Submitted:
25 May 2023
Posted:
26 May 2023
You are already at the latest version
Abstract
Human induced pluripotent stem cell (hiPSC)-derived neural cells have been used at the preclinical stage of drug development. As previously reported, hiPSC-derived neurons exhibit greater tolerance to excitotoxicity than that of primary cultures of rodent neurons; however, the underlying mechanisms remain unknown. We therefore investigated the functions of L-glutamate (L-Glu) transporters, the most important machinery used to maintain low extracellular L-Glu concentrations, in hiPSC-derived neural cells. We also clarified the contribution of each L-Glu transporter subtype. At 63 days in vitro (DIV), we detected neuronal circuit functions in hiPSC-derived neural cells by a microelectrode array system (MEA). Exposure to 100 μM L-Glu for 24 hrs did not affect the viability of these 63 DIV neural cells. Pharmacological inhibition of excitatory amino acid transporter 1 (EAAT1) and EAAT2 blocked almost 100% of L-Glu uptake. In this condition, L-Glu exposure dramatically decreased cell viability. These results suggest that in hiPSC-derived neural cells, EAAT1 and EAAT2 are predominant L-Glu transporters, and their uptake potentials are stronger than those of primary cultures of rodent neurons. Furthermore, hiPSC-derived neural cells may be useful for screening drugs that target L-Glu transporters.
Keywords:
excitotoxicity
; human induced pluripotent stem cell
; neuron
; astrocyte
; L-glutamate transporter
; EAAT1
; EAAT2
Introduction
Currently, a low number of new drugs are within the final steps in the drug development process, which requires approximately 10 years of research and more than $800 million [1]. The low success rate is partly due to safety problems and inappropriate therapeutic indices. Human induced pluripotent stem cell (hiPSC) technology is expected to provide a breakthrough to solve this problem. In 2006, Yamanaka et al. developed iPSC technology in which adult somatic cells were reprogrammed to pluripotent stem-like cells through the introduction of four genes (OCT4, SOX2, KLF4, MYC) [2]. These iPSC-derived tissue-specific cells are expected to bridge animal models and human tissues with multifactorial and multigenic features. In addition, hiPSC-derived neural cells are being used in safety assessment and drug screening [3] because one-third of safety issues in clinical trials were attributed to central nervous system (CNS)-related problems [4-6]. However, little information is available concerning the molecular functions of hiPSC-derived astrocytes. In a study using astrocytes prepared from acutely resected surgical tissue, human astrocytes were found to be larger, structurally more complex, and more diverse than those of rodents [7], suggesting that neuron-astrocyte interactions also exhibit differences.
In the CNS, there is a neuron-specific cell death mechanism called excitotoxicity, in which excessive concentrations of the extracellular excitatory neurotransmitter L-glutamate (L-Glu) activate N-methyl-D-aspartate receptors (NMDARs) and cause acute Ca2+ influx, leading to neuronal death [8]. The machinery necessary to reproduce excitotoxicity should therefore be installed in the safety assessment and drug screening systems for the CNS. To date, most in vitro preclinical studies have employed a primary culture of rodent neural cells and their equivalent cell line systems, in which excitotoxic neurons are reproduced by exposure to L-Glu at micromolar concentrations [9]. On the other hand, the excitotoxicity levels remain intermediate in hiPSC-derived neurons even when exposed to much higher concentrations of L-Glu. In this study, we focused on L-Glu transporter functions in hiPSC-derived neural cells. Under physiological conditions, L-Glu transporters (excitatory amino acid transporters: EAATs in humans) rapidly remove extracellular L-Glu in synaptic transmission and protect neurons from excitotoxicity [10, 11]. In the present study, we examined whether astrocytes were differentiated in hiPSC-derived neural cells and whether EAATs helped remove extracellular L-Glu in this culture system. Here, we showed that in hiPSC-derived neural cells, functional astrocytes are differentiated and express L-Glu transporters. Furthermore, the functions of L-Glu transporters are potent enough to remove exogenously applied L-Glu and prevent excitotoxicity.
Results
Astrocytes Are Differentiated in hiPSC-Derived Neural Cultures.
Here, we used commercially available hiPSC-derived neuronal precursors (XCL-1 neurons, XCell Science, Novato, USA). We first examined the expression of markers for stem cells, neurons, and astrocytes at the mRNA and protein levels at 0 (just before seeding), 14, and 63 days in vitro (DIV). The gene expression of NESTIN, a neuronal stem cell marker [12], was decreased with the culture period (Figure 1A-a1). The gene expression of MAP2, a neuronal marker [13], was transiently increased at 14 DIV and then decreased at 63 DIV (Figure 1A-a2). On the other hand, the gene expression levels of GFAP and S100β, astrocyte markers [14, 15], were increased with the culture period (Figure 1A-a3 and a4). When we performed western blotting analysis (Figure 1B) and immunocytochemistry (Figure 1C), similar tendencies were observed. As shown in western blotting data, the expression level of HuC/D, a neuronal marker [16], was slightly decreased with the culture period (Figure 1B-b1 and b2). Immunocytochemical data showed HuC/D+ neurons at 14 and 63 DIV (Figure 1C). On the other hand, the expression of GFAP and S100β was clearly increased with the culture period (Figure 1B-b1, b3 and b4) in western blotting. In the immunocytochemical data, an increase in the numbers of GFAP+ and S100β+ cells was clearly observed (Figure 1C). These results indicate that in hiPSC-neural cell culture, astrocytes are also differentiated, while stemness is decreased with the culture period.
Functional Maturation of hiPSC-Derived Neural Networks
We investigated the functional neuronal maturation of hiPSC-derived neurons over time in culture using a microelectrode array (MEA) system. As shown in the raster plot, network bursts with very low firing densities were observed for only some electrodes at 28 DIV. On the other hand, at 56 DIV, a high frequency of network bursts with high synchronous and high firing density was observed at all electrodes (Figure 2A-a1). The difference in firing density between 28 DIV and 56 DIV was more evident when observed with the array wide spike detection rate (AWSDR, Figure 2A-a2). Four parameters, i.e., number of wells with network activity, total spikes per min, number of network bursts (NBs) per min, and spikes in an NB, were compared to examine functional maturation over time between 14, 21, 28, 42, and 56 DIV. NBs were detected after 28 DIV, and the number of wells with network activity increased with culture days (Figure 2A-a3). Total spikes per min, number of NBs per min, and spikes in an NB increased with culture days (Figure 2A-a4, a5 and a6). These results indicate that in hiPSC-derived neural cell culture, neural networks began to form at approximately 28 DIV and reached functional maturation by 56 DIV.
We also confirmed synaptic maturation at 63 DIV by immunocytochemical experiments. The presynaptic protein synapsin 1 (Syn1) puncta merged with vesicular glutamate transporter 2 (Vglut2) puncta (yellow arrowheads in Figure 2B-b1), indicating the maturation of the glutamatergic presynapse structure. Syn1 puncta were closely adjoined postsynaptic density protein 95 (PSD95) puncta (yellow arrowhead in Figure 2B-b2). These results indicate that glutamatergic synaptic structures were mature at 63 DIV. Taking the MEA data and the immunocytochemical data together, we judged that functional neural circuits had been formed by 63 DIV in hiPSC-derived neural cell culture.
The Roles of EAATs in the Sensitivity of hiPSC-Derived Neurons to Excitotoxicity.
