (b) COS-7 cells were transfected with the d270KD-EGFP expression vector for 24 h, stimulated with phosphate buffer (control) or 4 mM GA for 6 h, and then fixed. Nuclei were counterstained with DAPI and analyzed by fluorescence microscopy. Scale bars = 50 μm.
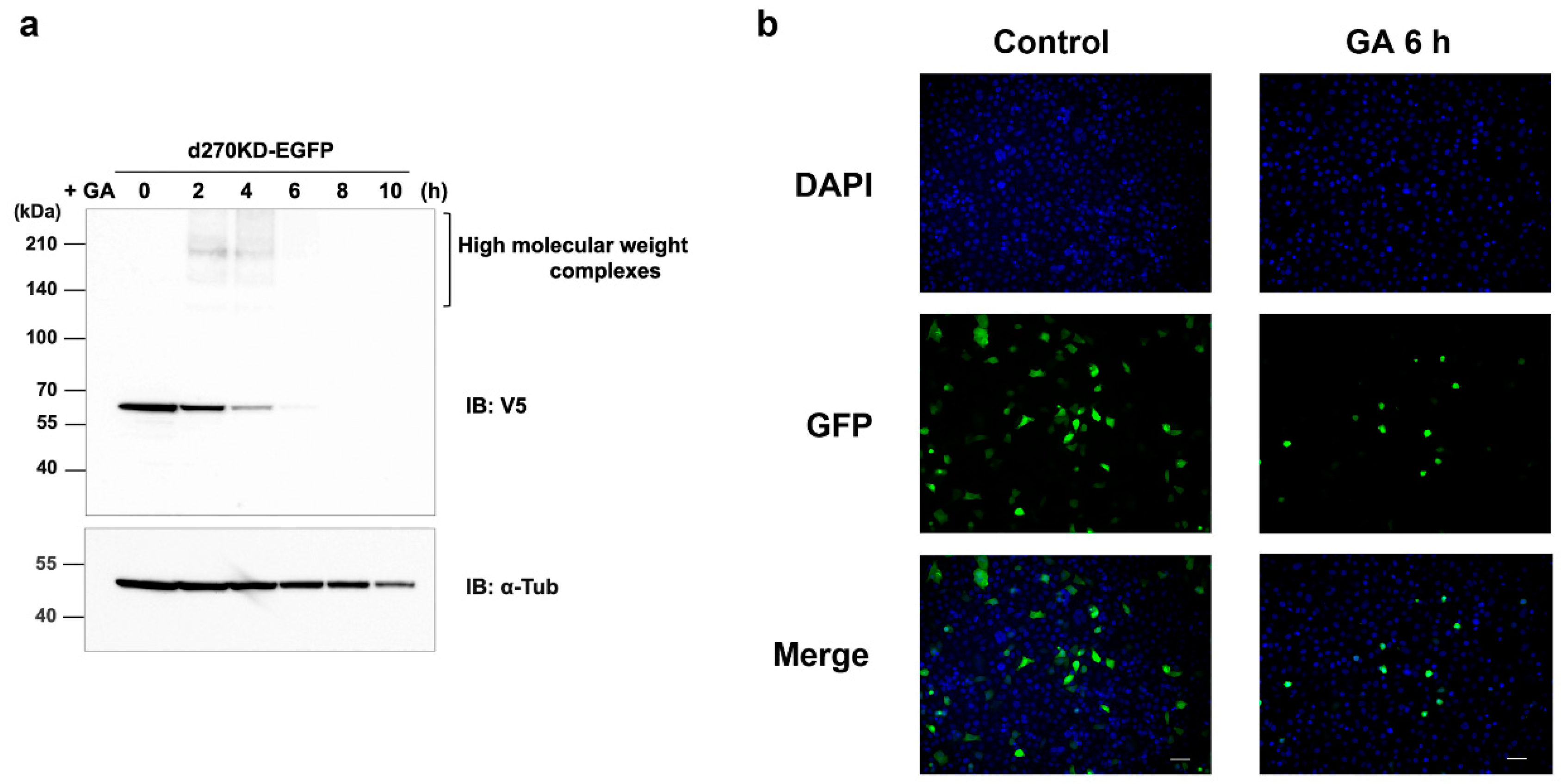
Figure 4.