When the extracellular L-Glu concentration is elevated beyond control, Ca2+ overload is induced in neurons through NMDAR and neuronal death, which is called ‘excitotoxicity’ [17-20]. Excitotoxicity has been reported to be the critical step in various kinds of CNS disorders [21, 22]. Using in vitro neuronal cultures in preclinical studies is therefore desirable for installing the machinery necessary for reproducing excitotoxicity. Although hiPSC-derived neural cell cultures have been developed for translational studies between preclinical and clinical studies, the information concerning the sensitivity of these cells to excitotoxicity is insufficient. We first examined the effects of 24 hr application of 100 µM L-Glu, the typical experimental condition that causes almost 100% neuronal death in primary culture of rodent neurons [23, 24] at 14 DIV and 63 DIV. The cell viability was quantified by the extent of MTT reduction [25]. As shown in Figure 3A-a1 and a2, a 24 hr application of 100 μM L-Glu generated no effects on the cell viabilities at both 14 and 63 DIV. L-Glu transporters remove the excitatory neurotransmitter L-Glu from the synaptic cleft just after release and maintain extracellular L-Glu concentration homeostasis [10, 11]. We therefore examined the contribution of EAATs to the tolerance of hiPSC-derived neural cell cultures to high concentrations of L-Glu. TFB-TBOA (TFB) blocks EAAT1-3 and mainly blocks EAAT1 and 2 at 30 nM (Table S1) [26]. When 30 nM TFB-TBOA was coapplied with 100 μM L-Glu for 24 hr at 14 DIV, it caused no additive effects on L-Glu alone (Figure 3B-b1). In contrast, at 63 DIV, coapplication of TFB and L-Glu significantly decreased MTT reduction compared with that of L-Glu alone (Figure 3B-b2). AP5 (100 µM), an NMDAR antagonist, almost completely abrogated the effects of TFB (Figure 3B-b2). These results indicate that L-Glu transporters are active in hiPSC-derived neural cell culture at 63 DIV; thus, we investigated the uptake potential of L-Glu transporters in this culture. We measured the extracellular concentrations of L-Glu at 0 min, 10 min, 20 min, 30 min, and 60 min after application at 100 µM (Figure 3C-c1). L-Glu concentration started to decrease immediately after the application, reaching 14.8±1.0 µM (n=3) 60 min later. We also investigated the contribution of each EAAT subtype pharmacologically using 100 µM UCPH-101 (UCPH, a selective EAAT1 inhibitor) [27], 300 µM dihydrokainic acid (DHK, a competitive EAAT2 inhibitor) [28], 30 nM TFB, and 10 µM WAY213613 (WAY, a potent competitive EAAT2 inhibitor with inhibition of EAAT1 and EAAT3 at 10 µM) [29]. The IC50 values for EAAT1-3 of these compounds are shown in Table S1. Figure 3C-c2 compares the inhibitory potentials of these inhibitors at 30 min of L-Glu uptake, and a 50% decrease in extracellular L-Glu was observed (Figure 3C-c1). The inhibition of UCPH, DHK, TFB, and WAY was 39.4±18.6% (n=5), 48.9±11.0% (n=5), 89.5±9.7% (n=5), and 80.5±17.5% (n=7), respectively. The effect of DHK was slightly stronger than that of UCPH, while the effect of TFB was much stronger than those two inhibitors. Because the effect of WAY is slightly weaker than that of TFB, the contribution of EAAT3 was small. These results strongly suggest that extracellular L-Glu was removed mainly by EAAT1 and EAAT2. We further confirmed the contributions of the L-Glu transporter subtype to L-Glu uptake in this hiPSC-derived neural cell culture by analyzing the correlation between the inhibitory potentials and the decrease in cell viabilities (MTT reductions) (Fig. 3D). The blue (UCPH), green (DHK), pink (WAY), and red (TK) dots were distributed from left to right. The inverse correlation between the inhibitory potentials and the decrease in viabilities was significant, as shown by Pearson’s correlation coefficient (PCC=-0.7110), further supporting that EAAT1 and EAAT2 play main roles in preventing excitotoxicity. In addition, the contribution of EAAT2 tended to be stronger than that of EAAT1.
The Expression of EAAT1 and EAAT2 in hiPSC-Derived Neural Cells.
Finally, we confirmed the expression of EAAT1 or EAAT2 in hiPSC-derived neural cell cultures at 14 and 63 DIV. qRT‒PCR analysis showed that the gene expression levels of EAAT1 (Figure 4A-a1) and EAAT2 (Figure 4A-a2) were increased during the culture period and that the expression of EAAT2 was more acutely increased than that of EAAT1. We also confirmed that the protein expression levels of EAAT1 and EAAT2 were culture period-dependent by western blotting (Figure 4B). We then investigated the cell types that expressed EAAT1 (Figure 4C-c1) or EAAT2 (Figure 4C-c2) at 63 DIV by immunocytochemistry. We determined the cell types based on the expression patterns of marker proteins, i.e., GFAP+Nestin+ for radial glial cells, GFAP+S100β+ for astrocytes, and HuC/D+ or MAP2+ for neurons. Representative images of the immunocytochemistry are shown in Figure 4C. EAAT1 was mainly expressed in radial glia and astrocytes (Figure 4C-c1), while EAAT2 was expressed in radial glia, astrocytes, and neurons (Figure 4C-c2). The hiPSC-derived neurons should be protected from excitotoxicity by these L-Glu transporters.
Discussion
Patterns of the Expression of Developmental Markers and L-Glu Transporters
In hiPSC-derived neural cell culture, the expression of MAP2 was increased at first, followed by an increase in the expression of GFAP and S100β, while the expression of nestin was gradually decreased. The gliogenesis of protoplasmic astrocytes is a late event in the human fetal period that occurs after neurogenesis [30], in which process radial glial cells play main roles [31, 32]. These results suggest that the cellular differentiation pattern in these hiPSC-derived neural cells follows CNS developmental steps. In vivo observations indicated that EAAT1 protein is expressed in radial glia in the periventricular zone [33] and in GFAP-positive astrocytes in the cerebral cortex [34], while EAAT2 protein is expressed in both neurons and astrocytes in the cerebral cortex [34]. The expression patterns of EAAT1 and EAAT2 were also consistent with those of the in vivo forebrain. Taken together, the results indicate that the hiPSC-derived neural cells used in this study are differentiated following the physiological developmental process.
EAAT Subtypes Contribute to Tolerance to Excitotoxicity in hiPSC-Derived Neurons
EAATs have 5 subtypes, EAAT1, 2, 3, 4, and 5. Our data show that EAAT1 and EAAT2 are the main L-Glu transporters that remove extracellular L-Glu in hiPSC-derived neural cell culture. It has been reported that when astrocytes are differentiated from iPSCs, over 2-3 months are needed to express EAAT1 [35-37]. Our data indicate that the astrocytes differentiated in the iPSC-derived neural cell culture express both functional EAAT1 and EAAT2 at 63 DIV, and these L-Glu transporters protect neurons from excitotoxicity caused by 100 µM L-Glu, the experimental condition that has been routinely used to cause almost 100% cell death in primary culture of rodent neurons [23, 24].
In this study, we detected EAAT1 expression mainly in astrocytes, while EAAT2 was expressed in both astrocytes and neurons. Rodent studies have shown that the protein expression levels of GLAST (EAAT1 in humans) and GLT1 (EAAT2 in humans) are dramatically increased during synaptogenesis beginning from E18 [38]. Although GLAST and GLT1 are astrocytic L-Glu transporters, GLT1 was also reported to be expressed in neurons [39]. A study using synaptosomes showed that neuronal GLT1 significantly contributes to L-Glu uptake [40]. Therefore, the expression pattern of EAAT1 and EAAT2 in hiPSC-derived neural cells is consistent with that in vivo. Furthermore, although GLT1 (EAAT2) accounts for ~90% of L-Glu uptake in the forebrain [41], cultured pure astrocytes express only GLAST (EAAT1) [40]. On the other hand, because the expression of both GLAST (EAAT1) and GLT1 (EAAT2) was detected in mixed cultures of astrocytes and neurons [40], the expression of GLT1 (EAAT2) may be regulated by soluble factors from neurons [42-45]. These mechanisms may be active in this hiPSC-derived neural cell culture. Regarding the significance of GLT1 (EAAT2) in L-Glu clearance at excitatory synapses, hiPSC-derived neural cells may provide useful neuropharmacological assays targeting L-Glu transporters, especially EAAT2.
The Significance of Our Data for Preclinical St udies
Before human stem cell-derived neurons appeared, in vitro toxicity and safety tests were performed using rodent primary cultures [46-49]. In these primary neuron cultures, exposure to 100 µM L-Glu caused severe excitotoxicity, thereby leading to almost 100% neuronal death in both neuronal culture and neuron-glia coculture. To date, although excitotoxicity is reproduced in hiPSC-derived neurons, the concentration of L-Glu used is very high (>1 mM), and the resulting cell damage is small. [50, 51]. In this study, when L-Glu transporters (mainly EAAT1 and 2) were inhibited, 100 µM L-Glu induced a significant decrease in the viability of hiPSC-derived neural cells. We also confirmed that the decrease in cell viability was mediated by NMDAR. In our previous report, NMDAR-mediated Ca2+ influx was also induced in hiPSC-derived neurons [51]. Our data are important for the application of hiPSC-derived neural cells in preclinical studies to characterize the mechanisms underlying the tolerance of hiPSC-derived neurons to excitotoxicity, i.e., the large contribution of L-Glu transporters. However, we should avoid discussing how species exhibit differences in sensitivity to excitotoxicity because the neuron-glia composition and developmental stage of each cell are completely different between primary cultures of rodent neurons and hiPSC-derived neural cells.
- Further utilization of hiPSC-derived neural cells in drug development
Astrocytes are involved in brain functions and the mechanisms of neurodegenerative diseases. One of the important homeostatic processes that astrocyte control involves controlling the extracellular concentration of L-Glu by L-Glu transporters. Deregulated expression of L-Glu transporters has been reported in various kinds of neurodegenerative diseases, such as Huntington’s disease (EAAT2 [52, 53]), amyotrophic lateral sclerosis (ALS) (EAAT2 [54, 55]), Alzheimer’s disease (AD) ([56, 57]) and neuropsychiatric diseases such as schizophrenia (SCZ) (EAAT2 [58]). EAAT2 accounts for ~90% of L-Glu uptake in the frontal cortex and prevents excitotoxicity. Because loss of EAAT2 functions leads to excitotoxic neuronal death in the above diseases, [52, 55, 58, 59], L-Glu transporters remain drug development targets for neuroprotection [60]. Ceftriaxone increases EAAT2 activity in rodent brains and has long been expected to be a breakthrough in the treatment of ALS. However, despite promising stage-2 efficacy data, stage-3 ceftriaxone in an ALS clinical study failed to show clinical efficacy [61]. One lessons learned from these cases is that in vitro preclinical assays with high human predictability should be employed. In this regard, our study suggests that hiPSC-derived neural cells may be promising candidates for preclinical in vitro tests with high human predictability.