Rapid proteolysis of d270KD-fused luciferase and its reduced activity upon a GA stimulation. (a) A d270KD fusion luciferase expression vector (d270KD-Luc) was transfected into HeLa cells and stimulated with 4 mM GA after 24 h. Cell lysates were collected at the indicated times and subjected to a Western blot analysis. (b) As in (a), d270KD-Luc was transfected into HeLa cells for 24 h and stimulated with GA at the indicated concentrations. Cell lysates collected at each indicated time were subjected to the luciferase assay. (c) HeLa cells were transfected with d270KD-Luc for 24 h and then pretreated with phosphate buffer or aminoguanidine (AG) at the indicated concentrations for 2 h. Cells were then stimulated with phosphate buffer or 2 mM GA for 6 h and lysed. Cell lysates were collected and luciferase activity was measured and expressed as 100 for the control. Each value was obtained from three independent experiments. Error bars indicate SEM. *p <0.01 versus 2 mM GA alone. (d) Cell lysates treated similarly to the experiment in (c) were subjected to a Western blot analysis using the antibodies indicated.
Figure 4.
Rapid proteolysis of d270KD-fused luciferase and its reduced activity upon a GA stimulation. (a) A d270KD fusion luciferase expression vector (d270KD-Luc) was transfected into HeLa cells and stimulated with 4 mM GA after 24 h. Cell lysates were collected at the indicated times and subjected to a Western blot analysis. (b) As in (a), d270KD-Luc was transfected into HeLa cells for 24 h and stimulated with GA at the indicated concentrations. Cell lysates collected at each indicated time were subjected to the luciferase assay. (c) HeLa cells were transfected with d270KD-Luc for 24 h and then pretreated with phosphate buffer or aminoguanidine (AG) at the indicated concentrations for 2 h. Cells were then stimulated with phosphate buffer or 2 mM GA for 6 h and lysed. Cell lysates were collected and luciferase activity was measured and expressed as 100 for the control. Each value was obtained from three independent experiments. Error bars indicate SEM. *p <0.01 versus 2 mM GA alone. (d) Cell lysates treated similarly to the experiment in (c) were subjected to a Western blot analysis using the antibodies indicated.
Figure 5.
Involvement of the proteasome pathway in the rapid degradation of d270KD by GA. (a) HeLa cells were transfected with the Flag-d270KD expression vector and cells were incubated 24 h later with 5 mM of a proteasome inhibitor (MG132) or DMSO (control) for 4 h. Cells were treated with 4 mM GA and cell lysates were collected at the indicated times for a Western blot analysis. (b) The GA stimulation increased the ubiquitination of d270KD-fused EGFP. The expression vector for the d270KD fusion EGFP (d270KD-EGFP) expression vector was transfected into HeLa cells for 48 h and cells were then treated with DMSO (control) or 5 µM MG132 for 3 h. Cells were collected after the stimulation with phosphate buffer or 4 mM GA for 3 h. Cell lysates were extracted under denaturing conditions, diluted in lysis buffer, and immunoprecipitated (IP) with an anti-V5 antibody. Whole cell lysates (WCL) and IP samples were subjected to a Western blot analysis using the indicated antibodies.
Figure 5.