Materials and Methods
Materials and Methods
All chemical compounds were purchased from FUJIFILM Wako Pure Chemical (Oosaka, Japan) unless otherwise stated. Protease inhibitor cocktail set1 (539131) was purchased from Calbiochem (Darmstadt, Land Hessen, Germany). Can Get SignalTM was purchased from Toyobo (Osaka, Japan). Stripping buffer was purchased from Thermo Fisher Scientific (Massachusetts, USA). Bromophenol blue sodium salt (BPB), bovine serum albumin (BSA), dimethyl sulfoxide (DMSO), β-nicotinamide adenine dinucleotide (β-NAD), dihydrokainic acid (DHK, an EAAT2-specific inhibitor), 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetra-zolium bromide (MTT), 1-methoxy-5-methyl-phenazinium methyl sulfate (MPMS), and sodium dodecyl sulfate (SDS) were purchased from Sigma‒Aldrich (Darmstadt, Germany). (3s)-3-[[3-[[4-(trifluoromethyl)benzoyl]amino]phenyl]methoxy]-l-aspartic acid (TFB-TBOA, TFB, a nonspecific EAAT inhibitor), D-(-)-2-amino-5-phosphonopentanoic acid (AP5, NMDAR antagonist), and N-[4-(2-bromo-4,5-difluorophenoxy)phenyl]-L-asparagine (WAY213613, a potent EAAT2 inhibitor) were purchased from TOCRIS (Minneapolis, USA). 2-Amino-5,6,7,8-tetrahydro-4-(4-methoxyphenyl)-7-(naphthalen-1-yl)-5-oxo-4H-chromene-3-carbonitrile (UCPH-101, UCPH, an EAAT1-specific inhibitor) was purchased from Abcam (Cambridge, UK). Bovine liver glutamate dehydrogenase (GLUD1) was purchased from Roche (Mannheim, Germany). Tris (hydroxymethyl) aminomethane (Tris–HCl) was purchased from Bio-Rad (California, USA). Ethylenediaminetetraacetate (EDTA) and ethylene glycol tetraacetate (EGTA) were purchased from Dojindo (Kumamoto, Japan). Goat serum was purchased from Vector Laboratories (California, USA). Donkey serum was purchased from Rockland Immunochemicals (Pennsylvania, USA).
Culture of hiPSC-Derived Neurons
Commercially available hiPSC-neurons were used in this study (XCL-1 neurons, XCell Science). The hiPSC-neurons were cultured according to the manufacturer’s instructions with modifications. Cells were plated in 48-well plastic plates, 8-well glass chambers (155409JP, Nunc, Massachusetts, USA), 16 channels per well across 24-well MEA plates (eco24, MED-Q2430M, Alpha Med Scientific, Oosaka, Japan), or 96-well plastic plates at a density of 3.0 × 105 cells/cm2 in neural maturation basal medium (NM-001-BM100, XCell Science) with neuron maturation supplement A (NM-001-SA100, XCell Science). Plates were precoated with polyethyleneimine (PEI) (For MEA, 0.005% for 10 min at 37 °C; for others, 0.025% for 1 hr at room temperature; P3413, Sigma‒Aldrich) and then coated with Matrix-511 (2.5 µg/ml for 3 hrs at 37 °C, 892011, Nippi, Tokyo, Japan). For WB, ICC and MEA cultures, a glass ring with a diameter of 3.4 mm (Ring-05, Iwaki, Shizuoka, Japan) was placed in the center of the well, and cell suspensions were seeded in the ring. Half of the medium was replaced every 2 days. After 8 days of culture, the medium was replaced with BrainPhysTM Neuronal medium with SM1 neuronal supplement (STEMCELL Technologies, Vancouver, Canada). Half of the medium was replaced every 4 days. All experiments using hiPSC-neurons were approved by the Research Ethics Committee of National Institute of Health Sciences (NIHs) in accordance with the Declaration of Helsinki.
Quantitative Real-Time Reverse Transcription Polymerase Chain Reaction (Real-Time PCR)
Total RNA was isolated from cells using TRIzol reagent (Sigma‒Aldrich). The amount of total RNA was quantified by measuring the OD260 using a Nanodrop spectrophotometer (Nanodrop, Thermo Fisher Scientific). Real-time PCR was performed using the QuantiTect SYBR Green RT‒PCR kit (Qiagen, Hilden, Germany) and an ABI PRISM 7900HT sequence detection system (Thermo Fisher Scientific) according to the manufacturer’s protocol. The reactions (20 μL) contained 5 ng of total RNA and 0.5 μM forward and reverse primers in the master mix solution. The data were analyzed with 7900 System SDS Software 2.2.2 (Thermo Fisher Scientific) by relative quantification using the comparative CT method. Relative changes in transcript levels were normalized to the mRNA levels of β-Actin.
The primer sequences were as follows: 5'-CCAAGACTGCCCTGGAAAC-3', 5'-CCTCCCTCTCCAAGGAAACA-3' (NESTIN), 5'-CTCAGCACCGCTAACAGAGG-3', 5'-CATTGGCGCTTCGGACAAG-3' (MAP2) 5'-AGGTCCATGTGGAGCTTGAC-3', 5'-GCCATTGCCTCATACTGCGT-3' (GFAP), 5'-TGGCCCTCATCGACGTTTTC-3', 5'-ATGTTCAAAGAACTCGTGGCA-3' (S100β), 5'-CATGTACGTTGCTATCCAGGC-3' 5'-CTCCTTAATGTCACGCACGAT-3' (β-Actin), 5'-ATGAGGATGTTACAGATGCTGG-3' 5'-CAGGATGGATGATGATGACAAT-3' (EAAT1), 5'-CTGTTGTCTCTCTGTTGAACG-3', 5'-CAACCACTTCTAAGTCCTTGATTG-3' (EAAT2).
Western Blotting
The cells were lysed with NP-40 lysis buffer [150 mM NaCl, 10 mM EDTA, 5 mM EGTA, 0.5% NP-40, and 0.5% sodium deoxycholate in 10 mM Tris–HCl buffer (pH 6.8)]. The protein concentration was measured using a BCA protein assay (Pierce™ BCA Protein Assay Kit, 23225, Thermo Fisher Scientific). The proteins (10 μg/lane) were resolved with SDS‒PAGE and transferred onto a PVDF membrane (Bio-Rad, California, USA). The membrane was blocked with 3% nonfat dry milk or Block Ace blocking solution (DS Pharma Biomedical, Osaka, Japan) for 1 hr at room temperature. The membrane was incubated with mouse anti-HuC/D monoclonal antibody (1:100, A21271, Thermo Fisher Scientific), mouse anti-GFAP monoclonal antibody (1:5000, MAB3402, Millipore, Hessen, Germany), rabbit anti-S100β polyclonal antibody (1:5000, A5971, Sigma‒Aldrich), rabbit anti-EAAT1 polyclonal antibody (1:1000, 5684, Cell Signaling, Massachusetts, USA), guineapig anti-EAAT2 polyclonal antibody (1:5000, AB1783, Chemicom), or mouse anti β-actin monoclonal antibody (1:5000, ab8226, Abcam) overnight at 4 °C followed by incubation with horseradish peroxidase-conjugated anti-mouse, anti-rabbit (1:20,000, Amersham Biosciences, Buckinghamshire, UK), or guineapig (1:50,000, Invitrogen, Massachusetts, USA) antibodies. The signals were scanned with an LAS3000 (Fujifilm, Tokyo, Japan) using an ECL western blot detection system (SuperSignalTM West Femto maximum Sensitivity Substrate, 34095, Thermo Fisher Scientific). Relative densities of bands were quantified using Multi Gauge software (FUJIFILM). Relative changes in expression were determined by normalization to β-actin.
Immunocytochemistry Scale
The cells were fixed with 4% PFA for 1 hr at room temperature. After a PBS wash was performed, the cells were immunostained using the AbScale clearing/labeling protocol [62]. The fixed cells were permeabilized and cleared with sequential incubation in multiple solutions, ScaleS0, ScaleA2, ScaleB4(0), and ScaleA2. Then, after deScaling by washing with PBS, the cells were incubated with primary antibodies for 3 days at 4 °C in an AbScale solution. Mouse anti-HuC/D monoclonal antibody (1:100, A21271, Thermo Fisher Scientific), chicken anti-GFAP polyclonal antibody (1:400, ab4674, Abcam), rabbit anti-S100β polyclonal antibody (1:500, ab52642, Abcam), goat anti-Vglut2 polyclonal antibody (1:500, Go-Af310-1, Frontier Institute, Hokkaido, Japan), rabbit anti-Syn1 polyclonal antibody (1:1000, AB1543, Chemicon), chicken anti-MAP2 polyclonal antibody (1:5000, ab5392, Abcam), mouse anti-PSD95 monoclonal antibody (1:500, 7E3-1B8, Thermo Fisher Scientific), mouse anti-EAAT1 monoclonal antibody (1:200, ab49643, Abcam), rabbit anti-Nestin polyclonal antibody (1:200, ABD69, Millipore), or guineapig anti-EAAT2 polyclonal antibody (1:1000, AB1783, Chemicon) were used. The cells were incubated with secondary antibodies conjugated to fluorochromes (1:500, Invitrogen) and Hoechst (1:200, Dojindo, Kumamoto, Japan) for 2 days at 4 °C. Before refixation with 4% PFA, cells were rinsed in an AbScale rinse solution. Finally, cells were optically cleared by incubation in ScaleS4. Fluorescent images of the cells were obtained by confocal microscopy (A1R, Nikon, Tokyo, Japan). The composition of the solution was as follows: ScaleS0 (a PBS(–) solution containing 20% D-(–)-sorbitol, 5% glycerol, 1 mM methyl-β-cyclodextrin, 1 mM γ-cyclodextrin, 1 mM N-acetyl-L-hydroxyproline, 3% DMSO, pH 7.2), ScaleA2 (10% glycerol, 4 M urea, 0.1% Triton X-100, pH 7.7), ScaleB4(0) (8 M urea, pH 8.4), AbScale (a PBS(–) solution containing 0.33 M urea, 0.1–0.5% Triton X-100, pH 7.2), AbScale rinse solution (a 0.1× PBS(–) solution containing 2.5% BSA, 0.05% (w/v) Tween-20), and ScaleS4 (40% D-(–)-sorbitol, 10% glycerol, 4 M urea, 15-25% DMSO, pH 7.9).