Involvement of the proteasome pathway in the rapid degradation of d270KD by GA. (a) HeLa cells were transfected with the Flag-d270KD expression vector and cells were incubated 24 h later with 5 mM of a proteasome inhibitor (MG132) or DMSO (control) for 4 h. Cells were treated with 4 mM GA and cell lysates were collected at the indicated times for a Western blot analysis. (b) The GA stimulation increased the ubiquitination of d270KD-fused EGFP. The expression vector for the d270KD fusion EGFP (d270KD-EGFP) expression vector was transfected into HeLa cells for 48 h and cells were then treated with DMSO (control) or 5 µM MG132 for 3 h. Cells were collected after the stimulation with phosphate buffer or 4 mM GA for 3 h. Cell lysates were extracted under denaturing conditions, diluted in lysis buffer, and immunoprecipitated (IP) with an anti-V5 antibody. Whole cell lysates (WCL) and IP samples were subjected to a Western blot analysis using the indicated antibodies.
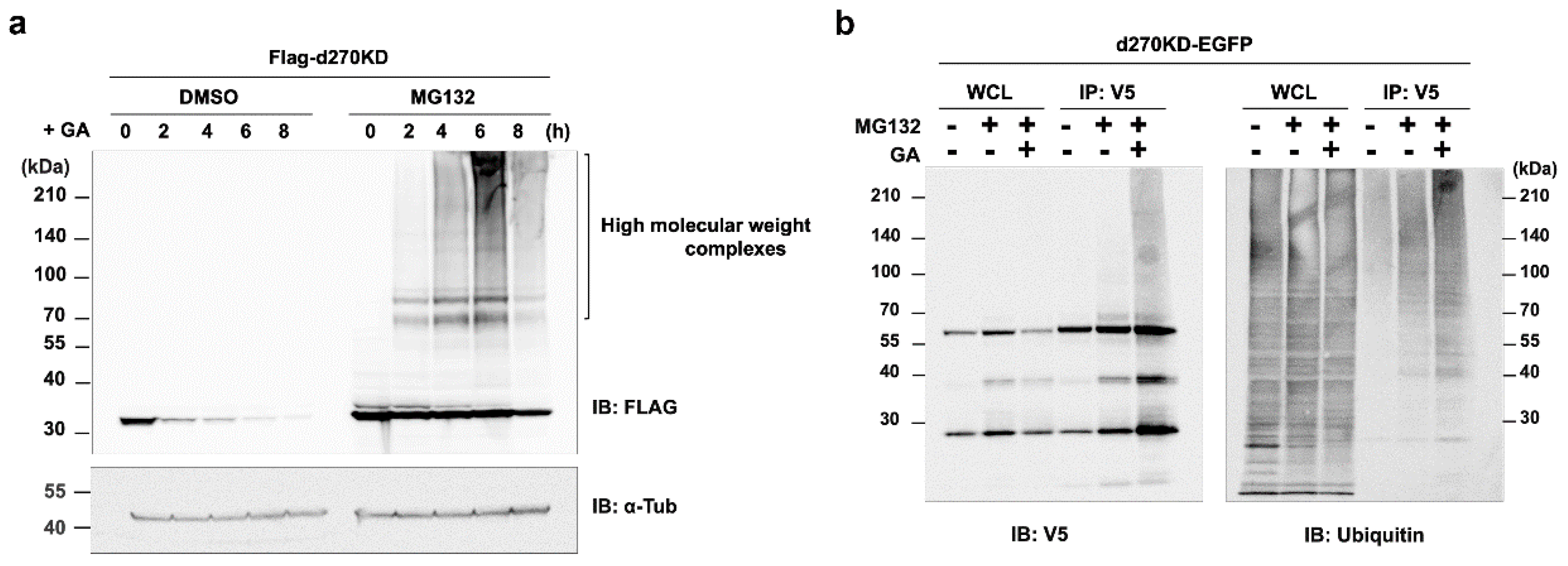
Figure 6.