MEA Recording and Data Analysis (Extracellular Recording, Burst Analysis)
Spontaneous extracellular field potentials were acquired at 37 °C under a 5% CO2 atmosphere using a 24-well MEA system (Presto, Alpha Med Scientific, Oosaka, Japan) at a sampling rate of 20 kHz/channels. Signals were high-pass filtered at 0.1 Hz and stored on a personal computer. The spikes in the acquired data were detected using the 100 Hz high-pass filter. Spontaneous firing was recorded at 14 DIV, 21 DIV, 28 DIV, 42 DIV, and 56 DIV. Electrophysiological activities were analyzed using Presto software (Alpha Med Scientific, Oosaka, Japan). A spike was counted when the extracellularly recorded signal exceeded a threshold of ±5.3 σ, where σ was the standard deviation of the baseline noise during quiescent periods. Network bursts (NBs) were detected using the 4-step method, which was described previously [63]. First, spikes separated by interspike intervals of 10 ms were attributed to the same NB. Second, datasets with a maximum number of spikes in the NB below 3 spikes/NB were eliminated from the analysis. Third, NBs separated by inter-NB intervals shorter than 300 ms were combined. Finally, an NB was defined when it contained more than 50 spikes/NB. All data are expressed as the mean ± standard deviation.
MTT Reduction Assays
Cell viability was determined by MTT reduction activity. MTT reduction activity was measured according to a previously described method [25, 59]. Briefly, hiPSC-neurons cultured in the presence or absence of EAAT inhibitor were exposed to L-Glu (100 μM) for 24 hrs. MTT was added to each well at 15 μg and incubated for 15 min at 37 °C. The medium in each well was carefully removed, and 50 µl of DMSO was added to dissolve the reaction product (MTT formazan). The amount of MTT formazan was determined by measuring the absorbance at 570 nm (test wavelength) and 655 nm (reference wavelength) with an iMarkTM microplate reader (Bio-Rad).
Measurement of the Extracellular L-Glu Concentration (L-Glu Uptake Assay)
The L-Glu concentration in the medium was measured according to a previously described method [59]. The medium in the 96-well plates was replaced with fresh medium containing 100 μM L-Glu. Twenty-five microliters of the medium per well was collected. The L-Glu concentration was measured by mixing the medium with 25 μL of substrate mixture [20 U/mL GLUD1, 2.5 mg/mL β-NAD, 0.25 mg/mL MTT, 100 μM MPMS, and 0.1% Triton X-100 in 0.2 M Tris–HCl buffer (pH 8.2)] and incubating at room temperature for 7 min. The reaction was stopped by adding 50 μL of stop solution [50% dimethylformamide and 20% SDS in purified water (DIRECT-Q, Millipore), pH 4.7]. The amount of MTT formazan was determined by measuring the absorbance at 570 nm (test wavelength) and 655 nm (reference wavelength) with an iMarkTM microplate reader (Bio-Rad). The extracellular L-Glu concentration was estimated from a standard curve constructed for each assay using cell-free medium containing known concentrations of L-Glu.
Drug Treatment
Stock solutions of 100 mM L-Glu, 25 mM DHK, and 100 mM AP5 in purified water and 50 mM TFB, 25 mM UCPH, and 100 mM WAY in DMSO were dissolved into the medium at the time of application. EAAT inhibitor and AP5 were incubated with the cells for 1 hr before the application of L-Glu (100 μM).
DMSO was used as a vehicle control. For the MTT reduction assay and L-Glu uptake activity assay, 0.4% DMSO, the maximum concentration used for drug dilution, was added to all test solutions.
Statistical Analysis
All data are expressed as the mean ± the SD. Statistical analyses were performed using Student’s t test or one-way measures analysis of variance (ANOVA) followed by Tukey’s post hoc test for multiple pairwise comparisons in GraphPad Prism (GraphPad Software, USA), as shown in the figure legends. In all the comparisons, the differences were considered statistically significant when P<0.05. All the experiments were repeated in triplicate, and the same results were obtained in all sessions.
The Pearson correlation coefficient (PCC) was used to assess the correlation between the strength of L-Glu uptake inhibition and the cell viability caused by EAAT inhibitors. The PCC was determined by linear regression analysis using GraphPad Prism. The correlation coefficient is a number between -1 and 1 that represents the strength and direction of the relationship between two variables.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. This research includes Supplemental Table S1.
Author Contributions
K.S. designed this work and wrote the paper. K.T. performed the experiments and analyzed the data. K.C. partly contributed to the experiments. I.S. and Y.I. analyzed the data and revised the paper. All authors approved the present version of the manuscript and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding
This research is supported in part by the Research Grant on Regulatory Harmonization and Evaluation of Pharmaceuticals, Medical Devices, Regenerative and Cellular Therapy Products, Gene Therapy Products, and Cosmetics from AMED, a grant for Research on Risks of Chemicals from the MHLW, Japan, awarded to K.S.
Institutional Review Board Statement
This study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Review Board and Ethics Committee of the National Institute of Health Sciences (protocol code #242 [2017.6.1-]).
Data Availability Statement
All data are contained within the manuscript.
Acknowledgments
We would like to thank the Consortium for Safety Assessment using Human iPS Cells (CSAHi) and International Life Sciences Institute (ILSI) Health and Environmental Sciences Institute (HESI) for the kind advice about the hiPSC neural culture and MEA recording.
Conflicts of Interest
The authors declare that they have no conflicts of interest with the contents of this article.
References
- Bonaventura, G.; Iemmolo, R.; Attaguile, G.A.; La Cognata, V.; Pistone, B.S.; Raudino, G.; D'Agata, V.; Cantarella, G.; Barcellona, M.L.; Cavallaro, S. iPSCs: A Preclinical Drug Research Tool for Neurological Disorders. International journal of molecular sciences 2021, 22. [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663-676. [CrossRef]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: a decade of progress. Nature reviews. Drug discovery 2017, 16, 115-130. [CrossRef]
- Cook, D.; Brown, D.; Alexander, R.; March, R.; Morgan, P.; Satterthwaite, G.; Pangalos, M.N. Lessons learned from the fate of AstraZeneca's drug pipeline: a five-dimensional framework. Nature reviews. Drug discovery 2014, 13, 419-431. [CrossRef]
- Onakpoya, I.J.; Heneghan, C.J.; Aronson, J.K. Post-marketing withdrawal of 462 medicinal products because of adverse drug reactions: a systematic review of the world literature. BMC medicine 2016, 14, 10. [CrossRef]
- Arrowsmith, J.; Miller, P. Trial watch: phase II and phase III attrition rates 2011-2012. Nature reviews. Drug discovery 2013, 12, 569. [CrossRef]
- Oberheim, N.A.; Takano, T.; Han, X.; He, W.; Lin, J.H.; Wang, F.; Xu, Q.; Wyatt, J.D.; Pilcher, W.; Ojemann, J.G.; et al. Uniquely hominid features of adult human astrocytes. J Neurosci 2009, 29, 3276-3287. [CrossRef]
- Michaelis, E.K. Molecular biology of glutamate receptors in the central nervous system and their role in excitotoxicity, oxidative stress and aging. Progress in neurobiology 1998, 54, 369-415. [CrossRef]
- Kritis, A.A.; Stamoula, E.G.; Paniskaki, K.A.; Vavilis, T.D. Researching glutamate - induced cytotoxicity in different cell lines: a comparative/collective analysis/study. Front Cell Neurosci 2015, 9, 91. [CrossRef]
- Danbolt, N.C. Glutamate uptake. Progress in neurobiology 2001, 65, 1-105.