Effects of the elimination of Mule/HUWE1 E3 ubiquitin ligase on the rapid degradation of d270KD stimulated by GA (a) HeLa cells were transfected with scrambled control siRNA (control) or Mule/HUWE1 siRNA, treated with cycloheximide (CHX; 20 μg/mL) 48 h later, and cell lysates were collected at the indicated times. The resulting cell lysates were subjected to a Western blot analysis with the indicated antibodies. (b, c) Scrambled control siRNA or Mule/HUWE1 siRNA was transfected into HeLa cells as in (a), and cells were transfected 24 h later with a full-length CHK1 expression vector fused with a Flag tag (b) or GFP-d270KD expression vector (c). Cells were treated 24 h later with cycloheximide (CHX; 20 μg/mL) (b) or 2 mM GA (c) for the indicated times, and cell lysates were collected for a Western blot analysis.
Figure 6.
Effects of the elimination of Mule/HUWE1 E3 ubiquitin ligase on the rapid degradation of d270KD stimulated by GA (a) HeLa cells were transfected with scrambled control siRNA (control) or Mule/HUWE1 siRNA, treated with cycloheximide (CHX; 20 μg/mL) 48 h later, and cell lysates were collected at the indicated times. The resulting cell lysates were subjected to a Western blot analysis with the indicated antibodies. (b, c) Scrambled control siRNA or Mule/HUWE1 siRNA was transfected into HeLa cells as in (a), and cells were transfected 24 h later with a full-length CHK1 expression vector fused with a Flag tag (b) or GFP-d270KD expression vector (c). Cells were treated 24 h later with cycloheximide (CHX; 20 μg/mL) (b) or 2 mM GA (c) for the indicated times, and cell lysates were collected for a Western blot analysis.
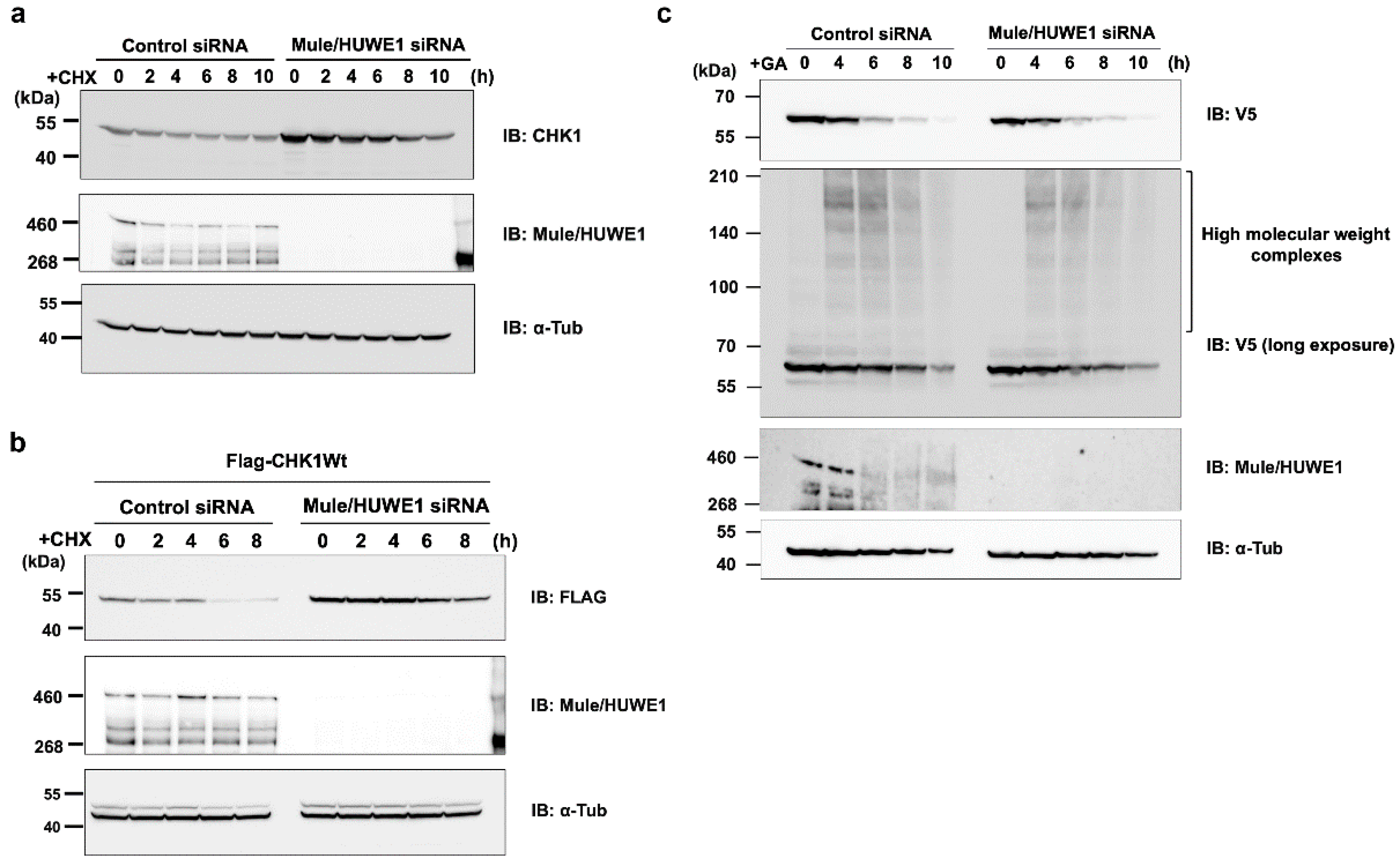
Figure 7.