- Zerangue, N.; Kavanaugh, M.P. Flux coupling in a neuronal glutamate transporter. Nature 1996, 383, 634-637. [CrossRef]
- Gilyarov, A.V. Nestin in central nervous system cells. Neurosci Behav Physiol 2008, 38, 165-169. [CrossRef]
- Sánchez, C.; Díaz-Nido, J.; Avila, J. Phosphorylation of microtubule-associated protein 2 (MAP2) and its relevance for the regulation of the neuronal cytoskeleton function. Progress in neurobiology 2000, 61, 133-168. [CrossRef]
- Zimmer, D.B.; Cornwall, E.H.; Landar, A.; Song, W. The S100 protein family: history, function, and expression. Brain research bulletin 1995, 37, 417-429. [CrossRef]
- Eng, L.F.; Ghirnikar, R.S.; Lee, Y.L. Glial fibrillary acidic protein: GFAP-thirty-one years (1969-2000). Neurochemical research 2000, 25, 1439-1451. [CrossRef]
- Akamatsu, W.; Okano, H.J.; Osumi, N.; Inoue, T.; Nakamura, S.; Sakakibara, S.; Miura, M.; Matsuo, N.; Darnell, R.B.; Okano, H. Mammalian ELAV-like neuronal RNA-binding proteins HuB and HuC promote neuronal development in both the central and the peripheral nervous systems. Proceedings of the National Academy of Sciences of the United States of America 1999, 96, 9885-9890.
- Lucas, D.R.; Newhouse, J.P. The toxic effect of sodium L-glutamate on the inner layers of the retina. AMA Arch Ophthalmol 1957, 58, 193-201. [CrossRef]
- Olney, J.W.; Ho, O.L.; Rhee, V. Cytotoxic effects of acidic and sulphur containing amino acids on the infant mouse central nervous system. Exp Brain Res 1971, 14, 61-76. [CrossRef]
- Olney, J.W.; Sharpe, L.G. Brain lesions in an infant rhesus monkey treated with monsodium glutamate. Science 1969, 166, 386-388. [CrossRef]
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623-634. [CrossRef]
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Archiv : European journal of physiology 2010, 460, 525-542. [CrossRef]
- Maragakis, N.J.; Rothstein, J.D. Glutamate transporters in neurologic disease. Archives of neurology 2001, 58, 365-370. [CrossRef]
- Rosenberg, P.A.; Amin, S.; Leitner, M. Glutamate uptake disguises neurotoxic potency of glutamate agonists in cerebral cortex in dissociated cell culture. J Neurosci 1992, 12, 56-61. [CrossRef]
- Choi, D.W.; Maulucci-Gedde, M.; Kriegstein, A.R. Glutamate neurotoxicity in cortical cell culture. J Neurosci 1987, 7, 357-368. [CrossRef]
- Abe, K.; Matsuki, N. Measurement of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction activity and lactate dehydrogenase release using MTT. Neurosci Res 2000, 38, 325-329.
- Shimamoto, K.; Sakai, R.; Takaoka, K.; Yumoto, N.; Nakajima, T.; Amara, S.G.; Shigeri, Y. Characterization of novel L-threo-beta-benzyloxyaspartate derivatives, potent blockers of the glutamate transporters. Mol Pharmacol 2004, 65, 1008-1015. [CrossRef]
- Abrahamsen, B.; Schneider, N.; Erichsen, M.N.; Huynh, T.H.; Fahlke, C.; Bunch, L.; Jensen, A.A. Allosteric modulation of an excitatory amino acid transporter: the subtype-selective inhibitor UCPH-101 exerts sustained inhibition of EAAT1 through an intramonomeric site in the trimerization domain. J Neurosci 2013, 33, 1068-1087. [CrossRef]
- Arriza, J.L.; Fairman, W.A.; Wadiche, J.I.; Murdoch, G.H.; Kavanaugh, M.P.; Amara, S.G. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci 1994, 14, 5559-5569.
- Dunlop, J.; McIlvain, H.B.; Carrick, T.A.; Jow, B.; Lu, Q.; Kowal, D.; Lin, S.; Greenfield, A.; Grosanu, C.; Fan, K.; et al. Characterization of novel aryl-ether, biaryl, and fluorene aspartic acid and diaminopropionic acid analogs as potent inhibitors of the high-affinity glutamate transporter EAAT2. Mol Pharmacol 2005, 68, 974-982. [CrossRef]
- Marín-Padilla, M. Prenatal development of fibrous (white matter), protoplasmic (gray matter), and layer I astrocytes in the human cerebral cortex: a Golgi study. J Comp Neurol 1995, 357, 554-572. [CrossRef]
- Noctor, S.C.; Flint, A.C.; Weissman, T.A.; Dammerman, R.S.; Kriegstein, A.R. Neurons derived from radial glial cells establish radial units in neocortex. Nature 2001, 409, 714-720. [CrossRef]
- Malatesta, P.; Hartfuss, E.; Götz, M. Isolation of radial glial cells by fluorescent-activated cell sorting reveals a neuronal lineage. Development (Cambridge, England) 2000, 127, 5253-5263. [CrossRef]
- Bar-Peled, O.; Ben-Hur, H.; Biegon, A.; Groner, Y.; Dewhurst, S.; Furuta, A.; Rothstein, J.D. Distribution of glutamate transporter subtypes during human brain development. J Neurochem 1997, 69, 2571-2580. [CrossRef]
- DeSilva, T.M.; Borenstein, N.S.; Volpe, J.J.; Kinney, H.C.; Rosenberg, P.A. Expression of EAAT2 in neurons and protoplasmic astrocytes during human cortical development. The Journal of comparative neurology 2012, 520, 3912-3932. [CrossRef]
- Lundin, A.; Delsing, L.; Clausen, M.; Ricchiuto, P.; Sanchez, J.; Sabirsh, A.; Ding, M.; Synnergren, J.; Zetterberg, H.; Brolen, G.; et al. Human iPS-Derived Astroglia from a Stable Neural Precursor State Show Improved Functionality Compared with Conventional Astrocytic Models. Stem cell reports 2018, 10, 1030-1045. [CrossRef]
- Santos, R.; Vadodaria, K.C.; Jaeger, B.N.; Mei, A.; Lefcochilos-Fogelquist, S.; Mendes, A.P.D.; Erikson, G.; Shokhirev, M.; Randolph-Moore, L.; Fredlender, C.; et al. Differentiation of Inflammation-Responsive Astrocytes from Glial Progenitors Generated from Human Induced Pluripotent Stem Cells. Stem cell reports 2017, 8, 1757-1769. [CrossRef]
- Krencik, R.; Weick, J.P.; Liu, Y.; Zhang, Z.J.; Zhang, S.C. Specification of transplantable astroglial subtypes from human pluripotent stem cells. Nature biotechnology 2011, 29, 528-534. [CrossRef]
- Holmseth, S.; Dehnes, Y.; Huang, Y.H.; Follin-Arbelet, V.V.; Grutle, N.J.; Mylonakou, M.N.; Plachez, C.; Zhou, Y.; Furness, D.N.; Bergles, D.E.; et al. The density of EAAC1 (EAAT3) glutamate transporters expressed by neurons in the mammalian CNS. J Neurosci 2012, 32, 6000-6013. [CrossRef]
- Furness, D.N.; Dehnes, Y.; Akhtar, A.Q.; Rossi, D.J.; Hamann, M.; Grutle, N.J.; Gundersen, V.; Holmseth, S.; Lehre, K.P.; Ullensvang, K.; et al. A quantitative assessment of glutamate uptake into hippocampal synaptic terminals and astrocytes: new insights into a neuronal role for excitatory amino acid transporter 2 (EAAT2). Neuroscience 2008, 157, 80-94. [CrossRef]
- Danbolt, N.C.; Furness, D.N.; Zhou, Y. Neuronal vs glial glutamate uptake: Resolving the conundrum. Neurochemistry international 2016. [CrossRef]
- Tanaka, K.; Watase, K.; Manabe, T.; Yamada, K.; Watanabe, M.; Takahashi, K.; Iwama, H.; Nishikawa, T.; Ichihara, N.; Kikuchi, T.; et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 1997, 276, 1699-1702.