All high-molecular-weight complexes of d270KD formed by a GA stimulation accumulate in the insoluble fraction of RIPA. (a) A Flag-d270KD expression vector was transfected into HeLa cells, and cells were stimulated 24 h later with 2 mM GA and collected at the indicated times. Cells were lysed in RIPA buffer and separated into a supernatant (RIPA-soluble fraction) and pellet (RIPA-insoluble fraction) by centrifugation. After the addition of SDS sample buffer to each fraction, samples from both fractions were analyzed by Western blotting using the antibodies indicated. (b) HeLa cells were treated with phosphate buffer (control) or 2 mM GA. After 8 h, cells were collected and lysed in RIPA buffer. Each cell lysate was separated into RIPA-soluble/-insoluble fractions in the same manner as in (a). Samples of both fractions were prepared by four independent experiments and subjected to a Western blot analysis using the indicated antibodies.
Figure 7.
All high-molecular-weight complexes of d270KD formed by a GA stimulation accumulate in the insoluble fraction of RIPA. (a) A Flag-d270KD expression vector was transfected into HeLa cells, and cells were stimulated 24 h later with 2 mM GA and collected at the indicated times. Cells were lysed in RIPA buffer and separated into a supernatant (RIPA-soluble fraction) and pellet (RIPA-insoluble fraction) by centrifugation. After the addition of SDS sample buffer to each fraction, samples from both fractions were analyzed by Western blotting using the antibodies indicated. (b) HeLa cells were treated with phosphate buffer (control) or 2 mM GA. After 8 h, cells were collected and lysed in RIPA buffer. Each cell lysate was separated into RIPA-soluble/-insoluble fractions in the same manner as in (a). Samples of both fractions were prepared by four independent experiments and subjected to a Western blot analysis using the indicated antibodies.