- Gegelashvili, G.; Danbolt, N.C.; Schousboe, A. Neuronal soluble factors differentially regulate the expression of the GLT1 and GLAST glutamate transporters in cultured astroglia. Journal of neurochemistry 1997, 69, 2612-2615. [CrossRef]
- Gegelashvili, G.; Dehnes, Y.; Danbolt, N.C.; Schousboe, A. The high-affinity glutamate transporters GLT1, GLAST, and EAAT4 are regulated via different signalling mechanisms. Neurochemistry international 2000, 37, 163-170. [CrossRef]
- Plachez, C.; Danbolt, N.C.; Récasens, M. Transient expression of the glial glutamate transporters GLAST and GLT in hippocampal neurons in primary culture. Journal of neuroscience research 2000, 59, 587-593. [CrossRef]
- Martinez-Lozada, Z.; Guillem, A.M.; Robinson, M.B. Transcriptional Regulation of Glutamate Transporters: From Extracellular Signals to Transcription Factors. Advances in pharmacology (San Diego, Calif.) 2016, 76, 103-145. [CrossRef]
- Lau, A.C.; Cui, H.; Tymianski, M. The use of propidium iodide to assess excitotoxic neuronal death in primary mixed cortical cultures. Methods in molecular biology (Clifton, N.J.) 2007, 399, 15-29. [CrossRef]
- Semkova, I.; Krieglstein, J. Neuroprotection mediated via neurotrophic factors and induction of neurotrophic factors. Brain research. Brain research reviews 1999, 30, 176-188. [CrossRef]
- Bullock, R.; Zauner, A.; Woodward, J.J.; Myseros, J.; Choi, S.C.; Ward, J.D.; Marmarou, A.; Young, H.F. Factors affecting excitatory amino acid release following severe human head injury. J Neurosurg 1998, 89, 507-518. [CrossRef]
- O'Byrne, M.; Tipton, K.; McBean, G.; Kollegger, H. Assessment of neurotoxicity and "neuroprotection". J Neural Transm Suppl 1997, 50, 153-164. [CrossRef]
- Bauersachs, H.G.; Bengtson, C.P.; Weiss, U.; Hellwig, A.; García-Vilela, C.; Zaremba, B.; Kaessmann, H.; Pruunsild, P.; Bading, H. N-methyl-d-aspartate Receptor-mediated Preconditioning Mitigates Excitotoxicity in Human Induced Pluripotent Stem Cell-derived Brain Organoids. Neuroscience 2022, 484, 83-97. [CrossRef]
- Sato, K.; Takahashi, K.; Shigemoto-Mogami, Y.; Chujo, K.; Sekino, Y. Glypican 6 Enhances N-Methyl-D-Aspartate Receptor Function in Human-Induced Pluripotent Stem Cell-Derived Neurons. Front Cell Neurosci 2016, 10, 259. [CrossRef]
- Wilton, D.K.; Stevens, B. The contribution of glial cells to Huntington's disease pathogenesis. Neurobiology of disease 2020, 143, 104963. [CrossRef]
- Garcia, V.J.; Rushton, D.J.; Tom, C.M.; Allen, N.D.; Kemp, P.J.; Svendsen, C.N.; Mattis, V.B. Huntington's Disease Patient-Derived Astrocytes Display Electrophysiological Impairments and Reduced Neuronal Support. Frontiers in neuroscience 2019, 13, 669. [CrossRef]
- Tyzack, G.; Lakatos, A.; Patani, R. Human Stem Cell-Derived Astrocytes: Specification and Relevance for Neurological Disorders. Current stem cell reports 2016, 2, 236-247. [CrossRef]
- Rothstein, J.D.; Van Kammen, M.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Annals of neurology 1995, 38, 73-84. [CrossRef]
- Fontana, I.C.; Souza, D.G.; Souza, D.O.; Gee, A.; Zimmer, E.R.; Bongarzone, S. A Medicinal Chemistry Perspective on Excitatory Amino Acid Transporter 2 Dysfunction in Neurodegenerative Diseases. Journal of medicinal chemistry 2023, 66, 2330-2346. [CrossRef]
- Malik, A.R.; Willnow, T.E. Excitatory Amino Acid Transporters in Physiology and Disorders of the Central Nervous System. International journal of molecular sciences 2019, 20. [CrossRef]
- Heider, J.; Vogel, S.; Volkmer, H.; Breitmeyer, R. Human iPSC-Derived Glia as a Tool for Neuropsychiatric Research and Drug Development. International journal of molecular sciences 2021, 22. [CrossRef]
- Takaki, J.; Fujimori, K.; Miura, M.; Suzuki, T.; Sekino, Y.; Sato, K. L-glutamate released from activated microglia downregulates astrocytic L-glutamate transporter expression in neuroinflammation: the 'collusion' hypothesis for increased extracellular L-glutamate concentration in neuroinflammation. Journal of neuroinflammation 2012, 9, 275. [CrossRef]
- Colombo, E.; Pascente, R.; Triolo, D.; Bassani, C.; De Angelis, A.; Ruffini, F.; Ottoboni, L.; Comi, G.; Martino, G.; Farina, C. Laquinimod Modulates Human Astrocyte Function and Dampens Astrocyte-Induced Neurotoxicity during Inflammation. Molecules 2020, 25. [CrossRef]
- Cudkowicz, M.E.; Titus, S.; Kearney, M.; Yu, H.; Sherman, A.; Schoenfeld, D.; Hayden, D.; Shui, A.; Brooks, B.; Conwit, R.; et al. Safety and efficacy of ceftriaxone for amyotrophic lateral sclerosis: a multi-stage, randomised, double-blind, placebo-controlled trial. The Lancet. Neurology 2014, 13, 1083-1091. [CrossRef]
- Hama, H.; Hioki, H.; Namiki, K.; Hoshida, T.; Kurokawa, H.; Ishidate, F.; Kaneko, T.; Akagi, T.; Saito, T.; Saido, T.; et al. ScaleS: an optical clearing palette for biological imaging. Nat Neurosci 2015, 18, 1518-1529. [CrossRef]
- Matsuda, N.; Odawara, A.; Katoh, H.; Okuyama, N.; Yokoi, R.; Suzuki, I. Detection of synchronized burst firing in cultured human induced pluripotent stem cell-derived neurons using a 4-step method. Biochemical and biophysical research communications 2018, 497, 612-618. [CrossRef]
Figure 1.
Astrocytes are differentiated in hiPSC-derived neural culture. (A) The significant decrease in the mRNA expression level of NESTIN (a1) and increase in the expression of MAP2 (a2), GFAP (a3) and S100β (a4) along with culture days were confirmed by real-time PCR. **P < 0.01, ***P < 0.0001 vs. DIV 0 group (n=3), Tukey’s test following one-way ANOVA. (B) Representative immunoblot at 14 and 63 DIV (b1). The expression level of HuC/D protein tended to decrease with culture days (b2). The protein expression levels of GFAP (b3) and S100β (b4) were increased over time. (D) hiPSC-neurons were immunostained with anti-HuC/D (blue), anti-GFAP (magenta) and anti-S100β (green) antibodies at 14 (c1) and 63 DIV (c2). Scale bar, 100 µm. Similar results were obtained in three independent experiments.
Figure 1.
Astrocytes are differentiated in hiPSC-derived neural culture. (A) The significant decrease in the mRNA expression level of NESTIN (a1) and increase in the expression of MAP2 (a2), GFAP (a3) and S100β (a4) along with culture days were confirmed by real-time PCR. **P < 0.01, ***P < 0.0001 vs. DIV 0 group (n=3), Tukey’s test following one-way ANOVA. (B) Representative immunoblot at 14 and 63 DIV (b1). The expression level of HuC/D protein tended to decrease with culture days (b2). The protein expression levels of GFAP (b3) and S100β (b4) were increased over time. (D) hiPSC-neurons were immunostained with anti-HuC/D (blue), anti-GFAP (magenta) and anti-S100β (green) antibodies at 14 (c1) and 63 DIV (c2). Scale bar, 100 µm. Similar results were obtained in three independent experiments.
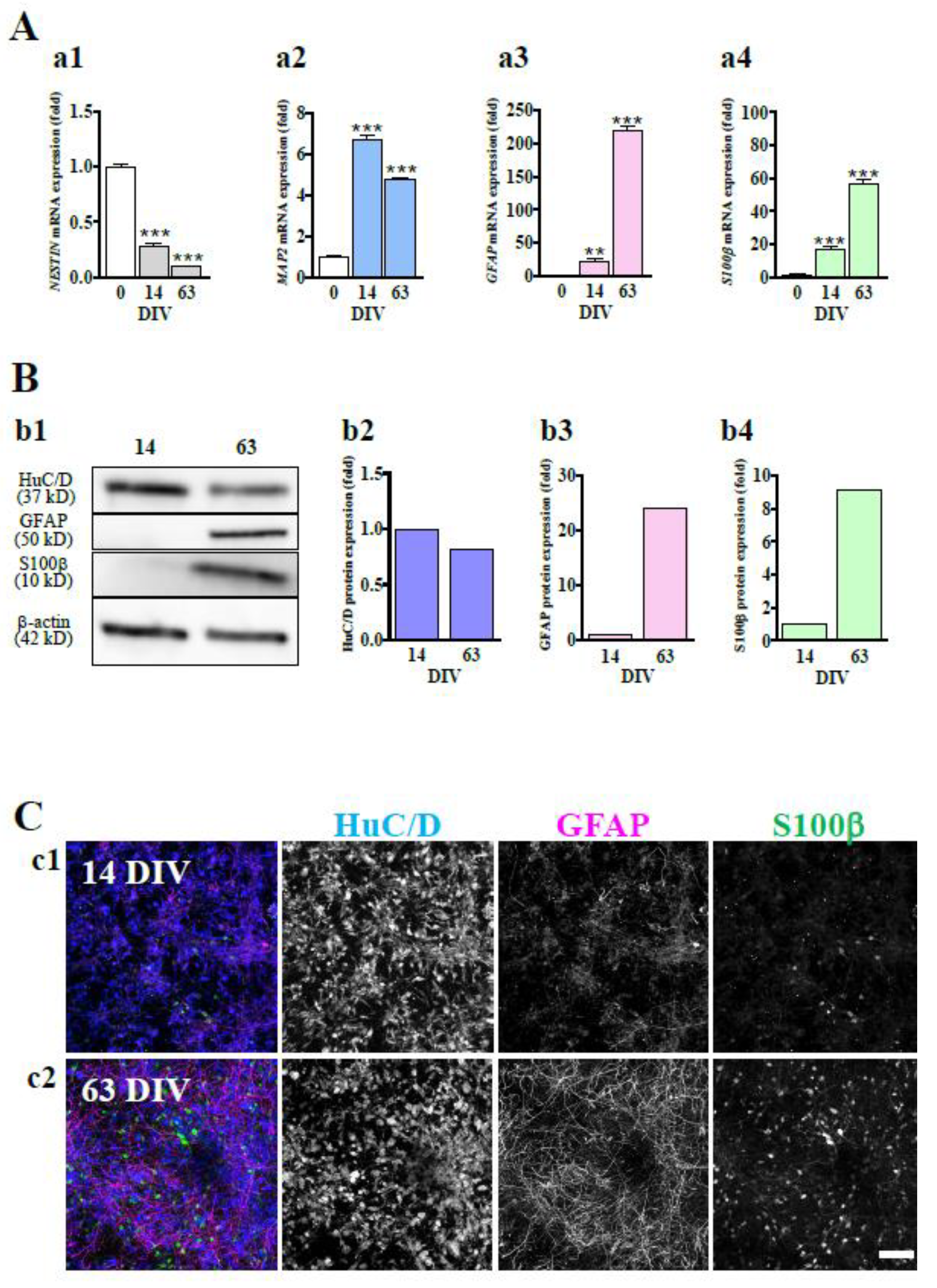
Figure 2.