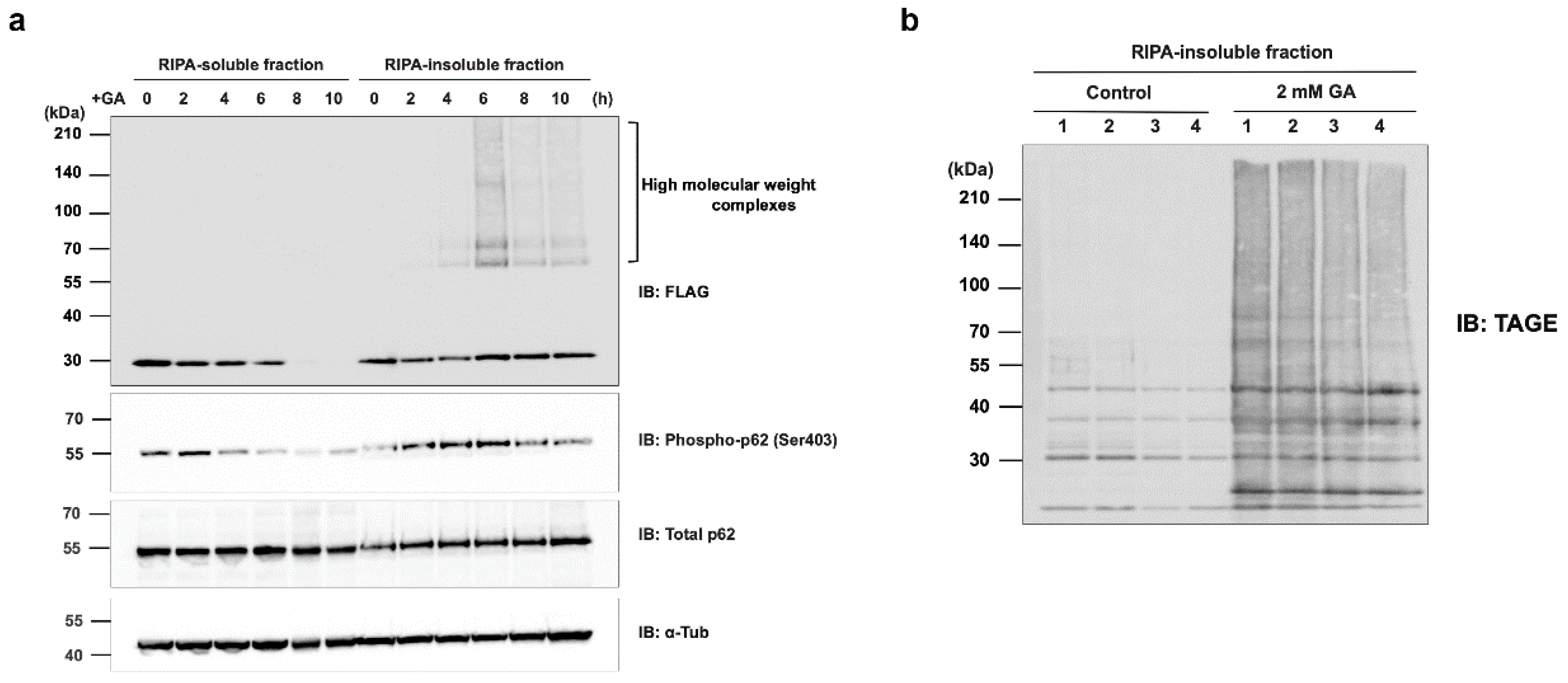
Figure 8.
Effects of the p62/SQSTM1 knockdown on the rapid GA-stimulated degradation of d270KD. HeLa cells were transfected with scrambled control siRNA or p62/SQSTM1 siRNA and transfected 24 h later with a Flag-d270KD expression vector. Cells were stimulated 24 h later with 2 mM GA for the indicated times. RIPA buffer was added to cells and cell lysates were separated into RIPA-soluble (a) and RIPA-insoluble (b) fractions by centrifugation. Each fraction was analyzed by Western blotting using the indicated antibodies.
Figure 8.
Effects of the p62/SQSTM1 knockdown on the rapid GA-stimulated degradation of d270KD. HeLa cells were transfected with scrambled control siRNA or p62/SQSTM1 siRNA and transfected 24 h later with a Flag-d270KD expression vector. Cells were stimulated 24 h later with 2 mM GA for the indicated times. RIPA buffer was added to cells and cell lysates were separated into RIPA-soluble (a) and RIPA-insoluble (b) fractions by centrifugation. Each fraction was analyzed by Western blotting using the indicated antibodies.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