Functional maturation of hiPSC-derived neural networks. (A) Spontaneous firing of hiPSC-derived neural networks and comparison of 4 parameters at 14, 21, 28, 42, and 56 DIV (n=72). The measurements were performed for 15 min. Raster plots for all 16 electrodes at 28 and 56 DIV (a1). Array wide spike detection rate (AWSDR, number of spikes/ms) at 28 and 56 DIV (a2). Network bursts (NBs) were detected after 28 DIV, and the number of wells with network activity increased with culture days (a3). Total spikes per min increased with culture days (a4). The number of network bursts (NBs) per min increased with culture days (a5). Spikes in an NB increased with culture days (a6). Data are expressed as the means + standard deviations. (B) b1, At 63 DIV, hiPSC-neurons were immunostained with anti-Vglut2 (green: vesicular glutamate transporter 2), anti-Syn1 (magenta: presynapse), and anti-MAP2 (blue: dendrite) antibodies. Yellow arrowheads indicate Vglut2-positive and Syn1-positive puncta on MAP2-positive fibers. b2, At 63 DIV, hiPSC-neurons were immunostained with anti-PSD95 (green: postsynapse), anti-Syn1 (magenta: presynapse), and anti-MAP2 (blue: dendrite) antibodies. Yellow arrowheads indicate PSD95-positive and Syn1-positive puncta on MAP2-positive fibers. Scale bar, 10 µm. Similar results were obtained in three independent experiments.
Figure 2.
Functional maturation of hiPSC-derived neural networks. (A) Spontaneous firing of hiPSC-derived neural networks and comparison of 4 parameters at 14, 21, 28, 42, and 56 DIV (n=72). The measurements were performed for 15 min. Raster plots for all 16 electrodes at 28 and 56 DIV (a1). Array wide spike detection rate (AWSDR, number of spikes/ms) at 28 and 56 DIV (a2). Network bursts (NBs) were detected after 28 DIV, and the number of wells with network activity increased with culture days (a3). Total spikes per min increased with culture days (a4). The number of network bursts (NBs) per min increased with culture days (a5). Spikes in an NB increased with culture days (a6). Data are expressed as the means + standard deviations. (B) b1, At 63 DIV, hiPSC-neurons were immunostained with anti-Vglut2 (green: vesicular glutamate transporter 2), anti-Syn1 (magenta: presynapse), and anti-MAP2 (blue: dendrite) antibodies. Yellow arrowheads indicate Vglut2-positive and Syn1-positive puncta on MAP2-positive fibers. b2, At 63 DIV, hiPSC-neurons were immunostained with anti-PSD95 (green: postsynapse), anti-Syn1 (magenta: presynapse), and anti-MAP2 (blue: dendrite) antibodies. Yellow arrowheads indicate PSD95-positive and Syn1-positive puncta on MAP2-positive fibers. Scale bar, 10 µm. Similar results were obtained in three independent experiments.
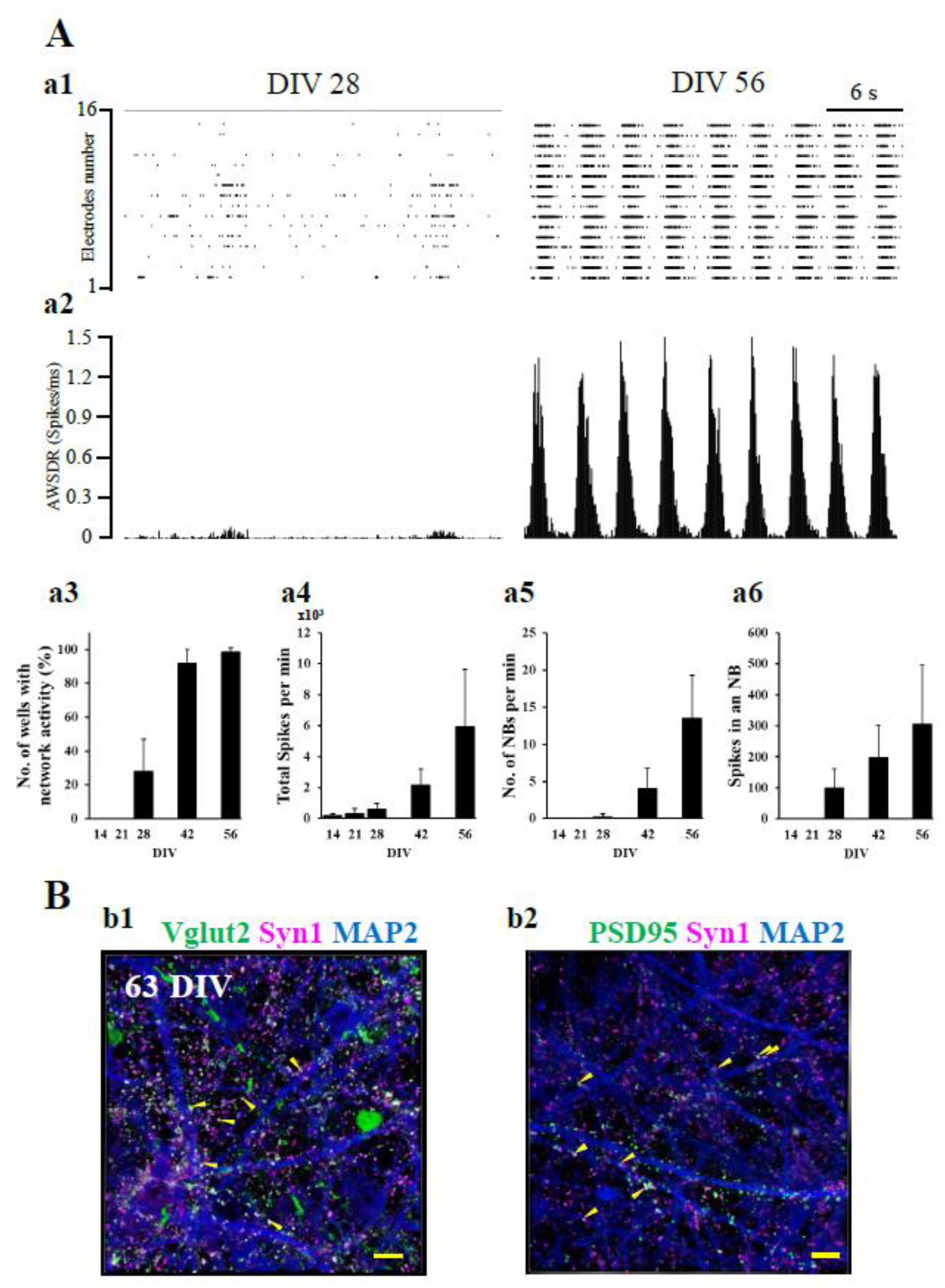
Figure 3.
Inhibition of EAATs increases the extracellular L-Glu concentration and leads to excitotoxicity at 63 DIV. (A) The effect of L-Glu alone on cell viability at 14 and 63 DIV. Cell viability was assessed using the MTT reduction assay. At 14 (a1) and 63 (a2) DIV, the application of L-Glu at 100 μM for 24 hrs did not change MTT reductions compared with the application of DMSO at 0.4% (Cont), which was used as a vehicle. Unpaired t test (n=6). Similar results were obtained in three independent experiments. (B) The effects of TFB-TBOA (TFB, nonspecific EAAT inhibitor, 30 nM) when coapplied with L-Glu on cell viability at 14 and 63 DIV. At 14 DIV, TFB caused no effect on MTT reductions (b1). On the other hand, at 63 DIV, TFB significantly decreased MTT reductions, and AP5 (NMDAR antagonist, 100 μM) blocked the decrease in MTT reductions by TFB (b2). Similar results were obtained in three independent experiments. ***P < 0.0001 vs. L-Glu(+)TFB(-)AP5(-) group, Tukey’s test following ANOVA. ###P < 0.0001 vs. L-Glu(+)TFB(+)AP5(-) group, Tukey’s test following ANOVA. n=4-6. (C) Identification of the contribution of specific EAATs to the decrease in exogenously applied L-Glu. c1. Change in the concentration of L-Glu in the medium ([L-Glu]out) after L-Glu (100 μM) was applied at 63 DIV. [L-Glu]out was nearly zero at 60 mins. n=3. Similar results were obtained in three independent experiments. c2. The effects of EAAT inhibitors on L-Glu uptake at 63 DIV. The effects of EAATs inhibitor on the decrease in [L-Glu]out were assessed at 30 mins, which was the time for the 50% decrease in [L-Glu]out from Figure 2C-c1. The percentage inhibition of EAAT inhibitors on L-Glu uptake activity was calculated as 100% of the decrease in extracellular L-Glu concentration in the absence of EAAT inhibitors. UCPH, DHK, WAY, or TFB inhibited L-Glu uptake. n=5-7. (D) Inverse correlation between the strength of L-Glu uptake inhibition and the cell viability caused by EAAT inhibitors. The Pearson correlation coefficient (PCC) = 0.7110. EAAT inhibitors are shown as follows: UCPH: blue circle; DHK: green circle, TFB: red circle and WAY: pink circle. Data are means ± SDs.
Figure 3.
Inhibition of EAATs increases the extracellular L-Glu concentration and leads to excitotoxicity at 63 DIV. (A) The effect of L-Glu alone on cell viability at 14 and 63 DIV. Cell viability was assessed using the MTT reduction assay. At 14 (a1) and 63 (a2) DIV, the application of L-Glu at 100 μM for 24 hrs did not change MTT reductions compared with the application of DMSO at 0.4% (Cont), which was used as a vehicle. Unpaired t test (n=6). Similar results were obtained in three independent experiments. (B) The effects of TFB-TBOA (TFB, nonspecific EAAT inhibitor, 30 nM) when coapplied with L-Glu on cell viability at 14 and 63 DIV. At 14 DIV, TFB caused no effect on MTT reductions (b1). On the other hand, at 63 DIV, TFB significantly decreased MTT reductions, and AP5 (NMDAR antagonist, 100 μM) blocked the decrease in MTT reductions by TFB (b2). Similar results were obtained in three independent experiments. ***P < 0.0001 vs. L-Glu(+)TFB(-)AP5(-) group, Tukey’s test following ANOVA. ###P < 0.0001 vs. L-Glu(+)TFB(+)AP5(-) group, Tukey’s test following ANOVA. n=4-6. (C) Identification of the contribution of specific EAATs to the decrease in exogenously applied L-Glu. c1. Change in the concentration of L-Glu in the medium ([L-Glu]out) after L-Glu (100 μM) was applied at 63 DIV. [L-Glu]out was nearly zero at 60 mins. n=3. Similar results were obtained in three independent experiments. c2. The effects of EAAT inhibitors on L-Glu uptake at 63 DIV. The effects of EAATs inhibitor on the decrease in [L-Glu]out were assessed at 30 mins, which was the time for the 50% decrease in [L-Glu]out from Figure 2C-c1. The percentage inhibition of EAAT inhibitors on L-Glu uptake activity was calculated as 100% of the decrease in extracellular L-Glu concentration in the absence of EAAT inhibitors. UCPH, DHK, WAY, or TFB inhibited L-Glu uptake. n=5-7. (D) Inverse correlation between the strength of L-Glu uptake inhibition and the cell viability caused by EAAT inhibitors. The Pearson correlation coefficient (PCC) = 0.7110. EAAT inhibitors are shown as follows: UCPH: blue circle; DHK: green circle, TFB: red circle and WAY: pink circle. Data are means ± SDs.
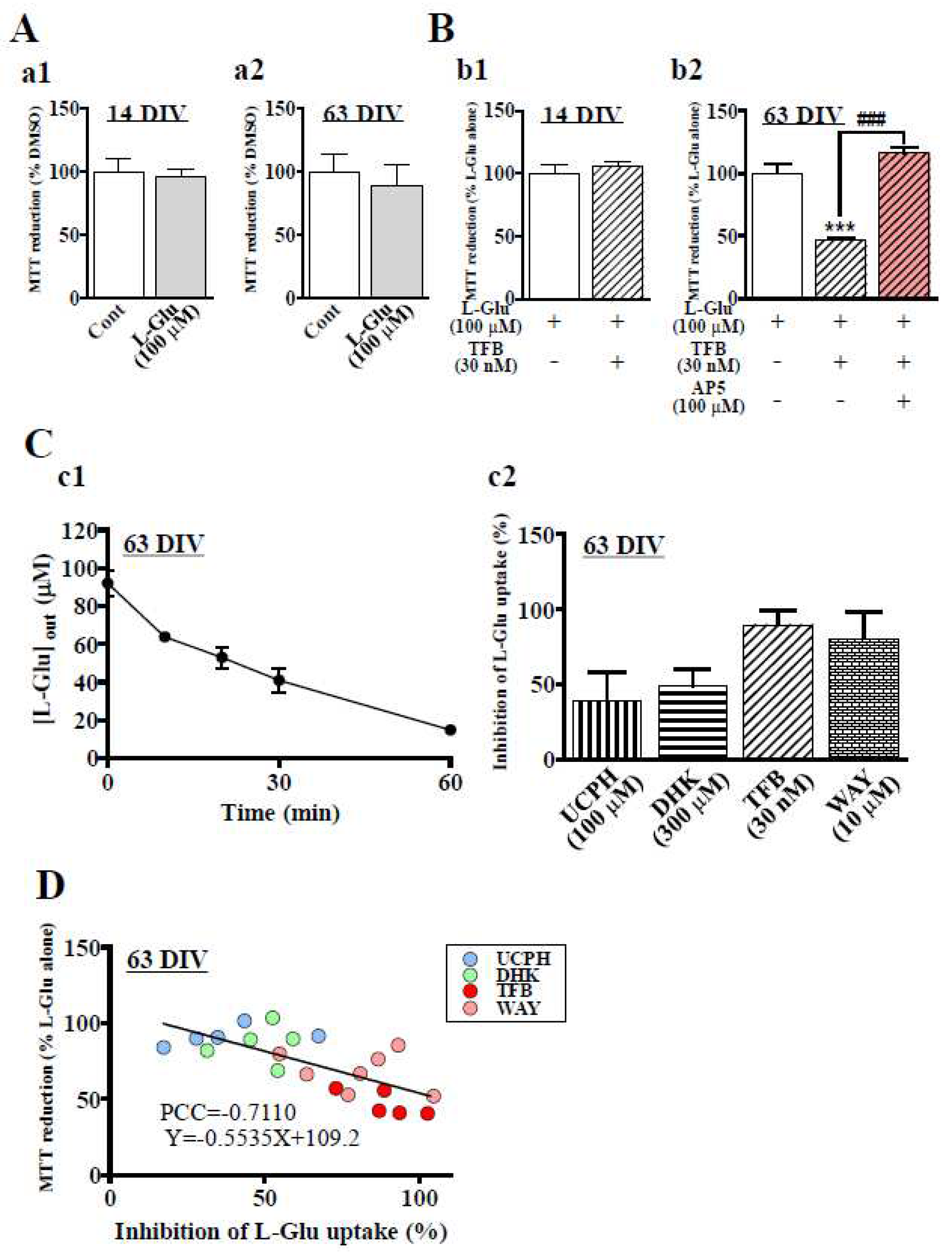
Figure 4.
Expression of EAAT1 and EAAT2 in hiPSC-derived neural cells. (A) The significant increase in the mRNA expression levels of EAAT1 (a1) and EAAT2 (a2) along with culture days was confirmed by qRT‒PCR. **P < 0.01, ***P < 0.0001 vs. DIV 0 group (n=3), Tukey’s test following ANOVA. (B) Representative immunoblot at 14 and 63 DIV (b1). The expression levels of EAAT1 (b2) and EAAT2 (b3) protein tended to increase with culture days. (C) Identification of cell types expressed EAAT1 and EAAT2 at 63 DIV. We used the following cell markers: GFAP+Nestin for radial glial cells, GFAP+S100β+ for astrocytes and HuC/D+ or MAP2+ for neurons. c1. EAAT1 was localized in radial glia (top) and astrocytes (bottom). c2. EAAT2 was localized in radial glia (top), neurons (middle) and astrocytes (bottom). Scale bar, 100 µm. Data are means ± SDs. Similar results were obtained in three independent experiments.
Figure 4.
Expression of EAAT1 and EAAT2 in hiPSC-derived neural cells. (A) The significant increase in the mRNA expression levels of EAAT1 (a1) and EAAT2 (a2) along with culture days was confirmed by qRT‒PCR. **P < 0.01, ***P < 0.0001 vs. DIV 0 group (n=3), Tukey’s test following ANOVA. (B) Representative immunoblot at 14 and 63 DIV (b1). The expression levels of EAAT1 (b2) and EAAT2 (b3) protein tended to increase with culture days. (C) Identification of cell types expressed EAAT1 and EAAT2 at 63 DIV. We used the following cell markers: GFAP+Nestin for radial glial cells, GFAP+S100β+ for astrocytes and HuC/D+ or MAP2+ for neurons. c1. EAAT1 was localized in radial glia (top) and astrocytes (bottom). c2. EAAT2 was localized in radial glia (top), neurons (middle) and astrocytes (bottom). Scale bar, 100 µm. Data are means ± SDs. Similar results were obtained in three independent experiments.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



